

Abstract
Pneumonia represents a major health care burden and Gram‐negative bacteria provide an increasing therapeutic challenge at least in part through the emergence of multidrug‐resistant strains. IL‐33 is a multifunctional cytokine belonging to the IL‐1 family that can affect many different cell types. We sought here to determine the effect of recombinant IL‐33 on the host response during murine pneumonia caused by the common Gram‐negative pathogen Klebsiella pneumoniae. IL‐33 pretreatment prolonged survival for more than 1 day during lethal airway infection and decreased bacterial loads at the primary site of infection and distant organs. Postponed treatment with IL‐33 (3 h) also reduced bacterial growth and dissemination. IL‐33‐mediated protection was not observed in mice deficient for the IL‐33 receptor component IL‐1 receptor‐like 1. IL‐33 induced a brisk type 2 response, characterized by recruitment of type 2 innate lymphoid cells to the lungs and enhanced release of IL‐5 and IL‐13. However, neither absence of innate lymphoid cells or IL‐13, nor blocking of IL‐5 impacted on IL‐33 effects in mice infected with Klebsiella. Likewise, IL‐33 remained effective in reducing bacterial loads in mice lacking B, T, and natural killer T cells. Experiments using antibody‐mediated cell depletion indicated that neutrophils and inflammatory monocytes were of importance for antibacterial defense. The capacity of IL‐33 to restrict bacterial growth in the lungs was strongly reduced in mice depleted of both neutrophils and inflammatory monocytes, but not in mice selectively depleted of either one of these cell types. These results suggest that IL‐33 boosts host defense during bacterial pneumonia by a combined effect on neutrophils and inflammatory monocytes. © 2020 The Authors. The Journal of Pathology published by John Wiley & Sons, Ltd. on behalf of The Pathological Society of Great Britain and Ireland.
Keywords: IL‐33, pneumonia, sepsis, Klebsiella, innate immunity, neutrophils, inflammatory monocytes
Introduction
Sepsis is the consequence of a dysregulated host response to an infection [1]. Sepsis represents a high socio‐economic and health care burden world‐wide [2], and reported case fatality rates remain above 20% even in high income countries [3]. Lower respiratory tract infection is the most common source of sepsis [4] and the world's leading infectious killer, responsible for an estimated 3 million deaths annually (World Health Organization 2016) [5, 6]. Klebsiella pneumoniae is an important Gram‐negative causative agent in pneumonia and sepsis, which recently has received much attention due to the emergence of multidrug‐resistant strains [7].
Interleukin (IL)‐33 is a pleiotropic mediator belonging to the IL‐1 family of cytokines [8, 9]. IL‐33 is expressed in the nuclei of many different cell types, where it likely functions as an inhibitor of pro‐inflammatory signaling through binding of nuclear factor (NF)‐κB subunit p65 and inhibiting expression of NF‐κB target genes. Consistent with a function as an alarmin, IL‐33 can be released from cells after injury or death by necrosis, and extracellular IL‐33 can activate MyD88‐dependent signaling by triggering the IL‐1 receptor‐like 1 (IL1RL1)/IL‐1 receptor‐associated protein (IL‐1RAcP) receptor complex. IL‐33‐responsive target cells include macrophages, monocytes, neutrophils, mast cells, eosinophils, T helper type 2 (Th2) cells, natural killer T (NKT) cells, natural killer (NK) cells, and innate lymphoid cells (ILCs) [8, 9].
While extracellular IL‐33 in particular has been implicated in the induction and effector phase of type 2 immune responses, such as during helminth infections, allergy, and asthma, more recent research indicates that administration of IL‐33 exerts protective effects in experimental sepsis [10]. Most investigations on the effects of IL‐33 made use of the cecal ligation and puncture (CLP) model, inducing acute polymicrobial abdominal sepsis, in which IL‐33 reduced mortality and improved bacterial clearance by mechanisms that involved neutrophils and γδ T and NK cells, but not type 2 cytokines [11, 12, 13, 14]. In contrast, in a systemic infection model induced by a lethal intravenous dose of Staphylococcus aureus, IL‐33 administration protected against lethality via type 2 ILC (ILC2)‐dependent type 2 cytokine (IL‐5 and IL‐13) production [15]. Reports on the effects of IL‐33 in pneumonia are more limited and variable. In pneumonia caused by Legionella pneumophila, IL‐33 increased (rather than decreased) bacterial loads and lethality through induction of type 2 cytokines [16]; in post‐influenza S. aureus pneumonia, IL‐33 diminished bacterial loads and mortality by an effect that did not require ILC2s [17]. In both pneumonia models, high bacterial doses were used, which – while causing lethality due to excessive inflammation – were cleared from the lungs [16, 17].
Here we studied the effect of recombinant IL‐33 in an established model of airway infection by K. pneumoniae [18, 19, 20], which is associated with a steadily evolving infection of the lungs, that subsequently disseminates to distant organs causing sepsis, allowing us to study the host response in the context of early protective innate immunity as well as the subsequent harmful consequences of aberrant immune reactions. We demonstrate that IL‐33 treatment enhances host defense during Klebsiella pneumonia via a mechanism that relies on IL1RL1 and in the lungs is dependent on the presence of neutrophils and inflammatory monocytes (IMs), while type 2 cytokines, B, T, NK cells or ILCs are not crucially involved herein.
Materials and methods
Mice
BALB/c and C57BL/6 mice were purchased form Charles River (Maastricht, The Netherlands). Il1rl1 −/− BALB/c mice and Il13 −/− C57BL/6 mice were kindly provided by Dr Andrew NJ McKenzie (MRC Laboratory of Molecular Biology, Cambridge, UK). Rag2 −/− BALB/c mice were kindly provided by Dr Karin de Visser (Dutch National Cancer Institute, Amsterdam, The Netherlands). Rag2 −/−/Il2rγc −/− BALB/c mice were kindly provided by Dr Kees Weijer (Amsterdam‐UMC, Amsterdam, The Netherlands). All animals were bred and maintained under specific pathogen‐free conditions at the local animal facility according to local legislation. All experiments were carried out with 8‐ to 10‐week‐old sex‐matched mice. The Institutional Animal Care and Use Committee of the Academic Medical Center approved all experiments.
Mouse model of Klebsiella pneumonia
The pneumonia model was induced as previously described [18, 19, 20]. In short, a virulent strain of K. pneumoniae serotype 2 (43816; ATCC, Rockville, MD, USA) was grown in TSB medium to log phase. Cell suspensions were washed and diluted in isotonic saline. Mice were anesthetized by inhaling isoflurane carried in oxygen and thereafter 50 μl of a suspension containing 1 × 104 colony‐forming units (CFU) of K. pneumoniae was inoculated intranasally. In neutrophil (anti‐Ly6G) and neutrophil and monocyte (anti‐Gr1)‐depleting experiments, mice were infected with a lower dose (~0.5 × 104 CFU) considering the higher susceptibility of the mice in this model.
Treatment of mice
Mice received 1 μg of recombinant murine IL‐33 (BioLegend, San Diego, CA, USA) or vehicle intravenously 24 and 1 h prior to the start of infection. In some experiments, IL‐33 was given as a single intravenous injection 3 or 24 h post‐inoculation. For antibody mediated‐depletion, the following antibodies were used: anti‐Gr1 antibody (clone RB6‐8C5; BioLegend) or its isotype control antibody anti‐phytochrome (clone AFRC Mac 5.1), and these were administered intraperitoneally (100 μg) 48 h prior to infection. Anti‐Ly6G antibody (clone 1A8; Bio X Cell, West Lebanon, NH, USA) was given intraperitoneally (250 μg) 48 and 1 h prior to infection. Anti‐CCR2 antibody (clone MC‐21) [21] or its isotype control (clone MC‐67; rat IgG2b kappa) were administered intraperitoneally (20 μg) starting 48 h prior to infection and repeated every 24 h until the end of the experiment. Anti‐IL‐5 antibody (clone TRFK5; Bioceros, Utrecht, The Netherlands) or isotype control (clone GL113; Bioceros) were administered intraperitoneally (500 μg) 24 and 1 h prior to infection. Anti‐CD90 (clone 30H12; Bio X Cell) or isotype control (clone LTF2; rat IgG2b; Bio X Cell) were injected twice intraperitoneally (250 μg) in Rag2 −/− at days −4 and −1, prior to bacterial inoculation.
Bacterial burden determination
At the indicated time points, lungs, spleen, and liver were harvested and placed in sterile tubes. Blood was withdrawn by cardiopuncture and collected in heparin tubes. Lungs, spleen, and liver were homogenized and dilutions of the lysates and blood were plated on agar‐blood plates for determination of CFUs as previously described [18, 19, 20].
Analysis of cytokine production
IL‐5 and IL‐13 concentrations in lung homogenate were determined by ELISA according to the manufacturer's instructions (R&D Systems, Minneapolis, MN, USA).
Flow cytometry
Lung tissues were diced and subsequently digested for 45 min at 37 °C with 5 mg/ml LiberaseTM (Sigma, St Louis, MO, USA) and 10 mg/ml DNase (Roche, Basel, Switzerland). Single‐cell suspensions were stained and analyzed using a FACS Canto II cytometer (Becton Dickinson, Franklin Lakes, NJ, USA). Antibodies were purchased from BioLegend unless otherwise listed. For monocytes and neutrophils, the following antibodies were used: Ly6G (1A8), CD45 (30‐F11; eBioscience, San Diego, CA, USA), CD11c (HL3; BD, San Jose, CA, USA), CD11b (M1/70; BD), Ly6C (AL‐21; BD), and IL1RL1 (DIH9). For ILC staining, the following antibodies were used: CD3 (145‐2C11), CD19 (6D5), Gr1 (RB6‐8C5), B220 (RA3‐6B2), Ter119 (TER‐119), FcaR1 (MAR‐1), CD49b (DX5; eBioscience), Il1rl1 (DIH9), CD90 (30‐H12), and CD45 (30‐F11). A Fixable Viability Dye kit (eBioscience) was used to exclude dead cells. Data were analyzed using FlowJo software (Tree Star Inc, Ashland, OR, USA).
Histopathology
Major basic protein staining was conducted as previously described [22] using an antibody kindly provided by Nancy Lee and James Lee (Mayo Clinic, Phoenix, AZ, USA). Slides were scanned using an Olympus Dot‐Slide virtual slide microscope (Olympus, Tokyo, Japan) and expression was quantified by digital image analysis using ImageJ (US National Institutes of Health, Bethesda, MD, USA).
Statistical analysis
Data are expressed as indicated in the figure legends. Differences were analyzed using the Mann–Whitney U‐test or t‐test. For survival studies, Kaplan–Meier analysis followed by log‐rank testing was performed. Analyses were performed using GraphPad Prism 7.03 (GraphPad Software, San Diego, CA, USA). p < 0.05 was considered statistically significant.
Results
IL‐33 improves outcome and limits bacterial growth and dissemination during Klebsiella pneumonia‐derived sepsis by an IL1RL1‐dependent mechanism
To obtain a first insight into the effect of IL‐33 on host defense during pneumonia‐derived sepsis, mice were intravenously injected with recombinant mouse IL‐33 (1 μg) or vehicle 24 and 1 h prior to infection, a treatment schedule previously shown to be protective in a model of CLP‐induced abdominal sepsis [11], and observed for 6 days. IL‐33 prolonged survival during this lethal infection: the median survival time in IL‐33‐treated wild‐type (WT) mice (91.4 h) was significantly longer than in vehicle‐treated WT mice (60.8 h) (Figure 1A). IL‐33‐mediated protection required the presence of the IL‐33 receptor, since it was not observed in IL‐33‐treated Il1rl1 −/− mice (median survival time 63.8 h).
Figure 1.
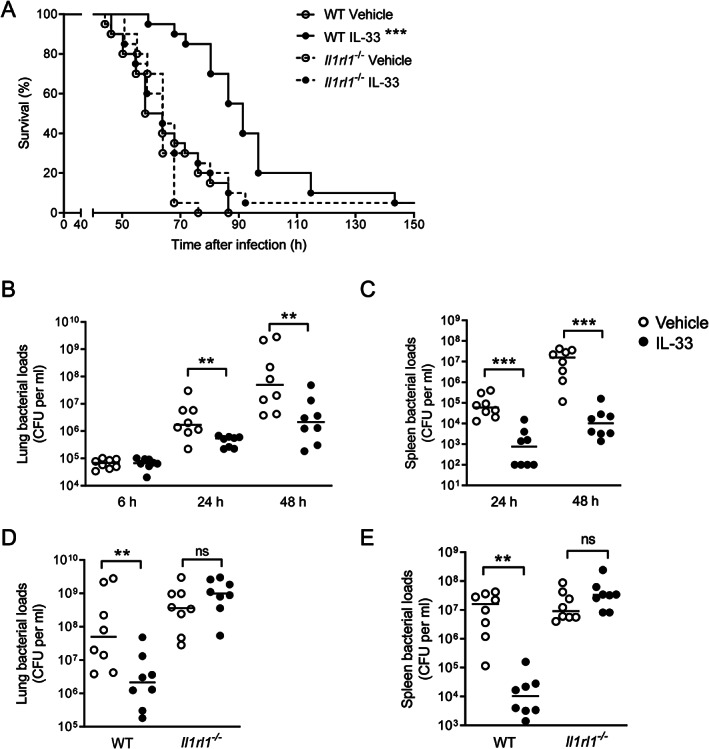
IL‐33 treatment improves host defense during Klebsiella pneumonia by an IL1RL1‐dependent mechanism. (A) Survival of vehicle or IL‐33‐treated WT and Il1rl1 −/− mice following K. pneumoniae inoculation (20 mice per group). ***p < 0.001 compared with all other experimental groups (Mantel–Cox test). (B, C) Bacterial loads in lung (B) and spleen (C) in vehicle and IL‐33‐treated WT mice 6, 24, and 48 h following induction of pneumonia. (D, E) Bacterial loads in lung (D) and spleen (E) from vehicle and IL‐33‐treated WT and Il1rl1 −/− mice 48 h following inoculation with K. pneumoniae. (B–E) Graphs show the median and every dot represents one individual mouse. Significances are indicated as **p < 0.01, ***p < 0.001, ns (not significant) from Mann–Whitney U‐tests. See also supplementary material, Figures S1 and S2.
To assess whether the beneficial effect of IL‐33 was associated with an improved antibacterial defense, bacterial loads were measured at the primary site of infection and distant organs at predefined early and late endpoints after infection prior to the occurrence of the first deaths. At 6 h after infection, bacterial loads in lungs (Figure 1B) were similar in IL‐33‐treated and control WT mice, while cultures in distant organs remained negative in both groups. At 24 and 48 h, mice given IL‐33 had much lower bacterial burdens at the primary site of infection (Figure 1B) and in blood (supplementary material, Figure S1A), which was accompanied by markedly lower bacterial counts in the spleen (Figure 1C) and liver (supplementary material, Figure S1B).
To determine whether this IL‐33 effect was dependent on the presence of the IL‐33 receptor, we treated Il1rl1 −/− and WT mice with IL‐33, using the exact same dosing schedule as in the first series of experiments, and measured bacterial loads 48 h after infection. While IL‐33‐treated WT mice again demonstrated strongly reduced bacterial counts at all body sites, such an effect was not seen in Il1rl1 −/− mice (Figure 1D,E and supplementary material, Figure S1C,D). Postponed IL‐33 treatment, given as a single injection at 3 h post‐infection (1 μg), remained effective in reducing bacterial loads in lungs and spleen measured 48 h after inoculation with Klebsiella; however, IL‐33 administered 24 h after infection did not modify bacterial burdens (supplementary material, Figure S2A,B).
Taken together, these data suggest that IL‐33 improves survival and antibacterial defense during Klebsiella pneumonia‐derived sepsis by an effect via IL1RL1.
IL‐5 or IL‐13 is not required for IL‐33‐induced improved antibacterial defense
Administration of IL‐33 increased the pulmonary levels of IL‐5 (Figure 2A) and IL‐13 (Figure 2B), especially within the first hours after the second IL‐33 injection, which is consistent with previous studies demonstrating that IL‐33 plays a key role in triggering type 2 immune responses [23, 24]. This induction of type 2 cytokines was accompanied by a marked increase in eosinophil numbers in the lung tissue of IL‐33‐treated mice, as determined by digital imaging and quantification of eosinophils in lung tissue slides stained with an antibody against major basic protein (supplementary material, Figure S3). To assess the potential roles of these cytokines in the IL‐33 effect on bacterial loads, we treated mice with an anti‐IL‐5 or control antibody prior to IL‐33 treatment and K. pneumoniae infection (Figure 2C,D). The biological activity of the anti‐IL‐5 antibody was illustrated by the fact that the increased eosinophil influx into lung tissue induced by IL‐33 treatment was prevented by anti‐IL‐5 administration (supplementary material, Figure S3A,B). Of interest, anti‐IL‐5 reduced bacterial loads in lungs relative to mice treated with the control antibody, suggesting that IL‐5 may impair rather than enhance local defense against Klebsiella. While IL‐33 did not further reduce bacterial burdens in the lungs of anti‐IL‐5‐treated mice (Figure 2C), IL‐33 strongly lowered bacterial counts in the spleen of these animals (Figure 2D). IL‐33 was equally effective in decreasing bacterial loads in the lungs (Figure 2E) and spleen (Figure 2F) of Il13 −/− and WT mice. While all previous experiments were carried out with BALB/c mice, Il13 −/− mice and littermate controls were on a C57BL/6 background, thereby demonstrating that the effect of IL‐33 does not rely on the genetic background of the mice. Taken together, these data suggest that the cytokines IL‐5 and IL‐13 are not essential for the effect of IL‐33 on antibacterial defense during Klebsiella pneumonia.
Figure 2.
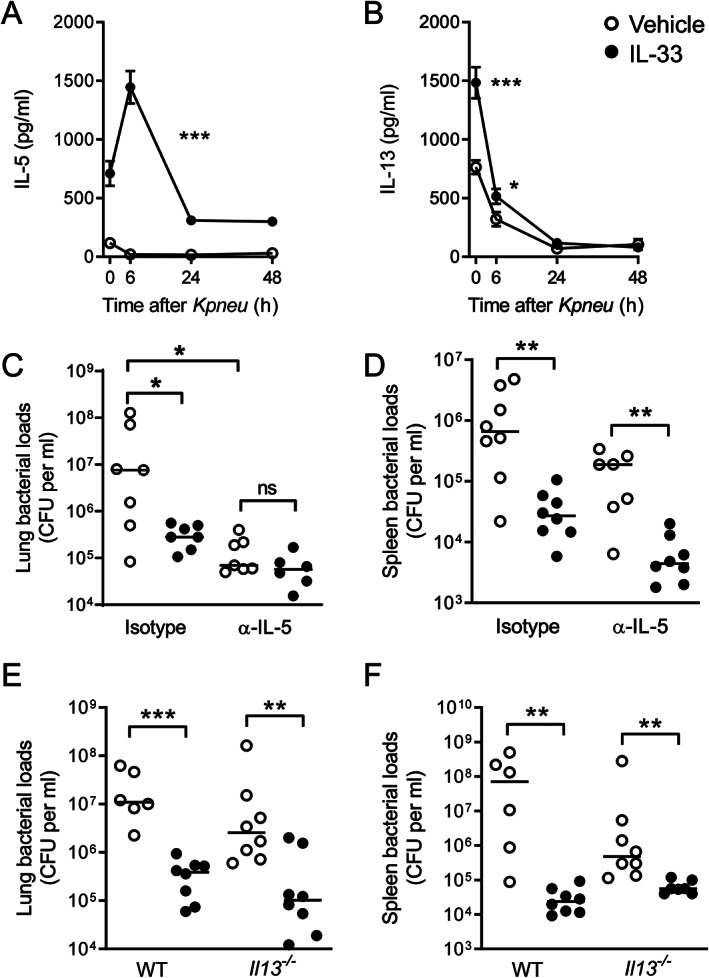
IL‐33‐mediated restriction of bacterial growth does not require IL‐5 or IL‐13. (A, B) Lung levels of IL‐5 (A) and IL‐13 (B) at the indicated time points after K. pneumoniae infection. Data are presented as mean (eight animals per group), error bars reflect the SEM, and significances are indicated as *p < 0.05, ***p < 0.001 from Mann–Whitney U‐tests. (C, D) Bacterial loads in lung (C) and spleen (D) 48 h after infection of mice treated with IL‐33 or vehicle together with an anti‐IL‐5 or isotype control antibody. (E, F) Bacterial loads in lung (E) and spleen (F) 48 h after infection of Il13 −/− and WT mice treated with IL‐33 or vehicle. (C–F) Graphs show the median and every dot represents one individual mouse. Significances are indicated as *p < 0.05, **p < 0.01, ***p < 0.001, ns (not significant) from Mann–Whitney U‐tests. See also supplementary material, Figure S3.
Lymphocyte depletion does not abrogate IL‐33‐induced enhanced antibacterial defense
Since IL‐33 improved host defense by an IL1RL1‐dependent mechanism, we next investigated the involvement of different IL1RL1‐expressing cells. Considering that a broad number of lymphoid cells express IL1RL1 [25, 26], we first assessed the effect of IL‐33 in Rag2 −/− mice, which lack B, T, and NKT cells [27]. IL‐33 remained effective in reducing the bacterial loads in the lungs (Figure 3A) and spleen (Figure 3B) of Rag2 −/− mice.
Figure 3.
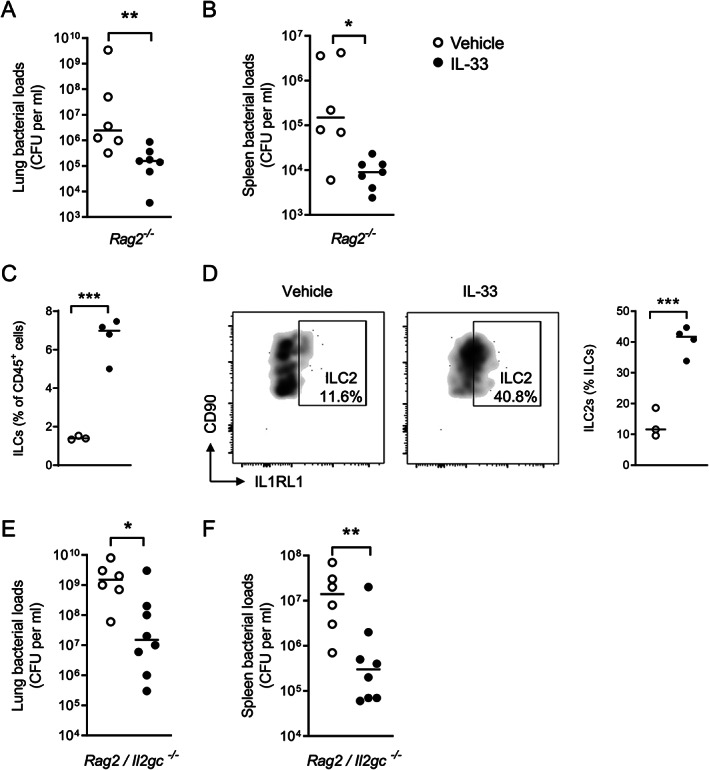
IL‐33‐mediated bacterial growth restriction does not depend on B, T, NKT or innate lymphoid cells. (A, B) Bacterial loads in lung (A) and spleen (B) 48 h after infection of Rag2 −/− mice treated with IL‐33 or vehicle. Graphs show the median and every dot represents one individual mouse. Significances are indicated as *p < 0.05, **p < 0.01 from Mann–Whitney U‐tests. Data from one experiment representative of two independent experiments with six to eight mice per group each. (C, D) Innate lymphoid cell populations in lungs are expanded after IL‐33 treatment (C) as a result of an increased number of IL1RL1+ ILC2s (D). Graphs show the mean and every dot represents one individual mouse. Significance is indicated as ***p < 0.001 from an unpaired t‐test. Histograms are representative of three or four individual mice. (E, F) Bacterial loads in lung (E) and spleen (F) 42 h after infection of Rag2/Il2gc −/− mice treated with IL‐33 or vehicle. Graphs show the median and every dot represents one individual mouse. Significances are indicated as *p < 0.05, **p < 0.01 from Mann–Whitney U‐tests. Data pooled from two independent experiments with three or four mice per group. See also supplementary material, Figure S4.
ILC2s are well‐established IL1RL1‐expressing target cells of IL‐33 [9, 25], which prompted us to investigate the potential contribution of this cell population to the beneficial effect of IL‐33. IL‐33 treatment increased the total frequency of ILCs, specifically IL1RL1+ ILCs, also defined as ILC2s [28], in lung tissue relative to vehicle‐treated mice (Figure 3C,D). IL‐33 remained effective in reducing bacterial loads in the lungs (Figure 3E) and spleen (Figure 3F) of Rag2 −/−/Il2rγc −/− mice, which are deficient in all lymphocytes, including ILCs [27], after infection with K. pneumoniae via the airways, suggesting that ILCs are not essential for the effect of IL‐33 in this model. Likewise, IL‐33 remained capable of lowering bacterial counts in Rag2 −/− mice lacking ILCs after treatment with a depleting monoclonal antibody specific for CD90 (supplementary material, Figure S4A,B). Taken together, these data indicate that lymphocytes, including ILCs, do not drive the effect of IL‐33 on antibacterial defense during Klebsiella pneumonia.
Neutrophils and inflammatory monocytes are required for IL‐33‐mediated enhanced antibacterial defense in the lung
Neutrophils and inflammatory monocytes (IMs) expressed IL1RL1 on their surface (supplementary material, Figure S5A,B) and therefore, both cell types are targets of IL‐33, confirming previous reports [8, 29, 30]. Both neutrophils and IMs have been implicated in antibacterial defense against lower respiratory tract infections caused by K. pneumoniae [31]. IL‐33 treatment increased the influx of both neutrophils (Figure 4A) and IMs (Figure 4B) in the lungs of uninfected mice when compared with vehicle treatment. IL‐33 also increased myeloperoxidase (MPO) concentrations in whole lung homogenates (Figure 4C), further supporting an effect on neutrophils. This prompted us to study the contribution of IMs and neutrophils to the effect of IL‐33 on bacterial growth and dissemination during Klebsiella pneumonia. To this end, we depleted both populations by administering an anti‐Gr1 monoclonal antibody (RB6‐8C5) or depleted only neutrophils by treatment with an Ly6G‐specific monoclonal antibody (1A8) as described previously [19, 32]. We confirmed the efficiency of cell depletion by FACS in blood at baseline (supplementary material, Figure S5C). Since we anticipated early mortality in neutropenic mice, we determined the bacterial loads in these experiments 24 h after infection with a 50% lower K. pneumoniae dose (~0.5 × 104 CFU). IL‐33 was still able to lower the bacterial loads in lungs (Figure 4D) but not in spleen (Figure 4E) of mice receiving the isotype antibody. Of note, this low inoculum resulted in positive cultures of spleens of only half of isotype control antibody‐treated mice, precluding establishment of an effect of IL‐33 on dissemination of the infection. As expected, anti‐Gr1 and anti‐Ly6G treatment resulted in much higher bacterial loads in the lungs (Figure 4D) and spleen (Figure 4E) compared with mice treated with a control antibody. The capacity of IL‐33 to reduce bacterial burdens in the lungs was much stronger in mice depleted of only neutrophils (by anti‐Ly6G administration) than in mice depleted of both neutrophils and IMs (by anti‐Gr1 administration), suggesting that IL‐33 mediates its effect on bacterial growth in the lungs at least in part through IMs (Figure 4D). In contrast, bacterial counts in the spleens of IL‐33‐treated anti‐Gr1 or anti‐Ly6G administered mice were not different, indicating that for an optimal bacterial growth‐limiting effect of IL‐33 in distant organs, IMs are not required (Figure 4E). To further examine the role of IMs in the bacterial growth‐limiting effect of IL‐33, we treated mice with an anti‐CCR2 antibody, which has been described to selectively deplete IMs [21]. Indeed, anti‐CCR2 depleted mice of IMs without impacting the numbers of neutrophils or patrolling monocytes (supplementary material, Figure S5D). Remarkably, IL‐33 was still able to reduce bacterial counts in the lungs (Figure 5A) and spleens (Figure 5B) of anti‐CCR2‐treated mice. These results taken together with the findings in mice selectively depleted of neutrophils or depleted of neutrophils and IMs suggest that IL‐33 mediates its effects on bacterial growth by a combined effect on neutrophils and IMs. Of note, mice given anti‐CCR2 not treated with IL‐33 showed higher bacterial loads in lungs and spleens when compared with mice given a control antibody, confirming the protective role of IMs during Klebsiella pneumonia [32].
Figure 4.
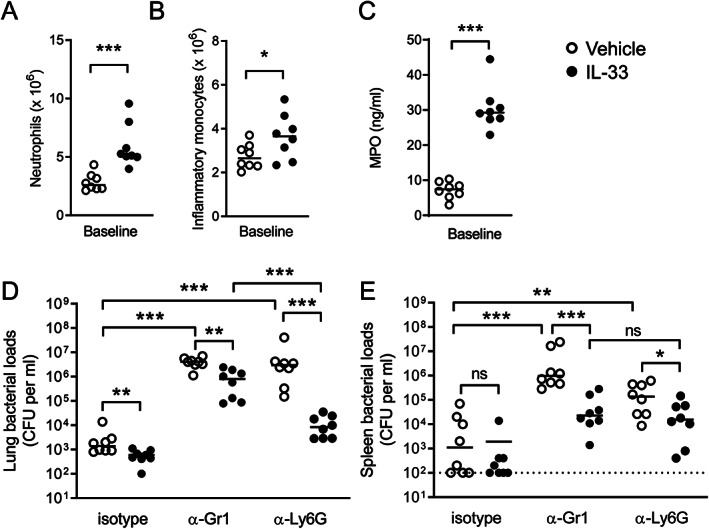
IL‐33‐mediated bacterial growth restriction in the lungs at least in part relies on inflammatory monocytes. (A, B) Neutrophil (A) and IM (B) numbers in lung tissue were evaluated by flow cytometry in whole lung directly before infection (baseline) after receiving vehicle or IL‐33 treatment. (C) MPO levels in lung before infection in vehicle and IL‐33‐treated mice. (D, E) Bacterial loads in lung (D) and spleen (E) of mice treated with IL‐33 or vehicle together with anti‐Gr1, anti‐Ly6G, or a control isotype antibody. Graphs show the median and every dot represents one individual mouse. Data in A–C are representative of two independent experiments with similar results. Significances are indicated as *p < 0.05, **p < 0.01, ***p < 0.001, ns (not significant) from Mann–Whitney U‐tests. See also supplementary material, Figure S5.
Figure 5.
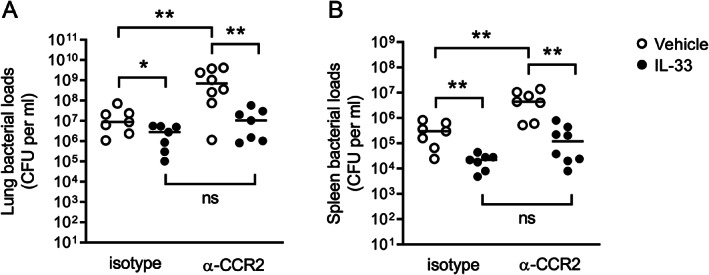
IL‐33‐mediated bacterial growth restriction remains intact in mice selectively depleted of inflammatory monocytes. (A, B) Bacterial counts in lung (A) and spleen (B) of mice depleted of inflammatory monocytes (anti‐CCR2) or given a control isotype antibody and treated with IL‐33 or vehicle. Significances are indicated as *p < 0.05, **p < 0.01, ns (not significant) from Mann–Whitney U‐tests. See also supplementary material, Figure S5.
Discussion
IL‐33 is a pleiotropic cytokine that can affect a variety of cell types involved in immune defense. Here we sought to determine the effect of IL‐33 treatment in a model of pneumonia caused by inoculation of a low dose of a common human pathogen via the airways and characterized by a steadily developing infection of the lower respiratory tract and gradual induction of local inflammation, only in later stages resulting in sepsis. This model differs substantially from sepsis models previously used to investigate the effects of IL‐33, which involved either acute polymicrobial fecal peritonitis in the context of a necrotic cecum induced by CLP or the administration of high bacterial doses causing acute lethal inflammation [10]. Our main findings are that IL‐33 administration improves survival and enhances host defense at the primary site of infection during Klebsiella pneumonia by a novel mechanism involving both neutrophils and IMs. Mechanisms previously reported to contribute to IL‐33 effects, i.e. type 2 cytokines, B cells, T cells, NKT cells or ILCs, were not required for IL‐33‐mediated improved antibacterial defense.
IL‐33 administration led to an influx of neutrophils and IMs into the lungs. Depletion of either neutrophils alone, IMs alone, or neutrophils and IMs resulted in increased bacterial loads. Previous studies documented distinct protective roles for neutrophils and IMs during pneumonia caused by Klebsiella, depending on the specific bacterial strain used [31, 32]. The current results obtained with antibodies that deplete either neutrophils, IMs, or neutrophils and IMs indicate that antibacterial defense against the Klebsiella strain used here (ATCC 43816) is mediated by neutrophils and IMs. Importantly, the bacterial growth‐restricting effect of IL‐33 in the lungs required both neutrophils and IMs, whereas in mice selectively depleted of either neutrophils or IMs the effect of IL‐33 remained intact. While a role for neutrophils in IL‐33‐mediated protection during bacterial infection has been reported previously [11, 17], our results identify IMs as an additional IL‐33 target in this context. IMs are derived from the bone marrow and are able to rapidly migrate into the bloodstream and infected tissues. IMs are pluripotent and have been shown to improve host defense against several infections [33]. Importantly, a recent study documented the capacity of IMs to ingest and kill Klebsiella after infection via the airways [32]. IL‐33 can activate a variety of pro‐inflammatory pathways in monocytes through interaction with IL1RL1, including mitogen‐activated protein kinases, TNF‐α receptor‐associated factor 6, and nuclear factor‐κB [30, 34], and a recent study has indicated that amongst different monocyte subsets, especially IMs express IL1RL1 and are responsive to IL‐33 [30]. Collectively, these data suggest that IL‐33 reduced Klebsiella numbers in the lung by IL1RL1‐dependent activation of both neutrophils and IMs recruited into the airways.
Two earlier studies reported on the effects of IL‐33 in murine pneumonia with paradoxical results [16, 17]. In a model of pneumonia caused by Legionella pneumophila, intranasal treatment with IL‐33 increased bacterial loads and lethality [16]. This detrimental effect was likely mediated through IL‐4 and/or IL‐13, considering that IL‐33 induced elevated levels of these cytokines and that IL‐33 did not impair host defense in Stat6‐deficient mice, which are severely hampered in their capacity to signal IL‐4 and IL‐13 effects [16]. The adverse outcome of IL‐33‐treated mice during Legionella pneumonia is remarkable in light of IL‐33‐mediated protection in other severe infection models [10] and may be related to the route of IL‐33 administration and the fact that mice are intrinsically resistant to this pathogen [35]. In a model of post‐influenza S. aureus pneumonia, intraperitoneal administration of IL‐33 reduced bacterial burdens in the lungs, which was associated with enhanced IL‐33‐induced recruitment of neutrophils [17]. IL‐33 remained effective in enhancing bacterial clearance in mice depleted of neutrophils by treatment with an anti‐Ly6G antibody (i.e. the same antibody used to deplete neutrophils in the current study). Nonetheless, the authors concluded that IL‐33 mediated its protective effect through an effect on neutrophils based on the fact that bacterial loads in IL‐33‐treated mice without prior neutrophil depletion were modestly lower than in IL‐33‐treated mice in which neutrophils were depleted [17]. Notably, however, neutrophil depletion per se caused a marked increase in bacterial burdens, and IL‐33 caused a much stronger decrease in bacterial counts in neutrophil‐depleted than in neutrophil‐competent mice [17], which is similar to our findings in anti‐Ly6G‐treated mice reported here, indicating that the IL‐33 effect does not rely solely on neutrophils. In agreement with our findings, IL‐33 remained effective in lowering the number of bacteria in Rag2/Il2rγc −/− mice, indicating the lack of requirement for ILC2s; the contribution of monocytes was not studied [17].
Several cell types have been implicated in the protective effects of IL‐33 during severe extrapulmonary infections. IL‐33 given intravenously 24 and 1 h prior to CLP protected against lethality, which was associated with an accelerated bacterial clearance [11]. The beneficial effect of IL‐33 was linked to enhanced neutrophil recruitment to the abdominal cavity, while IL‐33 did not impact IL‐4 or IL‐13 responses [11]. In another investigation using CLP to produce abdominal sepsis, IL‐33‐mediated protection was linked to induction of interferon‐γ by γδ T and NK cells [14]. Intratracheal administration of IL‐33 for 3 days prior to intravenous injection of a lethal dose of S. aureus reduced mortality [36], which was associated with a modest and transient reduction in bacterial loads in the liver and lungs, but not in the spleen, and which was dependent on IL‐5 and IL‐13 produced by ILC2s [15]. Depletion of eosinophils eliminated IL‐33‐mediated protection, indicating that ILC2‐derived type 2 cytokines induced their beneficial effect through induction of eosinophilia [15]. Of note, however, the effect of IL‐33 on bacterial loads was modest at best and transient, and an unlikely explanation for the protection against lethality. In contrast, our data demonstrate that IL‐33 administered to Rag2/Il2rγc −/− mice (lacking, among others, ILC2s) or to anti‐CD90 (which depletes ILCs)‐treated Rag2 −/− mice still reduced bacterial growth during Klebsiella pneumonia. In addition, despite the fact that IL‐33 triggered the release of IL‐5 and IL‐13, neither of them was required for the beneficial effect of IL‐33 in the K. pneumoniae pneumonia model. In an acute S. aureus peritonitis model, IL‐33 administration enhanced bacterial clearance and reduced mortality [37]. IL‐33 did not reduce bacterial burdens in mice treated with anti‐Gr1, letting the authors conclude that the IL‐33‐mediated protection was mediated by neutrophils [37]. However, as shown in the current study and previously [31], anti‐Gr1 is not specific for neutrophils but also depletes monocytes.
In conclusion, we studied here the effect of IL‐33 in a model of pneumonia‐derived sepsis caused by a low infectious dose of a clinically relevant Gram‐negative respiratory pathogen. IL‐33 improved host defense during Klebsiella pneumonia, as reflected by prolonged survival and reduced bacterial growth and dissemination. The protective effect of IL‐33 was dependent on IL1RL1 and the restriction of bacterial growth in the lungs at least in part was mediated through a combined effect on neutrophils and IMs. This study is the first to point at IMs as a target cell for IL‐33 to enhance host defense against infection.
Author contributions statement
DCB, AFdV and TvdP were responsible for conceptualization, and IRM, AFdV, DCB and TvdP for methodology. IRM validated the study. IRM and DCB carried out formal analysis. IRM, AFdV, DCB, IGL and JHB performed the investigations. SF, LB, SS, SK, MM and HS were responsible for resources. IRM, DCB and TvdP wrote the original draft of the manuscript. IRM, AFdV, DCB, IGL, JHB, HS, LB, SK, SF, CvV, SS and TvdP reviewed and edited the paper. Visualization was carried out by IRM and DCB. TvdP was responsible for funding acquisition.
Supporting information
Figure S1. IL‐33 treatment reduces bacterial loads in blood and liver during Klebsiella pneumonia by an IL1RL1‐dependent mechanism
Figure S2. Effect of delayed IL‐33 treatment on bacterial growth and dissemination during Klebsiella pneumonia
Figure S3. Anti‐IL‐5 treatment abrogates IL‐33‐induced eosinophil recruitment to the lungs
Figure S4. Anti‐CD90 treatment does not impact IL‐33‐mediated restriction of bacterial growth and dissemination
Figure S5. IL1RL1 expression and efficiency of antibody treatment in depleting neutrophils and inflammatory monocytes
Acknowledgements
We thank Joost Daalhuisen and Marieke ten Brink (Center for Experimental and Molecular Medicine, Amsterdam‐UMC) for their technical support during the animal experiments.
No conflicts of interest were declared.
References
- 1. van der Poll T, van de Veerdonk FL, Scicluna BP, et al. The immunopathology of sepsis and potential therapeutic targets. Nat Rev Immunol 2017; 17: 407–420. [DOI] [PubMed] [Google Scholar]
- 2. Rudd KE, Johnson SC, Agesa KM, et al. Global, regional, and national sepsis incidence and mortality, 1990–2017: analysis for the Global Burden of Disease Study. Lancet 2020; 395: 200–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fleischmann C, Scherag A, Adhikari NKJ, et al. Assessment of global incidence and mortality of hospital‐treated sepsis. Current estimates and limitations. Am J Respir Crit Care Med 2016; 193: 259–272. [DOI] [PubMed] [Google Scholar]
- 4. Angus DC, van der Poll T. Severe sepsis and septic shock. N Engl J Med 2013; 369: 840–851. [DOI] [PubMed] [Google Scholar]
- 5. Mizgerd JP. Respiratory infection and the impact of pulmonary immunity on lung health and disease. Am J Respir Crit Care Med 2012; 186: 824–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Global Health Estimates 2016: Deaths by Cause, Age, Sex, by Country and by Region, 2000‐2016. World Health Organization: Geneva, 2018. [Google Scholar]
- 7. Paczosa MK, Mecsas J. Klebsiella pneumoniae: going on the offense with a strong defense. Microbiol Mol Biol Rev 2016; 80: 629–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Martin NT, Martin MU. Interleukin 33 is a guardian of barriers and a local alarmin. Nat Immunol 2016; 17: 122–131. [DOI] [PubMed] [Google Scholar]
- 9. Cayrol C, Girard J‐P. Interleukin‐33 (IL‐33): a nuclear cytokine from the IL‐1 family. Immunol Rev 2018; 281: 154–168. [DOI] [PubMed] [Google Scholar]
- 10. Xu H, Turnquist HR, Hoffman R, et al. Role of the IL‐33–ST2 axis in sepsis. Mil Med Res 2017; 4: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Alves‐Filho JC, Sônego F, Souto FO, et al. Interleukin‐33 attenuates sepsis by enhancing neutrophil influx to the site of infection. Nat Med 2010; 16: 708–712. [DOI] [PubMed] [Google Scholar]
- 12. Li S, Zhu F‐X, Zhao X‐J, et al. The immunoprotective activity of interleukin‐33 in mouse model of cecal ligation and puncture‐induced sepsis. Immunol Lett 2016; 169: 1–7. [DOI] [PubMed] [Google Scholar]
- 13. Lv R, Zhao J, Lei M, et al. IL‐33 attenuates sepsis by inhibiting IL‐17 receptor signaling through upregulation of SOCS3. Cell Physiol Biochem 2017; 42: 1961–1972. [DOI] [PubMed] [Google Scholar]
- 14. Bao Q, Lv R, Lei M. IL‐33 attenuates mortality by promoting IFN‐gamma production in sepsis. Inflamm Res 2018; 67: 531–538. [DOI] [PubMed] [Google Scholar]
- 15. Krishack PA, Louviere TJ, Decker TS, et al. Protection against Staphylococcus aureus bacteremia‐induced mortality depends on ILC2s and eosinophils. JCI Insight 2019; 4: e124168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nascimento DC, Melo PH, Piñeros AR, et al. IL‐33 contributes to sepsis‐induced long‐term immunosuppression by expanding the regulatory T cell population. Nat Commun 2017; 8: 14919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Robinson KM, Ramanan K, Clay ME, et al. Novel protective mechanism for interleukin‐33 at the mucosal barrier during influenza‐associated bacterial superinfection. Mucosal Immunol 2018; 11: 199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. van Lieshout MHP, Anas AA, Florquin S, et al. Hematopoietic but not endothelial cell MyD88 contributes to host defense during gram‐negative pneumonia derived sepsis. PLoS Pathog 2014; 10: e1004368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Claushuis TAM, de Vos AF, Nieswandt B, et al. Platelet glycoprotein VI aids in local immunity during pneumonia‐derived sepsis caused by Gram‐negative bacteria. Blood 2018; 131: 864–876. [DOI] [PubMed] [Google Scholar]
- 20. Ding C, Scicluna BP, Stroo I, et al. Prekallikrein inhibits innate immune signaling in the lung and impairs host defense during pneumosepsis in mice. J Pathol 2020; 250: 95–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Brühl H, Cihak J, Plachý J, et al. Targeting of Gr‐1+,CCR2+ monocytes in collagen‐induced arthritis. Arthritis Rheum 2007; 56: 2975–2985. [DOI] [PubMed] [Google Scholar]
- 22. Daan de Boer J, Roelofs JJTH, de Vos AF, et al. Lipopolysaccharide inhibits Th2 lung inflammation induced by house dust mite allergens in mice. Am J Respir Cell Mol Biol 2013; 48: 382–389. [DOI] [PubMed] [Google Scholar]
- 23. Kondo Y, Yoshimoto T, Yasuda K, et al. Administration of IL‐33 induces airway hyperresponsiveness and goblet cell hyperplasia in the lungs in the absence of adaptive immune system. Int Immunol 2008; 20: 791–800. [DOI] [PubMed] [Google Scholar]
- 24. Sjöberg LC, Nilsson AZ, Lei Y, et al. Interleukin 33 exacerbates antigen driven airway hyperresponsiveness, inflammation and remodeling in a mouse model of asthma. Sci Rep 2017; 7: 4219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Griesenauer B, Paczesny S. The ST2/IL‐33 axis in immune cells during inflammatory diseases. Front Immunol 2017; 8: 475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dinarello CA. Overview of the IL‐1 family in innate inflammation and acquired immunity. Immunol Rev 2018; 281: 8–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Moro K, Koyasu S. Innate lymphoid cells, possible interaction with microbiota. Semin Immunopathol 2015; 37: 27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Moro K, Ealey KN, Kabata H, et al. Isolation and analysis of group 2 innate lymphoid cells in mice. Nat Protoc 2015; 10: 792–806. [DOI] [PubMed] [Google Scholar]
- 29. Du XX, Shi Y, Yang Y, et al. DAMP molecular IL‐33 augments monocytic inflammatory storm in hepatitis B‐precipitated acute‐on‐chronic liver failure. Liver Int 2018; 38: 229–238. [DOI] [PubMed] [Google Scholar]
- 30. Stojkovic S, Thulin Å, Hell L, et al. IL‐33 stimulates the release of procoagulant microvesicles from human monocytes and differentially increases tissue factor in human monocyte subsets. Thromb Haemost 2017; 117: 1379–1390. [DOI] [PubMed] [Google Scholar]
- 31. Xiong H, Carter RA, Leiner IM, et al. Distinct contributions of neutrophils and CCR2+ monocytes to pulmonary clearance of different Klebsiella pneumoniae strains. Infect Immun 2015; 83: 3418–3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Xiong H, Keith JW, Samilo DW, et al. Innate lymphocyte/Ly6Chi monocyte crosstalk promotes Klebsiella pneumoniae clearance. Cell 2016; 165: 679–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Murray PJ. Immune regulation by monocytes. Semin Immunol 2018; 35: 12–18. [DOI] [PubMed] [Google Scholar]
- 34. Mun SH, Ko NY, Kim HS, et al. Interleukin‐33 stimulates formation of functional osteoclasts from human CD14+ monocytes. Cell Mol Life Sci 2010; 67: 3883–3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ng GZ, Solomatina A, van Driel IR, et al. The mouse as a model for pulmonary Legionella infection. Methods Mol Biol 2019; 1921: 399–417. [DOI] [PubMed] [Google Scholar]
- 36. Krishack PA, Wang K, Rzhetsky A, et al. Preexisting type 2 immune activation protects against the development of sepsis. Am J Respir Cell Mol Biol 2017; 57: 628–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lan F, Yuan B, Liu T, et al. Interleukin‐33 facilitates neutrophil recruitment and bacterial clearance in S. aureus‐caused peritonitis. Mol Immunol 2016; 72: 74–80. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. IL‐33 treatment reduces bacterial loads in blood and liver during Klebsiella pneumonia by an IL1RL1‐dependent mechanism
Figure S2. Effect of delayed IL‐33 treatment on bacterial growth and dissemination during Klebsiella pneumonia
Figure S3. Anti‐IL‐5 treatment abrogates IL‐33‐induced eosinophil recruitment to the lungs
Figure S4. Anti‐CD90 treatment does not impact IL‐33‐mediated restriction of bacterial growth and dissemination
Figure S5. IL1RL1 expression and efficiency of antibody treatment in depleting neutrophils and inflammatory monocytes