

Abstract
Since the introduction of next‐generation sequencing, an increasing number of disorders have been discovered to have genetic etiology. To address diverse clinical questions and coordinate research activities that arise with the identification of these rare disorders, we developed the Human Disease Genes website series (HDG website series): an international digital library that records detailed information on the clinical phenotype of novel genetic variants in the human genome (https://humandiseasegenes.info/). Each gene website is moderated by a dedicated team of clinicians and researchers, focused on specific genes, and provides up‐to‐date—including unpublished—clinical information. The HDG website series is expanding rapidly with 424 genes currently adopted by 325 moderators from across the globe. On average, a gene website has detailed phenotypic information of 14.4 patients. There are multiple examples of added value, one being the ARID1B gene website, which was recently utilized in research to collect clinical information of 81 new patients. Additionally, several gene websites have more data available than currently published in the literature. In conclusion, the HDG website series provides an easily accessible, open and up‐to‐date clinical data resource for patients with pathogenic variants of individual genes. This is a valuable resource not only for clinicians dealing with rare genetic disorders such as developmental delay and autism, but other professionals working in diagnostics and basic research. Since the HDG website series is a dynamic platform, its data also include the phenotype of yet unpublished patients curated by professionals providing higher quality clinical detail to improve management of these rare disorders.
Keywords: clinical data, HDG, HDG website series, HPO, online resource, phenotype
1. INTRODUCTION
In the last decade, revolutionary advances in sequencing techniques have dramatically improved the diagnostic yield in clinical practice for patients with rare genetic disorders. For neurodevelopmental disorders such as intellectual disability, over 1,000 genes have been reported—leading to a diagnostic yield up to 56% for patients (De Ligt et al., 2012; Pfundt et al., 2017; Stojanovic et al., 2020). For other disorders such as autism, the diagnostic yield is lower (<15%) but the number of genes has dramatically grown over the last few years (Feliciano et al., 2019; O'Roak et al., 2012; Satterstrom et al., 2020; Stessman et al., 2017) with considerable gene overlap between the two disorders (Coe et al., 2019). For most recently identified genes, only a limited number of patients are known and/or reported, hampering a good assessment of the phenotypic spectrum of such novel disorders. Obtaining this spectrum is not only highly relevant for the patient and his/her family, but also for the professionals dealing with rare genetic disorders—from clinicians and molecular biologists to researchers, as the most up‐to‐date clinical information can assist them in generating accurate diagnosis and prognosis, but also opens new research questions. Moreover, it overcomes reporting bias as professionals are more likely to publish cases with either novel symptoms or novel mutations, which may skew the available information.
Currently, several online resources are available to help assess the clinical problems associated with a specific disorder or a syndrome, including GeneReviews (Adam et al., 2010) and Online Mendelian Inheritance in Man (OMIM; Amberger et al., 2014). However, these are primarily reviews based on published cases. Clinical data of individual, newly identified patients after a first publication on a novel gene are generally not included in these resources, simply because follow‐up papers of single cases are frequently not published. This significantly limits our knowledge on the clinical spectrum of a novel genetic disorder—especially when it is a rare disorder, as large patient series are hard to obtain. To address this issue of publication bias, we introduce the Human Disease Gene website series (HDG website series, https://humandiseasegenes.info/), a noncommercial collective initiative of the Human Genetics Department of the Radboud university medical center (Nijmegen, the Netherlands), the University of Washington (Seattle, USA), and the University of Adelaide (Adelaide, Australia).
The aim of the HDG website series is to have a central online resource in which the most up‐to‐date clinical information of patients with a defect in a specific gene is collected and accessible. The series consists of a specific website for each individual disease‐causing gene, curated by professionals worldwide who are experts on the disease‐gene relation presented. Each website not only provides general clinical and molecular overviews but also allows collection of phenotypic data on novel patients using standardized Human Phenotype Ontology (HPO)‐based terminology (Robinson et al., 2008), accessible through an easy online interface. In this article the structure, development and use of the HDG website series are described.
2. MATERIALS AND METHODS
2.1. Model and confidentiality
Each website in the series is moderated by a dedicated team of professionals, including at least one clinician and/or molecular biologist, hereafter called the moderators. As specialists, these moderators have adopted a gene of interest after which a gene website is created. The moderators are asked to supply content, such as information on the gene and associated syndrome (clinical and molecular characteristics, clinical management, publications and research collaborations). When the content is completed, the website is opened for collection of novel phenotypic data. In this phase, the moderators are asked to curate the website upon submission of novel patients by others.The phenotypic data are collected using an online form, “Upload clinical information”, which can be filled in by a clinician (after obtaining informed consent) within a couple of minutes (for example see: https://www.humandiseasegenes.info/arid1a/professionals/upload‐clinical‐information/). The data are stored using HPO terms in line with current guidelines on FAIR (Findable, Accessible, Intra‐operable, and Reusable) data repositions. In addition, the HDG website series is actively collaborating with GenIDA (https://genida.unistra.fr/), which collects phenotypic data directly from patients/caregivers. Hence, GenIDA (data from patients/caregivers) and the HDG website series (data from clinicians/professionals) work synergistically.
All data that are uploaded to a specific gene website are available to the moderators; the summary and anonymous overview of the clinical data are available to every visitor of the website in the “Graph and Chart” section of a gene website once the uploaded clinical data are reviewed and approved by the moderator to ensure quality of the data (for example see: https://www.humandiseasegenes.info/arid1a/graph-and-chart/).
2.2. Comparison to other databases
There are several databases providing information on the genetic variation of a certain disorder. To compare our efforts to other resources available with comparable purposes, we assessed several online databases/resources for the following characteristics (Table 1):
TABLE 1.
Comparison of several online resources for clinical/phenotypic data for rare genetic syndromes
| Source | Description and aim of database | Focus | Collecting unpublished patient data | Data stored in HPO | Easy display of collected data | Specialized moderators for each gene |
|---|---|---|---|---|---|---|
| LOVD (Fokkema et al., 2011) | Freely available web‐based software for the collection, display, and curation of DNA variants in locus‐specific databases. | Genotype/phenotype | + | ± | − | + |
| HGMD (Stenson et al., 2003) | An attempt to collate all known (published) gene lesions responsible for human inherited disease. | Genotype | − | − | − | − |
| DECIPHER (Firth et al., 2009) | An interactive web‐based database that incorporates a suite of tools designed to aid in the interpretation of genomic variants. | Genotype/phenotype | + | + | + | − |
| OMIM (Amberger et al., 2014) | A continuously updated catalog of human genes and genetic disorders and traits, with particular focus on the relationship between genetic variation and phenotypic expression. | Phenotype | − | ± | − | − |
| GeneReviews (Adam et al., 2010) | An international point‐of‐care resource for clinicians providing clinically relevant and medically actionable information for inherited conditions in a standardized format. | Phenotype | − | − | + | + |
| OrphaNet (Weinreich et al., 2008) | A unique resource, gathering and improving knowledge on rare diseases so as to improve the diagnosis, care and treatment of patients with rare diseases. | Phenotype | − | + | − | − |
| Monarch Initiative (Mungall et al., 2017) | An integrative data and analytic platform connecting phenotypes to genotypes across species, bridging basic and applied research with semantics‐based analysis. | Genotype/phenotype | + | + | − | − |
| ClinGen (Rehm et al., 2015) | A central resource that defines the clinical relevance of genes and variants for use in precision medicine and research. | Genotype | − | − | − | − |
| HDG website series | A platform providing up‐to‐date clinical data for novel rare genetic disorders, using published and unpublished data. | Genotype/phenotype | + | + | + | + |
The main focus (geno‐ or phenotype, or both) and the major reason the resource was established.
Whether the database/online resource is actively collecting data for unpublished patients and, thus, avoiding publication bias.
Whether the data are saved and accessible in HPO terms. This is important as it ensures reusability of data, even between databases.
If an easy display, preferably graphical, of (a summary of) collected data is available.
If there is manual curation of the data and, if so, if this is performed by a specialized team for each gene. This significantly improves the quality of the data.
3. RESULTS
3.1. Website series
The HDG website series launched in 2016. Since then, 424 genes have been adopted, and for 225 (53%), gene websites have already been created (data included till July 2020). Already, for almost half of the constructed genes websites (95/225, 42%) detailed phenotypic information is available on a special “Graph and Charts” page (for example see: https://www.humandiseasegenes.info/arid1a/graph-and-chart/) and the number of gene websites is growing swiftly (Figure 1). To illustrate this, out of the top 10 most‐frequently mutated genes in intellectual disability, discovered in the next‐generation sequencing era (from or after 2010), namely DDX3X, ANKRD11, KMT2A, ARID1B, DYNCH1H1, GRIN2B, PURA, DYRK1A, MED13L and ADNP (adopted from Kaplanis et al., 2019), nine genes (90%) have already been adopted and for six (60%) the website is completed with accurate and detailed phenotypic information of more patients than initially reported in the discovery paper. On average, detailed phenotypic information of 14.4 patients is available for all 95 completed websites. Of note, a full list of constructed gene websites and adopted genes, is provided in Supplemental Table 1 and https://www.humandiseasegenes.info/gene-websites/.
FIGURE 1.
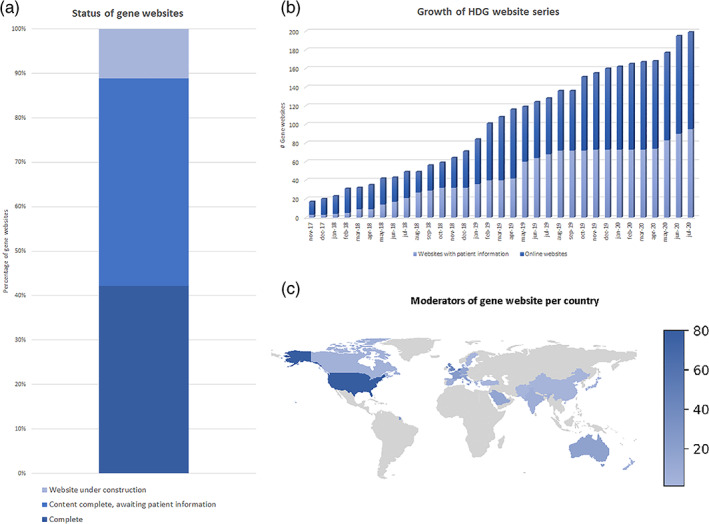
(a) The status of websites of the adopted genes. (b) The growth of the HDG website series in the last 3 years. (c) The countries from which the moderators of the gene website originate [Color figure can be viewed at wileyonlinelibrary.com]
The overall assessment of rare disorders for which HDG sites are available showed that of the 424 adopted genes, 286 (67%) are associated with either autism, mental retardation, intellectual disability or developmental delay. Systematic analyses of these terms in the clinical synopsis of OMIM shows that there are currently 1891 of such syndromes, indicating that 15% of all currently known neurodevelopmental disorders genes have been adopted for a disease‐specific HDG site.
3.2. HDG contents
Each gene website consists of four sections: Home, Information for clinicians, Information for parents, and Graph and Chart.
The homepage serves as a general introduction to the gene website created by the moderators. The information sections for clinicians show a summary of general knowledge of the specific syndrome: the clinical characteristics, molecular characteristics, and any recommendations for management that may apply. An overview of the current research projects and aims are available in this section as well to facilitate collaborations with professionals by creating a gene‐specific scientific community to advance research efforts. To add novel cases, an easy‐to‐use online form is created where clinicians can upload clinical data of an as of yet unpublished patient using HPO terms (Figure 2(a) and https://www.humandiseasegenes.info/hdgw/professionals/upload-clinical-information/). The section Graph and Chart (Figure 2(b),(c)) provides an at‐a‐glance overview of the collected phenotypic data so far. Directly following the moderators' curation and approval of submission of new patients, the patient's HPO terms are automatically integrated and added to the Graph and Chart section, thereby always providing the most up‐to‐date information of a specific syndrome (for example for IQSEC2 see: https://www.humandiseasegenes.info/iqsec2/graph-and-chart/).
FIGURE 2.
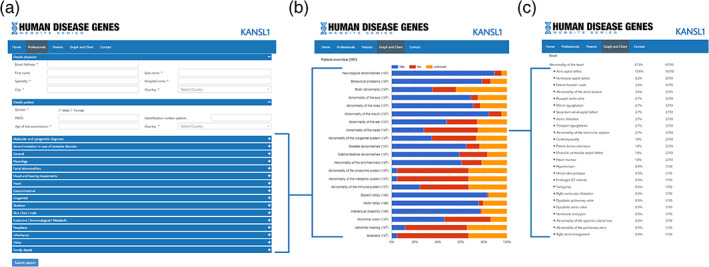
The collection and summary of phenotypic data in the HDG website series, showing in three screenshots. (a) Online form clinicians can use to easily upload new clinical information per patient to a gene website. After curation, this information is then added to the database and a summary can be easily accessed in the phenotypic fingerprint in the graph and chart section (b). Here, all specific symptoms are listed with their absolute and relative count (c). Of note, the example print screens provided show data of Koolen‐de Vries syndrome [Color figure can be viewed at wileyonlinelibrary.com]
The data are organized according to the organ systems—a system based on the hierarchical parent–child structure of the HPO terms. The graphical representation thereof not only makes it easier to obtain the information, but at the same time simplifies explaining the information to patients and caregivers.
Moreover, below this phenotype fingerprint all specific symptoms are displayed one by one—for example “Ventriculomegaly” or “Seizures”—with both the absolute and relative count of this symptom for this specific gene (Figure 2(c)). Finally, a map is shown displaying the origin (by country) of the submitted patients to visualize possible clustering of patients.
Another interesting observation is that 325 different moderators, based at clinical genetic centers and research institutes worldwide, have already adopted a gene or genes of interest (https://www.humandiseasegenes.info/moderators/) suggesting that the HDG initiative is achieving global coverage and concentrating expertise at an international scale (Figure 1(c)).
3.3. Comparison to other databases
Examples of available resources with comparable purposes include the open source Leiden Open Variation Database (LOVD, https://www.lovd.nl; Fokkema et al., 2011) and public and (commercial) professional domains of the Human Gene Mutation Database (HGMD; Stenson et al., 2003). Although the LOVD collects (basic) phenotypic data as well, it seems more focused on genotype and does not have a straightforward and detailed way of presenting clinical information. The well‐known DECIPHER database (Firth et al., 2009) collects phenotypic data in a similar manner and has a similar ability to display a graphical summary. Previously mentioned resources like OMIM (Amberger et al., 2014) and GeneReviews (Adam et al., 2010) have their main focus on phenotype: they aim to summarize the currently available literature of a specific syndrome as well as historical record of how the gene was discovered. The HDG website series focuses on phenotypic data of published and unpublished patients—the latter collected directly from clinicians—and thereby fills the gap between the information available in the scientific literature and the clinical data of unpublished patients (Table 1 displays a complete overview of this comparison between different databases).
4. DISCUSSION
When compared to the other currently available online resources (Table 1), the HDG website series adds value given its combination of several properties. First and foremost, the HDG website series builds a dynamic up‐to‐date resource of phenotypes and genotypes for clinicians, researchers and families. The data in the HDG website series are presented in an easy to understand graphical fashion, resulting in an up‐to‐date, at‐a‐glance, overview. Another strong suit is that the quality of data is ensured by the curation of a gene website by a team of experts on that specific gene leading to better and more accurate data. For instance, underreporting of symptoms in other databases, such as DECIPHER per patient, has been previously reported (van der Sluijs et al., 2018), possibly due to the method of data collection. This significantly hampers acquiring a clear and up‐to‐date overview of the whole phenotypic spectrum of novel gene defects, information that is not only crucial for counseling patients and their caregivers, but also for researchers looking for the breadth and depth of the clinical information.
As we have shown, the HDG website series is rapidly expanding and we hope to keep growing it until there is a specific gene website for each known, rare, disease‐causing gene. Although there has been tremendous discovery over the last 10 years, for some disorders such as autism spectrum disorder, a large number of new genes await discovery and much larger numbers of patients will need to be surveyed to prove their disease association (Feliciano et al., 2019). For these rarer disorders, phenotypic details will be even more critical. Going forward, we plan to make the genotype and phenotype of a specific patient viewable on the website—which for instance DECIPHER already has implemented.
Some genes can be related to different clinical syndromes or different forms of inheritance. It is up to the moderator team to decide whether there is such a clear geno‐ or phenotypic distinction to be made and, subsequently, a second different gene website for such a gene should be created. Two examples are SETBP1 and DEAF1, where for each gene two gene websites are available. For SETBP1 the gain‐of‐function mutations leading to the Schinzel Giedion syndrome (OMIM #269150) and the loss‐of‐function mutations leading to just a developmental delay (OMIM #616078) (see https://www.humandiseasegenes.info/setbp1geneschinzelgiedionsyndrome/ https://www.humandiseasegenes.info/setpb1geneautosomaldominantmentalretardation29/). For DEAF1 the HDG website series has distinct websites for the autosomal dominant form and the autosomal recessive form (OMIM #615828 resp # 617171) (see https://www.humandiseasegenes.info/deaf1autosomaldominant/ resp https://www.humandiseasegenes.info/deaf1autosomalrecessive/).
The difficulty that arises for recently discovered novel and rare genes (on which HDG is particularly focusing) is that the spectrum and possible multiple distinct syndromes are not known (yet). Therefore, we have made the moderator team responsible for deciding whether multiple gene websites should be created for a specific gene.
Next, we showcase three examples of gene websites and their added value to demonstrate the importance of the HDG website series. First, the ARID1B gene website (https://www.humandiseasegenes.info/ARID1B), which was created in 2018 and used to gather phenotypic data for a study. Mutations in ARID1B can lead to a scope of phenotypes ranging from the recognizable Coffin‐Siris syndrome to less specific intellectual disability (Hoyer et al., 2012; Santen et al., 2012). Because of this broad spectrum and the possibility of relatively mild symptoms in patients with mutations in ARID1B, one could argue that collecting high‐quality phenotypic data is even more important for this gene. Without knowledge of the whole spectrum of disease (which may not be captured in the published work), one could, erroneously, discard a possible pathogenic variant in ARID1B if the patient does not fit the Coffin‐Siris syndrome phenotype.
The benefit of the HDG website series for Coffin‐Siris syndrome has already been demonstrated by providing the full clinical spectrum by Van der Sluijs et al. (van der Sluijs et al., 2018). As moderators of this gene website, they utilized the ARID1B website to collect novel phenotypic data. With this strategy, information of 81 new, unpublished patients was collected, which made an extensive geno‐ and phenotype study possible. Presently, the phenotypic information of 170 patients with mutations in ARID1B is freely available on the ARID1B website. This example shows added value for not only clinicians, but also for clinicians/researchers.
Another two examples are the Koolen‐de Vries syndrome gene website (https://www.humandiseasegenes.info/kansl1/) and the gene website for patients with mutations in YY1 (https://www.humandiseasegenes.info/yy1/). Koolen‐de Vries syndrome is a multisystem disorder characterized by developmental delay, intellectual disability, distinctive facial features, hypotonia, epilepsy, amiable behavior, and congenital malformations in multiple other organ systems. It is caused by either a truncating mutation in KANSL1 or a by a 17q21.31 microdeletion (Koolen et al., 2006; Koolen & Vries, 2013; Sharp et al., 2006; Shaw‐Smith et al., 2006). In the current literature, 128 unique patients have been published for whom the phenotype has been clearly described (Barone et al., 2015; Ciaccio et al., 2016; Dornelles‐Wawruk et al., 2013; Dubourg et al., 2011; El Chehadeh‐Djebbar et al., 2011; Kavakli, 2018; Koolen et al., 2006, 2012, 2016; Koolen et al., 2008; Morgan et al., 2018; Nascimento et al., 2017; Sharkey et al., 2009; Terrone et al., 2012; Zollino et al., 2012).
The Koolen‐de Vries syndrome gene website currently shows the phenotypic data of 147 patients in a clear, graphical overview. Not only is there more data available than in the literature, all information is available in one place, which saves valuable clinician time looking to inform her/his patient. Clinical research is significantly easier in like manner with all the phenotypic data stored in HPO terms in one place.
The same goes for the final example, YY1 (named Gabriele‐de Vries syndrome). Mutations in this gene were associated with a syndromic form of intellectual disability in 2017 and, in total, 11 patients with a pathogenic variant in YY1 have been described in the literature (Gabriele et al., 2017; Morales‐Rosado & Kaiwar, 2018). The YY1 gene website, however, has currently collected data from 14 patients. Although three additional patients might not seem like much, symptoms such as a ventricular septal defect or hepatic fibrosis were described in these additional patients, which had not been reported before.
5. CONCLUSION
The Human Disease Gene (HDG) website series is a noncommercial initiative providing up‐to‐date clinical data for rare genetic syndromes, thus not only assisting clinicians but also other professionals in acquiring this essential information. Since these data also include the phenotype of unpublished patients, publication bias is avoided and considerably more information is available. Moreover, it covers data coming from clinical genetic centers and research groups worldwide, underscoring that the HDG initiative is a global grassroot initiative, leveraging expertise at an international scale. Those interested to adopt a gene as part of the HDG website series, please contact us at info@humandiseasegenes.com.
AUTHOR CONTRIBUTIONS
A.v.R., M.V., D.A.K., H.B., E.E., J.G., and B.B.A.d.V. initiated the design and development of the HDG website series. A.J.M.D., D.E.S., S.J., M.J.N.S., A.v.R., N.J., K.T., S.v.d.V., J.E., M.V., and B.B.A.d.V. maintained the website on a daily basis. A.J.M.D., L.E.L.M.V., and B.B.A.d.V. designed and discussed the experiments and the contents of the manuscript. All authors discussed the results and commented on the manuscript.
Supporting information
Appendix S1: Excel file 1, List of genes with a website and adopted genes.
ACKNOWLEDGMENTS
We are grateful to the moderators for their participation in curating the gene websites, as well as for the financial support of the Dutch Organization for Health Research and Development: ZON‐MW grants 912‐12‐109 (B.B.A.d.V. and L.E.L.M.V.), Donders Junior researcher grant 2019 (B.B.A.d.V. and L.E.L.M.V.) and the Dutch Research Council Aspasia grant 015.014.066 (L.E.L.M.V.). This work was supported, in part, by a grant from the US National Institutes of Health (NIH R01 MH101221 to E.E.E). The aims of this study contribute to the Solve‐RD project (C.G. and L.E.L.M.V.), which has received funding from the European Union's Horizon 2020 research and innovation program under grant agreement No. 779257. E.E.E. is an investigator of the Howard Hughes Medical Institute. Finally, we declare there is no conflict of interest.
Dingemans AJM, Stremmelaar DE, Vissers LELM, et al. Human disease genes website series: An international, open and dynamic library for up‐to‐date clinical information. Am J Med Genet Part A. 2021;185A:1039–1046. 10.1002/ajmg.a.62057
Funding information Donders Junior researcher grant 2019; Dutch Research Council Aspasia grant, Grant/Award Number: 015.014.066; European Union's Horizon 2020; US National Institutes of Health, Grant/Award Number: NIH R01 MH101221; ZON‐MW, Grant/Award Number: 912‐12‐109
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are openly available at the corresponding gene websites, www.humandiseasegenes.info.
REFERENCES
- Adam, M. P. , Ardinger, H. H. , Pagon, R. A. , Wallace, S. E. , Bean, L. J. H. , Stephens, K. , & Amemiya, A. (Eds.). (2010). GeneReviews. Seattle: University of Washington. [Google Scholar]
- Amberger, J. S. , Bocchini, C. A. , Schiettecatte, F. , Scott, A. F. , & Hamosh, A. (2014). OMIM.Org: Online Mendelian inheritance in man (OMIM®), an online catalog of human genes and genetic disorders. Nucleic Acids Research, 43(D1), D789–D798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barone, C. , Novelli, A. , Capalbo, A. , Del Grano, A. C. , Giuffrida, M. G. , Indaco, L. , & Bianca, S. (2015). An additional clinical sign of 17q21.31 microdeletion syndrome: Preaxial polydactyly of hands with broad thumbs. American Journal of Medical Genetics. Part A, 167(7), 1671–1673. [DOI] [PubMed] [Google Scholar]
- Ciaccio, C. , Dordoni, C. , Ritelli, M. , & Colombi, M. (2016). Koolen‐de Vries syndrome: Clinical report of an adult and literature review. Cytogenetic and Genome Research, 150(1), 40–45. [DOI] [PubMed] [Google Scholar]
- Coe, B. P. , Stessman, H. A. F. , Sulovari, A. , Geisheker, M. R. , Bakken, T. E. , Lake, A. M. ,. .. Eichler, E. E. (2019). Neurodevelopmental disease genes implicated by de novo mutation and copy number variation morbidity. Nature Genetics, 51(1), 106–116.doi: 10.1038/s41588-018-0288-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Ligt, J. , Willemsen, M. H. , Van Bon, B. W. M. , Kleefstra, T. , Yntema, H. G. , Kroes, T. , Vulto‐van Silfhout, A. T. , Koolen, D. A. , De Vries, P. , Gilissen, C. , & Others. (2012). Diagnostic exome sequencing in persons with severe intellectual disability. The New England Journal of Medicine, 367(20), 1921–1929. [DOI] [PubMed] [Google Scholar]
- Dornelles‐Wawruk, H. , Pic‐Taylor, A. , Rosenberg, C. , Krepischi, A. C. V. , Safatle, H. P. N. , Ferrari, I. , & Mazzeu, J. F. (2013). Complex phenotype associated with 17q21.31 microdeletion. Molecular Syndromology, 4(6), 297–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubourg, C. , Sanlaville, D. , Doco‐Fenzy, M. , Le Caignec, C. , Missirian, C. , Jaillard, S. , … Andrieux, J. (2011). Clinical and molecular characterization of 17q21.31 microdeletion syndrome in 14 French patients with mental retardation. European Journal of Medical Genetics, 54(2), 144–151. [DOI] [PubMed] [Google Scholar]
- El Chehadeh‐Djebbar, S. , Callier, P. , Masurel‐Paulet, A. , Bensignor, C. , Méjean, N. , Payet, M. , … Thauvin‐Robinet, C. (2011). 17q21.31 microdeletion in a patient with pituitary stalk interruption syndrome. European Journal of Medical Genetics, 54(3), 369–373. [DOI] [PubMed] [Google Scholar]
- Feliciano, P. , Zhou, X. , Astrovskaya, I. , Turner, T. N. , Wang, T. , Brueggeman, L. ,. .. Chung, W. K. (2019). Exome sequencing of 457 autism families recruited online provides evidence for autism risk genes. NPJ Genomic Medicine, 4, 19. doi: 10.1038/s41525-019-0093-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firth, H. V. , Richards, S. M. , Bevan, A. P. , Clayton, S. , Corpas, M. , Rajan, D. , … Carter, N. P. (2009). DECIPHER: Database of chromosomal imbalance and phenotype in humans using Ensembl resources. American Journal of Human Genetics, 84(4), 524–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fokkema, I. F. A. C. , Taschner, P. E. M. , Schaafsma, G. C. P. , Celli, J. , Laros, J. F. J. , & den Dunnen, J. T. (2011). LOVD v.2.0: The next generation in gene variant databases. Human Mutation, 32(5), 557–563. [DOI] [PubMed] [Google Scholar]
- Gabriele, M. , Vulto‐van Silfhout, A. T. , Germain, P.‐L. , Vitriolo, A. , Kumar, R. , Douglas, E. , … de Vries, B. B. A. (2017). YY1 Haploinsufficiency causes an intellectual disability syndrome featuring transcriptional and chromatin dysfunction. American Journal of Human Genetics, 100(6), 907–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyer, J. , Ekici, A. B. , Endele, S. , Popp, B. , Zweier, C. , Wiesener, A. , … Reis, A. (2012). Haploinsufficiency of ARID1B, a member of the SWI/SNF‐a chromatin‐remodeling complex, is a frequent cause of intellectual disability. American Journal of Human Genetics, 90(3), 565–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplanis, J. , Samocha, K. E. , Wiel, L. , Zhang, Z. , Arvai, K. J. , Eberhardt, R. Y. , Gallone, G. , Lelieveld, S. H. , Martin, H. C. , McRae, J. F. , Short, P. J. , Torene, R. I. , de Boer, E. , Danecek, P. , Gardner, E. J. , Huang, N. , Lord, J. , Martincorena, I. , Pfundt, R. , … Retterer, K. (2019). Integrating healthcare and research genetic data empowers the discovery of 28 novel developmental disorders. 10.1101/797787 [DOI]
- Kavakli, A. S. (2018). Anaesthesia and orphan disease: A child with Koolen‐de Vries syndrome. European Journal of Anaesthesiology, 35(12), 980–981. [DOI] [PubMed] [Google Scholar]
- Koolen, D. A. , DDD Study , Pfundt, R. , Linda, K. , Beunders, G. , Veenstra‐Knol, H. E. , … de Vries, B. B. A. (2016). The Koolen‐de Vries syndrome: A phenotypic comparison of patients with a 17q21.31 microdeletion versus a KANSL1 sequence variant. European Journal of Human Genetics, 24(5), 652–659. 10.1038/ejhg.2015.178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koolen, D. A. , & de Vries, B. B. A. (2013). KANSL1‐related intellectual disability syndrome. GeneReviews. University of Washington, Seattle. [Google Scholar]
- Koolen, D. A. , Kramer, J. M. , Neveling, K. , Nillesen, W. M. , Moore‐Barton, H. L. , Elmslie, F. V. , Toutain, A. , Amiel, J. , Malan, V. , Tsai, A. C.‐H. , Cheung, S. W. , Gilissen, C. , Verwiel, E. T. P. , Martens, S. , Feuth, T. , Bongers, E. M. H. F. , de Vries, P. , Scheffer, H. , Vissers, L. E. L. M. , … de Vries, B. B. A. (2012). Mutations in the chromatin modifier gene KANSL1 cause the 17q21.31 microdeletion syndrome. Nature Genetics, 44(6), 639–641. [DOI] [PubMed] [Google Scholar]
- Koolen, D. A. , Lisenka, E. L. , Pfundt, R. , de Leeuw, N. , Knight, S. J. L. , Regan, R. , … de Vries, B. B. A. (2006). A new chromosome 17q21.31 microdeletion syndrome associated with a common inversion polymorphism. Nature Genetics, 38(9), 999–1001. 10.1038/ng1853 [DOI] [PubMed] [Google Scholar]
- Koolen, D. A. , Sharp, A. J. , Hurst, J. A. , Firth, H. V. , Knight, S. J. L. , Goldenberg, A. , Saugier‐Veber, P. , Pfundt, R. , Vissers, L. E. L. M. , Destrée, A. , Grisart, B. , Rooms, L. , Van der Aa, N. , Field, M. , Hackett, A. , Bell, K. , Nowaczyk, M. J. M. , Mancini, G. M. S. , Poddighe, P. J. , … de Vries, B. B. A. (2008). Clinical and molecular delineation of the 17q21.31 microdeletion syndrome. Journal of Medical Genetics, 45(11), 710–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales‐Rosado, J. A. , & Kaiwar, C. (2018). A case of YY1‐associated syndromic learning disability or Gabriele‐de Vries syndrome with myasthenia gravis. American Journal of Medical Genetics Part A, 176, 2846–2849. [DOI] [PubMed] [Google Scholar]
- Morgan, A. T. , van Haaften, L. , van Hulst, K. , Edley, C. , Mei, C. , Tan, T. Y. , Amor, D. , Fisher, S. E. , & Koolen, D. A. (2018). Early speech development in Koolen de Vries syndrome limited by oral praxis and hypotonia. European Journal of Human Genetics: EJHG, 26(1), 75–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mungall, C. J. , McMurry, J. A. , Köhler, S. , Balhoff, J. P. , Borromeo, C. , Brush, M. , … Haendel, M. A. (2017). The monarch initiative: An integrative data and analytic platform connecting phenotypes to genotypes across species. Nucleic Acids Research, 45(D1), D712–D722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nascimento, G. R. , Pinto, I. P. , de Melo, A. V. , da Cruz, D. M. , Ribeiro, C. L. , da Silva, C. C. , … Minasi, L. B. (2017). Molecular characterization of Koolen De Vries syndrome in two girls with idiopathic intellectual disability from Central Brazil. Molecular Syndromology, 8(3), 155–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Roak, B. J. , Vives, L. , Girirajan, S. , Karakoc, E. , Krumm, N. , Coe, B. P. ,. .. Eichler, E. E. (2012). Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature, 485(7397), 246–250.doi: 10.1038/nature10989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfundt, R. , Del Rosario, M. , Vissers, L. E. L. M. , Kwint, M. P. , Janssen, I. M. , de Leeuw, N. , … Hehir‐Kwa, J. Y. (2017). Detection of clinically relevant copy‐number variants by exome sequencing in a large cohort of genetic disorders. Genetics in Medicine: Official Journal of the American College of Medical Genetics, 19(6), 667–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehm, H. L. , Berg, J. S. , Brooks, L. D. , Bustamante, C. D. , Evans, J. P. , Landrum, M. J. , … ClinGen . (2015). ClinGen–the clinical genome resource. The New England Journal of Medicine, 372, 2235–2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson, P. N. , Köhler, S. , Bauer, S. , Seelow, D. , Horn, D. , & Mundlos, S. (2008). The human phenotype ontology: A tool for annotating and analyzing human hereditary disease. American Journal of Human Genetics, 83(5), 610–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santen, G. W. E. , Aten, E. , Sun, Y. , Almomani, R. , Gilissen, C. , Nielsen, M. , … Kriek, M. (2012). Mutations in SWI/SNF chromatin remodeling complex gene ARID1B cause coffin‐Siris syndrome. Nature Genetics, 44(4), 379–380. [DOI] [PubMed] [Google Scholar]
- Satterstrom, F. K. , Kosmicki, J. A. , Wang, J. , Breen, M. S. , De Rubeis, S. , An, J. Y. ,. .. Buxbaum, J. D. (2020). Large‐scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell, 180(3), 568–584 e523. doi: 10.1016/j.cell.2019.12.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharkey, F. H. , Morrison, N. , Murray, R. , Iremonger, J. , Stephen, J. , Maher, E. , … Jackson, A. P. (2009). 17q21.31 microdeletion syndrome: Further expanding the clinical phenotype. Cytogenetic and Genome Research, 127(1), 61–66. [DOI] [PubMed] [Google Scholar]
- Sharp, A. J. , Hansen, S. , Selzer, R. R. , Cheng, Z. , Regan, R. , Hurst, J. A. ,. .. Eichler, E. E. (2006). Discovery of previously unidentified genomic disorders from the duplication architecture of the human genome. Nature Genetics , 38(9), 1038–1042.doi: 10.1038/ng1862 [DOI] [PubMed] [Google Scholar]
- Shaw‐Smith, C. , Pittman, A. M. , Willatt, L. , Martin, H. , Rickman, L. , Gribble, S. ,. .. Carter, N. P. (2006). Microdeletion encompassing MAPT at chromosome 17q21.3 is associated with developmental delay and learning disability. Nature Genetics , 38(9), 1032–1037.doi: 10.1038/ng1858 [DOI] [PubMed] [Google Scholar]
- Stenson, P. D. , Ball, E. V. , Mort, M. , Phillips, A. D. , Shiel, J. A. , Thomas, N. S. T. , … Cooper, D. N. (2003). Human gene mutation database (HGMD®): 2003 update. Human Mutation, 21(6), 577–581. [DOI] [PubMed] [Google Scholar]
- Stessman, H. A. , Xiong, B. , Coe, B. P. , Wang, T. , Hoekzema, K. , Fenckova, M. ,. .. Eichler, E. E. (2017). Targeted sequencing identifies 91 neurodevelopmental‐disorder risk genes with autism and developmental‐disability biases. Nature Genetics, 49(4), 515–526.doi: 10.1038/ng.3792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stojanovic, J. R. , Miletic, A. , Peterlin, B. , Maver, A. , Mijovic, M. , Borlja, N. , … Cuturilo, G. (2020). Diagnostic and clinical utility of clinical exome sequencing in children with moderate and severe global developmental delay/intellectual disability. Journal of Child Neurology, 35(2), 116–131. [DOI] [PubMed] [Google Scholar]
- Terrone, G. , D'Amico, A. , Imperati, F. , Carella, M. , Palumbo, O. , Gentile, M. , … Del Giudice, E. (2012). A further contribution to the delineation of the 17q21.31 microdeletion syndrome: Central nervous involvement in two Italian patients. European Journal of Medical Genetics, 55(8–9), 466–471. [DOI] [PubMed] [Google Scholar]
- van der Sluijs, P. J. , Jansen, S. , Vergano, S. A. , Adachi‐Fukuda, M. , Alanay, Y. , AlKindy, A. , … Santen, G. W. E. (2018). The ARID1B spectrum in 143 patients: From nonsyndromic intellectual disability to Coffin–Siris syndrome. Genetics in Medicine: Official Journal of the American College of Medical Genetics, 21(6), 1295–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinreich, S. S. , Mangon, R. , Sikkens, J. J. , Teeuw, M. E. , & Cornel, M. C. (2008). Orphanet: A European database for rare diseases. Nederlands Tijdschrift voor Geneeskunde, 152(9), 518–519. [PubMed] [Google Scholar]
- Zollino, M. , Orteschi, D. , Murdolo, M. , Lattante, S. , Battaglia, D. , Stefanini, C. , … Marangi, G. (2012). Mutations in KANSL1 cause the 17q21.31 microdeletion syndrome phenotype. Nature Genetics, 44(6), 636–638. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Excel file 1, List of genes with a website and adopted genes.
Data Availability Statement
The data that support the findings of this study are openly available at the corresponding gene websites, www.humandiseasegenes.info.