

Abstract
Tofacitinib is an oral, small molecule Janus kinase inhibitor for the treatment of ulcerative colitis (UC). We characterized tofacitinib pharmacokinetics in patients with moderate to severe UC, and the effects of covariates on variability in pharmacokinetic parameter estimates. Data were pooled from 1 8‐week phase 2 and 2 8‐week phase 3 induction studies, and a 52‐week phase 3 maintenance study (N = 1096). Population pharmacokinetic analysis was conducted using nonlinear mixed‐effects modeling. Potential predictors of apparent oral clearance (CL/F) and volume of distribution (V/F) were evaluated. The PK was described by a 1‐compartment model parameterized in terms of CL/F (26.3 L/hour [h]) and V/F (115.8 L), with first‐order absorption (Ka; 9.85 h−1) and lag time (0.236 h). The derived elimination half‐life was approximately 3.05 h. In the final model, baseline creatinine clearance, sex, and race (Asian vs non‐Asian) were significant covariates for CL/F; significant covariates for V/F were age, sex, and body weight; baseline albumin and baseline Mayo score were not significant covariates. CL/F between‐patient variability was estimated at 22%. Tofacitinib exposure did not change significantly over the duration of induction/maintenance treatment in patients with UC. Although statistically significant covariate effects on CL/F and V/F were observed, the magnitude of the effects are not clinically significant. Therefore, dose adjustment/restrictions for age, body weight, sex, race, or baseline disease severity are not required during tofacitinib treatment. ClinicalTrials.gov numbers: NCT00787202, NCT01465763, NCT01458951, NCT01458574.
Keywords: CP‐690,550 (tofacitinib); inflammatory bowel disease; Janus kinase (JAK) inhibitors; pharmacokinetics; ulcerative colitis
Ulcerative colitis (UC) is a chronic, relapsing, inflammatory disease, primarily affecting the colonic mucosa. 1 Clinical characteristics of UC include rectal bleeding, fecal urgency, and diarrhea. 1 Therapies used to treat UC include mesalamine (5‐aminosalicylic acid [5‐ASA]), corticosteroids, thiopurines (eg, azathioprine, 6‐mercaptopurine), tumor necrosis factor inhibitors (TNFi; eg, adalimumab, infliximab, golimumab), and integrin inhibitors (eg, vedolizumab). 1 , 2 , 3 , 4 , 5 However, many patients either do not respond to these therapies or do not have a sustained response; therefore, novel treatments with different mechanisms of action are required.
Tofacitinib is an oral, small molecule Janus kinase (JAK) inhibitor for the treatment of UC. Tofacitinib is a potent inhibitor of JAK1 and JAK3 with only moderate potency for JAK2 in in vitro assays, 6 and JAK1/3 inhibition by tofacitinib is proposed to block signaling for several cytokines integral to lymphocyte activation, proliferation, and function, including interleukin‐2, ‐4, ‐7, ‐9, ‐15, and ‐21. In addition, JAK1 inhibition may also attenuate signaling by interleukin‐6 and interferon gamma. 6 , 7 , 8 Therefore, tofacitinib treatment results in modulation of multiple aspects of the immune response associated with the pathogenesis of UC. 6 , 8 Previous investigations in healthy volunteers have found that tofacitinib is absorbed rapidly; plasma concentrations peak approximately 1 hour (h) after oral administration, and mean terminal‐phase half‐life (t½) is approximately 3.2 h. 9 Also, the majority of tofacitinib clearance (70%) is via hepatic metabolism (primarily via cytochrome P450 3A4), while only 30% of tofacitinib clearance is due to renal elimination. 9
The efficacy of tofacitinib 10 mg twice daily as induction therapy has been demonstrated, vs placebo, in a phase 2, 8‐week dose‐ranging induction study (A3921063) 10 and in 2 identical phase 3, placebo‐controlled, 8‐week UC induction studies (OCTAVE Induction 1 and 2). 11 Tofacitinib 5 and 10 mg twice daily have also demonstrated efficacy as maintenance therapy, vs placebo, in a phase 3, placebo‐controlled, 52‐week maintenance study (OCTAVE Sustain), 11 which enrolled responders from OCTAVE Induction 1 and 2. The long‐term safety and efficacy of tofacitinib 5 and 10 mg twice daily for patients with UC are being evaluated in an ongoing open‐label, long‐term extension study (OCTAVE Open). 12
Population pharmacokinetic (PK) analysis provides insight into the disposition properties of a drug under investigation; characterizes sources and predictors of its PK variability, along with pharmacodynamic end points; and can assist in determining the optimal drug dose in the target patient population. In addition, population PK analysis also allows for further assessment of the relationships between systemic exposure and pharmacodynamic responses, which can facilitate various stages of drug development, and this provides valuable insights for regulatory review, approval, and labeling.
In this analysis, we characterized the PK characteristics of tofacitinib in patients with moderate to severe UC using data collected from the tofacitinib UC clinical program, to identify intrinsic and extrinsic factors (demographics, prior and concomitant UC treatments, baseline disease activity) that impact the PK of tofacitinib in these patients.
Methods
Study Design and PK Assessments
Data were pooled from patients with UC treated with tofacitinib 0.5, 3, 5, 10, or 15 mg twice daily (immediate‐release formulation) enrolled in phase 2 and phase 3 studies in the tofacitinib UC clinical program. 10 , 11 The key features of the studies included in the analysis, including patient numbers and per‐protocol (nominal) PK sampling times, are shown in Table 1. Plasma samples were stored at approximately –20°C and tofacitinib concentrations determined using a validated analytical method, 13 with a 0.100 ng/mL limit of quantification. Patients with creatinine clearance <40 mL/min were excluded from all UC clinical trials per protocol.
Table 1.
Overview of Tofacitinib Phase 2 and Phase 3 Studies in Patients With UC Included in the Population PK Analysis
| Study Identifier | Design/Total Duration | Tofacitinib Treatment Groups | Number of Patients in Data Set a | Sampling Schedule |
|---|---|---|---|---|
| A3921063 (NCT00787202) 10 | 8‐week, phase 2 induction study of patients with moderately to severely active UC | 0.5 mg twice daily3 mg twice daily10 mg twice daily15 mg twice daily | 31333148 | Baseline (day 1) and week 8 (±3 days): predose b and 0.25, 0.5, 1, 2‐3 h after in‐clinic dose; week 2 (±3 days) c and week 4 (±3 days): c 2 samples taken >1 h apart |
| OCTAVE Induction 1 A3921094 (NCT01465763) 11 | 8‐week, d phase 3 induction study of patients with moderately to severely active UC. Patients had either previously failed or were intolerant to treatment with corticosteroids, immunosuppressants, and/or TNFi | 10 mg twice daily15 mg twice daily | 46815 e | Week 2 (±3 days): predose b and 0.5 h (0.25‐1.5 h) after in‐clinic dose; week 8 (±3 days): predose b and 2 h (1‐3 h) after in‐clinic dose |
| OCTAVE Induction 2 A3921095 (NCT01458951) 11 | 8‐week, d phase 3 induction study of patients with moderately to severely active UC. Patients had either previously failed or were intolerant to treatment with corticosteroids, immunosuppressants, and/or TNFi | 10 mg twice daily15 mg twice daily | 4225 e | Week 2 (±3 days): predose b and 0.5 h (0.25‐1.5 h) after in‐clinic dose; week 8 (±3 days): predose b and 2 h (1‐3 h) after in‐clinic dose |
| OCTAVE Sustain A3921096 (NCT01458574) 11 | 52‐week, f phase 3 maintenance study that enrolled clinical responders from OCTAVE Induction 1 and 2 | 5 mg twice daily10 mg twice daily | 191189 | Baseline (day 1) g and week 24 (±5 days): predose b and 2 h (1‐3 h) after dosing; week 8 (±5 days) and week 52 (±5 days): predose b and 0.5 h (0.25‐1.5 h) after dosing |
Abbreviations: h, hours; PK, pharmacokinetics; TNFi, tumor necrosis factor inhibitors; UC, ulcerative colitis.
Details of study sites for all phase 2 and phase 3 studies are shown in Table S1.
Patients in all studies included in this analysis were allowed to continue background UC treatments, but concomitant treatment with azathioprine, 6‐mercaptopurine, methotrexate, or TNFi was prohibited.
Patients receiving tofacitinib in induction studies who were reassigned to receive tofacitinib 5 mg or 10 mg twice daily in the maintenance study were counted only once in this analysis.
Predose sampling should have occurred 12 ± 2 h after the evening dose of study medication was taken and immediately before the in‐clinic dose of study medication.
Patients could take medication at home or at the study site; the first sample was taken upon arrival at the medical center.
The final efficacy assessment was at week 8, and the end of treatment was at week 9.
The 15 mg twice daily dose was removed from the study after subsequent protocol amendment; therefore, the final sample size for this dose was small.
The final efficacy assessment was at week 52, and the end of treatment was at week 53.
Week 8 assessment for patients who completed OCTAVE Induction 1 and 2 and achieved clinical response was considered as baseline value for Mayo score (day 1) in OCTAVE Sustain.
All studies were registered with ClinicalTrials.gov (NCT00787202, NCT01465763, NCT01458951, NCT01458574), approved by the institutional review boards and/or independent ethics committees at each of the investigational centers participating in the studies or a central institutional review board, and conducted in compliance with the Declaration of Helsinki and the International Conference on Harmonization Good Clinical Practice Guidelines. All patients provided written informed consent. All authors had access to the study data and reviewed and approved the final manuscript.
Data Analysis
A nonlinear mixed‐effects modeling (NONMEM) approach was used in this population PK analysis. The software packages NONMEM version 7.3 (ICON Development Solutions, Ellicott City, Maryland), and Perl‐speaks‐NONMEM version 4.2.0 as supporting software for the execution of NONMEM, were applied. R version 3.1.2 (R Development Core Team, Vienna, Austria) and R package Xpose4 version 4.5.3 were used for data handling, exploratory data analysis, and creation of graphs for presentation and reports.
The PK data were first transformed using a natural log transformation before data analysis. The analyses were conducted in the following steps: base structural model development, random‐effects model development, covariate model development, assessment of model adequacy, and assessment of model predictive performance (model validation). 14
The population PK of tofacitinib has been previously reported in patients with psoriasis and psoriatic arthritis. 15 , 16 A structural PK model for tofacitinib in patients with UC was previously reported in the PK data as part of an exposure‐response analysis in Study A3921063. 17 Based on prior experience of structural PK model evaluation in US patients, and noncompartment analysis of phase 1 PK data from healthy individuals, disposition of tofacitinib was expected to be monophasic. 9 , 18 , 19 Therefore, a 1‐compartment model parameterized in terms of apparent oral clearance (CL/F) and apparent volume of distribution (V/F), with first‐order (with or without lag time) absorption, was used as the base structural model. Estimation of a first‐order absorption rate constant (Ka) was supported by the availability of early serial PK samples after an in‐clinic dose of the study medication (Table 1); therefore, other absorption models, such as a zero‐order process, were not evaluated. Interindividual variabilities (IIV) on CL/F and on V/F were modeled using exponential variance models. IIV and interoccasion variability (IOV) were also evaluated with respect to Ka. Residual error was modeled as an additive term on the natural log scale, which is equivalent to a proportional error model on the linear scale. Since the impact of data collection errors on residual error is likely to be higher for drugs with short t½, such as tofacitinib, IIV and time of sample collection were evaluated with respect to residual error. IIV was evaluated to describe potential between‐subject differences in error related to the collection of actual dose and PK collection times in large multicenter patient studies. Estimation of 2 separate residual error terms for early and late postdose PK sample collection time points was also evaluated to address potential differences in data collection error, given that early postdose PK sample collection time points were mostly related to the in‐clinic dose time recorded at the site, whereas late postdose PK sample collection time points were related to the most recent postdose time recalled by patients in the study. A postdose time of 8 h was selected based on the distribution of PK data over time, to classify late (>8 h) samples that were mostly trough values, and early (≤8 h) samples that were mostly nontrough values. Population parameters, including fixed‐ and random‐effects parameters, were estimated using the first‐order conditional estimation with interaction method. 20
Predefined covariates and covariate parameter relationships were evaluated based on exploratory graphics, clinical interest, mechanistic plausibility, or prior knowledge from tofacitinib psoriasis and psoriatic arthritis studies. 15 , 21 Covariates with high correlation or collinearity (|r|>0.5) were introduced into the model separately and along with other covariates. Covariate selection was performed using a stepwise covariate modeling (SCM) approach. 22 , 23 , 24 The parameter‐covariate relationships were evaluated using stepwise forward‐inclusion backward‐deletion procedures, and inclusion and exclusion of a significant covariate was guided by the decrease in the objective function value (OFV) (–2 × log likelihood). A P value of .05 was chosen for inclusion (forward), and a stricter P value of .01 was used for exclusion (backward) of an additional covariate to find a parsimonious final model. All covariates included in the SCM were nested covariates under model parameters. Final inferences regarding the clinical relevance of covariate effects were based on parameter estimates from the final model and measures of estimated precision derived from the bootstrap of the final model.
The predefined covariates for inclusion in the analysis for CL/F (mL/min) were as follows: race (categorical), racial designation (categorical), sex (categorical), ethnicity (categorical), extent of disease (categorical), region (categorical), age (years), body weight (kg), baseline renal function (as assessed by creatinine clearance [mL/min]), baseline C‐reactive protein (mg/dL), baseline albumin (g/dL), baseline alanine transaminase (U/L), baseline aspartate transaminase (U/L), baseline alkaline phosphatase (U/L), baseline Mayo score, concomitant medication (oral corticosteroids and 5‐ASA [categorical]), status of prior TNFi exposure (categorical), and status of prior TNFi failure (categorical). The predefined covariates for inclusion in the analysis for V/F were body weight (kg), age (years), and sex (categorical). In the case of categorical covariates with >2 categories, each category (eg, race, which includes White, Black, Asian, and Other categories) was evaluated relative to the rest of the population for differences in parameter values; for example, CL/F for Asian patients was evaluated relative to the CL/F for the remaining population (non‐Asian).
The selected categorical covariates entered the model as 1 coefficient for each category. The selected continuous covariates were incorporated as power functions, normalized to the reference (approximate median) values in the data set.
Diagnostic plots were used for guiding model development. At all stages, visual inspection of plots of observed vs predicted values and observed vs individual predicted values—along with residual plots, shrinkage, and precision of the parameter estimates—were evaluated to assess model adequacy. Nonparametric bootstraps were performed for the base and final PK models, to obtain the final precision of the parameter estimates. Visual predictive checks (VPCs) were performed for the final PK models and were stratified by study, dose level, and visit occasion.
Results
Patient and PK Sample Disposition
In total, 1096 patients were included in the analysis, and patient demographics and baseline characteristics are summarized in Table 2. Additional details of the study populations have been reported previously. 10 , 11
Table 2.
Patient Demographics and Baseline Characteristics
| Total Patient Population (n = 1096) | ||||
|---|---|---|---|---|
| Continuous Covariates for the Patients in the Population PK Data Set at Baseline | ||||
| Covariate | Mean (SD) | Median | Range | Missing n (%) |
| Age, y | 41.3 (13.8) | 40 | 18‐80 | 0 |
| Body weight, kg | 73.6 (16.6) | 72 | 37‐154.5 | 0 |
| Height, cm | 171.5 (9.6) | 171.6 | 145.5‐199.0 | 0 |
| BMI, kg/m 2 | 25.0 (5.0) | 24.2 | 15.4‐54.6 | 0 |
| BCCL, mL/min a | 112.3 (30.3) | 108.7 | 40.8‐255.2 | 1 (0.1) |
| Serum creatinine, mg/dL | 0.9 (0.2) | 0.9 | 0.4‐1.6 | 1 (0.1) |
| CRP, mg/dL | 11.5 (18.7) | 4.7 | 0.1‐208.4 | 16 (1.5) |
| Albumin, g/dL | 4.2 (0.4) | 4.2 | 2.1‐5.4 | 1 (0.1) |
| AP, U/L | 71.4 (25.7) | 67.0 | 12.0‐393.0 | 1 (0.1) |
| ALT, U/L | 18.8 (13.2) | 15.5 | 5.0‐227.0 | 2 (0.2) |
| AST, U/L | 18.2 (8.5) | 17.0 | 7.0‐125.0 | 3 (0.3) |
| Mayo score b | 8.9 (1.5) | 9.0 | 3.0‐12.0 | 2 (0.2) |
| Categorical Covariates for the Patients in the Population PK Data Set at Baseline | ||
|---|---|---|
| Covariate | n | Ratio (%) |
| Sex | ||
| Male | 641 | 58.5 |
| Female | 455 | 41.5 |
| Race | ||
| White | 891 | 81.3 |
| Black | 11 | 1.0 |
| Asian | 119 | 10.9 |
| Other | 46 | 4.2 |
| Ethnicity | ||
| Hispanic | 37 | 3.4 |
| Non‐Hispanic | 887 | 80.9 |
| Smoking status | ||
| Never smoked | 694 | 63.3 |
| Current smoker | 62 | 5.7 |
| Ex‐smoker | 340 | 31.0 |
| History of alcohol use | 332 | 30.3 |
| TNFi‐naïve | 558 | 50.9 |
| Prior TNFi failure | 511 | 46.6 |
| Concomitant medications | ||
| 5‐ASA | 852 | 77.7 |
| Oral corticosteroids | 469 | 42.8 |
| Extent of disease | ||
| Proctosigmoiditis | 138 | 12.6 |
| Left‐sided colitis | 327 | 29.8 |
| Extensive colitis | 108 | 9.9 |
| Pancolitis | 376 | 34.3 |
| Proctitis | 1 | 0.1 |
Abbreviations: 5‐ASA, 5‐aminosalicylic acid; ALT, alanine transaminase; AP, alkaline phosphatase; AST, aspartate transaminase; BCCL, baseline creatinine clearance; BMI, body mass index; CRP, C‐reactive protein; PK, pharmacokinetics; SD, standard deviation; TNFi, tumor necrosis factor inhibitor.
Creatinine clearance <40 mL/min at induction was an exclusion criterion for the studies included in this analysis.
Considered a continuous variable for this analysis.
A total of 7231 observation records were collected. Approximately 4% of the total observations were reported as data records below the limit of quantification and were treated as missing and therefore not included in the PK analysis. In addition, predose PK samples collected from each patient at week 2 and week 4 visits in Study A3921063, corresponding to 519 of 7231 (7.2%) records, were excluded systematically based on evaluation of the data that suggested significant data collection errors at these visits, and 20 (0.3%) additional records were excluded across all protocols based on discrepancies with dosing information.
Base Model Results
The 1‐compartment base model with first‐order absorption with a lag time provided an adequate and unbiased description of the data, based on goodness‐of‐fit plots and other model diagnostics; base model structural parameter estimates had a relative standard error (RSE) of <8% across all estimated parameters. The typical estimates of CL/F and V/F from the base model were 25.0 L/h and 111 L, with RSEs of <2%, and the derived elimination t½ was approximately 3.05 h. In addition, Ka was estimated to be 9.9 h−1, with an RSE of 7.4%. The IIV estimate (% coefficient of variation [CV]) for CL/F and the IOV estimate (%CV) for Ka were 25.2% and 192.4%, respectively. The IIV for V/F could not be estimated independently, which is likely due to the limited information available from sparse PK data and the correlation between the random effects for CL/F and V/F. Therefore, the random effect for V/F was estimated by using a fixed‐effect scalar (θscale) on the random effect for CL/F, as shown in Equation 1 below:
| (1a) |
| (1b) |
Where:
CLi/Vi is the individual clearance/volume;
θTV is the population value;
and η,i is the individual patient‐specific variability term.
The estimation of Ka and at least 1 level of random effect (IOV) was supported by serial PK sampling performed early after in‐clinic dose of study medication on ≥2 separate occasions. In the phase 2 study, PK samples were collected per protocol from each of the 143 patients included in the analysis data set, at predose and 0.25, 0.5, 1, and 2–3 h after dosing at baseline and week 8; and 2 samples were taken >1 h apart at weeks 2 and 4. In phase 3 induction studies (N = 910), each patient contributed PK samples at predose and 0.5 h after dosing at baseline and week 2; and at predose and 2 h after dosing at week 8. In the maintenance study (N = 380), each patient contributed PK samples at predose and 2 h after dosing at baseline and week 24; and at predose and 0.5 h after dosing at weeks 8 and 52 (Table 1).
The estimated t½ in patients with UC was consistent with that observed previously in other patient populations, such as rheumatoid arthritis and psoriasis (Pfizer, data on file), as well as in healthy individuals. 18 , 19 In previous phase 1 studies in healthy individuals where dense PK sampling was used, tofacitinib demonstrated a t½ of approximately 3 h with a monoexponential elimination profile. 18 , 19
Time dependency on CL/F was tested in the base model using an exponential model for change in CL/F over duration of treatment (Equation 2):
| (2) |
Where:
θTVCL is the typical value of clearance;
OCCW is the numerical index of each occasion week;
and θtime is the estimated exponent characterizing the time‐effect on clearance.
Although the effect was statistically significant (∆OFV = −3.815 for 5% type I error), the estimate of the exponent was very small (–8.8947 × 10–4), indicating lack of a meaningful change in CL/F over the duration of treatment. Therefore, no time‐effect was included on CL/F for subsequent model development.
The correlation between individual CL/F estimates from the base model, and baseline albumin or baseline Mayo score, shown in Figure S1, indicated the absence of a trend between change in CL/F and changes in baseline albumin or baseline Mayo score. These disease‐specific covariates were further confirmed to be nonsignificant in the SCM. Therefore, these covariates were not included in the final model.
Figure 2.
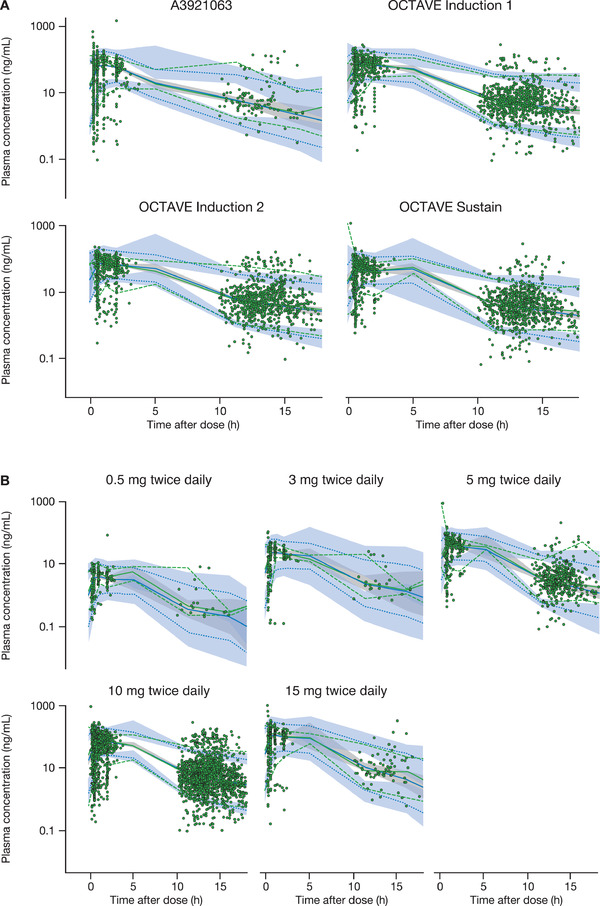
Prediction‐corrected VPCs for the final model stratified by (A) study and (B) dose. The green dashed lines represent the 90%CI of observed data. The green solid line is the median of observed data. The blue dashed lines represent the 90% predictive interval based on simulations. The blue solid line represents the median based on simulations. The blue or gray shaded area is the predicted 95%CI of upper limit, lower limit, or median (50%), based on simulations. CI, confidence interval; h, hours; VPC, visual predictive check.
Final Model Results
A 1‐compartment disposition model with first‐order absorption rate constant and an absorption lag time described the data adequately, with reasonable precision for parameter estimates. IIV for CL/F was modeled using an exponential variance model, and a scaling parameter was used to describe the IIV of V/F based on the IIV of CL/F. Both IIV and IOV were evaluated on Ka, and only IOV was included as a random effect on Ka based on significant improvement in model fit. Residual random effects were described with 2 proportional (additive on logarithmic scale) error models for trough (time after dose >8 h) and nontrough (time after dose ≤8 h) data.
The parameter estimates for the final model and bootstrap results, and the covariate parameter estimates, are shown in Table 3; Figure S2 demonstrates the goodness‐of‐fit for the final model. The typical estimates (95% confidence interval [CI] from bootstrap) of CL/F, V/F, Ka, and lag time from the final model were 26.3 (25.5–27.2) L/h, 115.8 (111.5–120.6) L, 9.85 (7.9–10.8) h−1, and 0.236 (0.218–0.238) h, respectively, for the typical reference individual (non‐Asian; male; body weight, 72.0 kg; 40.0 years of age; baseline creatinine clearance, 108.7 mL/min). The derived elimination t½ was approximately 3.05 h. The estimated t½ of tofacitinib in patients with UC is consistent with that in healthy individuals in a phase 1 study based on noncompartment analysis. 19 IIV estimate (%CV) for CL/F, and IOV estimate (%CV) for Ka, were 22.2% and 191.8%, respectively. η‐shrinkage for CL/F was estimated at 11.3% and η‐shrinkage for IOV, for the 6 occasions estimated, ranged from 46.9% to 87.4%. ε‐shrinkage was estimated at 3.9%.
Table 3.
Parameter and Covariate Parameter Estimates for the Population PK Model
| Parameter Estimates and Bootstrap Results | ||||||
|---|---|---|---|---|---|---|
| Final Model | Bootstrap a | |||||
| Estimates (RSE%) | IIV (RSE%) | IOV (RSE%) | Median (IIV%) | 95%CI | 95%CI for IIV | |
| CL/F, L/h | 26.3 (1.2) | 22.2 (7.7) | NA | 26.3 (22.2) | 25.5‐27.2 | 19.0‐24.5 |
| V/F, L | 115.8 (1.1) | NA | NA | 115.5 (NA) | 111.5‐120.6 | NA |
| Ka, h−1 | 9.9 (7.1) | NA | 191.8 (7.0) | 9.86 (191.9) b | 7.9‐10.8 | 179.0‐205.0 b |
| Proportional error, TAD ≤8 h, % | 41.6 (2.6) | 58.0 (6.3) | NA | 41.3 (58.0) | 38.6‐44.4 | 54.0‐62.2 |
| Proportional error, TAD >8 h, % | 68.9 (2.6) | 58.0 (6.3) | NA | 68.6 (58.0) | 64.1‐73.5 | 54.0‐62.2 |
| Lag time, h | 0.236 (0.52) | NA | NA | 0.236 (NA) | 0.218‐0.238 | NA |
| Scaling parameter c | 0.392 (7.19) | NA | NA | 0.399 (NA) | 0.305‐0.481 | NA |
| Covariate Parameter Estimates | ||||||
| PK Parameter | Covariate | Estimate d | RSE% | 95%CI e | ||
| CL/F | BCCL | 0.354 | 7.57 | 0.279‐0.426 | ||
| CL/F | Female | 0.868 | 1.86 | 0.829‐0.909 | ||
| CL/F | Asian | 0.932 | 2.16 | 0.888‐0.972 | ||
| V/F | Age | –0.116 | 15.05 | −0.173 to −0.065 | ||
| V/F | Female | 0.845 | 1.54 | 0.802‐0.887 | ||
| V/F | Body weight | 0.585 | 5.03 | 0.501‐0.674 | ||
Abbreviations: BCCL, baseline creatinine clearance; CI, confidence interval; CL/F, apparent oral clearance; h, hours; IIV, interindividual variability; IOV, interoccasion variability; Ka, absorption rate constant; NA, not available; PK, pharmacokinetics; RSE, relative standard error; TAD, time after dose; V/F, apparent volume of distribution.
The derived elimination half‐life was approximately 3.05 h.
621 in 1000 runs minimized successfully.
As IOV (%) and 95%CI for IOV, instead of IIV.
A scaling parameter was applied to describe IIV of V/F relative to IIV of CL/F, due to the lack of samples to describe peak and trough concentrations for phase 3 studies and a high estimated correlation between V/F and CL/F.
For continuous covariates, estimates were computed as power functions, normalized to the reference (approximate median) values in the data set; for categorical covariates, estimates were computed as a fraction of the reference category, with 1 estimated coefficient for each category.
95%CI for covariate parameter estimates were calculated based on the bootstrap method.
The scaling parameter to describe IIV of V/F relative to IIV of CL/F (applied due to the lack of samples to describe peak and trough concentrations for phase 3 studies and a high estimated correlation between V/F and CL/F) was 0.392 (95%CI: 0.305‐0.481). Residual variability for observations collected before or after 8 h after dose were 41.6% and 68.9%, respectively, with IIV estimated to be 58.0%.
Continuous covariates included in the final model (baseline body weight, age, and creatinine clearance) were incorporated as power functions, normalized to the reference (approximate median) values of 72.0 kg, 40.0 years of age, and 108.7 mL/min, respectively. Categorical covariates (sex and race defined as Asian vs non‐Asian) entered the model as 1 coefficient for each category.
Effect of Covariates
Covariates determined to have a statistically significant effect on CL/F were baseline creatinine clearance, sex, and race (Asian), and on V/F were body weight, sex, and age. Females were predicted to have a 13.2% lower CL/F compared with males. Asian patients were predicted to have a 6.8% lower CL/F compared with non‐Asian patients. A patient with a baseline creatinine clearance of 60 mL/min (minimal value designating mild impairment) was estimated to have a 19% lower CL/F compared to a patient with baseline creatinine clearance of 108.7 mL/min (median value in the analysis data set). V/F estimates for patients weighing 53 or 95 kg (corresponding to the 10th and 90th percentiles of the weight distribution in the PK data set) were approximately 16.4% lower or 17.6% higher, respectively, relative to a patient weighing 72.0 kg (median value). In addition, female patients were predicted to have a 15.5% lower V/F, compared with male patients, and V/F was estimated to be 7.7% lower for a patient aged 80 years, compared with that of a patient aged 40 years (median value).
To evaluate the effects of covariates on exposure metrics, and hence establish if tofacitinib dose adjustment would be recommended for specific subgroups of patients, steady‐state area under the concentration‐time curve (AUC), average concentration at steady state, maximum concentration at steady state (Cmax), and trough concentration at steady state were derived from the parameter estimates, generated from the estimation with the final model of 1000 nonparametric bootstrap data sets.
Data were then computed as ratios (with 90% bootstrap CI) for AUC and Cmax for each covariate of interest, relative to the reference patient.
For continuous covariates, the 10th and 90th percentile values from the population PK data set were used to calculate the expected magnitude of change at the tails of the distribution in this population.
The geometric means of AUC (%CV) for tofacitinib 5 and 10 mg twice daily were 211.3 ng·h·mL−1 (22.6) and 403.8 ng·h·mL−1 (24.6), respectively. The geometric means of Cmax (%CV) for tofacitinib 5 and 10 mg twice daily were 3.6 ng/mL (47.4) and 6.7 ng/mL (51.2), respectively.
The impact of covariates on the PK of tofacitinib 10 mg twice daily in patients with UC is presented in Figure 1, along with recommendations regarding dose adjustment. With the exception of baseline creatinine clearance, point estimates of AUC and Cmax ratios and the associated 90%CI were within the range of 80% to 125%, indicating that there were no clinically relevant differences in tofacitinib exposure over the range of covariates evaluated in the clinical studies. A patient with a creatinine clearance of 60 mL/min (minimal value designating mild impairment) was estimated to have a 19% lower CL/F compared to a patient with creatinine clearance of 108.7 mL/min (median value in the analysis data set). Dose adjustment is recommended in patients with impaired renal function in the current US tofacitinib prescribing information.
Figure 1.
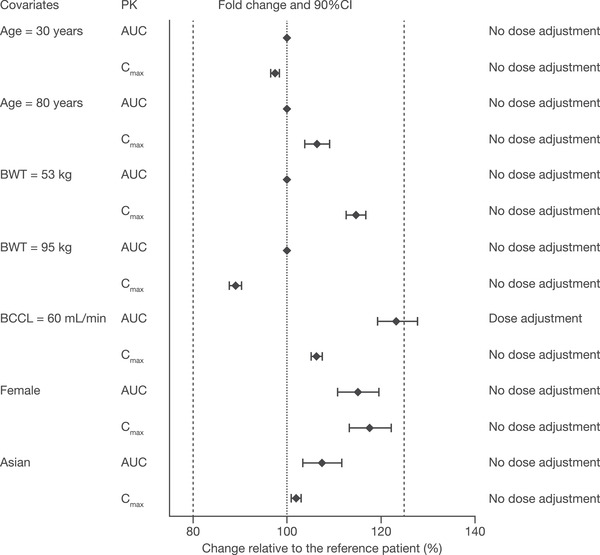
Impact of covariates on the PK of tofacitinib 10 mg twice daily in patients with UC.
The dashed lines represent the limits of a range from 80% to 125%. Data are presented as ratios (with 90%CI) for AUC and Cmax for each covariate of interest, relative to a reference patient (non‐Asian male; BWT, 72.0 kg; 40.0 years of age; baseline creatinine clearance, 108.7 mL/min). Weights of 53 kg and 95 kg are the 10th and 90th percentiles of BWT in this analysis data set. The current recommendation in US tofacitinib prescribing information is to reduce dose by half in patients with moderate and severe renal impairment. AUC, area under the concentration‐time curve over a dosing interval; BCCL, baseline creatinine clearance; BWT, body weight; CI, confidence interval; Cmax, maximum concentration at steady state; PK, pharmacokinetics; UC, ulcerative colitis.
Model Predictive Performance
Model evaluation using VPCs revealed that the final model provided a reliable description of the data. The simulated distributions matched the observed concentrations adequately, although some underprediction for peak values was observed for individuals with very rapid absorption. Although 2 different residual errors with an IIV on residual errors were introduced, the 95th percentile of observations around trough values was not always entirely covered by the 95%CI around the upper bound of the 90th prediction interval from the final model, especially for phase 3 studies. Similarly, the 5th percentile of observations for the absorption phase was also not always included by the 95%CI around the upper bound of the 90th prediction interval from the final model. Figure 2 shows the VPCs for the final model, stratified by study and dose.
Discussion
The objectives of this analysis were to describe tofacitinib PK in patients with moderate to severe UC and to estimate the effects of covariates on the variability in PK parameter estimates. A 1‐compartment model with first‐order absorption and elimination and an absorption lag was found to be an adequate and appropriate structural PK model to describe the PK of tofacitinib in a dose‐proportional manner. The typical elimination t½ in patients with UC was estimated in the model to be 3.05 h, and was consistent with previous population PK studies in other indications (Pfizer, data on file). Furthermore, these results indicated that tofacitinib plasma exposure, as measured by steady‐state AUC after tofacitinib 5 or 10 mg twice daily, was similar between UC patient populations and others such as rheumatoid arthritis (Pfizer, data on file) and psoriatic arthritis patient populations; 16 differences in geometric means were within 20%.
In patients with UC, tofacitinib exposure in individual patients did not change significantly over the duration of induction and maintenance treatment. A dose‐proportional increase in tofacitinib exposure was observed over the dose range (0.5‐15 mg twice daily) evaluated.
Based on the results of covariate analysis, tofacitinib does not require dose adjustment or restrictions for age, body weight, sex, or race (Asian vs non‐Asian) in the adult UC patient population, except in those with moderate or severe renal impairment, and this was consistent with the impact of covariates on the tofacitinib PK in other patient populations. 15 , 16 There was also no significant difference between the population with prior TNFi experience vs TNFi‐naïve patients, and no difference between patients with or without concomitant use of oral corticosteroids and 5‐ASA. Furthermore, baseline disease severity, as measured by baseline total Mayo score or extent of disease, was not a determinant of tofacitinib clearance. Additionally, baseline serum albumin was not a significant covariate, contrary to the findings of previous studies of TNFi (infliximab and adalimumab) in patients with UC and Crohn's disease, respectively, where lower levels of albumin predicted higher clearance rates 25 and poorer clinical outcomes. 26 , 27
Currently, the US tofacitinib prescribing information recommends reducing tofacitinib dose by half in patients with moderate and severe renal impairment. This recommendation is based on characterization of tofacitinib PK in healthy individuals, and those with mild, moderate, or severe renal impairment in phase 1 studies. 28 AUC values in individuals with mild, moderate, and severe renal impairment were 37%, 43%, and 123% higher than in healthy individuals, indicating that a 50% dose reduction in patients with moderate and severe renal impairment would achieve approximately the same exposure as in healthy individuals.
To date, the safety profile of tofacitinib has been reported to be generally similar to that of biological therapies. However, an increased risk of herpes zoster and venous thromboembolism has been observed, and incidence of thromboembolism in patients receiving tofacitinib, including patients with UC, has been reported. 29 , 30 , 31 , 32 , 33
In conclusion, the population PK of tofacitinib in patients with moderate to severe UC was adequately described by a 1‐compartment model parameterized in terms of CL/F, apparent V/F, and first‐order absorption with a lag time. Finally, dose adjustment or restrictions for age, body weight, sex, race, or baseline disease severity, to account for differences in exposure, are not required. This is based on <20% predicted difference in AUC or Cmax between a reference patient with a typical value and those with minimum or maximum values of each of the above patient‐specific factors in UC clinical trials.
Data Sharing Statement
Upon request, and subject to certain criteria, conditions, and exceptions (see https://www.pfizer.com/science/clinical‐trials/trial‐data‐and‐results for more information), Pfizer will provide access to individual deidentified participant data from Pfizer‐sponsored global interventional clinical studies conducted for medicines, vaccines, and medical devices (1) for indications that have been approved in the United States and/or European Union, or (2) in programs that have been terminated (ie, development for all indications has been discontinued). Pfizer will also consider requests for the protocol, data dictionary, and statistical analysis plan. Data may be requested from Pfizer trials 24 months after study completion. The deidentified participant data will be made available to researchers whose proposals meet the research criteria and other conditions, and for which an exception does not apply, via a secure portal. To gain access, data requestors must enter into a data‐access agreement with Pfizer.
Conflicts of Interest
S.W.M., R.X., C.S., and A.M. are employees and stockholders of Pfizer Inc. C.V., C.D., and K.I. were employees of Pfizer Inc at the time the study was conducted. W.J.S. reports consulting fees from AbbVie, Akros Pharma, Allergan, Ambrx Inc., Amgen, Ardelyx, Arena Pharmaceuticals, Atlantic Pharmaceuticals, Avaxia, Biogen, Boehringer Ingelheim, Bristol‐Myers Squibb, Celgene, Conatus, Cosmo Technologies, Escalier Biosciences, Ferring, Ferring Research Institute, Forward Pharma, Galapagos, Genentech, Gilead Sciences, Gossamer Biosciences, Immune Pharmaceuticals, Index Pharmaceuticals, Janssen, Kyowa Hakko Kirin Pharma, Lilly, MedImmune, Mesoblast, Miraca Life Sciences, Nivalis Therapeutics, Novartis, Nutrition Science Partners, Oppilan Pharma, Otsuka, Palatin, Paul Hastings, Pfizer Inc, Precision IBD, Progenity, Prometheus Laboratories, Qu Biologics, Regeneron, Ritter Pharmaceuticals, Robarts Clinical Trials (owned by University of Western Ontario), Salix, Seattle Genetics, Seres Therapeutics, Shire, Sigmoid Biotechnologies, Takeda, Theradiag, Theravance, TiGenix, Tillotts Pharma, UCB Pharma, Vascular Biogenics, and Vivelix; research grants from AbbVie, Amgen, Atlantic Healthcare Limited, Celgene/Receptos, Genentech, Gilead Sciences, Janssen, Lilly, and Takeda; payments for lectures/speakers bureau from AbbVie, Janssen, and Takeda; and he holds stock/stock options in Escalier Biosciences, Gossamer Biosciences, Oppilan Pharma, Precision IBD, Progenity, and Ritter Pharmaceuticals.
Funding
These studies were funded by Pfizer Inc.
Supporting information
Supporting Information
Acknowledgments
The authors thank the patients, investigators, and study teams who were involved in the tofacitinib UC clinical program. Medical writing support, under the guidance of the authors, was provided by Anthony G. McCluskey, PhD, CMC Connect, McCann Health Medical Communications and was funded by Pfizer Inc, New York, NY, USA, in accordance with Good Publications Practice guidelines (Ann Intern Med. 2015;163(6):461‐464).
Vong C, Martin SW, Deng C, et al. Population pharmacokinetics of tofacitinib in patients with moderate to severe ulcerative colitis. Clinical Pharmacology in Drug Development. 2020;00:00‐00. 10.1002/cpdd.899
References
- 1. Ordás I, Eckmann L, Talamini M, Baumgart DC, Sandborn WJ. Ulcerative colitis. Lancet. 2012;380(9853):1606‐1619. [DOI] [PubMed] [Google Scholar]
- 2. Harbord M, Eliakim R, Bettenworth D, et al. Third European evidence‐based consensus on diagnosis and management of ulcerative colitis. Part 2: current management. J Crohns Colitis. 2017;11(7):769‐784. [DOI] [PubMed] [Google Scholar]
- 3. Kornbluth A, Sachar DB, Practice Parameters Committee of the American College of Gastroenterology . Ulcerative colitis practice guidelines in adults: American College of Gastroenterology, Practice Parameters Committee. Am J Gastroenterol. 2010;105(3):501‐523. [DOI] [PubMed] [Google Scholar]
- 4. Hoffmann P, Krisam J, Stremmel W, Gauss A. Real‐world outcomes of vedolizumab therapy in ulcerative colitis and Crohn's disease at a tertiary referral center. Dig Dis. 2019;37(1):33‐44. [DOI] [PubMed] [Google Scholar]
- 5. Rubin DT, Ananthakrishnan AN, Siegel CA, Sauer BG, Long MD. ACG clinical guideline: ulcerative colitis in adults. Am J Gastroenterol. 2019;114(3):384‐413. [DOI] [PubMed] [Google Scholar]
- 6. Meyer DM, Jesson MI, Li X, et al. Anti‐inflammatory activity and neutrophil reductions mediated by the JAK1/JAK3 inhibitor, CP‐690,550, in rat adjuvant‐induced arthritis. J Inflamm (Lond). 2010;7:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rochman Y, Spolski R, Leonard WJ. New insights into the regulation of T cells by gamma(c) family cytokines. Nat Rev Immunol. 2009;9(7):480‐490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Danese S, Grisham MB, Hodge J, Telliez JB. JAK inhibition using tofacitinib for inflammatory bowel disease treatment: a hub for multiple inflammatory cytokines. Am J Physiol Gastrointest Liver Physiol. 2016;310(3):G155‐G162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dowty ME, Lin J, Ryder TF, et al. The pharmacokinetics, metabolism, and clearance mechanisms of tofacitinib, a Janus kinase inhibitor, in humans. Drug Metab Dispos. 2014;42(4):759‐773. [DOI] [PubMed] [Google Scholar]
- 10. Sandborn WJ, Ghosh S, Panés J, et al. Tofacitinib, an oral Janus kinase inhibitor, in active ulcerative colitis. N Engl J Med. 2012;367(7):616‐624. [DOI] [PubMed] [Google Scholar]
- 11. Sandborn WJ, Su C, Sands BE, et al. Tofacitinib as induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2017;376(18):1723‐1736. [DOI] [PubMed] [Google Scholar]
- 12. Lichtenstein GR, Loftus Jr EV, Wei SC, et al. DOP61 Tofacitinib, an oral, small‐molecule Janus kinase inhibitor, in the treatment of ulcerative colitis: analysis of an open‐label, long‐term extension study with up to 5.9 years of treatment. J Crohns Colitis. 2020;14(Suppl 1):S100‐S101. [Google Scholar]
- 13. Gupta P, Chow V, Wang R, et al. Evaluation of the effect of fluconazole and ketoconazole on the pharmacokinetics of tofacitinib in healthy adult subjects. Clin Pharm Drug Dev. 2013;3(1):72‐77. [DOI] [PubMed] [Google Scholar]
- 14. Byon W, Smith MK, Chan P, et al. Establishing best practices and guidance in population modeling: an experience with an internal population pharmacokinetic analysis guidance. CPT Pharmacometrics Syst Pharmacol. 2013;2:e51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ma GL, Xie R, Ito K, et al. Population pharmacokinetics of tofacitinib in adult patients with moderate to severe chronic plaque psoriasis. Abstract number 2977967 presented at the World Congress of Dermatology, Vancouver, Canada, June 8–13, 2015.
- 16. Xie R, Deng C, Wang Q, Kanik KS, Nicholas T, Menon S. Population pharmacokinetics of tofacitinib in patients with psoriatic arthritis. Int J Clin Pharmacol Ther. 2019;57(9):464‐473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mukherjee A, Hazra A, Smith MK, et al. Exposure‐response characterization of tofacitinib efficacy in moderate to severe ulcerative colitis: results from a dose‐ranging phase 2 trial. Br J Clin Pharmacol. 2018;84(6):1136‐1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Krishnaswami S, Boy M, Chow V, Chan G. Safety, tolerability, and pharmacokinetics of single oral doses of tofacitinib, a Janus kinase inhibitor, in healthy volunteers. Clin Pharm Drug Dev. 2015;4(2):83‐88. [DOI] [PubMed] [Google Scholar]
- 19. Krishnaswami S, Wang T, Yuan Y, et al. Single‐ and multiple‐dose pharmacokinetics of tofacitinib in healthy Chinese volunteers. Clin Pharmacol Drug Dev. 2015;4(5):395‐399. [DOI] [PubMed] [Google Scholar]
- 20. Beal S, Sheiner LB, Boeckmann AJ, Bauer RJ. NONMEM User's Guides (1989–2009). Ellicott City, MD: Icon Development Solutions; 2009. [Google Scholar]
- 21. Paul C, Bushmakin AG, Cappelleri JC, Mallbris L, Mamolo C. Do patients and physicians agree in their assessment of the severity of psoriasis? Insights from tofacitinib Phase 3 clinical trials. J Dermatolog Clin Res. 2015;3(3):1048. [Google Scholar]
- 22. Lindbom L, Pihlgren P, Jonsson EN. PsN‐Toolkit ‐ a collection of computer intensive statistical methods for non‐linear mixed effect modeling using NONMEM. Comput Methods Programs Biomed. 2005;79(3):241‐257. [DOI] [PubMed] [Google Scholar]
- 23. Lindbom L, Ribbing J, Jonsson EN. Perl‐speaks‐NONMEM (PsN) ‐ a Perl module for NONMEM related programming. Comput Methods Programs Biomed. 2004;75(2):85‐94. [DOI] [PubMed] [Google Scholar]
- 24.PsN4 website. Perl‐speaks‐NONMEM (PsN4) ‐ Documentation. https://uupharmacometrics.github.io/PsN/docs.html. Accessed June 1, 2020.
- 25. Fasanmade AA, Adedokun OJ, Ford J, et al. Population pharmacokinetic analysis of infliximab in patients with ulcerative colitis. Eur J Clin Pharmacol. 2009;65(12):1211‐1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sharma S, Eckert D, Hyams JS, et al. Pharmacokinetics and exposure‐efficacy relationship of adalimumab in pediatric patients with moderate to severe Crohn's disease: results from a randomized, multicenter, phase‐3 study. Inflamm Bowel Dis. 2015;21(4):783‐792. [DOI] [PubMed] [Google Scholar]
- 27. Fasanmade AA, Adedokun OJ, Olson A, Strauss R, Davis HM. Serum albumin concentration: a predictive factor of infliximab pharmacokinetics and clinical response in patients with ulcerative colitis. Int J Clin Pharmacol Ther. 2010;48(5):297‐308. [DOI] [PubMed] [Google Scholar]
- 28. Krishnaswami S, Chow V, Boy M, Wang C, Chan G. Pharmacokinetics of tofacitinib, a Janus kinase inhibitor, in patients with impaired renal function and end‐stage renal disease. J Clin Pharmacol. 2014;54(1):46‐52. [DOI] [PubMed] [Google Scholar]
- 29. Sandborn WJ, Panes J, D'Haens GR, et al. Safety of tofacitinib for treatment of ulcerative colitis, based on 4.4 years of data from global clinical trials. Clin Gastroenterol Hepatol. 2019;17(8):1541‐1550. [DOI] [PubMed] [Google Scholar]
- 30. Winthrop KL. The emerging safety profile of JAK inhibitors in rheumatic disease. Nat Rev Rheumatol. 2017;13(5):320. [DOI] [PubMed] [Google Scholar]
- 31. Winthrop KL, Melmed GY, Vermeire S, et al. Herpes zoster infection in patients with ulcerative colitis receiving tofacitinib. Inflamm Bowel Dis. 2018;24(10):2258‐2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sandborn WJ, Panés J, Sands BE, et al. Venous thromboembolic events in the tofacitinib ulcerative colitis clinical development programme. Aliment Pharmacol Ther. 2019;50(10):1068‐1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mease P, Charles‐Schoeman C, Cohen S, et al. Incidence of venous and arterial thromboembolic events reported in the tofacitinib rheumatoid arthritis, psoriasis and psoriatic arthritis development programmes and from real‐world data. Ann Rheum Dis. 2020;79(11):1400‐1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information