

Abstract
Photochemical processes, such as isomerizations and cycloadditions, have proven to be very useful in the construction of highly strained molecular frameworks. Photoinduced ring strain enables subsequent exergonic reactions which do not require the input of additional chemical energy and provides a variety of attractive synthetic options leading to complex structures. This review covers the progress achieved in the application of sequences combining excitation by ultraviolet light to form strained intermediates, which are further transformed to lower energy products in strain‐release reactions. As ring strain is considerable in small ring systems, photogenerated three‐ and four‐membered rings will be covered, mainly focusing on examples from 2000 to May 2020.
Keywords: green chemistry, photochemistry, ring strain, strain-release, transition-metal catalysis
Strain‐release‐driven reactions are an attractive extension of the synthetic toolbox. The strain energy contained particularly in photogenerated small ring systems provides efficient access to complex molecular scaffolds under very mild conditions. This review summarizes key strain‐release transformations of photogenerated three‐ and four‐membered ring intermediates promoted by Brønsted or Lewis acids as well as by metal‐ or photoredox catalysis. The progress made in such transformations not only concerns their sustainability but has also led to powerful methodologies suitable for the construction of natural products.
Introduction
The concept of ring strain, first described and intensely investigated by Adolf von Baeyer in 1885, has found broad application in organic synthesis. [1] In addition, a large number of natural products contain strained substructures. [2] Starting from the almost unstrained cyclohexane, contraction of the ring size builds ring strain energy dominated by angular (Baeyer) strain and torsional (Pitzer) strain (Figure 1), making three‐ and four‐membered rings ideal substrates for strain‐releasing chemistry. [3]
Figure 1.
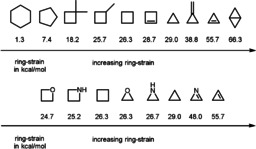
Ring strain energies of different (substituted) small‐ring‐systems and their hetero‐analogues. [5]
Although many of these small‐ring systems are commonly synthesized in thermal reactions, the shape of the energy hypersurface of electronically excited states offers unique possibilities in terms of both selectivity of the reaction and structural complexity of the accessible strained compounds. Numerous excellent reviews on light‐induced rearrangements and cycloadditions in the synthesis of small‐ring carbocycles, heterocyclic analogues as well as natural products, have been published recently, pointing out the high potential of these transformations. [4]
Under the premise of sufficiently high quantum yields, photochemistry can also be preferable over classical reactions in view of the concept of green chemistry. Photon‐absorption of UV‐B irradiation (λ≈300 nm) for example corresponds to an energy equivalent of 95 kcal mol−1 and therefore gives access to traceless valence isomerizations or photoinduced cycloadditions resulting in products that energetically are clearly located uphill of the starting materials. The use of energy‐transfer from photocatalysts excited by visible light to circumvent the use of UV radiation was also reviewed lately. It can also result in eco‐friendly transformations, although the amount of energy that can be transferred to the substrate during these processes is lower than in the case of direct excitation. [6] Although other examples also exist, the light energy is often transformed into a certain ring strain of the photoproducts, which can be used in (catalyzed) follow‐up reactions. As a potential drawback, high‐energy intermediates are frequently also prone to undergo thermal or photochemical secondary reactions, making it often necessary to directly transform them into thermodynamically stable products to obtain high yields.
Depending on the reactivity of the strained intermediate and the nature of the reaction partner, the intended follow‐up reactions can be performed without any added catalyst as well as under catalysis of Brønsted acids or bases or under Lewis acid catalysis. Recently, several groups also described (transition‐)metal and photoredox catalysis in follow‐up transformations of strained photoproducts. In all these examples, the inherent ring strain of the photogenerated intermediate serves as the driving force for the subsequent step(s). Surprisingly, in contrast to the rapidly growing number of transition metal‐ [7] as well as photoredox‐catalyzed [8] transformations of strained substrates in terms of C−H and C−C bond activations, only few of the examples incorporate strained photoproducts. The combination of photoinduced generation of strained intermediates with fast (catalyzed) follow‐up reactions could nevertheless be an advantageous general concept. The activation energy is injected into the molecule by photon absorption and circumvents the generation of low‐energy co‐products that for example drive classical C−C‐cross coupling reactions. Additionally, the photochemical generation of the high‐energy intermediates enables formal endergonic reactions between starting materials and follow‐up products to proceed (Figure 2).
Figure 2.
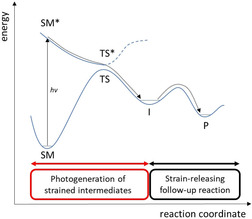
Feasible energy profile of a formal endergonic strain‐releasing reaction via a photogenerated high‐energy intermediate (I). (SM=starting material; TS=transition state; P=product).
Finally, the use of fast trapping reactions allows for the use of highly reactive intermediates in one‐pot procedures which cannot be isolated in acceptable yields as they readily react in secondary photochemically or thermally induced side reactions. Based on these considerations, this review will cover strain‐releasing follow‐up reactions of photogenerated small‐ring intermediates focusing on recent (2000–2020) examples from the literature, including our own work.
Three‐Membered Ring Systems
Examples of photogenerated three‐membered carbocycles in strain‐releasing chemistry are still rare. The picture changes drastically when it comes to the hetero‐analogues and especially to the nitrogen containing three‐membered ring‐systems. Both aziridines, in terms of highly reactive vinyl aziridines, and azirines are frequently used in such reaction sequences.
Aziridines
Aziridines are the simplest aza‐heterocycles and feature a significant ring strain as well as polarized carbon‐heteroatom bonds, which makes them very reactive in various kinds of transformations such as cycloadditions, rearrangements and isomerizations as well as ring‐opening reactions induced by a broad range of nucleophiles. [9]
Vinyl aziridines starting from pyridinium salts
Among the variously functionalized aziridines, vinyl aziridines have shown to be particularly versatile building blocks in organic synthesis. One of the first reports on the photogeneration of such compounds was published by the group of Kaplan in 1972, who described a photoinduced cyclization of N‐alkylpyridinium chloride 1 in aqueous solution to intermediately furnish the 6‐azabicyclo[3.1.0]hexenyl cation 4, which is readily transformed into an 6‐azabicyclo[3.1.0]hex‐3‐en‐2‐exo‐ol with the same structure as 5 upon spontaneous addition of water. [10] The addition was found to proceed stereoselectively via an attack from the convex face. The synthetic potential of pyridinium salt photochemistry was not recognized until the late 1990s, when Mariano and others developed numerous reaction sequences involving the highly strained bicyclic vinyl aziridinols or ethers (obtained by replacing water with alcohols as the nucleophiles) to several ring strain‐releasing follow‐up reactions in order to stereoselectively synthesize 3,4,5‐trisubstituted cyclopentenes in simple photoirradiation‐nucleophilic ring‐opening sequences. [11] The advantages of this reaction sequence could impressively be demonstrated in the total synthesis of several natural products as (+)‐castanospermine, [12] (+)‐mannostatin A, [13] (−)‐swainsonine [14] and (−)‐cephalotaxine. [15]
A representative example of such a sequence is the conversion of the photogenerated 4‐methoxy‐6‐propyl‐6‐azabicyclo[3.1.0]hex‐2‐ene 5 to the (3,4‐cis)(3,5‐trans)‐aminocyclopentenyl alcohol 3, reported by Mariano and co‐workers in 1996 (Scheme 1 a). Photoirradiation of N‐propylpyridinium chloride 1 in methanol followed by addition of sodium hydrogen carbonate furnished the bicyclic aziridine 5 as a single diastereomer. The subsequent ring‐opening of 5 with ethyl chloroformate stereoselectively generated the (3,4‐trans)(3,5‐cis)‐chlorocarbamate 6, which underwent azlactonization upon heating. The oxazolidone 7 was afterwards reduced to afford the (3,4‐cis)(3,5‐trans)‐cyclopentene 3. This lactonization‐reduction sequence enabled the stereoinversion at C‐3. [11a] In 1997, the Burger group reacted the photogenerated 6‐azabicyclo[3.1.0]hex‐3‐en‐2‐ol derivatives 11 with diiron nonacarbonyl followed by oxidative decomplexation to generate cis‐fused cyclopentene‐β‐lactams 9 (Scheme 1 b). Irradiation of N‐alkylpyridinium salt 8 under basic conditions again afforded the bicyclic aziridines 11 that were subsequently treated with diiron nonacarbonyl in THF to form the iron complex 12, which could be isolated in good yields. The authors expected [Fe(CO)4(THF)] to be the active carbonylation reagent, which was assumed to be in equilibrium with dinuclear starting material under the reaction conditions. The attack itself was found to stereoselectively occur from the endo‐face of the bicyclic vinyl aziridines 11. Subsequent CO insertion into the metal‐nitrogen bond and oxidative decomplexation with ceric ammonium nitrate (CAN) furnished the β‐lactams 9. [11b]
Scheme 1.
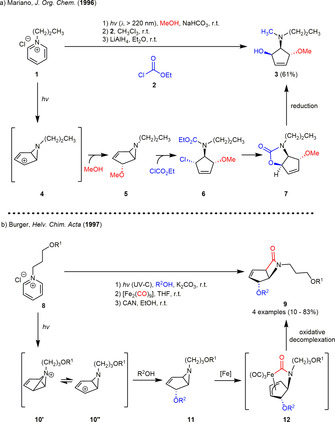
(a) Photocyclization‐aziridine ring‐opening sequences to construct the 3,4,5‐trisbustituted cyclopentene 3. (b) Photolysis of N‐alkylpyridinium salts to construct 6‐azabicyclo[3.1.0]hex‐3‐en‐2‐ol derivatives 11 as building blocks in the synthesis of bicyclic β‐lactams 9.
In 2001, the group of Simpson was able to extend the photoisomerization to 3‐alkoxypyridinium salt 13, which generated the 6‐alkyl‐4,4‐dimethoxy‐1‐methyl‐6‐azabicyclo[3.1.0]hex‐2‐enes 17 upon irradiation in aqueous methanol. The photogenerated intermediates 17 were subsequently reacted with the organocuprates 14 or 15 under Lewis acid catalysis (BF3⋅OEt2) to afford the (3,5‐trans)aminocyclopentenes 16 in moderate yields (Scheme 2 a). [16] The authors assumed the stereoselective generation of the trans‐isomer during this SN2’ reaction to be based on the steric bulk introduced by the Lewis acid coordinated to the nitrogen lone pair. An alternative explanation was proposed by Corey in 1984, who suggested that SN2’‐reactions of cuprates generally show predominantly anti‐stereoselectivity due to the fact that filled d‐orbitals on the copper can interact with both the π* antibonding orbital of the olefin and the σ* orbital of the carbon‐leaving group bond. [17] As already mentioned, the Mariano group used the latter concept in the synthesis of several natural products. The enantiopure starting material in all of these cases was synthesized starting from pyridine 19, which was incorporated in the photoisomerization‐ring‐opening sequence with subsequent acetylation of 3,5‐dihydroxy‐4‐aminocyclopentene 21. (Scheme 2 b). The extension of the sequence on unsubstituted pyridines required the use of aqueous perchloric acid solution as a solvent to generate the protonated pyridinium salt in situ during irradiation. The meso‐configurated triacetylated 3,4,5‐trisubstituted cyclopentene 20 could be enzymatically desymmetrized and directly used for the total synthesis of (+)‐mannostatin A. [13] Subsequent blocking of the hydroxy groups with different protecting groups and allylation of the Boc‐protected nitrogen led to precursor 22, which was for example successfully incorporated in an olefin metathesis for the total synthesis of (−)‐swainsonine. [14]
Scheme 2.
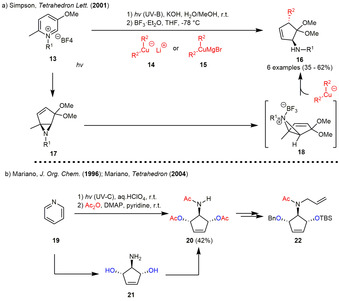
(a) Stereoselective addition of organocuprates to 6‐alkyl‐4,4‐dimethoxy‐1‐methyl‐6‐azabicyclo[3.1.0]hex‐2‐enes. (b) Photoisomerization‐ring‐opening sequence to generate enantiopure 3,4,5‐trisubstituted cyclopentene as starting material for the total synthesis of several natural products.
In 2005, Mariano also reported the extension of the shown methodology to 1,2‐fused pyridinium salts in order to enable an SN2’‐type ring‐opening reaction of the highly strained intermediates 27, 28 and 30 (Scheme 3 a). Irradiation of the tetrahydroquinolizinium salt 23 in aqueous perchloric acid solution furnished a mixture of the tricycles 27 and 28 (7:1). Subsequent SN2’ ring‐opening and peracetylation led to the formation of the [5.3.0]bicycle 24 and spiro‐bicycle 25 in low yield. When basic conditions were applied to the dihydroindolizinium salt 29 during irradiation, a remarkably regioselective photoisomerization yielded in the exclusive formation of the tricyclic allylic alcohol 30, which could be converted into the meso‐diester 31 in high yield (Scheme 3 b). [18]
Scheme 3.
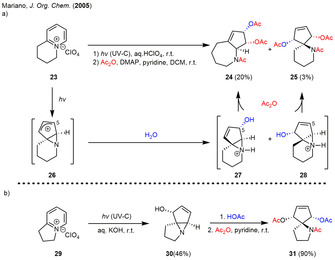
(a) Photoirradiation‐ring‐opening sequence of the tetrahydroquinolizinium salt 23 under acidic conditions. (b) Photoirradiation‐ring‐opening sequence of dihydroindolizinium salt 29 under basic conditions.
The group of Afonso also contributed to the photochemistry of pyridinium salts in the past years by applying flow‐conditions to the synthesis of the fused vinyl aziridines and the ring‐opened trisubstituted cyclopentenes. While the first reports basically focused on the transfer of the known procedures into flow‐conditions and the investigation of the nucleophile substrate scope for the subsequent ring‐opening of the 6‐alkyl‐6‐azabicyclo[3.1.0]hex‐2‐en‐4‐yl alcohols and ethers, the group was very recently able to demonstrate a palladium‐mediated ring‐opening reaction of the photogenerated 6‐alkyl‐6‐azabicyclo[3.1.0]hex‐2‐en‐4‐yl alcohols or ethers (36). By applying these conditions, the substrate scope of nucleophiles capable of the latter ring‐opening could be extended to 1,3‐dicarbonyls and analogues (33 and 34) in moderate to high yield. Additionally, the authors reported the structure of the products to indicate the nucleophilic attack to formally occur from the endo‐face of the vinyl aziridines 36 affording the syn‐SN2’‐products. Mechanistically, the palladium(0) catalyst was assumed to oxidatively add into the aziridine's C−N bond initially forming a palladium‐stabilized allylic zwitterion, in which the negative charge is located on nitrogen. The basicity of the anionic nitrogen was assumed to be high enough for the deprotonation of cyclic 1,3‐diketones 34, while a stronger external base (NaH) must be added for the acyclic analogues 33. The palladium catalyst is assumed to attack from the exo‐face forcing the deprotonated nucleophile to attack the allyl‐palladium complex 37 via an outer sphere C‐allylation from the former endo‐face. Ligand exchange liberates the 3,4,5‐trisubstituted cyclopentenes 35 in low to high yield (Scheme 4). [19]
Scheme 4.
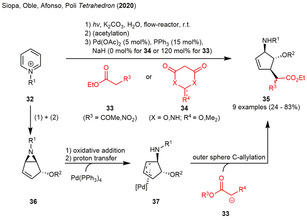
Palladium‐catalyzed nucleophilic ring‐opening of photogenerated vinylaziridines with 1,3‐dicarbonyl compounds leading to syn‐SN2’ products.
Terminal alkenyl tethered pyrroles as substrates
Another photochemical reaction leading to highly reactive polycyclic vinyl aziridines was described by the group of Booker‐Milburn in 2013. Irradiation of N‐butenyl substituted pyrroles 38 furnished the tricyclic aziridines 42 via two subsequent photoinduced isomerizations. The developed methodology could also be transferred to photoflow conditions providing the highly strained aziridines 42 in multi‐gram quantities (up to 22 gram per day). Mechanistically, excitation with a 254 nm low pressure Hg lamp results in an initial [2+2] cycloaddition of the terminal alkene with the C2−C3 bond of the pyrrole to give cyclobutane 41, which undergoes a second photoinduced fragmentation‐rearrangement reaction leading to tricycle 42. [20] The proximity of functional groups within the tricycle enables the cycloaddition and ring‐opening reactions. Treatment of vinyl aziridine 42 with a range of carbon‐nucleophiles and Pd(PPh3)4 via a Tsuji–Trost‐type SN2’ ring‐opening yielded in an inseparable mixture of diastereomers 39 and 40 (Scheme 5 a). The anti/syn ratio was reported to be highly dependent on the polarity of the solvent. Less polar solvents such as 1,4‐dioxane favored formation of the bicycle 39, while more polar solvents like acetonitrile or N,N‐dimethylformamide favored the formation of 40. The observation was explained by the change from an outer sphere mechanism (direct attack of the nucleophile on carbon—overall retention) to an inner sphere process (initial attack on metal—overall inversion) upon increasing the polarity of the solvent.
Scheme 5.
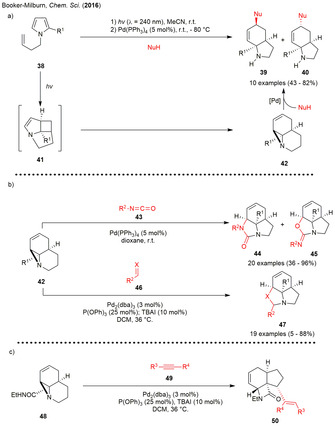
(a) Pd‐catalyzed ring‐opening of photogenerated tricyclic aziridines. (b) Pd‐catalyzed [3+2] cycloadditions of aziridines with isocycanates as well as aldehydes and imines. (c) Pd‐catalyzed addition/cyclisation sequence of photogenerated aziridine with internal alkynes.
The second part of the study involved [3+2] cycloadditions between the photogenerated aziridines 42 and a wide variety of aryl and sulfonyl isocyanates (43) under Pd0 catalysis to form the vinyl substituted cyclic urea compounds 44. Surprisingly, lower (1 mol %) catalyst loadings and catalyst‐free conditions led to a change in chemoselectivity, furnishing the cyclic imidates 45. The cycloaddition could also be extended to other dipolarophiles like alkenes, aldehydes, and imines to generate the tricyclic compounds 47 with high diastereoselectivity. For these sets of reactions, the use of triphenyl phosphite as a ligand was found to be crucial (Scheme 5 b). Finally, this approach was extended to generate the tricyclic fused β‐lactams 50 as a single diastereomer by palladium‐mediated reaction of amide 48 with the internal alkynes 49. The high reactivity of the photogenerated aziridines in this reaction could be impressively demonstrated by the generation of the products under catalyst‐free conditions at room temperature, although reaction times were long (Scheme 5 c). [21]
In 2017, the developed methodologies were successfully incorporated into the total synthesis of (±)‐3‐demethoxyerythratidinone in five steps starting from a simple pyrrole derivative. [22] Irradiation of pyrrole carboxylate 51 using the already introduced flow reactor produced gram quantities of the aziridine (±)‐55. The following Pd‐catalyzed Tsuji–Trost‐type reaction with acetic acid gave the SN2’ ring‐opened hexahydroindole (±)‐52 in excellent yield and as single diastereomer. Further manipulations resulted in (±)‐3‐demethoxyerythratidinone 53 (Scheme 6).
Scheme 6.
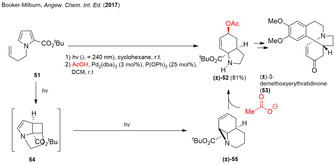
Pd‐catalyzed ring‐opening of photogenerated tricyclic aziridines in the total synthesis of (±)‐3‐demethoxyerythratidinone.
2H‐Azirines
2H‐Azirines are commonly used in strain‐release chemistry as they occupy both high ring strain (vide supra) as well as a polarized internal imine function that serves as reaction center for most of the follow‐up reactions. 2H‐Azirines are commonly synthesized using the Neber rearrangement as well as the thermal or photochemical decomposition of vinyl azides, which proceeds via a highly reactive nitrene intermediate. [23]
2H‐Azirines starting from vinylnitrenes
The photochemical synthesis of 2H‐azirines from vinyl azides in those cases are carried out using UV‐ or visible‐light irradiation depending on the substitution pattern of the starting material. In 2014, the Yoon group reported a general visible‐light photosensitization approach to form 2H‐azirines from vinyl azides under very mild conditions. Catalytic amounts (1 mol %) of [Ru(dtbbpy)3](PF6)2 were used as energy transfer agent enabling UV‐absorbing substrates to be formed using visible light as well as generally accelerating the 2H‐azirine formation from all kinds of substrates. [24] Based on this fast and scalable isomerization, several procedures were developed that are incorporating the photogenerated 2H‐azirines in strain‐releasing chemistry.
In the same year, the groups of Lu and Xiao described a photoredox‐catalyzed formal [3+2] cycloaddition of 2H‐azirines bearing alkyl and aryl substituents with electron‐poor alkynes forming highly substituted pyrroles in an overall redox‐neutral process (Scheme 7). [25] The authors were able to demonstrate the replacement of the azirine starting material with the corresponding vinyl azides furnishing pyrroles 59 in similar yield. Further control experiments pointed towards the high potential of Fukuzumi's acridinium‐based photocatalyst 57 both in terms of energy as well as of electron transfer. Mechanistically, the 2H‐azirine 60 is generated by means of an energy transfer from the excited photocatalyst. Afterwards, the 2H‐azirines 60 are oxidized by the strongly oxidizing photocatalyst leading to a ring opening under scission of the C−C bond. Addition of the resulting azaallenyl radical cation 61 to an alkyne results in intermediate 62. The catalytic cycle is closed by the single‐electron‐reduction of the radical cation 62, which finally leads to the formation of pyrrole 59 by cyclization and aromatization (Scheme 7).
Scheme 7.
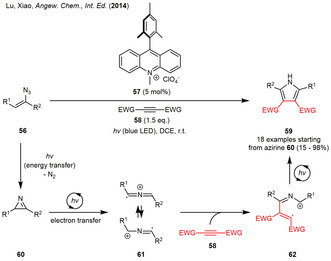
Photogenerated 2H‐azirines in the photoredox‐mediated formal [3+2] cycloaddition with alkynes forming highly substituted pyrroles.
Although the authors did not incorporate the photogeneration of azirine 60 in succeeding studies, several other groups were able to extend the demonstrated methodology to other reaction partners furnishing oxazoles, [26] 2‐acyloxazoles, [27] 2,5‐dihydro imidazoles, [26] 2,5‐dihydroisoxazoles, [28] 1,2,4‐triazoles [29] and 2,4,5‐trisubstituted pyrroles. [30]
In the following years, the Maurya group described several examples which combined the sensitized generation of 2H‐azirines with further follow‐up reactions. The first examples were published in 2016 and 2017, where the photocatalyst [Ru(bpy)3](PF6)2 was employed both as energy and electron transfer catalyst furnishing 1,3‐diazabicyclo[3.1.0]hexanes in moderate to good yields and excellent diastereoselectivity (Scheme 8). [31]
Scheme 8.
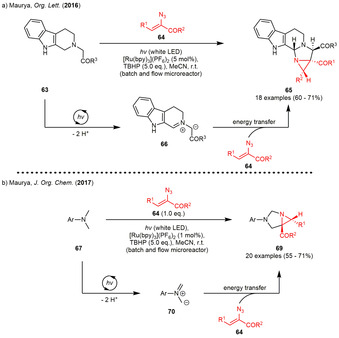
Photogenerated 2H‐azirines as dipolarophiles in [3+2] cycloadditions with photoredox‐generated 1,3‐dipoles forming (a) fused β carboline derivatives and (b) 1,3 diazabicyclo[3.1.0]hexanes.
The dual catalysis enabled by a single photocatalyst gives access to [3+2] cycloadditions of the photoredox‐/tert‐butylhydroperoxide (TBHP) generated dipoles 66 and 70 to 2H‐azirines as the dipolarophiles. The dipole formation was proposed to involve two subsequent SET‐oxidations of the tertiary amine alternating with hydroxide‐induced deprotonation. TBHP was assumed to serve as the terminal oxidant in this process, liberating the required hydroxide ions upon reductive O−O‐bond scission. The photogenerated 2H‐azirine on the other hand acts as a dipolarophile and is again produced in situ by energy transfer from the excited photocatalyst.
In the first example, 1,2,3,4‐tetrahydro‐β‐carboline derivatives (63) were incorporated into the previously described [3+2] cycloaddition yielding in highly complex 1,3‐diazabicyclo[3.1.0]hexanes 65 as single diastereomers. The authors were also able to transfer the reaction into a flow microreactor in order to decrease the reaction time while simultaneously increasing the yield by 16 % absolute. [31a] Basically the same reaction conditions could be used for [3+2] cycloadditions between photogenerated 2H‐azirines and azomethine ylides, which are generated from N,N‐dimethylanilines 67 with the photoredox/TBHP system described above. Again, a large variety of aromatic substituents on the vinyl azide substrate as well as halogen and alkyl substituents on the aniline starting materials furnished the 1,3‐diazabicyclo[3.1.0]hexanes 69 in moderate to good yield. The authors were again able to increase the yield by 19 % absolute through application of photoflow conditions. [31b]
The group also described intra‐ and intermolecular reactions of the photogenerated 2H‐azirines with phenolic hydroxy groups (Scheme 9). In 2017, an intramolecular approach was described starting from α‐azido‐γ‐(2‐hydroxyaryl)chalcones 71 and yielding 2‐aminobenzofuran 72 (Scheme 9 a) in moderate to high yields through action of p‐toluenesulfonic acid (p‐TSA). In these examples, the authors were able to induce the azirine formation without sensitization, although reaction times increased using the uncatalyzed approach. The mechanism proceeds via the intramolecular addition of the phenolic hydroxy group to the C=N bond of the photogenerated azirine 73 forming the [3.1.0]bicycle 74. This bicycle was sometimes found to be the main product when no acid was added to the reaction mixture. The product formation was presumed to be induced by an [1,2]‐acyl‐shift after acid‐catalyzed ring‐opening of the aziridine 74. [32] When the phenolic hydroxy group was replaced by a cyclic tertiary amine, the chemoselectivity changed and β‐stilbenes 77 were formed as the major products in moderate to good yields (Scheme 9 b). Moreover, no C=N addition products were observed. Instead, an intramolecular ring opening of the photogenerated azirine was proposed, forming zwitterion 79. The zwitterion was presumed to undergo a [1,2]‐acyl‐shift leading to the β‐stilbene 77 after tautomerization. [33] In 2019, an intermolecular acid‐free approach using phenols was described forming (fused) indoles in good yields (Scheme 9 c). The photocatalyst [Ru(bpy)3](PF6)2 was again used in the transformation in order to accelerate the 2H‐azirine formation. The reaction was reported to involve an initial proton transfer to the azirine enabling a nucleophilic attack of the enolate fragment inducing a dearomatization of the former phenol moiety. The formed β‐ketoaziridines 83 furnish the indole derivative 82 by ring‐opening and subsequent cyclization. [34] The latter concept was also adopted by other groups. In 2019, the Handlon group developed a metal‐free base‐induced imidazole synthesis starting from vinyl azides and cyanamides in moderate to excellent yields (Scheme 10 a). During the optimization, the authors also compared thermal and photochemical conditions for the generation of the 2H‐azirine intermediates. While the yields were slightly lower for most of the photoinduced examples, the photochemical reactions could expectably be carried out at room temperature. [35]
Scheme 9.
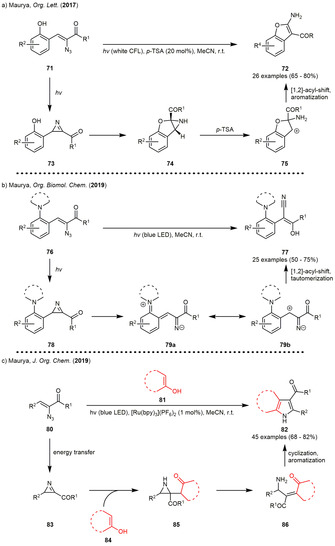
Photogenerated 2H‐azirines in (un)catalyzed strain‐releasing chemistry forming heterocycles and other rearrangement products.
Scheme 10.
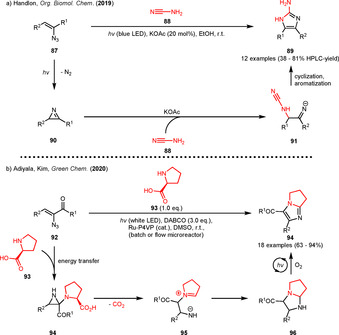
Photogenerated 2H‐azirines as intermediates in the strain‐releasing synthesis of (fused) imidazoles.
Mechanistically, the authors proposed a nucleophilic attack of the deprotonated cyanamide on C‐2 of the 2H‐azirine producing the iminyl anion 91. The anion 91 is assumed to cyclize, furnishing the imidazole product 89 after protonation and tautomerization. Based on other reports, the reaction might be more likely to proceed via an addition to the C=N bond, transprotonation, ring‐opening/ring closure and tautomerization or through intermediate formation of a [2.1.0]bicycle. [35]
Recently, the groups of Adiyala and Kim were also able to combine photoredox catalysis with the sensitized generation of 2H‐azirines in order to synthesize fused imidazoles 94 starting from α‐azidochalcones 92 and proline (93) (Scheme 10 b). [36] An immobilized ruthenium(III)‐polyvinylpyridine complex employed both under batch as well as under flow microreactor conditions afforded the fused imidazoles in good to excellent yields while tolerating a variety of mostly electron‐rich aromatic substituents on the α‐azidochalcones 92. The authors assumed the reaction to proceed via an off‐cycle addition of the amine moiety of proline into the C=N double bond of the photogenerated 2H‐azirine followed by ring‐opening, decarboxylation and cyclization giving the saturated analogue 96. Photoredox catalysis with oxygen as terminal oxidant is presumed to subsequently enable aromatization to imidazole 94 via two SET‐oxidations alternating with superoxide induced deprotonation. [36]
2‐Acylazirines from arylated isoxazoles
In 1967, the group of Singh described the photoisomerization of isoxazoles under UV irradiation leading to thermodynamically stable oxazoles. The isomerization itself is known to proceed via the formation of the highly strained and metastable 2‐acylazirines, that are further converted into the oxazoles upon continued irradiation. [37] Therefore, the Opatz group developed one‐pot procedures to trap the photoproducts in fast thermal follow‐up reactions in order to suppress the yield‐limiting oxazole formation.
In 2014, a metal‐free three‐component synthesis of tetrasubstituted imidazoles (100) was described which employs a base induced condensation of the photogenerated 2‐acylazirines (101) with α‐(alkylideneamino)‐nitriles 102 (Scheme 11). The reaction partners were also generated in situ from the corresponding aldehydes 98 and α‐aminonitriles 99 by means of a condensation reaction. Both aryl and alkyl substituents on all of the substrates could be incorporated in this transformation in moderate to high yield. Mechanistic studies and in‐silico calculations suggested the reaction to proceed via an addition of the deprotonated α‐(alkylideneamino)nitrile to the C=N double bond of the photogenerated 2‐acylazirine 101. A 5‐endo‐trig cyclization and cyanide elimination furnish the [3.1.0]bicycle 104, which is converted to imidazole 100 via proton elimination, ring‐opening and reprotonation of the formed enolate. [38] The Opatz group then focused on the use of metal‐catalysis for the downstream reactions of the photogenerated 2‐acylazirines. In 2016, a cobalt‐catalyzed condensation reaction between 1,3‐diketones 105 and photogenerated 2‐acylazirines 101 was reported. Using this methodology, tetrasubstituted pyrroles could be synthesized in moderate to excellent yields depending on the nature of the alkyl‐ or aryl‐substituents on the starting isoxazole (Scheme 12 a). [39]
Scheme 11.
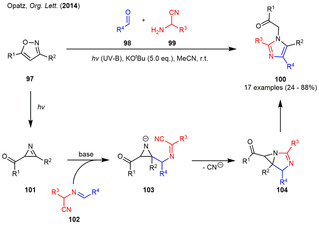
Photogenerated 2‐acylazirines in the metal‐free synthesis of tetrasubstituted imidazoles.
Scheme 12.
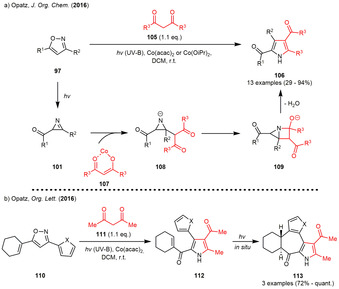
Photogenerated 2‐acylazirines in (a) metal‐catalyzed synthesis of tetrasubstituted pyrroles and (b) photoinduced vinylogous Nazarov cyclization when α,β‐unsaturated isoxazoles were incorporated.
The mechanism was proposed to involve an addition of the 1,3‐diketone‐cobalt chelate 107 to the C=N bond of the 2‐acylazirine followed by an 4‐exo‐trig cyclization forming the [2.1.0]bicycle 109. Ring‐opening and water elimination furnish the tetrasubstituted pyrrole 106. The latter concept could also be extended to α,β‐unsaturated isoxazoles. The resulting α,β‐unsaturated pyrroles instantly underwent a unexpected electrocyclization upon irradiation furnishing the vinylogous Nazarov products 113 (Scheme 12 b). [40]
In 2019, Opatz and co‐workers were able to demonstrate a metal‐catalyzed dimerization reaction of the photogenerated 2‐acylazirines 101 (Scheme 13).
Scheme 13.
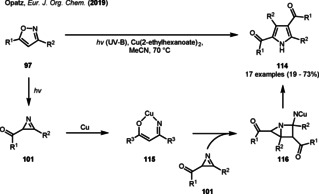
Photogenerated 2‐acylazirines in the metal‐catalyzed dimerization of tetrasubstituted imidazoles.
By employing a simple copper catalyst at elevated temperatures, aryl substituted isoxazoles were readily transformed into dimerized tetrasubstituted pyrroles (114) under UV‐irradiation in moderate to high yield. Mechanistically, the copper catalyst was presumed to oxidatively add into the C−N single bond of a 2‐acylazirine initially. Ring enlargement forms the copper chelate 115, which adds to the C=N double bond of another 2H‐azirine. The tetrasubstituted pyrrole 114 was assumed to be subsequently furnished by 4‐exo‐trig cyclization, ring opening of the formed [2.1.0]bicycle 116 to the corresponding 2H‐pyrrole and a 1,5‐sigmatropic H‐shift. [41] Recently, the Nakamura group also adopted the isoxazole/oxazole photoisomerization for a catalyst‐free synthesis of 5‐hydroxyimidazolines 118 by reacting the photogenerated 2‐acylazirines 119 with an excess of a primary amine under mediation of water (Scheme 14).
Scheme 14.
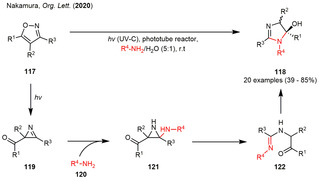
Photogenerated 2‐acylazirines in the catalyst‐free synthesis of 5 hydroxyimidazolines.
No further solvent was required in these examples. Additionally, the reaction times could be drastically reduced compared to the previous examples by performing the reactions in a thin FEP tubing wrapped around a UV‐C lamp. The products were isolated in moderate to excellent yields with the reaction tolerating several aromatic substituents on the isoxazole as well as the employment of various aliphatic amines. The authors were also able to further transform the synthesized imidazolines 118 into their unsaturated imidazole analogues with TFAA as a dehydrating agent. Mechanistically, the primary amine adds to the C=N double bond of the photogenerated 2‐acylazirine forming aziridine 121, which readily undergoes ring‐opening and isomerization to ketone 122. Cyclization and tautomerization finally yield in the 5‐hydroxyimidazoline derivatives 118. [42]
Four‐Membered Small‐Ring Systems
As the ring strain energies of the four‐membered cycloalkanes and heteroanalogues are very similar to those of their previously discussed three‐membered analogues (see Figure 1), it is not very surprising that they are frequently used in ring strain releasing chemistry as well as natural product synthesis.[ 3e , 7a , 7c ] Four‐membered ring systems can be easily accessed using for example, photochemical [2+2] cycloadditions starting from readily available precursors and therefore making them versatile intermediates in the synthesis of complex frameworks. [43] In earlier years, these photogenerated intermediates were widely applied in strain‐release methodologies under uncatalyzed as well as under Lewis‐ or Brønsted acidic conditions for natural product synthesis. Recently their unique properties and reactivities were also incorporated in more demanding transformations. The most prominent [2+2] photoproducts are cyclobutanes (derived from alkenes and enones), cyclobutenes (derived from alkenes and alkynes) and oxetanes (prepared in Paternò–Büchi reactions). Since the chemistry of oxetanes was recently reviewed elsewhere,[ 3b , 3c ] this section will focus on ring strain releasing chemistry of photogenerated cyclobutanes and cyclobutenes. The described examples mainly focus on [2+2] photocycloaddition‐fragmentation strategies using a wide variety of fragmentation conditions and therefore complement the well‐known medium‐ring annulation procedures. [44]
(Fused) cyclobutanes and cyclobutenes from enones
The use of photogenerated cyclobutanes (derived from two alkenes) and especially of α‐ketocylobutanes (derived from an α,β‐unsaturated ketone and an alkene) in natural product synthesis is well‐established. For example, Pirrung demonstrated a Cargill rearrangement [45] as a follow‐up reaction on the photogenerated cyclobutane 127 in 1979. The addressed target (±)‐isocomene (126) belongs to a family of tricyclic angular triquinane sesquiterpenes initially isolated from the plant Isocoma wrigthii. [46] The tricyclo[6.3.0.01,6]undecanone intermediate 127 was produced from an intramolecular [2+2] photocycloaddition of enone 123 and directly transformed into the tricyclic ketones 124 and 125 (Scheme 15). The Cargill reaction was afterwards induced by para‐toluenesulfonic acid (p‐TSA) to generate the cyclobutylcarbinyl cation 128 in situ, which readily underwent two [1,2]‐alkyl‐shifts furnishing the tricyclic ketones 124 (path a) and 125 (path b). The natural product 126 could afterwards be generated by methyl addition and elimination on ketone 125. [47]
Scheme 15.
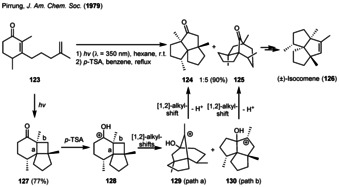
Total synthesis of (±)‐isocomene via a combination of intramolecular [2+2] photocycloaddition and subsequent Cargill rearrangement as the key step.
A more recent example of a (Lewis acid‐promoted) skeletal rearrangement of photogenerated cyclobutanes in natural product synthesis was described by Kakiuchi and co‐workers in 2004. Lewis acid‐catalyzed ring expansion of the photogenerated fused α‐keto‐exo‐methylencyclobutane 135 yields in the triquinane structure 133, [48] which has previously been converted to the natural product (±)‐pentalenene (134) by the group of Paquette. [49] Among a variety of Lewis‐ or Brønsted acids such as boron trifluoride etherate (BF3⋅OEt2) or triflic acid (TfOH), only titanium(IV)‐chloride (TiCl4) afforded the desired product 133 in excellent yield (Scheme 16). The carbonyl group of the photogenerated cyclobutane 135 was selectively addressed by the titanium catalyst in the presence of the highly reactive exo‐methylene moiety, which is attacked by Brønsted acids instead. Under these conditions, ring‐opening selectively occurs under C1‐C6 scission furnishing cation 136, which readily undergoes a [1,2]‐hydride‐ shift and cyclization yielding in the tricyclic triquinane structure 133. [48]
Scheme 16.
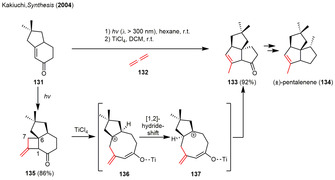
Allene‐enone [2+2] photocycloaddition and Lewis acid‐catalyzed rearrangement towards in the formal total synthesis of (±)‐pentalenene.
The combination of the ring strain of cyclobutanes with the reactivity of the α‐keto group was also employed in radical reactions. In 1996, the group of Crimmins described a free radical ring‐expansion reaction via fragmentation of fused α‐ketocyclobutanes 140, which were generated by means of a stereoselective intramolecular [2+2] cycloaddition of the enone and terminal alkene moiety of substrate 138. Removal of the methoxymethyl ether (MOM) protecting group followed by the Barton‐McCombie sequence (thioacylation and subsequent free radical reduction) afforded the ring‐expanded carbocycles 139 in moderate to excellent yield. The regioselective bond scission was assumed to result from the stabilization of the radical intermediate by the two neighboring carbonyl groups. The authors were also able to extend the sequence to different medium‐sized fused bicycles by introducing different ring sizes and tethered terminal alkene chains into the substrate (Scheme 17). [50]
Scheme 17.
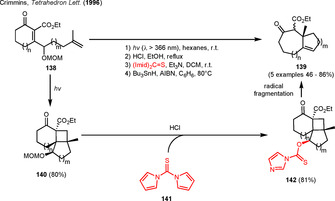
Photocycloaddition‐free radical fragmentation sequence in the synthesis of medium‐sized bicycles.
In order to introduce an alkene moiety into the extended bicyclic ring system 144, the groups of Shipe and Sorensen described a different approach in the enantioselective synthesis of the guanacastepene diterpenes A and E. Again, an intramolecular enone‐olefin [2+2] photocycloaddition furnished the strained α‐ketocyclobutane 145. Ring opening of 145 was introduced using a remote single electron transfer reduction of the α‐carbonyl by samarium diiodide. Subsequent ring‐opening and trapping of the samarium enolate with phenylselenenyl bromide (146) afforded the organoselenide 147. Elimination after oxidation with meta‐chlorperbenzoic acid (m‐CPBA) finally afforded the tricyclic compound 144 (Scheme 18). [51]
Scheme 18.
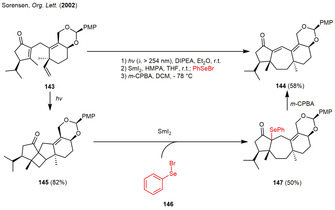
Radical fragmentation of a photogenerated cyclobutane in the synthesis of (+) guanacastepene A and E.
(Fused) cyclobutanes from vinylogous amides
The extension of the previously discussed photocycloaddition/fragmentation sequence to vinylogous amides/enaminones leads to nitrogen‐containing ring systems and is therefore frequently used in natural product synthesis for the construction of alkaloid structures. Based on the first contribution of Tamura and co‐workers in 1975, [52] several groups adopted the photocycloaddition/retro‐Mannich sequence in order to synthesize various natural products like sesquiterpenes, [53] taxane, [54] mesembrine, [55] vindorosine, [56] manzamine A, [57] koumine, [58] (±)‐coerulescine, [59] and peduncularine. [60] As the sequence was also incorporated in several methodological publications,[ 59 , 61 ] only selected more recent examples of the topic will be described below in order to showcase the reactivity of the photoadducts.
The Winkler group described numerous reaction sequences building on the ring‐opening of photogenerated ring systems as key steps and incorporated these reactions in the synthesis of several natural products. In 2001, the group reported a photocyclization of the Boc‐prototected vinylamide 148 furnishing the [2.1.1]bicycle 151 (Scheme 19 a). The preferred formation of the [2.1.1]‐ over the [2.2.0]bicyclic system was assumed to be based on kinetic reasons (“rule of five”). Subsequent thermal decomposition and acid‐catalyzed cyclization furnished the [3.2.1]bicycle 149 in excellent yield. The transfer of the developed methodology to a cyclohexenyl derivative afforded tricycle 150, which can be viewed as the core of the carbon skeleton of the hetisine alkaloids. [61b] In 2006, the methodology was further adopted in the total synthesis of the natural product peduncularine. [60]
Scheme 19.
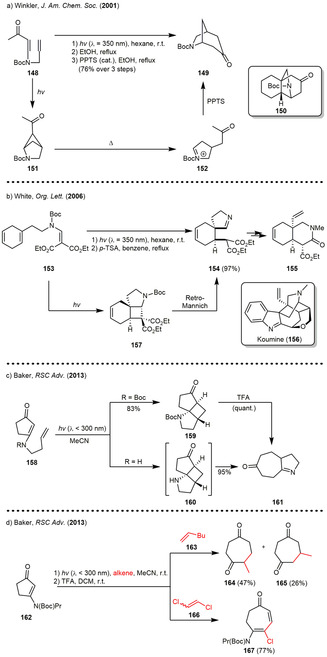
Photocyclization/retro‐Mannich sequences in (a) the synthesis of the tricyclic core of the hetisine alkaloids, (b) the total synthesis of koumine (c) the comparison between unprototected (R=H) and protected (R=Boc) vinylamide precursors and (d) intermolecular sequences.
In the same year, White employed β‐phenethylamine 153 in the synthesis of the spiropyrroline 154, which was used as key intermediate for the construction of the tetracyclic core of the indolenine alkaloid koumine (156) (Scheme 19 b). An intramolecular photocycloaddition of β‐aminoalkylidene malonate 153 furnished cyclobutane 157 in almost quantitative yield. Upon removal of the Boc‐protecting group with trifluoroacetic acid (TFA), tricyclic cyclobutane 157 spontaneously underwent a retro‐Mannich fragmentation forming spiroimine 154. The latter product was isolated as a single stereoisomer indicating both the concerted mechanism of the photocycloaddition as well as the stereospecificity of the retro‐Mannich reaction. [58]
In 2013, the Baker group described a variety of both intra‐ and intermolecular [2+2] cycloadditions of alkenes with vinylamides 158 and 162 followed by retro‐Mannich reactions leading to fused 1‐pyrrolines, diketones, ketoimines or conjugated enaminones (Scheme 19 c and d). In terms of intramolecular reactions, the group was able to confirm earlier observations[ 52 , 61a , 62 ] of such sequences, which pointed out, that the tricyclic fused cyclobutanes 159/160 can only be isolated if the amine was Boc‐protected, whereas both deprotection as well as irradiation of the unprotected vinylamide resulted in spontaneous retro‐Mannich‐type ring enlargement reactions of the fused cyclobutane 160 yielding in tricyclic 1‐pyrroline 161. In these examples, the authors were able to suppress the retro‐Mannich reaction by sodium borohydride‐induced reduction of the keto function of the tricyclic cyclobutane 159. In the same publication, vinylogous amides 162 not tethered with terminal alkenes could be incorporated in intermolecular [2+2] cycloaddition‐retro‐Mannich sequences using different alkenes in order to synthesize 1,4‐diketocycloheptanes 164/165 or conjugated enaminone 167 depending on the alkene used.
de Mayo and related reactions
A significant contribution to the field of photochemistry was made by the de Mayo group in 1962, who described the synthesis of 1,5‐diketones through a two‐step process involving the [2+2] photocycloaddition of an enolizable 1,3‐dicarbonyl compound with an olefin forming a cyclobutanol intermediate, which is readily fragmented via a retro‐aldol reaction yielding in 1,5‐dicarbonyl compounds. [63] The sequence is frequently incorporated in organic chemistry and especially in natural product total synthesis. Some of the recent examples are shown in Scheme 20 and will be discussed below.
Scheme 20.
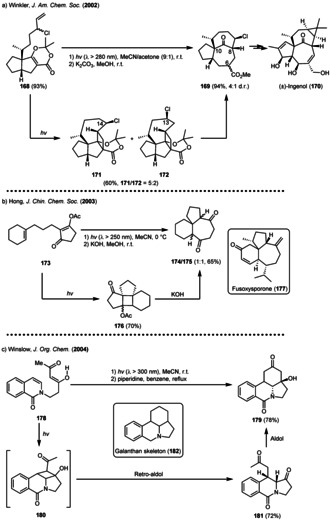
de Mayo‐type reactions in the natural product synthesis of (a) (±) ingenol (b) fusoxysporone and (c) the galanthan skeleton.
In 2002, Winkler and co‐workers reported the first total synthesis of (±)‐ingenol 170, a complex diterpenoid originally isolated from Euphorbia ingens. [64] The authors were able to establish the unusual C‐8/C‐10 „inside‐outside“ stereochemistry of 170 by using a dioxenone photocycloaddition/fragmentation sequence. An intramolecular [2+2] photocycloaddition of the allylic chloride 168 gave cyclobutane 171 together with its C‐13‐chloro‐analogue 172 (171/172 = 5:2). Fragmentation of cyclobutane 171 with methanolic potassium carbonate finally yielded in the ring‐enlarged tricycle 169 in 40 % yield over two steps. Tricycle 169 could be subsequently converted to (±)‐ingenol 170 (Scheme 20 a). [65]
In 2003, the group of Hong described a photochemical access towards the tricyclic core of fusoxysporone 177, [66] a viscidane‐type diterpene first isolated from liquid cultures of the fungus Fusarium oxysporum in 1992. [67] The authors irradiated the β‐acetoxy‐substituted enone 173 yielding in cyclobutane 177. Treatment with aqueous KOH in methanol afforded a racemic mixture of the desired tricyclic diones 174 and 175 (Scheme 20 b). Another example was reported by Minter and Winslow, who used a de Mayo‐approach towards the synthesis of the galanthan tetracyclic skeleton 182. [68] To this end, isocarbostyril 178 was irradiated in acetonitrile to give 181 in 72 % yield. Mechanistically, a remarkable regioselective intramolecular [2+2] photocycloaddition afforded the corresponding cyclobutane 180, which subsequently underwent a retro‐aldol fragmentation yielding in tricycle 181. A piperidine‐mediated aldol cyclization gave galanthan derivative 181 as single diastereomer, as the C1‐carbonyl is selectively attacked from the endo‐face (Scheme 20 c).
Cyclobutanes as Norrish–Yang products
A different approach for the synthesis of highly reactive cyclobutanes are the Norrish–Yang reactions, which proceed via an H‐atom transfer (HAT) after excitation of a carbonyl compound containing abstractable γ‐hydrogen atoms. The formed biradicals usually recombine to cyclobutanols. Although Norrish–Yang conditions are frequently used in photochemical reactions leading to cyclic compounds and natural products, [69] the use of these carbocyclic photoadducts as strain‐releasing intermediates are rare. Nevertheless, in 2014 the Murakami group described a ring expansion reaction of orthocyclophanes 183 to the more strained metacyclophanes 184. The described reaction combines a light‐induced Norrish‐Yang type cyclization yielding in cyclobutanol (±)‐185, which was afterwards incorporated in a subsequent rhodium‐mediated C−C activation reaction. The example demonstrates that the use of an initial endergonic photocyclization reaction enables a formal endergonic reaction sequence. Mechanistically, excitation of the carbonyl group using UV‐irradiation formed the already mentioned 1,4‐biradicals that readily generated o‐quinodimethanes 184 as mixture of diastereomers. The cyclobutanol (±)‐185 was only generated from the (Z)‐isomer by means of a 8π‐electrocyclization, whereas the (E)‐isomer having an inward‐oriented hydroxy group was more likely to undergo a 1,5‐proton shift to regenerate 183. Deprotonation and ligand exchange of the rhodium‐hydroxide complex with the photogenerated cyclobutanol (±)‐185 generated rhodium benzocyclobutenolate 186, in which the benzene ring likely π‐coordinated to rhodium. The following β‐carbon elimination of the C(ipso)−C(sp3) bond formed arylrhodium complex 187. Finally, protonation furnished the metacyclophane 184 (Scheme 21 a). The arylrhodium intermediate could also be incorporated into a subsequent conjugated addition with methyl vinyl ketone 190 giving rise to the stereospecific synthesis of metacyclophanes (±)‐191, when bulky substituents such as alkyltrimethylsilane, benzyl or phenoxymethyl were used as R1. The authors were able to show that irradiation of precursor 188 gave tricyclic benzocyclobutenol (±)‐189 as only diastereomer because of steric and kinetic (vide supra) reasons (Scheme 21 b). The rhodium‐mediated ring‐opening was assumed to occur under central‐to‐planar chirality transfer as the coordinating benzene facilitates a site‐selective migration of the ipso‐sp2‐carbon onto the rhodium center. The bulky COD‐ligand was assumed to prohibit the ring flipping across the newly expanded ansa‐chain afterwards. [70]
Scheme 21.
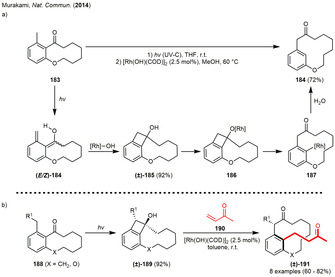
Rhodium‐catalyzed ring‐enlargement reaction via a Norrish–Yang‐cyclization/metal‐catalyzed fragmentation sequence in the synthesis of (a) metacyclophane and (b) substituted metacyclophane generated by a subsequent C−H activation.
Azetidines as Norrish–Yang products
In recent years, Norrish–Yang reactions were also incorporated into the synthesis of azetidines as highly reactive photogenerated intermediates. As azetidines almost possess the same ring strain energy as their lighter aziridine homologues, it is not surprising that they are also suitable as synthetically valuable intermediates capable of undergoing various ring opening transformations. [71]
In 2013, the Murakami group reported the synthesis of substituted indoles in moderate to high yield by combination of a sunlight‐induced Norrish–Yang reaction with a subsequent rhodium‐catalyzed ring‐opening‐cyclization sequence (Scheme 22). The 2‐(N‐aryl‐N‐methylamino) acetophenones 192 were irradiated in a Pyrex flask equipped with a previously exposed sunlight collector for 8 h on a sunny day. The subsequent addition of the rhodium catalyst followed by treatment with aqueous HCl led to the formation of indoles 193 in moderate to good yields. Irradiation of α‐aminoketone 192 resulted in the expected Norrish–Yang photoproduct 195. Rhodium‐mediated ring‐opening of the 3‐azetidinol 195 and subsequent 1,4‐rhodium shift furnished intermediate 197, which underwent an intramolecular addition to the carbonyl group. The generated hydroxyindoline 198 was aromatized using acidic conditions yielding in a variety of indoles 193 in moderate to good yield (Scheme 22 a). The authors reported the sequence to be highly regioselective when unsymmetrically substituted α‐aminoketones 192 were used. [72]
Scheme 22.
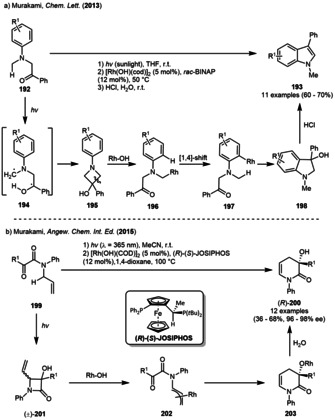
Photogenerated azetidinols as intermediates in the rhodium‐mediated (a) synthesis of indole derivatives and (b) enantioselective preparation of 1,4‐dihydropyridin‐2‐ones.
In 2015, the Murakami group described a related approach starting from N‐allylglyoxylamides 199 to construct (3R)‐3‐hydroxy‐1,4‐dihydropyridin‐2‐ones (200) in moderate yield but excellent enantioselectivity. The sequence was again based on the combination of a Norrish–Yang photogeneration of azetidinol (±)‐201 with a rhodium‐catalyzed ring‐opening‐cyclization follow‐up reaction. The group was able to perform the reaction in an enantioselective fashion by using the chiral (R)‐(S)‐JOSIPHOS ligand (Scheme 22 b). The irradiation of the glyoxylamides 199 with a blue LED lamp generated the four‐membered lactam (±)‐201 as a mixture of diastereomers. The subsequent rhodium‐mediated reaction using a combination of [Rh(OH)(COD)]2 and chiral (R)‐(S)‐JOSIPHOS ligand furnished 1,4‐dihydropyridin‐2‐ones (R)‐200 with excellent ee. Mechanistically, deprotonation and ligand exchange of the rhodium catalyst followed by ring‐opening afforded the metal‐allyl‐complex 202. Rhodium‐catalyzed nucleophilic addition to the α‐carbonyl furnished the six‐membered metal‐bound‐lactam 203. Addition of water gave (3R)‐3‐hydroxy‐1,4‐dihydropyridin‐2‐ones (R)‐200. [72]
In 2019, the Baxendale group began to contribute to the field of photogenerated azetidinol intermediates by applying photoflow conditions in their synthesis. [73] The group was able to incorporate the photogenerated azetidinols 207 in a Ritter‐type‐reaction with several aliphatic and aromatic nitriles followed by an acid‐catalyzed rearrangement reaction furnishing oxazolines 206 in moderate to excellent yield (Scheme 23). Mechanistically, irradiation of sulfonamides 204 under photo‐flow conditions furnished the Norrish–Yang products 207. Acid‐induced elimination of water followed by addition to the nitrile and hydrolysis of the nitrilium‐ion gave amide 208 that undergoes a strain‐releasing ring‐enlargement rearrangement furnishing oxazolines 206 via nucleophilic attack of the carbonyl at the polarized proximal C−N bond. Applying the same reaction conditions, the authors were also able to introduce oxygen and sulfur‐based nucleophiles in order to synthesize 2,3‐dihydrobenzofurans and sulfur‐substituted azetidines. [74]
Scheme 23.
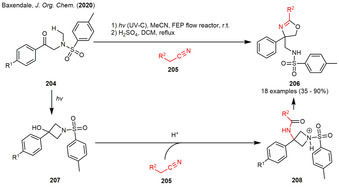
Photogenerated N‐tosylated azetidinols in a Ritter‐type reaction furnishing oxazolines.
Very recently, the groups of Musaev, Yeung, and Sarpong reported the use of photogenerated azetidinol 212 in a hitherto unknown palladium‐catalyzed C−C cross coupling reaction with aryl and alkenyl bromides as well as triflates to generate α‐functionalized piperidines 211 (Scheme 24).
Scheme 24.
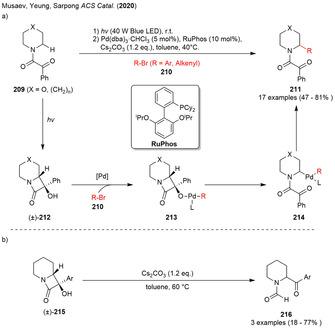
Photogenerated N‐fused bicyclo‐β‐lactams in (a) the palladium‐mediated α‐functionalization of piperidines and analogues as well as (b) base‐mediated α‐acetylation of piperidines.
Irradiation of the phenylglyoxylamides 209 in the solid state with blue LEDs lead the formation of N‐fused bicyclo‐β‐lactams (±)‐212 through a Norrish‐Yang reaction. The azetidinols (±)‐212 were subsequently incorporated in the palladium‐mediated α‐functionalization reaction as masked nucleophiles using aryl and alkenyl bromides and triflates as cross‐coupling partners. Optimization studies showed the use of RuPhos as a ligand to be crucial for obtaining decent yields of the desired product 211. The authors reported a plausible rection mechanism consistent with the proposal by Uemura in 1999. [75] Oxidative addition of the palladium(0)‐catalyst in the aryl bromide formed a palladium(II) complex. Deprotonation and ligand exchange furnished intermediate 213, which underwent β‐C−C cleavage in order to generate α‐palladated piperidine 214. The catalytic cycle was closed by reductive elimination liberating the α‐arylated product 211 and the active catalyst (Scheme 24 a). The phenylglyoxyl directing group could be removed after the reaction under basic conditions. [76]
Additionally, a control experiment without the palladium catalyst starting from azetidinols (±)‐215 revealed a Cs2CO3‐promoted rearrangement yielding in α‐acylated piperidines 216. Product formation was assumed to be the result of base‐induced cleavage of the distal bond of the azetidinols (±)‐215. Both experimental as also computational evidence pointed towards the importance of the cesium counterion for this transformation (Scheme 24 b). [76]
Conclusions and Outlook
Photochemical reactions contribute to the emerging field of sustainable chemistry, since the driving force of these transformations is introduced into the molecule by photon absorption and therefore can provide very mild and potentially eco‐friendly reaction conditions. The high excitation energy transferred in particular by UV irradiation leads to high energy intermediates and products. Accordingly, the so called uphill‐photochemistry enables photoinduced isomerizations and cycloadditions leading to a versatile toolbox of strained three‐ and four‐membered photoproducts. Impressive progress in this field was made by developing efficient photoflow conditions for the synthesis of such strained photoproducts. The latter can subsequently be subjected to numerous strain‐releasing reactions, in which their inherent ring strain energy serves as a driving force facilitating very mild and sustainable conditions for the overall sequences compared to the generation of low‐energy co‐products in classical bond activation procedures. This review features recent contributions to the field of photogenerated strained intermediates. While earlier reports clearly focused on acid‐ or uncatalyzed approaches, recent contributions open the field to metal‐catalyzed C−C activations in order to access structurally complex products. The use of very potent photocatalysts in terms of both energy transfer as well as photoinduced electron transfer (PET) might provide further innovations with respect to sustainability by applying visible light for such transformations. The progress made in this area has so far led to powerful methodologies suitable for the construction of natural products or biologically active molecules. The exceptional reactivity and structural features of photogenerated strained ring systems will likely lead to broader applications in various areas of the chemical sciences. In strained photoproducts, single bonds which are difficult to activate in other settings can be broken and suitable catalysts can take control over these processes. Classical synthetic methods often achieve bond activations or bond forming processes through the generation of low‐energy co‐products (e.g. metal halides or other salts as in classical cross‐couplings) which must either be recycled or disposed of. In contrast, photoactivation/strain‐release can even produce final products of higher energy content than the starting materials without the generation of any other potentially undesired molecular entities. Its inherent sustainable nature holds promises for more environmentally benign synthetic approaches and thus fits well into the general context of current research activities worldwide.
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
Adriana Luque received her bachelor's degree in chemistry at UIS (Colombia) in 2013. After obtaining her master's degree at UNAM (Mexico) in 2016, she started working as a graduate student in the Opatz lab at JGU Mainz. Her current research interests focus on catalyzed ring‐opening reactions of stable photoproducts.

Biographical Information
Jan Paternoga graduated at the University of Mainz (B.Sc in 2015 and honoured M.Sc in 2017). During his studies he received a DAAD scholarship for visiting the lab of Prof. Winnik at the University of Toronto. Currently, he is working on his PhD in the group of Prof. Opatz. His research interests focus on telescoping or one‐pot strain‐releasing sequences of photoproducts.

Biographical Information
Till Opatz graduated from Frankfurt University in 1997 and completed his PhD at JGU Mainz with Prof. Horst Kunz in 2001. After a postdoctoral stay in Utrecht, he returned to JGU for his habilitation. In 2007, he was appointed professor at Hamburg University and returned again to JGU as a full professor in 2010. His research focuses on new synthetic methods, bioactive compounds, natural products and sustainable chemistry.

Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft (Grant Op90/12‐1). Open access funding enabled and organized by Projekt DEAL.
A. Luque, J. Paternoga, T. Opatz, Chem. Eur. J. 2021, 27, 4500.
References
- 1. Baeyer A., Ber. Dtsch. Chem. Ges. 1885, 18, 2269–2281. [Google Scholar]
- 2.
- 2a. Blunt J. W., Copp B. R., Munro M. H., Northcote P. T., Prinsep M. R., Nat. Prod. Rep. 2011, 28, 196–268; [DOI] [PubMed] [Google Scholar]
- 2b. Heterocycles in Natural Product Synthesis (Eds: Majumdar K. C., Chattopadhyay S. K.), Wiley-VCH, Weinheim, 2011; [Google Scholar]
- 2c. Chen D. Y.-K., Pouwer R. H., Richard J.-A., Chem. Soc. Rev. 2012, 41, 4631–4642; [DOI] [PubMed] [Google Scholar]
- 2d. Gulder T., Baran P. S., Nat. Prod. Rep. 2012, 29, 899–934; [DOI] [PubMed] [Google Scholar]
- 2e. Fan Y.-Y., Gao X.-H., Yue J.-M., Sci. China Chem. 2016, 59, 1126–1141. [Google Scholar]
- 3.
- 3a. Namyslo J. C., Kaufmann D. E., Chem. Rev. 2003, 103, 1485–1538; [DOI] [PubMed] [Google Scholar]
- 3b. Ahmad S., Yousaf M., Mansha A., Rasool N., Zahoor A. F., Hafeez F., Rizvi S. M. A., Synth. Commun. 2016, 46, 1397–1416; [Google Scholar]
- 3c. Bull J. A., Croft R. A., Davis O. A., Doran R., Morgan K. F., Chem. Rev. 2016, 116, 12150–12233; [DOI] [PubMed] [Google Scholar]
- 3d. Badenock J. C., Prog. Heterocycl. Chem. Vol. 30, Elsevier, 2018, pp. 43–76; [Google Scholar]
- 3e. Anaya J., Sánchez R. M., in Prog. Heterocycl. Chem. Vol. 30, Elsevier, 2018, pp. 77–108; [Google Scholar]
- 3f. Rotstein B. H., Zaretsky S., Rai V., Yudin A. K., Chem. Rev. 2014, 114, 8323–8359. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Lefebvre C., Fortier L., Hoffmann N., Eur. J. Org. Chem. 2020, 1393–1404; [Google Scholar]
- 4b. Kärkäs M. D., J. A. Porco Jr , Stephenson C. R., Chem. Rev. 2016, 116, 9683–9747; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4c. D′auria M., Racioppi R., Molecules 2013, 18, 11384–11428; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4d. Kaur N., J. Heterocycl. Chem. 2019, 56, 1141–1167. [Google Scholar]
- 5.
- 5a. Dudev T., Lim C., J. Am. Chem. Soc. 1998, 120, 4450–4458; [Google Scholar]
- 5b. Bach R. D., Dmitrenko O., J. Org. Chem. 2002, 67, 2588–2599; [DOI] [PubMed] [Google Scholar]
- 5c. Khoury P. R., Goddard J. D., Tam W., Tetrahedron 2004, 60, 8103–8112; [Google Scholar]
- 5d. Bach R. D., Dmitrenko O., J. Am. Chem. Soc. 2006, 128, 4598–4611; [DOI] [PubMed] [Google Scholar]
- 5e. Padwa A., in Adv. Heterocycl. Chem. Vol. 99, Elsevier, 2010, pp. 1–31. [Google Scholar]
- 6. Zhou Q. Q., Zou Y. Q., Lu L. Q., Xiao W. J., Angew. Chem. Int. Ed. 2019, 58, 1586–1604; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 1600–1619. [Google Scholar]
- 7.For recent review articles on transition-metal-catalyzed activation of strained substrates, see:
- 7a. Souillart L., Cramer N., Chem. Rev. 2015, 115, 9410–9464; [DOI] [PubMed] [Google Scholar]
- 7b. Wang F., Yu S., Li X., Chem. Soc. Rev. 2016, 45, 6462–6477; [DOI] [PubMed] [Google Scholar]
- 7c. Fumagalli G., Stanton S., Bower J. F., Chem. Rev. 2017, 117, 9404–9432; [DOI] [PubMed] [Google Scholar]
- 7d. Shah T. A., De P. B., Pradhan S., Banerjee S., Punniyamurthy T., Chem. Asian J. 2019, 14, 4520–4533; [DOI] [PubMed] [Google Scholar]
- 7e. Seiser T., Cramer N., Org. Biomol. Chem. 2009, 7, 2835–2840; [DOI] [PubMed] [Google Scholar]
- 7f. Kanazawa J., Uchiyama M., Synlett 2019, 30, 1–11. [Google Scholar]
- 8.For recent review articles on photoredox chemistry in general and strained substrates in particular, see:
- 8a. Xuan J., He X.-K., Xiao W.-J., Chem. Soc. Rev. 2020, 49, 2546–2556; [DOI] [PubMed] [Google Scholar]
- 8b. Prier C. K., Rankic D. A., MacMillan D. W., Chem. Rev. 2013, 113, 5322–5363; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8c. Romero N. A., Nicewicz D. A., Chem. Rev. 2016, 116, 10075–10166. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Stanković S., D′hooghe M., Catak S., Eum H., Waroquier M., Van Speybroeck V., De Kimpe N., Ha H.-J., Chem. Soc. Rev. 2012, 41, 643–665; [DOI] [PubMed] [Google Scholar]
- 9b. Ohno H., Chem. Rev. 2014, 114, 7784–7814; [DOI] [PubMed] [Google Scholar]
- 9c. Nielsen I. M. B., J. Phys. Chem. A 1998, 102, 3193–3201. [Google Scholar]
- 10. Kaplan L., Wilzbach K. E., Pavlik J. W., J. Am. Chem. Soc. 1972, 94, 3283. [Google Scholar]
- 11.
- 11a. Ling R., Yoshida M., Mariano P. S., J. Org. Chem. 1996, 61, 4439–4449; [DOI] [PubMed] [Google Scholar]
- 11b. Glarner F., Thornton S. R., Scharer D., Bernardinelli G., Burger U., Helv. Chim. Acta 1997, 80, 121–127; [Google Scholar]
- 11c. Zou J., Mariano P. S., Photochem. Photobiol. Sci. 2008, 7, 393–404; [DOI] [PubMed] [Google Scholar]
- 11d. Sowmiah S., Esperança J. M. S. S., Rebelo L. P. N., Afonso C. A. M., Org. Chem. Front. 2018, 5, 453–493; [Google Scholar]
- 11e. Damiano T., Morton D., Nelson A., Org. Biomol. Chem. 2007, 5, 2735–2752. [DOI] [PubMed] [Google Scholar]
- 12. Zhao Z., Song L., Mariano P. S., Tetrahedron 2005, 61, 8888–8894. [Google Scholar]
- 13. Ling R., Mariano P. S., J. Org. Chem. 1998, 63, 6072–6076. [DOI] [PubMed] [Google Scholar]
- 14. Song L., Duesler E. N., Mariano P. S., J. Org. Chem. 2004, 69, 7284–7293. [DOI] [PubMed] [Google Scholar]
- 15. Zhao Z., Mariano P. S., Tetrahedron 2006, 62, 7266–7273. [Google Scholar]
- 16. Penkett C. S., Simpson I. D., Tetrahedron Lett. 2001, 42, 1179–1181. [Google Scholar]
- 17. Corey E. J., Boaz N. W., Tetrahedron Lett. 1984, 25, 3063–3066. [Google Scholar]
- 18. Zhao Z. M., Duesler E., Wang C. H., Guo H., Mariano P. S., J. Org. Chem. 2005, 70, 8508–8512. [DOI] [PubMed] [Google Scholar]
- 19. Oliveira J. A. C., Kiala G., Siopa F., Bernard A., Gontard G., Oble J., Afonso C. A. M., Poli G., Tetrahedron 2020, 131182. [Google Scholar]
- 20. Maskill K. G., Knowles J. P., Elliott L. D., Alder R. W., Booker-Milburn K. I., Angew. Chem. Int. Ed. 2013, 52, 1499–1502; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 1539–1542. [Google Scholar]
- 21. Blackham E. E., Knowles J. P., Burgess J., Booker-Milburn K. I., Chem. Sci. 2016, 7, 2302–2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Blackham E. E., Booker-Milburn K. I., Angew. Chem. Int. Ed. 2017, 56, 6613–6616; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 6713–6716. [Google Scholar]
- 23.
- 23a. Neber P., Friedolsheim A. V., Justus Liebigs Ann. Chem. 1926, 449, 109–134; [Google Scholar]
- 23b. Smolinsky G., J. Am. Chem. Soc. 1961, 83, 4483–4484. [Google Scholar]
- 24. Farney E. P., Yoon T. P., Angew. Chem. Int. Ed. 2014, 53, 793–797; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 812–816. [Google Scholar]
- 25. Xuan J., Xia X.-D., Zeng T.-T., Feng Z.-J., Chen J.-R., Lu L.-Q., Xiao W.-J., Angew. Chem. Int. Ed. 2014, 53, 5653–5656; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 5759–5762. [Google Scholar]
- 26. Zeng T.-T., Xuan J., Ding W., Wang K., Lu L.-Q., Xiao W.-J., Org. Lett. 2015, 17, 4070–4073. [DOI] [PubMed] [Google Scholar]
- 27. Chen L., Li H., Li P., Wang L., Org. Lett. 2016, 18, 3646–3649. [DOI] [PubMed] [Google Scholar]
- 28. Cai B.-G., Chen Z.-L., Xu G.-Y., Xuan J., Xiao W.-J., Org. Lett. 2019, 21, 4234–4238. [DOI] [PubMed] [Google Scholar]
- 29. Wang H., Ren Y., Wang K., Man Y., Xiang Y., Li N., Tang B., Chem. Commun. 2017, 53, 9644–9647. [DOI] [PubMed] [Google Scholar]
- 30. Karki B. S., Devi L., Pokhriyal A., Kant R., Rastogi N., Chem. Asian J. 2019, 14, 4793–4797. [DOI] [PubMed] [Google Scholar]
- 31.
- 31a. Chandrasekhar D., Borra S., Nanubolu J. B., Maurya R. A., Org. Lett. 2016, 18, 2974–2977; [DOI] [PubMed] [Google Scholar]
- 31b. Borra S., Chandrasekhar D., Adhikary S., Rasala S., Gokulnath S., Maurya R. A., J. Org. Chem. 2017, 82, 2249–2256. [DOI] [PubMed] [Google Scholar]
- 32. Borra S., Chandrasekhar D., Khound S., Maurya R. A., Org. Lett. 2017, 19, 5364–5367. [DOI] [PubMed] [Google Scholar]
- 33. Borra S., Borkotoky L., Newar U. D., Kalwar A., Das B., Maurya R. A., Org. Biomol. Chem. 2019, 17, 5971–5981. [DOI] [PubMed] [Google Scholar]
- 34. Borra S., Chandrasekhar D., Newar U. D., Maurya R. A., J. Org. Chem. 2019, 84, 1042–1052. [DOI] [PubMed] [Google Scholar]
- 35. Man L., Copley R. C., Handlon A. L., Org. Biomol. Chem. 2019, 17, 6566–6569. [DOI] [PubMed] [Google Scholar]
- 36. Adiyala P. R., Jang S., Vishwakarma N. K., Hwang Y.-H., Kim D.-P., Green Chem. 2020, 22, 1565–1571. [Google Scholar]
- 37.
- 37a. Singh B., Ullman E. F., J. Am. Chem. Soc. 1967, 89, 6911–6916; [Google Scholar]
- 37b. Singh B., Zweig A., Gallivan J., J. Am. Chem. Soc. 1972, 94, 1199–1206. [Google Scholar]
- 38. Pusch S., Opatz T., Org. Lett. 2014, 16, 5430–5433. [DOI] [PubMed] [Google Scholar]
- 39. Pusch S., Kowalczyk D., Opatz T., J. Org. Chem. 2016, 81, 4170–4178. [DOI] [PubMed] [Google Scholar]
- 40. Pusch S., Schollmeyer D., Opatz T., Org. Lett. 2016, 18, 3043–3045. [DOI] [PubMed] [Google Scholar]
- 41. Paternoga J., Opatz T., Eur. J. Org. Chem. 2019, 7067–7078. [Google Scholar]
- 42. Morita T., Fuse S., Nakamura H., Org. Lett. 2020, 22, 3460–3463. [DOI] [PubMed] [Google Scholar]
- 43.
- 43a. Lee-Ruff E., Mladenova G., Chem. Rev. 2003, 103, 1449–1483; [DOI] [PubMed] [Google Scholar]
- 43b. Xu Y., Conner M. L., Brown M. K., Angew. Chem. Int. Ed. 2015, 54, 11918–11928; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 12086–12097. [Google Scholar]
- 44. Iriondo-Alberdi J., Greaney M. F., Eur. J. Org. Chem. 2007, 4801–4815. [Google Scholar]
- 45. Cargill R. L. B., Maurice E., Siebert A. E., Dorn J., J. Org. Chem. 1965, 30, 3647–3650. [Google Scholar]
- 46. Zalkow L. H., Harris R. N., Vanderveer D., Bertrand J. A., J. Chem. Soc. Chem. Commun. 1977, 456–457. [Google Scholar]
- 47. Pirrung M. C., J. Am. Chem. Soc. 1979, 101, 7130–7131. [Google Scholar]
- 48. Kakiuchi K., Morimoto T., Horiguchi T., Yamada K., Tsutsumi K., Kurosawa H., Synthesis 2004, 2004, 753–756. [Google Scholar]
- 49. Paquette L. A. A., Gary D., J. Am. Chem. Soc. 1983, 105, 7358–7363. [Google Scholar]
- 50. Crimmins M. T., Huang S. J., GuiseZawacki L. E., Tetrahedron Lett. 1996, 37, 6519–6522. [Google Scholar]
- 51.
- 51a. Shipe W. D., Sorensen E. J., Org. Lett. 2002, 4, 2063–2066; [DOI] [PubMed] [Google Scholar]
- 51b. Shipe W. D., Sorensen E. J., J. Am. Chem. Soc. 2006, 128, 7025–7035. [DOI] [PubMed] [Google Scholar]
- 52. Tamura Y., Ishibashi H., Hirai M., Kita Y., Ikeda M., J. Org. Chem. 1975, 40, 2702–2710. [Google Scholar]
- 53. Amougay A., Pete J.-P., Piva O., Tetrahedron Lett. 1992, 33, 7347–7350. [Google Scholar]
- 54. Swindell C. S., Patel B. P., Tetrahedron Lett. 1987, 28, 5275–5278. [Google Scholar]
- 55. Winkler J. D., Muller C. L., Scott R. D., J. Am. Chem. Soc. 1988, 110, 4831–4832. [Google Scholar]
- 56. Winkler J. D., Scott R. D., Williard P. G., J. Am. Chem. Soc. 1990, 112, 8971–8975. [Google Scholar]
- 57. Winkler J. D., Axten J. M., J. Am. Chem. Soc. 1998, 120, 6425–6426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. White J. D., Ihle D. C., Org. Lett. 2006, 8, 1081–1084. [DOI] [PubMed] [Google Scholar]
- 59. White J. D., Li Y., Ihle D. C., J. Org. Chem. 2010, 75, 3569–3577. [DOI] [PubMed] [Google Scholar]
- 60. Ragains J. R., Winkler J. D., Org. Lett. 2006, 8, 4437–4440. [DOI] [PubMed] [Google Scholar]
- 61.
- 61a. Vogler B., Bayer R., Meller M., Kraus W., Schell F. M., J. Org. Chem. 1989, 54, 4165–4168; [Google Scholar]
- 61b. Kwak Y.-S., Winkler J. D., J. Am. Chem. Soc. 2001, 123, 7429–7430; [DOI] [PubMed] [Google Scholar]
- 61c. Roupany A. J. A., Baker J. R., RSC Adv. 2013, 3, 10650–10653; [Google Scholar]
- 61d. Lutteke G., AlHussainy R., Wrigstedt P. J., Hue B. T. B., de Gelder R., van Maarseveen J. H., Hiemstra H., Eur. J. Org. Chem. 2008, 925–933. [Google Scholar]
- 62. Swindell C. S., deSolms S. J., Springer J. P., Tetrahedron Lett. 1984, 25, 3797–3800. [Google Scholar]
- 63. de Mayo P., Takeshita H., Sattar A. B. M. A., Proc. Chem. Soc. 1962, 119. [Google Scholar]
- 64. Hecker E., Cancer Res. 1968, 28, 2338–2349. [PubMed] [Google Scholar]
- 65. Winkler J. D., Rouse M. B., Greaney M. F., Harrison S. J., Jeon Y. T., J. Am. Chem. Soc. 2002, 124, 9726–9728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Hong B. C., Chen S. H., Kumar E. S., Lee G. H., Lin K. J., J. Chin. Chem. Soc. 2003, 50, 917–926. [Google Scholar]
- 67. Abraham W. R., Hanssen H. P., Tetrahedron 1992, 48, 10559–10562. [Google Scholar]
- 68. Minter D. E., Winslow C. D., J. Org. Chem. 2004, 69, 1603–1606. [DOI] [PubMed] [Google Scholar]
- 69.
- 69a. Bach T., Hehn J. P., Angew. Chem. Int. Ed. 2011, 50, 1000–1045; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 1032–1077; [Google Scholar]
- 69b. Dalal A., Khanna R., Kumar D., Jindal P., Chaudhary A., Kamboj R. C., Curr. Org. Chem. 2015, 19, 2156–2195. [Google Scholar]
- 70. Ishida N., Sawano S., Murakami M., Nat. Commun. 2014, 5, 3111. [DOI] [PubMed] [Google Scholar]
- 71. Bott T. M., West F. G., Heterocycles 2012, 84, 223–264. [Google Scholar]
- 72. Ishida N., Necas D., Masuda Y., Murakami M., Angew. Chem. Int. Ed. 2015, 54, 7418–7421; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 7526–7529. [Google Scholar]
- 73. Ruggeri M., Dombrowski A. W., Djuric S. W., Baxendale I. R., ChemPhotoChem 2019, 3, 1212–1218. [Google Scholar]
- 74. Ruggeri M., Dombrowski A. W., Djuric S. W., Baxendale I. R., J. Org. Chem. 2020, 85, 7276–7286. [DOI] [PubMed] [Google Scholar]
- 75. Nishimura T., Uemura S., J. Am. Chem. Soc. 1999, 121, 11010–11011. [Google Scholar]
- 76. Roque J. B., Kuroda Y., Jurczyk J., Xu L. P., Ham J. S., Gottemann L. T., Roberts C. A., Adpressa D., Sauri J., Joyce L. A., Musaev D. G., Yeung C. S., Sarpong R., ACS Catal. 2020, 10, 2929–2941. [DOI] [PMC free article] [PubMed] [Google Scholar]