

Summary
The genes required for host‐specific pathogenicity in Fusarium oxysporum can be acquired through horizontal chromosome transfer (HCT). However, it is unknown if HCT commonly contributes to the diversification of pathotypes.
Using comparative genomics and pathogenicity phenotyping, we explored the role of HCT in the evolution of F. oxysporum f. sp. fragariae, the cause of Fusarium wilt of strawberry, with isolates from four continents.
We observed two distinct syndromes: one included chlorosis (‘yellows‐fragariae’) and the other did not (‘wilt‐fragariae’). All yellows‐fragariae isolates carried a predicted pathogenicity chromosome, ‘chrY‐frag’, that was horizontally transferred at least four times. chrY‐frag was associated with virulence on specific cultivars and encoded predicted effectors that were highly upregulated during infection. chrY‐frag was not present in wilt‐fragariae; isolates causing this syndrome evolved pathogenicity independently. All origins of F. oxysporum f. sp. fragariae occurred outside of the host’s native range.
Our data support the conclusion that HCT is widespread in F. oxysporum, but pathogenicity can also evolve independently. The absence of chrY‐frag in wilt‐fragariae suggests that multiple, distinct pathogenicity chromosomes can confer the same host specificity. The wild progenitors of cultivated strawberry (Fragaria × ananassa) did not co‐evolve with this pathogen, yet we discovered several sources of genetic resistance.
Keywords: convergent evolution, Fragaria × ananassa, Fusarium wilt, horizontal chromosome transfer, pathogen resistance, strawberry
Introduction
Fusarium oxysporum is a species complex containing many host‐specific pathogenic forms (called formae speciales). Pathogenic strains of F. oxysporum cause devastating crop losses worldwide, but most formae speciales have poorly characterised evolutionary origins. This includes F. oxysporum f. sp. fragariae (Fof), which causes Fusarium wilt of strawberry (strawberry = Fragaria × ananassa Duchesne ex Rozier). This pathogen was first reported in eastern Australia in 1962 and soon thereafter in Japan (1969) and South Korea (1974) (Winks & Williams, 1965; Okamoto et al., 1970; Kim et al., 1982). New reports of this disease surged globally during the early 2000s, including in major fruit production regions in California (2006) and Spain (2014) (Borrero et al., 2017; Henry et al., 2017). The evolutionary processes leading to the emergence of Fof remain uncharacterised, and it is unknown if the expanding geographic range of this pathogen resulted from dispersal or independent evolution of pathogenicity.
Although the exchange of DNA through meiotic recombination has never been documented for F. oxysporum (Taylor et al., 2015), genetic exchange can occur via horizontal chromosome transfer (HCT), a process involving the transfer of ‘accessory’ chromosomes (Ma et al., 2010). F. oxysporum chromosomes can be categorised into two types: (1) 11 conserved chromosomes that are present in all strains; and (2) a variable number of ‘accessory’, or ‘lineage‐specific’, chromosomes that can be acquired via HCT (Ma et al., 2010). For HCT to occur, two germinating spores fuse, a haploid nucleus from each of the two strains fuse, and one strain’s core chromosomes are systematically lost (Shahi et al., 2016). In plant pathogenic strains of F. oxysporum, accessory chromosomes that are acquired via HCT may have the genes required for host‐specific pathogenicity (i.e. a pathogenicity chromosome). However, while the process of HCT is understood, there are few documented examples of HCT in nature, and it is unknown how frequently HCT has contributed to the diversity of strains that cause disease on a particular host.
Many formae speciales, including Fof (Nagarajan et al., 2006; Paynter et al., 2016; Henry et al., 2017), are polyphyletic and HCT provides an attractive explanation for this diversity. Where HCT occurred, virulence would be determined by the same genes, that is those residing on a shared pathogenicity chromosome (Ma et al., 2010; van Dam et al., 2017; Li et al., 2020). However, given the vast genetic diversity observed in F. oxysporum (O’Donnell et al., 2009), it is plausible that the same host specificity could be conferred by different repertoires of genes on nonhomologous pathogenicity chromosomes. These pathogenicity determinants could have accrued through independent processes of mutation (such as transposon activity, DNA copy error, etc.) and/or the acquisition of novel alleles through HCT. If true, polyphyly in a forma specialis could result from independently evolved genetic determinants of pathogenicity, not HCT alone.
Isolates with different genetic determinants of host specificity could also cause distinct disease phenotypes. Such phenotypic distinctions have been observed, for example F. oxysporum f. sp. melonis was differentiated into isolates that cause chlorosis (‘yellows’) or that only cause wilting (‘wilts’) (Jacobson & Gordon, 1988). It is not yet known if the ‘yellows’ and ‘wilt’ phenotypes of F. oxysporum f. sp. melonis resulted from independently evolved pathogenicity chromosomes. If so, the mechanisms responsible for inducing disease may differ, and genetic resistance may not function against both mechanisms.
Deployment of genetically resistant cultivars has been the most effective strategy for management of Fusarium wilt in many crops. All characterised host resistance (R) genes for F. oxysporum wilt pathogens function by recognising pathogen ‘avirulence’ proteins and subsequently initiating defence responses (Ori et al., 1997; Joobeur et al., 2004; Diener & Ausubel, 2005; Lv et al., 2014; Catanzariti et al., 2016). Mutations in pathogen avirulence genes can render the resulting protein imperceptible by the cognate host resistance protein. When these mutations occur, the pathogen is once again able to cause disease. This ‘gene‐for‐gene’ interaction is used to categorise pathogens into different ‘races’ based on the reaction of cultivars with different susceptibility phenotypes. Of the few, well documented avirulence genes in F. oxysporum, all are carried on pathogenicity chromosomes (Ma et al., 2010; Catanzariti et al., 2016; Schmidt et al., 2016). Therefore, the efficacy of resistance genes may not extend to strains that do not share the same pathogenicity chromosome.
The objective of the present study was to characterise the evolutionary processes responsible for the emergence and diversification of the pathogen causing Fusarium wilt of strawberry. It has been unknown if the expanding geographic range of this pathogen resulted from dispersal of a clonal strain, whether horizontal transfer of a pathogenicity chromosome occurred, and/or if pathogenicity evolved independently on multiple continents. To identify the processes that resulted in the emergence of Fof we: (1) sampled isolates at a global scale, including historic isolates from the sites of first reports and more recent outbreaks; (2) investigated the evolutionary relationships between representative isolates from each country with comparative genomics/transcriptomics; and (3) assessed the co‐evolutionary framework of host and pathogen with comprehensive host resistance phenotyping. Here, we show that understanding the processes underlying pathogen diversification is important for the early identification of novel races and the development and deployment of genetic resistance to Fusarium wilt.
Materials and Methods
Isolate collection and initial characterisation
Corresponding authors from 15 publications reporting Fof in seven countries were contacted for the isolates used in their studies. Isolates were additionally obtained from the Queensland Herbarium culture collection (Australia) and the National Agricultural and Food Research Organisation Genebank (Japan). Isolate diversity was maximised by selecting representatives of the broadest variation in date of collection, region of origin, and genetically distinct groups (Supporting Information Dataset S1).
To reduce redundancy in subsequent experiments, we identified clonally related isolates by multilocus genotyping and a phenotypic test of clonality based on somatic compatibility. Isolates were determined to be compatible if complementary nitrate nonutilising mutants formed wild‐type growth when co‐cultured on a medium lacking reduced nitrogen (Correll et al., 1987). Wild‐type growth indicates that intra‐isolate hyphal fusion occurred, allowing for complementation and effective utilisation of nitrate (Correll et al., 1987). We sequenced the translation elongation factor 1‐α (EF‐1α) and intergenic spacer (IGS) loci of all isolates as previously described by Henry et al. (2017). Isolates with the same two‐locus haplotype that were somatically compatible were considered to be clonally related.
Pathogenicity assays with cultivars ‘Sweet Ann’ and ‘Ventana’
To confirm the reported characterisation as forma specialis fragariae, we tested pathogenicity on strawberry for 31 putative Fof isolates that represented the observed spatiotemporal and genotypic diversity of clonal groups from each country. We additionally tested the pathogenicity of seven putative nonpathogenic F. oxysporum isolates from strawberry fields. Pathogenicity tests were conducted with cultivars that are known to be susceptible (‘Sweet Ann’) and resistant (‘Ventana’) to Californian strains of Fof (Pincot et al., 2018). For each isolate, two fully colonised 100 cm potato dextrose agar (PDA) plates were blended with sterile, de‐ionised water and mixed into #1 Sunshine mix (Sun Gro Horticulture, MA, USA) in a 10 × 10 × 20 cm pot (L × W × H). A single plant that had broken dormancy was then planted in each pot (n = 4 per cultivar). Noninoculated PDA plates were used for a negative control. This experiment was conducted twice.
Plants were maintained in a growth chamber with 12 h : 12 h photoperiod, 28°C (high) : 20°C (low) temperature settings for 8 wk. Disease severity was assessed at 4, 5, 6, 7 and 8 wk post inoculation (wpi) using a 1–5 ordinal rating system (Fig. S1). For isolates that did not cause chlorosis, ratings were based on the following symptoms: 1, no symptoms; 2, 0–25% of leaves wilted; 3, 25–50% of leaves wilted; 4, more than 50% of leaves wilted; and 5, dead (all leaves wilted/brown). For isolates that caused chlorosis, the rating scale was: 1, no symptoms; 2, mild stunting; marginal chlorosis; 3, youngest leaves fully chlorotic; 4, outer leaves chlorotic, some wilting observed; and 5, dead (all leaves were brown). At 8 wpi, or the time of plant mortality, four 2.5‐cm sections from the base of petioles of each plant were surface sterilised with 1% sodium hypochlorite for 2 min and plated onto Komada’s medium to confirm the recovery of the pathogen (Henry et al., 2017).
Differences in symptom severity at 8 wpi on ‘Sweet Ann’ were compared between chlorosis‐causing (yellows‐fragariae), wilt‐only causing (wilt‐fragariae) isolates. The proportion of ‘Sweet Ann’ plants that were dead at 8 wpi was calculated for each isolate, with data pooled across both experiments (n = 8). Plant mortality proportions were arcsin square root transformed to achieve a normal distribution (package: stats, function: ‘qqnorm’), and Welch’s t‐test was used to test the hypothesis that yellows‐fragariae and wilt‐fragariae isolate groups differed in mean mortality proportions at 8 wpi (package: stats, function: t.test, R v.3.5.2) (R Core Team, 2013).
Whole genome sequencing and assembly
To investigate the evolutionary relationships between Fof isolates, we generated whole genome assemblies for representatives of all clonal groups of this pathogen present in each country (35 total), 12 nonpathogenic isolates from strawberry plants or field soil, and one isolate of F. oxysporum f. sp. mori (the cause of Fusarium wilt of blackberry; Dataset S1; Table S1). DNA was extracted from lyophilised mycelia or conidia using a phenol–chloroform extraction protocol (Kaur et al., 2017). Libraries were prepared and sequenced on an Illumina NextSeq or HiSeq (150 bp, paired end) at the University of California, Davis DNA Technologies Core Facility or at the Michigan State University Genomics Core Facility. SMRTBell libraries were additionally prepared for GL1080 and GL1381, size selected to fragments greater than 15 kbp, and sequenced on a Pacific Biosciences (PacBio) RSII at the DNA Technologies Core Facility (Davis, CA). The ProxiMeta high‐throughput chromatin conformation capture (Hi‐C) library preparation kit (Phase Genomics; Seattle, WA) was used to prepare libraries from GL1381 and GL1080 germlings. Hi‐C libraries were sequenced on an Illumina NovaSeq system with 150‐bp paired‐end reads.
Genomes were assembled for 48 isolates from 150‐bp paired‐end Illumina reads using de novo A5 assembler software (Tritt et al., 2012). Chromosome‐level assemblies were additionally generated for isolates GL1080 and GL1381. First, PacBio reads were assembled by the HGAP3 pipeline in Smrtanalysis (v.2.3.0) with default parameters (Chin et al., 2013) and error corrected with pilon (v.1.23) from Bowtie2‐mapped Illumina reads (v.2.3.4.1; Langmead & Salzberg, 2012; Walker et al., 2014). These assemblies were then scaffolded with Hi‐C library reads (113 and 49 million reads for GL1080 and GL1381) using the Proximo proprietary software from Phase Genomics, Inc. (Seattle, WA, USA). Scaffolded assemblies were gap‐filled by PBJelly (v.15.8.24) with proofread error‐corrected PacBio reads (v.2.14.0; English et al., 2012; Hackl et al., 2014).
Core genome phylogenetic analysis
We analysed the core genome phylogenetic relationships between the 48 new assemblies and 91 previously published F. oxysporum genome assemblies (n = 139 total). Busco (v.2.0) was run for every genome, and 2718 orthologues were identified as single copy in every genome (Simao et al., 2015). Each of the 2718 genes were aligned with Muscle (v.3.8), and the alignments were concatenated (Edgar, 2004; Simao et al., 2015). The maximum likelihood phylogeny of the concatenated alignment was determined by RAxML (v.8.2.12) using the general time reversible model with gamma correction and 1000 bootstrap replicates (Stamatakis, 2014). Phylograms were visualised by Ggtree (v.1.14.6; Yu et al., 2017).
Gene and transposon annotation
The in vitro and in planta transcriptome of F. oxysporum f. sp. lycopersici 4287 has been sequenced previously (GenBank accession: GCA_000149955.2; van Dam et al., 2016). We used Hisat2 (v.2.1.0) to align these reads to the reference genome (GenBank accession: GCA_000149955.2) with a maximum intron length of 6 kbp (Kim et al., 2019). Cufflinks (v.2.2.1) was used to assemble and merge transcripts (Trapnell et al., 2010). Assembled transcripts were used as evidence by codingquarry (v.2.0) to generate the gene models used during annotation (Testa et al., 2015).
Using these pretrained gene models, we annotated all other genomes included in this analysis with CodingQuarry (n = 139 genomes, in ‘default’ and ‘pathogen mode’). Secreted proteins were predicted by signalp4.1 (run mode = ‘best’) and effectorp v.1.0 and v.2.0 were used for effector prediction (Nielsen, 2017; Sperschneider et al., 2018). Repeatmodeler (v.1.0.11) and Repeatmasker (v.4.0.8) were used to annotate transposable elements (Tarailo‐Graovac & Chen, 2009). Miniature impala transposons were identified by TIRmite (v.1.1.3) run four separate times with TIR alignments that collectively represent the breadth of miniature impala diversity (TIRmite: https://github.com/Adamtaranto/TIRmite; ‘mimp_finder.py’ from Repertoire v.6: https://github.com/pmhenry/Repertoire_v6).
Read mapping and coverage analysis
We identified conserved sequences in Fof by mapping reads to the GL1080 and GL1381 chromosome‐level genomes. Raw reads were filtered with htstream to remove predicted PCR duplicates (hts_superdeduper), remove Phi‐X sequences (hts_Seqscreener), trim adapters (hts_Adaptertrimmer), trim ends to an average quality value of ‘20’ (hts_qwindowtrim), and remove Ns from the ends of reads (hts_NTrimmer). To ensure consistent read depth when mapping, 5.5 Gbp of quality‐filtered reads were extracted for each isolate (c. 100× coverage; reformat.sh from BBTools). These reads were aligned with Bwa‐mem (v.0.7.17‐r1188; Li & Durbin, 2009). Bedtools (v.2.29.0) function ‘genomecov’ calculated the coverage of reads at each position in the genome, and regions with coverage greater than 9 were considered ‘present’ (Quinlan & Hall, 2010). To identify conserved sequences, coverage files were compared by Bedtools ‘intersect ‐v’. To identify unique sequences, we used Bedtools ‘subtract’.
Variant calling and SNP analysis
Because BUSCO genes were not evenly distributed among the chromosomes, we used variant calling to investigate the phylogeny of specific chromosomes and test for linkage disequilibrium. We called variants with Freebayes (v.1.3.1) using the following parameters: ‘‐‐ploidy 1 ‐‐min‐base‐quality 20 ‐‐min‐coverage 10 ‐‐min‐alternate‐fraction 0.4 ‐‐haplotype‐length 0’ (Garrison & Marth, 2012). The output was filtered by Vcftools to remove indels, require at least 70% of samples to have a genotype call, and exclude sites with minor allele count less than 2. The resulting SNPs were filtered by Bedtools ‘intersect’ to remove SNPs that were not in regions with read coverage by all yellows‐fragariae isolates. We used the vcflib function ‘vcfrandomsample’ to randomly extract SNPs from core chromosomes. Genotype calls were extracted into fasta formatted files by the ‘allele‐seq’ function of vcfriend (https://github.com/pmhenry/VCFriend). Maximum likelihood phylogenetic analysis was conducted with raxml (v.8.2.12) correcting for ascertainment bias with the following parameters: ‘‐m ASC_GTRCAT ‐V ‐‐asc‐corr = lewis’ (Stamatakis, 2014). Statistical support for each phylogeny was tested with 1000 bootstrap replicates.
For the index of association test of linkage disequilibrium, variants were called by Freebayes with a minimum minor allele frequency of 3 and no ‘‐‐haplotype‐length’ limit. For this analysis, we used a clone‐corrected dataset that included 11 distinct genotypes of yellows‐fragariae. Variants were randomly subsampled with the Vcflib ‘vcfrandomsample’ to a rate of 0.05. The GL1080 chromosome‐level assembly was used as the reference. Linkage disequilibrium was assessed by the R package poppr function ‘ia()’ with 999 permutations (Kamvar et al., 2015).
Transcriptomic experiments
To determine if genes on predicted pathogenicity chromosomes are differentially expressed during plant infection, we sequenced the in vitro and in planta transcriptome of a yellows‐fragariae isolate. Tissue‐cultured strawberry plants (cultivar ‘Camarosa’) were inoculated with a spore suspension of isolate GL1381 (n = 10 plants) as previously described, planted into twice‐autoclaved sand and maintained in a growth chamber as previously described. Roots from five plants at each timepoint (6 d and 13 d post inoculation) were gently removed from sand, washed in sterile, de‐ionised water and flash frozen in liquid nitrogen. For an in vitro control, GL1381 mycelia were scraped from PDA at 72 h post inoculation and flash frozen in liquid nitrogen. RNA was extracted from these tissues with the 3% CTAB #1 protocol described in Yu et al. (2012). RNA was further purified by Ambion Turbo DNase treatment and the Zymo Research RNA Clean and Concentrate kit. RNA integrity was assessed with an Agilent Bioanalyser before 3′ QuantSeq library preparation (Lexogen, Inc.) with unique molecular indices. Libraries were sequenced on an Illumina NovaSeq (150 bp paired‐end) to a minimum depth of 5 million reads per library.
Transcriptome quantification and analysis
Each read’s unique molecular index (UMI) was extracted with the ‘extract’ function from UMI tools (v.1.0.0; Smith et al., 2017). Reads were filtered with HTStream and aligned with Star (v.2.6.1a) to the GL1381 chromosome‐level assembly (Dobin et al., 2013). The aligned reads were deduplicated with UMI tools (Li et al., 2009). Because the library preparation method selected primarily for sequences in 3′ un‐translated regions (UTRs), we modified the count function in htseq (v.0.6.1p1; Anders et al., 2015) so that if a read aligned to overlapping UTR and CDS annotations from different genes, the transcript would be counted for the UTR‐aligned gene. This program is available from: https://github.com/pmhenry/Publications/tree/master/Frag_Fof_TAGseq_Jenner_2020/HTSeq‐TAG‐Counts. Our modified ‘count’ function was used to generate read counts per gene from an annotation file where a 1000‐bp UTR was added at the 3′ end of each gene. The program to add UTR annotations is available from: https://github.com/pmhenry/Publications/blob/master/Global_Fof_Henry_2020/TAGseq_gtf_annotation/tag_annotation.py.
Differential expression was evaluated in R using the edger package (Robinson et al., 2010). Genes were filtered by filterbyexpr, counts were transformed by Voom, and linear models fit with Lmfit. In planta samples from 6 d and 13 d post inoculation were contrasted against in vitro samples. Genes with an adjusted P‐value less than or equal to 0.05 and a log‐fold change greater than 1.5 or less than −1.5 were considered to be differentially expressed.
Principal component analysis
In order to select diverse cultivars for resistance phenotyping, we analysed the genotypic diversity of available strawberry germplasm. These strawberry accessions had previously been genotyped on the Affymetrix® FanaSNP 50K Array (Hardigan et al., 2020). Genotypes were called using the Affymetrix® Axiom Analysis Suite (v.4.0.1), and samples with a call‐rate greater than 89% were retained for further analysis (Pincot et al., 2018). The ‘A.mat’ function from the R package rrblup was used to construct a realised additive genetic relationship (A) matrix, filtering SNPs for a minor allele frequency of 0.05 and a maximum missing rate of 0.80 in the process (Endelman, 2011). Principal component analysis was run on the A matrix using the ‘prcomp’ command found in the base stats package (R v.3.5.2; R Core Team, 2013).
Pathogenicity assays for cultivar differentials
We tested a panel of 25 diverse strawberry accessions against six strains of Fof that differed in their evolutionary origin or pathogenicity chromosome type. Conidia were harvested from isolates grown on PDA or Kerr’s broth (Kerr, 1962). Bare‐root transplants were submerged in 5 × 106 spores ml−1 of 0.1% water agar for 7 min before planting (Henry et al., 2017). Pathogenicity assays with isolates MAFF727510, F79, BRIP62122 and AMP132 were conducted in a growth chamber with the settings indicated above. Strawberry accessions were also tested for resistance to isolate AMP132 in a field at the University of California, Davis Plant Pathology Farm (Methods S1). Isolates GL1315 and GL1381 were tested in a glasshouse in Salinas, California with 30°C (high) : 20°C (low) temperature settings. Each experiment was conducted at least twice, contained noninoculated controls, and two inoculated plants per accession × isolate combination. A small number of accession × isolate combinations had only one replicate in some experiments due to plant availability (Dataset S1). Least‐squares means were generated using the lme4 (v.1.1‐18‐1) and emmeans packages (v.1.3.0; Bates et al., 2015; Lenth, 2016; Lenth, 2018). Models were fit for each isolate with ‘accession’ as a fixed effect and ‘experiment’ and ‘replication’ as random effects.
Results
Isolate collection and characterisation
We obtained DNA or cultures of 85 F. oxysporum isolates from Australia, Japan, South Korea, Spain, and California (Dataset S1). Twelve isolates did not cause symptoms on either cultivar. Of these, F74 and F79 were confirmed to be pathogenic on the cultivar Splendor, as previously reported (Borrero et al., 2017). Isolate NRF0806 was still considered Fof based on reported pathogenicity to other cultivars (Suga et al., 2013) and somatic compatibility with pathogenic isolates. Nine isolates did not meet these criteria and were therefore not considered to be forma specialis fragariae.
We considered any isolate that caused symptoms in both experiments to be pathogenic, and assumed (based on past research) that clones of pathogenic isolates were also pathogenic (Henry et al., 2017). Given these criteria, we confirmed that 69 isolates were Fof. These isolates comprised 8 SCGs and 10 EF‐1α/IGS haplotypes (Dataset S1; Fig. S2; Table S1). One EF‐1α/IGS haplotype contained two SCGs (Fig. S2); 11 genotypes of Fof were therefore distinguished based on combined SCG and two‐locus haplotypes.
Fof isolates cause two symptom phenotypes and can overcome FW1‐mediated resistance
We observed two, distinct symptom phenotypes caused by Fof. Both phenotypic groups caused wilting and death. However, all isolates in two SCGs from Queensland in eastern Australia and an SCG from Spain caused little or no chlorosis (Fig. 1); we named these isolates ‘wilt‐fragariae’ (Fig. 1a; Notes S1). All other pathogenic isolates, originating in Japan, South Korea, California and Western Australia caused severe chlorosis; we named these isolates ‘yellows‐fragariae’ (Fig. 1a; Notes S1). Spanish wilt‐fragariae isolates were pathogenic to ‘Splendor’, but not to ‘Sweet Ann’ or ‘Ventana’. Results did not differ significantly between experiments, so data were pooled in subsequent analyses (P = 0.1, df = 1 using Kruskal–Wallis rank sum test). Yellows‐fragariae isolates caused greater disease severity than Australian wilt‐fragariae on the susceptible cultivar Sweet Ann; the mortality of ‘Sweet Ann’ was higher at 8 wpi among plants inoculated with yellows‐fragariae than with Australian wilt‐fragariae (Welch’s t‐test; P < 0.001, df = 10; Fig. 1b,c; R Core Team, 2013).
Fig. 1.
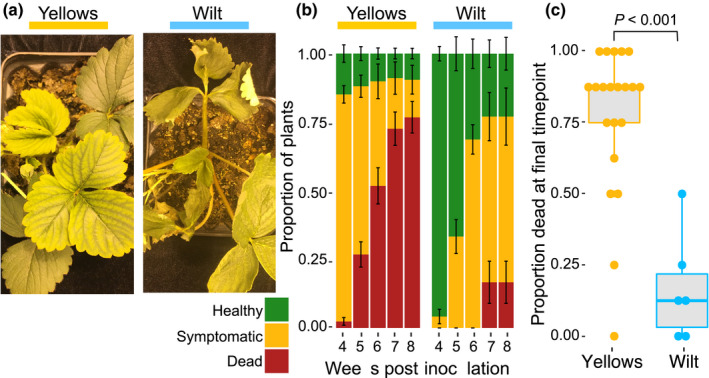
Symptoms and disease severity induced by Fusarium oxysporum f. sp. fragariae. (a) The photograph on the left shows typical chlorosis symptoms caused by a yellows‐fragariae isolate (N‐18462) on new (inner) leaves observed 3 wk post inoculation. Symptoms caused by a wilt‐fragariae isolate (BRIP62122) are shown on the right. (b) Disease severity at 4–8 wk post inoculation. The mean proportion of plants (cultivar ‘Sweet Ann’) that are healthy (green), symptomatic (yellow), or dead (red) for yellows‐ (n = 21) and wilt‐fragariae (n = 6) isolates are reported. Proportions were calculated from the pooled results of two experiments in which four plants were inoculated per isolate (n = 8 plants total). The two experiments did not differ significantly in disease severity by a Kruskal–Wallis rank sum test (df = 1, P = 0.1). Error bars depict one standard error. (c) Boxplot depicting the proportion of ‘Sweet Ann’ plants that were dead by 8 wk post inoculation for each yellows‐ and wilt‐fragariae isolate. Individual points correspond to each isolate in the yellows‐ and wilt‐fragariae groups, for which there are 21 and 6, respectively. In each boxplot, the middle line corresponds to the median value, upper and lower bars correspond to first and third quartiles, respectively, and whiskers correspond to 1.5 times the interquartile range. The two symptom groups had significantly different proportions of dead plants per isolate at 8 wk post inoculation in a Kruskal–Wallis rank sum test (df = 2; P < 0.001).
Five isolates consistently caused disease on the cultivar Ventana, which is heterozygous for the dominant Fusarium wilt resistance gene, FW1 (Pincot et al., 2018). FW1‐resistance breaking isolates included four yellows‐fragariae (MAFF727510, Fo160618, Fo160403 and SK1) and one wilt‐fragariae isolate (BRIP62122). For each isolate, Wilcoxon rank sum tests showed no difference in virulence on the cultivars Ventana and Sweet Ann (P ≥ 0.24). All isolates that caused disease on FW1‐resistant cultivars were classified as ‘race 2’, whereas isolates that caused disease on ‘Sweet Ann’ but not ‘Ventana’ were classified as ‘race 1’. Spanish wilt‐fragariae isolates did not receive a ‘race’ classification because they caused disease on ‘Splendor’ but not the other two cultivars, and the genetics of resistance to these isolates in ‘Sweet Ann’ is unknown.
Whole genome sequencing and assembly
Assemblies for all isolates generated from 150‐bp paired‐end reads had a high level of gene completeness; between 97.6% and 98.2% of Busco genes were present and single copy (n = 3725 total searched; Dataset S1). For both of the GL1080 and GL1381 reference assemblies, >99% of the total genome length was assembled into 14 chromosome‐sized scaffolds (Tables S2–S4; Fig. S3). In each assembly, 11 scaffolds corresponded to conserved chromosomes identified in the reference strain, F. oxysporum f. sp. lycopersici 4287 (Tables S2–S4; Fig. S3; Ma et al., 2010). Each assembly additionally contained three accessory chromosomes that were not present in F. oxysporum f. sp. lycopersici 4287.
Fof is polyphyletic within F. oxysporum clade 2
To determine the core genome phylogeny of Fof isolates, we conducted maximum likelihood phylogenetic analysis on a 4.97 Mbp concatenated alignment of 2718 conserved, single copy orthologues (Stamatakis, 2014). Strong bootstrap support was observed for the three previously reported F. oxysporum clades (Figs 2, S4; O’Donnell et al., 1998). We further resolved clade 2 into two subgroups (2A and 2B) with high bootstrap support (Figs 2, S4). Only Spanish wilt‐fragariae isolates (called ‘W3’) were associated with clade 2A; all other wilt‐ and yellows‐fragariae were in clade 2B (Figs 2, S4). Within clade 2B, yellows‐fragariae isolates were in eight monophyletic clades (Y1–Y8) and Australian wilt‐fragariae isolates were in two monophyletic clades (W1, W2; Figs 2, S4).
Fig. 2.
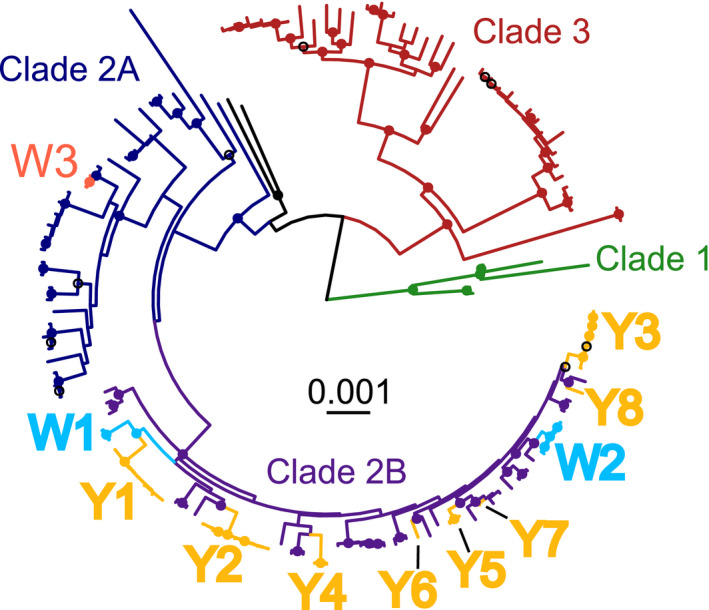
Core genome phylogeny of the Fusarium oxysporum species complex with labels for monophyletic clades of Fusarium oxysporum f. sp. fragariae. Single copy orthologous genes (n = 2718) from 139 F. oxysporum genomes were aligned by Muscle and concatenated into a single, c. 5.5 Mbp alignment. Maximum likelihood phylogenetic comparisons were conducted by RAxML using the general time reversible model with gamma correction and 1000 bootstrap replicates. Branches are colour‐coded by F. oxysporum f. sp. fragariae symptom group (Spanish wilt‐fragariae = W3, Australian wilt‐fragariae = W1‐W2, and yellows‐fragariae = Y1‐Y8) and by core genome phylogenetic clades. Nodes with a filled circle have bootstrap support > 90%, a black open circle indicates bootstrap support > 70%. Monophyletic groups of yellows‐ and wilt‐fragariae are annotated as Y1–Y8 and W1–W3, respectively.
A putative mobile pathogenicity chromosome was identified in all yellows‐fragariae isolates
We hypothesised that HCT occurred between yellows‐fragariae strains because: (1) all isolates in this group caused the distinct chlorosis phenotype; and (2) at least one isolate from each monophyletic group (Y1–Y8) possessed forma specialis‐specific PCR‐detection loci that were absent in wilt‐fragariae isolates (Dataset S1; Suga et al., 2013; Burkhardt et al., 2019). To identify chromosomes that were putatively horizontally transferred, we mapped quality‐filtered, 150‐bp paired‐end Illumina reads to the two reference genomes and identified one accessory contig in each genome that had large regions that were conserved among all yellows‐fragariae isolates. These contigs also contained the yellows‐fragariae PCR‐detection loci and had the characteristics of previously discovered F. oxysporum mobile pathogenicity chromosomes: they were enriched with DNA transposons, carried most (58–65%) of the genome’s total miniature impala (mimp) transposons, and contained the only secreted‐in‐xylem (SIX) effector gene homologue (SIX6) (Figs 3, S3; Ma et al., 2010; Schmidt et al., 2013; van Dam et al., 2017; Li et al., 2020). Based on these results, they were hypothesised to be pathogenicity chromosomes: chrY‐frag.T1381 and chrY‐frag.T1080.
Fig. 3.
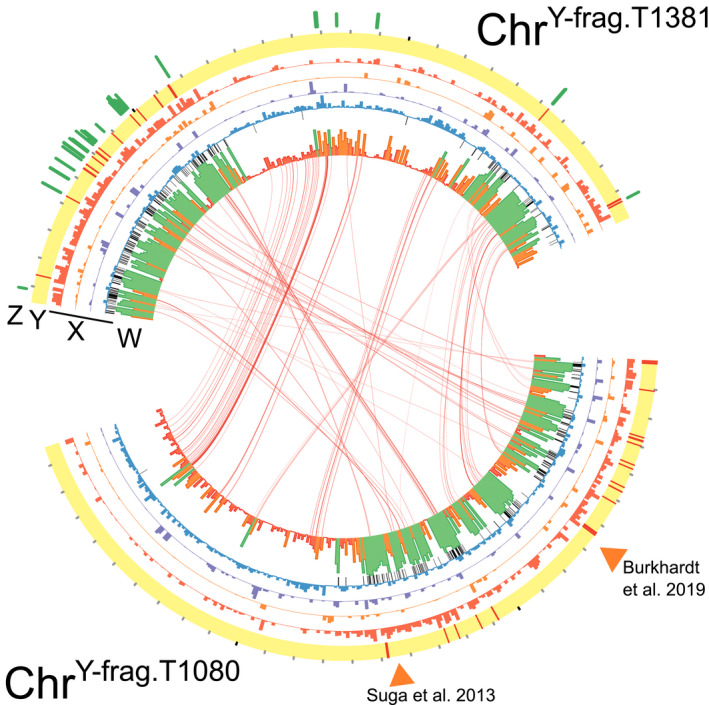
Synteny, sequence conservation, and differential expression of genes on chrY‐frag.T1381 and chrY‐frag.T1080. (z) For GL1381, the log‐fold change of in planta upregulated genes is shown by green bars on a scale from 0 to 15. (y) Red lines indicate the position of miniature impala transposable elements. Black tic marks occur every 100 kbp on the outside of the ideogram. (x) The proportion of each 10 kbp window covered by DNA transposons (red), LTR transposons (orange), LINEs (purple), or predicted genes (blue). Black lines indicate the position of sequences that are conserved in yellows‐fragariae and are in no other F. oxysporum clade 2B isolate. (w) The proportion of each 10 kbp window with coverage in all yellows‐fragariae isolates; green ≥ 0.66, 0.6 > orange ≥ 0.34, red < 0.33. Links. Reciprocal best Blast hits between predicted genes in the yellows‐fragariae conserved regions of the two chromosomes are connected by red bands. Orange triangles indicate the positions of yellows‐fragariae‐specific detection loci identified by Suga et al. (2013) and Burkhardt et al. (2019).
chrY‐frag.T1080 and chrY‐frag.T1381 carried homologous sequences that were highly conserved among yellows‐fragariae isolates. We identified 1.36 Mbp of chrY‐frag.T1381 and 1.29 Mbp of chrY‐frag.T1080 that were conserved in all yellows‐fragariae isolates (Fig. 3; Datasets S2, S3). In these regions, there were 140 and 143 predicted genes in chrY‐frag.T1381 and chrY‐frag.T1080, respectively. Based on reciprocal best Blast hit matching, 82 of these genes were homologous (with sequence identity and coverage thresholds of 0.8; Fig. 3). chrY‐frag.T1381 and chrY‐frag.T1080 contained 98.9% and 92.5% (respectively) of sequences that were conserved in yellows‐fragariae and absent in all other clade 2B isolates (Fig. 3; Table S5). No other clade 2B isolate had complete coverage of the 1.36 Mbp of chrY‐frag.T1381 and 1.29 Mbp of chrY‐frag.T1080 that were conserved among yellows‐fragariae isolates (Table S5). Wilt‐fragariae isolates from eastern Australia shared at most 27% of conserved chrY‐frag sequences, and this supports the hypothesis that wilt‐ and yellows‐fragariae have different genetic determinants of pathogenicity (Table S5). These data indicated that, while not identical, chrY‐frag.T1080 and chrY‐frag.T1381 contained homologous sequences that are highly conserved within yellows‐fragariae.
chrY‐frag was horizontally transferred at least four times
The phylogeny of chrY‐frag.T1381 and chrY‐frag.T1080 provided compelling evidence for at least four HCT events. The phylogenies of chrY‐frag.T1381 and chrY‐frag.T1080 were nearly identical and distinct from the core chromosome phylogenies (Figs 4, S5). Branch lengths in the phylogenies of chrY‐frag.T1381 and chrY‐frag.T1080 indicated that at least six variants of this chromosome exist among yellows‐fragariae, called: T1, T2, T3, T1381, T1080 and T1080B (Fig. 4). Phylogenetic incongruence provided evidence for a minimum of four HCT events: chrY‐frag.T1080 transferred between Y1 and Y6; chrY‐frag.T1381 transferred twice between Y2, Y3 and Y4; and chrY‐frag.T1 transferred between Y2 and Y7 (Figs 4, S6).
Fig. 4.
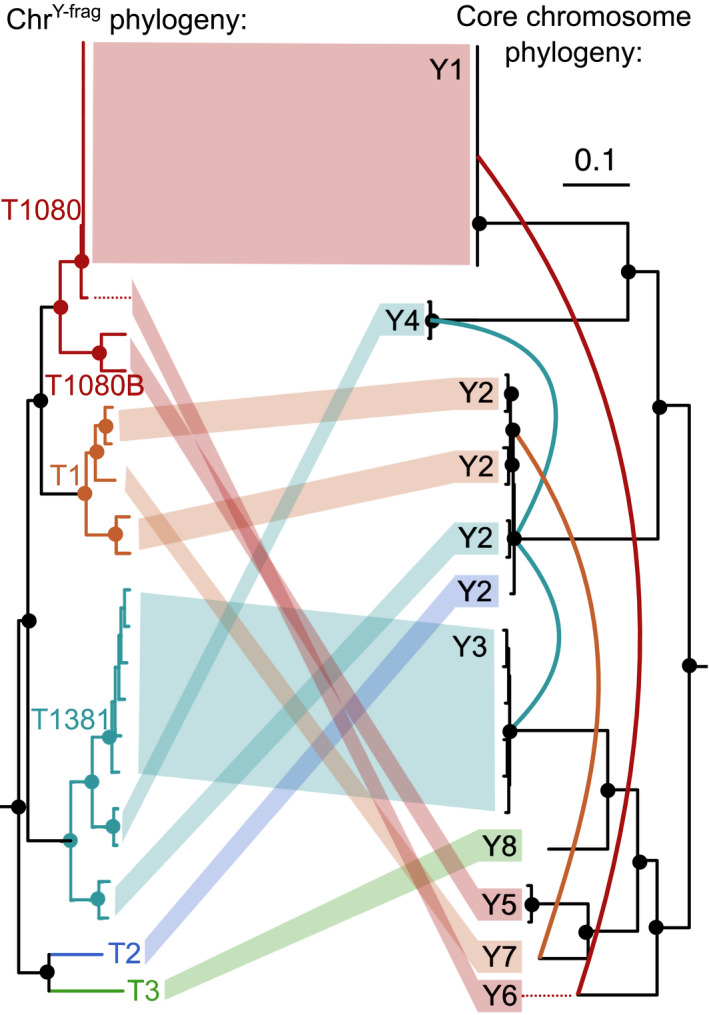
Incongruence between the phylogeny of chrY‐frag.T1080 and core chromosomes. Single nucleotide polymorphisms (SNPs) were called by Freebayes (v.1.3.1) on chrY‐frag.T1080 and 11 core chromosomes from all 27 yellows‐fragariae isolates. Genotype calls were concatenated for both datasets (chrY‐frag and combined core chromosomes), and the maximum likelihood phylogeny was inferred by RAxML with correction for ascertainment bias. Nodes with a filled circle have 100% bootstrap support. The chrY‐frag type (T1080, T1080B, T1381, T1, T2 and T3) is colour‐coded and indicated adjacent to its corresponding clade. The monophyletic yellows‐fragariae groups (Y1‐Y8) are indicated next to their corresponding branches on the core genome phylogeny. Four predicted HCT events are indicated with solid, coloured lines between branches of the core genome phylogenetic tree.
These predicted HCT events were further supported by differences in read coverage of chrY‐frag.T1381 and chrY‐frag.T1080 by putative recipients. For example, GL1080 is in the Y1 group, but isolate Fo160609 (Y6) had 99% coverage of the 2.96 Mbp chrY‐frag.T1080 and 98.1% identical alleles at SNP sites (Figs S7, S8). Similarly, GL1381 is in the Y3 group, but >97% coverage and high sequence similarity of chrY‐frag.T1381 were observed for both Y4 isolates (NRF0833 and NRF0995; >98% identical) and two Y2 isolates (MAFF305557 and MAFF305558; >87.7% identical; Figs S7, S9). Two other Y2 isolates (GL1315 and GL1268) had >90% identical SNPs with a Y7 isolate (MAFF744009) and a nearly identical coverage pattern for chrY‐frag.T1381 and chrY‐frag.T1080, suggesting that chrY‐frag.T1 was shared between these strains (Figs S7–S9). By contrast, other yellows‐fragariae isolates had coverage for 70–86% of different chrY‐frag types, and other clade 2B isolates had less than 75% coverage.
Although F. oxysporum is considered to be asexual, we looked for evidence of meiosis with an index of association test of linkage equilibrium. We conducted the index of association test on a clone‐corrected dataset of yellows‐fragariae isolates and discovered strong signatures of linkage disequilibrium on all chromosomes, including chrY‐frag (P = 0.001 for all chromosomes; Fig. S10). This indicates that meiosis is unlikely to explain the presence of chrY‐frag in diverse F. oxysporum genotypes.
chrY‐frag was probably polyphyletic before the first report of Fusarium wilt of strawberry
Phylogeny and sequence similarity suggested that chrY‐frag was present in multiple F. oxysporum genotypes before Fusarium wilt of strawberry was observed in Japan circa 1969 (Okamoto et al., 1970). Within 20 yr of the first report, the isolates MAFF305557 (Y2), MAFF744009 (Y7) and MAFF727510 (Y8) were each recovered with distinct chrY‐frag types: T1381, T1 and T3, respectively (Fig. 4). If the level of divergence between chrY‐frag.T1381 in Y3 and Y2 genotypes is typical for genetic drift over c. 20 yr (Figs 4, S7, S9), then all types of chrY‐frag could not have descended from a common ancestor after the observation of this pathogen in 1969.
chrY‐frag carries multiple effectors that are highly expressed during infection
GL1381’s in planta transcriptome suggests that the expression of genes on chrY‐frag is associated with strawberry root infection. In total, 31 genes on chrY‐frag.T1381 were differentially expressed in planta at 6 d or 13 d post inoculation compared with in vitro (adjusted P‐value < 0.05, log‐fold change > 1.5 or < −1.5) (Fig. 3; Dataset S1; Table S6). These genes were located in conserved regions among yellows‐fragariae and were highly upregulated, with a 4.3–14.8 log‐fold change in expression at 13 d post inoculation (Fig. 3; Dataset S1). Using effectorp (v.1 and/or v.2), 12 of the 31 upregulated genes on chrY‐frag.T1381 were predicted to be effectors, including a homologue of SIX6 (Dataset S1; Sperschneider et al., 2018). Eight of these genes were present in all yellows‐fragariae isolates. By contrast, only eight genes on lineage‐specific chromosomes LS2 (2.70 Mbp) or LS3 (2.21 Mbp) were differentially expressed in planta, despite these chromosomes having a combined length (4.91 Mbp) that is almost double that of chrY‐frag.T1381 (2.75 Mbp) (Dataset S1; Fig. S3). No differentially expressed genes on LS2 or LS3 were conserved among all yellows‐fragariae isolates (Dataset S1). The observed expression pattern supports the hypothesis that chrY‐frag.T1381 carries the genes necessary for host‐specific pathogenicity.
Differential host responses are associated with the presence of chrY‐frag
Comparative genomics revealed that chrY‐frag was only present in yellows‐fragariae isolates; this chromosome was largely missing in strains of wilt‐fragariae (Table S5). Given the transcriptomic results, we postulated that chrY‐frag conferred common virulence/avirulence factors among the yellows‐fragariae isolates. If true, host susceptibility would be more similar for yellows‐fragariae isolates than between isolates of yellows‐ and wilt‐fragariae.
Therefore, we hypothesised that isolates of yellows‐fragariae from distinct monophyletic groups would have a similar cultivar host range, despite differences in core genome phylogeny. To test our hypothesis, we determined the phenotypic response of 25, diverse strawberry genotypes to four yellows‐fragariae isolates (representing Y1/T1080, Y2/T1, Y3/T1381 and Y8/T3) and two wilt‐fragariae isolates (representing W2 and W3). We used geographic, historic, and genotypic data (inferred from the octoploid Affymetrix® FanaSNP 50k array) to maximise diversity in the strawberry accessions tested (Hardigan et al., 2020). Ultimately, we selected 23 F. × ananassa hybrids and two F. virginiana ecotypes (Fig. 5a; Datasets S1, S4).
Fig. 5.
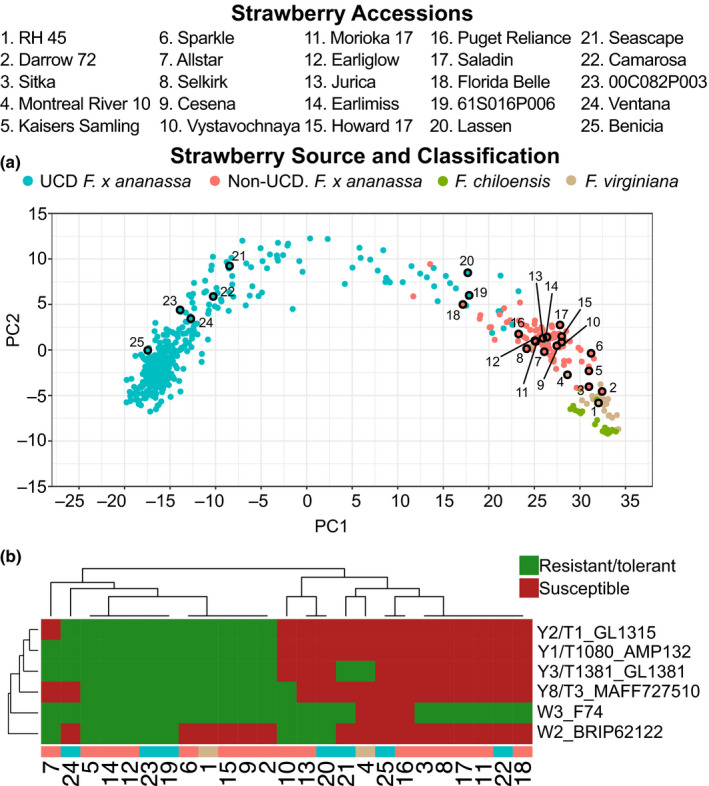
Genetic relatedness and resistance/susceptibility phenotypes of diverse strawberry accessions inoculated with Fusarium oxysporum f. sp. fragariae. (a) Principal component analysis (PCA) of 501 strawberry accessions estimated from the genotypes of 31 212 SNP markers assayed with the Affymetrix® FanaSNP 50K Array. The source and taxonomic classification of each accession is colour‐coded; ‘UCD’ indicates cultivars developed at the University of California, Davis, and ‘Non‐UCD’ indicates cultivars that were developed at other breeding programmes. Numbers correspond to the 25 cultivars phenotyped for resistance to six isolates of F. oxysporum f. sp. fragariae; their aliases are provided above the figure. (b) Disease phenotype of 25 strawberry accessions (columns) tested for resistance to six isolates of F. oxysporum f. sp. fragariae (rows). Disease severity was rated on a 1 to 5 ordinal scale, where 1 = symptomless and 5 = dead. Isolate cultivar combinations that had a least‐squares mean disease severity score greater than or equal to 2.5 were considered ‘susceptible’ (red), and below 2.5 ‘resistant/tolerant’ (green).
Consistent with our hypothesis, we observed that the presence of chrY‐frag is more predictive of cultivar susceptibility than core genome phylogeny. For example, the Y3 and Y8 core genomes are more closely related to W2 than Y1 and Y2 (Fig. 2) but have greater similarity in cultivar host range with the yellows‐fragariae isolates (Figs 5b, S11). Between 80–96% of the strawberry accessions were consistently resistant or susceptible between yellows‐fragariae isolates (Fig. S11). By contrast, only 52–68% of disease phenotypes were consistent between any yellows‐ and wilt‐fragariae isolate (Figs 5b, S11). Therefore, cultivar host range is largely associated with the presence or absence of chrY‐frag.
Discussion
Using isolates from multiple continents that were collected over time, including at the first reports of Fusarium wilt until present, we reconstruct the evolutionary and dispersal history of Fof. Of the two distinct syndromes we observed, yellows‐fragariae isolates caused greater disease severity and were more widespread than wilt‐fragariae isolates. Our data suggested that genes on chrY‐frag conferred the pathogenicity phenotype to yellows‐fragariae isolates, and horizontal transfer of chrY‐frag has led to a polyphyletic distribution of this chromosome. The characterisation of chrY‐frag as a mobile pathogenicity chromosome is supported by the conservation of chrY‐frag among yellows‐fragariae, in planta expression patterns, the association between its presence and host resistance/susceptibility phenotypes, and characteristic features that it shares with previously identified mobile pathogenicity chromosomes (Ma et al., 2010; Schmidt et al., 2013; van Dam et al., 2017; Li et al., 2020). We harnessed natural variation in the pathogen population to discover this chromosome; it was the only lineage‐specific chromosome present in eight core genome phylogenetic groups of yellows‐fragariae. chrY‐frag was horizontally transferred at least four times, resulting in the diversification of this pathogen group. Experimental transfer of this chromosome in vitro and characterisation of recipients’ pathogenicity phenotypes would provide an additional test of our hypothesis that HCT occurred among yellows‐fragariae strains.
Pathogenicity in Australian and Spanish wilt‐fragariae isolates appears to have evolved independently from yellows‐fragariae, without chrY‐frag (Notes S1, S2). This inference is primarily supported by: (1) sequences on chrY‐frag that were conserved among yellows‐fragariae were mostly absent in wilt‐fragariae (Table S5); and (2) we discovered no evidence of sympatry between wilt‐ and yellows‐fragariae, which would have been necessary for HCT to occur (Figs S11, S12). Furthermore, among isolates classified as wilt‐fragariae, pathogenicity on strawberry appears to have evolved independently in both Spain and Australia. We observed no overlap in genotypic groups between these countries that could have provided evidence for dispersal. Both Spanish and Australian wilt‐fragariae isolates are likely to possess other pathogenicity chromosomes that were not identified in the present study.
Other formae speciales have been classified into ‘yellows’ and ‘wilt’ syndromes (Edel‐Hermann & Lecompte, 2019). To our knowledge, this is the first time that ‘yellows’ and ‘wilt’ syndromes have been shown to be caused by independently evolved pathogen genotypes that do not share a pathogenicity chromosome. Our results confirmed that disease phenotyping can facilitate the identification of evolutionarily distinct groups within a forma specialis.
Host resistance is a critical tool for disease management in many Fusarium wilt pathosystems, including Fusarium wilt of strawberry (Koike & Gordon, 2015). Here, we showed that isolates with a common pathogenicity chromosome have similar cultivar host ranges, despite differences in core genome phylogeny. Therefore, differentiating between pathogen genotypes that emerged from transfer of a common pathogenicity chromosome versus a different pathogenicity chromosome has important implications for the identification and deployment of host resistance.
We observed a unique pattern of resistance/susceptibility for all six Fof genotypes we tested for pathogenicity on 25 diverse strawberry accessions (Fig. 5b). This result suggested that there were undiscovered gene‐for‐gene interactions that could ultimately demonstrate that each of these Fof genotypes is a different race. Here we propose race designations only when a differential interaction between the pathogen and a specific R‐gene has been documented, following the commonly used convention (Bus et al., 2011). In strawberry, only one Fusarium wilt resistance locus, FW1, has been identified (Pincot et al., 2018). We report for the first time the existence of Fof ‘race 2’ isolates that overcome FW1‐mediated resistance. The existence of this new race underscores the need to identify novel sources of resistance and breed them into commercially available cultivars.
Fortunately, our data suggested that numerous sources of resistance are available in hybrid cultivars and wild ecotypes. This is an unexpected finding, because we did not discover evidence of host–pathogen co‐evolution. Fragaria × ananassa is an allo‐octoploid (2n = 8x = 56) resulting from chance hybridisation between F. chiloensis (L. Miller) and F. virginiana (Duchesne), c. 300 yr before present (Darrow, 1966; Edger et al., 2019). The native ranges of the wild octoploid progenitors span diverse biomes across North and South America (Staudt, 1988). The origins of Fof in Europe, Asia and Australia were separated by oceans from the natural range of these octoploid progenitors. This suggests that Fof did not co‐evolve with its host and emerged from a host shift or host jump after the introduction of F. × ananassa. Therefore, F. chiloensis and F. virginiana should lack co‐evolved defences that are specific to the Fof isolates we recovered. Consistent with this expectation, we found a wild F. virginiana ecotype that was susceptible to all pathogen genotypes (Fig. 5b). Why F. chiloensis and F. virginiana carry genes that confer resistance to a noncoevolved pathogen remains unclear. It is possible that genetic resistance evolved in response to genes in sympatric microorganisms that have homologues in Fof.
The selection pressures that led to the assembly of pathogenicity‐enabling genes on chrY‐frag remain unknown. Presumably, proto‐pathogenicity chromosomes have been under selection for millennia in natural ecosystems. Given their role in plant infection, pathogenicity chromosomes could have initially evolved to facilitate asymptomatic, systemic colonisation of native plants (Gordon & Martyn, 1997). These strains could have emerged as pathogens through host shifts/jumps after the introduction of naïve agricultural crops, as has been postulated here for Fof and previously for formae speciales vasinfectum and cubense (Davis et al., 1996; Ploetz & Pegg, 1997; Wang et al., 2004).
Regardless of chrY‐frag’s evolutionary origin, strains carrying this chromosome would have been amplified by widespread cultivation of strawberries. Our data suggested that this chromosome was present in multiple core genome backgrounds before the first report of Fusarium wilt in Japan. However, commercial strawberry production began in Japan and South Korea in the early 20th century, long before the first observation of the disease in 1969 (Yoshida, 2013; Lee, 2014). If chrY‐frag already existed in natural ecosystems, it could have taken several decades for strawberries to be planted in a field with a chrY‐frag‐carrying strain. Furthermore, initial inoculum densities were likely to be low, and many successive crops may have been required for disease to become apparent. Circumstances such as these could account for the delay between the introduction of strawberry agriculture and the first report of disease in 1969. Once abundant in strawberry production fields, isolates with chrY‐frag were dispersed between Japan, South Korea, California and Western Australia, presumably on infected nursery plants (Notes S2; Fig. S6; Okamoto et al., 1970; Nam et al., 2011; Pastrana et al., 2019). By contrast, we found no evidence that wilt‐fragariae strains from eastern Australia or Spain have spread to other countries since their emergence.
We showed that HCT promoted diversification of Fof, but pathogenic genotypes also emerged independently, without receiving the same pathogenicity chromosome. These data supported the hypothesis that HCT is prevalent in F. oxysporum and has contributed to the evolution of many economically important plant pathogenic forms. The predicted pathogenicity chromosome chrY‐frag is likely to have been present in multiple phylogenetic groups before Fusarium wilt of strawberry was observed in Japan. Knowledge of past HCT events informed an efficient approach to testing for host resistance; we found that the cultivar host range was more similar for isolates that shared a pathogenicity chromosome than for independently evolved pathogen genotypes. Geographic isolation suggests that all pathogenic lineages lack a history of co‐evolution with F. × ananassa and its wild progenitors. However, we identified demographically and genetically diverse sources of resistance to this pathogen. Our results highlight the potential for comparative genomics of naturally diverse populations to reveal the evolutionary forces driving pathogen emergence and diversification.
Author Contributions
PMH, DDAP, SJK and TRG designed research; PMH, DDAP, BNJ, CB, MA and M‐HN performed research; PMH, DDAP, LE analysed data; PMH wrote the paper; all authors read and revised the paper before submission.
Supporting information
Dataset S1 Isolate metadata, genome metadata, GenBank/SRA accessions, raw ordinal scores for Fusarium wilt resistance phenotypes at final timepoints (corresponding to Fig. 5), differentially expressed accessory genome genes in GL1381, and the metadata and least‐squares means for all Fragaria spp. x isolate combinations tested in the differential resistance panel.
Dataset S2 A ‘.bed’ formatted file with the coordinates of sequences in the GL1381 genome that are conserved among all 27 yellows‐fragariae isolates tested.
Dataset S3 A ‘.bed’ formatted file with the coordinates of sequences in the GL1080 genome that are conserved among all 27 yellows‐fragariae isolates tested.
Dataset S4 Genotype matrix for all Fragaria spp. analysed in this study.
Fig. S1 Pictures corresponding to disease ratings for wilt‐ and yellows‐fragariae.
Fig. S2 Overview of two‐locus phylogeny, somatic compatibility, and pathogenicity phenotypes on cv ‘Sweet Ann’ for isolates used in this study.
Fig. S3 Whole genome comparison of coding sequences, transposon density and yellows‐fragariae sequence conservation in GL1381 and GL1080 reference assemblies.
Fig. S4 Core genome phylogeny of the Fusarium oxysporum species complex with isolate labels based on 2718 single copy orthologous genes.
Fig. S5 Comparison of the phylogeny inferred from single nucleotide polymorphisms in yellows‐fragariae conserved regions of chrY‐frag.T1080 and chrY‐frag.T1381.
Fig. S6 Predicted events of horizontal chromosome transfer (HCT) and dispersal of yellows‐fragariae core and chrY‐frag genotypes.
Fig. S7 Distance matrices based on the proportion of identical genotypes at single nucleotide polymorphic sites (SNPs) on A) chrY‐frag.T1381 and B) chrY‐frag.T1080.
Fig. S8 Coverage of GL1080 accessory chromosomes (chrY‐frag.T1080, lineage‐specific #2 (LS2), and lineage‐specific #3 (LS3)) by short, high‐fidelity reads from yellows‐fragariae isolates.
Fig. S9 Coverage of GL1381 accessory chromosomes (chrY‐frag.T1381, lineage‐specific #2 (LS2), and lineage‐specific #3 (LS3)) by short, high‐fidelity reads from yellows‐fragariae isolates.
Fig. S10 Index of association tests of single nucleotide polymorphisms (SNPs) on core and accessory chromosomes from a clone‐corrected yellows‐fragariae population.
Fig. S11 The proportion of identical disease phenotypes (resistant vs susceptible) between six isolates of Fusarium oxysporum f. sp. fragariae.
Fig. S12 World map of isolates characterised phenotypically and with whole genome sequencing for this study.
Methods S1 Detailed methods for experiments testing resistance to isolate AMP132 in a field near Davis, CA.
Notes S1 Additional notes on symptoms caused by yellows‐ and wilt‐fragariae.
Notes S2 A partial reconstruction of the emergence, dispersal and diversification of F. oxysporum f. sp. fragariae.
Table S1 Summary of isolates characterised in this study.
Table S2 Assembly stats for scaffolded GL1080 and GL1381 genomes.
Table S3 Chromosome and unplaced scaffold lengths and stats for the GL1381 assembly.
Table S4 Chromosome and unplaced scaffold lengths and stats for the GL1080 assembly.
Table S5 The coverage of regions on chrY‐frag that are conserved among yellows‐fragariae isolates by F. oxysporum clade 2B isolates.
Table S6 Sequencing and mapping statistics for transcriptomic experiments with GL1381.
Please note: Wiley Blackwell are not responsible for the content or functionality of any Supporting Information supplied by the authors. Any queries (other than missing material) should be directed to the New Phytologist Central Office.
Acknowledgements
We thank Sam Koehler, Sukhwinder Kaur, Lia Lopez, Michelle Haugland, Mariel Munji, Samuel Brinker, Polly Goldman, and Shawn Melendy for providing technical assistance, and Dr Matthew Settles for providing general guidance on bioinformatics. Dr Haruhisa Suga, Michelle Paynter, Dean Beasley and Dr Youn‐Sig Kwak are thanked for sending isolates of F. oxysporum f. sp. fragariae. We thank Joshua Puckett and the University of California, Davis Foundation Plant Services for providing tissue culture‐derived strawberry plants for transcriptomics experiments. Thanks to our funding sources: the National Science Foundation Graduate Research Fellowship Program, the California Strawberry Commission, National Institute of Food and Agriculture (NIFA) Specialty Crops Research Initiative (#2017‐51181‐26833), pilot grants from the University of California, Davis, Bioinformatics and DNA Technologies Core Facilities, and Ministerio de Ciencia, Innovación y Universidades of Spain Project, Ref: RTI2018‐094537‐B‐I00. The authors have no conflicts of interest to declare regarding the research conducted or publication of this article.
Data availability
Information on isolates, genes of interest and disease phenotypes are included in Datasets S1–S4. GenBank accession codes and assembly statistics are listed in Dataset S1. All raw reads and assemblies for this project are available from BioProject accessions PRJNA578477 and PRJNA606324. Any additional data will be made available upon request.
References
- Anders S, Pyl PT, Huber W. 2015. HTSeq: a python framework to work with high‐throughput sequencing data. Bioinformatics 31: 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates DM, Mäechler M, Bolker B, Walker S. 2015. Fitting linear mixed‐effects models using lme4 . Journal of Statistical Software 67: 1–48. [Google Scholar]
- Borrero C, Bascon J, Gallardo MA, Orta MS, Aviles M. 2017. New foci of strawberry Fusarium wilt in Huelva (Spain) and susceptibility of the most commonly used cultivars. Scientia Horticulturae 226: 85–90. [Google Scholar]
- Burkhardt A, Henry PM, Koike ST, Gordon TR, Martin F. 2019. Detection of Fusarium oxysporum f. sp. fragariae from infected strawberry plants. Plant Disease 103: 1006–1013. [DOI] [PubMed] [Google Scholar]
- Bus VGM, Rikkerink EHA, Caffier V, Durel C‐E, Plummer KM. 2011. Revision of the nomenclature of the differential host‐pathogen interactions of Venturia inaequalis and Malus . Annual Review of Phytopathology 49: 391–413. [DOI] [PubMed] [Google Scholar]
- Catanzariti A‐M, Do HTT, Bru P, de Sain M, Thatcher LF, Rep M, Jones DA. 2016. The tomato I gene for Fusarium wilt resistance encodes an atypical leucine‐rich repeat receptor‐like protein whose function is nevertheless dependent on SOBIR1 and SERK3/BAK1. The Plant Journal 89: 1195–1209. [DOI] [PubMed] [Google Scholar]
- Chin C‐S, Alexander DH, Marks P, Klammer AA, Drake J, Heiner C, Clum A, Copeland A, Huddleston J, Eichler EE et al. 2013. Nonhybrid, finished microbial genome assemblies from long‐read SMRT sequencing data. Nature Methods 10: 563–571. [DOI] [PubMed] [Google Scholar]
- Correll JC, Klittich CJR, Leslie JF. 1987. Nitrate nonutilizing mutants of Fusarium oxysporum and their use in vegetative compatibility tests. Phytopathology 77: 1640–1646. [Google Scholar]
- van Dam P, Fokkens L, Ayukawa Y, van der Gragt M, ter Horst A, Brankovics B, Houterman PM, Arie T, Rep M. 2017. A mobile pathogenicity chromosome in Fusarium oxysporum for infection of multiple cucurbit species. Scientific Reports 7: 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dam P, Fokkens L, Schmidt SM, Linmans JHJ, Kistler HC, Ma L, Rep M. 2016. Effector profiles distinguish formae speciales of Fusarium oxysporum . Environmental Microbiology 18: 4087–4102. [DOI] [PubMed] [Google Scholar]
- Darrow GM. 1966. The strawberry: history, breeding and physiology. New York, NY, USA: Holt, Rinehart & Winston. [Google Scholar]
- Davis RD, Moore NY, Kochman JK. 1996. Characterisation of a population of Fusarium oxysporum f. sp. vasinfectum causing wilt of cotton in Australia. Australian Journal of Agricultural Research 47: 1143–1156. [Google Scholar]
- Diener AC, Ausubel FM. 2005. RESISTANCE TO FUSARIUM OXYSPORUM 1, a dominant Arabidopsis disease‐resistance gene, is not race specific. Genetics 171: 305–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zalenski C, Jha S, Batut P, Chaisson M, Gingeras TR. 2013. STAR: ultrafast universal RNA‐seq aligner. Bioinformatics 29: 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edel‐Hermann V, Lecompte C. 2019. Current status of Fusarium oxysporum formae speciales and races. Phytopathology 109: 512–530. [DOI] [PubMed] [Google Scholar]
- Edgar R. 2004. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research 32: 1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edger PP, Poorten TJ, VanBuren R, Hardigan MA, Colle M, McKain MR, Smith RD, Teresi SJ, Nelson ADL, Wai CM et al. 2019. Origin and evolution of the octoploid strawberry genome. Nature Genetics 51: 541–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endelman JB. 2011. Ridge regression and other kernels for genomic selection with R package rrBLUP. Plant Genome 4: 250–255. [Google Scholar]
- English AC, Richards S, Han Y, Wang M, Vee V, Qu J, Qin X, Muzny DM, Reid JG, Worley KC et al. 2012. Mind the gap: upgrading genomes with pacific biosciences RS long‐read sequencing technology. PLoS ONE 7: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrison E, Marth G. 2012. Haplotype‐based variant detection from short‐read sequencing. arXiv preprint 1207.3907v2: 1‐9.
- Gordon TR, Martyn RD. 1997. The evolutionary biology of Fusarium oxysporum . Annual Review of Phytopathology 35: 111–128. [DOI] [PubMed] [Google Scholar]
- Hackl T, Hedrich R, Schultz J, Förster F. 2014. proovread: large‐scale high‐accuracy PacBio correction through iterative short read consensus. Bioinformatics 30: 3004–3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardigan MA, Feldmann MJ, Lorant A, Bird KA, Famula R, Acharya C, Cole GS, Edger PP, Knapp SJ. 2020. Genome synteny has been conserved among the octoploid progenitors of cultivated strawberry over millions of years of evolution. Frontiers in Plant Science 10: 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry PM, Kirkpatrick SC, Islas CM, Pastrana AM, Yoshisato JA, Koike ST, Daugovish O, Gordon TR. 2017. The population of Fusarium oxysporum f. sp. fragariae, cause of Fusarium wilt of strawberry, in California. Plant Disease 101: 550–556. [DOI] [PubMed] [Google Scholar]
- Jacobson DJ, Gordon TR. 1988. Vegetative compatibility and self‐incompatibility within Fusarium oxysporum f. sp. melonis . Phytopathology 78: 668–672. [Google Scholar]
- Joobeur T, King J, Nolin SJ, Thomas CE, Dean RA. 2004. The Fusarium wilt resistance locus Fom‐2 of melon contains a single resistance gene with complex features. The Plant Journal 39: 283–297. [DOI] [PubMed] [Google Scholar]
- Kamvar ZN, Brooks JC, Grünwald NJ. 2015. Novel R tools for analysis of genome‐wide population genetic data with emphasis on clonality. Frontiers in Genetics 6: 208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur S, Pham QA, Epstein L. 2017. High quality DNA from Fusarium oxysporum conidia suitable for library preparation and long read sequencing with PacBio. doi: 10.17504/protocols.io.i8ichue [DOI]
- Kerr A. 1962. The root rot‐Fusarium wilt complex of peas. Australian Journal of Biological Sciences 16: 55–69. [Google Scholar]
- Kim D, Paggi JM, Park C, Bennett C, Salzberg SL. 2019. Graph‐based genome alignment and genotyping with HISAT2 and HISAT‐genotype. Nature Biotechnology 37: 907–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim CH, Seo HD, Cho WD, Kim SB. 1982. Studies on varietal resistance and chemical control to the wilt of strawberry caused by Fusarium oxysporum . Korean Journal of Plant Protection 21: 61–67. [Google Scholar]
- Koike ST, Gordon TR. 2015. Management of Fusarium wilt of strawberry. Crop Protection 73: 67–72. [Google Scholar]
- Langmead B, Salzberg SL. 2012. Fast gapped‐read alignment with Bowtie 2. Nature Methods 9: 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee WS. 2014. Production of ever‐bearing strawberry and production technology in Korea. Acta horticulturae 1049: 561–564. [Google Scholar]
- Lenth R. 2016. Least‐squares means: the R package lsmeans. Journal of Statistical Software 69: 1–33. [Google Scholar]
- Lenth R. 2018. emmeans: Estimated marginal means, aka least‐squares means . [WWW document] URL https://cran.r‐project.org/web/packages/emmeans/index.html [Accessed 1 September 2020].
- Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics 25: 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Holmer N, Marth G, Abecasis G, Durbin R. 2009. The sequence alignment map format and SAMtools. Bioinformatics 25: 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Fokkens L, van Dam P, Rep M. 2020. Related mobile pathogenicity chromosomes in Fusarium oxysporum determine host range on cucurbits. Molecular Plant Pathology 21: 761–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv H, Fang Z, Yang L, Zhang Y, Wang Q, Liu Y, Zhuang M, Yang Y, Xie B, Liu B et al. 2014. Mapping and analysis of a novel candidate Fusarium wilt resistance gene FOC1 in Brassica oleracea . BMC Genomics 15: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, van der Does HC, Borkovich KA, Coleman JJ, Daboussi M, Di Pietro A, Dufresne M, Freitag M, Grabherr M, Henrissat B et al. 2010. Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium . Nature 464: 367–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagarajan G, Kang SW, Nam MH, Song JY, Yoo SJ, Kim HG. 2006. Characterization of Fusarium oxysporum f. sp. fragariae based on vegetative compatibility group, random amplified polymorphic DNA and pathogenicity. Plant Pathology Journal 22: 222–229. [Google Scholar]
- Nam MH, Kang YJ, Lee IH, Kim HG, Chun C. 2011. Infection of daughter plants by Fusarium oxysporum f. sp. fragariae through runner propagation of strawberry. Korean Journal of Horticultural Science and Technology 29: 273–277. [Google Scholar]
- Nielsen H. 2017. Predicting secretory proteins with SignalP. In: Kihara D, ed. Protein function prediction: methods in molecular biology. New York, NY, USA: Humana Press, 59–73. [DOI] [PubMed] [Google Scholar]
- O’Donnell K, Gueidan C, Sink S, Johnston PR, Crous PW, Glenn A, Riley R, Zitomer NC, Colyer P, Waalwijk C et al. 2009. A two‐locus DNA sequence database for typing plant and human pathogens within the Fusarium oxysporum species complex. Fungal Genetics and Biology 46: 936–948. [DOI] [PubMed] [Google Scholar]
- O’Donnell K, Kistler HC, Cigelnik E, Ploetz R. 1998. Multiple evolutionary origins of the fungus causing Panama disease of banana: Concordant evidence from nuclear and mitochondrial gene genealogies. Proceedings of the National Academy of Sciences, USA 95: 2044–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto H, Fujii S, Kato K, Yoshioka A. 1970. A new strawberry disease ‘Fusarium wilt’. Plant Protection 24: 231–235. [Google Scholar]
- Ori N, Eshed Y, Paran I, Presting G, Aviv D, Tanksley S, Zamir D, Fluhr R. 1997. The I2C family from the wilt disease resistance locus I2 belongs to the nucleotide binding, leucine‐rich repeat superfamily of plant resistance genes. The Plant Cell 9: 521–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastrana AM, Watson DC, Gordon TR. 2019. Transmission of Fusarium oxysporum f. sp. fragariae through stolons in strawberry plants. Plant Disease 103: 1249–1251. [DOI] [PubMed] [Google Scholar]
- Paynter ML, Czislowski E, Herrington ME, Aitken EAB. 2016. Differences in pathogenicity, genetic variability, and cultivar responses among isolates of Fusarium oxysporum from strawberry in Australia. American Society of Horticultural Science 141: 645–652. [Google Scholar]
- Pincot DDA, Poorten TJ, Hardigan MA, Harshman JM, Acharya CB, Cole GS, Gordon TR, Stueven M, Edger PP, Knapp S. 2018. Genome‐wide association mapping uncovers Fw1, a dominant gene conferring resistance to Fusarium wilt in strawberry. G3: Genes . Genomes, Genetics 8: 1817–1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ploetz R, Pegg K. 1997. Fusarium wilt of banana and Wallace’s line: Was the disease originally restricted to his Indo‐Malayan region? Australasian Plant Pathology 26: 239–249. [Google Scholar]
- Quinlan AR, Hall IM. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team . 2013. R: A language and environment for statistical computing. v.3.5.2. Vienna, Austria: R Foundation for Statistical Computing. [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK. 2010. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26: 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt SM, Houterman PM, Schreiver I, Ma L, Amyotte S, Chellappan B, Boeren S, Takken FLW, Rep M. 2013. MITEs in the promoters of effector genes allow prediction of novel virulence genes in Fusarium oxysporum . BMC Genomics 14: 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt SM, Lukasiewicz J, Farrer R, van Dam P, Bertoldo C, Rep M. 2016. Comparative genomics of Fusarium oxysporum f. sp. melonis reveals the secreted protein recognized by the Fom‐2 resistance gene in melon. New Phytologist 209: 307–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahi S, Beerens B, Bosch M, Linmans JHJ, Rep M. 2016. Nuclear dynamics and genetic rearrangement in heterokaryotic colonies of Fusarium oxysporum . Fungal Genetics and Biology 91: 20–31. [DOI] [PubMed] [Google Scholar]
- Simao FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. 2015. BUSCO: assessing genome assembly and annotation completemess with single‐copy orthologs. Bioinformatics 31: 3210–3212. [DOI] [PubMed] [Google Scholar]
- Smith T, Heger A, Sudbery I. 2017. UMI‐tools: Modeling sequencing errors in unique molecular identifiers to improve quantification accuracy. Genome Research 27: 491–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperschneider J, Dodds PN, Gardiner DM, Singh KB, Taylor JM. 2018. Improved prediction of fungal effector proteins from secretomes with EffectorP 2.0. Molecular Plant Pathology 19: 2094–2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post‐analysis of large phylogenies. Bioinformatics 30: 1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staudt G. 1988. The species of Fragaria, their taxonomy and geographical distribution. Acta horticulturae 265: 23–34. [Google Scholar]
- Suga H, Hirayama Y, Morishima M, Suzuki T, Kageyama K, Hyakumachi M. 2013. Development of PCR primers to identify Fusarium oxysporum f. sp. fragariae . Plant Disease 97: 619–625. [DOI] [PubMed] [Google Scholar]
- Tarailo‐Graovac M, Chen N. 2009. Using RepeatMasker to identify repetitive elements in genomic sequences. Current Protocols in Bioinformatics 4: 1–14. [DOI] [PubMed] [Google Scholar]
- Taylor JW, Hann‐Soden C, Branco S, Sylvain I, Ellison CE. 2015. Clonal reproduction in fungi. Proceedings of the National Academy of Sciences, USA 112: 8901–8908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Testa AC, Hane JK, Ellwood SR, Oliver RP. 2015. CodingQuarry: highly accurate hidden Markov model gene prediction in fungal genomes using RNA‐seq transcripts. BMC Genomics 16: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Willians B, Pertea G, Mortazavi A, Kwan G, van Baren J, Salzberg SL, Wold B, Pachter L. 2010. Transcript assembly and quantification by RNA‐Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature Biotechnology 28: 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tritt A, Eisen JA, Facciotti MT, Darling AE. 2012. An integrated pipeline for de Novo assembly of microbial genomes. PLoS ONE 7: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker BJ, Abeel T, Shea T, Priest M, Abouelliel A, Sakthikumar S, Cuomo CA, Zeng Q, Wortman J, Young SK et al. 2014. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 9: e112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Brubaker CL, Burdon JJ. 2004. Fusarium species and Fusarium wilt pathogens associated with native Gossypium populations in Australia. Mycological Research 108: 35–44. [DOI] [PubMed] [Google Scholar]
- Winks BL, Williams YN. 1965. A wilt of strawberry caused by a new form of Fusarium oxysporum . Queensland Journal of Agricultural and Animal Science 22: 475–479. [Google Scholar]
- Yoshida Y. 2013. Strawberry production in Japan: History and progress in production technology and cultivar development. International Journal of Fruit Science 13: 103–113. [Google Scholar]
- Yu D, Tang H, Zhang Y, Yu H, Chen Q. 2012. Comparison and improvement of different methods of RNA isolation from strawberry. Journal of Agricultural Science 4: 51–56. [Google Scholar]
- Yu G, Smith D, Zhu H, Guan Y, Lam TT. 2017. ggtree: an R package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods in Ecology and Evolution 8: 28–36. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Dataset S1 Isolate metadata, genome metadata, GenBank/SRA accessions, raw ordinal scores for Fusarium wilt resistance phenotypes at final timepoints (corresponding to Fig. 5), differentially expressed accessory genome genes in GL1381, and the metadata and least‐squares means for all Fragaria spp. x isolate combinations tested in the differential resistance panel.
Dataset S2 A ‘.bed’ formatted file with the coordinates of sequences in the GL1381 genome that are conserved among all 27 yellows‐fragariae isolates tested.
Dataset S3 A ‘.bed’ formatted file with the coordinates of sequences in the GL1080 genome that are conserved among all 27 yellows‐fragariae isolates tested.
Dataset S4 Genotype matrix for all Fragaria spp. analysed in this study.
Fig. S1 Pictures corresponding to disease ratings for wilt‐ and yellows‐fragariae.
Fig. S2 Overview of two‐locus phylogeny, somatic compatibility, and pathogenicity phenotypes on cv ‘Sweet Ann’ for isolates used in this study.
Fig. S3 Whole genome comparison of coding sequences, transposon density and yellows‐fragariae sequence conservation in GL1381 and GL1080 reference assemblies.
Fig. S4 Core genome phylogeny of the Fusarium oxysporum species complex with isolate labels based on 2718 single copy orthologous genes.
Fig. S5 Comparison of the phylogeny inferred from single nucleotide polymorphisms in yellows‐fragariae conserved regions of chrY‐frag.T1080 and chrY‐frag.T1381.
Fig. S6 Predicted events of horizontal chromosome transfer (HCT) and dispersal of yellows‐fragariae core and chrY‐frag genotypes.
Fig. S7 Distance matrices based on the proportion of identical genotypes at single nucleotide polymorphic sites (SNPs) on A) chrY‐frag.T1381 and B) chrY‐frag.T1080.
Fig. S8 Coverage of GL1080 accessory chromosomes (chrY‐frag.T1080, lineage‐specific #2 (LS2), and lineage‐specific #3 (LS3)) by short, high‐fidelity reads from yellows‐fragariae isolates.
Fig. S9 Coverage of GL1381 accessory chromosomes (chrY‐frag.T1381, lineage‐specific #2 (LS2), and lineage‐specific #3 (LS3)) by short, high‐fidelity reads from yellows‐fragariae isolates.
Fig. S10 Index of association tests of single nucleotide polymorphisms (SNPs) on core and accessory chromosomes from a clone‐corrected yellows‐fragariae population.
Fig. S11 The proportion of identical disease phenotypes (resistant vs susceptible) between six isolates of Fusarium oxysporum f. sp. fragariae.
Fig. S12 World map of isolates characterised phenotypically and with whole genome sequencing for this study.
Methods S1 Detailed methods for experiments testing resistance to isolate AMP132 in a field near Davis, CA.
Notes S1 Additional notes on symptoms caused by yellows‐ and wilt‐fragariae.
Notes S2 A partial reconstruction of the emergence, dispersal and diversification of F. oxysporum f. sp. fragariae.
Table S1 Summary of isolates characterised in this study.
Table S2 Assembly stats for scaffolded GL1080 and GL1381 genomes.
Table S3 Chromosome and unplaced scaffold lengths and stats for the GL1381 assembly.
Table S4 Chromosome and unplaced scaffold lengths and stats for the GL1080 assembly.
Table S5 The coverage of regions on chrY‐frag that are conserved among yellows‐fragariae isolates by F. oxysporum clade 2B isolates.
Table S6 Sequencing and mapping statistics for transcriptomic experiments with GL1381.
Please note: Wiley Blackwell are not responsible for the content or functionality of any Supporting Information supplied by the authors. Any queries (other than missing material) should be directed to the New Phytologist Central Office.
Data Availability Statement
Information on isolates, genes of interest and disease phenotypes are included in Datasets S1–S4. GenBank accession codes and assembly statistics are listed in Dataset S1. All raw reads and assemblies for this project are available from BioProject accessions PRJNA578477 and PRJNA606324. Any additional data will be made available upon request.