

Abstract
Multi-photon polymer crosslinking evolves as the core process behind high-resolution additive microfabrication with soft materials for implantable/wearable electronics, tissue engineering, micro-robotics, biosensing, drug delivery, etc. Electrons and soft X-rays, in principle, can offer even higher resolution and printing rates. However, these powerful lithographic tools are difficult to apply to vacuum incompatible liquid precursor solutions used in continuous additive fabrication. In this work, using biocompatible hydrogel as a model soft material, we demonstrate high-resolution in-liquid polymer crosslinking using scanning electron and X-ray microscopes. The approach augments the existing solid state electron/X-rays lithography and beam induced deposition techniques with a wider class of possible chemical reactions, precursors and functionalities. We discuss focused beam crosslinking mechanism, the factors affecting the ultimate feature size and layer-by-layer printing possibilities. The potential of this technology is demonstrated on a few practically important applications such as in-liquid encapsulation of nanoparticles for plasmonic sensing and interfacing of viable cells with hydrogel electrodes.
Keywords: electron beam induced crosslinking, X-rays induced crosslinking, 3D hydrogel printing, tissue engineering, gel electronics
Graphical Abstract
Modern additive fabrication of 3-dimensional (3D) micron to centimeters size constructs made of polymers and soft materials has immensely benefited from the development of photo-curable formulations suitable for optical photolithography,1 holographic,2 and stereolithography3 methods. Recent implementation of multiphoton laser polymerization and its coupling with advanced irradiation schemes4–8 has drastically improved the writing rates and resolution which now approaches 100 nm range.8
Alternatively, traditional electron beam lithography and its variations such as Electron‐Beam Chemical Lithography etc.9 rely on tightly focused electron beams and high interaction cross-section of 0.1keV÷10 keV electrons with the matter and has been routinely used for complex patterning of polymer resists,10 self-assembled monolayers11, 12 and dried gel films13–16 with up to few nanometers accuracy. Additive microfabrication of complex structures using focused electron beam induced deposition (3D-FEBID) has been demonstrated only recently (see perspective by Winkler et al.17 and references therein). This vacuum technique relies on electron beam induced dissociation of gaseous metal-containing precursors at the surface. The requirement for high-resolution printing, however, caps the allowable precursor pressure at the growth front rate which, in turn, limits the achievable printing speed. This challenge, in principle, can be resolved using nonvolatile liquids or precursor solutions with (quasi-) stable and replenishable liquid-vacuum interface.18 The fabrication of fine carbonaceous, metal and oxide structures has been demonstrated in these reports. However, the chemical composition of the structures in both approaches remains to be restricted by the number of available precursors or nonvolatile liquid formulations and still suffers from co-reactants impurities. FEBID from the liquid phase solutions using through-the-membrane19–23 (also called liquid cell scanning electron microscopy (LSEM)24, 25) approach in principle can offer greater materials selection, faster deposition rates and high purity of deposits and potentially be used for high resolution additive manufacturing.26
Similarly, a significant progress has been made in Deep X-ray lithography,27, 28 direct writing with zone plate focused X-ray beams for precise29 and chemically selective30 fabrication of high aspect ratio microstructures. Reduced radiation damage within so-called “water window”31 has spurt wide biomedical X-ray spectroscopy, microscopy and tomography research32 including material processing e.g. gels related controlled swelling33 and polymerization inside live systems,34 particles encapsulations35 and high aspect ratio structures fabrication.36 The potential of focused X-rays for additive fabrication trough deposition from gas phase precursors37 or from liquid solutions38 is now well recognized and is becoming an active area of research.
In this communication, we demonstrate that polymerization and crosslink chemical reactions can be initiated inside the liquid with focused electron beams. The beam writing has been done through the thin electron/X-rays transparent membranes separating high-vacuum equipment from the volatile precursor solution. Using a hydrogel as a model soft material, we were able to perform in-liquid direct writing with sub 100 nm resolution. We define the range of the experimental parameters controlling the writing rate, resolution and gel feature sizes and show that the diffusion of radiolytic radicals needs to be invoked to explain the observed systematic differences between the model predictions and experimentally obtained feature sizes. We further demonstrate the versatility of the approach by using focused soft X-rays with variable photon energies for gel crosslinking with chemical and spatial selectivity and discuss the key similarities and differences in 3D gel patterning mechanism between electrons and soft X-rays. Finally, we show that that crosslinking in hydrogel solutions takes place at very low radiation doses, below the viability threshold for biological cells, and high spatial resolution, thus, allowing for numerous applications in tissue micro-engineering, drugs delivery, biosensing, bio-electronics, etc.39
RESULTS AND DISCUSSION:
To deliver focused electron or soft X-ray beams to vacuum-incompatible liquid solutions and for patterning and imaging in liquids, we designed fluidic (and closed) chambers equipped with 30 nm to 50 nm thin silicon nitride (SiN) membranes that isolate the liquid solution from the vacuum of the microscope (Figure 1a for electrons and Figure 1b for X-rays).
Figure 1.
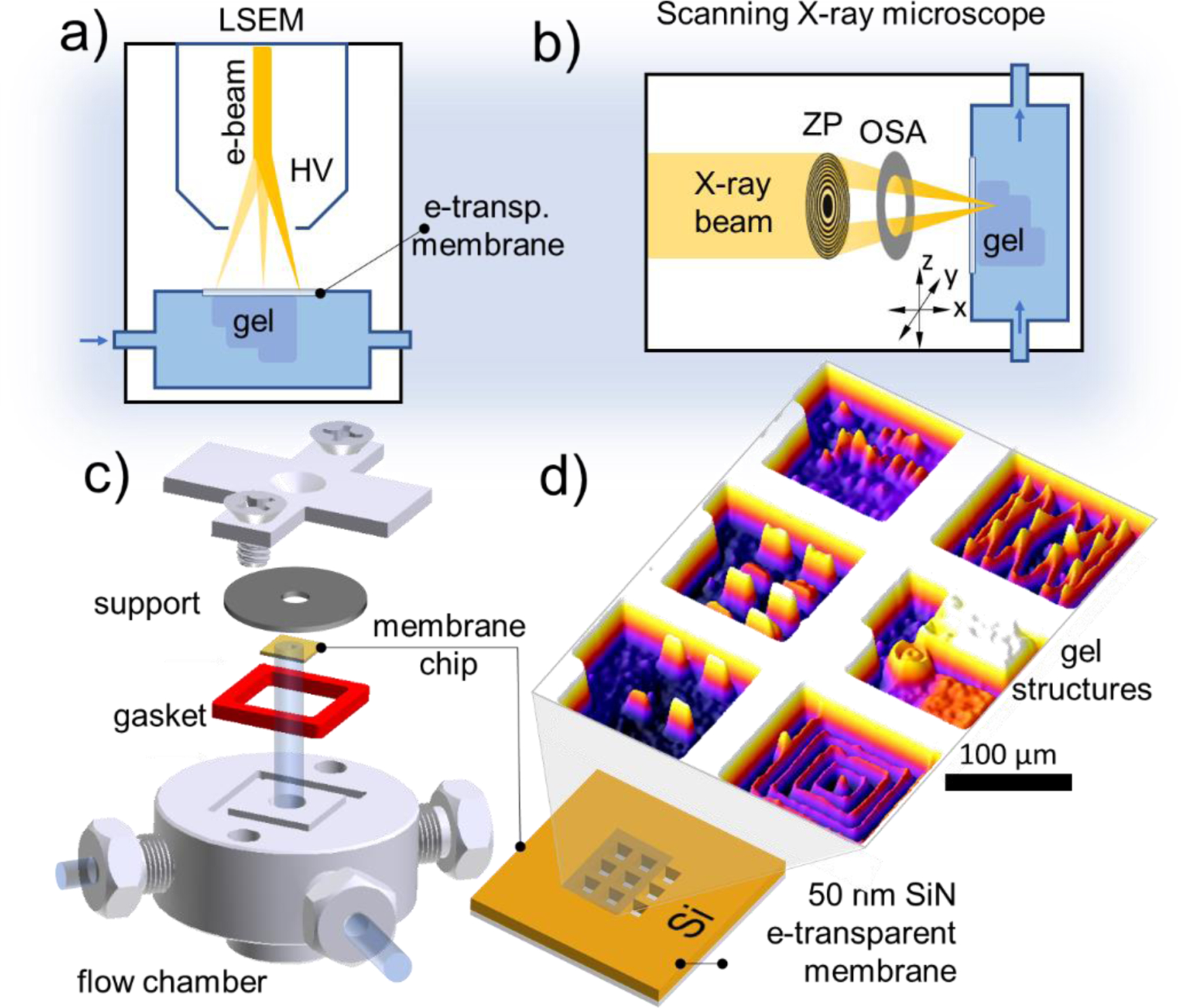
a) Liquid cell SEM (LSEM) setup for spatially resolved in-liquid hydrogel curing. High vacuum (HV) inside the microscope is protected with SiN membrane. b) Focused soft X-ray hydrogel curing in the liquid phase using zone plate (ZP) based optics. OSA stands for order sorting aperture; c) the basic design and parts of the liquid sample chamber used in this study; d) 3D surface plot of an optical image of gel structures electron beam printed on the liquid-facing side of selected six SiN windows
Figures 1c, d show an SEM compatible chamber used in this study for electron focused beam-induced polymer crosslinking in volatile solutions. The chamber for X-ray studies contained ca. 5 microliters of enclosed hydrogel precursor solution and was smaller in size. Both chambers were equipped with a SiN/Si chip patterned with an array of nine 50 nm thick and 100 µm × 100 µm wide SiN suspended membranes capable of withstanding a 1 Bar (105 Pa) pressure differential. This single-use exchangeable SiN/Si chip is vacuum-sealed against the body of the fluidic (or enclosed) cell. The interior of the chamber was filled with poly(ethylene glycol) diacrylate (PEGDA) 20 % w/v aqueous solution. Nine identical membrane windows were used for writing multiple features and combinatorial data collection within a single experiment. As an example (Figure 1d), arrays of rectangular and fine linear structures were printed on individual membrane windows by varying only one of the parameters: beam energy, irradiation intensity, step-size or dwell time while keeping others locked. After rinsing off the uncured solution with water, the dimensions of the cross-linked stable gel structures were inspected in the hydrated state using atomic force microscopy (AFM) and optical profilometry, and by SEM in dry state. By comparing the height of the same objects in their hydrated and dry state the gel’s average vertical swelling ratio was estimated as a function of the polymer molecular mass, concentration of the solution and typical electron beam irradiation conditions (see SI Figure S2). Such calibrations were later used to estimate the size of the dried-up hydrated gel objects without directly measuring them.
Focused beam induced crosslinking process in polymer aqueous solutions
We start by describing the the processes that govern polymerization in the studied system. Upon entering the PEGDA aqueous solution, the electrons experience a cascade of scattering events, which slow down and broaden the beam thus creating a droplet-like highly excited interaction volume where the crosslinking of the PEGDA molecules into an insoluble gel matrix takes place (Figure 2a, right facet). The radiation-induced crosslinking in polymer solutions was studied since 1950-s and occurs via two mechanisms:40 i) directly, by activating the reactive groups in the polymer solute with primary or secondary electrons; and ii) indirectly, by electron beam-induced solvent radiolysis that generates a variety of radicals promoting crosslinking. The partitioning between these two reaction channels depends on multiple parameters such as the solute concentration, molecular weight of the polymer, beam energy, its intensity, radical diffusion coefficients etc. These interdependences added a great flexibility to the method and have been well studied for highly penetrating ionizing radiations with spatially homogenous excitation and diffusional profiles (see recent review41 and references therein). Compared to broad beam irradiation conditions, focused electron or X-ray beams generate highly localized, spatially inhomogeneous radiolitic volumes for which the radiation chemistry and kinetics are less explored. To shed light on the chemical processes occurring inside the excitation volume we conducted a comparative side-to-side µ-Raman analysis of irradiated and pristine PEDGA regions. Raman spectra in the Figure 2b reveal quick quenching of the 1410 cm–1, 1640 cm–1 C=C stretching bands upon electron irradiation. The latter is expected42 for completely crosslinked PEGDA due to abstraction of hydrogen atoms and opening the carbon double bonds at the acrylate end groups by the radiolitic (predominantly OH·) radicals.43 This process is shown on the left side of Figure 2b. Note, that the C=O bonds are affected to a lesser extent, since they are significantly stronger and do not directly participate in cross-linking. The decrease in the C=O Raman signature could be due to the radical-induced reduction of C=O to C-OH, proceeding in parallel to cross-linking.
Figure 2.
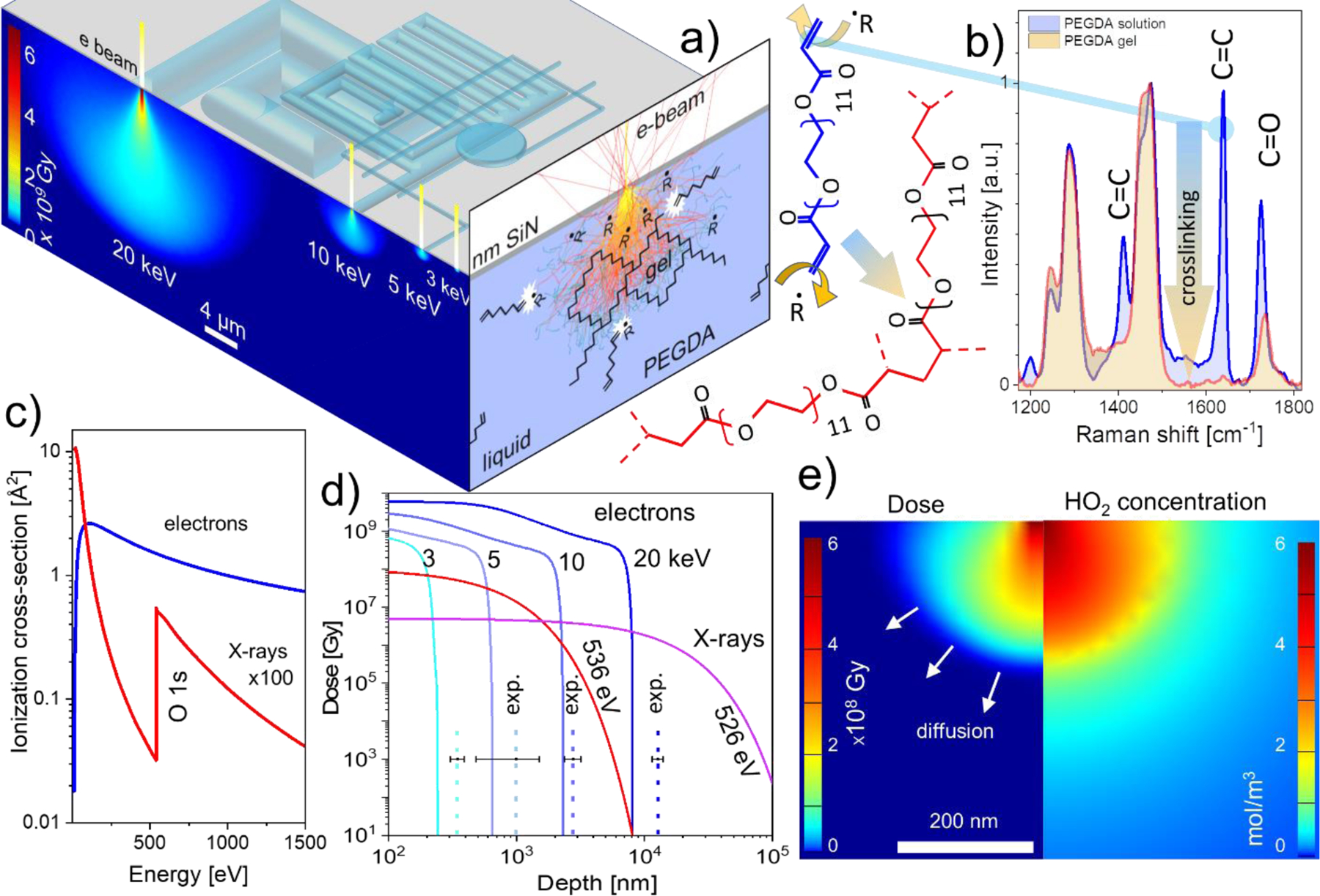
a) Top facet: the principle of electron beam gelation in liquid polymer solution through 50 nm thick SiN membrane. Lower energy electrons generate smaller excitation volume and therefore finer feature size. Left facet: spatial distribution of the energy deposition by electron beams in water calculated using Monte Carlo simulations. Right facet depicts the concept of direct and indirect (through radiolytic radicals R) PEGDA polymer crosslinking; b) Left: structure of PEGDA 700 (blue) with terminal C=C bonds that are attacked by the beam-generated radicals leading to cross-linking of the PEGDA molecules into polymeric chains (red). Right: vanishing of C=C signatures in PEGDA solution upon irradiation with ca. 106 Gy dose, indicating complete crosslinking of the precursor molecules. c) The comparison of energy dependence of the electron and soft X-ray ionization cross sections of liquid water; d) MC simulated absorbed energy doses as a function of depth for electrons (100 pA current, 1 ms dwell time) and for X-rays (2·109 photons/s and 10 ms dwell time). Dashed vertical lines depict experimentally observed hydrated feature sizes created using the same exposure parameters. Error bars represent standard deviations from three separate experiments. e) Calculated energy deposited distribution for 3 keV 200 pA electron beam (left panel) and the corresponding distribution of hydroperoxyl radical concentration (right panel)
When soft X-rays are used to trigger crosslinking, the net effect is similar, although it proceeds via different electronic excitation/relaxation pathways.30, 44, 45 Briefly, X-ray photon ionize/excites the valence and core electrons of the solute and solution molecules. The relaxation of core hole proceeds primarily via emission of Auger electrons.46 After such a de-excitation, the energy is effectively absorbed by the liquid via the same inelastic electron scattering mechanism as described above for electron beam induced crosslinking. The major differences between the electron and soft X-ray induced crosslinking, therefore, are due to their ionization cross-sections as can be seen from the liquid water example (Figure 2c). The values of liquid water photoionization cross-section for soft X-rays (100 eV to 2000 eV) are on the average ca. 100 times smaller than for the few keV electrons. Therefore, such X-rays can penetrate significantly deeper into the precursor solution forming gel features with larger aspect ratios and lower crosslinking densities (see SI Figure S2c). In addition, the electron ionization cross-section of water is a smooth function of energy (Figure 2c), thus the range of electrons in water solution and printed feature size always increase with energy. In the case of soft X-rays however, the photoionization cross-section sharply increases at the onset of the O1s core level excitations (Figure 2c, red curve). This chemical selectivity of the X-rays range in the solution will be used to control the aspect ratio of the printed features and chemically selective encapsulation of objects.
Since the crosslinking in diluted PEGDA solutions proceeds primarily via interaction with radiolytic radicals, their diffusion beyond the electron-sample interaction volume can be the feature size determining factor. To evaluate the effect of radical diffusion on crosslinking we compared the experimental feature sizes the e-beam printed in a 20 % w/v PEGDA aqueous solution with the modeled ones applying the same irradiation conditions. Monte Carlo (MC) simulated spatial distributions of the radiation dose deposited in aqueous solution through a 50 nm thick SiN membrane are depicted in Figure 2d for a few different energies and a 5 nm wide electron beam. Assuming the critical energy dose for PEGDA gelation being within the range 103 Gy41 ÷106 Gy13, the expected height of stable hydrated objects ranges from ca. 200 nm for 3 kV to ca. 8 micrometers for 20 keV electron beams (Figure 2d).
However, the experimental data on thicknesses of hydrated features obtained under the same conditions (dashed lines in the Figure 2d and Figures S1, S4 SI) are noticeably and systematically larger than the depths at the onset of gelation critical density obtained from Monte Carlo simulations. The latter indicates that indirect crosslinking via a runaway diffusion of radiolytic activators may indeed be responsible for the observed size increase of the printed features. To support this observation further, we adapted a kinetic radiolysis model (previously developed for environmental SEM47 and in-liquid transmission electron microscopy48, 49) to our SEM conditions (see details in SI). The numerical simulations based on this model predict a short lifetime and therefore a small runaway diffusion length (< 200 nm) for the most abundant crosslinking OHˑ radical outside the electron beam interaction volume, thus implying that there can exist other radicals with larger lifetimes that can, in principle, diffuse to longer distances before reacting out completely. One of the possible crosslinking agents particularly abundant in water under aerated conditions is hydroperoxyl radical.43 Figure 2e compares the energy dose distribution upon water irradiation with a 3 keV focused beam (left panel) with the corresponding HO2· concentration profile (right panel). As can be seen, HO2· crosslinking activator concentration remains high, well beyond the zone defined by the electron dose gelation threshold and therefore may account for the systematically increased size of the printed features.
Printing Controls
We now discuss the main experimental parameters that can be tuned to control the size, shape and sharpness of the features within the individual printed layer. In the raster scanning mode these are: electron beam energy (E), dwell time (τD) at a pixel location, step-size (L) during the scan and exposure dose D per pixel defined as 𝐷 = 𝐼𝐵𝜏𝐷𝑛⁄𝐿2 where IB is an electron (photon) beam current (intensity) and n is number of scans.
Figure 3a–g shows exemplary 3D structures printed using in-liquid crosslinking by electrons (a-e) and soft X-rays (f, g). The primary energy of electrons determines the range of electrons in the solvent and therefore the height profiles of the printed structure ranging from ca. 100 nm to ca. 10 µm while varying beam energy from 3 keV to 20 keV (Figure 3b, 3d). The dynamic range of heights exceeding 100 aspect ratio was achieved via beam energy variation, however, the sharpness of the features drops concomitantly with energy. The comparable aspect ratios of 3D structures can be obtained via varying only the dwell time (compare Figures 3d and 3e). Different from the beam energy variations, which requires refocusing, the facile and instant modulation of the dwell time and step-size allows the fabrication of the high-aspect ratio microstructures, (e.g. a flagella-like object in Figure 3c) in a single run with a writing speed of ca. 5 mm/s. Note that similar structures have been used for locomotion in liquids after being functionalized with magnetic nanoparticles.50
Figure 3.
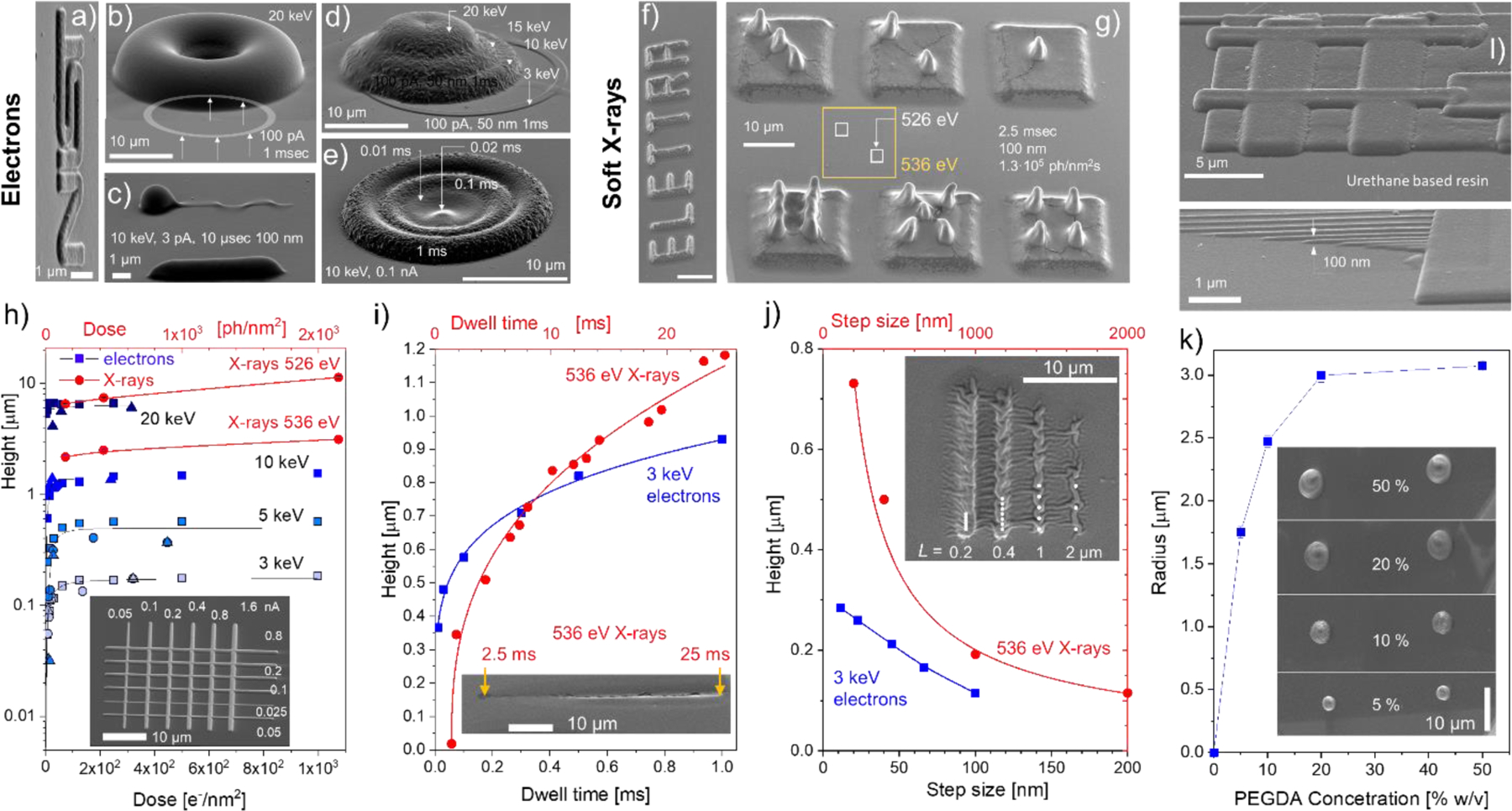
SEM images of 3D features example patterned using focused electron beam a) – e) and soft X-rays f), g). The numbers inside indicate energy, beam currents (photons intensities), dwell times and step sizes used. a) NIST logo is written with 3 keV electrons (100 pA, 1ms dwell time) using 10 nm pixel size. b) Donut-like gel feature made by writing 1 μm wide ring with a radius of 10 μm. c) Single line flagella-like structure, where the head blob is due to beam parking (elevated dwell time). d) Dome structures formed out of four overlapping coaxial rings by varying electron beam energy and dose e) for every ring. f) ELETTRA logo printed with 536 eV 150 nm wide X-ray beam with 25 ms dwell time. Photon flux ca 2·107 ph/nm2 s and 100 nm step size. g) Dices printed with two photon energies: 13 µm base squares- 536 eV (yellow color coding) and small 2.5 µm squares 526 eV (white color coding), h)-j) SEM measured heights of the dry rectangular and linear gel features as a function of exposure dose h), dwell time i) and step size j) for different energies of electrons (blue) and X-rays (red) beams. The insets show SEM image of gel structure written with variable beam current h), dwell time along its length using 536 eV X-rays i), and step-size L using 536 eV X-rays and 3 keV electrons. k) Size dependence of the grown features on the PEGDA concentration (exposure parameters: 15 keV, 40 pA, 3 s dwell time). l) In-liquid e-beam induced crosslinking of urethane-based resin. Bottom panel demonstrates the writing of very thin (ca. 100 nm wide/tall) lines. Exposure conditions were: 3 keV, 2 pA, 30 s dwell time.
The effect of the beam intensity and writing sequence on the linear feature size and morphology can be seen in the inset of Figure 3h. As discussed below, the feature size rapidly increases with the beam intensity and then saturates. This is a result of the increase and saturation of the crosslink density inside the excitation volume similarly to the radiation induced crosslinking in a dry state.13 Feature size can be controlled effectively via printing with different X-ray photon energies just below and above the element specific absorption edge (e.g. O1s Figure 2b). The example in the Figure 3g shows that this can be performed even through an already printed feature.
Compared to printing with electrons, the aspect ratio of the X-ray-induced structures can be appreciably larger similar to the deep X-ray lithography results on solid films. In addition, the crosslink density of X-ray printed hydrogel structures was noticeably lower compared to their e-beam counterparts reflecting significant surface rippling of the features upon drying. Figures 3h–j quantifies the measured heights of the crosslinked dried gel features as a function of each one of these parameters with others being unchanged. In addition to the apparent increase of the feature size with electron beam energy (Figure 3h), the height variation with exposure dose has a characteristic fast rise followed by saturation behavior, which was also commonly observed in dry films. In accordance with prior reports, the critical dose threshold (on the order of 1 e−/nm2)13 in our case is required for through-membrane crosslinking of a stable gel structure. While the dose increase does not affect the dimensions of the interaction volume in water, it expands the boundary at which the critical concentration of crosslinking radicals can be maintained. The same is valid for the feature heights increase with the dwell-time (Figure 3i).
Step-size becomes a rather important parameter when the beam is rastered across the sample surface. By increasing the step-size and therefore the pixel area, one can tune the overlap between the interaction volumes (diffusion zones) of individual adjacent voxels, thus change the effective thickness (and width) of the printed feature. An important distinction between the electron and soft X-ray beam writing used in this study is the size of the probe: electron beam had a diameter of ca. 5 nm, while for X-rays it was ca. 150 nm in our setups. On the other hand, the effective diameter of the interaction volume of a few keV electron beam is appreciably larger (see Figure 2a) compared to the one for soft X-rays. Therefore, for X-rays, the formation of corrugated/ discontinuous patterns can be observed as soon as the step-size becomes larger than 150 nm (see inset in Figure 3j). On the contrary, few keV electron beams generate continuous patterns, for step-size values even larger than 100 nm.
Feature size and crosslink density of hydrogels prepared by focused radiation can effectively be controlled by variation of the monomer concertation in the solution. Figure 3k shows size dependence of e-beam written features on the PEGDA 700 concentration in water. As expected for a diffusion-controlled cross-linking mechanism, the feature sizes increase with concertation. In diluted solutions the crosslink reaction rate (and written feature size) increases linearly with the precursor concentration (see Eq. 8 in Ref.41). As concentration increases, the reaction rate saturates, being limited by the more complex processes of radiation damage (scission) of precursor and polymer molecules, hampered diffusion, etc. At very high concentrations (not used here), a drop in the reaction rate is expected due to an increase in solution viscosity and solubility limit.41
Finally, in-liquid crosslinking can be generalized to other materials. Figure 3l displays SEM images of e-beam written features of crosslinked urethane-based resin as an example of a different chemical compound that can be used for the proposed method, confirming its wide applicability. With this material, lines as thin as 100 nm wide have been drawn.
To summarize: for printing applications, dwell-time and step-size are useful independent irradiation parameters to control the size and crosslink density of raster printed gel structures. Unlike the beam energy and intensity, these two parameters are decoupled in the SEM controls and can be tuned during the scanning without refocusing. In the case of the e-beam printing, the lateral and longitudinal resolution of the resultant features are coupled and are proportional to each other (Figure 2a). As one intensifies any of the parameters: beam energy, intensity, dwell time, both the width and height of the pattern increase. Finally, the smallest feature size and maximum resolution for SEM based gelation can be achieved via lowering the energy of electrons that can penetrate through the SiN window. We were able to routinely write ca. 150 nm thin and 100 nm wide gel lines through a 50 nm SiN window using 3 keV electron beam energy (see Fig. S1 of SI). Sub-100 nm features are attainable if thinner membranes are used.
We now discuss the particularities of in-liquid direct writing that can led to the layer-by-layer printing with electron beams. Let us consider sequentially crosslinked features 1 and 2 (Figure 4a) which overlap and thus have double-exposed region (DER) as a “second layer”. One can expect (as in the case of the solid resist grayscale lithography51) that the DER should have an increased thickness compared to the parent’s structures. Since the feature size depends on the accumulated dose (see Figure 3h), this height difference should vary from its maximum value at very low doses to zero at doses above ca. 102 e−/nm2. Moreover, the size and shape of the resultant structure (including DER) should not depend on the writing sequence (i.e. shape12= shape21). To verify this assumption for in-liquid crosslinking, the layer-by-layer test log-pile structures (Figure 4b) have been printed in PEGDA solution and studied as a function of writing sequence, electron energy, and dose.
Figure 4.
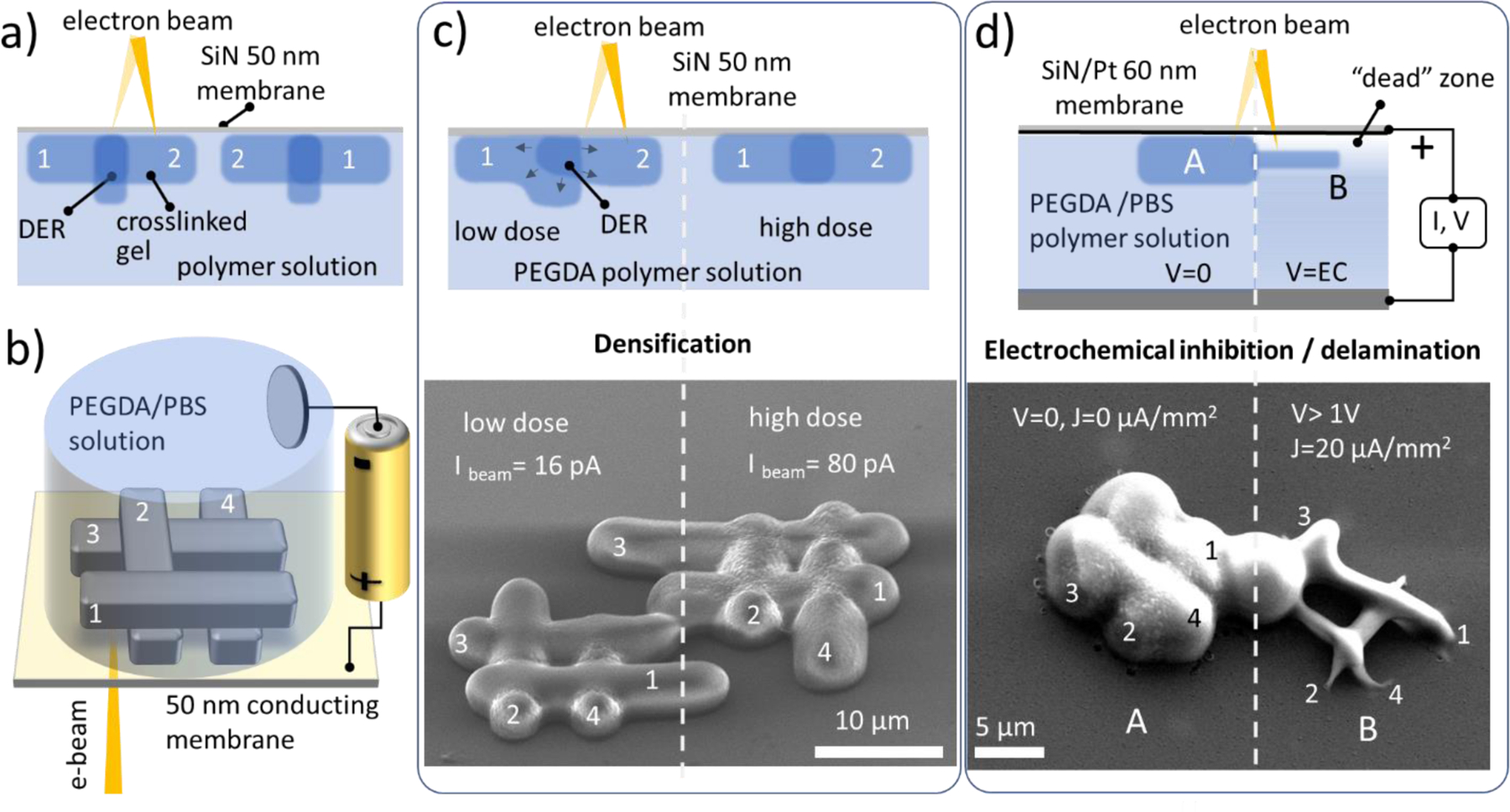
Layer-by-layer beam writing tests of overlapping structures in liquid PEGDA polymer solutions. a) An expected sequential writing of the overlapping structures in liquid solutions. b) Model log-pile structure printed with and without anodic potential on the SiN /Pt electrode; c) The morphology of the e-beam printed PEGDA structure depends on the writing sequence 1–2-3–4 for low dose exposure and is independent for saturated crosslinking density; d) Electrochemical size and adhesion control of the in-liquid crosslinked structures: panel A no electrochemical bias (V) is applied. Panel B, with applied cathodic potential V and current density of ca. 20 µA/mm2. Drastic reduction of the feature size and its footprint can be observed.
SEM images in Figure 4c show log-pile structures patterned side-by-side with low (left) and high (right side) doses. As expected, due to complete crosslinking along the length of the structures, the high-dose structure does not show any noticeable increase of the thickness of the nods compared to the thickness of the regular bar. The morphology of the structure printed in liquid with a low dose, however, exhibits a noticeable dependence on the writing sequence and does not follow the expected commutative exposure rule characteristic of solid resists (e.g. shape23≠shape43). We assume that in-liquid electron beam writing proceeds in an essentially different way compared to solid resist and the mechanical stress induced by densification of the DER upon sequential writing leads to mechanical lifting and eventually delamination of the prior-printed construct (Figure 4c left panel).
Delamination of the printed layer from the optical windows has been a speed limiting factor of the first bottom-up 3D laser printers. This has been resolved via implementation of continuous liquid interface production technology which employed quenching of the polymer crosslinking at the liquid-window interface with a controlled through-the-window delivery of molecular oxygen inhibitor.52 Along these lines, electrochemically driven pH variations in PEG solutions have be used to reduce the crosslinking density of hydrogel to improve their cellular adhesion and permeability.53 Here we explore the role of electrochemically-generated inhibitors on the size and adhesion of the electron beam printed structures. For that, the supporting SiN membrane was coated with a few tens of nanometers thick Pt layer defined as the working electrode in a two-electrode electrochemical cell filled with PEGDA / PBS aqueous electrolyte. The anodic potential of the working electrode was adjusted to near the onset of the water splitting reaction (Figures 4b and S8 of SI). SEM images in Figure 4d depict two log-pile constructs printed using the same irradiation parameters but with (right panel) and without (left pane) anodic potential applied to the conducting membrane with respect to the counter electrode. The drastic reduction of the size and adhesion of the printed construct (right panel) can be observed, which we explain by the electrochemical increase of the near electrode concertation of the crosslinking inhibitors like O2 that hamper beam induced crosslinking process.54 Cathodic potential, on the other hand, leads to electrochemical polymerization of the PEGDA solution at the membrane electrode and overwhelms the beam-induced prints (see Figure S8 SI). The electrochemical quenching /promoting of the PEGDA crosslinking not only provides an additional control of the e-beam printed feature size but has a direct consequence for the additive fabrication of overlying structures in a layer-by-layer manner.
Application and Device Examples
Composite hydrogels as plasmonic microsensors
The range of hydrogels applications became significantly broadened via synthesis of composite formulations, which allows for the rational engineering of their optical, electrical, mechanical and magnetic properties for numerous applications. Fabrication of composite organic hydrogels can be broadly classified into two methods: (i) in situ ones where functional inclusions (e.g. nanoparticles) or precursors are premixed in the prepolymer solution and become stabilized in the hydrogel during the crosslinking process, whereas (ii) ex situ techniques typically involve an inclusion impregnation process, applied after the crosslinking of the host matrix. In situ encapsulation offers the advantages of embedding objects independently of their size, homogeneously across the bulk, while the post-crosslinking impregnation depends on surface-to-bulk diffusion of chemicals into the gel’s matrix which is often hampered for the objects larger than gel’s mesh size. Figure 5a (panels 1and 2) shows the principle of in-liquid entrapping of nanoparticles and via focused e-beam induced cross-linking of nanoparticles suspension. Due to facile coupling of the mechanical optical, chemical and electrical properties of such composite gels to external stimuli they become a common platform for numerous sensor designs.55 Figure 5b depicts a setup of a plasmonic humidity microsensor e-beam printed out of PEGDA/50 nm Au nanoparticles suspension. A broadband light source excites the localized surface plasmon resonances (LSPR) in the selected gel-imbedded nanoparticle assembly (see inset in the Figure 5c). The scattered light is collected in the reflection dark field mode and the shift of the LSPR maximum λm is determined spectroscopically as a function of the humidity level above the sensor (see details in Fig. S7). The observed blue shift of the λm with increase of the humidity level (Figure 5c) is presumably due to a combination of two factors: reduction of the gel refractive index with humidity56 and weakening of the dipole-dipole coupling between the adjacent nanoparticles in the aggregate (which scales as (D/d)3) upon gel matrix swelling, here D is Au NP diameter and d is the interparticle distance.57 The size of the e-beam gelated composite in this particular study was ca. 100 µm ×100 µm × 5 µm, however the plasmonic hot spots were located inside Au NP assembly that is only ca. 500 nm wide (see insets in Figure 5c).
Figure 5.
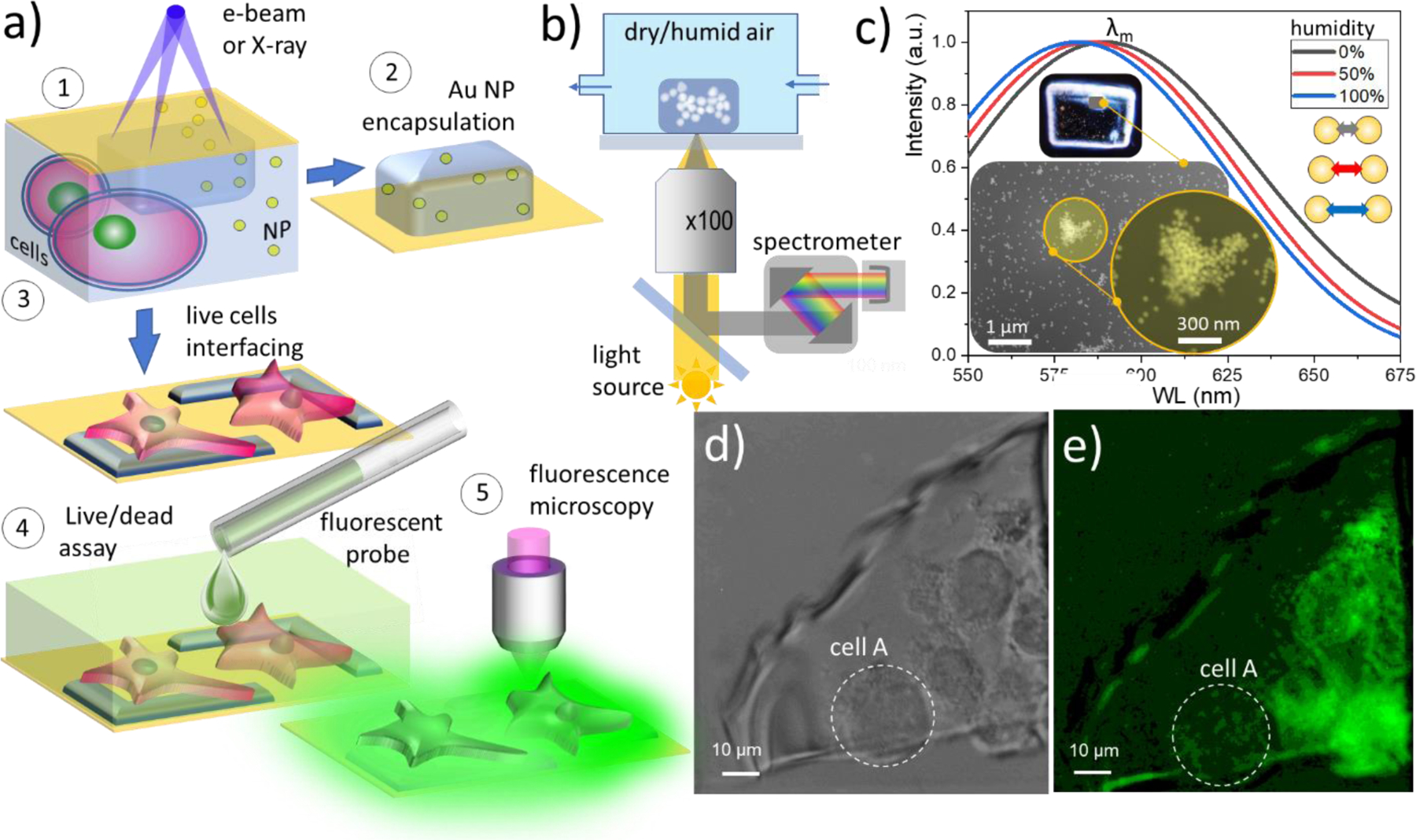
Potential applications of the e-beam and X-ray in liquid patterned hydrogel structures. a) The principle of e-beam induced encapsulation and interconnecting of micro- and nanoobjects using in liquid cured gel. (1) Irradiation and in-liquid patterning of the nanoparticles (or cells) hydrogel precursor suspension through the SiN membrane, followed with (2) or (3) microscopic and spectroscopic characterization in a dry or wet stage. (4) Hydrogel interfaced cells treated with a fluorescent probe for viability tests. (5) Fluorescence microscopy of biological objects interfaced with a gel. b) Setup for excitation and recording of the LSPR signal from e-beam printed Au NP/PEGDA composite hydrogel plasmonic sensor as a function of ambient humidity. c) Normalized reflectance signal collected from PEGDA incapsulated 50 nm Au assembly as a function of the humidity. Insets show dark-field optical image of e-printed composite hydrogel pack and SEM images of Au NP assembly where signal was collected from. d) Optical microscopy image of the hydrogel after electron beam irradiation of cell-laden PEGDA solution. e) Fluorescence microscopy image of gel interfaced cells stained with viability indicator (calcein green dye). Dashed line depicts a necrotic cell.
Cellular interfacing with hydrogel patches
The fields, such as computer-brain interfacing with neural probes or cardiac cellular electrophysiology, rely on development of soft microelectrode arrays that match mechanical chemical and electrical properties of the individual live cells. PEG-based hydrogels are prospective soft interface electrode materials due to their biocompatibility,58 adjustable elastic moduli and ionic conductance (see recent review59 and references therein). Sub-micron indexing of life cells with PEGDA microelectrodes can in principle be done using aforementioned multiphoton laser printing.60 However, to compensate for a small polymerization cross-section and yet preserve sufficient reaction rate inside the mesoscopic voxel, high concentrations of potentially cytotoxic photo-initiators are required which may compromise the biocompatibility of the electrodes.
Here we employ in-liquid gel focused electron (X-ray) beam crosslinking technique for cellular interfacing. In addition to highest lateral resolution of the membrane patching and flexible electrodes patterning, the advantage such an approach lays in a nanometers depth control of the electron range and crosslinking density across the irradiated patch such as desired degree of electrical and gentle mechanical contact can be achieved at the very cellular membrane-gel electrode interface. The key question arises whether PEGDA contact formation can be administered on a live cell before its membrane irreversibly degrades. The extent of radiation damage of the biological objects (cells) during electron beam-induced PEGDA crosslinking is not well known and generated reactive radiolytic species like OH·, O−, and H2O2 at high concentrations are known to be detrimental to the cells. Prior electron microscopy studies of biological specimens in the wet environment reported a range of critical dose values spanning from ca. 10−3 e–/nm2 to ca. 102 e–/nm2 that are considered acceptable for live mammalian cells, yeasts and other microorganisms. Such a wide margin manifests a fundamental challenge of high-resolution electron microscopy of live cells as well as the variance in live/dead criteria applied (see refs.61–65 and discussion therein). As we showed above, PEGDA crosslinking threshold dose is on the order of 1 e−/nm2 in our setup and can be reduced by at least an order of magnitude if higher molecular weight PEG is used.13 Since the imaging during crosslinking (which usually leads to high irradiation dose) is not a requirement, the encapsulation of live microorganisms using our in-liquid lithography can be feasible. The test process flow is depicted in Figure 5a, panels 1, 3, 4. Once a prepolymer solution with premixed and proximal to SiN membrane live cells was exposed to electrons, the crosslinked gel with trapped/interfaced cells was tested with standard calcein-AM cell viability assay.66 In this test, live cells uptake the non-fluorescent calcein-AM ester. Inside the living cell, ester reacts with cytosolic esterases, which convert it into a green-fluorochrome: calcein to which cellular membrane is not permeable. Bright-field optical image in Figure 5 d shows a SiN window containing cell-laden PEGDA solution after exposure to 10 keV primary beam with an average exposure dose of 8 e−/nm2 (absorbed dose ca 3 × 106 Gy). Fluorescent microscopy image of gel-immobilized cells in the Figure 5e complements Figure 5d and indicates (i) that cells B-E do produce fluorescent calcein after encapsulation; (ii) calcein distribution remains confined within the cellular borders indicating the integrity of cellular membrane and (iii) some of the cells (cell A in the Figure 5e) appear dark implying its necrosis, while the rest of the cells apparently survive the interfacing procedure. We speculate that although the local absorption dose at the point of beam incidence can be few orders of magnitude higher than the cell’s lethal dose limit, the successful interfacing of the life cells with PEGDA electrode is due to fact that the radiation damage is mainly localized within a submicron gap of solution between the cellular and SiN membranes (Figures 2a, c, d, and 5 a). Thus, the cells that are poorly adhered or floating in solution ca micron away from the SiN membrane see a significantly diluted load of radiolitic species. Moreover, the majority of the reactive species produced by the beam becomes scavenged during gel crosslinking process, which, therefore effectively reduce the concentration of toxic species seen by the cells. This is a promising result which, however, must be considered as preliminary observation and is a subject of ongoing studies.
CONCLUSIONS
We have demonstrated spatially controlled crosslinking inside liquid polymer solutions employing scanning electron and soft X-ray microscopy. The method enables fast (seconds) patterning of millimeters large complex constructs in liquids with the thickness variations and feature size ranging from ca. 100 nm to a few micrometers (ca. 100 nm to a few tens of micrometers in case of X-rays). Different from the lithography, the liquid environment, in principle, allows for layer-by-layer additive fabrication with the resolution and/or writing speed comparable (or even better) with the current state of the art optical and electron-based 3D printing methods. The possible applications of this technique were demonstrated through gel interfacing of live cells, fabrication of a composite hydrogel plasmonic device and 3D printing of model hydrogel structures with a sub-micrometer resolution. We estimated the threshold dose required for electron beam-induced PEGDA cross-linking in a liquid state to be on the order of 1 e−/nm2 and explored the effect of diffusion of radiolitic species and other experimentally tunable parameters such as electrons (or X-rays) energy, beams intensity, exposure time, electrochemically driven pH value and etc. on the resolution and size of the features formed. High spatial resolution printing of a large class of hydrogels precursor solutions in the liquid state can also be extended to gas phase polymerization and offers advantages in shape, size and precision compared to traditional dry gel lithography and can complement the existing multiphoton polymerization methods. The proposed technology can be implemented in any high vacuum, environmental SEMs, atmospheric SEMs, synchrotrons or laboratory-based X-ray microscopes. The tunability of X-ray energy at synchrotrons offers an additional opportunity to conduct element specific 3D gel printing in solutions relevant to biomedical, soft micro-robotics, electrochemical and other applications. Moreover, the combination of our method with the recently proposed implosive fabrication technique67 can, in principle, result in nanometer-scale 3D printing. In addition to the limitations common to any layer-by-layer printing, the remaining challenge is reliable delamination of the printed layer from the membrane, which is required for continuous printing. While the latter is a work in progress, the aforementioned challenge is not a principal one and finite and arbitrary shaped millimeter or even centimeter size objects can in principle be fabricated with submicron resolution via curing the open liquid surface directly using atmospheric SEM chamber-less setups.68
METHODS
Hydrogel Synthesis and Printing
Most of the tests have been performed with a stagnant liquid setup where ca. 5–10 µL of 20 % w/v PEGDA (average molar mass 0.7 kg/mol, no initiator) aqueous solution was drop-casted on to Si chip with an array of nine 50 nm thick SiN membranes. The chip was bonded to supporting plate and sealed with vacuum-tight (flow) chamber with silicone rubber gasket. Inside SEM, liquid prepolymer solution was patterned through the SiN window using focused electron beam with a known amount of dose at every pixel. The experiment was repeated in another part of the 100 µm × 100 µm window or at the different SiN window using a different set of irradiation parameters. After e-beam exposure, the chip was taken out of the chamber and gently rinsed in DI water to remove the uncured solution. This leaves an array of printed gel features adhered to SiN membranes. The latter were inspected in a hydrated state with AFM or optical microscopy/profilometry and/or in a dry state using SEM, AFM, EDS, XPS, µ-Raman and other characterization tools. Printing with soft X-rays was performed at ESCA microscopy beamline at ELETTRA equipped with zone plate optics capable to focus monochromatized light to a spot 150 nm in diameter. The undulator and monochromator have been set to operate either at 526 eV or at 536 eV with the photon flux in the order of 109 ph/s at 150 nm wide focal spot. The chamber equipped with the same chip with nine SiN membranes array was filled with PEGDA solution, sealed and scanned in front of the beam in a pre-programmed path to generate a required pattern. Electrochemical delamination tests have been made using PBS-based 20 % w/v PEDGA solution.
Composite Hydrogels
Gold nanoparticles (50 nm in dia.) suspension in water was pre-concentrated by centrifuging (2000 r/min, 5 min) and was subsequently extracted and mixed with a 20 % w/v PEGDA solution. The prepared composite mixture was irradiated in the liquid phase with an electron or soft X-ray beams through 50 nm SiN membrane. The chip with the printed composite gel structures was then developed in water and subsequently analyzed as described in the article and Supporting material.
Live Cells Interfacing and Proliferation Tests
Caco-2 cells were thawed and cultured in DMEM (Dulbecco’s Modification of Eagle’s medium) with 4.5 g/L glucose and L-glutamine without sodium pyruvate for a few days. These were subsequently washed in phosphate-buffered saline solution (PBS) and DMEM. Lifting off process was carried out using 2 ml of 0.05 % (w/v) Trypsin-EDTA and left for 5 min to10 min until the attached cells become mobile on the slide. Neutralization of Trypsin is done by adding an equal volume of growth medium. The obtained cell suspension is concentrated by centrifuge. The cell concentrate was added to the PBS-based 20 % w/v PEDGA solution and cells were allowed to adhere to the SiN membrane. For viability tests after irradiation, the crosslinked gel with encapsulated cells was rinsed in the growth medium for cell and exposed to calcein green dye in the growth medium for 1 hr. Inside the live cells, the non-fluorescent calcein is converted into green-fluorescent calcein via de-esterification of the acetoxymethyl group by the esterases only produced by a live cell.
Modeling Details
A stack of 50 nm SiN and 20-micron thick water layer was modeled with the electron beam incident on the SiN membrane. The Monte Carlo (MC) simulations (described in the SI section) generated the trajectories and corresponding energy deposited (Gy). The parameters used to generate Figure 2 a) were as follows: 6.25·105 electrons for a 5 nm beam diameter for 3 keV, 5 keV, 10 keV, and 20 keV primary beam energy. The energy deposition results in Gy for 6.25·105 electrons were scaled depending on the current value to obtain the rate of energy deposition (Gy/s) and fed into the radiolysis kinetics model (described in more details in the SI section). To generate results shown in Figure 2d), the CFD model was executed for 3 keV primary beam, and currents current values: 50 pA, 85 pA, 125 pA, 160 pA, 200 pA and 215 pA.
Supplementary Material
ACKNOWLEDGMENTS:
T.G. and E.S. acknowledge support under the Cooperative Research Agreement between the University of Maryland and the National Institute of Standards and Technology Center for Nanoscale Science and Technology, Award 70NANB14H209, through the University of Maryland. Authors are thankful to Dr. A. Vladar, Dr. D. DeLongchamp, Dr. P. Haney, Dr. J. Villarrubia (all at NIST), Prof. Dr. M. Zharnikov (Univ. of Heidelberg), Prof. G. Kolmakov (CUNY) for constructive feedback on the manuscript and to Dr. D. Perez (NIST), Dr. A. Agrawal (NIST) for the help with profilometry and photonic measurements.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI:. Details on the estimates on the structures’ height, X-ray and electron exposure doses, MC simulations, as well as details on the kinetic model, SEM imaging of composite gels, Raman analysis, humidity measurements, and electrochemical delamination are provided.
An earlier version of this manuscript can be found online as: T. Gupta; E. Strelcov; G. Holland; J. Schumacher; Y. Yang; M. Esch; V. Aksyuk; P. Zeller; M. Amati; L. Gregoratti; A. Focused Electron and X-ray Beam Crosslinking in Liquids for Nanoscale Hydrogels 3D Printing and Encapsulation. 2019, 1904.01652. arXiv. https://arxiv.org/abs/1904.01652 (accessed 08 18, 2020).
Publisher's Disclaimer: NIST Disclaimer: Certain commercial equipment, instruments, materials or products are identified in this paper to foster understanding. Such identification does not imply recommendation or endorsement by the National Institute of Standards and Technology, nor does it imply that the materials or equipment identified are necessarily the best available for the purpose.
Competing financial interests
The authors declare no competing financial interests.
REFERENCES
- 1.Revzin A; Russell RJ; Yadavalli VK; Koh W-G; Deister C; Hile DD; Mellott MB; Pishko MV, Fabrication of Poly (ethylene Glycol) Hydrogel Microstructures Using Photolithography. Langmuir 2001, 17, 5440–5447. [DOI] [PubMed] [Google Scholar]
- 2.Akselrod G; Timp W; Mirsaidov U; Zhao Q; Li C; Timp R; Timp K; Matsudaira P; Timp G, Laser-Guided Assembly of Heterotypic Three-Dimensional Living Cell Microarrays. Biophys. J 2006, 91, 3465–3473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Melchels FP; Feijen J; Grijpma DW, A Review on Stereolithography and Its Applications in Biomedical Engineering. Biomaterials 2010, 31, 6121–6130. [DOI] [PubMed] [Google Scholar]
- 4.Maruo S; Nakamura O; Kawata S, Three-Dimensional Microfabrication with Two-Photon-Absorbed Photopolymerization. Opt. Lett 1997, 22, 132–134. [DOI] [PubMed] [Google Scholar]
- 5.Li L; Gattass RR; Gershgoren E; Hwang H; Fourkas JT, Achieving Λ/20 Resolution by One-Color Initiation and Deactivation of Polymerization. Science 2009, 324, 910–913. [DOI] [PubMed] [Google Scholar]
- 6.Scott TF; Kowalski BA; Sullivan AC; Bowman CN; McLeod RR, Two-Color Single-Photon Photoinitiation and Photoinhibition for Subdiffraction Photolithography. Science 2009, 324, 913–917. [DOI] [PubMed] [Google Scholar]
- 7.Andrew TL; Tsai H-Y; Menon R, Confining Light to Deep Subwavelength Dimensions to Enable Optical Nanopatterning. Science 2009, 324, 917–921. [DOI] [PubMed] [Google Scholar]
- 8.Saha SK; Wang D; Nguyen VH; Chang Y; Oakdale JS; Chen S-C, Scalable Submicrometer Additive Manufacturing. Science 2019, 366, 105–109. [DOI] [PubMed] [Google Scholar]
- 9.Zharnikov M; Grunze M, Modification of Thiol-Derived Self-Assembling Monolayers by Electron and X-Ray Irradiation: Scientific and Lithographic Aspects. J. Vac. Sci. Technol., B: Microelectron. Nanometer Struct.-Process., Meas., Phenom 2002, 20, 1793–1807. [Google Scholar]
- 10.del Campo A; Arzt E, Fabrication Approaches for Generating Complex Micro-and Nanopatterns on Polymeric Surfaces. Chem. Rev 2008, 108, 911–945. [DOI] [PubMed] [Google Scholar]
- 11.Ballav N; Schilp S; Zharnikov M, Electron-Beam Chemical Lithography with Aliphatic Self-Assembled Monolayers. Angew. Chem., Int. Ed 2008, 47, 1421–1424. [DOI] [PubMed] [Google Scholar]
- 12.Nottbohm CT; Turchanin A; Beyer A; Golzhauser A, Direct E-Beam Writing of 1 nm Thin Carbon Nanoribbons. J. Vac. Sci. Technol., B: Microelectron. Nanometer Struct.-Process., Meas., Phenom 2009, 27, 3059–3062. [Google Scholar]
- 13.Krsko P; Sukhishvili S; Mansfield M; Clancy R; Libera M, Electron-Beam Surface-Patterned Poly (ethylene Glycol) Microhydrogels. Langmuir 2003, 19, 5618–5625. [Google Scholar]
- 14.Tirumala VR; Divan R; Ocola LE; Mancini DC, Direct-Write E-Beam Patterning of Stimuli-Responsive Hydrogel Nanostructures. J. Vac. Sci. Technol., B: Microelectron. Nanometer Struct.-Process., Meas., Phenom 2005, 23, 3124–3128. [Google Scholar]
- 15.Kolodziej CM; Maynard HD, Electron-Beam Lithography for Patterning Biomolecules at the Micron and Nanometer Scale. Chem. Mat 2012, 24, 774–780. [Google Scholar]
- 16.Meyerbröker N; Zharnikov M, Modification and Patterning of Nanometer-Thin Poly (ethylene Glycol) Films by Electron Irradiation. ACS Appl. Mater. Interfaces 2013, 5, 5129–5138. [DOI] [PubMed] [Google Scholar]
- 17.Winkler R; Fowlkes JD; Rack PD; Plank H, 3D Nanoprinting via Focused Electron Beams. J. Appl. Phys 2019, 125, 210901. [Google Scholar]
- 18.Fisher JS; Kottke PA; Kim S; Fedorov AG, Rapid Electron Beam Writing of Topologically Complex 3D Nanostructures Using Liquid Phase Precursor. Nano Lett 2015, 15, 8385–8391. [DOI] [PubMed] [Google Scholar]
- 19.Thiberge S; Zik O; Moses E, An Apparatus for Imaging Liquids, Cells, and Other Wet Samples in the Scanning Electron Microscopy. Rev. Sci. Instr 2004, 75, 2280–2289. [Google Scholar]
- 20.Donev EU; Hastings JT, Electron-Beam-Induced Deposition of Platinum from a Liquid Precursor. Nano Lett 2009, 9, 2715–2718. [DOI] [PubMed] [Google Scholar]
- 21.Kolmakova N; Kolmakov A, Scanning Electron Microscopy for In Situ Monitoring of Semiconductor−Liquid Interfacial Processes: Electron Assisted Reduction of Ag Ions from Aqueous Solution on the Surface of TiO2 Rutile Nanowire. J. Phys. Chem. C 2010, 114, 17233–17237. [Google Scholar]
- 22.Suga M; Nishiyama H; Konyuba Y; Iwamatsu S; Watanabe Y; Yoshiura C; Ueda T; Sato C, The Atmospheric Scanning Electron Microscope with Open Sample Space Observes Dynamic Phenomena in Liquid or Gas. Ultramicroscopy 2011, 111, 1650–1658. [DOI] [PubMed] [Google Scholar]
- 23.Jensen E; Købler C; Jensen PS; Mølhave K, In Situ SEM Microchip Setup for Electrochemical Experiments with Water Based Solutions. Ultramicroscopy 2013, 129, 63–69. [DOI] [PubMed] [Google Scholar]
- 24.Kolmakov A, Membrane Based Environmental Cells for SEM in Liquids. In Liquid Cell Electron Microscopy, Ross FM, Ed. Cambridge University Press: Cambridge, 2016; pp 78–105. [Google Scholar]
- 25.Donev EU; Bresin M; Hastings JT, Nanoscale Deposition and Etching of Materials Using Focused Electron Beams and Liquid Reactants. In Liquid Cell Electron Microscopy, Ross FM, Ed. Cambridge University Press: 2016; p 291. [Google Scholar]
- 26.Gupta T; Kolmakov A, 3-Dimensional Hydrogel Printing via Electron Crosslinking. Microsc. Microanal 2018, 24, 348–349. [Google Scholar]
- 27.Ehrfeld W; Lehr H, Deep X-Ray-Lithography for the Production of 3-Dimensional Microstructures from Metals, Polymers and Ceramics. Radiat. Phys. Chem 1995, 45, 349–365. [Google Scholar]
- 28.Malek CK; Saile V, Applications of Liga Technology to Precision Manufacturing of High-Aspect-Ratio Micro-Components and -Systems: A Review. Microelectron. J 2004, 35, 131–143. [Google Scholar]
- 29.Leontowich AFG; Hitchcock AP; Watts B; Raabe J, Sub-25-nm Direct Write (Maskless) X-Ray Nanolithography. Microelectron. Eng 2013, 108, 5–7. [Google Scholar]
- 30.Wang J; Stover HDH; Hitchcock AP, Chemically Selective Soft X-Ray Direct-Write Patterning of Multilayer Polymer Films. J. Phys. Chem. C 2007, 111, 16330–16338. [Google Scholar]
- 31.Sayre D; Kirz J; Feder R; Kim D; Spiller E, Potential Operating Region for Ultrasoft X-Ray Microscopy of Biological Materials. Science 1977, 196, 1339–1340. [DOI] [PubMed] [Google Scholar]
- 32.Jacobsen C, Soft X-Ray Microscopy. Trends Cell Biol 1999, 9, 44–47. [DOI] [PubMed] [Google Scholar]
- 33.Fujii S; Armes SP; Araki T; Ade H, Direct Imaging and Spectroscopic Characterization of Stimulus-Responsive Microgels. JACS 2005, 127, 16808–16809. [DOI] [PubMed] [Google Scholar]
- 34.Tseng S-J; Chien C-C; Liao Z-X; Chen H-H; Kang Y-D; Wang C-L; Hwu Y; Margaritondo G, Controlled Hydrogel Photopolymerization inside Live Systems by X-Ray Irradiation. Soft Matter 2012, 8, 1420–1427. [Google Scholar]
- 35.Wang C-H; Liu C-J; Wang C-L; Hua T-E; Obliosca JM; Lee K; Hwu Y; Yang C-S; Liu R-S; Lin H-M, Optimizing the Size and Surface Properties of Polyethylene Glycol (PEG)–Gold Nanoparticles by Intense X-Ray Irradiation. J. Phys. D: Appl. Phys 2008, 41, 195301. [Google Scholar]
- 36.Tirumala V; Divan R; Mancini D; Caneba G, Fabrication of High-Aspect-Ratio Hydrogel Microstructures. Microsyst. Technol 2005, 11, 347–352. [Google Scholar]
- 37.Späth A; Tu F; Vollnhals F; Drost M; Calderón SK; Watts B; Fink RH; Marbach H, Additive Fabrication of Nanostructures with Focused Soft X-Rays. RSC Adv 2016, 6, 98344–98349. [Google Scholar]
- 38.Ma Q; Moldovan N; Mancini DC; Rosenberg RA, Synchrotron-Radiation-Induced, Selective-Area Deposition of Gold on Polyimide from Solution. APL 2000, 76, 2014–2016. [Google Scholar]
- 39.Hoffman AS, Hydrogels for Biomedical Applications. Adv. Drug Deliv. Rev 2012, 64, 18–23. [DOI] [PubMed] [Google Scholar]
- 40.Alexander P; Charlesby A, Effect of X‐Rays and Γ‐Rays on Synthetic Polymers in Aqueous Solution. J. Pol. Sci 1957, 23, 355–375. [Google Scholar]
- 41.Dispenza C; Alessi S; Spadaro G, Radiation Processing of Polymers in Aqueous Media. In Applications of Ionizing Radiation in Materials Processing, Sun Y; Chmielewski A G., Eds. Institute of Nuclear Chemistry and Technology Warszawa, 2017; pp 291–326. [Google Scholar]
- 42.Tan G; Wang Y; Li J; Zhang S, Synthesis and Characterization of Injectable Photocrosslinking Poly (ethylene Glycol) Diacrylate Based Hydrogels. Polym. Bull 2008, 61, 91–98. [Google Scholar]
- 43.Rosiak J; Ulański P, Synthesis of Hydrogels by Irradiation of Polymers in Aqueous Solution. Radiat. Phys. Chem 1999, 55, 139–151. [Google Scholar]
- 44.Cazaux J, A Physical Approach to the Radiation Damage Mechanisms Induced by X-Rays in X-Ray Microscopy and Related Techniques. J. Microsc 1997, 188, 106–124. [Google Scholar]
- 45.Beetz T; Jacobsen CJ, Soft X-Ray Radiation-Damage Studies in PMMA Using a Cryo-STXM. J. Sync. Radiat 2003, 10, 280–283. [DOI] [PubMed] [Google Scholar]
- 46.Slavicek P; Kryzhevoi NV; Aziz EF; Winter B, Relaxation Processes in Aqueous Systems upon X-Ray Ionization: Entanglement of Electronic and Nuclear Dynamics. J. Phys. Chem. Lett 2016, 7, 234–243. [DOI] [PubMed] [Google Scholar]
- 47.Royall C; Thiel B; Donald A, Radiation Damage of Water in Environmental Scanning Electron Microscopy. J. Microsc 2001, 204, 185–195. [DOI] [PubMed] [Google Scholar]
- 48.Schneider NM; Norton MM; Mendel BJ; Grogan JM; Ross FM; Bau HH, Electron–Water Interactions and Implications for Liquid Cell Electron Microscopy. J. Phys. Chem. C 2014, 118, 22373–22383. [Google Scholar]
- 49.Gupta T; Schneider NM; Park JH; Steingart D; Ross FM, Spatially Dependent Dose Rate in Liquid Cell Transmission Electron Microscopy. Nanoscale 2018, 10, 7702–7710. [DOI] [PubMed] [Google Scholar]
- 50.Dreyfus R; Baudry J; Roper ML; Fermigier M; Stone HA; Bibette J, Microscopic Artificial Swimmers. Nature 2005, 437, 862. [DOI] [PubMed] [Google Scholar]
- 51.Kim J; Joy D; Lee S-Y, Controlling Resist Thickness and Etch Depth for Fabrication of 3D Structures in Electron-Beam Grayscale Lithography. Microelectron. Eng 2007, 84, 2859–2864. [Google Scholar]
- 52.Tumbleston JR; Shirvanyants D; Ermoshkin N; Janusziewicz R; Johnson AR; Kelly D; Chen K; Pinschmidt R; Rolland JP; Ermoshkin A, Continuous Liquid Interface Production of 3D Objects. Science 2015, 347, 1349–1352. [DOI] [PubMed] [Google Scholar]
- 53.Simona BR; Hirt L; Demkó L; Zambelli T; Vörös J; Ehrbar M; Milleret V, Density Gradients at Hydrogel Interfaces for Enhanced Cell Penetration. Biomater. Sci 2015, 3, 586–591. [DOI] [PubMed] [Google Scholar]
- 54.Decker C; Jenkins AD, Kinetic Approach of Oxygen Inhibition in Ultraviolet-and Laser-Induced Polymerizations. Macromol 1985, 18, 1241–1244. [Google Scholar]
- 55.Buenger D; Topuz F; Groll J, Hydrogels in Sensing Applications. Prog. Polym. Sci 2012, 37, 1678–1719. [Google Scholar]
- 56.Bilen B; Skarlatos Y; Aktas G; Inci MN; Dispinar T; Kose MM; Sanyal A, In Situ Measurement of Humidity Induced Changes in the Refractive Index and Thickness of Polyethylene Glycol Thin Films. J. Appl. Phys 2007, 102, 073534. [Google Scholar]
- 57.Muri H; Hjelme D, Lspr Coupling and Distribution of Interparticle Distances between Nanoparticles in Hydrogel on Optical Fiber End Face. Sensors 2017, 17, 2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Harris JM, Poly (ethylene Glycol) Chemistry: Biotechnical and Biomedical Applications Springer Science & Business Media: New York, 2013. [Google Scholar]
- 59.Yuk H; Lu B; Zhao X, Hydrogel Bioelectronics. Chem. Soc. Rev 2019, 48, 1642–1667. [DOI] [PubMed] [Google Scholar]
- 60.Ovsianikov A; Malinauskas M; Schlie S; Chichkov B; Gittard S; Narayan R; Löbler M; Sternberg K; Schmitz K-P; Haverich A, Three-Dimensional Laser Micro-and Nano-Structuring of Acrylated Poly (ethylene Glycol) Materials and Evaluation of Their Cytoxicity for Tissue Engineering Applications. Acta Biomater 2011, 7, 967–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Thiberge S; Nechushtan A; Sprinzak D; Gileadi O; Behar V; Zik O; Chowers Y; Michaeli S; Schlessinger J; Moses E, Scanning Electron Microscopy of Cells and Tissues under Fully Hydrated Conditions. PNAS 2004, 101, 3346–3351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Maruyama Y; Ebihara T; Nishiyama H; Suga M; Sato C, Immuno Em–Om Correlative Microscopy in Solution by Atmospheric Scanning Electron Microscopy (Asem). J. Struc. Bio 2012, 180, 259–270. [DOI] [PubMed] [Google Scholar]
- 63.de Jonge N; Peckys DB, Live Cell Electron Microscopy Is Probably Impossible. ACS Nano 2016, 10, 9061–9063. [DOI] [PubMed] [Google Scholar]
- 64.Kennedy E; Nelson EM; Tanaka T; Damiano J; Timp G, Live Bacterial Physiology Visualized with 5 nm Resolution Using Scanning Transmission Electron Microscopy. ACS Nano 2016, 10, 2669–2677. [DOI] [PubMed] [Google Scholar]
- 65.Kennedy E; Nelson EM; Damiano J; Timp G, Gene Expression in Electron-Beam-Irradiated Bacteria in Reply to “Live Cell Electron Microscopy Is Probably Impossible”. ACS Nano 2017, 11, 3–7. [DOI] [PubMed] [Google Scholar]
- 66.Wang XM; Terasaki PI; Rankin GW Jr; Chia D; Zhong HP; Hardy S, A New Microcellular Cytotoxicity Test Based on Calcein Am Release. Hum. Immunol 1993, 37, 264–270. [DOI] [PubMed] [Google Scholar]
- 67.Oran D; Rodriques SG; Gao R; Asano S; Skylar-Scott MA; Chen F; Tillberg PW; Marblestone AH; Boyden ES, 3D Nanofabrication by Volumetric Deposition and Controlled Shrinkage of Patterned Scaffolds. Science 2018, 362, 1281–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nguyen K; Holtz M; Muller D, Airsem: Electron Microscopy in Air, without a Specimen Chamber. Microsc. Microanal 2013, 19, 428–429. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.