

Summary
Allopolyploidisation merges evolutionarily distinct parental genomes (subgenomes) into a single nucleus. A frequent observation is that one subgenome is ‘dominant’ over the other subgenome, often being more highly expressed.
Here, we ‘replayed the evolutionary tape’ with six isogenic resynthesised Brassica napus allopolyploid lines and investigated subgenome dominance patterns over the first 10 generations postpolyploidisation.
We found that the same subgenome was consistently more dominantly expressed in all lines and generations and that >70% of biased gene pairs showed the same dominance patterns across all lines and an in silico hybrid of the parents. Gene network analyses indicated an enrichment for network interactions and several biological functions for the Brassica oleracea subgenome biased pairs, but no enrichment was identified for Brassica rapa subgenome biased pairs. Furthermore, DNA methylation differences between subgenomes mirrored the observed gene expression bias towards the dominant subgenome in all lines and generations. Many of these differences in gene expression and methylation were also found when comparing the progenitor genomes, suggesting that subgenome dominance is partly related to parental genome differences rather than just a byproduct of allopolyploidisation.
These findings demonstrate that ‘replaying the evolutionary tape’ in an allopolyploid results in largely repeatable and predictable subgenome expression dominance patterns.
Keywords: Brassica napus, DNA methylation, genomic shock, hybridisation, polyploidy, rapeseed, subgenome dominance
Introduction
Hybridisation among closely related species is a widespread and recurrent evolutionary process (Arnold & Meyer, 2006; Mallet, 2007; Soltis & Soltis, 2009). By merging the genomes of independently evolved species into a single nucleus, hybridisation creates a unique opportunity for immense variability that natural selection can act upon in subsequent generations (Anderson & Stebbins, 1954; Rieseberg et al., 2003). Hybridisation is known to produce transgressive phenotypes, including heterosis and novel phenotypic variation not observed in the parents (Pires et al., 2004; Dittrich‐Reed & Fitzpatrick, 2013). However, the hybridisation of highly diverged genomes, particularly those with different base chromosome numbers, can also lead to chromosome pairing issues during meiosis, which greatly reduces fertility. Proper bivalent pairing of homologous chromosomes in such interspecific hybrids can be restored through whole genome duplication (i.e. polyploidisation), resulting in the formation of an allopolyploid species (Charron et al., 2019). This may in part explain the high prevalence of polyploidy across flowering plants (Leitch & Leitch, 2008; Van de Peer et al., 2009e Peer et al., 2009) including the multiple origins of various naturally established allopolyploids (e.g. Tragopogon mirus and Tragopogon miscellus; Soltis et al., 2012), Mimulus peregrinus; Vallejo‐Marín et al., 2015), and Elymus caninus, (Yan & Sun, 2012). Several cultivated species, including rapeseed (Brassica napus; Chalhoub et al., 2014), cotton (Gossypium hirsutum; Wendel & Cronn, 2003), and wheat (Triticum aestivum; International Wheat Genome Sequencing Consortium, 2014), among many others (Leitch & Leitch, 2008), are also allopolyploids.
Genome‐scale analyses of recent and ancient allopolyploids led to the discovery that one of the parental species’ genomes (referred to as subgenomes) often exhibits greater gene retention (Thomas, 2006), more tandem gene duplications (Edger et al., 2019), higher gene expression (Schnable et al., 2011) and lower DNA methylation (Woodhouse et al., 2014). Collectively this phenomenon is referred to as ‘subgenome dominance’. The expression bias associated with subgenome dominance has the potential to greatly impact the observed phenotypic variation within a crop or species. In maize, the dominant subgenome contributes more to trait heritability than the nondominant subgenome (Simon et al., 2017) and in strawberry, the dominant subgenome largely controls several biological pathways related to agriculturally valuable traits like fruit flavour, colour, and aroma (Edger et al., 2019). A previous study demonstrated that subgenome dominance at the gene expression level can occur at the moment of interspecific hybridisation and increases over subsequent generations in the allopolyploid (Edger et al., 2017). This finding agrees with theoretical work of transcription factor binding and regulatory mismatch that predicts increasing subgenome dominance over generations in newly established allopolyploids (Bottani et al., 2018). Pre‐existing differences between parental genomes have been shown to influence these observed subgenome dynamics in allopolyploids (Buggs et al., 2014; Kryvokhyzha et al., 2019). For example, analyses of diverse allopolyploid species have revealed that gene expression differences among subgenomes mirrors differences in transposable element (TE) densities in flanking regions surrounding genes (Freeling et al., 2012; Cheng et al., 2016; Edger et al., 2019). These findings collectively suggest that subgenome dominance may be largely predetermined based on subgenome differences in certain genomic features including TE densities. This tendency for subgenome expression dominance patterns to mirror parental differences in some species has been termed ‘parental legacy’ (Buggs et al., 2014).
Given that gene expression level dominance may occur instantly following the initial hybridisation event (Edger et al., 2017), resynthesised allopolyploids are the ideal system to investigate the establishment and escalation of subgenome dominance. Few studies have used multiple independently derived resynthesised allopolyploids to investigate subgenome dominance (Chagué et al., 2010; Combes et al., 2015; Hao et al., 2017; Wu et al., 2018; Gaebelein et al., 2019; Li et al., 2019). It remains unclear the extent to which the emergence of subgenome expression dominance is a result of pre‐existing characteristics of the diploid progenitors or independent and nonrecurrent events during polyploid formation. In other words, if we were to replay the tape of life (Gould, 1991) will multiple independently established allopolyploids consistently exhibit the same patterns of subgenome expression dominance (e.g. towards the same subgenome)?
Here we analysed subgenome dominance in six independent resynthesised allopolyploid Brassica napus (2n = 4x = 38) lines formed by hybridising two doubled haploid parents from the progenitor species Brassica rapa (AA; 2n = 2x = 20) and Brassica oleracea (CC; 2n = 2x = 18) (Song et al., 1995). The crop B. napus was formed between 7500 and 12 500 yr ago and is widely grown present‐day as an oilseed crop (rapeseed), vegetable fodder crop (rutabaga) and vegetable crop (Siberian kale) (Chalhoub et al., 2014; An et al., 2019). The strengths of the B. napus polyploid system include not only having high‐quality reference genomes for both diploid progenitors and B. napus, but also being closely related to the model plant Arabidopsis thaliana, allowing for the integration of diverse genomic and bioinformatic resources (Cheng et al., 2012; Chalhoub et al., 2014; Parkin et al., 2014; Koenig & Weigel, 2015). Furthermore, a previous analysis of the B. napus reference genome identified a greater number of retained genes in the B. oleracea (BnC) subgenome compared to the B. rapa (BnA) subgenome, though the overall number of genes lost was small (Chalhoub et al., 2014). This is consistent with patterns observed in older allopolyploids that exhibit subgenome dominance, where the dominant subgenome retains more genes (Bird et al., 2018). Lastly, because the resynthesised B. napus lines were made with doubled haploids, each of the independent lines started out genetically identical (Ren et al., 2017). This permitted us to examine and compare the establishment of subgenome dominance across independently derived polyploid lines without the added influence of allelic variation segregating between different lines. We surveyed gene expression and methylation dynamics in each of the six resynthesised polyploid lines over 10 generations, which allowed with RNA‐seq and Bisulfite‐seq data to characterise gene expression and methylation differences between high confidence homoeologs over the first 10 generations. This permitted us to assess the variability of subgenome dominance during the earliest stages after polyploid formation.
Materials and Methods
Plant growth, tissue collection, library prep
The resynthesised B. napus allopolyploid lines (CCAA) were obtained from a previous study (Xiong et al., 2011). Plants were grown under 23°C : 20°C, 16 h : 8 h, day : night cycles in a growth chamber. True leaf three was collected from all plants within 1 h, starting at 10 am (4 h into the day) and immediately flash‐frozen in liquid nitrogen. Leaves were split in half for RNA and DNA isolation. Total RNA and DNA was isolated using the respective KingFisher Pure Plant kits (Thermo Fisher Scientific, Waltham, MA, USA) and quantified using a Qubit 3 fluorometer (Thermo Fisher Scientific). DNA and RNA libraries were prepared using the KAPA HyperPrep and mRNA HyperPrep kit protocols, respectively (KAPA Biosystems, Roche, USA). Bisulfite conversion was performed using the EZ DNA Methylation Kit (Zymo, Irvine, CA, USA). All libraries were submitted to a genomics facility (Beijing Nuohe Zhiyuan Technology Corp., Beijing, China) and sequenced with paired‐end 150‐bp reads on an Illumina HiSeq 4000 system.
In silico reference genome construction
Paired‐end 150‐bp Illumina reads for the doubled haploid Brassica rapa accession IMB‐218, were aligned to the Brassica rapa R500 reference genome using Bowtie2 v.2.3.4.1 (Langmead & Salzberg, 2012) on default settings with the flag ‘‐‐very‐sensitive‐local’. The resulting alignment files were sorted and had read groups added with Picardtools v.2.8.1. Single nucleotide polymorphisms (SNPs) were called between the R500 reference and the IMB‐218 alignment using Gatk v.3.5.0 Unified Genotyper, filtered to only include homozygous SNPs, and a new fasta reference was made using Gatk v.3.5.0 FastaAlternativeReferenceMaker. This IMB‐218 reference genome was concatenated to the B. oleracea TO1000 reference genome to create an in silico reference genome for B. napus matching the two progenitor used in our study.
Homoeologous exchange analysis
Paired‐end 150‐bp genomic Illumina reads were filtered with Trimmomatic v.0.33 (Bolger et al., 2014) to remove Illumina TruSeq3 adapters. Trimmed reads were aligned to the in silico B. napus reference genome with Bowtie2 v.2.3.4.1(Langmead & Salzberg, 2012) on default settings with the flag ‘‐‐very‐sensitive‐local’. BAM files were sorted using Bamtools (Barnett et al., 2011) for use in downstream analyses.
The MCScan toolkit (Tang et al., 2008) was used to identify syntenic, homologous gene pairs (syntelogs) between Brassica rapa (reference genome R500) and Brassica oleracea (reference genome TO1000; Parkin et al., 2014). In the synthetic polyploid these can be thought of as syntenic homoeologs. BED files based on the chromosome and start/stop position information for each subgenome were generated. For all 18 samples (6 individuals × 3 generations) read depths for the A subgenome (BnA) syntenic homoeologs were determined in Bedtools (Quinlan & Hall, 2010) with Bedcov using the R500 syntelog BED file and for the C subgenome (BnC) using the TO1000 syntelog BED file. In R v.3.4.1, read depths for each syntenic homoeolog were normalised to reads per million for subgenome of origin and the ratio of reads mapping to a syntenic homoeolog compared to the overall read mapping for a syntenic homoeolog pair was averaged over a window of 50 genes with a step of one gene.
Homoeologous exchanged regions were identified by calculating average read depth for the BnC subgenome along a sliding window of 170 (85 upstream and downstream) genes and step size of one. If 10 or more consecutive genes had read depths within a preselected range it was called a homoeologous exchange. Regions 0 ≤ read depth < 0.2 were predicted to be in a 0BnC‐to‐4BnA ratio, 1BnC‐to‐3BnA was predicted for 0.2 ≤ read depth < 0.4, 2BnC‐to‐2BnA was predicted for 0.4 ≤ read depth < 0.6, 3BnC‐to‐1BnA for read depth between 0.6 ≤ read depth < 0.8 and 4BnC‐to‐0BnA for read depth between 0.8 ≤ read depth < 1.
RNA‐seq analysis
Raw RNA‐seq reads were filtered using Trimmomatic v.0.33 (Bolger et al., 2014) to remove Illumina TruSeq3 adapters and mapped to the in silico reference using Star v.2.6.0 (Dobin et al., 2013) on default settings. Transcripts were quantified in transcripts per million (TPM) from RNA‐seq alignments using Stringtie v.1.3.5 (Pertea et al., 2015). Because the syntelogs in the progenitor genomes are in the subgenomes of the synthetic polyploids, they can be thought of as syntenic homoeologs. To avoid dosage imbalance, only syntenic homoeologs determined to be at a 2 : 2 dosage balance were analysed for homoeolog expression bias. Additionally, to remove lowly expressed genes that might be noise, syntenic homoeologs were only kept if the total TPM of the pair was > 10. Syntenic homoeolog pairs with log2 fold change (FC) > 3.5 were called BnC biased, and < 3.5 were called BnA biased. This cutoff follows the practice of Woodhouse et al. (2014) who used a log FC cutoff of 2 to determine homoeolog expression bias, however to more confidently reduce false positives a higher FC cutoff of 3.5 was used. Because the lack of subgenome dominance would follow a normal distribution in which deviations from 0 FC are equal in either direction, a chi‐squared goodness of fit test was carried out to test for normality. The R package upsetr was used to identify and plot syntenic homoeologs shared by all lines for a given generation. Intermediate files and code to reproduce plots can be found at https://doi.org/10.5061/dryad.h18931zjr. For each generation, Arabidopsis thaliana orthologues were identified for genes showing the same subgenome bias in all six lines and the progenitors and were investigated for Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment (Ashburner et al., 2000; Kanehisa & Goto, 2000) in the STRING PPI network (Szklarczyk et al., 2017) using the online STRING network search application. STRING also calculated and reported average node degree, clustering coefficients and enrichment for network interactions.
DNA methylation analysis
Whole genome bisulfite sequencing (WGBS) data was mapped to the combined in silico reference genome using Methylpy v.1.3.8 (Schultz et al., 2015); using Cutadapt v.2.3 (Martin, 2011) for adaptor trimming, Bowtie2 v.2.3.5 (Langmead & Salzberg, 2012) for alignment, and Picard tools v.2.20.2 for marking duplicates. The chloroplast genome is unmethylated in plants and was used as an internal control for calculating the nonconversion rate of bisulfite treatment, the percentage of unmethylated sites that fail to be converted to uracil (Lister et al., 2008). Methylpy accounts for this nonconversion in calling methylated sites.
When the parental WGBS data were mapped to the combined genome (TO1000 + R500), a small fraction of reads of each sample mapped to the other subgenome, c. 1.3% TO1000 to B. rapa and c. 6.1% IMB218 to B. oleracea. We compared results from mapping of the parental data to either the combined genome or their own respective genome. There was little difference in DNA methylation levels or patterns for either parent and we therefore concluded that the impact of this mismapping was insignificant. As a further control, we created a ‘mock’ allopolyploid in silico. The TO1000 data were randomly downsampled to an equal number of read pairs as for IMB218. The two datasets were combined and mapped to the combined genome to mimic an in silico allopolyploid. DNA methylation levels in this ‘mock’ allopolyploid were either about half‐way between the two parents for the whole genome or nearly identical to their respective parent at a subgenome level. If DNA methylation in the resynthesised lines is simply a combination of both parent’s methylomes, then we expected global DNA methylation in the resynthesised lines to be similar to this combined mock dataset. Deviation from this pattern would indicate global remodelling of DNA methylation.
Genome‐wide levels of DNA methylation and DNA methylation metaplots were analysed as previously described (Niederhuth et al., 2016) using Python v.3.7.3, Pybedtools v.0.8 (Dale et al., 2011), and Bedtools v.2.25.0 (Quinlan & Hall, 2010). Genome‐wide DNA methylation levels were calculated for each sequence context (CG, CHG, and CHH) using the weighted methylation level (Schultz et al., 2012), which accounts for sequencing coverage. For gene metaplots, cytosines from 2 kilobase (kb) upstream, 2 kb downstream and within the gene/TE body were extracted. For gene bodies, only cytosines in coding sequences were used, as the presence of TEs in introns and problems of proper UTR annotation can obscure DNA methylation at start/stop sites and introduce misleadingly high levels of DNA methylation (Niederhuth et al., 2016). Each of these three regions (upstream, gene body, and downstream) were then divided into 20 windows and the weighted methylation level for each window calculated and averaged for all genes. for long‐terminal repeat (LTR) metaplots, the same analysis was performed, except all cytosines within the LTR body were included. plot were made in R v.3.6.0 (R Core team, 2018) using Ggplot2 (Wickham, 2009). All code and original analysed data and plots are available on github (https://github.com/niederhuth/replaying‐the‐evolutionary‐tape‐to‐investigate‐subgenome‐dominance).
Results
Homoeolog expression bias
This population of resynthesised polyploids provided a unique opportunity to examine if the same subgenome would repeatedly exhibit subgenome expression dominance. Gene expression in leaves was surveyed using RNA‐seq for 16 of the 18 resequenced individuals (six lines and three generations). Library construction failed for two individuals, therefore these were not able to be included in this analysis. However, representative generations from all six lines were included in these sets of analyses. Samples were aligned to an in silico polyploid reference genome. We restricted gene expression analyses to genomic regions with balanced gene dosage (2 : 2; AA : CC) identified using genome resequencing data for each individual to reduce the confounding factor of dosage changes in regions that have undergone homoeologous exchange. Expression patterns of the six lines were also compared with the parental B. rapa and B. oleracea genotypes to test if expression differences may exist among the diploid progenitors.
The mean expression bias (log2 FC BnC expression/BnA expression) for homoeologs in balanced (2 : 2) regions ranged from 0.12 to 1.16 (median −0.50 to 0.96), with 15 of 16 individuals having mean expression bias significantly > 0 (one‐way Wilcoxon‐Mann–Whitney test, P < 2.2e−16; Fig. 1, Supporting Information Figs S1–S5). These results suggested a transcriptome‐wide bias toward the BnC subgenome, however the magnitude of the expression bias was smaller than that observed previously in other allopolyploids (Edger et al., 2017). The homoeolog expression bias between the parents was also significantly > 0 in these balanced regions. Comparing the bias difference between the parental lines and the synthetic polyploids revealed that only 5 of the 16 were significantly different from the parents (two‐way Wilcoxon–Mann–Whitney test, P < 0.001).
Fig. 1.
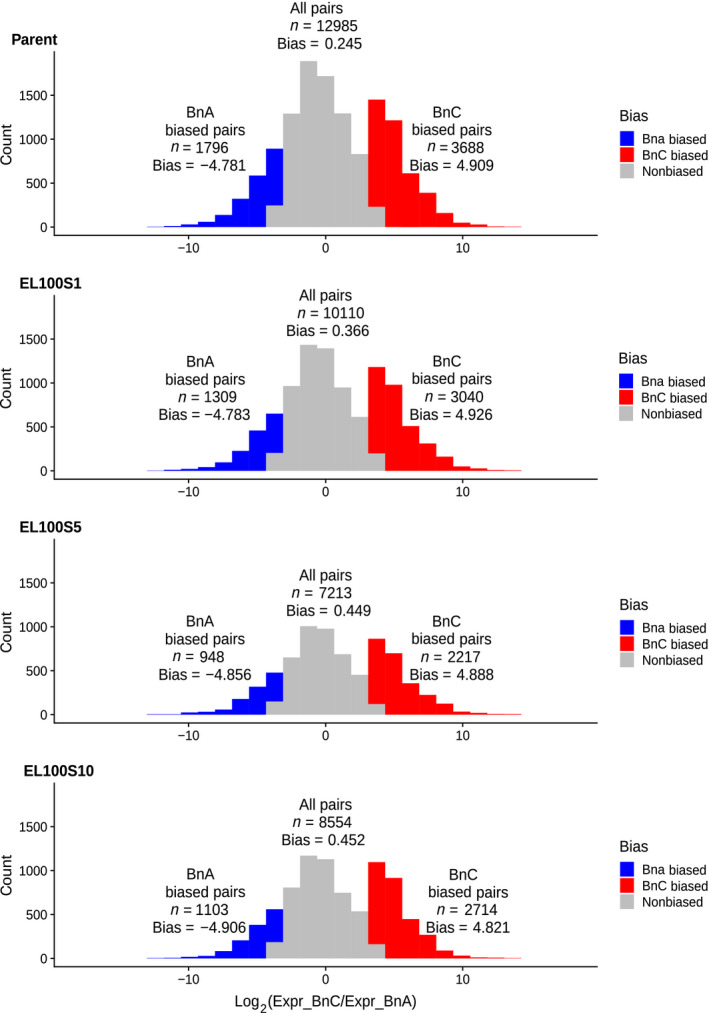
Homoeolog expression bias in resynthesised Brassica napus. Distribution of homoeolog bias in the parent and three generations of line EL100. Red regions indicate BnC‐biased homoeologs with log2 expression fold change > 3.5 and blue regions indicate BnA‐biased homoeologs with log2 expression fold change < −3.5.
We examined homoeolog pairs that were biased toward either subgenome (log2 FC > |3.5|) and found that there were significantly more BnC‐biased homoeolog pairs than expected in all individuals (χ2‐test, χ2 > 170, df = 1, P < 2.2e−16; Figs 1, S1–S5; Table S1). This indicated that there was a bias in homoeolog expression on a gene‐by‐gene basis towards the BnC subgenome. This BnC homoeolog bias was consistent across generations. We also found that homoeolog bias in the synthetic polyploids was significantly different than existing expression bias in the parents for 7 of 16 individuals (χ2 test, χ2 > 12.459, df = 2, P < 0.003125; Figs 1, S1–S5; Table S2). In five of those seven individuals, there were more BnC‐biased homoeologs than expected based on expression biases of the progenitor genomes. A bimodal distribution was observed when comparing BnA and BnC subgenome expression, the rightmost distribution being largely due to homoeolog pairs with a lack of BnA expression.
Next, we investigated whether individual gene pairs were biased in the same direction across the six lines. Due to the stochastic nature of homoeologous exchanges, dosage of a gene pair may differ between lines. To adjust for this, we first looked only at genes found in 2 : 2 dosage for all lines in a generation, resulting in 6917, 3574 and 2252 homoeologous pairs for generations S1, S5 and S10, respectively. The reduction in 2 : 2 homeologs over time suggested that homoeologous exchange is a dynamic and variable process across independent lines and that it reaches some stability or equilibrium over successive generations. The frequency of homoeologous exchanges across each chromosome of this synthetic B. napus population was previously reported based on cytogenetic analyses (Xiong et al., 2011). A majority of BnC‐biased gene pairs was biased toward the BnC subgenome in all synthetic lines for each generation and in the parents (S1 = 1806 (75%), S5 = 772 (71%), and S10 = 602 (70%); Fig. 2a–c). In the 1st generation, 36 gene pairs (1.5%) were uniquely dominant in only the parents and 32 (1.3%) were dominant in all six synthetic lines but not the parent (Fig. 2a). In the 5th and 10th generation, there was a similar number of BnC dominant homoeologs that were dominant in only the parents (S5 = 17 (1.5%), S10 = 14 (1.6%)) and dominant in all six lines but not the parent (S5 = 17 (1.5%), S10 = 13 (1.5%); Fig. 2c). Similar patterns were observed for BnA‐biased homoeologs with most genes showing similar bias in all lines across generations and the parents (S1 = 698 (61%), S5 = 401 (58%), S10 = 221 (51%), respectively, Fig. S6a–c), and a consistently low number of genes biased in all six lines but not the parents in each generation (S1 = 28 (2.5%), S5 = 15 (2.2%), S10 = 10 (2.3%)) and only biased in the parents (S1 = 41 (3.6%), S5 = 22 (3.2%), S10 = 18 (4.1%); Fig. S6a–c).
Fig. 2.
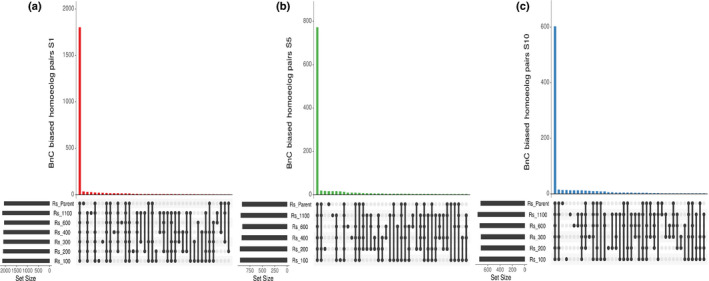
Common shared biased homoeolog pairs in resynthesised Brassica napus. UpSet plot showing BnC‐biased homoeologs pairs for generations 1 (a), 5 (b), and 10 (c) that are shared or unique among comparisons across all six lines for the three sampled generations. This analysis was restricted only to homoeolog pairs in 2 : 2 balance in all six lines.
Lastly, we performed GO (Ashburner et al., 2000), KEGG pathway (Kanehisa & Goto, 2000) and network enrichment analyses in the Arabidopsis thaliana protein–protein interaction network (Szklarczyk et al., 2017) to determine if Arabidopsis thaliana homologues of BnC and BnA‐biased gene pairs were enriched with certain biological functions. Placing biased homoeologs in a network context allowed us to determine the extent to which the genes interact with each other, the average number of connections a gene has with other genes (average node degree), and the extent to which genes are clustered together in the network. In all generations, BnC‐biased gene pairs showed enrichment for interactions in the PPI network in the 1st, 5th and 10th generations (all three generations; P < e−16) with high average node degree (S1 = 20.6, S5 = 12.1, S10 = 7.99) and clustering coefficient (S1 = 0.386, S5 = 0.385, S10 = 0.385). GO terms for BnC‐biased gene pairs were highly enriched with core metabolic processes, including photosynthesis and organellar and ribosomal functions. For example, in the 1st generation, 263 genes were annotated as being active in the chloroplast (Table S3). BnC‐biased gene pairs were also significantly overrepresented in KEGG pathways annotated for amino acid biosynthesis, MAPK signalling and photosynthesis (Tables S3–S5). The enrichment for organellar functions for BnC‐biased pairs may be expected, given that the BnC subgenome is the maternal progenitor of the resynthesised lines. This suggests that subgenome dominance may, in part, be related to maintenance of balanced nuclear–organellar interactions. By contrast, BnA‐biased gene pairs showed no enrichment for any GO terms or KEGG pathways. However, there was an observed enrichment for interactions in the PPI network for BnA‐biased gene pairs in generations 1 (P = 9.75e−07) and 5 (P = 0.00681), but not generation 10 (P = 0.16). Lower network statistics were also observed for average node degree (S1 = 1.13, S5 = 0.912, S10 = 0.338) and average clustering coefficient (S1 = 0.270, S5 = 0.235, S10 = 0.209) for BnA‐biased gene pairs. In summary, subgenome expression dominance in our resynthesised Brassica napus was biased towards a set of highly interconnected BnC genes that were enriched for a wide variety of biological processes.
DNA methylation
Cytosine methylation is involved in defining regions of chromatin, silencing TEs, maintaining genome integrity and can affect gene expression. DNA methylation itself is shaped by factors such as gene expression and the underlying DNA sequence (Niederhuth & Schmitz, 2017). To understand how DNA methylation evolves following polyploidy and may contribute to subgenome dominance, DNA methylation was assessed using WGBS in the parents and allopolyploids (Cokus et al., 2008; Lister et al., 2008). Three sequence contexts are generally recognised for DNA methylation in plants: dinucleotide CG (or CpG), and trinucleotide CHG and CHH (where H = A, T or C). Methylation of these contexts is established and maintained by different molecular pathways and their associations with gene expression differs based on the pattern of DNA methylation (Niederhuth & Schmitz, 2017). Coding sequences methylated in all three contexts (CG, CHG and CHH) are associated with transcriptional silencing. By contrast, so‐called gene body methylated (gbM) genes exhibited relatively high and broad expression patterns. GbM is a specific pattern of CG‐only DNA methylation in gene bodies, with depletion at the transcriptional start site (TSS) and termination site. Finally, unmethylated genes showed variable expression patterns and were often developmentally and environmentally responsive genes. Regardless of context, depletion of methylation around the TSS appears to be important for transcription as even CG methylation in this region is associated with lower expression.
In line with current conceptual models of subgenome dominance (Woodhouse et al., 2014; Garsmeur et al., 2014; Bird et al., 2018; Alger and Edger, 2020), we might expect to see generally lower methylation, especially in the CHH context, upstream of genes or in TEs in the dominant subgenome (Edger et al., 2017, 2019; Renny‐Byfield et al., 2017; Zhao et al., 2017). The predicted mechanism at play is that silencing of TEs through the RNA‐directed DNA methylation pathway affects expression of nearby genes, as part of the trade‐off described by Hollister and Gaut (2009). Although as noted above, these are not the only methylation patterns that can explain expression differences among homoeolog pairs.
In all methylation contexts, the BnC subgenome progenitor showed higher methylation levels than the BnA subgenome progenitor (Figs 3, 4). For genes, CG methylation levels are predominately at mid‐parent levels, but appear to increase over time, most notably upstream and downstream of gene bodies. By the 10th generation, some lines showed upstream and downstream CG methylation closer to the BnC progenitor, while others showed methylation levels in line with the mid‐parent level. CG methylation of LTR retrotransposons showed a similar trend as the genes, with 1st generation methylation levels matching mid‐parent levels and increasing over time, particularly in the LTR body (Figs 3, 4). In the 1st generation, CHG methylation fell between the mid‐parent value and the BnA progenitor for both genes and LTRs. In the 5th and 10th generations, CHG methylation progressively increased to more reflect the mid‐parent value. As with CG methylation, at the 10th generation, we saw two clusters, one with CHG methylation levels above the mid‐parent value and one with methylation below the mid‐parent value in the upstream and downstream regions of genes (Figs 3, 4). CHH methylation patterns were even more striking. In genes and LTRs, the 1st generation shows methylation levels between the mid‐parent and the BnC progenitor, with methylation increasing and surpassing the BnC progenitor levels in the 5th and 10th generation (Figs 3, 4). As with CG and CHG methylation, increased variation in the flanking regions by the 10th generation is observed, however all lines still showed a consistently higher methylation than the BnC progenitor.
Fig. 3.
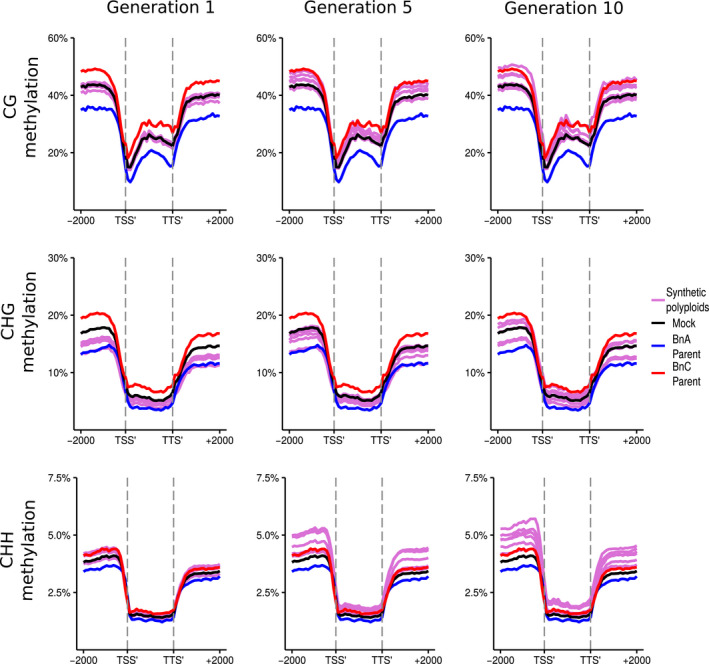
DNA methylation of genes in resynthesised Brassica napus. Metaplots of CG, CHG and CHH mean weighted methylation of all annotated gene models for generations 1, 5 and 10 assessed using Bisulfite‐seq. The six resynthesised lines are shown in purple, the Brassica oleracea progenitor is shown in red, Brassica rapa progenitor in blue, and the in silico mock polyploid in black. Methylation levels are shown for the transcription start site (TSS′), gene body, transcription termination site (TTS′) and 2 kb upstream and downstream of the TSS and TTS.
Fig. 4.
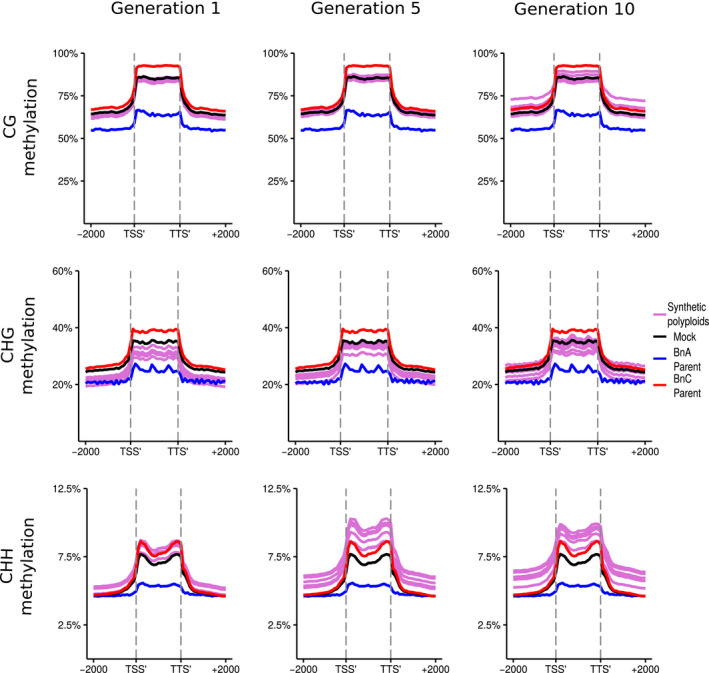
DNA methylation of long‐terminal repeat (LTR) TEs in resynthesised Brassica napus. Metaplots of CG, CHG and CHH mean weighted methylation of all annotated LTR TEs for generations 1, 5 and 10 assessed using Bisulfite‐seq. The six resynthesised lines are shown in purple, the Brassica oleracea progenitor is shown in red Brassica rapa progenitor in blue, and the in silico mock polyploid in black. Methylation levels are shown for the transcription start site (TSS'), gene body, transcription termination site (TTS') and 2 kb upstream and downstream of the TSS and TTS.
Analysed by subgenome, methylation levels in the resynthesised lines were on average lower for the BnA subgenome than the BnC subgenome for every methylation context and generation observed (Figs 5, 6). Additionally, there was a more pronounced increase in CG, CHG, and CHH methylation levels at the boundaries of flanking regions and the LTR body for the BnC subgenome compared with the BnA subgenome (Figs 7 ,8). Syntenic genes are those that have remained in identical positions in the respective subgenomes, identified based on the collinearity of annotated genes across a region in the genome. Comparing syntenic genes to nonsyntenic genes, CG, CHG and CHH methylation levels in gene bodies were both higher and more variable for nonsyntenic genes. Differences in the changes in gene methylation levels across generations were also observed. From generations 1–10, individuals showed increased CG methylation in gene bodies and flanking regions for BnC genes, with some exceeding progenitor methylation levels (Fig. 5), but little increase was observed for BnA genes (Fig. 6). In genic regions on the BnC subgenome, CG methylation increase occurred primarily for nonsyntenic genes (Fig. S8), however in the flanking regions, CG methylation increases occurred for both syntenic and nonsyntenic genes (Figs S7–S10). For CHG sites, average methylation levels in the 1st generation, samples were below progenitor levels, both in the bodies and in flanking regions of genes, and increased in subsequent generations with some lines equal to the progenitor level on the BnA subgenome and some exceeding the progenitor level on the BnC subgenome (Figs 5, 6). Syntenic genes showed both less variability and lack of increase across generations in CHG gene body methylation compared with nonsyntenic genes for both the BnA and BnC subgenomes, while patterns for flanking regions of genes were largely the same for syntenic and nonsyntenic genes (Figs S7–S10). For LTRs, the same pattern of methylation increase over generations was observed, however LTRs on the BnC subgenome do not exceed progenitor methylation levels, while some lines showed BnA subgenome methylation exceeding the progenitor (Figs 7, 8). CHH methylation showed the most striking differences across generations. First generation allopolyploids showed flanking and gene body CHH methylation on the BnC subgenome that was slightly higher than the BnC progenitor methylation levels and BnA subgenome methylation equal to or below the BnA progenitor (Figs 5, 6). In subsequent generations, CHH methylation in the flanking regions and gene bodies increased to surpass the progenitors on both BnA and BnC subgenomes, with the BnC subgenome showing the largest increase in methylation levels over time (Figs 5, 6). As with CG and CHG methylation, syntenic genes did not show an increase in gene body CHH methylation, while nonsyntenic genes did. CHH methylation of LTRs started around or below progenitor levels and increased to exceed progenitor methylation levels in both BnA and BnC subgenomes by the 5th and 10th generations, although the increase was much larger for the BnC subgenome (Figs 7, 8).
Fig. 5.
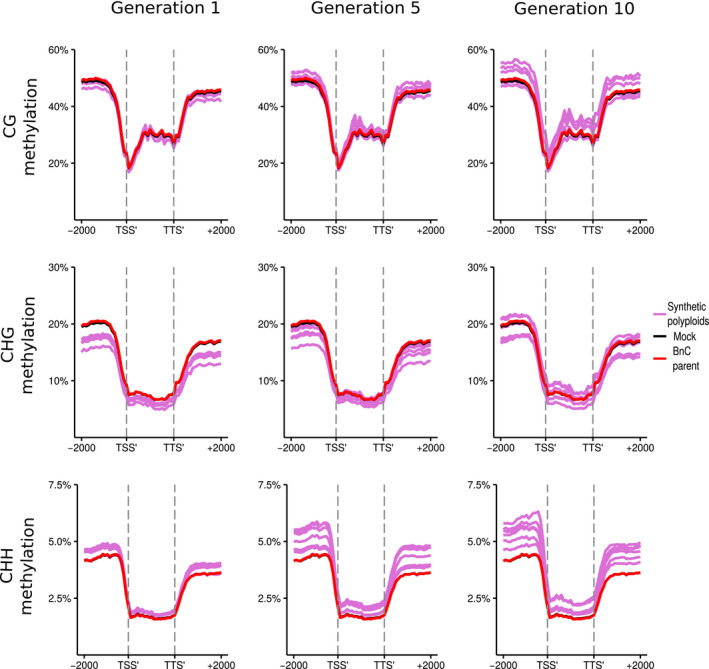
DNA methylation of BnC subgenome genes in resynthesised Brassica napus. Metaplots of CG, CHG and CHH mean weighted methylation of all gene models on the BnC subgenome for generations 1, 5 and 10 assessed using Bisulfite‐seq. The six resynthesised lines are shown in purple, the Brassica oleracea progenitor is shown in red and in silico mock polyploid in black as a visual control for cross‐mapping errors. Methylation levels are shown for the transcription start site (TSS′), gene body, transcription termination site (TTS′) and 2 kb upstream and downstream of the TSS and TTS.
Fig. 6.
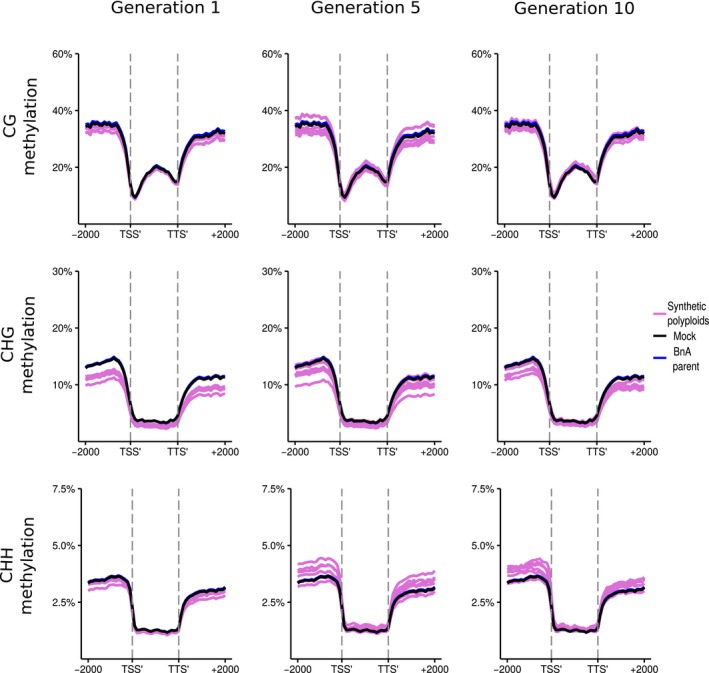
DNA methylation of BnA subgenome genes in resynthesised Brassica napus. Metaplots of CG, CHG and CHH mean weighted methylation of all gene models on the BnA subgenome for generations 1, 5 and 10 assessed using Bisulfite‐seq. The six resynthesised lines are shown in purple, the Brassica rapa progenitor is shown in blue and in silico mock polyploid in black as a visual control for cross‐mapping errors. Methylation levels are shown for the transcription start site (TSS′), gene body, transcription termination site (TTS′) and 2 kb upstream and downstream of the TSS and TTS.
Fig. 7.
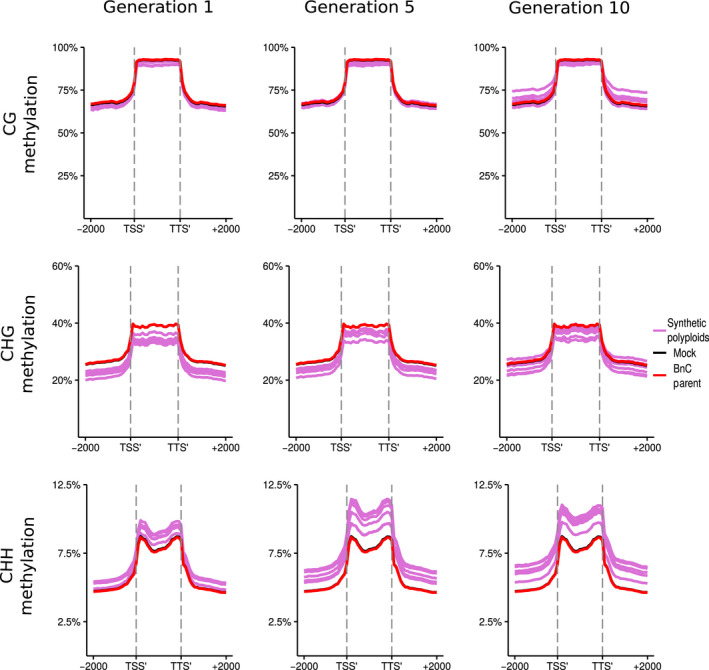
DNA methylation of BnC subgenome long‐terminal repeat (LTR) TEs in resynthesised Brassica napus. Metaplots of CG, CHG and CHH mean weighted methylation of all LTR TEs on the BnC subgenome for generations 1, 5 and 10 assessed using Bisulfite‐seq. The six resynthesised lines are shown in purple, the Brassica oleracea progenitor is shown in red and in silico mock polyploid in black as a visual control for cross‐mapping errors. Methylation levels are shown for the transcription start site (TSS′), gene body, transcription termination site (TTS′) and 2 kb upstream and downstream of the TSS and TTS.
Fig. 8.
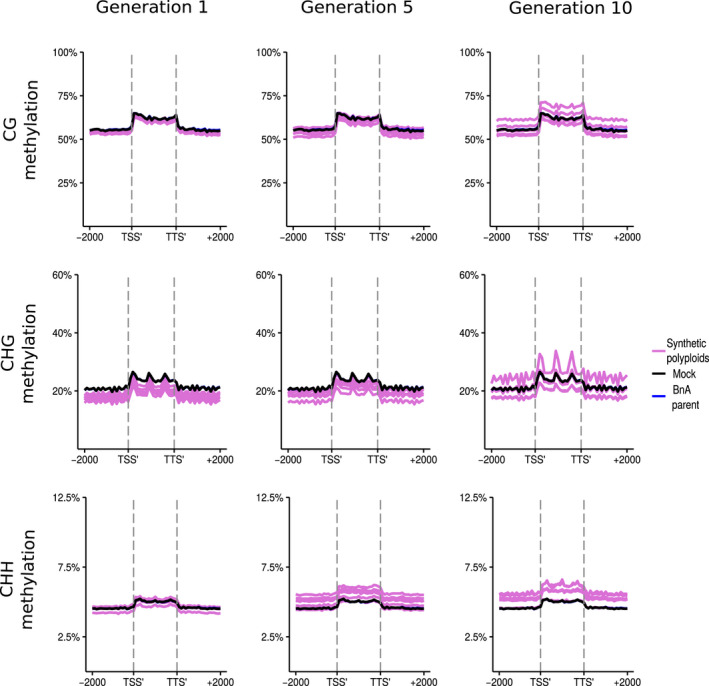
DNA methylation of BnA subgenome long‐terminal repeat (LTR) transposable elements in resynthesised Brassica napus. Metaplots of CG, CHG and CHH mean weighted methylation of all LTR transposable elements on the BnA subgenome for generations 1, 5 and 10 assessed using Bisulfite‐seq. The six resynthesised lines are shown in purple, the Brassica rapa progenitor is shown in blue and in silico mock polyploid in black as a visual control for cross‐mapping errors. The data here show unique and unexpected oscillations for CHG methylation, which may be tracking nucleosomes, but are unlikely to be an artefact as CG and CHH methylation is unaffected. Methylation levels are shown for the transcription start site (TSS′), gene body, transcription termination site (TTS′) and 2 kb upstream and downstream of the TSS and TTS.
To investigate the potential roles and patterns of DNA methylation related to homoeolog expression bias, we focused on homoeologs identified in 2 : 2 dosage with biased expression in the previous analyses. We compared homoeologs on both subgenomes with different homoeolog expression bias patterns to identify methylation patterns associated with expression bias. For clarity, each subgenome is coloured in a consistent way as in Fig. 1 (BnA is blue, BnC is red). We observed that homoeolog pairs that had BnC‐biased expression showed higher CG, CHG and CHH methylation in the flanking regions of genes (Fig. 9). Furthermore, BnC homoeologs shown to be more highly expressed than their BnA partner had lower CG methylation levels at the TSS, (Fig. 9, top right). While the reciprocal patterns were not observed for dominantly expressed BnA homoeologs at the TSS (Fig. 9, top left), differences in methylation patterns were observed in the flanking regions of these genes which were not observed for dominantly expressed BnC homoeologs. Homoeologs on the BnC subgenome more highly expressed than their BnA partner also showed lower CHH and CHG methylation levels in coding sequences. These patterns were not observed for the more highly expressed homoeologs on the BnA subgenome. No clear DNA methylation pattern differences were observed to explain higher expression for homoeologs on the BnA subgenome than their BnC partners.
Fig. 9.
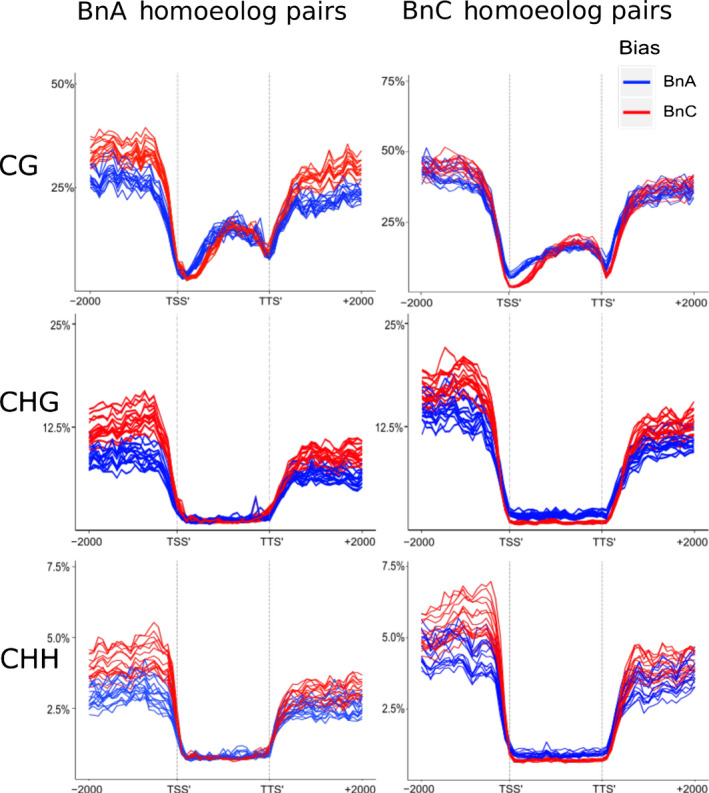
DNA Methylation of biased homoeologs in resynthesised Brassica napus. Average weighted methylation for homoeolog pairs in 2 : 2 dosage for all 16 lines assessed using Bisulfite‐seq. BnC‐biased expressed homoeologs in red and BnA‐biased expressed homoeologs in blue. Results are shown for methylation in CG, CHG and CHH contexts. Methylation levels are shown for the transcription start site (TSS′), gene body, transcription termination site (TTS′) and 2 kb upstream and downstream of the TSS and TTS.
Discussion
Subgenome expression bias is consistent and mirrors expression differences of the diploid progenitor species
We observed a significant expression bias towards the maternal Brassica oleracea (BnC) subgenome in all six resynthesised lines. We saw c. 70% of biased homoeolog pairs showing the same bias relationship in all six lines and in the parents. This further bolsters the claim that subgenome expression dominance may be predominately inherited from progenitors rather than an outcome of interspecific hybridisation and whole genome duplication (Buggs et al., 2014). However, given that 7 of 16 individuals had a proportion of BnC‐biased homoeologs that significantly deviated from parental expectations, and that 30% or more of BnC‐biased homoeolog pairs were not shared by all resynthesised lines and/or the parental genomes, there appears to be some variability in the extent to which homoeologous pairs are biased towards or away from the BnC subgenome.
The observed excess of BnC‐biased pairs is also reflective of the pattern identified in natural Brassica napus cultivar Darmor‐bzh (Chalhoub et al., 2014), but runs counter to observations of more BnA‐biased pairs genome‐wide and in genes related to cyto‐nuclear interactions in other resynthesised Brassica napus lines (Wu et al., 2018; Ferreira de Carvalho et al., 2019). The differences between the results presented here are likely, in part, to be due to B. rapa (BnA) being used as the maternal parent in the other two studies, while B. oleracea (BnC) is the maternal parent in this study. Furthermore, differences in methodology may have affected the results obtained in these other studies. For example, we controlled for homoeologous exchanges that altered gene dosage by excluding genomic regions that were not in a balanced 2 : 2 ratio between both parents. Gene dosage is known to produce dosage‐dependent expression changes (Conant et al., 2014; Lloyd et al., 2018). Furthermore, the differences between our study and these two previous studies (Wu et al., 2018; Ferreira de Carvalho et al., 2019) could, in part, be due to genetic differences in the accessions used to resynthesise Brassica napus. For example, previous work in autopolyploid Arabidopsis thaliana showed that parental genotype affected transcriptomic changes upon duplication (Yu et al., 2010). The current model to explain subgenome expression dominance patterns is related to methylated TE density differences near genes in parental genomes (Bird et al., 2018), and that gene and TE content are known to be highly variable within a species (Golicz et al., 2016; Anderson et al., 2019). Due to this intraspecific variation, the progenitor genomes may differ in such a way as to yield variable, and even opposite subgenome dominance patterns in different resynthesised allopolyploid lines.
This proposed explanation may seem to contradict the metaphor of ‘replaying the tape of life’ or the general determinism of subgenome dominance, however our model of subgenome dominance is based around aspects of genotypes rather than assumptions about fixed interspecies genomic differences. If subgenome dominance was due to ‘genomic shock’ of interspecific hybridisation and genome doubling, we might expect individual genotypes to exhibit variable subgenome dominance outcomes (i.e. chance of either subgenome being dominant). However, if subgenome dominance is due to genotype‐specific genomic features, we can reliably predict the subgenome dominance based on the parental genomes. The results presented here strongly suggested that an intermediate position holds true. Subgenome expression dominance in these resynthesised allopolyploids is strongly dependent upon progenitor genomic features, and crosses from the same genotypes seem to result in predictable and repeatable subgenome dominance patterns for a majority of genes in a given tissue type and environment. However, there is a sizeable proportion of genes that are variable between lines, suggesting that dynamic processes are also occurring and affecting homoeolog expression.
GO and pathway enrichment analyses of the Arabidopsis thaliana protein–protein interaction network revealed that Arabidopsis thaliana homologs of BnC‐biased homoeologs are highly enriched with primary metabolic and organellar functions, while homologs of BnA‐biased homoeologs were not enriched with any known biological functions. Additionally, the protein–protein interaction networks constructed from BnC‐biased homoeologs were more highly interconnected and clustered compared with BnA‐biased homoeologs. However, there was still observed enrichment for interactions in the PPI network for BnA‐biased gene pairs in generations 1 (P = 9.75e−07) and 5 (P = 0.00681), but not generation 10 (P = 0.16). These results suggested that the hundreds of BnC‐biased homoeologs are, to a greater extent, regulating vital cellular functions. The enrichment of organellar functions towards the BnC subgenome, which is the maternally contributed subgenome in our resynthesised lines, may implicate cyto‐nuclear interactions as a driving force of subgenome dominance (Sharbrough et al., 2017). One potential explanation is that genes on the maternal genome need to be expressed to interact with the plastid and mitochondria and paternal gene copies need to be silenced, and this is carried out by genomic imprinting. However, this is not definitive until similar analyses are repeated with reciprocal crosses. To further investigate the potential role of needing to maintain proper cyto‐nuclear interactions, we compared our results with those of Ferreira de Carvalho et al. (2019) who analysed 110 nuclear‐encoded components of plastid protein complexes and failed to find maternal progenitor homoeolog expression bias in their resynthesised Brassica napus. Of these 110 genes, we found 41 genes biased toward the BnC subgenome in all six lines in generation 1, 24 in generation 5 and 14 in generation 10. None of the 110 genes were biased towards the BnA subgenome in any individual across the six lines. However, because our analyses are genome‐wide rather than just focused on the 110 gene subset, we were able to identify a BnC subgenome bias towards the organelles based on the thousands of known nuclear‐encoded organellar genes (Savage et al., 2013) as indicated by the 263 BnC‐biased genes with GO cellular component annotation for the chloroplast in the 1st generation.
Homoeolog DNA methylation patterns are consistent, are correlated with observed expression biases and partially reflect differences in progenitors
We also identified consistent DNA methylation pattern differences between subgenomes. These differences appear to be a combination of inherited progenitor methylation patterns and methylation changes following interspecific hybridisation and polyploidisation. Globally, CG, CHG and CHH methylation of genes and LTR retrotransposons are higher in the dominant BnC subgenome than in the BnA subgenome, similar to results from natural B. napus (Chaloub et al., 2014). These methylation patterns were also present when comparing diploid progenitor epigenomes. The observed methylation responses are possibly due to ‘genomic shock’ following interspecific hybridisation, however without F1 hybrids we cannot rule out that it is a response to genome doubling rather than hybridisation. When we investigated the methylation level differences of biased expressed homoeologs, we saw that BnC homoeologs that were more highly expressed than their BnA partner showed markedly lower CG methylation at the TSS and lower CHG and CHH methylation across the coding sequence. These DNA methylation patterns were each associated with higher gene expression (Niederhuth et al., 2016). However, it remains unclear whether these methylation pattern differences are the causal mechanism or are possibly just the result of expression differences of biased homoeologs.
The methylation patterns observed here appear to diverge from expectations based on the conceptual model outlined in Bird et al. (2018) and with observations in other polyploid systems that exhibit subgenome expression dominance. This model expects spill‐over from methylation of TEs to affect neighbouring gene expression via RNA‐directed DNA methylation pathways, predicting that subgenomes with lower TE density and lower methylation of TEs and/or upstream of genes will be dominantly expressed (Bird et al., 2018). For example, in Mimulus peregrinus resynthesised and natural polyploids, the dominant subgenome had lower TE density and tended to have equal or lower expression than the nondominant subgenome (Edger et al., 2017). Edger et al. (2017) highlighted that TEs on the dominant subgenome do not return to parental levels of CHH methylation, while they do on the nondominant subgenome and argued that this lower‐than‐parental methylation contributed to subgenome dominance. In maize, the dominant subgenome has consistently been observed to have lower methylation upstream of genes, in particular lower CHH methylation around 500 bp upstream of the TSS. (Renny‐Byfield et al., 2017; Zhao et al., 2017). By contrast here, we found higher CHH methylation upstream for both the BnC subgenome on average and for BnC‐biased homoeologs (Figs 5, 7, 9). There are several possible explanations for this. Regions of high CHH methylation, called ‘CHH islands’, have been found to be associated with highly expressed genes in maize (Gent et al., 2013; Li et al., 2015) and other species (Niederhuth et al., 2016). It was proposed that this could serve as a boundary to reinforce TE silencing (Li et al., 2015). Alternatively, it was recently shown in Arabidopsis thaliana that highly methylated CHH regions near genes recruited a protein complex that enhanced transcription and counteracted the deleterious effects of neighbouring TEs on genes (Harris et al., 2018). It has yet to be demonstrated that this mechanism operates in other species, but it could explain paradoxical associations of high proximal CHH methylation with higher gene expression. Both of these cases provide alternatives to the current subgenome expression dominance model and potentially explain why higher expression and CHH methylation levels were observed on the dominant subgenome of our resynthesised lines.
Additionally, there are some patterns in common between our results and previous investigations of subgenome expression dominance. In Mimulus peregrinus, there is markedly lower CG methylation at the TSS on the dominant subgenome, similar to what we observed with BnC homoeologs with higher expression than their BnA partner. Additionally, when Renny‐Byfield et al. (2017) compared higher and lower expressed paralogues, regardless of subgenome of origin, they observed lower CG, CHG and CHH methylation at the TSS and across the coding sequence for the higher expressed paralogues. However, this lower expression was also seen upstream of the TSS, unlike our observations in resynthesised B. napus. These results may be due to particular genomic features of the progenitors.
For example, in soybean where there is no observable subgenome expression dominance, the paralogues from the two subgenome groups showed no differences in the distance to the nearest TE or in methylation levels (Zhao et al., 2017). However, few genes in the soybean genome have a TE nearby (<1 kb), but more highly expressed soybean genes tended to be further away from TEs (Zhao et al., 2017). These results suggested that multiple processes contributed to subgenome expression dominance, especially in genomes in which genes do not have TEs closely flanking upstream regions. This may also help to explain the observed spectrum of subgenome dominance in different species. For example, maize, Mimulus peregrinus, Brassica rapa, garden strawberry (Fragaria x ananassa), and others exhibited strong transcriptome‐wide subgenome expression dominance, where genes on the dominant subgenome are, on average, significantly more highly expressed than the nondominant subgenome, and significantly more gene pairs that are biased toward the dominant subgenome. In addition, soybean exhibited neither transcriptome‐wide bias, nor differences in how many gene pairs are biased to each subgenome. In our resynthesised Brassica napus, as well as natural B. napus, there is little if any transcriptome‐wide bias, but significant bias in the number of homoeolog pairs that are biased toward the BnC subgenome. This may be a promising new avenue to further dissect the role of the various DNA methylation contexts as the underlying mechanism(s) of subgenome expression dominance.
While many of the methylation differences between subgenomes appear to be inherited from the diploid progenitor genomes, we also identified methylation patterns that appeared to have been repeatedly modified following interspecific hybridisation and/or polyploidy. CHH methylation of both BnA and BnC genes and LTR retrotransposons showed transgressive hypermethylation patterns, first matching parental methylation levels in the 1st generation, and then surpassing parental methylation levels in 5th and 10th generations. This increase in LTR methylation over parental and mid‐parent value in later generations matched previous observations in resynthesised B. napus that LTR‐derived 21 and 24 nucleotide (nt) siRNAs are transgressively expressed in later generations (Martinez Palacios et al., 2019). Additionally, for the BnC subgenome, CG methylation also appeared to be transgressively hypermethylated in the later generations. The functional effect, if any, of hypermethylation at CG sites on driving subgenome expression dominance remains poorly understood and should also be the focus of future studies.
Conclusions
By analysing genomic, transcriptomic and epigenomic datasets of six isogenic resynthesised polyploids, we were able to observe repeated patterns of subgenome expression dominance across multiple plays of the ‘evolutionary tape’. Collectively, results from this study suggested that subgenome expression dominance in this B. napus system was largely, but not exclusively, based on pre‐existing parental gene expression differences and related to nuclear genome interactions with organelles. Similarly, we observed subgenome methylation patterns consistent with pre‐existing differences between parental epigenomes, although many cases of transgressive hypermethylation suggested that the epigenome was not as constrained by parental legacy. The observed DNA methylation differences between subgenomes were distinct from expectations of conceptual models and some previous findings. However, these patterns were consistent with patterns and mechanisms that could explain the observed expression bias, some not previously reported in cases of subgenome expression dominance to our knowledge. The causal role of DNA methylation patterns, particularly the pre‐existing differences among the diploid progenitor genomes, in affecting subgenome expression differences remains poorly understood. Future progress in these areas will bring us closer to answering looming questions on the underlying mechanisms of subgenome dominance and the connections between subgenome expression and DNA methylation differences in polyploid genomes.
Author contributions
KAB, ZX, RV and PPE planned and designed the research. KAB, CH, SO, MG, JCP, ZX, RV and PPE performed data analysis, collection and/or interpretation. KAB, RV and PPE wrote the manuscript. KAB, CH, SO, MG, JCP, ZX, RV and PPE reviewed and edited the manuscript.
Supporting information
Figs S1–S5 Homoeolog bias in the parents and across generations of the five lines.
Fig. S6 Common shared biased homoeolog pairs.
Fig. S7 DNA methylation of BnC syntenic genes.
Fig. S8 DNA methylation of BnA syntenic genes.
Fig. S9 DNA methylation of BnC nonsyntenic genes.
Fig. S10 DNA methylation of BnA nonsyntenic genes.
Table S1 homoeolog expression bias chi‐squared table.
Table S2 homoeolog expression bias vs parent chi‐squared table.
Table S3 Enrichment analyses of BnC‐biased gene pairs in the 1st generation.
Table S4 Enrichment analyses of BnC‐biased gene pairs in the 5th generation.
Table S5 Enrichment analyses of BnC‐biased gene pairs in the 10th generation.
Please note: Wiley Blackwell are not responsible for the content or functionality of any Supporting Information supplied by the authors. Any queries (other than missing material) should be directed to the New Phytologist Central Office.
Acknowledgements
This work was supported by MSU‐AgBioResearch funding to PPE and RV, USDA‐NIFA HATCH 1009804 and NSF‐IOS PGRP 2029959 to PPE, NSF‐GRFP DGE‐1424871 to KAB and National Key Research and Developmental Program of China (2016YFD0100202) and National Natural Science Foundation of China (31871239 and 31471173) to ZX.
Contributor Information
Zhiyong Xiong, Email: xiongzy2003@aliyun.com.
Robert VanBuren, Email: vanbur31@msu.edu.
Patrick P. Edger, Email: edgerpat@msu.edu.
Data availability
Raw data from this project are available on the NCBI Sequence Read Archive (SRA) Project PRJNA577908. Intermediate files and code to reproduce plots can be found at https://doi.org/10.5061/dryad.h18931zjr.
References
- Alger EI, Edger PP. 2020. One subgenome to rule them all: underlying mechanisms of subgenome dominance. Current Opinion in Plant Biology 54: 108–113. [DOI] [PubMed] [Google Scholar]
- An H, Qi X, Gaynor ML, Hao Y, Gebken SC, Mabry ME, McAlvay AC, Teakle GR, Conant GC, Barker MS et al. 2019. Transcriptome and organellar sequencing highlights the complex origin and diversification of allotetraploid Brassica napus. Nature Communications 10: 2878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson E, Stebbins GL. 1954. Hybridization as an evolutionary stimulus. Evolution 8: 378–388. [Google Scholar]
- Anderson SN, Stitzer MC, Brohammer AB, Zhou P, Noshay JM, Hirsch CD, Ross‐Ibarra J, Hirsch CN, Springer NM. 2019. Transposable elements contribute to dynamic genome content in maize. The Plant Journal 100: 1052–1065. [DOI] [PubMed] [Google Scholar]
- Arnold ML, Meyer A. 2006. Natural hybridization in primates: one evolutionary mechanism. Zoology 109: 261–276. [DOI] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Michael Cherry J, Davis AP, Dolinski K, Dwight SS, Eppig JT et al. 2000. Gene Ontology: tool for the unification of biology. Nature Genetics 25: 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnett DW, Garrison EK, Quinlan AR, Strömberg MP, Marth GT. 2011. BamTools: a C++ API and toolkit for analyzing and managing BAM files. Bioinformatics 27: 1691–1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird KA, VanBuren R, Puzey JR, Edger PP. 2018. The causes and consequences of subgenome dominance in hybrids and recent polyploids. New Phytologists 220: 87–93. [DOI] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottani S, Zabet NR, Wendel JF, Veitia RA. 2018. Gene expression dominance in allopolyploids: hypotheses and models. Trends in Plant Science 23: 393–402. [DOI] [PubMed] [Google Scholar]
- Buggs RJA, Wendel JF, Doyle JJ, Soltis DE, Soltis PS, Coate JE. 2014. The legacy of diploid progenitors in allopolyploid gene expression patterns. Philosophical Transactions of the Royal Society of London. Series B: Biological Sciences 369: 20130354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chagué V, Just J, Mestiri I, Balzergue S, Tanguy A‐M, Huneau C, Huteau V, Belcram H, Coriton O, Jahier J et al. 2010. Genome‐wide gene expression changes in genetically stable synthetic and natural wheat allohexaploids. New Phytologist 187: 1181–1194. [DOI] [PubMed] [Google Scholar]
- Chalhoub B, Denoeud F, Liu S, Parkin IAP, Tang H, Wang X, Chiquet J, Belcram H, Tong C, Samans B et al. 2014. Plant genetics. Early allopolyploid evolution in the post‐Neolithic Brassica napus oilseed genome. Science 345: 950–953. [DOI] [PubMed] [Google Scholar]
- Charron G, Marsit S, Hénault M, Martin H, Landry CR. 2019. Spontaneous whole‐genome duplication restores fertility in interspecific hybrids. Nature Communications 10: 4126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng F, Sun C, Wu J, Schnable J, Woodhouse MR, Liang J, Cai C, Freeling M, Wang X. 2016. Epigenetic regulation of subgenome dominance following whole genome triplication in Brassica rapa . New Phytologist 211: 288–299. [DOI] [PubMed] [Google Scholar]
- Cheng F, Wu J, Fang L, Sun S, Liu B, Lin K, Bonnema G, Wang X. 2012. Biased gene fractionation and dominant gene expression among the subgenomes of Brassica rapa . PLoS ONE 7: e36442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cokus SJ, Feng S, Zhang X, Chen Z, Merriman B, Haudenschild CD, Pradhan S, Nelson SF, Pellegrini M, Jacobsen SE. 2008. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 452: 215–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Combes M‐C, Hueber Y, Dereeper A, Rialle S, Herrera J‐C, Lashermes P. 2015. Regulatory divergence between parental alleles determines gene expression patterns in hybrids. Genome Biology and Evolution 7: 1110–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conant GC, Birchler JA, Pires JC. 2014. Dosage, duplication, and diploidization: clarifying the interplay of multiple models for duplicate gene evolution over time. Current Opinion in Plant Biology 19: 91–98. [DOI] [PubMed] [Google Scholar]
- Dale RK, Pedersen BS, Quinlan AR. 2011. Pybedtools: a flexible Python library for manipulating genomic datasets and annotations. Bioinformatics 27: 3423–3424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittrich‐Reed DR, Fitzpatrick BM. 2013. Transgressive hybrids as hopeful monsters. Evolutionary Biology 40: 310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. 2013. STAR: ultrafast universal RNA‐seq aligner. Bioinformatics 29: 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edger PP, Poorten TJ, VanBuren R, Hardigan MA, Colle M, McKain MR, Smith RD, Teresi SJ, Nelson ADL, Wai CM et al. 2019. Origin and evolution of the octoploid strawberry genome. Nature Genetics 51: 541–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edger PP, Smith R, McKain MR, Cooley AM, Vallejo‐Marin M, Yuan Y, Bewick AJ, Ji L, Platts AE, Bowman MJ et al. 2017. Subgenome dominance in an interspecific hybrid, synthetic allopolyploid, and a 140‐year‐old naturally established neo‐allopolyploid monkeyflower. Plant Cell 29: 2150–2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira de Carvalho J, Lucas J, Deniot G, Falentin C, Filangi O, Gilet M, Legeai F, Lode M, Morice J, Trotoux G et al. 2019. Cytonuclear interactions remain stable during allopolyploid evolution despite repeated whole‐genome duplications in Brassica. The Plant Journal 98: 434–447. [DOI] [PubMed] [Google Scholar]
- Freeling M, Woodhouse MR, Subramaniam S, Turco G, Lisch D, Schnable JC. 2012. Fractionation mutagenesis and similar consequences of mechanisms removing dispensable or less‐expressed DNA in plants. Current Opinion in Plant Biology 15: 131–139. [DOI] [PubMed] [Google Scholar]
- Gaebelein R, Schiessl SV, Samans B, Batley J, Mason AS. 2019. Inherited allelic variants and novel karyotype changes influence fertility and genome stability in Brassica allohexaploids. New Phytologist 223: 965–978. [DOI] [PubMed] [Google Scholar]
- Garsmeur O, Schnable JC, Almeida A, Jourda C, D’Hont A, Freeling M. 2014. Two evolutionarily distinct classes of paleopolyploidy. Molecular Biology and Evolution 31: 448–454. [DOI] [PubMed] [Google Scholar]
- Gent JI, Ellis NA, Guo L, Harkess AE, Yao Y, Zhang X, Dawe RK. 2013. CHH islands: de novo DNA methylation in near‐gene chromatin regulation in maize. Genome Research 23: 628–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golicz AA, Bayer PE, Barker GC, Edger PP, Kim H, Martinez PA, Chan CKK, Severn‐Ellis A, McCombie WR, Parkin IAP et al. 2016. The pangenome of an agronomically important crop plant Brassica oleracea . Nature Communications 7: 13390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould SJ. 1991. Wonderful life—the Burgess shale and the nature of history. New York, NY, USA: WW. Norton & Company. [Google Scholar]
- Hao M, Li A, Shi T, Luo J, Zhang L, Zhang X, Ning S, Yuan Z, Zeng D, Kong X et al. 2017. The abundance of homoeolog transcripts is disrupted by hybridization and is partially restored by genome doubling in synthetic hexaploid wheat. BMC Genomics 18: 149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris CJ, Scheibe M, Wongpalee SP, Liu W, Cornett EM, Vaughan RM, Li X, Chen W, Xue Y, Zhong Z et al. 2018. A DNA methylation reader complex that enhances gene transcription. Science 362: 1182–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollister JD, Gaut BS. 2009. Epigenetic silencing of transposable elements: a trade‐off between reduced transposition and deleterious effects on neighboring gene expression. Genome Research 19: 1419–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Wheat Genome Sequencing Consortium . 2014. A chromosome‐based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science 345: 1251788. [DOI] [PubMed] [Google Scholar]
- Kanehisa M, Goto S. 2000. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Research 28: 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenig D, Weigel D. 2015. Beyond the thale: comparative genomics and genetics of Arabidopsis relatives. Nature Reviews Genetics 16: 285–298. [DOI] [PubMed] [Google Scholar]
- Kryvokhyzha D, Salcedo A, Eriksson MC, Duan T, Tawari N, Chen J, Guerrina M, Kreiner JM, Kent TV, Lagercrantz U et al. 2019. Parental legacy, demography, and admixture influenced the evolution of the two subgenomes of the tetraploid Capsella bursa‐pastoris (Brassicaceae). PLoS Genetics 15: e1007949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. 2012. Fast gapped‐read alignment with Bowtie 2. Nature Methods 9: 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitch AR, Leitch IJ. 2008. Genomic plasticity and the diversity of polyploid plants. Science 320: 481–483. [DOI] [PubMed] [Google Scholar]
- Li N, Xu C, Zhang A, Lv R, Meng X, Lin X, Gong L, Wendel JF, Liu B. 2019. DNA methylation repatterning accompanying hybridization, whole genome doubling and homoeolog exchange in nascent segmental rice allotetraploids. New Phytologist 223: 979–992. [DOI] [PubMed] [Google Scholar]
- Li Q, Gent JI, Zynda G, Song J, Makarevitch I, Hirsch CD, Hirsch CN, Dawe RK, Madzima TG, McGinnis KM et al. 2015. RNA‐directed DNA methylation enforces boundaries between heterochromatin and euchromatin in the maize genome. Proceedings of the National Academy of Sciences, USA 112: 14728–14733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister R, O'Malley RC, Tonti‐Filippini J, Gregory BD, Berry CC, Millar AH, Ecker JR. 2008. Highly integrated single‐base resolution maps of the epigenome in Arabidopsis. Cell 133: 523–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd A, Blary A, Charif D, Charpentier C, Tran J, Balzergue S, Delannoy E, Rigaill G, Jenczewski E. 2018. Homoeologous exchanges cause extensive dosage‐dependent gene expression changes in an allopolyploid crop. New Phytologist 217: 367–377. [DOI] [PubMed] [Google Scholar]
- Mallet J. 2007. Hybrid speciation. Nature 446: 279–283. [DOI] [PubMed] [Google Scholar]
- Martin M. 2011. Cutadapt removes adapter sequences from high‐throughput sequencing reads. EMBnet.journal 17: 10. [Google Scholar]
- Martinez Palacios P, Jacquemot MP, Tapie M, Rousselet A, Diop M, Remoué C, Falque M, Lloyd A, Jenczewski E, Lassalle G et al. 2019. Assessing the response of small RNA populations to allopolyploidy using resynthesized Brassica napus Allotetraploids. Molecular Biology and Evolution 36: 709–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niederhuth CE, Bewick AJ, Ji L, Alabady MS, Kim KD, Li Q, Rohr NA, Rambani A, Burke JM, Udall JA et al. 2016. Widespread natural variation of DNA methylation within angiosperms. Genome Biology 17: 194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niederhuth CE, Schmitz RJ. 2017. Putting DNA methylation in context: from genomes to gene expression in plants. Biochimica et Biophysica Acta (BBA) ‐ Gene Regulatory Mechanisms 1860: 149–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkin IAP, Koh C, Tang H, Robinson SJ, Kagale S, Clarke WE, Town CD, Nixon J, Krishnakumar V, Bidwell SL et al. 2014. Transcriptome and methylome profiling reveals relics of genome dominance in the mesopolyploid Brassica oleracea. Genome Biology 15: R77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertea M, Pertea GM, Antonescu CM, Chang T‐C, Mendell JT, Salzberg SL. 2015. StringTie enables improved reconstruction of a transcriptome from RNA‐seq reads. Nature Biotechnology 33: 290–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pires JC, Zhao J, Schranz ME, Leon EJ, Quijada PA, Lukens LN, Osborn TC. 2004. Flowering time divergence and genomic rearrangements in resynthesized Brassica polyploids (Brassicaceae). Biological Journal of the Linnaean Society 82: 675–688. [Google Scholar]
- Quinlan AR, Hall IM. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team . 2018. R: a language and environment for statistical computing Vienna, Austria: The R Foundation. [WWW document] URL http://www.r‐project.org/. [Google Scholar]
- Ren J, Wu P, Trampe B, Tian X, Lübberstedt T, Chen S. 2017. Novel technologies in doubled haploid line development. Plant Biotechnology Journal 15: 1361–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renny‐Byfield S, Rodgers‐Melnick E, Ross‐Ibarra J. 2017. Gene fractionation and function in the ancient subgenomes of maize. Molecular Biology and Evolution 34: 1825–1832. [DOI] [PubMed] [Google Scholar]
- Rieseberg LH, Raymond O, Rosenthal DM, Lai Z, Livingstone K, Nakazato T, Durphy JL, Schwarzbach AE, Donovan LA, Lexer C. 2003. Major ecological transitions in wild sunflowers facilitated by hybridization. Science 301: 1211–1216. [DOI] [PubMed] [Google Scholar]
- Savage LJ, Imre KM, Hall DA, Last RL. 2013. Analysis of essential Arabidopsis nuclear genes encoding plastid‐targeted proteins. PLoS ONE 8: e73291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnable JC, Springer NM, Freeling M. 2011. Differentiation of the maize subgenomes by genome dominance and both ancient and ongoing gene loss. Proceedings of the National Academy of Sciences, USA 108: 4069–4074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz MD, He Y, Whitaker JW, Hariharan M, Mukamel EA, Leung D, Rajagopal N, Nery JR, Urich MA, Chen H et al. 2015. Human body epigenome maps reveal noncanonical DNA methylation variation. Nature 523: 212–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz MD, Schmitz RJ, Ecker JR. 2012. ‘Leveling’ the playing field for analyses of single‐base resolution DNA methylomes. Trends in Genetics 28: 583–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharbrough J, Conover JL, Tate JA, Wendel JF, Sloan DB. 2017. Cytonuclear responses to genome doubling. American Journal of Botany 104: 1277–1280. [DOI] [PubMed] [Google Scholar]
- Soltis DE, Buggs RAJ, Barbazuk WB, Chamala S, Chester M, Gallagher JP, Schnable PS, Soltis PS. 2012. The early stages of polyploidy: rapid and repeated evolution in Tragopogon. In: Soltis PS, Soltis DE, eds. Polyploidy and genome evolution. Berlin/Heidelberg, Germany: Springer, 271–292. [Google Scholar]
- Soltis PS, Soltis DE. 2009. the role of hybridization in plant speciation. Annual Review of Plant Biology 60: 561–588. [DOI] [PubMed] [Google Scholar]
- Song K, Lu P, Tang K, Osborn TC. 1995. Rapid genome change in synthetic polyploids of Brassica and its implications for polyploid evolution. Proceedings of the National Academy of Sciences, USA 92: 7719–7723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D, Morris JH, Cook H, Kuhn M, Wyder S, Simonovic M, Santos A, Doncheva NT, Roth A, Bork P et al. 2017. The STRING database in 2017: quality‐controlled protein‐protein association networks, made broadly accessible. Nucleic Acids Research 45: D362–D368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H, Wang X, Bowers JE, Ming R, Alam M, Paterson AH. 2008. Unraveling ancient hexaploidy through multiply‐aligned angiosperm gene maps. Genome Research 18: 1944–1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas BC. 2006. Following tetraploidy in an Arabidopsis ancestor, genes were removed preferentially from one homeolog leaving clusters enriched in dose‐sensitive genes. Genome Research 16: 934–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallejo‐Marín M, Buggs RJ, Cooley AM, Puzey JR. 2015. Speciation by genome duplication: repeated origins and genomic composition of the recently formed allopolyploid species Mimulus peregrinus. Evolution 69: 1487–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van de Peer Y, Maere S, Meyer A. 2009. The evolutionary significance of ancient genome duplications. Nature Reviews Genetics 10: 725–732. [DOI] [PubMed] [Google Scholar]
- Wendel JF, Cronn RC. 2003. Polyploidy and the evolutionary history of cotton. Advances in Agronomy 78: 139. [Google Scholar]
- Wickham H. 2009. ggplot2: elegant graphics for data analysis. Springer Science & Business Media. [Google Scholar]
- Woodhouse MR, Cheng F, Pires JC, Lisch D, Freeling M, Wang X. 2014. Origin, inheritance, and gene regulatory consequences of genome dominance in polyploids. Proceedings of the National Academy of Sciences, USA 111: 5283–5288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Lin L, Xu M, Chen P, Liu D, Sun Q, Ran L, Wang Y. 2018. Homoeolog expression bias and expression level dominance in resynthesized allopolyploid Brassica napus . BMC Genomics 19: 586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Z, Gaeta RT, Pires JC. 2011. Homoeologous shuffling and chromosome compensation maintain genome balance in resynthesized allopolyploid Brassica napus . Proceedings of the National Academy of Sciences, USA 108: 7908–7913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan C, Sun G. 2012. Multiple origins of allopolyploid wheatgrass Elymus caninus revealed by RPB2, PepC and TrnD/T genes. Molecular Phylogenetics and Evolution 64: 441–451. [DOI] [PubMed] [Google Scholar]
- Yu Z, Haberer G, Matthes M, Rattei T, Mayer KF, Gierl A, Torres‐Ruiz RA. 2010. Impact of natural genetic variation on the transcriptome of autotetraploid Arabidopsis thaliana . Proceedings of the National Academy of Sciences, USA 107: 17809–17814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao M, Zhang B, Lisch D, Ma J. 2017. Patterns and consequences of subgenome differentiation provide insights into the nature of paleopolyploidy in plants. Plant Cell 29: 2974–2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figs S1–S5 Homoeolog bias in the parents and across generations of the five lines.
Fig. S6 Common shared biased homoeolog pairs.
Fig. S7 DNA methylation of BnC syntenic genes.
Fig. S8 DNA methylation of BnA syntenic genes.
Fig. S9 DNA methylation of BnC nonsyntenic genes.
Fig. S10 DNA methylation of BnA nonsyntenic genes.
Table S1 homoeolog expression bias chi‐squared table.
Table S2 homoeolog expression bias vs parent chi‐squared table.
Table S3 Enrichment analyses of BnC‐biased gene pairs in the 1st generation.
Table S4 Enrichment analyses of BnC‐biased gene pairs in the 5th generation.
Table S5 Enrichment analyses of BnC‐biased gene pairs in the 10th generation.
Please note: Wiley Blackwell are not responsible for the content or functionality of any Supporting Information supplied by the authors. Any queries (other than missing material) should be directed to the New Phytologist Central Office.
Data Availability Statement
Raw data from this project are available on the NCBI Sequence Read Archive (SRA) Project PRJNA577908. Intermediate files and code to reproduce plots can be found at https://doi.org/10.5061/dryad.h18931zjr.