

Abstract
A high degree of charge heterogeneity is an unfavorable phenomenon commonly observed for therapeutic monoclonal antibodies (mAbs). Removal of these impurities during manufacturing often comes at the cost of impaired step yields. A wide spectrum of posttranslational and chemical modifications is known to modify mAb charge. However, a deeper understanding of underlying mechanisms triggering charged species would be beneficial for the control of mAb charge variants during bioprocessing. In this study, a comprehensive analytical investigation was carried out to define the root causes and mechanisms inducing acidic variants of an immunoglobulin G1‐derived mAb. Characterization of differently charged species by liquid chromatography–mass spectrometry revealed the reduction of disulfide bonds in acidic variants, which is followed by cysteinylation and glutathionylation of cysteines. Importantly, biophysical stability and integrity of the mAb are not affected. By in vitro incubation of the mAb with the reducing agent cysteine, disulfide bond degradation was directly linked to an increase of numerous acidic species. Modifying the concentrations of cysteine during the fermentation of various mAbs illustrated that redox potential is a critical aspect to consider during bioprocess development with respect to charge variant control.
Keywords: acidic charge variants, bioprocess optimization, cysteine, product quality, redox‐sensitive modifications
1. INTRODUCTION
With more than 300 molecules currently in development for diverse medical applications, monoclonal antibodies (mAbs) have emerged as the leading class of biopharmaceuticals (Shukla et al., 2017). Product heterogeneity of therapeutic mAbs is commonly observed, as they are exposed to numerous factors during bioprocessing, such as media components, fermentation conditions and downstream processing (Gandhi et al., 2012; Hossler et al., 2015; Kishishita et al., 2015; Xie et al., 2016). These conditions result in product‐related impurities, which must be closely controlled and monitored during the manufacturing process, as mandated by regulatory authorities (ICH Q 6 B).
Charge heterogeneity in mAbs is considered a critical quality attribute, as it can alter efficacy and pharmacokinetics (Boswell et al., 2010; Dakshinamurthy et al., 2017). Due to the shifted isoelectric point (pI), mAb variants show differential elution behavior on cation exchange chromatography (CEX), where more negatively charged variants elute first as an acidic peak group (APG), followed by the main peak and the more positively charged basic peak group (BPG). Modifications responsible for charge variations are of chemical and structural nature and have been reviewed extensively (Chung et al., 2018; Du et al., 2012; Liu et al., 2008). The origin of acidic species is generally traced back to chemical modifications such as deamidation (Harris et al., 2001), oxidation of side chains (Yan et al., 2009), cysteinylation (Banks et al., 2008), glycosylations (Yan et al., 2009), glycations (Miller et al., 2011), sialylation (Khawli et al., 2010; Lyubarskaya et al., 2006), as well as fragmentation (Yan et al., 2009). Contributions to basic species include C‐terminal Lys variants (Moorhouse et al., 1997), N‐terminal pyroGlu cyclization (Lyubarskaya et al., 2006; Moorhouse et al., 1997), succinimide formation resulting from Asp isomerization (Harris et al., 2001; Yan et al., 2009), and aggregation (Khawli et al., 2010).
The structure of immunoglobulin G (IgG) consists of two heavy chains (HCs) and two light chains (LCs) stabilized by four interchain disulfide bonds (Milstein, 1966; Pinck & Milstein, 1967). The locations of these interchain disulfide bonds are specific to the IgG subclass. For IgG1, the most frequent therapeutic mAb format, the HCs are connected in their hinge regions by two bonds, and the constant domain of each HC (CH1) is linked to the constant domain of the LC (CL) via one more bond (Figure 1). An additional 12 domain‐specific intrachain disulfide bonds and noncovalent interactions contribute to the correct folding of the IgG (Edelman et al., 1969; Frangione et al., 1968).
Figure 1.
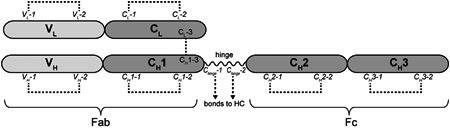
Schematic representation of an immunoglobulin G1 (IgG1) half molecule. Domains are labeled in bold and cysteines are shown in italic. The antigen‐binding fragment (Fab) consists of the variable (VL) and constant (CL) domains of the light chain, and the variable (VH) and constant (CH1) domains of the heavy chain. The Fab fragment is connected via the hinge region to the crystallizable fragment (Fc), consisting of the constant domains CH2 and CH3 of the heavy chain. A total of 16 disulfide bonds stabilize the native IgG1 structure, as represented by dashed lines connecting the cysteines involved
It is known that changes in the disulfide bond network influence structural integrity of mAbs and can alter their chromatographic behavior (Dillon et al., 2008; Wypych et al., 2008). Modifications which result in acidic elution on CEX include trisulfide bonds, disulfide bond scrambling, and unbound cysteines resulting in free thiol (sulfhydryl) groups (Dillon et al., 2008; Schuurman et al., 2001; Wypych et al., 2008; Yoo et al., 2003). The IgG2 and IgG4 subclasses are most frequently affected by these modifications, but some have also been reported for IgG1 (Bloom et al., 1997; Gevondyan et al., 2006; Gu et al., 2010; Schauenstein et al., 1986; Seibel et al., 2017; Wang et al., 2018). On average, one IgG1 molecule contains 0.1–1.1 free cysteines (Gevondyan et al., 2006). Unpaired cysteines are typically located in the constant domains of IgG1s, but have been reported for variable domains (Chumsae et al., 2009; Xiang et al., 2009). Cysteinylation, the result of oxidation of a free thiol with a free cysteine molecule, can impair the pharmacological properties of IgGs (Banks et al., 2008).
Unpaired cysteines can originate from insufficient formation of disulfide bridges during protein folding and assembly, as well as the degradation of intact bonds. A higher degree of free thiols is reported for cysteines involved in intrachain as compared to interchain disulfide bonds (Chumsae et al., 2009; Xiang et al., 2009). Cysteines involved in intrachain bonds are protected from degradation as they are buried between the β‐sheets of the IgG fold, indicating that these unpaired cysteines must originate from incomplete formation during the production process (Amzel & Poljak, 1979; Kikuchi et al., 1986). In particular, the redox environment during fermentation may be critical for a correct disulfide bond network (Chaderjian et al., 2005; Handlogten et al., 2017; Kshirsagar et al., 2012). This is supported by the observation that the addition of copper sulfate to cell culture facilitates the formation of an intact intrachain disulfide bridge by acting as an oxidative agent (Chaderjian et al., 2005). Accordingly, interchain disulfide bonds are reduced by the thioredoxin and glutathione pathway during fermentation (Handlogten et al., 2017).
The importance of controlling disulfide bridge conversions and resulting charge variants during bioprocessing is highlighted by reports where a decrease in potency has been linked to free thiols, in particular in the variable domains (Harris, 2005; Ouellette et al., 2010). Destabilizing effects of incomplete disulfides have been observed for some cases, but they do not introduce substantial changes to the IgG structure (Kikuchi et al., 1986; McAuley et al., 2008). Although there is general consensus on chemical and structural modifications contributing to charge variants, there is a lack of understanding of the underlying mechanisms responsible for acidic variants (Chung et al., 2018). Previously published reports comparing CEX fractions by bioanalytical techniques have shown that the origin of mAb charge variants is heterogeneous and that combinations of protein modifications can complicate their characterization (Griaud et al., 2017; Miao et al., 2017; Ponniah et al., 2015). This study aimed to fully characterize the root cause of charged species of an IgG1 mAb. By characterizing individual charged mAb species, the generation of acidic mAb1 species was associated to ruptured and modified disulfide bonds. These modifications are induced by cysteine, as demonstrated by in vitro experiments. In cultivation experiments, it was shown that the media component cysteine triggers changes in mAb charge profile and alters the media redox potential. The cysteine concentration in cell culture media may thus be crucial for the control of mAb charge during bioprocessing.
2. MATERIALS AND METHODS
2.1. Materials
All mAbs investigated in this study are IgG1 with pIs of 8.5–8.9. mAb1, mAb2, and mAb3 are IgG1 and mAb4 is an IgG1‐derived antibody, all produced in Chinese hamster ovary (CHO) cells. All chemicals and reagents used in this study were high‐performance liquid chromatography (HPLC) grade purchased from Sigma‐Aldrich Chemie GmbH.
2.2. mAb1 purification by protein A affinity chromatography
mAb1 was purified from cell culture free fluid using an in‐house‐packed MabSelect SuRe column, operated on an ÄKTA avant 25 using UNICORN 7.0 (GE Healthcare GmbH). The protein was eluted in 50 mM acetate pH 3.5. The eluted product pool was neutralized to pH 5.6 and stored at –70 °C until further use.
2.3. Treatment of mAb1 samples with cysteine, S‐sulfocysteine, cystine, and glutathione
Intact and fractionated mAb1 were incubated with cysteine (n = 4), S‐sulfocysteine (n = 3), and cystine (n = 3) at a protein concentration of 2 g/L in 10 mM acetate at pH 5.6. Glutathione incubations of intact mAb1 with 2.5 mM GSH and 2.5 mM GSSG (n = 2) were carried out at a protein concentration of 2 g/L in 50 mM Tris at pH 8.2. Cysteine, S‐sulfocysteine, GSH, and GSSG stock solutions were prepared in Milli‐Q® (Merck‐Millipore) at 100X and added to the mAb to reach the respective final concentrations. Before further analysis, the samples were rebuffered into 10 mM sodium phosphate pH 6.8 using 7 kDa molecular weight cut‐off ZebaTM Spin Desalting Columns (Thermo Fisher Scientific Inc.) to remove the reagents.
2.4. Preparation of mAb1 charge variants
Preparative separation of the mAb charge variants was carried out on a 9 × 250 mm ProPac™ WCX‐10 (Thermo Fisher Scientific Inc.) using an ÄKTA avant 25 and UNICORN 7.0 (GE Healthcare GmbH). mAb1 fractions were eluted using a linear NaCl gradient and pooled to obtain the charge variants as shown in Figure S1. As a control, all fractions of one run were pooled to obtain a sample of all charge variants exposed to the same conditions as the fractions. The charge variants and control sample were concentrated to around 2–5 g/L and rebuffered to 10 mM sodium phosphate pH 6.8 using 10 kDa molecular weight cut‐off Amicon® Ultra‐15 centrifugal filter units (Sigma‐Aldrich Chemie GmbH). In total, the fractionation experiment was carried out twice, and all resulting samples were checked by weak cation exchange chromatography (WCX) and capillary electrophoresis (CE). Biophysical properties (circular dichroism [CD], nano differential scanning fluorimetry [nano‐DSF], size exclusion chromatography [SEC]) were determined from one run in duplicates.
2.5. Capillary electrophoresis (CE)
Protein size variants were quantified via microfluidic CE using the LabChip GXII instrument and LabChip GX 5.3 software (PerkinElmer). Samples were prepared according to the manufacturer's specifications of the Protein Express Assay Reagent Kit, and separated on Protein Express Assay LabChip using the HT Protein Express 200 run (PerkinElmer).
2.6. Analytical separation and quantification of charge variants
Analytical separation and quantification of mAb1, mAb2, and mAb3 charge variants was achieved by analytical CEX, operated on Alliance HPLC systems (Waters). mAb1 and mAb3 were separated on a 4 × 250 mm ProPac WCX‐10 column and mAb2 was separated on a 4 × 250 mm MabPacTM SCX‐10 column (Thermo Fisher Scientific Inc). System control and data analysis was performed using EMPOWER 3 (Waters). mAb4 charge variant analysis was performed by capillary zone electrophoresis using a PA 800 Plus (Beckman Coulter).
2.7. Analytical size‐exclusion chromatography (SEC)
Analytical separation and quantification of mAb1 high molecular weight (HMW), intact (main), and low molecular weight species (LMW) was achieved on a 4.6 × 30 mm TSKgel UP‐SW3000 (Tosoh Bioscience LLC) using an Acquity UPLC system (Waters). System control and data analysis was performed using EMPOWER 3 (Waters).
2.8. Thermal unfolding by nano‐differential scanning fluorimetry (nano‐DSF)
Protein stability was assessed via thermal unfolding detected by nano‐DSF on the Prometheus NT.48 (NanoTemper Technologies GmbH). Protein samples were measured using a temperature ramp from 45°C to 85°C. System control and data analysis were performed using PR.ThermControl (NanoTemper Technologies GmbH).
2.9. Far UV CD spectroscopy
Far UV CD measurements were acquired on a JASCO spectrometer at protein concentrations ranging from 1.5 to 2.0 g/L. Spectra were recorded in millidegrees (m°).
2.10. Combined endopeptidase Lys‐C/tryptic digest with LC‐MS/MS analysis for quantification of free thiols, cysteinylated, and glutathionylated thiols
Samples were subjected to a combined Lys‐C/tryptic digest as described previously (Seibel et al., 2017). For analysis of free thiols, half of the Lys‐C/tryptic digest was reduced by addition of 4.4 µl 0.64 M Tris‐(2‐carboxyethyl)‐phosphin (Roth) and incubation for 30 min at 37°C. Subsequently, 8 µl 20% formic acid to stop the digestion and analyzed by liquid chromatography–mass spectrometry (LC‐MS/MS).
Sample measurement was conducted as described previously (Seibel et al., 2017). For analysis of disulfide connectivity, reduced and nonreduced data sets from the same sample were analyzed using PepFinderTM (Thermo Fisher Scientific Inc.) and manually reviewed (n = 1). Extracted ion chromatograms (EICs) of each dipeptide were generated using the two or three most intense charge states. The total peak area of all EIC peaks containing disulfide linked peptides was set to 100% and the relative amount of each disulfide linked peptide calculated serving for inter‐sample comparison. “Free” thiols were relatively quantified using the peak areas of the EICs of the NEM‐modified (“free thiol”) peptide and the reduced peptide as 100% value. Cysteinylated and glutathionylated peptides were quantified using the nonreduced peptide map data and the EIC peak areas of NEM‐alkylated (“free thiol”), the cysteinylated, the glutathionylated peptide and dipeptide as 100% value. For relative quantification of “free” cysteines, cysteine‐containing tryptic peptides were selected that showed good chromatographic and mass spectrometric properties for both, the alkylated and the nonalkylated cysteine‐containing tryptic peptide.
2.11. LC‐MS analysis of deglycosylated intact, reduced and IdeS digested samples
For determination of the intact mass (n = 1), the antibody was deglycosylated with PNGaseF (R&D Systems) at 37°C over night according to manufacturer's instructions. For measurement of the reduced antibody samples, deglycosylated samples were treated with 8 M guanidine hydrochloride and Tris(2‐carboxyethyl)phosphine hydrochloride for 30 min at 37°C. For cleavage in the hinge region, antibody samples were incubated with IdeS enzyme (Fabricator, Genovis) for 2 h at 37°C according to manufacturer's instructions. Samples were analyzed by LC‐MS using a linear organic solvent gradient on C4 column (Acquity BEH300 C4, 1 mm, 50 mm, 1.7 µm; 150 µl/min, 75°C, 1.6 µg on column) and a Quadrupole time‐of‐flight mass spectrometer (MAXIS; Bruker Daltonics).
2.12. Cell and culture method
Three different CHO‐K1‐GS derived cell lines expressing different mAbs (mAb2, mAb3, mAb4) were thawed with a ThawSTAR automated cell thawing system (MedCision Inc). Precultures were cultivated in shake flasks of adequate size at 36.5°C, 5% CO2, and 120 rpm in Multitron incubators. Cultures were subcultivated every two or three days by diluting an appropriate volume of cell culture liquid with fresh medium in a new shake flask before the inoculation of the ambr15TM bioreactors.
2.13. Bioreactors (ambr15)
Cells were cultivated in the Advanced Microscale Bioreactor System (ambr15; Sartorius AG) in a 14‐day fed‐batch culture. The seed density for all cell lines was 0.7E6 cells/ml. The cell culture conditions were maintained at a temperature of 34.5°C, DO setpoint of 50%, pH of 6.95 ± 0.25 and an agitation speed of 1000 rpm over the complete cultivation time. Total and viable cell numbers were monitored by a Cedex analyzer (Roche Innovatis GmbH), while metabolic parameters such as glucose, lactate, and ammonium were measured by the Konelab. For harvest, the cell culture broth was centrifuged at 7100g and 4°C for 15 min in a KR4i centrifuge (Thermo Fisher Scientific Inc). The supernatant was filtered through a syringe (BD Plastipak 20 ml; BD) using 0.7 µm prefilters (Ministart NML GF hydrophilic; Satorius Stedim Biotech GmbH) and 0.22 µm main filters (Ministart Ophthalsart; Satorius Stedim Biotech GmbH) and stored at −70°C until further processing. The redox potential was measured after harvest and filtration by using the EasyFerm Plus ORP Arc 120 (Hamilton) probe according to manufacturer's description.
Cultivation experiments testing 0 g/L and 3 g/L cysteine in the process media were fermented in duplicates. The duplicates of mAb4 were pooled prior charge variant analysis. The effect of cysteine concentrations in the process medium and boli was conducted using an I‐optimal DoE design. All models were checked to contain no aliasing for discussed effects, for consistency and statistical power.
3. RESULTS
3.1. Heterogeneous charge does not impact biophysical mAb1 structure and stability
The main focus of this study was to identify the root cause of acidic charge variants of an IgG1 mAb (mAb1). When separated by analytical WCX, 25% of acidic variants were observed for mAb1. The APG consisted of three overlapping peaks (Figure 2a). To understand the root cause of the acidic variant formation, mAb1 was fractionated to yield eight APG fractions (A–H), one main peak fraction (I) and two BPG (BPG; J–K) fractions (Figure S1). These sequential small volume fractions were collected to stepwise characterize the full APG spectrum. A control sample was prepared by pooling all fractions of an individual preparative run. The purity of each fraction was confirmed by reinjecting the fractions onto the WCX column, immediately after buffer exchange (Figure 2a), and after 1 week of storage at 4°C (Figure S2). The charge variants were characterized for their biophysical and structural properties, as well as stability. Analytical SEC demonstrated a rather uniform distribution of LMW species (Figure 2b; Table S1). LMWs observed by analytical SEC originate from backbone fragmentation. Merely the most acidic fraction A contains slightly elevated levels of LMWs, which is expected from the literature (Du et al., 2012). HMW species, on the other hand, were more dominant in the most basic species K. The structural integrity of the charge variants was analyzed by far UV CD spectroscopy (Figure 2c). The minima at 219–220 nm and maxima at 202 nm observed for all samples were indicative for dominant β‐sheet structural elements, and local minima at 209–210 nm implied α‐helical contributions, as would be expected for a mAb (Buijs et al., 1996). The distinct overlap between the spectra suggested similar secondary structural elements and thus highly comparable structures in the individual charge fractions. The stability of the charge variants was assessed by nano‐DSF, which revealed changes in intrinsic Trp fluorescence upon thermal unfolding (Figure 2d; Table S1). Two melting temperatures (T m) were detected, corresponding to unfolding events of individual mAb domains, as well as the onset temperature for scattering, indicating aggregation. The CH2 and Fab domains unfolded at 69.4–70.0°C and 74.9–75.1°C, respectively. Overall, no differences in thermal unfolding or aggregation onset were found between the fractions, implying that charge heterogeneity does not affect the stability of mAb1.
Figure 2.
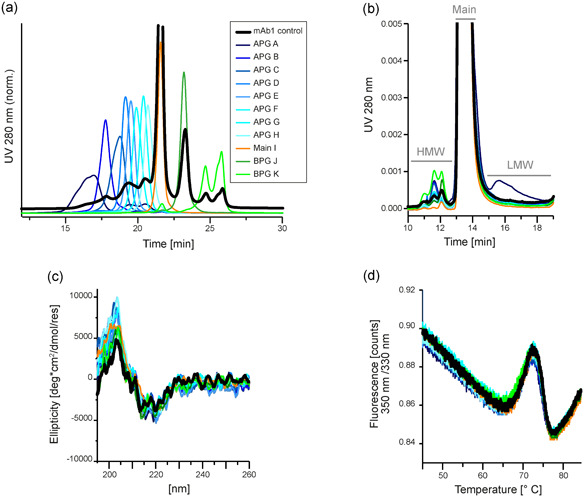
Characterization of weak cation exchange chromatography (WCX)‐fractionated monoclonal antibody 1 (mAb1) indicates biophysical stability of mAb1 charge variants. mAb1 was separated by cation exchange chromatography and individual charge fractions were collected and characterized. The fractions include species from the acidic peak group A–H, the main species I, and species from the basic peak group J–K. As a control, fractions were pooled again to result in a sample including all charge variants, which was subjected to the same conditions as the fractions. (a) Purity of the collected fractions was verified by reinjection of the samples onto the WCX column. The fractions were thus suitable for further characterization. (b) Separation of the fractions by analytical size‐exclusion chromatography (SEC) revealed the size distribution within the charge variants. All charged species contain monomeric mAb1. The level of high molecular weight (HMW) species were enhanced in more basic fractions, and low molecular weight (LMW) species are found only in acidic fractions. Full data of the SEC measurements is shown in Table S1. (c) Circular dichroism spectroscopy showed no differences between the collected fractions, and thus indicated structural integrity of all mAb1 charge variants. (d) Nano differential scanning fluorimetry (nano‐DSF) measurements of the collected fractions resulted in highly comparable profiles and melting temperatures, verifying that protein stability is sustained in mAb1 charge variants. Full data of the nano‐DSF measurements is shown in Table S1 [Color figure can be viewed at wileyonlinelibrary.com]
3.2. Modified disulfide bonds contribute to acidic antibody species
To investigate the nature of the acidic variants, the individual fractions were analyzed by nonreducing CE, which allows a characterization of antibody integrity. Noncomplete mAb species originating from broken interchain disulfide bonds migrate as separate peaks on the CE under nonreducing (native) conditions. These species include the free LC, free HC, half‐molecules (HL), two interconnected HCs lacking LCs (HH), and mAbs missing one LC (2H1L). In contrast to analytical SEC where these species would elute with the main peak, nonreducing CE is a valuable technique to evaluate the degradation of interchain disulfide bonds. Nonreducing CE demonstrated an increase of noncomplete mAbs in the more acidic fractions (Figure 3a; Table S1). In particular, the LC and 2H1L species were accumulated in early eluting acidic fractions (up to 8.2% of LC), showing increased interchain disulfide bond rupture in acidic fractions.
Figure 3.
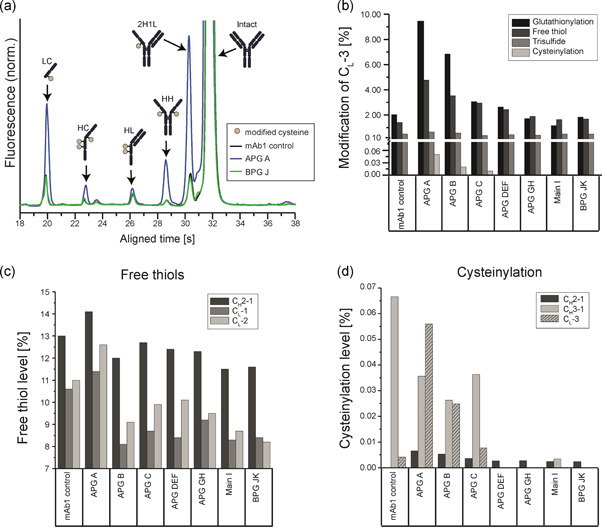
Analysis of monoclonal antibody 1 (mAb1) cysteines revealed that rupture of intra‐ and intermolecular mAb1 disulfide bonds contribute to acidic charge variants. (a) The mAb1 charge variants were analyzed by nonreducing capillary electrophoresis (CE) to identify mAb1 size variants resulting from interchain disulfide bond degradation (n = 2). Full CE data for all size variants is shown in Table S1. (b–d) The mAb1 charge variants were further analyzed by a liquid chromatography–mass spectrometry (LC‐MS) peptide mapping approach to quantify modifications of different mAb1 cysteine residues (n = 1). (b) LC‐MS measurement of mAb1 charge variants was performed to quantify the relative levels of glutathionylations, free thiols, trisulfides, and cysteinylations for the CL‐3 residue, which is the cysteine residue in the light chain typically forming an interchain disulfide bond to the heavy chain. (c,d) Using LC‐MS peptide mapping, mAb1 cysteines typically involved in intrachain disulfide bonds in native mAb1 were analyzed. The levels of free thiol content (c) and cysteinylation (d) were quantified to investigate rupture and modification on intramolecular disulfide bonds. The cysteinylation content in CH3‐1 in the mAb1 control is presumably an outlier. In addition, the levels of cysteinylation are shown for CL‐3 (d) [Color figure can be viewed at wileyonlinelibrary.com]
Peptide mapping LC‐MS measurements revealed that modifications of the C‐terminal cysteine (CL‐3) of the LC are responsible for the abundance of free LC found in acidic fractions (Figure 3b). In the most acidic fraction APG A, relative levels of glutathionlyations (9.3%), free thiols (4.8%), trisulfide bonds (0.67%), and also cysteinylations (0.05%), although to a lower extent, were found. In contrast to glutathionylations, free thiols and cysteinylations, which accumulate in acidic fractions, trisulfide bonds showed a constant distribution in all fractions, and therefore do not seem to contribute to acidic elution.
Next, the status of additional cysteines forming intramolecular bridges, which cannot be observed by CE, was investigated in the different mAb1 charge variants. In the peptide‐map LC‐MS approach, it was possible to analyze further mAb1 cysteines, which showed sufficient chromatographic separation and mass spectrometric properties. Regarding free thiols, the cysteines CH2‐1, CL‐1, and CL‐2 (see Figure 3 for reference) could be analyzed and the experiment revealed, that the number of unpaired cysteines was increased in APG fractions with respect to the main peak and fractions from the BPG (Figure 3c). Intriguingly, the most acidic eluting APG A fraction contained the highest number of free thiols. With respect to cysteinylation, further three cysteines showed a comparable trend, namely cysteinylation in the APG, alhough at low absolute levels (Figure 3d). These investigated cysteines included two residues typically forming intrachain disulfide bonds in the HC (CH2‐1 and CH3‐1). Additionally, the cysteine involved in the interchain disulfide bond between the LC and 2H1L (CL‐3, also shown in Figure 3b) is shown for comparison.
In‐line with CL‐3 (Figure 3b), the presented data demonstrated that other cysteines are affected by modifications, although at lower levels. Taken together, these data strongly support that disulfide bond rupture and possible subsequent modification contribute to acidic variants. Yet, disulfide scrambling, another typical disulfide bond modification which can contribute to acidic variant formation, was not detectable by LC‐MS (Table S2). In addition to the redox‐sensitive modifications well known posttranslational modifications were found including N‐acetylneuraminic acid, high mannose, lysine glycation, and N‐terminal pyroglutamate formation (Figure S3). These modifications were distributed throughout the charge variants as expected from the literature (Chung et al., 2018; Du et al., 2012; Liu et al., 2008).
The peptide‐mapping results indicate that redox‐sensitive modifications are distributed throughout the mAb1 structure, however, only individual cysteines had been analyzed and most cysteines were not visible in this approach. To further locate the modification‐sites, IdeS digestion of the charge variants was carried out, and the resulting F(ab')2 and Fc fragments were subjected to intact LC‐MS measurements. Heterogeneous peaks and degradation were observed for the F(ab')2 fragment (Figure S4b), while the Fc fragment appeared as a homogeneous peak (Figure S4a), pinpointing the majority of cysteinylations and glutathionylations, as well as glycations, to the F(ab')2 part of mAb1. It should be noted that this experiment can provide only a qualitative trend and no quantitative information for all cysteines.
3.3. In vitro cysteine treatment induces acidic variants by breaking disulfide bonds and introducing cysteinylations
To prove that disulfide bond breakage contributes to acidic mAb1 species, in vitro experiments were conducted by adding cysteine to mAb1 and analyzing the resulting charge species (Figure 4). Cysteine treatment did promote increased levels of acidic peaks in mAb1, in a concentration‐dependent manner (Figure 4a). Incubation with cysteine induced a uniform increase in APG levels, rather than enhancing particular peaks. BPG levels did slightly decrease, as well as the Main species, indicating that both were converted into more acidic species. In control experiments, intact mAb1 and the Main I species were incubated with S‐sulfocysteine and cystine, nonreducing derivatives of cysteine (Figure S5). In contrast to cysteine, the control substances did not trigger changes in charge distribution, as shown by the WCX profiles.
Figure 4.
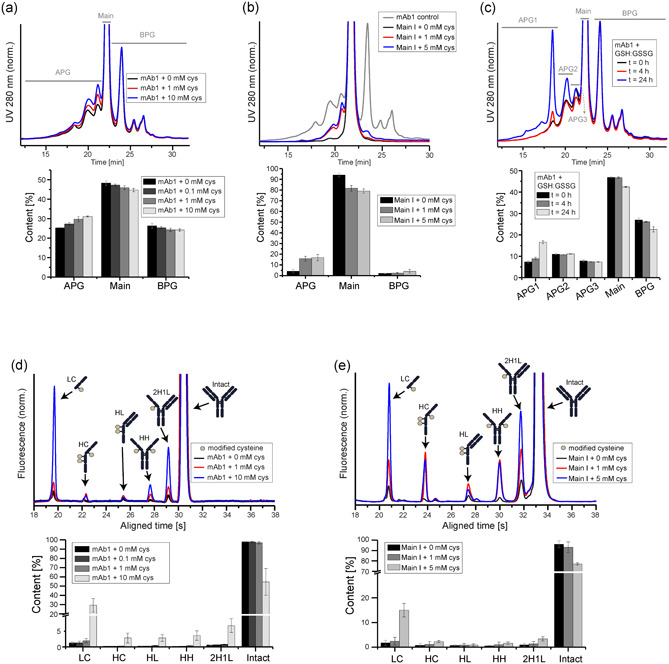
In vitro treatment of monoclonal antibody 1 (mAb1) with cysteine causes degradation of intermolecular mAb1 disulfide bonds. (a) Nonfractionated, intact mAb1 was treated with increasing cysteine concentrations (n = 4) for 2 h and subsequently analyzed by analytical weak cation exchange chromatography (WCX) to track changes in the charge variant profile. (b) Likewise, the fractionated, isolated mAb1 fraction Main I was treated with increasing cysteine concentrations for 2 h and analyzed by WCX to study the impact of cysteine incubation on acidic peak group formation (n = 4). As a control, the nonfractionated mAb1 is shown in gray. (c) WCX was carried out for mAb1 upon incubation with glutathione (GSH:GSSG (1:1)) for 0, 4, and 24 h (n = 2). (d,e) Nonreducing capillary electrophoresis was conducted after a 2 h incubation with increasing cysteine concentrations to observe the effect of cysteine on the rupture of intermolecular disulfide bonds (n = 4). This experiment was carried out forboth the nonfractionated, intact mAb1 (d), as well as on the fractionated, isolated mAb1 fraction Main I (e) [Color figure can be viewed at wileyonlinelibrary.com]
These results indicate that incubation with cysteine is sufficient to generate the full APG profile of mAb1. This observation is further supported by an experiment, in which cysteine was added to the purified main species (referred to as Main I), which was free from acidic variants prior incubation. Remarkably, cysteine treatment caused the formation of multiple acidic species representing the full APG profile (Figure 4b). Analogs to cysteine, glutathione treatment caused a change in the mAb1 charge pattern, as illustrated by WCX (Figure 4c). However, while cysteine induced a uniform increase of the acidic peak group, glutathione targeted predominantly one specific signal, which enhanced the particular variant eluting at 18.5 min.
As expected, the reducing characteristic of cysteine led to the generation of mAb1 species eluting more acidic on the WCX. The resulting species of the non‐fractionated mAb1 incubation with cysteine were further characterized by CE analysis, which demonstrated an increase of numerous variants, such as LC, HC, HH, and 2H1L after cysteine treatment of the nonfractionated mAb1 (Figure 4d). This data further supports that native mAb1 contains significant amounts of variants, which are the result of intermolecular disulfide bond reduction. The same trend was observed for the purified mAb1 Main I peak after cysteine incubation (Figure 4e). Cysteine concentrations of 1 mM triggered a more significant increase in APG levels in the Main I sample (3.4% to 14.4%) as compared to the nonfractionated mAb1 (25.1% to 28.9%). This result supports that in nonfractionated mAb1, the existing acidic variants are, to some extent, already composed of species induced by disulfide bond rupture. Thus, it required higher concentrations of cysteine (10 mM) to induce considerable disulfide bond breakage and modification to observe changes in the APG profile of nonfractionated mAb1, in comparison to the Main I species, where these pre‐existent species were not present.
An LC‐MS peptide‐mapping analysis was performed to study the type of modifications introduced by in vitro incubation with cysteine to additional mAb1 cysteine residues. Several mAb1 cysteines showed sufficient chromatographic separation and mass spectrometric properties for analysis. The analyzed mAb1 cysteine residues included those involved in intramolecular disulfide bonds, which did not appear in CE analysis. Free thiol levels were slightly increased by incubation with cysteine for three intramolecular bonded cysteines (CH2‐1, CL‐1, CL‐2; see Figure 1 for reference) (Figure 5a), thus showing the same trend as observed for intermolecular cysteines by CE (Figure 4d). In addition to the reduction to free thiols, intramolecular disulfide bonds of further cysteines (CH1‐1, CH2‐1, and CH3‐1) were susceptible to cysteinylation, in particular at high cysteine concentrations (Figure 5b). The same trends were observed for the interchain disulfide bonded CL‐3 residue.
Figure 5.
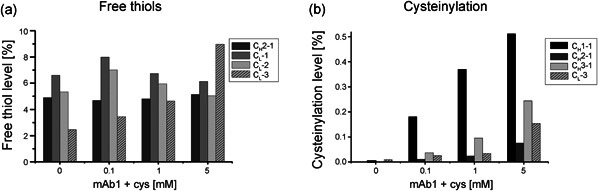
Intramolecular monoclonal antibody 1 (mAb1) disulfide bonds are disintegrated and modified by in vitro cysteine incubation. Liquid chromatography–mass spectrometry peptide‐mapping was conducted on mAb1 treated with increasing cysteine concentrations for 2 h. The peptide mapping approach allowed to study the state of several cysteines typically involved in intramolecular disulfide bonds in native mAb1. The levels of free thiols (a) and cysteinylation (b) were quantified to investigate rupture and modification on intramolecular disulfide bonds induced by incubation with cysteine (n = 1). In addition, levels were detected for CL‐3, a cysteine residue involved in an interchain disulfide bond in native mAb1
3.4. Cysteine changes the redox potential during fermentation and shapes mAb charge profile
Cysteine is an important amino acid and required for stabilizing cell viability in long‐term cultivations as well as in elevating final product concentrations (data not shown). In previous experiments of this study, it had been shown that cysteine induces acidic mAb variants in vitro. To study the effect of cysteine during cell cultivation different amounts of cysteine were added to fermentation of three different mAbs (Figure 6). Usually cysteine is part of the production medium and the feed medium, but it can also be given as an independent bolus during cultivation. When adding cysteine to the production medium, a trend towards lower redox potentials (favoring a reducing environment) was observed after 14 days of cultivation (Figure 6a). Simultaneously, APG levels increased in a molecule‐independent manner. For mAb2 and mAb3, enhanced APG levels correlated with increased cysteine concentrations and lower redox potentials.
Figure 6.
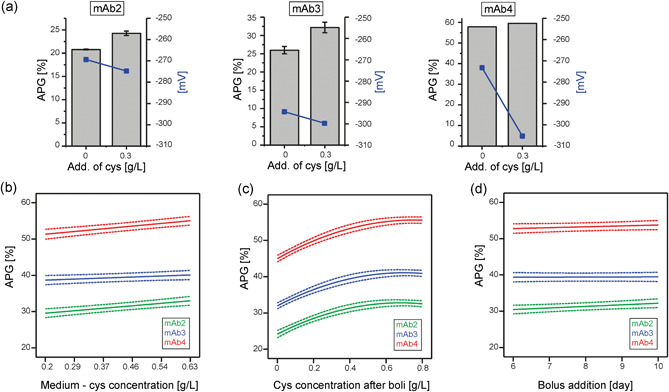
During fermentation, the media component cysteine can influence the redox potential and impact the resulting monoclonal antibody (mAb) charge profile. (a) Three mAbs were cultivated at 0 and 3 g/L cysteine concentrations in the process media (n = 2), and the acidic peak group (APG) levels, as well as the redox potential (plotted on the right y‐axis), were detected at the end of the fermentations (mAb4 was pooled prior charge variant analysis). (b–d) DoE of three mAbs and the resulting APG profiles after addition of cysteine at different stages of fermentation. The model function of the main effects are depicted including the 95% confidence intervals (dotted lines). (b) APG levels as a result of different cysteine concentrations in the process medium (c) APG levels as a result of different cysteine concentrations in the bolus addition (d) APG levels as a result of the day of cysteine‐bolus addition [Color figure can be viewed at wileyonlinelibrary.com]
Varying cysteine concentrations in the production medium and bolus additions on different cultivation days were investigated (Figure 6b–d). While increased levels of cysteine in the production medium only contributed to a moderate elevation of APG levels (Figure 6b), large bolus addition of cysteine had a more pronounced effect on APG generation (Figure 6c). Acidic species increased at low cysteine concentrations added as bolus to the cultivation and eventually reached a plateau at higher cysteine levels. This effect was observed for all mAbs, although to different extents. Intriguingly, in contrast to concentration, the time point of bolus addition did not contribute to charge variant generation. Thus, addition at any point during fermentation resulted in full APG manifestation (Figure 6d).
4. DISCUSSION
4.1. Redox‐sensitive modifications are a common feature of acidic variants of mAb1
A defective disulfide bond network has been reported to introduce different charge to mAbs (Bloom et al., 1997; Gevondyan et al., 2006; Gu et al., 2010; Schauenstein et al., 1986; Seibel et al., 2017; Wang et al., 2018). Although mAb charge variants have been characterized and described in the literature, insights to the underlying mechanisms of their generation would be beneficial in controlling mAb charge variants in industrial production processes. The current study provides an in‐depth investigation of the origin, impact and control of acidic species of an IgG1 (mAb1). When separated by WCX, the APG level accounts for 25% of the total mAb1 charge population (Figure 2a). In the current study, eight mAb1 charge variants were separated, collected, and analyzed for their biophysical properties and modifications. Despite the different charge, the mAb1 species were comparable in terms of stability and structural integrity, as demonstrated by thermal unfolding and CD spectroscopy (Figure 2c,d). This data implies that mAb1 modifications leading to changes in charge do not affect stability and thus may be irrelevant for drug safety and efficacy. These findings are in contrast to an earlier study, where high levels of acidic species were associated with changes in the biophysical properties and bioactivity of an IgG (Banks et al., 2008). In the earlier report, the origin of these changes was traced back to cysteinylation of an unpaired cysteine in the complementary‐determining region (CDR). It should be noted, that the investigated mAb1 in the current study does not contain cysteine residues in the CDR. Thus, the location of the affected cysteine, the availability of partner‐cysteine for pairing, and the degree of modification seem to determine the criticality towards structure, stability, and activity.
Further investigation of the isolated acidic species by nonreducing CE and LC‐MS revealed that degraded and modified disulfide bonds contribute considerably to the acidic species found in mAb1 (Figure 3). Noncomplete antibodies (e.g., missing LCs) were observed by nonreducing CE for acidic variants (Figure 3a) but not by analytical SEC (Figure 2b), thus, the noncomplete antibodies were most likely a result of degradation of disulfide bonds, leading to chain dissolution in nonreducing CE. The reduction of an interchain disulfide bond would not cause the LC to elute separately from the remaining mAb under the native conditions of SEC measurements, but the presence of SDS in the nonreducing CE measurement leads to separate migration of the LC.
Cysteine residues involved in inter‐ and intrachain disulfide bonds were modified in acidic mAb1 species, as shown by LC‐MS (Figure 3b–d). The C‐terminal cysteine of the LC which forms a disulfide bond to the HC has been described as being vulnerable to modifications (Wang et al., 2018). In acidic species, this cysteine contained considerable modifications, as compared to more basic species. These modifications included the reduction to free thiols, as well as cysteinylations and glutathionylations (Figure 3b).
4.2. Modified cysteine disulfide bonds are distributed throughout the mAb structure
As revealed by a peptide‐mapping LC‐MS approach, affected cysteine residues in acidic species seem to be distributed to all mAb1 domains and include intra‐ and interchain disulfide bonded cysteines (Figure 3b–d). Detailed site‐specific relative quantification by LC‐MS was not feasible for all cysteine residues due to poor resolution and mass spectrometric behavior. Yet, a significant amount of redox‐sensitive modifications contributing to acidic character originated from cysteines located in the F(ab')2 region of mAb1, as demonstrated by intact LC‐MS measurements of nonreduced IdeS‐digested mAb1 (Figure S4). This is in line with earlier reports stating that IgG1s feature unpaired cysteines in their variable domains (Chumsae et al., 2009; Xiang et al., 2009). Results of the current study suggest that such cysteines are susceptible to cysteinylation and glutathionylation, in addition to being in a reduced oxidation state.
4.3. The media component cysteine induces and enhances the full acidic variant profile
Cysteines of an intact intrachain disulfide bond in a natively folded antibody are more protected from degradation and modification, as compared to those involved interchain disulfide bonds (Amzel & Poljak, 1979; Kikuchi et al., 1986), The occurrence of nonintact intrachain disulfide bonds in mAb1 indicates that the disulfide network is not properly formed during the production process. Cysteine as a media component contributes to the redox‐environment during fermentation, which is critical for a correctly formed disulfide network (Chaderjian et al., 2005; Handlogten et al., 2017). As cysteinylation was a modification detected by LC‐MS, the influence of cysteine on mAb1 was simulated in in vitro experiments. Indeed, the addition of cysteine alone was able to increase the number of acidic species (Figure 4a) concomitantly with disulfide bond degradation (Figure 4d) and residue‐specific cysteinylation (Figure 5b), providing a strong link between disulfide bond rupture and the incidence of acidic species.
Cysteine incubation of the purified main peak of mAb1 strikingly resulted in the reproduction of the entire APG profile on a WCX column, featuring all peaks found in the control sample (Figure 4b). Thus, multiple acidic variants can be generated from “from scratch”. The generation of all three major acidic peaks via in vitro incubation with cysteine implies that analogously, the media component cysteine may contribute considerably to mAb AGP levels during cell culture, in combination with further complex factors described previously (Chaderjian et al., 2005; Handlogten et al., 2017). Further, the findings provide insight into the mechanism of cysteine‐induced APG formation. Modifications introduced by cysteine include cysteinylations as well as reduction of cysteines to free thiols resulting in non‐complete antibodies. Thus, the WCX profile of native mAb1 must consist of various variants caused by disulfide bond degradation and potential subsequent modification. Further possible effects such as disulfide bond shuffling and mispairings were not detected for mAb1 (Table S2). Yet, posttranslational modifications also contributed to mAb1 acidic species formation, as seen from LC‐MS (Figure S3) and as expected from the literature (Harris et al., 2001; Khawli et al., 2010; Lyubarskaya et al., 2006; Miller et al., 2011; Moorhouse et al., 1997; Yan et al., 2009), but did not explain the entire panel of acidic species observed.
Incubation of mAb1 with glutathione resulted in the increase of specific acidic peaks (Figure 4c), instead of an uniform increase of the entire APG, as in the case of cysteine. Thus, glutathione and cysteines may operate with different specificities. While the cysteine‐induced mAb1 species were heterogeneous and characteristic for the full APG profile, glutathione seemed to generate a specific species, presumably a glutathionylated mAb1 LC. This assumption is supported by the observation of high degrees of glutathionylation found in the LC (Figure 3b). Furthermore, glutathione has been reported to disintegrate interchain disulfide bonds during fermentation (Handlogten et al., 2017). In comparison to glutathione, the mechanism of cysteine during in vitro treatment is less specific, as supported by the higher number of cysteine residues affected by cysteinylations (Figure 5b). Hypothetically, glutathione targets CL‐3 specifically, due to its solvent exposed location at the C‐terminus of the LC. Steric hindrance may prevent glutathione from reacting with more solvent‐protected cysteines. Furthermore, cysteine is often ranked as a hydrophobic residue (Poole, 2015), and may exert higher binding affinities to free cysteine, as they share the same hydrophobic properties, in comparison to glutathione. This may serve as an additional explanation why a higher number of mAb1 cysteines are modified by free cysteine than free glutathione.
As cysteine contributed significantly to the APG profile in vitro, its influence was investigated in fermentation experiments. Indeed, cultivation at increased cysteine concentrations in the process media enhanced the APG levels of three different mAbs (Figure 6a,b). A product‐specific contribution of this effect may need to be taken into account, as not all investigated mAbs were affected to the same extent. In addition, APG generation was found to be dependent on cysteine concentration, as well as on the route of cysteine addition (i.e., bolus addition had the most impact; Figure 6c,d). Based on the in vitro data, it can be assumed that cysteine causes the rupture of disulfide bonds and introduction of cysteinylations during fermentation. The formation of trisulfide bonds has previously been linked to cysteine in the feed (Kshirsagar et al., 2012), and may additionally contribute to acidic species.
The media component cysteine is subjected to complex sulfur metabolism during cell culture (Ali et al., 2019). Cysteine has been shown to oxidize to cystine over 28 days in chemically defined media, yet 20% of the originally added substance remains in its reduced cysteine state (Krattenmacher et al., 2019). Thus, although it is not expected that the added absolute cysteine concentrations during cell culture reflect the levels present throughout fermentation, it can be assumed that the relative levels correlate. Cysteine therefore exerts a concentration‐dependent effect on acidic species, which is highly relevant from an applied, bioprocessing perspective. An impact of cystine during cell culture is not expected, based on in vitro experiments (Figure S5).
4.4. Formation of acidic variants is most likely driven by redox reactions induced by cysteine in fermentation media
The modification of mAb disulfide bonds by cysteine may be driven by two possible mechanisms (Figure 7): the oxidized disulfide bond can undergo a SN2 substitution resulting in cysteinylations (1), alternatively, the cysteines of an oxidized disulfide bond will first be reduced to free thiols by a favorable redox potential and subsequently be cysteinylated by excess cysteine in solution (2). Numerous results from this study support the occurrence of free thiols and highlight the significance of the right redox potential for an intact disulfide bond network, thereby supporting the second scenario. Firstly, S‐sulfocysteine and cystine, nonredox active derivatives of cysteine, were not able to alter mAb1 WCX profiles (Figure S5). Secondly, free thiols were detected in highly acidic mAb1 species (Figure 3b,c). Thirdly, in cultivation experiments of mAb2 and mAb3, the addition of cysteine to the production medium was not only associated with an increase in acidic variants, but also a decrease in redox potential, favorable for disulfide bond reduction (Figure 6a). Although SN2 substitution by cysteine in the media may occur, the mechanism via redox potential is more likely, as it contributes to acidic species associated with defective disulfide bonds.
Figure 7.
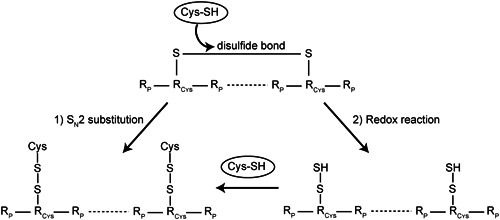
Possible mechanisms of cysteine‐induced cysteinylation of monoclonal antibody disulfide bonds. (1) SN2 substitution of intact disulfide bonds resulting in cysteinylation. (2) Redox reaction leads to a reduction of disulfide bonds followed by cysteinylation of the free thiols. Results of the study point to the second scenario as the most likely mechanism
The findings reported in this study illustrate the APG‐promoting effect of cysteine in cell culture media for an IgG1 resulting in disulfide bond degradation and modification. Elevated levels of acidic species are undesirable during bioprocessing as they impair step yields. The demonstrated data highlights cysteine concentration in cell culture media as a potential point of interference for manipulating mAb charge during the production process. Specifically, careful monitoring of cysteine as a media component and thus regulation of the redox potential during fermentation may enable a control of charge heterogeneity.
AUTHOR CONTRIBUTIONS
Elke Prade and Ingo H. Gorr designed the research and wrote the manuscript with input from all authors. Anne Zeck measured and analyzed the liquid chromatography–mass spectrometry data. Fabian Stiefel and Andreas Unsoeld designed and analyzed the cell culture experiments. David Mentrup performed the sample preparation and analytical characterization of fractionated and intact mAb1 and analyzed the data. Elke Prade and Erik Arango Gutierrez supervised the mAb1 wet lab activities. All authors discussed the results.
Supporting information
Supporting information.
ACKNOWLEDGMENTS
Boehringer Ingelheim Pharma GmbH & Co. KG. funded all research of this manuscript. The authors are especially grateful to Göran Hübner, Nils Drechsler and Sarah Lergenmüller for scientific discussions and input on analytical data, as well as Oliver Burkert for providing the opportunity for circular dichroism spectroscopic measurements. Further, the authors would like to thank Michelle Ahlers‐Hesse for technical support in purification and in vitro experiments, Elena Bollgoen and Chris Schubert for their contributions in the cell culture experiments, as well as Amadine Calvet for measurements of redox potentials.
Prade E, Zeck A, Stiefel F, et al. Cysteine in cell culture media induces acidic IgG1 species by disrupting the disulfide bond network. Biotechnology and Bioengineering. 2021;118:1091–1104. 10.1002/bit.27628
REFERENCES
- Ali, A. S. , Raju, R. , Kshirsagar, R. , Ivanov, A. R. , Gilbert, A. , Zang, L. , & Karger, B. L. (2019). Multi‐omics study on the impact of cysteine feed level on cell viability and mAb production in a CHO bioprocess. Biotechnology Journal, 14(4):e1800352. 10.1002/biot.201800352 [DOI] [PubMed] [Google Scholar]
- Amzel, L. M. , & Poljak, R. J. (1979). Three‐dimensional structure of immunoglobulins. Annual Review of Biochemistry, 48, 961–997. 10.1146/annurev.bi.48.070179.004525 [DOI] [PubMed] [Google Scholar]
- Banks, D. D. , Gadgil, H. S. , Pipes, G. D. , Bondarenko, P. V. , Hobbs, V. , Scavezze, J. L. , Kim, J. , Jiang, X. R. , Mukku, V. , & Dillon, T. M. (2008). Removal of cysteinylation from an unpaired sulfhydryl in the variable region of a recombinant monoclonal IgG1 antibody improves homogeneity, stability, and biological activity. Journal of Pharmaceutical Sciences, 97(2), 775–790. 10.1002/jps.21014 [DOI] [PubMed] [Google Scholar]
- Bloom, J. W. , Madanat, M. S. , Marriott, D. , Wong, T. , & Chan, S. Y. (1997). Intrachain disulfide bond in the core hinge region of human IgG4. Protein science: A publication of the Protein. Society, 6(2), 407–415. 10.1002/pro.5560060217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boswell, C. A. , Tesar, D. B. , Mukhyala, K. , Theil, F. P. , Fielder, P. J. , & Khawli, L. A. (2010). Effects of charge on antibody tissue distribution and pharmacokinetics. Bioconjugate Chemistry, 21(12), 2153–2163. 10.1021/bc100261d [DOI] [PubMed] [Google Scholar]
- Buijs, J. , Norde, W. , & Lichtenbelt, J. W. T. (1996). Changes in the secondary structure of adsorbed IgG and F(ab‘)2 studied by FTIR spectroscopy. Langmuir, 12(6), 1605–1613. 10.1021/la950665s [DOI] [Google Scholar]
- Chaderjian, W. B. , Chin, E. T. , Harris, R. J. , & Etcheverry, T. M. (2005). Effect of copper sulfate on performance of a serum‐free CHO cell culture process and the level of free thiol in the recombinant antibody expressed. Biotechnology Progress, 21(2), 550–553. 10.1021/bp0497029 [DOI] [PubMed] [Google Scholar]
- Chumsae, C. , Gaza‐Bulseco, G. , & Liu, H. (2009). Identification and localization of unpaired cysteine residues in monoclonal antibodies by fluorescence labeling and mass spectrometry. Analytical Chemistry, 81(15), 6449–6457. 10.1021/ac900815z [DOI] [PubMed] [Google Scholar]
- Chung, S. , Tian, J. , Tan, Z. , Chen, J. , Lee, J. , Borys, M. , & Li, Z. J. (2018). Industrial bioprocessing perspectives on managing therapeutic protein charge variant profiles. Biotechnology and Bioengineering, 115(7), 1646–1665. 10.1002/bit.26587 [DOI] [PubMed] [Google Scholar]
- Dakshinamurthy, P. , Mukunda, P. , Prasad Kodaganti, B. , Shenoy, B. R. , Natarajan, B. , Maliwalave, A. , Halan, V. , Murugesan, S. , & Maity, S. (2017). Charge variant analysis of proposed biosimilar to Trastuzumab. Biologicals, 46, 46–56. 10.1016/j.biologicals.2016.12.006 [DOI] [PubMed] [Google Scholar]
- Dillon, T. M. , Ricci, M. S. , Vezina, C. , Flynn, G. C. , Liu, Y. D. , Rehder, D. S. , Plant, M. , Henkle, B. , Li, Y. , Deechongkit, S. , Varnum, B. , Wypych, J. , Balland, A. , & Bondarenko, P. V. (2008). Structural and functional characterization of disulfide isoforms of the human IgG2 subclass. The Journal of Biological Chemistry, 283(23), 16206–16215. 10.1074/jbc.M709988200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du, Y. , Walsh, A. , Ehrick, R. , Xu, W. , May, K. , & Liu, H. (2012). Chromatographic analysis of the acidic and basic species of recombinant monoclonal antibodies. mAbs, 4(5), 578–585. 10.4161/mabs.21328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelman, G. M. , Cunningham, B. A. , Gall, W. E. , Gottlieb, P. D. , Rutishauser, U. , & Waxdal, M. J. (1969). The covalent structure of an entire gammaG immunoglobulin molecule. Proceedings of the National Academy of Sciences of the United States of America, 63(1), 78–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frangione, B. , Milstein, C. , & Franklin, E. C. (1968). Intrachain disulphide bridges in immunoglobulin G heavy chains. The Fc fragment. The Biochemical Journal, 106(1), 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi, S. , Ren, D. , Xiao, G. , Bondarenko, P. , Sloey, C. , Ricci, M. S. , & Krishnan, S. (2012). Elucidation of degradants in acidic peak of cation exchange chromatography in an IgG1 monoclonal antibody formed on long‐term storage in a liquid formulation. Pharmaceutical Research, 29(1), 209–224. 10.1007/s11095-011-0536-0 [DOI] [PubMed] [Google Scholar]
- Gevondyan, N. M. , Volynskaia, A. M. , & Gevondyan, V. S. (2006). Four free cysteine residues found in human IgG1 of healthy donors. Biochemistry. Biokhimii͡a (Moscow, Russia), 71(3), 279–284. [DOI] [PubMed] [Google Scholar]
- Griaud, F. , Denefeld, B. , Lang, M. , Hensinger, H. , Haberl, P. , & Berg, M. (2017). Unbiased in‐depth characterization of CEX fractions from a stressed monoclonal antibody by mass spectrometry. MAbs, 9, 820–830. 10.1080/19420862.2017.1313367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu, S. , Wen, D. , Weinreb, P. H. , Sun, Y. , Zhang, L. , Foley, S. F. , Kshirsagar, R. , Evans, D. , Mi, S. , Meier, W. , & Pepinsky, R. B. (2010). Characterization of trisulfide modification in antibodies. Analytical Biochemistry, 400(1), 89–98. 10.1016/j.ab.2010.01.019 [DOI] [PubMed] [Google Scholar]
- Handlogten, M. W. , Zhu, M. , & Ahuja, S. (2017). Glutathione and thioredoxin systems contribute to recombinant monoclonal antibody interchain disulfide bond reduction during bioprocessing. Biotechnology and Bioengineering, 114(7), 1469–1477. 10.1002/bit.26278 [DOI] [PubMed] [Google Scholar]
- Harris, R. J. (2005). Heterogeneity of recombinant antibodies: Linking structure to function. Developments in biologicals, 122, 117–127. [PubMed] [Google Scholar]
- Harris, R. J. , Kabakoff, B. , Macchi, F. D. , Shen, F. J. , Kwong, M. , Andya, J. D. , & Chen, A. B. (2001). Identification of multiple sources of charge heterogeneity in a recombinant antibody. Journal of Chromatography B: Biomedical Sciences and Applications, 752(2), 233–245. [DOI] [PubMed] [Google Scholar]
- Hossler, P. , Wang, M. , McDermott, S. , Racicot, C. , Chemfe, K. , Zhang, Y. , Chumsae, C. , & Manuilov, A. (2015). Cell culture media supplementation of bioflavonoids for the targeted reduction of acidic species charge variants on recombinant therapeutic proteins. Biotechnology Progress, 31(4), 1039–1052. 10.1002/btpr.2095 [DOI] [PubMed] [Google Scholar]
- Khawli, L. A. , Goswami, S. , Hutchinson, R. , Kwong, Z. W. , Yang, J. , Wang, X. , Yao, Z. , Sreedhara, A. , Cano, T. , Tesar, D. B. , Nijem, I. , Allison, D. E. , Wong, P. Y. , Kao, Y. H. , Quan, C. , Joshi, A. , Harris, R. J. , & Motchnik, P. (2010). Charge variants in IgG1: Isolation, characterization, in vitro binding properties and pharmacokinetics in rats. MAbs, 2(6), 613–624. 10.4161/mabs.2.6.13333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi, H. , Goto, Y. , & Hamaguchi, K. (1986). Reduction of the buried intrachain disulfide bond of the constant fragment of the immunoglobulin light chain: Global unfolding under physiological conditions. Biochemistry, 25(8), 2009–2013. [DOI] [PubMed] [Google Scholar]
- Kishishita, S. , Nishikawa, T. , Shinoda, Y. , Nagashima, H. , Okamoto, H. , Takuma, S. , & Aoyagi, H. (2015). Effect of temperature shift on levels of acidic charge variants in IgG monoclonal antibodies in Chinese hamster ovary cell culture. Journal of Bioscience and Bioengineering, 119(6), 700–705. 10.1016/j.jbiosc.2014.10.028 [DOI] [PubMed] [Google Scholar]
- Krattenmacher, F. , Heermann, T. , Calvet, A. , Krawczyk, B. , & Noll, T. (2019). Effect of manufacturing temperature and storage duration on stability of chemically defined media measured with LC‐MS/MS. Journal of Chemical Technology & Biotechnology, 94(4), 1144–1155. 10.1002/jctb.5861 [DOI] [Google Scholar]
- Kshirsagar, R. , McElearney, K. , Gilbert, A. , Sinacore, M. , & Ryll, T. (2012). Controlling trisulfide modification in recombinant monoclonal antibody produced in fed‐batch cell culture. Biotechnology and Bioengineering, 109(10), 2523–2532. 10.1002/bit.24511 [DOI] [PubMed] [Google Scholar]
- Liu, H. , Gaza‐Bulseco, G. , Faldu, D. , Chumsae, C. , & Sun, J. (2008). Heterogeneity of Monoclonal Antibodies. Journal of Pharmaceutical Sciences, 97(7), 2426–2447. 10.1002/jps.21180 [DOI] [PubMed] [Google Scholar]
- Lyubarskaya, Y. , Houde, D. , Woodard, J. , Murphy, D. , & Mhatre, R. (2006). Analysis of recombinant monoclonal antibody isoforms by electrospray ionization mass spectrometry as a strategy for streamlining characterization of recombinant monoclonal antibody charge heterogeneity. Analytical Biochemistry, 348(1), 24–39. 10.1016/j.ab.2005.10.003 [DOI] [PubMed] [Google Scholar]
- McAuley, A. , Jacob, J. , Kolvenbach, C. G. , Westland, K. , Lee, H. J. , Brych, S. R. , Rehder, D. , Kleemann, G. R. , Brems, D. N. , & Matsumura, M. (2008). Contributions of a disulfide bond to the structure, stability, and dimerization of human IgG1 antibody CH3 domain. Protein Science, 17(1), 95–106. 10.1110/ps.073134408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao, S. , Xie, P. , Zou, M. , Fan, L. , Liu, X. , Zhou, Y. , Zhao, L. , Ding, D. , Wang, H. , & Tan, W. S. (2017). Identification of multiple sources of the acidic charge variants in an IgG1 monoclonal antibody. Applied Microbiology and Biotechnology, 101(14), 5627–5638. 10.1007/s00253-017-8301-x [DOI] [PubMed] [Google Scholar]
- Miller, A. K. , Hambly, D. M. , Kerwin, B. A. , Treuheit, M. J. , & Gadgil, H. S. (2011). Characterization of site‐specific glycation during process development of a human therapeutic monoclonal antibody. Journal of Pharmaceutical Sciences, 100(7), 2543–2550. 10.1002/jps.22504 [DOI] [PubMed] [Google Scholar]
- Milstein, C. (1966). The disulphide bridges of immunoglobulin kappa‐chains. The Biochemical Journal, 101(2), 338–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moorhouse, K. G. , Nashabeh, W. , Deveney, J. , Bjork, N. S. , Mulkerrin, M. G. , & Ryskamp, T. (1997). Validation of an HPLC method for the analysis of the charge heterogeneity of the recombinant monoclonal antibody IDEC‐C2B8 after papain digestion. Journal of Pharmaceutical and Biomedical Analysis, 16(4), 593–603. [DOI] [PubMed] [Google Scholar]
- Ouellette, D. , Alessandri, L. , Chin, A. , Grinnell, C. , Tarcsa, E. , Radziejewski, C. , & Correia, I. (2010). Studies in serum support rapid formation of disulfide bond between unpaired cysteine residues in the VH domain of an immunoglobulin G1 molecule. Analytical Biochemistry, 397(1), 37–47. 10.1016/j.ab.2009.09.027 [DOI] [PubMed] [Google Scholar]
- Pinck, J. R. , & Milstein, C. (1967). Disulphide bridges of a human immunoglobulin G protein. Nature, 216(5118), 941–942. [DOI] [PubMed] [Google Scholar]
- Ponniah, G. , Kita, A. , Nowak, C. , Neill, A. , Kori, Y. , Rajendran, S. , & Liu, H. (2015). Characterization of the acidic species of a monoclonal antibody using weak cation exchange chromatography and LC‐MS. Analytical Chemistry, 87(17), 9084–9092. 10.1021/acs.analchem.5b02385 [DOI] [PubMed] [Google Scholar]
- Poole, L. B. (2015). The basics of thiols and cysteines in redox biology and chemistry. Free Radical Biology and Medicine, 80, 148–157. 10.1016/j.freeradbiomed.2014.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schauenstein, E. , Dachs, F. , Reiter, M. , Gombotz, H. , & List, W. (1986). Labile disulfide bonds and free thiol groups in human IgG. I. Assignment to IgG1 and IgG2 subclasses. International Archives of Allergy and Applied Immunology, 80(2), 174–179. [DOI] [PubMed] [Google Scholar]
- Schuurman, J. , Perdok, G. J. , Gorter, A. D. , & Aalberse, R. C. (2001). The inter‐heavy chain disulfide bonds of IgG4 are in equilibrium with intra‐chain disulfide bonds. Molecular Immunology, 38(1), 1–8. [DOI] [PubMed] [Google Scholar]
- Seibel, R. , Maier, S. , Schnellbaecher, A. , Bohl, S. , Wehsling, M. , Zeck, A. , & Zimmer, A. (2017). Impact of S‐sulfocysteine on fragments and trisulfide bond linkages in monoclonal antibodies. MAbs, 9(6), 889–897. 10.1080/19420862.2017.1333212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla, A. A. , Wolfe, L. S. , Mostafa, S. S. , & Norman, C. (2017). Evolving trends in mAb production processes. Bioengineering & Translational Medicine, 2(1), 58–69. 10.1002/btm2.10061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, S. , Liu, A. P. , Yan, Y. , Daly, T. J. , & Li, N. (2018). Characterization of product‐related low molecular weight impurities in therapeutic monoclonal antibodies using hydrophilic interaction chromatography coupled with mass spectrometry. Journal of Pharmaceutical and Biomedical Analysis, 154, 468–475. 10.1016/j.jpba.2018.03.034 [DOI] [PubMed] [Google Scholar]
- Wypych, J. , Li, M. , Guo, A. , Zhang, Z. , Martinez, T. , Allen, M. J. , Fodor, S. , Kelner, D. N. , Flynn, G. C. , Liu, Y. D. , Bondarenko, P. V. , Ricci, M. S. , Dillon, T. M. , & Balland, A. (2008). Human IgG2 antibodies display disulfide‐mediated structural isoforms. The. Journal of Biological Chemistry, 283(23), 16194–16205. 10.1074/jbc.M709987200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang, T. , Chumsae, C. , & Liu, H. (2009). Localization and quantitation of free sulfhydryl in recombinant monoclonal antibodies by differential labeling with 12C and 13C iodoacetic acid and LC‐MS analysis. Analytical Chemistry, 81(19), 8101–8108. 10.1021/ac901311y [DOI] [PubMed] [Google Scholar]
- Xie, P. , Niu, H. , Chen, X. , Zhang, X. , Miao, S. , Deng, X. , Liu, X. , Tan, W. S. , Zhou, Y. , & Fan, L. (2016). Elucidating the effects of pH shift on IgG1 monoclonal antibody acidic charge variant levels in Chinese hamster ovary cell cultures. Applied Microbiology and Biotechnology, 100(24), 10343–10353. 10.1007/s00253-016-7749-4 [DOI] [PubMed] [Google Scholar]
- Yan, B. , Steen, S. , Hambly, D. , Valliere‐Douglass, J. , Bos, T. V. , Smallwood, S. , Yates, Z. , Arroll, T. , Han, Y. , Gadgil, H. , Latypov, R. F. , Wallace, A. , Lim, A. , Kleemann, G. R. , Wang, W. , & Balland, A. (2009). Succinimide formation at Asn 55 in the complementarity determining region of a recombinant monoclonal antibody IgG1 heavy chain. Journal of Pharmaceutical Sciences, 98(10), 3509–3521. 10.1002/jps.21655 [DOI] [PubMed] [Google Scholar]
- Yoo, E. M. , Wims, L. A. , Chan, L. A. , & Morrison, S. L. (2003). Human IgG2 can form covalent dimers. Journal of Immunology, 170(6), 3134–3138. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information.