

Abstract
In metazoan tissues, cells decide their fates by sensing positional information provided by specialized morphogen proteins. To explore what features are sufficient for positional encoding, we asked whether arbitrary molecules (e.g., green fluorescent protein or mCherry) could be converted into synthetic morphogens. Synthetic morphogens expressed from a localized source formed a gradient when trapped by surface-anchoring proteins, and they could be sensed by synthetic receptors. Despite their simplicity, these morphogen systems yielded patterns reminiscent of those observed in vivo. Gradients could be reshaped by altering anchor density or by providing a source of competing inhibitor. Gradient interpretation could be altered by adding feedback loops or morphogen cascades to receiver cell response circuits. Orthogonal cell-cell communication systems provide insight into morphogen evolution and a platform for engineering tissues.
Development of multicellular organisms requires precise spatial control of cell fate. Morphogens are molecules that provide positional information. They diffuse from a source to form a concentration gradient that is interpreted by neighboring cells (1–4). In metazoans, a small set of specialized molecules, including sonic hedgehog (Shh), Wnt, fibroblast growth factor (FGF), and bone morphogenetic protein (BMP)–transforming growth factor–β (TGFβ) family members, serve as morphogens (5). Reconstitution of morphogen signaling in vitro is a powerful approach to understand how morphogens encode positional information (6, 7). To define the minimal requirements for a functional morphogen, we asked whether it is possible to construct a synthetic morphogen signaling system that functions orthogonally to endogenous morphogens. Orthogonal morphogen signaling would enable the systematic exploration of patterning circuits, free from confounding cross-talk with endogenous systems.
To create synthetic morphogens, we modified the recently developed synthetic Notch (synNotch) system to detect user-defined soluble factors. SynNotch receptors are a modular platform for engineering orthogonal juxtacrine signaling (8), composed of an extracellular recognition domain [e.g., nanobody or single chain antibody (scFv)], the Notch core regulatory domain, and an intracellular transcriptional domain (e.g., TetR-VP64). When an anti–green fluorescent protein (GFP) synNotch receptor recognizes membrane-tethered GFP on a neighboring sender cell, the synNotch core undergoes cleavage, which releases its intracellular domain to enter the nucleus to activate target gene expression. Soluble GFP does not activate synNotch because exposure of the cleavage site requires the mechanical force of a membrane-tethered ligand (8). Here, we reengineered the synNotch system to detect soluble molecules by tethering the diffusible ligand to a complementary engineered anchor cell (Fig. 1A). In the case of GFP, we used two noncompetitive anti-GFP nanobodies: One serves as the anchor binding domain, and the other serves as the receptor binding domain (Fig. 1B). We designed this diffusible synNotch system with anti-GFP LaG2 nanobody fused to a transmembrane domain as the anchor molecule and anti-GFP LaG17 nanobody as the recognition domain of the synNotch receptor (fig. S1A). These nanobodies recognize different sites on GFP (9). SynNotch receiver cells were only activated by soluble GFP in the presence of anchor cells (Fig. 1C and fig. S1, B and C).
Fig. 1. Turning arbitrary proteins into synthetic morphogens.
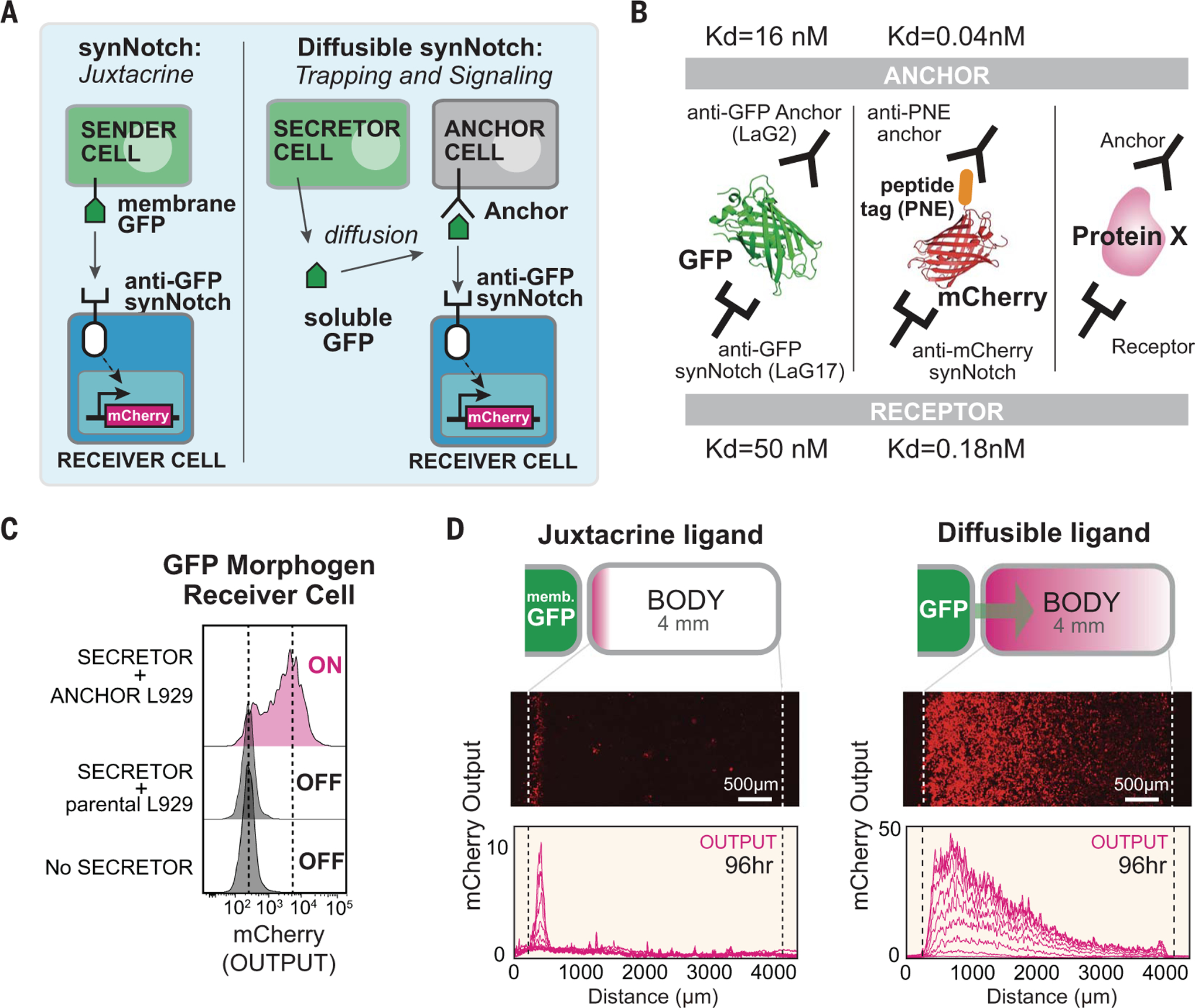
(A) SynNotch receptors detect juxtacrine signals (e.g., membrane-tethered GFP). In the diffusible synNotch system, soluble GFP is produced from a secretor cell, then trapped by anti-GFP anchor protein, and finally presented to anti-GFP synNotch on a receiver cell. (B) Multiple arbitrary proteins with two recognition sites could be converted into synthetic morphogens (see fig. S3 for construction of mCherry-PNE peptide morphogen). Kd, dissociation constant. (C) Testing diffusible GFP synNotch system in L929 mouse fibroblasts. Anchor cell expresses anti-GFP LaG2 anchor protein. Receiver cell expresses anti-GFP LaG17 synNotch (induces mCherry reporter). 1 × 104 GFP-secreting cells, 0.5 × 104 anchor cells, and 0.5 × 104 receiver cells were cultured overnight, and mCherry induction in receiver cells was measured by flow cytometry. (D) Juxtacrine versus diffusible GFP signaling gradient. Left pole has 3 × 104 sender cells, and right body has 1.5 × 104 cells (100% receiver cells for juxtacrine; 50:50 anchor:receiver cells for diffusible GFP; see fig. S2, A and B). Images were taken by incucyte system over 4 days when system reached steady state (movie S1). Individual lines show mCherry intensity every 12 hours. memb., membrane.
To test whether this soluble synNotch system allowed GFP to function as a morphogen, we reconstituted an in vitro model of L929 fibroblast cells organized into a pole and a body (fig. S2A) (culture well divided by insert wall; see materials and methods). The pole was composed of GFP secretor cells and the body of mixed anchor and receiver cells. To minimize disruptive convective flow, we used a solidified media containing 1% agarose (fig. S2B). We observed the formation of a long-range gradient (~4 mm) of both secreted GFP and synNotch reporter gene expression (mCherry) in contrast to the narrow band of activation observed when pole cells expressed membrane-tethered GFP (Fig. 1D and fig. S2C). The GFP gradient formed within ~24 hours, whereas induced mCherry reporter expression reached steady state in ~96 hours (fig. S2, C and D, and movie S1). We found that another bivalently recognized protein—a fusion protein of mCherry with a PNE peptide (mCherry-PNE)—could also be recognized by an analogous anchor-receptor system (fig. S3 and supplementary text), which further supports the idea that arbitrary proteins can be converted into morphogens (Fig. 1B).
To investigate how the shape of a synthetic morphogen gradient could be modulated, we systematically perturbed different interaction parameters. Using the LaG2 anti-GFP nanobody as an anchor protein, we tested how anchor protein density can regulate gradient shape. We generated four different levels of anchor protein density by varying both the anchor protein expression level and the anchor cell number in the body. More specifically, we constructed high- or low-expressing anchor cells and mixed them at 50% (a 1:1 ratio) with receiver cells in the body (Fig. 2A and fig. S4). We also created a combination anchor-receiver L929 cell, which was used at 100% in the body, yielding an approximately twofold higher density of anchor proteins for a given expression level (fig. S4). These studies showed that when the overall density of anchor protein was reduced, the maximal signal amplitude close to the pole decreased, but the signal range extended a longer distance as morphogen diffused further before being trapped. The importance of anchor density as a determinant of morphogen gradient shape is consistent with trends predicted by a simple computational model (fig. S5B).
Fig. 2. Systematic control over distance range of synthetic morphogen gradient.
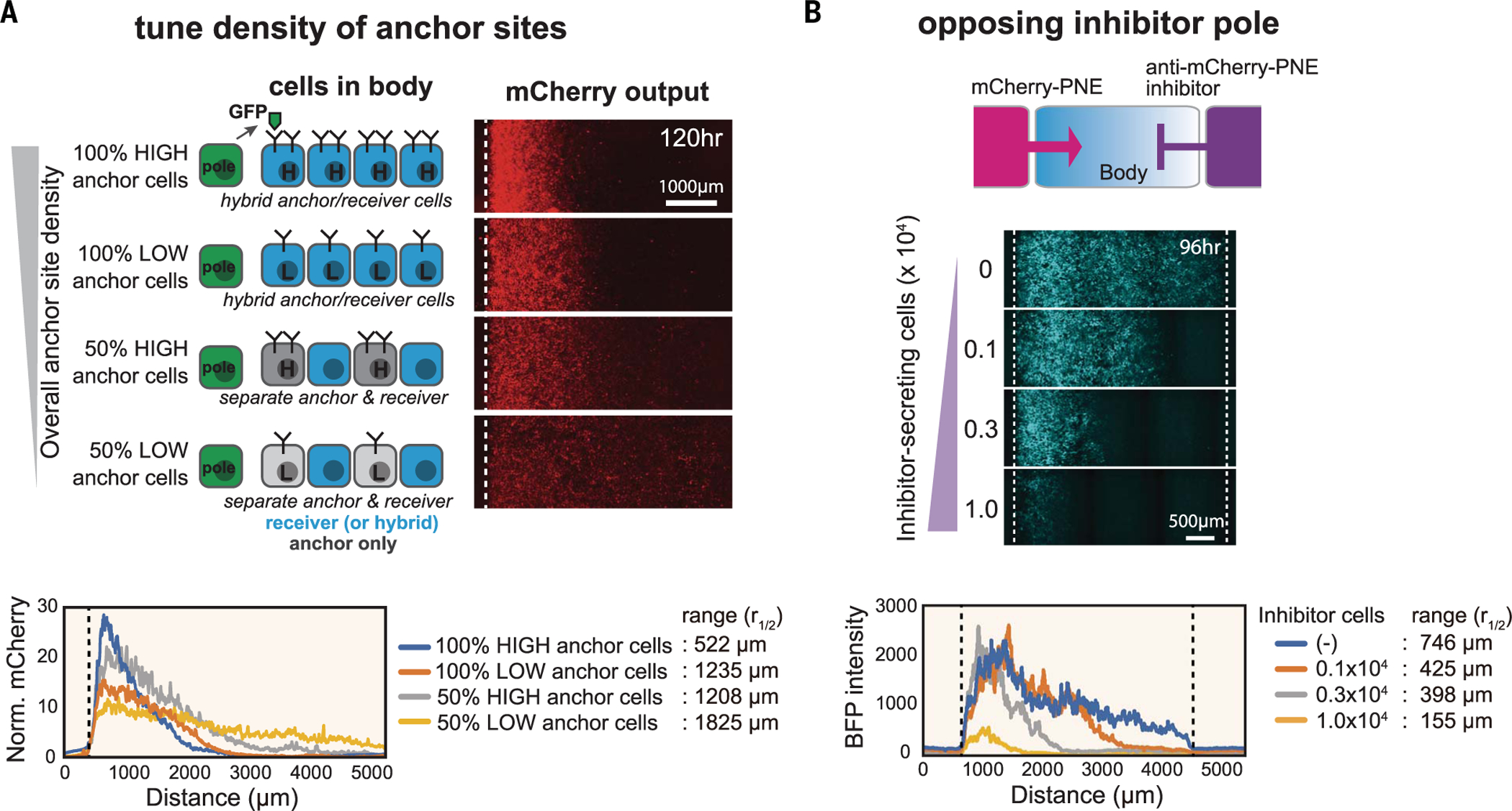
(A) Anchor density can tune synthetic GFP morphogen gradient shape and signaling range. We constructed bodies with four different densities of LaG2-anchor by using two types of variations: anchor expression level (high or low) and fraction of cells in the body that express anchor (100 or 50%). See materials and methods for details. Norm., normalized. (B) Controlling signaling range of mCherry-PNE morphogen with inhibitor. We used a three-well insert wall to build three regions: morphogen pole, body, and inhibitor pole. The morphogen pole has a mixture of 1 × 104 mCherry-PNE–secreting cells and 2 × 104 parental L929 cells. The body has a 1.5 × 104 mixture of anti-PNE anchor cells and anti-mCherry synNotch receiver cells (50:50 ratio). The inhibitor pole has cells expressing anti–mCherry-PNE inhibitor (total cell number: 3 × 104 of cells; varying number of inhibitor cells: 0, 0.1 × 104, 0.3 × 104, or 1.0 × 104; remaining cells were parental L929). BFP output was quantified by In Cell Analyzer 6000 at day 4 (see fig. S6 for GFP inhibitor analysis).
The spatial distribution of morphogens can also be regulated by antimorphogen inhibitors. For example, during Xenopus embryogenesis, BMP is secreted from a ventrally located pole, whereas Chordin—a BMP-binding inhibitor— is secreted from an opposing pole (10, 11). Such antagonism by an inhibitor is a common theme observed in development. Inspired by activator-inhibitor opposition, we designed a three-region configuration on the culture dish: a central body surrounded by a morphogen-secreting pole and an inhibitor-secreting pole on opposite sides (Fig. 2B; see fig. S6, A and B, and materials and methods for development of morphogen inhibitor). We tested both morphogen and inhibitor sets (GFP and mCherry-PNE) in the double-pole system. When the GFP inhibitor was secreted from the opposing pole, we observed a reduced amplitude of the activation gradient generated by the GFP morphogen (fig. S6C). The mCherry-PNE inhibitor showed a somewhat different pattern, reducing the signaling range of the activation gradient generated by mCherry-PNE morphogen with smaller effects on amplitude (Fig. 2B and supplementary text). Together, these results show that the signaling range and amplitude of a synthetic morphogen gradient can be tuned to generate a variety of shapes similar to those seen in embryos.
In natural morphogen systems, receiver cells can interpret a morphogen gradient in diverse, higher-order ways. We explored several mechanisms to reshape how the body cells interpret the synthetic morphogen gradient, taking advantage of the flexibility with which synNotch signaling can be engineered to drive any genetically encoded payload. We particularly focused on engineering intercellular positive and negative feedback loops among receiver cells (Fig. 3).
Fig. 3. Reshaping morphogen interpretation with positive or negative feedback.
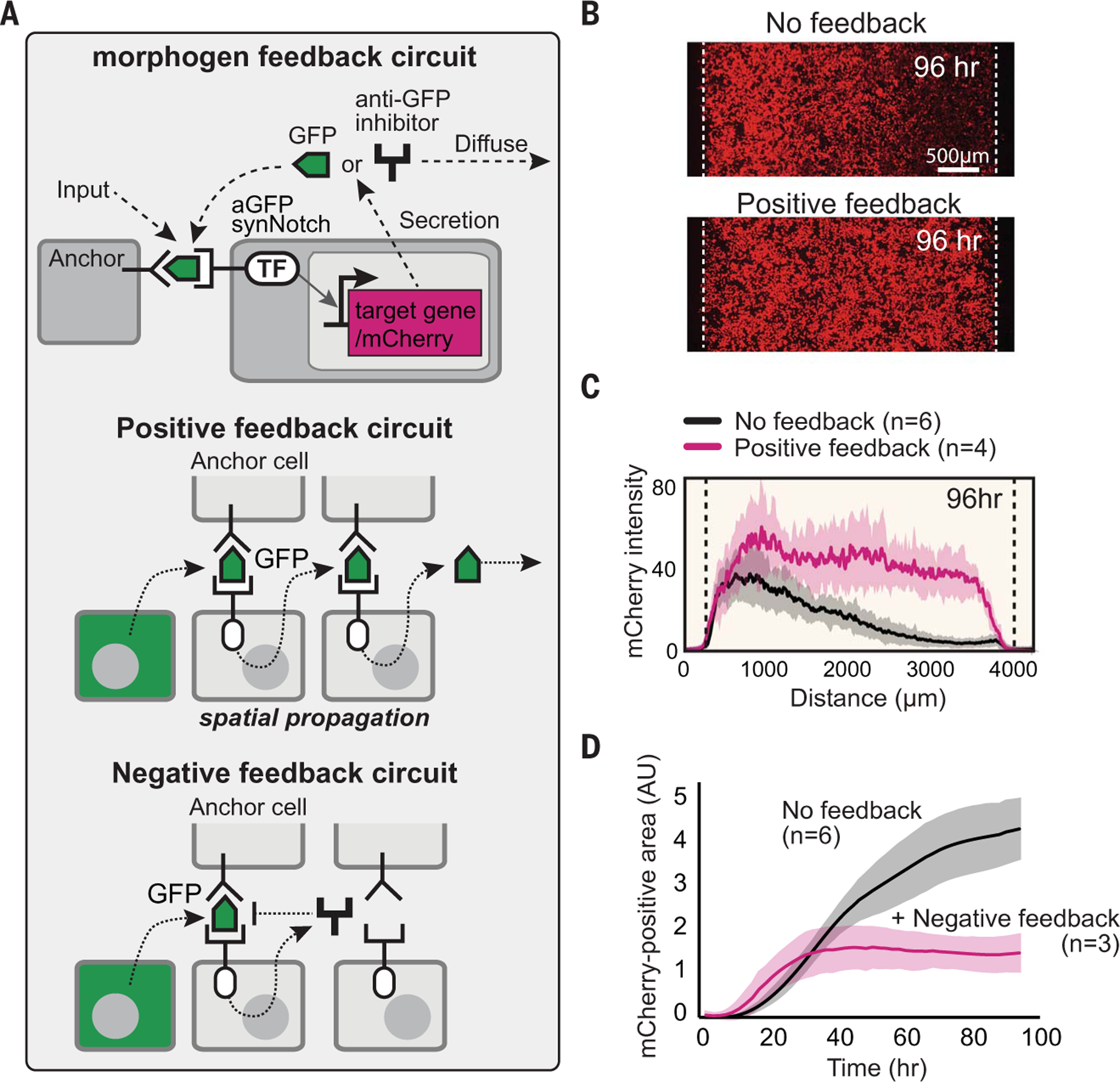
(A) In a positive feedback circuit, GFP morphogen activates receiver cells to induce the secretion of more GFP. In a negative feedback circuit, GFP morphogen induces the expression of antimorphogen inhibitor by receiver cells. TF, transcription factor. (B) Comparison of mCherry output in the body with and without positive feedback at 96 hours (see fig. S7C for time course). The pole has 3 × 104 GFP-secreting cells; the body has a 1.5 × 104 mixture of anchor cells and receiver cells engineered with a positive feedback circuit (50:50 ratio). Images were taken by incucyte system for 4 days (movie S1). (C) Activity gradient profiles at 96 hours, with and without positive feedback. Shaded area shows SD from multiple experiments. (D) The mCherry-positive area (integral of top plots) plotted over time shows that the body with negative feedback reaches steady state faster than it does without feedback. AU, arbitrary units.
We first designed an intercellular positive feedback circuit in which the receiver cells sense the GFP morphogen and, in response, induce expression of more GFP (Fig. 3A). Cells with this positive feedback circuit did not trigger spontaneous activation with the anchor cells when engineered with sufficiently low background expression of GFP (fig. S7A). By contrast, when a body of positive feedback receiver cells (with anchor cells) was placed next to a pole of GFP-producing cells, the positive feedback circuit caused rapid, high-amplitude spatial propagation of morphogen signal and reporter gene activation across the entire body (Fig. 3, B and C; fig. S7C; and movie S1).
We also constructed intercellular negative feedback receiver cells, in which the receiver cells sense the GFP morphogen and, in response, produce the soluble GFP inhibitor (Fig. 3A and fig. S7B). When negative feedback receiver cells (mixed with anchor cells) were placed in the body, the maximal amplitude of the response gradient was notably dampened. The negative feedback activity gradient reached a stable steady state far more rapidly (~30 versus 70 hours for negative feedback versus nonfeedback cells) (Fig. 3D and fig. S7D). Such stable gradient formation is similar to that which is observed in response to natural morphogens (e.g., wingless or hedgehog), with negative feedback pathways involving self-induced morphogen degradation, endocytosis, or inhibition (6, 12). In these cases, the common principle of morphogen-induced negative feedback is thought to lead to higher levels of negative regulation in the vicinity of the morphogen source but lower levels of negative regulation at a greater distance from the source, which yields a more extended and stable gradient.
We have created a modular toolkit of intercellular signaling components that can flexibly reshape morphogen production and interpretation. We wanted to test whether these modules could be combined to program higher-order pattern formation, such as the formation of multiple distinct segments within a body plan (Fig. 4). We first asked whether we could create a body plan with two distinct domains (activated and unactivated). To do so, we combined the positive feedback morphogen circuit with a counteracting inhibitor pole on the opposite side of the body (Fig. 4A). This circuit yielded two distinct domains: receiver cells close to morphogen source triggered positive feedback to drive strong synNotch activation, whereas signal activation close to the inhibitor source was blocked, which lead to a nonlinear, switch-like transition from active to inactive domains (Fig. 4B). By changing the number of GFP inhibitor–secreting cells, we could tune the widths of the domains (fig. S8, A and B).
Fig. 4. Combining synthetic morphogen interpretation circuits to engineer multidomain spatial patterns.
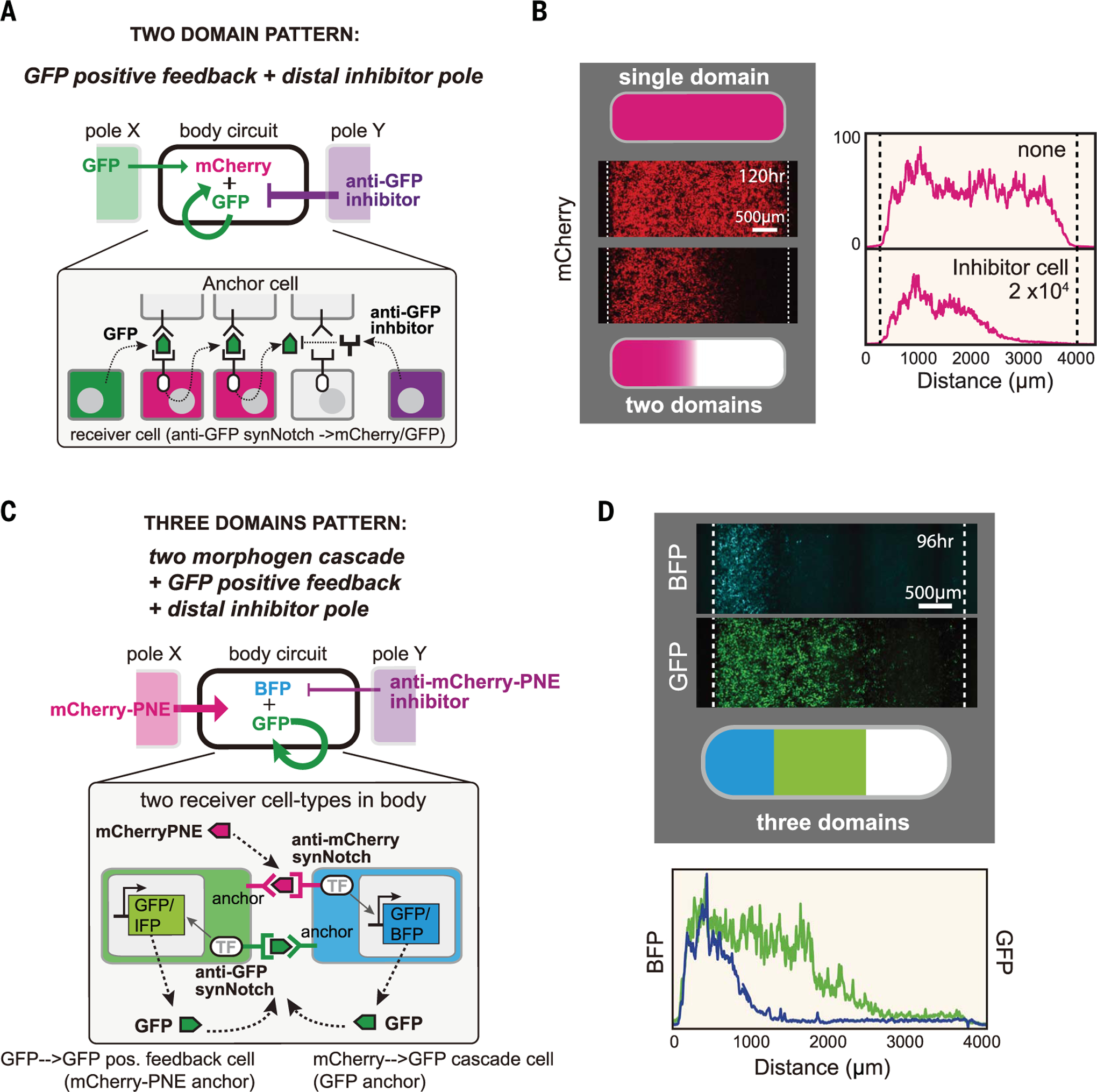
(A and B) Programming two-domain pattern by combining positive feedback circuit with opposing morphogen and inhibitor poles. Morphogen pole (X) has 3 × 104 GFP-secreting cells; the body has a 50:50 mixture of anchor cells and receiver cells with positive feedback (used in Fig. 3B) (1.5 × 104 total cells); and the inhibitor pole (Y) has 2 × 104 anti-GFP inhibitor–secreting cells. Images were taken by incucyte at 120 hours (movie S1). See fig. S8B for variant circuits. (C and D) Programming three-domain pattern. We combined the two-morphogen cascade and positive feedback circuit with opposing morphogen and inhibitor poles. The body contains two types of cells: Cell A expresses anti-mCherry synNotch that induces BFP reporter and GFP morphogen (mCherry-PNE→GFP cascade), and cell B expresses anti-GFP LaG17 synNotch that induces expression of GFP morphogen (GFP→GFP positive feedback). See materials and methods for details. Image was taken at 96 hours by In Cell Analyzer 6000 (movie S2). IFP, infrared fluorescent protein.
Finally, we set out to engineer a circuit that produces a three-domain body, akin to Wolpert’s classic French Flag pattern (2). We designed a circuit that incorporates a two-morphogen cascade, positive feedback, and opposing pole inhibition (Fig. 4C). In this circuit, the left pole secretes mCherry-PNE morphogen. The body contains two types of uniformly mixed receiver cells (fig. S8C): receiver A cells that sense mCherry-PNE to induce the expression of a blue fluorescent protein (BFP) reporter and GFP morphogen (mCherry-PNE→GFP two-morphogen cascade) and receiver B cells that sense GFP to induce the secretion of GFP (GFP→GFP positive feedback) (cells A and B are engineered to serve as anchors for each other). To oppose morphogen signaling, the right pole secretes the anti–mCherry-PNE inhibitor to sharpen the initial morphogen gradient. With this composite circuit, we observe the robust formation of three distinct domains (Fig. 4D): a BFP+GFP+ domain closest to the mCherry-PNE morphogen pole; a BFP−GFP+ middle domain (where the GFP+ region is extended by GFP→GFP positive feedback); and, furthest (closest to the inhibitor pole), a BFP−GFP− domain (fig. S8D and movie S2). This toolkit of synthetic cell-cell communication components can be used to write spatial programs capable of encoding multiple, distinct body domains.
Although evolution has relied on a relatively small set of specialized morphogen families, we find that arbitrary proteins with no known history of functioning as morphogens can be converted into effective morphogens if they are deployed with a complementary system of receptors, anchoring interactions, and inhibitors. These synthetic morphogens differ from natural morphogens in that they explicitly require a distinct anchoring protein to constrain their distribution and mediate synNotch receptor activation. Whereas natural morphogens often function autonomously, most participate in weak tethering interactions that are analogous to anchoring—whether interacting with cell surface proteoglycans, extracellular matrix, or cell membranes through lipid modifications (fig. S9A) (13–15). These tethering interactions are proposed, in many cases, to constrain signaling range and to prevent the leakage of morphogens (16, 17) (supplementary text and fig. S10).
These synthetic morphogen platforms can program positional information without cross-talk to endogenous signaling pathways. Thus, it may be possible to deploy them in vivo as inert tools to probe or redirect development. Related studies (18) have shown that an analogous synthetic GFP morphogen can function in vivo in Drosophila. Thus, these synthetic morphogen systems could be used to facilitate controlled forward engineering of tissues and organs, both in a native-like or a modified fashion. We found that the diffusible synNotch system functioned in other cell types, including immune cells (fig. S11), which provides further possibilities as to how such synthetic signaling systems could be deployed to shape spatially controlled functions in vivo.
Supplementary Material
ACKNOWLEDGMENTS
We thank A. McMahon for Shh-GFP and L. Morsut, K. Roybal, J. Brunger, N. Frankel, and members of the Lim laboratory for discussion and assistance. We also thank members of the University of California San Francisco Center for Systems and Synthetic Biology and the NSF Center for Cellular Construction.
Funding: This work was supported by the Human Frontiers of Science Program (HFSP); the Senri Life Science Foundation; the Kato Memorial Bioscience Foundation; JSPS KAKENHI grant no. 20K15828 and World Premier International Research Center Initiative (WPI), MEXT, Japan (to S.T.); a Ruth L. Kirschstein National Research Service Award (NRSA) Individual Postdoctoral Fellowship F32DK123939 (to W.L.M.); the NSF DBI-1548297 Center for Cellular Construction; the DARPA Engineered Living Materials program; and the Howard Hughes Medical Institute (to W.A.L.). T.J.H. and O.D.K. were supported by NIH R01-DE028496 and R35-DE026602.
Footnotes
Competing interests: W.A.L. and S.T. have a financial interest in Gilead Biosciences. W.A.L. and S.T. are inventors on a patent application (PCT/US2016/019188) held by the Regents of the University of California that covers binding synthetic Notch receptors.
Data and materials availability: All data are available in the main manuscript and supplementary materials. All plasmids developed in this study will be deposited at Addgene (www.addgene.org/Wendell_Lim/), where they will be publicly available.
SUPPLEMENTARY MATERIALS
science.sciencemag.org/content/370/6514/327/suppl/DC1
Materials and Methods
Supplementary Text
Figs. S1 to S11
Table S1
References (19–27)
MDAR Reproducibility Checklist
Movies S1 and S2
REFERENCES AND NOTES
- 1.Turing AM, Phil. Trans. R. Soc. Lond. B 237, 37–72 (1952). [Google Scholar]
- 2.Wolpert L, J. Theor. Biol 25, 1–47 (1969). [DOI] [PubMed] [Google Scholar]
- 3.Rogers KW, Schier AF, Annu. Rev. Cell Dev. Biol 27, 377–407 (2011). [DOI] [PubMed] [Google Scholar]
- 4.Lander AD, Science 339, 923–927 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tabata T, Takei Y, Development 131, 703–712 (2004). [DOI] [PubMed] [Google Scholar]
- 6.Li P et al. , Science 360, 543–548 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sekine R, Shibata T, Ebisuya M, Nat. Commun 9, 5456 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morsut L et al. , Cell 164, 780–791 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fridy PC et al. , Nat. Methods 11, 1253–1260 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.De Robertis EM, Kuroda H, Annu. Rev. Cell Dev. Biol 20, 285–308 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bier E, De Robertis EM, Science 348, aaa5838 (2015). [DOI] [PubMed] [Google Scholar]
- 12.Eldar A, Rosin D, Shilo B-Z, Barkai N, Dev. Cell 5, 635–646 (2003). [DOI] [PubMed] [Google Scholar]
- 13.Yan D, Lin X, Cold Spring Harb. Perspect. Biol 1, a002493 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Parchure A, Vyas N, Mayor S, Trends Cell Biol. 28, 157–170 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang Y, Wang X, Wohland T, Sampath K, eLife 5, e13879 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Müller P, Rogers KW, Yu SR, Brand M, Schier AF, Development 140, 1621–1638 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kornberg TB, Guha A, Curr. Opin. Genet. Dev 17, 264–271 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stapornwongkul KS, de Gennes M, Cocconi L, Salbreux G, Vincent J-P, Science 370, 321–327 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.