

Abstract
The Ir‐catalyzed conversion of prochiral tert‐cyclobutanols to β‐methyl‐substituted ketones proceeds under comparably mild conditions in toluene (45–110 °C) and is particularly suited for the enantioselective desymmetrization of β‐oxy‐substituted substrates to give products with a quaternary chirality center with up to 95 % ee using DTBM‐SegPhos as a chiral ligand. Deuteration experiments and kinetic isotope effect measurements revealed major mechanistic differences to related RhI‐catalyzed transformations. Supported by DFT calculations we propose the initial formation of an IrIII hydride intermediate, which then undergoes a β‐C elimination (C−C bond activation) prior to reductive C−H elimination. The computational model also allows the prediction of the stereochemical outcome. The Ir‐catalyzed cyclobutanol cleavage is broadly applicable but fails for substrates bearing strongly coordinating groups. The method is of particular value for the stereo‐controlled synthesis of substituted chromanes related to the tocopherols and other natural products.
Keywords: asymmetric catalysis, C−C bond activation, cyclobutanols, deuteration, iridium
Iridium behaves differently! Unexpectedly, the Ir‐catalyzed cleavage of cyclobutanols to give β‐chiral ketones proceeds through a different mechanism as compared to related Rh‐catalyzed reactions. This was shown by deuteration experiments, kinetic isotope effect measurements and DFT calculations. The developed method for the catalytic enantioselective desymmetrization is of particular value in the synthesis of chromane‐related natural products from readily accessible spirocyclobutanols.

Introduction
The transition metal‐catalyzed activation of C−C single bonds has opened some unconventional strategies for the atom‐economic synthesis of complex molecules.[ 1 , 2 ] In many cases cyclobutane derivatives have been employed as substrates because the energy gain (22–26 kcal mol−1) associated with the opening of a strained four‐membered ring [3] represents a strong driving force. Mechanistically, the cleavage of cyclobutanes through metal‐catalyzed C−C bond activation can follow different pathways. For instance, cyclobutanones can undergo oxidative addition to RhI[4] or Pd0[5] to form reactive five‐membered metallacycles, which can then further react downstream in different ways as reported by Murakami et al. [6] Also other cyclobutane derivatives, such as biphenylenes, [7] cyclobutenediones, or cyclobutenones, can be activated by different transition metals (e.g., Ni, Pt, Rh, or Ru). [8] Furthermore, Ag [9] and Mn [10] reagents are able to induce the ring‐opening of cyclobutanols via radical processes (homolytic C−C bond cleavage).
From a synthetic point of view Pd‐ [11] and Rh‐ [12] ‐catalyzed transformations of tert‐cyclobutanols to ring‐opened ketones are of particular interest because such reactions can be exploited for the enantioselective synthesis of β‐substituted carbonyl compounds if prochiral substrates are employed in the presence of a chiral metal catalyst (Scheme 1).
Scheme 1.

Selected metal‐catalyzed cyclobutanol cleavage reactions according to the groups of Uemura, [13a] Murakami, [14a] and Cramer. [16]
As a first impressive example, Uemura and co‐workers reported the enantioselective Pd‐catalyzed reaction of cyclobutanols of type 1 into γ‐arylated products (2) in the presence of the chiral ferrocene‐derived ligand L1. [13a] In contrast, the groups of Murakami[ 14a , 15 ] and Cramer[ 16 , 17 ] used Rh catalysts to achieve the enantioselective conversion of prochiral cyclobutanols to β‐methyl‐substituted carbonyl compounds (such as 4 or 5) in the presence of SegPhos ligands L2 or L3, respectively. Extensive mechanistic studies (including deuterium‐labeling experiments) suggested these transformations to proceed according to the general mechanism shown in Scheme 2. At first, the cyclobutanol substrate (6) is supposed to react with the catalyst to form a Rh cylcobutanolate 8, which then undergoes a β‐C elimination as the key ring‐opening step. In agreement to the outcome of deuteration experiments, the resulting alkyl‐Rh intermediate 9 then isomerizes to a more stable Rh enolate 10 via 1,3‐hydrogen shift. Final hydrolysis of 10 then closes the catalytic cycle and affords the (α‐deuterated) product 7.[ 14 , 16 ]
Scheme 2.

General mechanism of the Rh‐catalyzed cleavage of cyclobutanols as suggested by the groups of Murakami [14a] and Cramer [15] based on deuteration studies.
In the course of our research into the stereoselective synthesis of α‐tocopherol [18] we recently discovered and exploited an Ir‐catalyzed stereo‐controlled ring opening of spirocyclobutanols of type 11 to establish the quaternary stereocenter with the desired absolute configuration (Scheme 3). [19] Interestingly, no stereoinduction could be achieved under Rh‐catalysis in this case. In contrast, the Ir‐catalyzed reaction afforded the product 12 with very high enantiomeric (ee) or diastereomeric excess (de; up to 99:1, depending on the nature of the group R) in the presence of DTBM‐SegPhos (L3) as a chiral ligand.
Scheme 3.
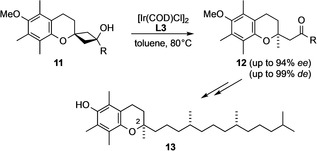
Ir‐catalyzed cleavage of spirocyclobutanols as a key step of our total synthesis of (2R)‐α‐tocopherol. [19]
Although the iridium‐based methodology enabled us to complete the total synthesis of (2R)‐α‐tocopherol (13), we were wondering about the differences between the Rh‐ and the Ir‐catalyzed processes. As only very few examples for Ir‐catalyzed C−C bond activation have been reported in the literature [20] (without any synthetic application [21] except our above‐mentioned tocopherol synthesis), we felt challenged to further explore the enantioselective Ir‐catalyzed cyclobutanol cleavage both mechanistically and with respect to its application scope. We here report the results of our study, which indeed revealed fundamental mechanistic differences between the Ir‐ and the Rh‐catalyzed cyclobutanol fragmentation pathways and additionally pinpoints the substrate scope and further synthetic applications of the Ir‐catalyzed methodology.
Results and Discussion
Initial experiments
Using the spirocyclobutanol 11 a [19] as a model substrate, we first reinvestigated different conditions for the metal‐mediated ring‐opening reaction to demonstrate the pronounced reactivity differences between the Rh‐ and the Ir‐based catalysts (Table 1). In all cases, a solution of the substrate and the catalyst precursors (metal salt and ligand) in toluene was stirred under argon atmosphere for 30–60 min at room temperature before the mixture was heated to the specified temperature and the conversion was monitored by means of TLC. Although the catalyst generated in situ from [Rh(COD)Cl]2 (cod=1,5‐cyclooctadiene) and rac‐BINAP proved to be completely inactive, the expected product 12 a was cleanly formed upon addition of Cs2CO3 as a base (Table 1, entries 1, 2). Remarkably, the use of the hydroxy complex [Rh(COD)OH]2 as the rhodium source also resulted in a smooth conversion without the necessity of a base additive (entry 3). However, virtually no enantioselectivity was observed under Rh catalysis when rac‐BINAP was replaced by either (R)‐BINAP or (R)‐DTBM‐SegPhos (ent‐L3), the latter corresponding to the original conditions of Seiser and Cramer (entry 4). [16] In contrast, the Ir‐based catalyst generated from the chloride salt [Ir(COD)Cl]2 and rac‐BINAP were found to be active even without a base additive. Moreover, a dramatic ligand acceleration was observed upon replacing BINAP by the (R)‐DTBM‐SegPhos ligand in the Ir‐catalyzed reaction. In this case, the desired transformation proceeded smoothly already at 70 °C to give the product (R)‐12 a in 98 % isolated yield and with 93 % ee (entry 6). [19]
Table 1.
Rh‐ versus Ir‐catalyzed cleavage of cyclobutanol 11 a.
|
| ||||||
|---|---|---|---|---|---|---|
|
Entry |
Catalyst ([mol %]) |
Ligand ([mol %]) |
T [°C] |
Base |
Yield [%] |
ee [%][a] (conf) |
|
1[b] |
[Rh(COD)Cl]2 (2.5) |
rac‐BINAP (6.0) |
110 |
– |
– |
– |
|
2[b] |
[Rh(COD)Cl]2 (5.0) |
rac‐BINAP (12.0) |
110 |
Cs2CO3 |
93 |
– |
|
3 |
[Rh(COD)OH]2 (5.0) |
(R)‐BINAP (10.0) |
110 |
– |
92[c] |
<2 |
|
4 |
[Rh(COD)OH]2 (5.0) |
ent‐L3 (10.0) |
110 |
– |
74[c] |
<2 |
|
5 |
[Ir(COD)Cl]2 (2.0) |
rac‐BINAP (6.0) |
100 |
– |
32[c] |
– |
|
6 |
[Ir(COD)Cl]2 (1.0) |
ent‐L3 (3.0) |
70 |
– |
98 |
93 (R) |

[a] Determined by GC (FID) on a chiral phase. [b] The cis diastereomer of the alcohol was used. [c] Conversion as determined by GC‐MS.
Mechanistic studies
The experiments summarized in Table 1 indicate the Ir‐ and the Rh‐catalyzed reactions to follow different mechanistic pathways. The fact that the Rh‐catalyzed reaction either essentially requires a base additive or the employment of the hydroxy complex as catalyst precursor suggested the formation of a rhodium cyclobutanolate intermediate of type 8 according to the established mechanism shown in Scheme 2. However, we were puzzled by the question why the Ir‐catalyzed reaction proceeds smoothly in the absence of a base under „acidic“ conditions using the chloro complex for the in situ generation of the active catalyst. We hypothesized that the primary intermediate 14 formed by coordination of the cyclobutanol substrate to the IrI‐catalyst does not lead to a cyclobutanolate complex 13 (related to 8) in the absence of a base (Scheme 4). Instead, it appeared feasible that the iridium center in 14 might undergo oxidative addition (O−H bond activation) to generate an IrIII hydride complex of type 15, [22] which (as a 16 valence electron intermediate) could then be involved in the subsequent C−C bond activation step.
Scheme 4.

Possible reactions of the supposed primary Ir intermediate 14. Base‐mediated generation of a cyclobutanolate 13 versus formation of an IrIII hydride intermediate 15 by oxidative addition into the O−H bond.
We started our experimental investigation of the mechanism of the Ir‐catalyzed reaction with a deuteration experiment. For this purpose, the substrate D‐11 a was prepared by O‐deuteration of 11 a either by partitioning between D2O/EtOAc (ca. 60 % D) or by treatment of an ethereal solution of 11 a with 1.5 equiv of nBuLi followed by quenching the resulting lithium alkoxide with D2O/DCl (ca. 70 % D). The success of the O‐deuteration was confirmed by IR analysis (see the Supporting Information). The reaction of D‐11 a under the proven conditions then proceeded cleanly (Scheme 5) to afford the ring‐opened ketone D‐12 a with the deuterium label located at the newly formed (angular) methyl group according to NMR analysis (see the Supporting Information). Only a minor degree of deuteration (≤10 %) was also detected at the terminal α‐carbonyl position.
Scheme 5.

Selective deuteriation of the angular methyl group in the Ir‐catalyzed conversion of D‐11 a.
The outcome of this experiment (Scheme 5) unambiguously proves the Ir‐catalyzed process to mechanistically differ from the Rh‐catalyzed reaction as virtually no deuteration occurred under Ir catalysis at the methylene position next to the keto function (compare Scheme 2). Thus, an 1,3‐hydrogen shift leading to a metal enolate, as a characteristic feature of the Murakami/ Cramer mechanism, could be excluded. Also, these authors never observed any deuteration of the newly formed methyl group during their studies of the Rh‐catalyzed cyclobutanol cleavage.[ 14 , 16 ]
Based on our experimental results we devised the mechanism shown in Scheme 6 for the Ir‐catalyzed transformation. This mechanism starts with the oxidative addition of the IrI‐complex into the O−D bond of D‐11 a leading to the IrIII‐hydride intermediate 16. Now, the iridium center is supposed to activate the adjacent C−C bond to induce a β‐carbon elimination via a transition state (TS) of type TS(16–17). The resulting IrIII‐alkyl‐ intermediate 17 finally undergoes reductive elimination to release the product D‐12 a under regeneration of the IrI‐catalyst.
Scheme 6.

Proposed mechanism for the Ir‐catalyzed cleavage of cyclobutanol D‐11 a that takes into account the specific deuteration outcome.
When the Ir‐catalyzed reaction of either 11 a or D‐11 a was performed in the presence of excess D2O (in toluene/D2O=4:1), the product D‐12 a again contained a (single) deuterium atom at the angular methyl group—in agreement with the proposed mechanism. In this case, however, the α‐carbonyl methyl group was completely deuterated as well while still almost no deuteration (<10 % D) was observed at the methylene group. This indicates the additional α‐deuteration to occur at the stage of the ketone product (D‐12 a) via kinetically controlled enolization, preferentially to the terminal position.
To support the hypothesis that an iridium hydride species is involved in the (rate‐determining) key step of the proposed mechanism we decided to also investigate the kinetic isotope effect (KIE) of the reaction. For this purpose, we performed four parallel reactions (two with H‐11 a and two with D‐11 a) and monitored the reaction rates by means of NMR. To minimize the experimental error, these reactions were carried out very carefully under absolutely identical conditions as follows: A stock solution containing [Ir(COD)Cl]2 and the chiral ligand L3 in dry toluene was stirred for 60 min at room temperature before equal amounts of this solution were transferred by syringe to the four reaction vials containing the substrate (H/D‐11 a) to give a 0.12 m solution in toluene. After heating the stirred reaction mixtures to 73 °C, small samples were taken after 30, 60, 90, 120, and 180 min. Two of the four reactions were stirred for another 90 min to ensure full conversion. The taken samples were immediately filtered through a tiny plug of silica and analyzed by 1H NMR spectroscopy. The degree of conversion was calculated based on the integral changes of four selected signals: Product signals at 2.79 ppm (d, 1 H) and 1.86 ppm (m, 1 H) and signals of the starting material at 2.31 ppm (m, 2 H) and 2.01 ppm (t, 2 H). The results of these measurements are depicted in Figure 1 and clearly reveal that the deuterated cyclobutanol D‐11 a reacts slower than the non‐deuterated cyclobutanol 11 a, which indicates a significant kinetic isotope effect.
Figure 1.

Determination of the H/D kinetic isotope effect (KIE) for the Ir‐catalyzed transformation of 11 a to 12 a by time‐resolved monitoring of the conversion of the deuterated and the non‐deuterated substrate in four parallel experiments. A KIE of 2.4 was calculated from the ratio of the initial reaction rates (indicated by lines) and consideration of the deuteration degree (70 %).
To quantify the kinetic isotope effect, the rate constants for H (k H) and D (k D) were calculated by determining the slope of the line for the initial reaction rate (Figure 1). From the average value of k H (0.872 min−1) and k D (0.520 min−1) a KIE of 1.68 was calculated for the Ir‐catalyzed cyclobutanol cleavage. Taking a deuteration degree of 70 % for D‐11 a into account, the corrected KIE calculates to 2.4. This corresponds to a primary kinetic isotope effect and supports our mechanistic proposal (Scheme 6) that a O−H (or O−D) bond activation is involved (even as a rate‐determining step) in the catalytic cycle.
To probe the role of the chloride ligand and in particular whether it possibly dissociates from iridium during the catalytic process, we added varying amounts of AgOTf to the reaction mixture and monitored the conversion of 11 a into 12 a under standard conditions (4 mol % [Ir(COD)Cl]2, 12 mol % L3, toluene, 73 °C). While addition of 2 mol % of AgOTf had no significant effect, the reaction was much slower upon addition of 8 mol % and completely inhibited in the presence of an excess of AgOTf (40 mol %). This may be a hint that the chloride ligand plays a certain role; however, oxidation of the IrI‐catalyst by AgI would also cause inhibition of the reaction. Therefore, a cationic Ir‐complex cannot be fully excluded.
Computational investigations
To shed additional light on the proposed mechanism of the Ir‐catalyzed cyclobutanol cleavage we performed DFT (PW6B95D3) computations. [23] Using a simplified test system (with L=PH3) the theoretic analysis confirmed the feasibility of the proposed mechanism (Figure 2). The calculations suggest the oxidative addition of the iridium center to the O−H bond of the cyclobutanol (14 to 15) to be the step with the highest activation energy (E A=26.8 kcal mol−1). This is in accordance with the experimentally found KIE of=2.4 as the activation energy of the β‐C elimination step (16.2 kcal mol−1) is significantly lower. The final reductive C−H elimination (E A=23.5 kcal mol−1) leads to a complex, which, according to the calculations, dissociates without any barrier to liberate the product and the catalyst.
Figure 2.

Results of a DFT computational study of the mechanism of the Ir‐catalyzed cyclobutanol fragmentation using a simplified model system (PW6B95D3 /6‐311G* (C,H,O,P,Cl) /SDD(+ECP)(Ir)). Gibbs energies (unscaled, 298.15 K, 1 bar, in Hartree's) and relative reaction (E r) and activation (E a) energies (kcal mol‐1) are given.
Understanding enantioselectivity
Although the absolute (S)‐configuration of the product 12 a, prepared by Ir‐catalyzed fragmentation of 11 a in the presence of (S)‐DTBM‐SegPhos (L3) as a chiral ligand, had been unambiguously assigned by its conversion into (2R)‐α‐tocopherol, [19] we felt challenged to rationalize the stereochemical outcome. For this reason, we took a closer look at the β‐carbon elimination as the stereo‐determining step of the catalytic cycle. [24] A first configurational analysis (supported by DFT calculations) revealed that four types of transition states (TS) can be distinguished, which are all characterized by a pseudo‐octahedral coordination geometry of the iridium center with the bidentate P,P‐ligand in maximum distance to the activated C−C bond (Figure 3). Two of these transition states lead to the (S)‐ and the other two to the (R)‐product, and in both series the hydride and chloride ligands are oriented either cis or trans to each other.
Figure 3.

Formal configurational analysis of possible transition states of the IrIII‐mediated C−C bond activation as the stereo‐determining step.
Orienting DFT calculations on a small model system (see the Supporting Information) suggested transition structures with trans‐oriented H and Cl ligands and the O−C−C‐substrate atoms aligned in‐plane with the P2Ir ring being energetically most favored for electronic reasons (Figure 4).
Figure 4.

DFT computations (PW6B95D3/6–311G**(C,H,O,P,Cl)/SDD (+ECP, Ir)‐SCRF(toluene)//ONIOM(B97D3/SDD(+ECP,Ir), D95 (C,H,O,P,Cl):PM6) of a simplified model system show the energetically most favorable transition structures with a trans‐orientation of H and Cl ligands and an in‐plane alignment of the P−P−IrIII plane and the metal‐bound O−C−C unit of the substrate.
To prepare for the computation of the competing transition states (leading to the different enantiomers of 12 a) at a higher level of theory, the conformation of the axially chiral ligand (L3) coordinated to the iridium metal center was analyzed. As shown in Figure 5, two of the P‐bound aryl groups adopt an axial and the other two an equatorial position. The axial P‐aryl groups are fixed in coplanar orientation to the adjacent benzodioxole moieties of the biaryl unit, whereas the equatorial P‐aryl groups were found to be conformationally more flexible and able to intensely interact with the substrate. All in all, the (S)‐DTBM‐SegPhos‐iridium unit was found to adopt a right‐turning C2‐symmetric propeller shape.
Figure 5.

Schematic view of the conformation of the C2‐symmetric (S)‐DTBM‐SegPhos ligand (L3) coordinated to the iridium center.
To limit the number of conformations, transition‐state optimizations were performed initially only with an in‐in‐in‐in orientation of the methoxy groups, which can either point towards the adjacent aryl unit (inwards) or away from it (outwards). Further calculations to rationalize the origins of the enantioselectivity were then performed on the complete system generated from cyclobutanol 11 a and (S)‐DTBM‐SegPhos‐IrCl. [19] The quantitative energetic analysis then revealed a clear preference for the (S)‐enantiomeric transition structure with a calculated ee of 95 % (for details see the Supporting Information). [24] This result, which is in excellent agreement with the experimental facts, can be „explained“ as follows. In the most favorable (S) transition state both the favorable anti H−Cl orientation and the favorable alignment of the involved C−C−O unit with the P2Ir‐plane are in harmony with an optimal co‐planar alignment of the equatorial P‐aryl groups with the substrate (Figure 6).
Figure 6.

Most stable (S)‐TS showing an H‐Cl‐trans orientation and a favorable in‐plane alignment of the C−C‐O‐IrIIIP2 moiety, in harmony with a co‐planar orientation of the substrate's aryl unit and an equatorial P‐aryl group.
In contrast, in the lowest energy (R)‐transition state (Figure 7) such a co‐planar alignment of an equatorial P‐aryl group with the substrate is only possible at the expense of an energetically unfavorable out‐of‐plane orientation of the P2Ir and the C−C−O units (H−Cl cis) (Figure 7).
Figure 7.
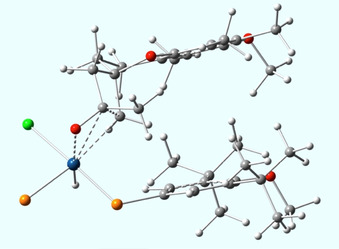
Most stable (R)‐TS with a H−Cl cis orientation and an unfavorable out‐of‐plane position of the P2Ir(III) and the C−C−O unit, enforced by a co‐planar arrangement of the substrate's aryl unit and an equatorial P‐aryl group.
Substrate scope
To explore the substrate scope of the Ir‐catalyzed cyclobutanol cleavage we initially used the easily accessible spirocyclobutanone 18 as a platform to prepare a variety of potential substrates of type 11 through addition of organometallic reagents. The results of the (mainly unoptimized) reactions are summarized in Table 2. Interestingly, the diastereoselectivity was found to depend on both the reagent and the solvent used. For instance, the reaction of 18 with MeMgBr in Et2O afforded selectively the trans product 11 a while a mixture of 11 a and its cis diastereomer 11’a was formed either with the same reagent in THF or with methyllithium in Et2O. As a general trend, we found that Grignard reagents in THF (except iso‐Pr‐MgCl and allyl‐MgBr) react with 18 in a cis‐selective fashion while trans‐products are favored in diethyl ether. This behavior might result from a different aggregation of the reagents in the different solvents. [25] Noteworthy, the branched Grignard reagent iso‐Pr‐MgCl afforded the addition product only in low yield because mainly reduction of the carbonyl group occurred in this case to yield a 1:1 mixture of diastereomeric alcohols (11/11’ with R=H). Fortunately, the cis‐ and trans‐diastereomers could be separated by column chromatography in all cases (Table 2).
Table 2.
Synthesis of cyclobutanols of type 11/11’.[a]
|
| ||||
|---|---|---|---|---|
|
Entry |
Reagent |
Solvent |
Yield[b] (11) [%] trans |
Yield[b] (11’) [%] cis |
|
1 |
MeLi |
Et2O |
17 |
53 |
|
2 |
MeMgBr |
Et2O |
93 |
– |
|
3 |
MeMgBr |
THF |
∼50[c] |
∼50[c] |
|
4 |
PhMgCl |
THF |
13 |
49 |
|
5 |
BnMgBr |
THF |
– |
13[d] |
|
6 |
vinyl‐MgBr[e] |
THF |
9 |
85 |
|
7 |
2‐butenyl‐MgBr |
THF |
12 |
27 |
|
8 |
propargyl‐MgBr |
Et2O |
71 |
24 |
|
9 |
TMS‐propargyl‐MgBr[f] |
Et2O |
62[g] |
46[g] |
|
10 |
1‐hexynyl‐Li[h] |
THF |
– |
78 |
|
11 |
iso‐propyl‐MgCl |
THF |
17 |
– |
|
12 |
allyl‐MgBr |
THF |
49 |
41 |

[a] The relative configuration of the products (cis/trans) was determined by means of 1H NMR (NOE) or X‐ray crystallography in the case of 11 a (Ref. [19]), 11’i and 11’n (see the Supporting Information); unless otherwise noted, the reactions were performed at −78 °C [b] Isolated yield. [c] Ratio determined by 1H NMR spectroscopy of the crude product mixture. [d] Low yield due to low quality of the Grignard reagent used. [e] Addition at −100 °C. [f] Addition at −40 °C. [g] The product still contained traces of solvent. [h] Prepared in situ from 1‐hexyne and nBuLi.
Additional substrates of type 11/11’ were prepared from the readily accessible vinylated compound 11’i (Scheme 7). Hydroboration of 11’i with 9‐BBN (9‐borabicyclo[3.3.1]nonane) and subsequent oxidation with H2O2 [26] gave the diol 19’, which in turn could easily be protected selectively at the primary OH group to give substrates 11’f (OAc), 11’g (OMOM), and 11’h (OBn).
Scheme 7.

Synthesis of cyclobutanols 19’ and 11’d. Reagents and conditions: a) see Table 2, entry 2; b) 9‐BBN, THF, 0 °C to RT, 5 h, then NaOH, H2O2, RT, 24 h; c) Ph‐I, Pd2(dba)3 (dba=dibenzylideneacetone) (5 mol %), PPh3, K3PO4, Me4NHCO2, DMF, 81 °C, 20 h.
In a similar fashion the trans‐isomer 19 (obtained from 11 i) was used to prepare the TBS‐protected (TBS=tert‐butyldimethylsilyl) substrate 11 e (structures shown in Scheme 8). We also employed the vinyl‐substituted cyclobutanol 19’ to prepare the substrate 11’d with a phenylethyl sidechain through Pd‐catalyzed reductive Heck reaction. [27] Remarkably, this transformation proceeded smoothly to afford the product 11’d in high yield without any significant Pd‐mediated cyclobutanol cleavage (as described by Uemura and co‐workers, [13a] compare Scheme 1).
Scheme 8.

Enantioselective conversion of various substrates of type 11 or 11’ in the Ir‐catalyzed cyclobutanol cleavage. Standard conditions: 5 mol % [Ir(COD)Cl]2, 15 mol % L3, 0.1 m solution of substrate in dry toluene, 110 °C (color change). [a] 1 mol % cat., 3 mol % L3, 70 °C; [b] 9 mol % cat., 26 mol % L3, toluene/H2O 4:1; [c] 10 mol % cat., 30 mol % L3; [d] 7 mol % cat., 19 mol % L3; [e] 85–95 °C; [f] 7 mol % cat., 23 mol % L3; [g] 8 mol % cat., 24 mol % L3; (for details see the Supporting Information).
With the various spirocyclobutanol substrates in our hands, the stage was set for the investigation of their performance in the Ir‐catalyzed ring opening. As a first important result we found that (using the same chiral ligand L3) the diastereomers 11 a and 11’a afforded the products with opposite absolute configuration (Scheme 8). However, in contrast to the trans‐spirocyclobutanol 11 a, which yields the methyl ketone 12 a with high enantioselectivity, the corresponding cis‐diastereomer 11’a gave the product ent‐12 a with only 18 % ee (non‐optimized). Nevertheless, other cis‐configurated substrates, that is, 11’b–11’d, gave rise to the expected products (12 b–12 d) with satisfying enantioselectivity.
Interestingly, the investigation of the three cis‐cyclobutanols 11’b–11’d with different spacer lengths between the phenyl group and the cyclobutanol unit showed that the reactivity drops with an increasing bulk of the side chain. Actually, the phenyl‐substituted substrate 11’b proved to be rather unreactive and the addition of water [16] was required to achieve at least a decent yield (44 %). In the case of 11’c, the catalyst load had to be increased to ensure a high yield. The enantioselectivity of the reaction of 11’c to 12 c increased from 78 % to 87 % ee upon lowering the temperature to 100 °C but at the expense of conversion (34 % yield). The bulky iso‐propyl‐substituted substrate (prepared according to Table 2, entry 11) did not react at all under the standard conditions. In contrast, the 2‐oxy‐ethyl substituted substrates, especially the TBS‐protected (11 e) and the benzyl‐protected (11’h) compounds, reacted smoothly to give the products 12 e and 12 h, respectively, in high yield and enantioselectivity (92–93 % ee). The corresponding acetyl‐protected substrate 11’f, however, proved to be completely unreactive and the MOM‐derivative 11’g only reacted very slowly, and unreacted starting material could be partly reisolated. Possibly, the catalysis is inhibited in the latter cases by coordination of the Ir atom to the polar functional groups.
While the examples given in Scheme 8 illustrate a fair scope of the method, a number of substrates with unsaturated sidechains failed to undergo the expected Ir‐catalyzed cyclobutanol cleavage (Scheme 9). For instance, the vinyl‐cyclobutanol 11’i and the related higher substituted allylic alcohol 11’k mainly afforded the dienes 20 and 21, respectively, possibly via formation of a π‐allyl‐Ir intermediate and subsequent β‐H elimination. Chiral GC analysis indicated that both of these compounds were formed as racemic mixtures. While the unprotected terminal alkyne 11 m only delivered a mixture of unreacted starting material and some unidentified side products (even after 20 h), the corresponding TMS‐protected alkyne 11 l quantitatively afforded 22 as the product of an Ir‐catalyzed 5‐endo‐dig cyclization. Noteworthy, the TIPS‐protected alkyne related to 11 l (not shown), the alkyne 11’n, and also both diastereomers of the allyl‐substituted cyclobutanol 11 o/11’o showed no conversion under the standard conditions, possibly due to inactivation of the catalyst through formation of a cyclic resting state formed by β‐insertion of the O‐bound Ir‐hydride to the unsaturated side chain.
Scheme 9.

Substrates of type 11/11’ that did not undergo Ir‐catalyzed cyclobutanol cleavage.
To further probe the scope of the Ir‐based methodology employing simpler prochiral cyclobutanols lacking the spirochromane moiety we converted the easily accessible cyclobutanone 23 [19] through addition of alkyl Grignard reagents and functional group manipulation into the substituted tertiary cyclobutanols 26 and 28 (Scheme 10). Noteworthy, the addition of MeMgBr in Et2O to the ketone 27 proceeds diastereoselectively to afford the trans‐product 28 in high yield as the only isolated product.
Scheme 10.

Synthesis of cyclobutanols 26 and 28. Reagents and conditions: a) nBuMgCl, THF, −78 °C, 1 h; b) NaOH, EtOH, 30 °C, 24 h; c) TBSOTf, 2,6‐lutidine, CH2Cl2, 0 °C to RT, 2.5 h; d) DMP, CH2Cl2, 0 °C to RT, 2 h; e) TBSOTf, 2,6‐lutidine, CH2Cl2, 0 °C to RT, 1.5 h; f) MeMgBr (3 m in Et2O), Et2O, −78 °C, 1 h.
Under the proven conditions of the Ir‐catalyzed cyclobutanol cleavage both of these substrates (26 and 28) afforded the corresponding chiral ketones (29 and 30, respectively) with high yields and good enantioselectivity (Scheme 11). Noteworthy, when 28 was treated with the corresponding Rh(OH) catalyst under the conditions of Seiser and Cramer, [16] a 2:1 mixture of the enone 31 and its non‐conjugated isomer 32 was formed, probably through elimination of TBS‐OH from the primary product 30. This again proves the advantage of our Ir‐based protocol for the enantioselective cleavage of 3‐oxy‐substituted cyclobutanols.
Scheme 11.

Ir‐ and Rh‐catalyzed cleavage of cyclobutanols 26 and 28.
Enantioselective synthesis of chromanes related to natural products
As already mentioned above, the starting point of the present study was our synthesis of α‐tocopherol (13) (Scheme 3) and in particular the discovery that the enantioselective opening of the prochiral spirocyclobutanol 11 a to the methylketone 12 a could be efficiently achieved using an Ir catalyst, whereas the related Rh‐based protocol only afforded the racemic product. In a similar fashion, the Ir‐catalyzed reaction of the more elaborated substrate 11 q afforded the α‐tocopherol precursor 12 q with virtually complete stereocontrol (Scheme 12). [19]
Scheme 12.

Ir‐catalyzed key step of our total synthesis of α‐tocopherol.
Against this background, we asked ourselves whether the methodology could be applied also to the synthesis of other tocopherol‐related compounds such as the antimalarial chromane natural product 33 recently isolated from Koeberlinia spinosa, which displays an interesting activity against the malaria parasite plasmodium falciparum (IC50=24 μm). (Figure 8). [28]
Figure 8.

Structure of an antiplasmodial chromane 33 isolated from Koeberlinia spinosa.
To probe the installation of an unsaturated (enone) sidechain, we used the substrate 11 r, which was obtained by TBS deprotection of an intermediate of our α‐tocopherol synthesis. [19] In this case (Scheme 13), the trisubstituted olefin in the side chain was well tolerated and the desired desymmetrized ketone 12 r was obtained in excellent yield (98 %) and diastereoselectivity (dr=98.5:1.5; determined by HPLC after transformation into α‐tocopherol methyl ether, see the Supporting Information).
Scheme 13.

Synthesis of the enone 34 (as a model compound related to 33) through cyclobutanol fragmentation and subsequent acid‐mediated double bond isomerization.
Treatment of the β,γ‐enone 12 r with trifluoromethane sulfonic acid in dichloromethane resulted in the migration of the double bond to give of the more stable conjugated enone 34 as a separable mixture of E and Z‐isomers (Scheme 13). Although E‐34 already displays some characteristic structural features of the natural product 33, we decided to also probe the Ir‐catalyzed cyclobutanol opening employing the spirochromane 38 with an aromatic substitution pattern related to 33 (Scheme 14). For this purpose, the literature‐known building blocks 35 [29] and 36 [19] were first fused to 37 in a Friedel–Cafts‐related condensation. While the use of BF3⋅Et2O as a Lewis acid [19] was not successful in this case, the desired reaction took place in the presence of an excess (4 equiv) of methane sulfonic acid in dichloromethane to afford 37 in 38 % yield as a mixture of cis and trans isomers. Subsequent saponification of the ester moiety, oxidation, [30] and reaction of the resulting ketone with MeMgBr in diethylether then cleanly afforded the trans‐cyclobutanol 38 as the desired desymmetrization precursor. And much to our satisfaction, the Ir‐catalyzed ring opening then proceeded smoothly under the proven conditions to give the ketone 39 in high yield and with excellent enantioselectivity (95 % ee).
Scheme 14.

Synthesis of the model chromane 39 (related to 33).
The expected absolute (S)‐configuration of the chiral 2,2‐disubstitued chromane 39 was confirmed by X‐ray crystallography (Figure 9). [31]
Figure 9.

Structure of 39 in the crystalline state.
Conclusions
We have demonstrated that the Ir‐catalyzed conversion of prochiral tert‐cyclobutanols proceeds under comparably mild conditions to afford β‐methyl‐substituted ketones in a variety of cases. In the presence of DTBM‐SegPhos as a chiral ligand the products are formed with up to 95 % ee. Our protocol appears to be particularly suited for the enantioselective desymmetrization [32] of prochiral β‐oxy‐substituted cyclobutanols that fail to react in a similar fashion under Rh catalysis. And indeed, deuteration experiments and kinetic isotope effect measurements revealed major mechanistic differences to related RhI‐catalyzed transformations. Based on the experimental data, we derived a plausible mechanism that involves the initial formation of an IrIII hydride intermediate by oxidative addition of IrI into the O−H bond of the cyclobutanol substrate. In the key C−C bond activating step, the four‐membered ring is cleaved by β‐C elimination, and the catalytic cycle is closed by reductive C−H elimination. This mechanism is supported by DFT calculations, and the computational analysis of competing transition states of the enantioselectivity‐determining β‐carbon elimination step even allowed the prediction of the stereochemical outcome. Although simple tert‐cyclobutanols such as 28 could be successfully employed as well, the developed protocol is of particular value for the stereo‐controlled synthesis of 2,2‐disubstituted chromanes related to natural products such as α‐tocopherol. Thus, we are optimistic that the Ir‐catalyzed cyclobutanol cleavage will find future application also in other laboratories. At least, it opens a new chapter in the use of Ir‐catalyzed reactions in natural product synthesis [21] and also compliments existing methods for the catalytic ring opening of cyclic alcohols to generate ketones with a (quaternary) chirality center in β‐position.[ 1 , 12b , 13b , 17 , 33 ] Furthermore, the protocol may find application in the preparation of selectively deuterated (or even tritium‐labeled) compounds. [34]
Experimental Section
General procedure for the Ir‐catalyzed cyclobutanol cleavage: A glass vial was charged under argon with [Ir(COD)Cl]2 and (S)‐DTBM‐SegPhos and the vial was sealed with a septum. After injection of a 0.1 m solution of the respective cyclobutanol in dry toluene at RT the solution was first stirred for 1.5 h and then heated to 85–110 °C. The reaction progress was monitored by TLC. Noteworthy, successful reactions were always associated with a color change of the solution from yellow–orange to dark red. Once the starting material was fully consumed (or nor further conversion was detected), the mixture was cooled to RT and a few milligrams of QuadraSil AP were added. After stirring for 30 min the mixture was filtered over a short pad of silica and all volatiles were removed under reduced pressure. The crude product was finally subjected to column chromatography to yield the ketone product (12) as a colorless oil.
(S)‐1‐(6‐Methoxy‐2,5,7,8‐tetramethylchroman‐2‐yl)propan‐2‐one (12 a): According to the general procedure, a solution of 150 mg (0.543 mmol) of cyclobutanol 11 a, 3.7 mg (5.51 μmol, 1 mol %) of [Ir(COD)Cl]2 and 20.0 mg (16.96 μmol, 3 mol %) of (S)‐DTBM‐SegPhos in 4.5 mL of dry toluene was heated for 18 h to 70 °C to give 142 mg (0.514 mmol, 95 %) of 12 a (92 % ee) after purification by column chromatography (SiO2, cHex/EtOAc 12:1). C17H24O3 (M=276.38 g mol−1). =1.04° (c=0.58 in CHCl3); 1H NMR (499 MHz, CDCl3): δ=3.63 (s, 3 H), 2.79 (d, 2 J H,H=14.0 Hz, 1 H), 2.64 (d, 2 J H,H=14.0 Hz, 1 H), 2.61–2.53 (m, 2 H), 2.22 (s, 3 H), 2.19 (s, 3 H), 2.14 (s, 3 H), 2.09 (s, 3 H), 1.96 (dt, 2 J H,H=13.9 Hz, 3 J H,H=7.0 Hz, 1 H), 1.86 (dt, 2 J H,H=13.6 Hz, 3 J H,H=6.7 Hz, 1 H), 1.35 ppm (s, 3 H); 13C NMR (125 MHz, CDCl3): δ=208.0, 150.0, 147.1, 128.3, 126.2, 123.0, 117.4, 74.2, 60.6, 52.8, 32.4, 31.6, 24.4, 20.7, 12.7, 12.1, 11.8 ppm; FTIR (ATR) =1707 (m), 1457 (m), 1404 (m), 1253 (s), 1090cm−1 (s); GC‐MS [t R]=9.932 min, m/z=276 ([M]+, 74), 243 (16), 219 (19), 203 (41), 179 (100), 135 (14), 91 (11), 43 % (18); HRMS (ESI): calcd 299.16177 [M+Na]+; found 299.16167.
2‐(6‐Methoxy‐2,5,7,8‐tetramethylchroman‐2‐yl)‐1‐phenylethan‐1‐one (12 b): According to the general procedure, a solution of 12 mg (35.4 μmol) of cyclobutanol 11’b, 2.2 mg (3.27 μmol, 9 mol %) of [Ir(COD)Cl]2 and 10.9 mg (9.24 μmol, 26 mol %) of (S)‐DTBM‐SegPhos in 0.4 mL of dry toluene and 0.1 mL of water was heated for 20 h to 110 °C to give 5.3 mg (15.7 μmol, 44 %) of 12 b (92 % ee) after purification by preparative TLC (SiO2, cHex/EtOAc 7:1). C22H26O3 (M=338.45 g mol−1). =8.30° (c=0.27 in CHCl3); 1H NMR (500 MHz, CDCl3): δ=7.94–7.92 (m, 2 H), 7.55–7.52 (m, 1 H), 7.42–7.39 (m, 2 H), 3.62 (s, 3 H), 3.38 (d, 2 J H,H=14.5 Hz, 1 H), 3.13 (d, 2 J H,H=14.5 Hz, 1 H), 2.63 (t, 3 J H,H=6.9 Hz, 2 H), 2.16–2.10 (m, 1 H), 2.14 (s, 6 H), 2.00–1.95 (m, 1 H), 1.84 (s, 3 H), 1.43 ppm (s, 3 H); 13C NMR (125 MHz, CDCl3): δ=199.0, 150.0, 147.2, 138.1, 133.1, 128.8, 128.5, 128.2, 126.1, 123.4, 117.5, 74.9, 60.6, 47.1, 31.6, 24.8, 20.8, 12.7, 11.9, 11.8 ppm; FTIR (ATR) =1676 (m), 1449 (m), 1254 (m), 1090 cm−1 (s); GC‐MS [t R]=11.668 min, m/z=338 ([M]+, 56), 305 (18), 218 (18), 203 (42), 179 (68), 135 (18), 105 (100), 91 (12), 77 (43), 44 % (12). HRMS (ESI): calcd 339.19547 [M+H]+; found 339.19585; calcd 361.17742 [M+Na]+; found 361.17750.
1‐(6‐Methoxy‐2,5,7,8‐tetramethylchroman‐2‐yl)‐3‐phenylpropan‐2‐one (12 c): According to the general procedure, a solution of 11 mg (31.2 μmol) of cyclobutanol 11’c, 2.1 mg (3.12 μmol, 10 mol %) of [Ir(COD)Cl]2 and 11.1 mg (9.41 μmol, 30 mol %) of (S)‐DTBM‐SegPhos in 0.4 mL of dry toluene was heated for 20 h to 110 °C to give 10.0 mg (28.4 μmol, 91 %) of 12 c (78 % ee) after purification by preparative TLC (SiO2, cHex/EtOAc 5:1). C23H28O3 (M=352.47 g mol−1). =−21.85° (c=0.18 in CHCl3); 1H NMR (500 MHz, CDCl3): δ=7.31–7.28 (m, 2 H), 7.25–7.23 (m, 1 H), 7.13–7.12 (m, 2 H), 3.82 (d, 2 J H,H=15.3 Hz, 1 H), 3.75 (d, 2 J H,H=15.3 Hz, 1 H), 3.64 (s, 3 H), 2.83 (d, 2 J H,H=14.2 Hz, 1 H), 2.65 (d, 2 J H,H=14.2 Hz, 1 H), 2.60–2.47 (m, 2 H), 2.20 (s, 3 H), 2.13 (s, 6 H), 2.00–1.94 (m, 1 H), 1.88–1.83 (m, 1 H), 1.36 ppm (s, 3 H); 13C NMR (125 MHz, CDCl3): δ=206.8, 150.1, 147.1, 134.2, 129.7, 128.8, 128.3, 127.1, 126.2, 123.0, 117.5, 74.5, 60.6, 51.9, 50.6, 31.6, 24.6, 20.7, 12.7, 12.2, 11.8 ppm; FTIR (ATR) =1714 (m), 1454 (m), 1403 (m), 1253 (s), 1088 cm−1 (s); GC‐MS [t R]=11.828 min, m/z=352 ([M]+, 62), 219 (20), 203 (19), 179 (57), 135 (13), 91 (100), 65 % (15); HRMS (ESI): calcd. 375.19307 [M+Na]+; found 375.19326.
1‐(6‐Methoxy‐2,5,7,8‐tetramethylchroman‐2‐yl)‐4‐phenylbutan‐2‐one (12 d): According to the general procedure, a solution of 11 mg (30.0 μmol) of cyclobutanol 11’d, 1.5 mg (2.23 μmol, 7 mol %) of [Ir(COD)Cl]2 and 6.7 mg (5.68 μmol, 19 mol %) of (S)‐DTBM‐SegPhos in 0.5 mL of dry toluene was heated for 2 h to 100 °C and 2 h to 110 °C to give 10 mg (27.3 μmol, 91 %) of 12 d (84 % ee) after purification by column chromatography (SiO2, cHex/EtOAc 35:1). C24H30O3 (M=366.50 g mol−1). =−27.58° (c=0.33 in CHCl3); 1H NMR (500 MHz, CDCl3): δ=7.27–7.24 (m, 2 H), 7.18–7.14 (m, 3 H), 3.63 (s, 3 H), 2.91–2.77 (m, 4 H), 2.74 (d, 2 J H,H=14.0 Hz, 1 H), 2.61 (d, 2 J H,H=14.0 Hz, 1 H), 2.60–2.50 (m, 2 H), 2.17 (s, 3 H), 2.13 (s, 3 H), 2.05 (s, 3 H), 1.97–1.92 (m, 1 H), 1.87–1.82 (m, 1 H), 1.33 ppm (s, 3 H); 13C NMR (125 MHz, CDCl3): δ=208.8, 150.0, 147.1, 141.2, 128.6, 128.5, 128.3, 126.2, 126.2, 123.0, 117.4, 74.4, 60.6, 51.9, 46.5, 31.6, 29.7, 24.6, 20.7, 12.7, 12.1, 11.8 ppm; FTIR (ATR) =2927 (m), 1711 (m), 1454 (s), 1403 (m), 1252 (s), 1088 (s), 1062 (m), 1009 (m), 699 cm−1 (m); GC‐MS [t R]=12.293 min, m/z=366 ([M]+, 100), 257 (13), 234 (18), 219 (26), 203 (32), 179 (79), 105 (32), 91 % (62); HRMS (ESI): calcd. 389.20872 [M+Na]+; found 389.20879.
4‐[(tert‐Butyldimethylsilyl)oxy]‐1‐(6‐methoxy‐2,5,7,8‐tetramethyl‐chroman‐2‐yl)butan‐2‐one (12 e): According to the general procedure, a solution of 15 mg (35.7 μmol) of cyclobutanol 11 e, 1.3 mg (1.94 μmol, 5 mol %) of [Ir(COD)Cl]2 and 6.5 mg (5.51 μmol, 15 mol %) of (S)‐DTBM‐SegPhos in 0.5 mL of dry toluene was heated for 2 h to 85 °C, 1 h to 90 °C and 1 h to 95 °C to give 15 mg (35.7 μmol, 99 %) of 12 e (92 % ee) after purification by column chromatography (SiO2, cHex/EtOAc 50:1). C24H40O4Si (M=420.67 g mol−1). 1 H NMR (500 MHz, CDCl3): δ=3.86 (t, 3 J H,H=6.3 Hz, 2 H), 3.63 (s, 3 H), 2.80 (d, 2 J H,H=14.5 Hz, 1 H), 2.76–2.64 (m, 3 H), 2.60–2.53 (m, 2 H), 2.19 (s, 3 H), 2.13 (s, 3 H), 2.09 (s, 3 H), 2.02–1.96 (m, 1 H), 1.90–1.84 (m, 1 H), 1.35 (s, 3 H), 0.85 (s, 9 H), 0.02 (s, 3 H), 0.02 ppm (s, 3 H). 13 C NMR (125 MHz, CDCl3): δ=208.7, 150.0, 147.1, 128.3, 126.1, 123.1, 117.5, 74.3, 60.5, 58.8, 52.4, 47.6, 31.5, 26.0, 24.6, 20.7, 18.3, 12.7, 12.1, 11.8, −5.3 ppm. FT‐IR (ATR) [cm−1]=2953 (m), 2930 (m), 2887 (m), 2857 (m), 1713 (m), 1462 (m), 1404 (m), 1254 (s), 1090 (s), 836 (m), 777 (m). GC‐MS [tR]=11.834 min, m/z (%)=420 ([M]+, 13), 219 (100), 203 (13), 179 (18), 145 (6), 115 (5), 91 (6), 75 (10), 41 (6). HRMS (ESI): calcd 443.25881 [M + Na]+; Found 443.25887. =9.74° (c=0.38 in CHCl3).
1‐(6‐Methoxy‐2,5,7,8‐tetramethylchroman‐2‐yl)‐4‐(methoxy‐methoxy)butan‐2‐one (12 g) According to the general procedure, a solution of 15 mg (42.8 μmol) of cyclobutanol 11’g, 2.0 mg (2.98 μmol, 7 mol %) of [Ir(COD)Cl]2 and 12.0 mg (10.2 μmol, 23 mol %) of (S)‐DTBM‐SegPhos in 0.6 mL of dry toluene was heated for 4.5 h to 110 °C to give 7 mg (20.0 μmol, 47 %) of 12 g (83 % ee) after purification by column chromatography (SiO2, cHex/EtOAc 9:1). C20H30O5 (M=350.46 g mol−1). =3.87° (c=0.16 in CHCl3); 1H NMR (500 MHz, CDCl3): δ=4.59 (s, 2 H), 3.78 (t, 3 J H,H=6.2 Hz, 2 H), 3.63 (s, 3 H), 3.34 (s, 3 H), 2.87–2.73 (m, 3 H), 2.69 (d, 2 J H,H=14.3 Hz, 1 H), 2.61–2.57 (m, 2 H), 2.19 (s, 3 H), 2.14 (s, 3 H), 2.09 (s, 3 H), 2.01–1.96 (m, 1 H), 1.90–1.85 (m, 1 H), 1.36 ppm (s, 3 H); 13C NMR (125 MHz, CDCl3): δ=207.7, 150.0, 147.1, 128.3, 126.2, 123.0, 117.5, 96.7, 74.3, 62.7, 60.5, 55.4, 52.3, 44.8, 31.5, 24.5, 20.7, 12.7, 12.1, 11.8 ppm. FTIR (ATR) =1714 (m), 1457 (m), 1253 (s), 1151 (m), 1111 (s), 1089 (s), 1059 (s), 1041 (s), 1017 cm−1 (m); GC‐MS [t R]=11.273 min, m/z=350 ([M]+, 100), 288 (15), 255 (13), 219 (49), 203 (50), 179 (52), 135 (14), 91 (15), 45 % (29); HRMS (ESI): calcd. 373.19855 [M+Na]+; found 373.19859.
4‐(Benzyloxy)‐1‐(6‐methoxy‐2,5,7,8‐tetramethylchroman‐2‐yl)‐butan‐2‐one (12 h) According to the general procedure, a solution of 20 mg (50.4 μmol) of cyclobutanol 11’h, 2.7 mg (4.01 μmol, 8 mol %) of [Ir(COD)Cl]2 and 14.0 mg (11.87 μmol, 24 mol %) of (S)‐DTBM‐SegPhos in 0.6 mL of dry toluene was heated for 4 h to 110 °C to give 11 mg (27.7 μmol, 55 %) of 12 h (93 % ee) after purification by column chromatography (SiO2, cHex/EtOAc 10:1). C25H32O4 (M=396.53 g mol−1). =−3.64° (c=0.17 in CHCl3). 1H NMR (600 MHz, CDCl3): δ=7.34–7.27 (m, 5 H), 4.49 (s, 2 H), 3.72 (td, 3 J H,H=6.3 Hz, 4 J H,H=4.1 Hz, 2 H), 3.63 (s, 3 H), 2.86–2.74 (m, 3 H), 2.69 (d, 2 J H,H=14.4 Hz, 1 H), 2.62–2.52 (m, 2 H), 2.18 (s, 3 H), 2.13 (s, 3 H), 2.08 (s, 3 H), 2.00–1.96 (m, 1 H), 1.88–1.84 (m, 1 H), 1.35 ppm (s, 3 H); 13C NMR (150 MHz, CDCl3): δ=208.0, 150.0, 147.1, 138.3, 128.5, 128.3, 127.8, 127.8, 126.2, 123.1, 117.5, 74.3, 73.3, 65.4, 60.5, 52.3, 44.9, 31.5, 24.6, 20.7, 12.7, 12.1, 11.8 ppm; FTIR (ATR): =1711 (m), 1454 (m), 1403 (m), 1252 (m), 1088 (s), 1062 (m), 1007 (m), 736 (m), 698 cm−1 (m); GC‐MS [t R]=12.929 min, m/z=396 ([M]+, 97), 281 (16), 255 (12), 217 (78), 203 (46), 179 (53), 135 (14), 105 (19), 91 (100), 77 % (27); HRMS (ESI): calcd. 419.21928 [M+Na]+; found 419.21948.
2‐[(tert‐Butyldimethylsilyl)oxy]octan‐4‐one (29): According to the general procedure, a solution of 30 mg (0.116 mmol) of cyclobutanol 26, 2.4 mg (3.57 μmol, 3 mol %) of [Ir(COD)Cl]2 and 12.5 mg (10.60 μmol, 9 mol %) of (S)‐DTBM‐SegPhos in 0.6 mL of dry toluene was heated for 4 h to 100 °C to give 24 mg (0.093 mmol, 80 %) of 29 (70 % ee, determined after deprotection of the alcohol) after purification by column chromatography (SiO2, cHex/EtOAc 100:1). C14H30O2Si (M=258.48 g mol−1). =23.79° (c=0.33 in CHCl3). 1H NMR (500 MHz, CDCl3): δ=4.32–4.26 (m, 1 H), 2.63 (dd, 2 J H,H=15.0 Hz, 3 J H,H=7.3 Hz, 1 H), 2.44–2.41 (m, 2 H), 2.38 (dd, 2 J H,H=15.0 Hz, 3 J H,H=5.1 Hz, 1 H), 1.57–1.50 (m, 2 H), 1.34–1.27 (m, 2 H), 1.16 (d, 3 J H,H=6.1 Hz, 3 H), 0.90 (t, 3 J H,H=7.4 Hz, 3 H), 0.85 (s, 9 H), 0.06 (s, 3 H), 0.02 ppm (s, 3 H); 13C NMR (125 MHz, CDCl3): δ=210.3, 65.8, 52.4, 44.5, 25.9, 25.7, 24.2, 22.4, 18.1, 14.0, −4.4, −4.8 ppm; FTIR (ATR): =1715 (m), 1254 (m), 1134 (m), 1060 (m), 1040 (m), 1005 (m), 993 (m), 835 (s), 809 (m), 776 cm−1 (s); GC‐MS [t R]=7.009 min, m/z=243 (2), 201 (91), 157 (100), 115 (12), 101 (18), 75 (88), 57 (22), 41 % (46); HRMS (EI): calcd. 201.1311 [M‐tBu]+; found 201.14.
4‐[(tert‐Butyldimethylsilyl)oxy]‐4‐methyloctan‐2‐one (30): According to the general procedure, a solution of 45 mg (0.165 mmol) of cyclobutanol 28, 2.2 mg (3.28 μmol, 2 mol %) of [Ir(COD)Cl]2 and 11.7 mg (9.92 μmol, 6 mol %) of (S)‐DTBM‐SegPhos in 0.8 mL of dry toluene was heated for 5.5 h to 100 °C to give 44 mg (0.161 mmol, 98 %) of 30 (85 % ee) after purification by column chromatography (SiO2, cHex/EtOAc 80:1). C15H32O2Si (M=272.50 g mol−1). =28.41° (c=0.21 in CHCl3); 1H NMR (500 MHz, CDCl3): δ=2.61 (d, 2 J H,H=13.6 Hz, 1 H), 2.48 (d, 2 J H,H=13.6 Hz, 1 H), 2.19 (s, 3 H), 1.57–1.53 (m, 2 H), 1.39–1.20 (m, 4 H), 1.31 (s, 3 H), 0.90 (t, 3 J H,H=7.1 Hz, 3 H), 0.87 (s, 9 H), 0.10 (s, 3 H), 0.09 ppm (s, 3 H); 13C NMR (125 MHz, CDCl3): δ=208.7, 75.6, 55.0, 43.0, 32.8, 28.1, 26.6, 26.1, 23.3, 18.4, 14.2, −1.7 ppm; FTIR (ATR) =2957 (m), 2930 (m), 1712 (m), 1253 (m), 1075 (m), 1028 (m), 1005 (m), 834 (s), 772 cm−1 (s); GC‐MS [t R]=6.757 min, m/z=239 (8), 215 (15), 157 (33), 132 (10), 115 (53), 75 (79), 57 % (100); HRMS (ESI): calcd. 215.1467 [M‐tBu]+; found 215.17.
(R,E )‐1‐[(S)‐6‐Methoxy‐2,5,7,8‐tetramethylchroman‐2‐yl]‐4,8,12‐trimethyltridec‐4‐en‐2‐one (12 r): According to the general procedure, a solution of 54 mg (0.118 mmol) of cyclobutanol 11 r, 4.0 mg (5.95 μmol, 5 mol %) of [Ir(COD)Cl]2 and 21.0 mg (17.81 μmol, 15 mol %) of (S)‐DTBM‐SegPhos in 0.8 mL of dry toluene was heated for 2 h to 85 °C, 1 h to 90 °C and 0.5 h to 95 °C to give 53 mg (0.116 mmol, 98 %) of 12 q (97 % de) after purification by column chromatography (SiO2, cHex/EtOAc 80:1). The diastereomeric purity was determined after conversion to α‐tocopherol methyl ether (see Supporting Information). C30H48O3 (M=456.71 g mol−1). =29.4° (c=0.36 in CHCl3); 1H NMR (500 MHz, CDCl3): δ=5.18–5.15 (m, 1 H), 3.63 (s, 3 H), 3.14 (d, 2 J H,H=14.8 Hz, 1 H), 3.10 (d, 2 J H,H=14.8 Hz, 1 H), 2.81 (d, 2 J H,H=14.4 Hz, 1 H), 2.62–2.53 (m, 3 H, H‐1’b), 2.19 (s, 3 H), 2.14 (s, 3 H), 2.09 (s, 3 H), 2.04–1.96 (m, 3 H, H‐3a), 1.89–1.84 (m, 1 H), 1.60 (s, 3 H), 1.52 (sept., 3 J H,H=6.7 Hz, 1 H), 1.35 (s, 3 H), 1.40–1.19 (m, 5 H), 1.15–1.05 (m, 4 H), 0.87–0.85 ppm (m, 9 H, H‐2’’); 13C NMR (125 MHz, CDCl3): δ=208.3, 150.0, 147.2, 130.6, 128.5, 128.2, 126.1, 123.0, 117.5, 74.4, 60.5, 55.9, 50.1, 39.5, 37.3, 36.9, 32.6, 31.5, 28.1, 25.8, 24.9, 24.6, 22.9, 22.8, 20.7, 19.7, 16.5, 12.7, 12.1, 11.8 ppm; FTIR (ATR): =2951 (s), 2926 (s), 2869 (m), 1713 (m), 1458 (s), 1404 (m), 1253 (s), 1089 cm−1 (s); GC‐MS [t R]=13.671 min, m/z=456 ([M]+, 40), 234 (23), 219 (100), 203 (90), 179 % (92); HRMS (ESI): calcd. 479.34957 [M+Na]+; found 479.34946.
(S)‐1‐(6‐Methoxy‐2,5,7,8‐tetramethylchroman‐2‐yl)propan‐2‐one (12 a): According to the general procedure, a solution of 150 mg (0.543 mmol) of cyclobutanol 11a, 3.7 mg (5.51 μmol, 1 mol %) of [Ir(COD)Cl]2 and 20.0 mg (16.96 μmol, 3 mol %) of (S)‐DTBM‐SegPhos in 4.5 mL of dry toluene was heated for 18 h to 70 °C to give 142 mg (0.514 mmol, 95 %) of 12 a (92% ee) after purification by column chromatography (SiO2, cHex/EtOAc 12:1). C17H24O3 (M=276.38 g mol−1). =1.04° (c=0.58 in CHCl3); 1H NMR (499 MHz, CDCl3): δ=3.63 (s, 3 H), 2.79 (d, 2 J H,H=14.0 Hz, 1 H), 2.64 (d, 2 J H,H=14.0 Hz, 1 H), 2.61–2.53 (m, 2 H), 2.22 (s, 3 H), 2.19 (s, 3 H), 2.14 (s, 3 H), 2.09 (s, 3 H), 1.96 (dt, 2 J H,H=13.9 Hz, 3 J H,H=7.0 Hz, 1 H), 1.86 (dt, 2 J H,H=13.6 Hz, 3 J H,H=6.7 Hz, 1 H), 1.35 ppm (s, 3H ); 13C NMR (125 MHz, CDCl3): δ=208.0, 150.0, 147.1, 128.3, 126.2, 123.0, 117.4, 74.2, 60.6, 52.8, 32.4, 31.6, 24.4, 20.7, 12.7, 12.1, 11.8 ppm; FTIR (ATR) =1707 (m), 1457 (m), 1404 (m), 1253 (s), 1090 cm−1 (s); GC‐MS [t R]=9.932 min, m/z=276 ([M]+, 74), 243 (16), 219 (19), 203 (41), 179 (100), 135 (14), 91 (11), 43 % (18); HRMS (ESI): calcd. 299.16177 [M+Na]+; found 299.16167.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Support of this work by the University of Cologne is gratefully acknowledged. We sincerely thank Thomas Netscher from DSM Nutritional Products for the determination of the enantiomeric purity of samples of α‐tocopherol methyl ether by means of HPLC. We also thank Nils Schlörer and the local NMR facility for the excellent service and help in context of NMR‐based ee measurements. Additionally, we thank the computing center of the University of Cologne (RRZK) for providing CPU time on the DFG‐funded supercomputer CHEOPS. Open access funding enabled and organized by Projekt DEAL.
F. Ratsch, J. P. Strache, W. Schlundt, J.-M. Neudörfl, A. Adler, S. Aziz, B. Goldfuss, H.-G. Schmalz, Chem. Eur. J. 2021, 27, 4640.
References
- 1.
- 1a. Souillart L., Cramer N., Chem. Rev. 2015, 115, 9410–9464; [DOI] [PubMed] [Google Scholar]
- 1b. Xia Y., Lu G., Liu P., Dong G., Nature 2016, 539, 546–550; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1c. Ruhland K., Eur. J. Org. Chem. 2012, 2683–2706; [Google Scholar]
- 1d. Souillart L., Parker E., Cramer N., Asymmetric Transformations via C−C Bond Cleavage, published in: C−C Bond Activation, Vol. 346 (Ed.: Dong G.), Springer-Verlag, 2014, pp 163–193; [DOI] [PubMed] [Google Scholar]
- 1e. Cleavage of Carbon-Carbon Single Bonds by Transition Metals (Eds.: Murakami M., Chatani N.), Wiley-VCH, 2016. [Google Scholar]
- 2. Seiser T., Cramer N., Org. Biomol. Chem. 2009, 7, 2835–2840. [DOI] [PubMed] [Google Scholar]
- 3. Khoury P. R., Goddard J. D., Tam W., Tetrahedron 2004, 60, 8103–8112. [Google Scholar]
- 4. Murakami M., Amii H., Ito Y., Nature 1994, 370, 540–541. [Google Scholar]
- 5.
- 5a. Ishida N., Ikemoto W., Murakami M., Org. Lett. 2012, 14, 3230–3232; [DOI] [PubMed] [Google Scholar]
- 5b. Ishida N., Ikemoto W., Murakami M., J. Am. Chem. Soc. 2014, 136, 5912–5915. [DOI] [PubMed] [Google Scholar]
- 6. Murakami M., Takahashi K., Amii H., Ito Y., J. Am. Chem. Soc. 1997, 119, 9307–9308. [Google Scholar]
- 7. Jones W. D., Mechanistic Studies of Transition Metal-Mediated C−C Bond Activation, published in: C−C Bond Activation, Vol. 346 (Ed.: Dong G.), Springer-Verlag, Berlin, Heidelberg: 2014, pp 1–32. [DOI] [PubMed] [Google Scholar]
- 8. Xu T., Dermenci A., Dong G., Transition Metal-Catalyzed C−C Bond Activation of Four-Membered Cyclic Ketones, published in: C−C Bond Activation, Vol. 346 (Ed.: Dong G.), Springer, Berlin, Heidelberg, 2014, pp 233–258. [DOI] [PubMed] [Google Scholar]
- 9. Zhao X. H., Fan X., Yu J., Zhu C., J. Am. Chem. Soc. 2015, 137, 3490–3493. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Huan L., Zhu C., Org. Chem. Front. 2016, 3, 1467–1471; [Google Scholar]
- 10b. Ren R., Zhao H., Huan L., Zhu C., Angew. Chem. Int. Ed. 2015, 54, 12692–12696; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 12883–12887. [Google Scholar]
- 11.
- 11a. Ziadi A., Correa A., Martin R., Chem. Commun. 2013, 49, 4286–4288; [DOI] [PubMed] [Google Scholar]
- 11b. Chen L., Sun F.-N., Sun Y.-L., Xu Z., Zheng Z.-J., Cui Y.-M., Cao J., Xu L.-W., Adv. Synth. Catal. 2018, 360, 411–415; [Google Scholar]
- 11c. Wang Q., Chen R., Lou J., Zhang D. H., Zhou Y.-G., Yu Z., ACS Catal. 2019, 9, 11669–11675. [Google Scholar]
- 12.
- 12a. Xia Y., Liu Z., Liu Z., Ge R., Ye F., Hossain M., Zhang Y., Wang J., J. Am. Chem. Soc. 2014, 136, 3013–3015; [DOI] [PubMed] [Google Scholar]
- 12b. Masarwa A., Weber M., Sarpong R., J. Am. Chem. Soc. 2015, 137, 6327–6334. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Matsumura S., Maeda Y., Nishimura T., Uemura S., J. Am. Chem. Soc. 2003, 125, 8862–8869; for a recent application of the Pd-catalyzed opening of cyclobutanols in total synthesis, see: [DOI] [PubMed] [Google Scholar]
- 13b. Kerschgens I., Rovira A. R., Sarpong R., J. Am. Chem. Soc. 2018, 140, 9810–9813. [DOI] [PubMed] [Google Scholar]
- 14.
- 14a. Matsuda T., Shigeno M., Murakami M., J. Am. Chem. Soc. 2007, 129, 12086–12087; for a more recent mechanistic study on Rh-catalyzed stereoselective C−C/C−H activation of tert-cyclobutanols, see also:17877354 [Google Scholar]
- 14b. Yu H., Wang C., Yang Y., Dang Z.-M., Chem. Eur. J. 2014, 20, 3839–3848. [DOI] [PubMed] [Google Scholar]
- 15.
- 15a. Matsuda T., Shigeno M., Makino M., Murakami M., Org. Lett. 2006, 8, 3379–3381; [DOI] [PubMed] [Google Scholar]
- 15b. Ishida N., Nakanishi Y., Murakami M., Angew. Chem. Int. Ed. 2013, 52, 11875–11878; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 12091–12094; [Google Scholar]
- 15c. Yada A., Fujita S., Murakami M., J. Am. Chem. Soc. 2014, 136, 7217–7220. [DOI] [PubMed] [Google Scholar]
- 16. Seiser T., Cramer N., J. Am. Chem. Soc. 2010, 132, 5340–5341. [DOI] [PubMed] [Google Scholar]
- 17.
- 17a. Cramer N., Seiser T., Synlett 2011, 4, 449–460; [Google Scholar]
- 17b. Cramer N., Seiser T., Quaternary Stereogenic Centers by Enantioselective β-Carbon Eliminations from tert-Cyclobutanols, published in Asymmetric Synthesis: More Methods and Applications (Eds.: Christmann M., Bräse S.), Wiley-VCH, Weinheim, 2012, pp 55–59; [Google Scholar]
- 17c. Souillart L., Cramer N., Chem. Sci. 2014, 5, 837–840. [Google Scholar]
- 18.
- 18a. Termath A. O., Sebode H., Schlundt W., Stemmler R. T., Netscher T., Bonrath W., Schmalz H.-G., Chem. Eur. J. 2014, 20, 12051–12055; [DOI] [PubMed] [Google Scholar]
- 18b. Termath A. O., Velder J., Stemmler R. T., Netscher T., Bonrath W., Schmalz H.-G., Eur. J. Org. Chem. 2014, 3337–3340. [Google Scholar]
- 19. Ratsch F., Schlundt W., Albat D., Zimmer A., Neudörfl J.-M., Netscher T., Schmalz H.-G., Chem. Eur. J. 2019, 25, 4941–4945. [DOI] [PubMed] [Google Scholar]
- 20.
- 20a. Crabtree R. H., Dion R. P., Gibboni D. J., McGrath D. V., Holt E. M., J. Am. Chem. Soc. 1986, 108, 7222–7227; [Google Scholar]
- 20b. Murakami M., Itami K., Ubukata M., Tsuji I., Ito Y., J. Org. Chem. 1998, 63, 4–5; [DOI] [PubMed] [Google Scholar]
- 20c. Nishimura T., Yoshinaka T., Nishiguchi Y., Maeda Y., Uemura S., Org. Lett. 2005, 7, 2425–2427; [DOI] [PubMed] [Google Scholar]
- 20d. Tashiro S., Yamada M., Shionoya M., Angew. Chem. Int. Ed. 2015, 54, 5351–5354; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 5441–5444; [Google Scholar]
- 20e. Yu J., Yan H., Zhu C., Angew. Chem. Int. Ed. 2016, 55, 1143–1146; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 1155–1158. [Google Scholar]
- 21.
- 21a. Yuan C., Liu B., Org. Chem. Front. 2018, 5, 106–131; [Google Scholar]
- 21b. Chen P., Wu Y., Zhu S., Jiang H., Ma Z., Org. Chem. Front. 2018, 5, 132–150. [Google Scholar]
- 22.
- 22a. Ladipo F. T., Kooti M., Merola J. S., Inorg. Chem. 1993, 32, 1681–1688; [Google Scholar]
- 22b. Blum O., Milstein D., J. Am. Chem. Soc. 1995, 117, 4582–4594; [Google Scholar]
- 22c. Tani K., Iseki A., Yamagata T., Angew. Chem. Int. Ed. 1998, 37, 3381–3383; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 1998, 110, 3590–3592; [Google Scholar]
- 22d. Kläring P., Pahl S., Braun T., Penner A., Dalton Trans. 2011, 40, 6785–6791; [DOI] [PubMed] [Google Scholar]
- 22e. Crestani M. G., Steffen A., Kenwright A. M., Batsanov A. S., Howard J. A. K., Marder T. B., Organometallics 2009, 28, 2904–2914; for related Rh-studies see: [Google Scholar]
- 22f. Yuwen J., Jiao Y., Brennessel W. W., Jones W. D., Inorg. Chem. 2016, 55, 9482–9491. [DOI] [PubMed] [Google Scholar]
- 23.M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, D. J. Fox, Gaussian 16, Revision B.01, Gaussian, Inc., Wallingford CT, 2016.
- 24.The origin of the enantioselectivity was analyzed by full geometry optimizations and energetic comparison of diastereomorphic transition structures of the β-elimination (C−C-bond activation) of IrIIIP(S)-DTBM-SEGHOS)(H)(Cl)-substrate complexes. For simplification, no intermediates or pre-equilibria were considered, assuming Curtin–Hammett conditions (cf. Eliel E. L., Wilen S. H., Doyle M. P., Basic Organic Stereochemistry, Wiley, 2001). All transition structures were fully geometry optimized without restrains employing a two-layer ONIOM(B97D3/SDD(+ECP Ir), D95 (C,H,O,P,Cl):PM6) model (see below). For the frequency computations, entropic quasi-harmonic corrections according to Grimme with a frequency cut-off value of 100.0 wavenumbers were applied, employing the python program GoodVibes. Single-point energies in the solvent toluene on these geometries were obtained using the PW6B95D3 functional, the 6–311G** (C H O P Cl) and SDD(+ECP, Ir) basis sets and reaction field calculations using the integral equation formalism model (SCRF, PCM). All computations and thermochemical analyses were performed at 343.15 K (70 °C). Dispersion corrections (S. Grimme, Chem. Eur. J. 2012, 18, 9955-9964) were implemented in both applied DFT methods (B97D3, PW6B95D3). [Google Scholar]
- 25.
- 25a. Peltzer R. M., Gauss J., Eisenstein O., Cascella M., J. Am. Chem. Soc. 2020, 142, 2984–2994; [DOI] [PubMed] [Google Scholar]
- 25b. Mori T., Kato S., J. Phys. Chem. A 2009, 113, 6158–6165. [DOI] [PubMed] [Google Scholar]
- 26. Brown H. C., Chen J. C., J. Org. Chem. 1981, 46, 3978–3988. [Google Scholar]
- 27. J. A. Gurak Jr , Engle K. M., ACS Catal. 2018, 8, 8987–8992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Presley C. C., Valenciano A. L., Fernández-Murga M. L., Du Y., Shanaiah N., Cassera M. B., Goetz M., Clement J. A., Kingston D. G. I., J. Nat. Prod. 2018, 81, 475–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Singh U. S., Scannell R. T., An H., Carter B. J., Hecht S. M., J. Am. Chem. Soc. 1995, 117, 12691–12699. [Google Scholar]
- 30. Guérin C., Bellosta V., Guillamot G., Cossy J., Org. Lett. 2011, 13, 3534–3537. [DOI] [PubMed] [Google Scholar]
- 31.Deposition numbers 2042320 (for 11′i), 2042317 (for 11′o), 2042322 (for 11′n), 2042319 (for rac-20), 2042321 (for trans-37), 2042314 (for cis-37), 2042315 (for S25), 2042318 (for 38), and 2042316 (for 39) contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.
- 32. Feng J., Holmes M., Krische M. J., Chem. Rev. 2017, 117, 12564–12580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yayla H. G., Wang H., Tarantino K. T., Orbe H. S., Knowles R. R., J. Am. Chem. Soc. 2016, 138, 10794–10797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Atzrodt J., Derdau V., Fey T., Zimmermann J., Angew. Chem. Int. Ed. 2007, 46, 7744–7765; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 7890–7911. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary