

Abstract
α‐Synuclein oligomers are thought to have a pivotal role in sporadic and familial Parkinson's disease (PD) and related α‐synucleinopathies, causing dysregulation of protein trafficking, autophagy/lysosomal function, and protein clearance, as well as synaptic function impairment underlying motor and cognitive symptoms of PD. Moreover, trans‐synaptic spread of α‐synuclein oligomers is hypothesized to mediate disease progression. Therapeutic approaches that effectively block α‐synuclein oligomer‐induced pathogenesis are urgently needed. Here, we show for the first time that α‐synuclein species isolated from human PD patient brain and recombinant α‐synuclein oligomers caused similar deficits in lipid vesicle trafficking rates in cultured rat neurons and glia, while α‐synuclein species isolated from non‐PD human control brain samples did not. Recombinant α‐synuclein oligomers also increased neuronal expression of lysosomal‐associated membrane protein‐2A (LAMP‐2A), the lysosomal receptor that has a critical role in chaperone‐mediated autophagy. Unbiased screening of several small molecule libraries (including the NIH Clinical Collection) identified sigma‐2 receptor antagonists as the most effective at blocking α‐synuclein oligomer‐induced trafficking deficits and LAMP‐2A upregulation in a dose‐dependent manner. These results indicate that antagonists of the sigma‐2 receptor complex may alleviate α‐synuclein oligomer‐induced neurotoxicity and are a novel therapeutic approach for disease modification in PD and related α‐synucleinopathies.
Keywords: autophagy, functional assay, lysosomal‐associated membrane protein‐2A, Parkinson's disease, progesterone receptor membrane component 1, RRID:AB_1603277, RRID:AB_2109656, RRID:AB_2533900, RRID:AB_2629502, RRID:AB_2877641, RRID:AB_571049, RRID:RGD_1566440, TMEM97
The protein α‐synuclein is central to Parkinson's disease. For the first time, we report that α‐synuclein from Parkinson's patients brain samples caused signaling deficits in brain cells, and novel drug candidates known to block the sigma‐2 receptor complex reversed these deficits.
Significance.
Oligomeric α‐synuclein proteins found in Parkinson's disease patient brain tissue cause neuron dysfunction, and therapeutic approaches effectively targeting them are urgently needed. For the first time, this study demonstrates that recombinant and Parkinson's patient‐derived α‐synuclein cause similar lipid vesicle trafficking deficits in neurons, while α‐synuclein species isolated from non‐Parkinson's human control brain samples do not. α‐Synuclein oligomers also upregulate lysosomal‐associated membrane protein‐2A (LAMP‐2A), a protein critical to chaperone‐mediated autophagy. A broad search of existing drug candidates revealed that antagonists of the sigma‐2 receptor complex were the most effective at blocking α‐synuclein oligomer‐induced trafficking deficits and LAMP‐2A upregulation. These drug candidates may represent a novel therapeutic approach against Parkinson's neuronal dysfunction and neurodegenerative disorders caused by α‐synuclein oligomer‐mediated toxicity.
1. INTRODUCTION
Parkinson's disease (PD) is a neurodegenerative disorder characterized by dysfunction in motor control, diminished autonomic functions, and non‐motor symptoms including cognitive loss (Aarsland et al., 2017; Mhyre et al., 2012). The hallmark histopathology that defines PD is intracytoplasmic inclusions called Lewy bodies, which contain high concentrations of the protein α‐synuclein in a predominantly beta sheet fibrillar conformation (Spillantini et al., 1997). α‐Synuclein is a 140 amino acid protein found in presynaptic terminals throughout the brain that has a role in controlling the movement of presynaptic vesicles and their fusion with synaptic membranes (Burré et al., 2014; Diao et al., 2013; Meade et al., 2019). In aging and disease, however, cumulative insults such as fatty acid lipid binding (Karube et al., 2008; Narayanan & Scarlata, 2001; Perrin et al., 2001), metal ions (Deas et al., 2016), oxidative stress (Esteves et al., 2009), acidosis (Meade et al., 2019), and endoplasmic reticulum (ER) stress (Jiang et al., 2010; Scheper & Hoozemans, 2015) can modulate the structure and form of endogenous α‐synuclein, resulting in aggregated species such as fibrils and oligomers, which are associated with Parkinson's pathology (Bernal‐Conde et al., 2020; Meade et al., 2019; Roberts et al., 2015; Wong & Krainc, 2017). Additionally, post‐translational modifications of α‐synuclein identified in the brains of individuals with PD, dementia with Lewy bodies, or Alzheimer's disease accelerate the aggregation of α‐synuclein into cytotoxic soluble oligomers (Barrett & Greenamyre, 2015; Luth et al., 2015; Meade et al., 2019; Paleologou et al., 2009; Tsigelny et al., 2008).
α‐Synuclein oligomers specifically, not the monomeric or fibril forms of α‐synuclein peptides, have been found to disrupt intracellular trafficking (Auluck et al., 2010; Chai et al., 2013; Hunn et al., 2015; Jang et al., 2010), disrupt normal calcineurin function (Martin et al., 2012), increase intracellular calcium levels (Bernal‐Conde et al., 2020; Martin et al., 2012), halt normal autophagy (Martinez‐Vicente et al., 2008; Wang et al., 2016), and cause synapse dysfunction and loss (Choi et al., 2015; Diógenes et al., 2012; Scott et al., 2010). The transsynaptic spread of extracellular α‐synuclein oligomers is hypothesized to underlie disease progression and correlates with Braak staging of PD (Hassink et al., 2018; Henderson et al., 2019) as well as Lewy body and synaptic pathology in neurons (Hansen & Li, 2012).
Currently there are no effective disease modifying therapeutics for PD and related synucleinopathies such as multiple system atrophy and dementia with Lewy bodies. Therapeutics that can effectively stop oligomer‐induced toxicity have the potential to treat the motor and cognitive symptoms resulting from synaptic dysfunction and prevent the spread of oligomer‐induced pathology during disease progression. Our goal was to identify anti‐α‐synuclein oligomer drug candidates by screening compounds for the ability to rescue α‐synuclein oligomer‐induced deficits in the target population: primary neurons.
We identified recombinant full‐length α‐synuclein protein oligomer preparations suitable for screening compound libraries that replicate the toxic effects of Parkinson's patient brain‐derived oligomers, using assays that measure two key aspects of cellular function known to be disrupted by α‐synuclein oligomers: intracellular lipid vesicle trafficking (Izzo, Staniszewski, et al., 2014) and chaperone‐mediated autophagy.
Treatment of mature primary hippocampal/cortical neuronal and glial cultures (21 days in vitro; DIV) with recombinant α‐synuclein oligomers as well as α‐synuclein oligomer species isolated from brain samples from individuals with PD, but not non‐PD age‐matched control individuals, resulted in lipid vesicle trafficking deficits. Treatment of neuronal cultures with recombinant α‐synuclein oligomers also upregulated the expression of lysosomal‐associated membrane protein‐2A (LAMP‐2A), a protein critically required for chaperone‐mediated autophagy. This is the first report demonstrating that recombinant α‐synuclein oligomers have a similar functional impact as PD patient brain‐derived α‐synuclein oligomers.
We then screened several libraries of small molecule compounds, including the NIH Clinical Collection to identify compounds capable of blocking recombinant α‐synuclein oligomer‐induced lipid vesicle trafficking deficits. Unexpectedly, the most effective compounds were selective sigma‐2 receptor allosteric antagonists, which blocked these deficits in a dose‐dependent manner. These compounds also blocked recombinant α‐synuclein oligomer‐induced LAMP‐2A upregulation. Molecular interactions between sigma‐2 receptor component proteins progesterone receptor membrane component 1(PGRMC1) and transmembrane protein 97 (TMEM97), α‐synuclein, and proteins that control vesicular tracking and autophagy (such as LC3B) may form the basis for these observations. Importantly, and for the first time, these data indicate that small molecule selective sigma‐2 receptor complex antagonists can impact a critical modulator in the α‐synuclein signaling cascade and stop oligomer‐induced deficits. Inhibitors that modulate sigma‐2 receptors may be therapeutic against oligomeric α‐synuclein‐induced neuronal dysfunction in PD and other α‐synucleinopathies.
2. MATERIALS AND METHODS
2.1. Neuronal cultures
All procedures were approved by the Institutional Animal Care and Use and Committee at Cognition Therapeutics (CogRx) and were in compliance with the Office of Laboratory Animal Welfare and the Guide for the Care and Use of Laboratory Animals, Eighth Edition. Hippocampal/cortical cultures were prepared from Sprague‐Dawley (Research Resource Identifier, RRID:RGD_1566440) embryonic rat brain as previously described (Izzo, Staniszewski, et al., 2014). Briefly, dissociated E18 hippocampal and cortical cells were plated at a density of 4.66 × 104 cells per cm2 in 384‐well poly‐d‐lysine coated plates (Greiner, Monroe, NC, USA) in serum‐free Neurobasal Media (Life Technologies, Carlsbad, CA, USA) supplemented with B27 (Life Technologies), Glutamax (Life Technologies), and antibiotics (penicillin 50 units/ml and streptomycin 50 mg/ml, Life Technologies). Cultures were maintained at 37°C in 5% CO2 with weekly media change for 3 weeks (21 DIV) prior to experimentation. These mixed cultures of hippocampal plus cortical neurons and glia were used for all in vitro experiments described. Healthy cultures typically contain 20%–35% microtubule‐associated protein 2 (MAP2)‐positive neurons (See Immunocytochemistry Assay below; MAP2; 1:5,000, 0.2 mg/ml, Millipore, catalog #AB5543, RRID:AB_571049). This defined media also prevents glial overgrowth by inhibiting glial cell division (Brewer et al., 1993). We previously characterized the glial population in these cultures based on the nuclear morphology visualized by the DNA‐binding dye (4′,6‐diamidino‐2‐phenylindole; DAPI) (Izzo, Staniszewski, et al., 2014). Approximately 27% of MAP2‐negative glial cells have a normal symmetrical nuclear morphology, with the remaining cells having an abnormal nuclear morphology and bright DAPI staining typical of fragmented and condensed chromatin, likely corresponding to unhealthy or dying glial cells (See supplemental figure 1 in: Izzo, Staniszewski, et al., 2014). The healthy glial population was further characterized by subtype based on protein expression. At 21 DIV, 36% ± 7% were OLIG2‐positive oligodendrocytes (OLIG2, 1:500, 0.3 mg/ml, Sigma‐Aldrich, St. Louis, MO, USA, catalog number ABN899, RRID:AB_2877641), and 7% ± 2% were astrocytes that expressed high levels of glial fibrillary acidic protein (GFAP; 1:500, 0.2 mg/ml, R&D Systems, Minneapolis, MN, USA, catalog number AF2594, RRID:AB_2109656) with labeled projections coming from the cell bodies, with the remainder likely microglia.
2.2. Oligomer preparation
Preparation of recombinant α‐synuclein oligomers: α‐synuclein oligomers were prepared as previously described (Martin et al., 2012) using Aβ oligomer to seed oligomerization of α‐synuclein monomers. To make Aβ oligomer seeds, synthetic human Aβ 1‐42 peptide (California Peptide Inc, American Peptide Company, Sunnyvale, CA, USA, cat #641‐15) was dissolved in 1,1,1,3,3,3‐hexa‐fluoro‐2‐propanol (HFIP) to remove secondary structure, and evaporated to a film at room temperature for 20 min using N2 gas. The film was dissolved in anhydrous dimethyl sulfoxide (DMSO; Sigma Aldrich, St. Louis, MO, USA, catalogue number D2650) and diluted to 100 µM with cold basal Medium Eagle media (BME, Life Technology, catalogue #21010) followed by incubation at 4°C for 24 hr to initiate oligomer formation. The resulting oligomer preparations were centrifuged at 16,000× g to remove any insoluble fibrils. Recombinant, human, wild‐type α‐synuclein was obtained from rPeptide (Bogart, GA, USA) and resuspended at 2 mg/ml in sterile water (Millipore, Burlington, MA, USA). Aβ oligomer preparation (1.78 µl) was added to 250 µl of α‐synuclein solution and stirred at room temperature for 20 min using a magnetic stir bar to form α‐synuclein oligomers. This stock preparation, containing 138 µM α‐synuclein and 714 nM Aβ was immediately diluted into Neurobasal media for treatment of cell cultures at the indicated final concentration (expressed as total α‐synuclein concentration). In all experimental conditions, the concentration of the Aβ seed was 1/193 of the indicated concentrations of α‐synuclein. For experiments with monomeric α‐synuclein, fresh peptide solution (2 mg/ml recombinant human wild‐type α‐synuclein in sterile water) was diluted directly in Neurobasal media prior to addition to cultures.
While many preparations of oligomeric α‐synuclein have been described in the literature, not all have demonstrated an impact on synaptic function (a tractable therapeutic intervention point, and therefore the focus of our studies). The method of preparing α‐synuclein oligomers used in these studies (vs. using α‐synuclein monomers or fibrils to seed oligomer formation) has been shown to effectively inhibit CREB phosphorylation and activate calcineurin in organotypic brain slices, as well as cause evoked memory impairments in mice that received acute intracerebroventricular injections (Martin et al., 2012).
2.3. Trafficking assay
Vesicular trafficking was measured using an adaptation of previously published methods (Yuanbin Liu & Schubert, 1997) as described (Izzo, Staniszewski, et al., 2014; Izzo, Xu, et al., 2014). Neurons were treated with α‐synuclein preparations and incubated for 24 hr at 37°C in 5% CO2. Tetrazolium salts (3‐(4,5‐dimethylthiazol‐2yl)‐2,5diphenyl tetrazolium bromide (MTT), Roche Molecular Biochemicals, Mannheim, Germany) were added to a final concentration of 0.75 mM and incubated at 37°C for 40–60 min. Time of incubation varied from 60 to 90 min to maximize the window between vehicle‐ and oligomer‐treated cells. Vesicular formazan remaining in cells was quantified using absorbance spectrometry (590 nm with 690 nm subtracted) following extraction with 1.6% Tween‐20. All compounds were tested in quadruplicate wells for each concentration in at least eight replicate experiments with data from all experiments pooled for analysis with means ± standard deviation (SD).
2.4. Immunoprecipitation of human non‐PD control‐ and PD patient‐derived α‐synuclein
Deidentified age‐ and gender‐matched PD and non‐PD control human cortex donor brain tissue was obtained through the Neurobiobank Brain and Tissue Repository (NBB), provided by McLean Hospital's Harvard Brain Tissue Resource Center and the Human Brain and Spinal Fluid Resource Center of the Veterans Affairs Greater Los Angeles Healthcare System under the Institutional Review boards of those institutions. Donor patients (n = 7) included six females and one male, aged 81–90 years, and diagnosed with PD ranging from early stage to late Braak and Del Tredici stage 4 (staging was unknown for two patients). Postmortem interval of PD tissue collection ranged from 5 hr 25 min to 18 hr 18 min. Donor non‐PD control individuals (n = 4) included three females and one male, aged 79 to 86 years. Postmortem interval of control tissue collection ranged from 18.5 to 29.05 hr. Human brain tissue (1.00 ± 0.01 g per immunoprecipitation) was homogenized (Dounce glass homogenizer) 1:7 (w/v) in ice‐cold buffer (20 mM Tris, 137 mM NaCl, pH 7.6 with 1 mM ethylenediaminetetraacetic acid (EDTA) and 1 mg/ml protease inhibitor cocktail Sigma P8340). The resulting suspension was then manually homogenized with 10 strokes of pestle‐A followed by 10 strokes of pestle‐B of a glass Dounce homogenizer set and then sonicated in an ice‐cold water bath. Centrifugation of the homogenate for 10 min at 4,500 rpm was performed to remove large cellular debris followed by ultracentrifugation of the supernatant for 1 hr at 36,500 rpm. Pre‐clearing of the supernatant using Protein A/G agarose was then performed followed by immunoprecipitation using a211‐agarose columns to isolate native α‐synuclein along with various isoforms and oligomeric species. Elution of α‐synuclein from the a211‐agarose columns was performed using heated 6 N guanidine HCl solutions followed by desalting steps using 10 kDa NMWCO spin columns to exchange the samples to phosphate‐buffered saline and frozen at −80°C until use. Samples were thawed a single time for application to neuronal cell cultures without dilution. For western blot analyses, samples were diluted 1:1 in sodium dodecyl sulfate (SDS) loading buffer and 30 µl added to gels. Due to the extremely limited amounts of patient brain samples, protein quantification was not performed, however, equal gram weights of PD and non‐PD brain tissue samples were prepared identically and compared side‐by‐side in the same experiment. These methods have been published previously as a poster (Silky et al., 2016).
2.5. Western blot analysis of patient‐derived α‐synuclein
α‐Synuclein preparations were run on 4%–15% Tris‐HCl nondenaturing gels (BioRad, Hercules, CA, USA) and electrophoresed in Tris‐glycine buffer (25 mM Tris Base, 192 mM glycine, pH 8.3) containing 0.1% SDS at 125 V for 120 min. The preparations were then transferred to a nitrocellulose membrane, probed with syn211 mouse monoclonal antibody to α‐synuclein (1 µg/ml, diluted 1:1,000, Abcam, Burlingame, MA, USA, catalog number ab80627, RRID:AB_1603277) followed by secondary goat anti‐mouse horse radish peroxidase‐conjugated secondary antibody (diluted 1:10,000, 1 mg/ml, Millipore, catalog number AP308P) and visualized with chemiluminescent detection (Immuno‐Star WesternC Kit, BioRad). Integrated gel band intensity (integrated density value, IDV) quantification following background correction was performed using the Alpha View FluorChem software program (Protein Simple, San Jose, CA, USA).
2.6. High‐throughput screening for anti‐α‐synuclein oligomer therapeutics
Unbiased screening approaches were utilized to identify potential therapeutics against α‐synuclein oligomer. The National Institutes of Health (NIH) Clinical Collection (NCC) small molecule drug collection (provided through the NIH Small Molecule Repository and obtained through Evotec (San Francisco, CA)) was screened in the trafficking assay at a single concentration (1 µM) in triplicate wells; drugs were considered effective if α‐synuclein oligomer‐induced membrane trafficking deficits were blocked (>80%) without having an effect on the rate of membrane trafficking when dosed in the absence of α‐synuclein oligomers (<20%). Dose–response analysis was performed for effective compounds (8‐point dilution range of 0.5–20 µM) and tested for dose‐dependent block of membrane trafficking deficits. This assay performs with a z′ factor = 0.91. z′ factors are a statistical calculation in high‐throughput screening used to warrant the use of a specific treatment, and z′ factors between 0.5 and 1.0 are considered excellent, indicating strong reliability and repeatability of the assay (Zhang et al., 1999). Bioinformatics pathway analysis (Metacore, Clarivate Analytics, Philadelphia, PA, USA) was employed to identify proteins and shared signaling pathways targeted by effective drugs. Additionally, we screened a proprietary library of CNS drug‐like small molecules that were previously used to identify therapeutic candidates that block Aβ oligomer‐induced vesicle trafficking deficits in mature, primary rat hippocampal, and cortical cultures (Izzo, Staniszewski, et al., 2014).
2.7. Radioligand binding
Radioligand competition assays to determine affinity for sigma‐2 receptors were performed at Eurofins Cerep SA (Le Bois l'Évêque, France) in membranes from human lymphocyte cell lines (Jurkat cells) using 25 nM [3H]1,3‐di(2‐tolyl) guanidine in the presence of 1 μM (+)‐pentazocine. Non‐specific binding was defined in the presence of 10 μM haloperidol (Ganapathy et al., 1999).
2.8. Immunocytochemistry
To determine immunolocalization of α‐synuclein oligomer, rat hippocampal/cortical primary cultures were treated for 24 hr with α‐synuclein oligomer (0.5 µM). Representative images were captured by fixing with 3.75% formaldehyde for 15 min, blocking with 5% normal goat serum, permeabilizing with 0.5% Triton X‐100, and incubating with primary antibodies for α‐synuclein oligomer (ASYO5; 1:1,000, 1 mg/ml, Agrisera, Vännäs, Sweden, catalog number AS13 2718, RRID:AB_2629502) and MAP2 (1:5,000, 0.2 mg/ml), then fluorescently labeled secondary antibodies (1:1,000, 2 mg/ml, Invitrogen, Carlsbad, CA, USA).
To assess the ability of test compounds to reduce the neuronal localization of α‐synuclein oligomers and recover LAMP‐2A expression, cultures were treated by adding oligomers 60 min prior to the addition of compounds, followed by 24 hr incubation at 37°C. Cells were fixed with 3.75% formaldehyde for 15 min, blocked with 5% normal goat serum, permeabilized with 0.5% Triton X‐100, and incubated with primary antibodies for α‐synuclein oligomer (1:1,000, 1 mg/ml ASYO5, Agrisera), MAP2 (1:5,000, 0.2 mg/ml), LAMP‐2A (1:400, 1 mg/ml, Thermo Scientific, Waltham, MA, USA, catalog number 51‐2200, RRID:AB_2533900), and then fluorescently labeled secondary antibodies (1:1,000, 2 mg/ml, Invitrogen).
Images were acquired on a ThermoFisher CellInsight CX7 platform automated microscope with a 20×, 0.75 NA objective and analyzed using ThermoFisher/Cellomics Neuronal Profiling bioapplication set to measure punctate labeling of peptide preparations along MAP2‐labeled neurites. For each replicate experiment, at least 100 neurons were sampled from four replicate wells for each experimental condition (400 to 500 neurons per experimental condition). All data presented for α‐synuclein associated with neurons represent total intensity of α‐synuclein label in neurite spots per neuron, in relative fluorescent units (RFU), unless otherwise indicated.
2.9. Statistical tests
For image analysis, at least 100 neurons were sampled using unbiased automated algorithms from four replicate wells for each experimental condition (400 to 500 neurons per experimental condition). The number of replicates from separate cell culture preparations is reported for each experiment. The number of replicates were determined a priori to attain statistical power of 80% and p values less than 0.05 were considered to be statistically significant (determined using G*Power software, Heinrich Heine University, Düsseldorf, Germany) (Faul et al., 2007). Statistical significance was determined for non‐linear curve fitting by an extra sum of squares F‐test using Prism (GraphPad Software, San Diego, California, USA). A D'Agostino‐Pearson normality test was used prior to all statistical analyses. No blinding or randomization of experimental groups was conducted. Outliers were not removed.
3. RESULTS
3.1. α‐Synuclein oligomer exposure resulted in membrane trafficking deficits in cultured primary neurons
We examined α‐synuclein oligomer effects in rat primary neurons, grown for 21 DIV, which contain a mixture of both MAP2‐positive neurons and various glial cell subtypes (Izzo, Staniszewski, et al., 2014) and express the full complement of synaptic proteins characteristic of neurons in the mature brain (Torre & Nicholls, 1998).
We and others have previously reported that the MTT assay can be used to measure changes in membrane trafficking rates induced by pathogenic oligomers (Hong et al., 2007; Izzo, Staniszewski, et al., 2014; Kreutzmann et al., 2010; Liu & Schubert, 1997). Significant increases in the rate of exocytosis of intracellular vesicles caused by pathogenic Aβ oligomers were observed using this assay, and the increased exocytosis rate is associated with a loss of synapses in neurons (Izzo, Xu, et al., 2014). Disruption of membrane trafficking in neurodegenerative diseases leads to the loss of neuronal function and subsequent loss of neurons (Hunn et al., 2015; Tong et al., 2004).
In the MTT assay, yellow tetrazolium salts are endocytosed by cells and reduced to insoluble purple formazan in the endosomal pathway. Following exocytosis, the insoluble formazan precipitates in aqueous media, appearing as needle‐shaped crystals on the plasma membrane. Here we show that at 60 min after MTT exposure, reduced cargo dye (formazan) was visualized within intracellular vesicles in vehicle‐treated cultures (Figure 1a), while cultures treated with α‐synuclein oligomer for 24 hr exhibited predominantly exocytosed dye crystals (Figure 1b). This indicates that α‐synuclein oligomers accelerate the rate of exocytosis. Assaying membrane trafficking as a function of α‐synuclein concentration, we found that α‐synuclein oligomers and high concentrations of α‐synuclein monomer caused dose‐dependent membrane trafficking deficits by increasing the rate of exocytosis in primary neurons (Figure 1c, wild‐type α‐synuclein oligomers (half maximal effective concentration (EC50) = 212 nM) and α‐synuclein monomer (EC50 = 938 nM). Together, these data demonstrate that α‐synuclein oligomers caused significant trafficking deficits when compared with vehicle at all concentrations >30 nM (p < 0.0500, Student's t test, n = 8), and α‐synuclein monomer caused deficits at concentrations >250 nM (p < 0.0500, Student's t test, n = 8) (Figure 1c, f = 6.821 (1, 792), n = 8, p < 0.0010). Side by side comparison of oligomeric and monomeric preparations of recombinant full‐length α‐synuclein in western blots (Figure 2a) revealed that most of the protein remained in monomeric form (~17 kDa) in both preparations, and both preparations contain higher weight oligomers (>30 kDa) as well. The oligomeric preparation, however, contained a higher concentration of these >30 kDa species compared with the monomeric preparation. As is typically the case for a variety of proteins, only a small percentage of the recombinant monomer protein oligomerizes during the incubation process; most of it remains in the monomeric form. Similarly, for aggregation‐prone proteins, freshly prepared monomer can also rapidly form low concentrations of oligomers, especially under denaturing gel chromatography conditions. This widely recognized phenomenon is also observed when oligomers are made from other synthetic or recombinant proteins, such as Aβ 1‐42 (Izzo, Staniszewski, et al., 2014). The functional impact of oligomers that do form is profound, however; despite this small increase in α‐synuclein oligomer concentration, recombinant oligomers are much more potent at inhibiting intracellular lipid vesicle trafficking than monomer, corresponding to a measurable fourfold difference in EC50 potency in the trafficking assay (Figure 1c).
FIGURE 1.
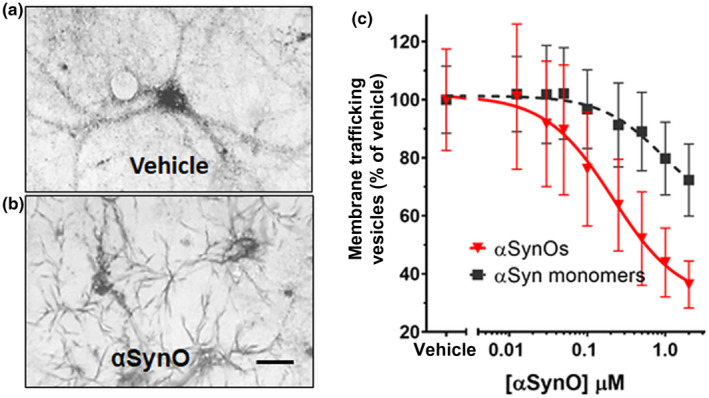
α‐synuclein oligomers cause trafficking deficits in vitro. Intracellular vesicles were labeled with reduced cargo dye (formazan) in vehicle‐treated cultures (a). In contrast, reduced formazan was trafficked out of the cell more rapidly (b) following addition of recombinant α‐synuclein oligomer (1 µM, total α‐synuclein concentration). Size bar = 20 microns. The EC50 was calculated by determining the percentage of dye in vesicles as a function of α‐synuclein concentration. α‐synuclein oligomers (red triangles) dose‐dependently induced deficits in vesicle trafficking (EC50 = 212 nM) (c), while α‐synuclein monomer only induced trafficking deficits at concentrations >1.0 µM (c, black squares; EC50 = 938 nM). Representative images (a and b) were taken 60 min following exposure to MTT formazan. Data points represent means ± SD for three replicate experiments from separate cell culture preparations [Color figure can be viewed at wileyonlinelibrary.com]
FIGURE 2.

Comparison of recombinant α‐synuclein oligomers with α‐synuclein isolated from PD patient brain samples. Denatured western blot comparing recombinant α‐synuclein oligomer (αsynO) with α‐synuclein monomer (αsyn Mono) labeled with α‐synuclein antibody (syn211) reveals that both preparations contain monomer (~17 kDa) and higher weight oligomers (>30 kDa), however, the oligomeric preparation contains a higher concentration of these >30 kDa species compared with the monomeric preparation (a). A total of 1 µg of synthetic α‐synuclein protein was loaded in each lane in (a). Denatured western blots of immunoprecipitated PD patient frontal cortex samples demonstrated heterogeneous populations of α‐synuclein assemblies (b). Western blots of α‐synuclein immunoprecipitated from six different PD patients (lanes 2–7) and two non‐PD control subjects (lanes 1 and 8) exhibit major bands >100 kDa. Using a longer exposure time, a 36 kDa doublet band can be observed in the bottom half of the blot in PD patient samples (lanes 2–7) that is absent in non‐PD controls (lanes 1 and 8, quantified in c), corresponding with α‐synuclein oligomeric species (Ardah et al., 2014; Tsika et al., 2010; Winner et al., 2011); full gel is shown in Figure S1). (c) Measurement of integrated gel band intensity (integrated density value, IDV) in the bracketed range shown in b using FluoroChem reveals substantial differences in low molecular weight range oligomer concentrations between PD and non‐PD brains
3.2. Recombinant α‐synuclein oligomer and PD patient‐derived α‐synuclein samples contained similar low molecular weight species that were absent in non‐PD control samples
Previous studies indicate that extreme non‐physiological conditions, such as high detergent and temperature, are required to isolate appreciable concentrations of α‐synuclein from postmortem PD‐patient brain tissue (Deramecourt et al., 2006; Garcia‐Esparcia et al., 2015; Kellie et al., 2014; Paleologou et al., 2009). To avoid such conditions, we explored a number of syn211 antibody‐mediated immunoprecipitation methods to maximize the recovery of α‐synuclein protein from PD patient postmortem brain tissue, and chose elution with 6 M guanidine HCl as the optimal condition, balancing physiological relevance and recovery concentrations.
We compared the protein size (molecular weight) ranges of PD patient brain‐derived α‐synuclein species and recombinant α‐synuclein oligomer via western blot under nondenaturing conditions using antibody syn211 (Figure 2). Freshly prepared α‐synuclein oligomer consisted of protein sizes ranging from 17 to 100 kDa with notable low molecular weight bands at 36 and 17 kDa (Figure 2a). These band sizes are consistent with the α‐synuclein oligomer size range previously reported (Ardah et al., 2014; Luk et al., 2009; Recasens et al., 2014; Tong et al., 2010; Tsika et al., 2010; Winner et al., 2011). Similar protein size ranges were detected in PD patient brain‐derived α‐synuclein, but notably, low molecular weight bands (17 kDa, corresponding to monomer and 36 kDa, corresponding to dimer) were not present in non‐PD control subjects (Figure 2b). This result is in agreement with previous reports stating that the α‐synuclein dimer, which has an important role in α‐synuclein oligomer aggregation and PD pathology (Medeiros et al., 2017; Roostaee et al., 2013) is present at higher levels in PD patients than non‐PD controls (Papagiannakis et al., 2018). Additional high molecular weight bands (>75 kDa) were consistently observed, but to variable extents, in both PD patients and non‐PD control subjects (Figure 2b). Density quantification indicates greater protein density in 10–36 kDa bands in tissue from PD patients relative to non‐PD patients (Figure 2c).
3.3. α‐Synuclein oligomers isolated from PD patient brains and recombinant α‐synuclein oligomers caused similar lipid vesicle trafficking deficits in vitro
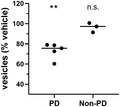
We compared the effects of PD patient brain‐derived α‐synuclein and recombinant wild‐type α‐synuclein oligomers on membrane trafficking rates using the MTT assay (Figure 3). PD patient brain‐derived α‐synuclein species (Figure 3) and recombinant α‐synuclein oligomers (1 µM, Figure 1) induced similar deficits in membrane trafficking rate compared with a vehicle control. In contrast, α‐synuclein isolated from the brains of non‐PD control subjects (Figure 3) induced no change in membrane trafficking rate compared with vehicle control. Thus, both recombinant α‐synuclein oligomers and PD patient brain‐derived α‐synuclein species, but not α‐synuclein species derived from non‐PD control brains, impact cellular membrane trafficking rates. Because the only difference in α‐synuclein species detected between PD and non‐PD brain on western blots (Figure 2) were the higher concentration of oligomers contained in PD brains, it is likely that the impact on trafficking observed is due to oligomers.
FIGURE 3.

PD patient brain‐derived α‐synuclein oligomers induce membrane trafficking deficits in vitro. Compared with vehicle‐treated cells, α‐synuclein immunoprecipitated from brain samples from multiple individuals with PD caused membrane trafficking deficits (**p ≤ 0.0100); α‐synuclein from brain samples from individuals without PD did not (n.s., not significantly different from vehicle‐treated cells). One‐way ANOVA with Dunnett's test for multiple comparisons. Each data point represents the mean value for an individual patient determined in quadruplicate from three separate cell preparations; undiluted samples were added to the assay wells. Black lines indicate group means
3.4. Small molecule compounds inhibited membrane trafficking deficits caused by α‐synuclein oligomers in vitro
To investigate the potential of brain‐penetrant small molecule therapeutic approaches, we screened for compounds capable of minimizing or eliminating α‐synuclein oligomer‐associated membrane trafficking deficits. The screening method was based on the MTT assay and optimized for performance in 384‐well microtiter plates with automated liquid handling robotics for compound formatting and assay plate replication as previously reported (Izzo, Staniszewski, et al., 2014). We set an acceptable window for oligomer‐induced effects with a 50%–80% loss of normal membrane trafficking rates due to treatment with α‐synuclein oligomers, compared with vehicle treatment. We used this assay to screen the NIH Small Molecule Repository (725 compounds that have been tested in phase I‐III clinical trials: full list shown in Table S1). Drugs were screened at a single assay concentration of 1 µM following the addition of α‐synuclein oligomers for 24 hr, in quadruplicate wells replicated in three experiments from independent cell primary cell preparations (n = 3 experiments) with greater than 80% reversal of α‐synuclein oligomer effects set as a hit. Statistical analysis indicated that these conditions had a power of 95% (GPower software, (Faul et al., 2007)) to detect false negatives. Control wells consisted of α‐synuclein oligomers (1 µM) and vehicle treatment without test compounds. Of the 725 compounds, 17 compounds significantly blocked α‐synuclein oligomer‐induced membrane trafficking deficits. Of these, compounds with limited brain penetration or significant expected toxicity (such as oncology or anti‐fungal therapeutics) were eliminated, and the resulting six hits were assayed for membrane trafficking effects across an 8‐point dilution range (0.5–20 µM, Figure 4). Bioinformatics pathway analyses (MetaCore, Clarivate Analytics, Philadelphia, PA, USA) indicated that these compounds impact the retinoic acid pathway, which has previously been implicated in PD (Esteves et al., 2015; Takeda et al., 2014), confirming the disease relevance of this screening assay. Three of six compounds were weakly active (Figure 4a–c), two compounds blocked α‐synuclein oligomer‐induced membrane trafficking deficits at concentrations lower than 2.5 µM (lovastatin EC50 < 1 µM, carbamazepine EC50 = 2.2 µM, Figure 4d,e), and one compound (thioridazine) was cytotoxic at concentrations above 1 µM (Figure 4f).
FIGURE 4.
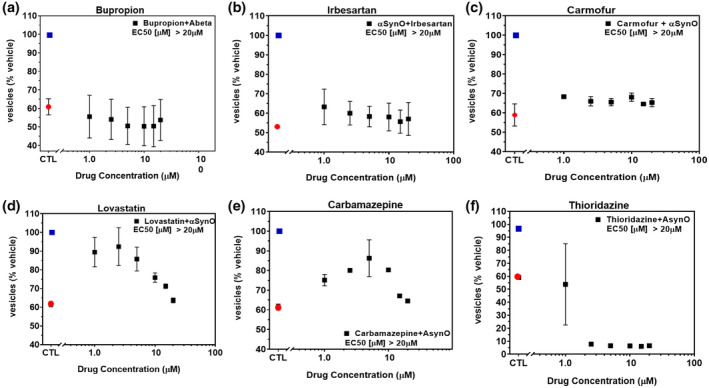
NIH Clinical Collection (NCC) small molecules inhibit α‐synuclein oligomer‐induced trafficking deficits. Treatment of primary neurons with α‐synuclein oligomer (1.0 μM final concentration) caused significant deficits in membrane trafficking rates (red dot) when compared with vehicle alone (blue dot, p < 0.0001, One‐way ANOVA, Dunnett's multiple comparisons). Seven hundred and twenty‐five NIH small molecules were tested in single‐point screens (data not shown) for their ability to block oligomer‐induced deficits and a subset of hit compounds (a–f) were tested in a range of doses with select examples shown above. Addition of (a) bupropion, (b) irbesartan, or (c) carmofur weakly inhibited α‐synuclein oligomer‐induced trafficking deficits. (d) Lovastatin and (e) carbamazepine inhibited deficits at low concentrations but were toxic at concentrations >5 µM. (f) Thioridazine was cytotoxic at concentrations >1 µM. All data points represent means ± SD for four to six replicate experiments in separate cell culture preparations [Color figure can be viewed at wileyonlinelibrary.com]
We then screened a proprietary CogRx library of high affinity sigma‐2 allosteric antagonists to identify therapeutic candidates capable of blocking oligomer‐induced trafficking deficits. Several hits were obtained (Figure 5); the affinities of these compounds at the sigma‐2 receptor complex are in the low nanomolar range: CT1978 (Kd = 9.2 nM), CT1970 (Kd = 1.8 nM), CT2168 (Kd = 2 nM), CT1681 (Kd = 12 nM), CT1696 (Kd = 11 nM), and CT1925 (Kd = 18 nM). When dosed against α‐synuclein oligomers, all of them inhibited α‐synuclein oligomer‐induced trafficking deficits in a dose‐dependent manner (CT1978 (EC50 = 1.48 μM), CT1970 (EC50 = 6.50 μM), CT2168 (EC50 = 1.5 μM), CT1681 (EC50 = 9.44 μM), CT1696 (EC50 = 15.53 μM), and CT1925 (EC50 = 8.95 μM) Figure 5a–f, respectively).
FIGURE 5.

α‐Synuclein oligomer‐induced trafficking deficits are blocked by CogRx sigma‐2 receptor antagonist small molecule therapeutic candidates. (a–f) Treatment of primary neurons with α‐synuclein oligomer (1.0 μM final concentration) caused significant deficits in membrane trafficking rates (red dot) when compared with vehicle alone (blue dot, p < 0.0001, One‐way ANOVA, Dunnett's multiple comparisons). CogRx small molecule sigma‐2 antagonists blocked the effect of recombinant α‐synuclein oligomer by >80%, in a dose‐dependent manner (p < 0.0010 for greatest dose versus α‐synuclein alone, One‐way ANOVA, Dunnett's multiple comparisons), while having no effect on membrane trafficking when dosed without the addition of α‐synuclein oligomer (data not shown). All data are means ± SD for 6 replicate experiments in separate cell culture preparations [Color figure can be viewed at wileyonlinelibrary.com]
3.5. α‐Synuclein oligomer‐induced LAMP‐2A expression increase is blocked by CogRx small molecule sigma‐2 allosteric antagonist therapeutic candidates
Misfolded proteins and damaged cells are normally degraded and cleared via autophagy pathways regulated by the sigma‐2 receptor complex (Mir et al., 2013); breakdown of these pathways is implicated in PD pathology (Cuervo & Wong, 2014; Xilouri et al., 2009, 2016; Yue et al., 2009), resulting in build up of α‐synuclein and other proteins in neurons (Spillantini et al., 1997). Chaperone‐mediated autophagy via LAMP‐2A is specifically responsible for α‐synuclein degradation in neurons and turnover of α‐synuclein monomers (Sala et al., 2016; Vogiatzi et al., 2008) and LAMP‐2A expression is dysregulated in PD (Mak et al., 2010; Orenstein et al., 2013; Sala et al., 2016). Disruption in chaperone‐mediated autophagy with an inhibitor or RNA interference to LAMP‐2A can lead to accumulation of α‐synuclein in neuronal cells (Vogiatzi et al., 2008). Because of these links between chaperone‐mediated autophagy, sigma‐2 receptors, LAMP‐2A, α‐synuclein, and PD, we examined whether LAMP‐2A expression was affected by exogenous α‐synuclein oligomers in vitro, and whether sigma‐2 antagonists could impact this. We quantified the intensity of LAMP‐2A immunofluorescence associated with primary neuronal cells co‐labeled for MAP2 and α‐synuclein oligomer (Figure 6). Neurons treated with α‐synuclein oligomers (0.5 μM) exhibited increased LAMP‐2A immunolabeling compared with vehicle (Figure 6a,b). Sigma‐2 receptor antagonist compounds CT1978 and CT2168, which actively blocked α‐synuclein oligomer‐induced membrane trafficking deficits (Figure 5), blocked the α‐synuclein oligomer‐induced increase in LAMP‐2A expression (Figure 6c,d). Because the CogRx compounds are known to be specific antagonists at the sigma‐2 receptor complex, these results confirm an important role for the sigma‐2 receptor complex in the regulation of LAMP‐2A‐mediated autophagy pathways, and suggest that sigma‐2 receptor antagonists may have therapeutic potential in PD.
FIGURE 6.

CogRx sigma‐2 receptor antagonists block α‐synuclein oligomer‐induced autophagy dysregulation. Neuronal cultures were treated with a low concentration (0.5 µM) of recombinant α‐synuclein oligomer for 1 hr followed by CogRx compounds for 24 hr. Cells were fixed and immunolabeled to visualize MAP2‐positive neuron expression of LAMP‐2A and α‐synuclein oligomer (antibody ASYO5). LAMP‐2A expression was quantified by measuring the relative fluorescent units of puncta spots per neuron and normalized to a vehicle control. Vehicle wells demonstrated endogenous expression of LAMP‐2A (a). α‐Synuclein oligomers exhibit punctate distribution on neurons and increased LAMP‐2A expression by > 75% (b). Treatment with CogRx compounds CT1978 (representative image, c) and CT2168 decreased α‐synuclein oligomer (α‐SynO) puncta intensity and LAMP‐2A puncta count per neuron, more closely resembling vehicle control wells (d). Data points represent means ± SD for four replicate experiments. (**p < 0.0100, ANOVA with Dunnett's test for multiple comparisons; n.s., not significantly different compared with vehicle‐treated cells.) [Color figure can be viewed at wileyonlinelibrary.com]
4. DISCUSSION
The protein α‐synuclein has a crucial role in PD and related synucleinopathies. Mutations in the α‐synuclein gene (SNCA) encoding mutant α‐synuclein forms such as A30P and A53T lead to familial early‐onset PD. Both mutant forms of α‐synuclein bind more strongly (two‐ to sixfold) to chaperone‐mediated autophagy receptor LAMP‐2A than does wild‐type α‐synuclein, but do not translocate into the lysosomal lumen, impairing degradation of other substrates (Cuervo et al., 2004) and shifting cellular disposal pathways to upregulate secretion of protein into the extracellular space. A variety of age‐related insults such as oxidative stress (Esteves et al., 2009) impact wild‐type α‐synuclein structure and associated function, leading to protein accumulation and subsequent oligomerization. α‐Synuclein amplifies the redox consequences of mitochondrial dysfunction in dopaminergic neurons (Van Laar et al., 2020). α‐Synuclein oligomers are the most toxic structural form of the protein (Karpinar et al., 2009), triggering autophagy/lysosomal dysregulation, synaptic dysfunction, mitochondrial disruption, and ER and oxidative stress, and secretion into extracellular fluid leading to transsynaptic spread and disease progression (Fields et al., 2019).
The development of novel therapeutic approaches that alleviate neuronal dysfunction and progression of PD pathology caused by α‐synuclein oligomers is an urgent unmet medical need (Fields et al., 2019; Shihabuddin et al., 2018). Cellular models using disease‐relevant α‐synuclein oligomers that replicate patient brain‐derived oligomer toxicity on target cell populations (neurons and glia) can be an effective platform for identifying potential therapeutics.
To establish such models, we began by identifying a method for generating recombinant full‐length α‐synuclein oligomers that produced oligomers that replicate the toxicity of patient brain‐derived species. Many such methods of generating α‐synuclein oligomers from wild‐type or modified protein have been published (Benner et al., 2008; Choi et al., 2013; Danzer et al., 2007; Yanying Liu et al., 2011; Outerio et al., 2009; Yu et al., 2010). Oligomers generated by seeding wild‐type full length recombinant α‐synuclein protein with extremely low concentrations of Aβ 1‐42 oligomers (thought to act as templates to promote oligomerization of α‐synuclein; Mandal et al., 2006; Martin et al., 2012; Masliah et al., 2001; Tsigelny et al., 2008)) have been reported to cause signaling deficits at low concentrations. Here for the first time, the effects of recombinant α‐synuclein oligomers made with this method were compared with Parkinson's patient brain‐derived α‐synuclein oligomer species effects on neurons and glia in primary culture. Both oligomer preparations disrupted normal membrane trafficking in a similar manner, whereas oligomers isolated from non‐PD age‐matched control brains with identical methods did not. This suggests that recombinant α‐synuclein oligomers made using this method are disease relevant and appropriate for use in compound screening models of the disease process in vitro, with the much less readily available patient brain‐derived oligomers used to confirm results obtained with recombinant oligomers.
Comparison of recombinant α‐synuclein oligomers with human‐derived α‐synuclein species using western blot revealed low molecular weight species in both the recombinant α‐synuclein oligomer and PD patient brain‐derived α‐synuclein samples, but not non‐PD control samples. Consistent with previous reports, these low molecular weight α‐synuclein oligomeric species potently induce changes in trafficking and autophagy consistent with disease pathology (Tsika et al., 2010; Winner et al., 2011). Similarly, low molecular weight α‐synuclein species have been shown to disrupt synaptic vesicle fusion and transmission (Medeiros et al., 2017). Notably, the human brain‐derived α‐synuclein preparation described here was shown for the first time to yield α‐synuclein protein species that caused trafficking deficits. Future studies will be required to characterize recombinant and PD patient brain‐derived oligomers in more detail with larger numbers of patient brain samples. Evidence indicates that soluble extracellular α‐synuclein oligomers can be transmitted between neighboring cells, which is thought to be the mechanism of the spread of disease pathology (Domert et al., 2016). Addition of exogenous recombinant α‐synuclein oligomers to primary neurons in culture may model this aspect of PD pathology in addition to intracellular effects. α‐Synuclein monomer had reduced effects on membrane trafficking deficits when compared with oligomers, an important functional difference between the two structural forms that may provide insight into early stages of disease development.
Cellular assays that measure processes disrupted in disease in primary neurons are also important for translational modeling of disease. We chose to use assays that measure two key aspects of neuronal function known to be disrupted by α‐synuclein oligomers: intracellular lipid vesicle trafficking (Izzo, Staniszewski, et al., 2014) and chaperone‐mediated autophagy. Intracellular lipid vesicle trafficking‐dependent processes are disrupted in PD and are thought to underlie cognitive and motor deficits (Alvarez‐Erviti et al., 2011; Ben Gedalya et al., 2009; Chai et al., 2013; Jang et al., 2010; Lee et al., 2005), stemming from α‐synuclein's central role in normal soluble N‐ethylmaleimide‐sensitive fusion attachment protein receptor (SNARE)‐mediated vesicle trafficking and regulation of synaptic vesicle exocytosis (Burré et al., 2014; Diao et al., 2013; Hunn et al., 2015). We used a previously established assay of intracellular lipid vesicle trafficking rate (Izzo, Staniszewski, et al., 2014; Izzo, Xu, et al., 2014) to model this process; this assay has been used to identify disease‐modifying Alzheimer's therapeutic candidates.
Chaperone‐mediated autophagy is one of several processes that degrade and remove altered proteins in response to cell stress or toxic material (Martinez‐Vicente et al., 2008; Vogiatzi et al., 2008), and regulates amino acid recycling and protein quality control (Martinez‐Vicente & Cuervo, 2007; Sala et al., 2016). The lysosomal receptor LAMP‐2A controls the translocation of substrates to the lysosomal lumen, and the rate of chaperone‐mediated autophagy is regulated by the assembly and disassembly of the LAMP‐2A translocation complex (Sala et al., 2016). LAMP‐2A function is pivotal to PD disease progression (Cuervo & Wong, 2014; Mak et al., 2010; Martinez‐Vicente & Cuervo, 2007; Sala et al., 2016; Vogiatzi et al., 2008). Disruption of chaperone‐mediated autophagy with a LAMP‐2A inhibitor can lead to accumulation of α‐synuclein in neuronal cells (Vogiatzi et al., 2008). α‐Synuclein and LAMP‐2A co‐immunoprecipitate in culture (Vogiatzi et al., 2008) and overexpressed wild‐type or mutant α‐synuclein binds to LAMP‐2A and disrupts internalization and degradation (Cuervo et al., 2004; Fonseca et al., 2015; Roberts & Brown, 2015; Sala et al., 2016). These and other data lead to the hypothesis that increasing LAMP‐2A levels is a compensatory mechanism to increase the autophagic response to toxic α‐synuclein oligomers (Mak et al., 2010; Orenstein et al., 2013). In agreement with this, the present results indicate that neurons treated with recombinant α‐synuclein oligomers significantly upregulate LAMP‐2A protein expression.
To identify small molecule drug candidates capable of blocking recombinant α‐synuclein oligomer‐induced toxicity, we screened several compound libraries (including the NIH Clinical Collection) in the previously established lipid vesicle trafficking rate assay. Unexpectedly, potent and specific sigma‐2 receptor antagonists (Izzo, Staniszewski, et al., 2014; Izzo, Xu, et al., 2014) most effectively dose‐dependently blocked α‐synuclein oligomer‐induced deficits in membrane trafficking, and also inhibited LAMP‐2A upregulation by oligomers. The effective inhibitor compounds identified within the NIH Clinical Collection partially reduced the effects of α‐synuclein oligomers at low concentrations, but then were cytotoxic themselves at higher concentrations. In contrast, the small molecule sigma‐2‐specific CogRx inhibitors were effective at low concentrations and did not exhibit cytotoxicity at similar higher concentrations. This is the first demonstration that allosteric antagonists of the sigma‐2 receptor complex have anti‐α‐synuclein oligomer effects.
One possible molecular basis for these observations is the direct role that sigma‐2 receptor complex component proteins PGRMC1 (Riad et al., 2018, 2020; Xu et al., 2011) and TMEM97 (Alon et al., 2017) have in both intracellular lipid vesicle trafficking and autophagy. PGRMC1 contains several immunoreceptor tyrosine‐based activation motif consensus sequences and binds directly to several vesicle trafficking regulatory proteins including N‐ethylmaleimide‐sensitive factor, microtubule‐associated proteins 1A/1B light chain 3B, ultra‐violet radiation resistance‐associated gene (UVRAG), unc‐51‐like autophagy activating kinase 2, autophagy‐related 5, and RB1‐inducible coiled‐coil protein 1 (Behrends et al., 2010; Mir et al., 2013). PGRMC1 regulates trafficking of a wide variety of transmembrane receptors between subcellular compartments and the plasma membrane and is required to stabilize these receptors in the plasma membrane including epidermal growth factor receptor (Ahmed et al., 2010), membrane progesterone receptor α (Thomas et al., 2014), glucagon‐like peptide receptor type 1 (Zhang et al., 2014), netrin receptor (UNC‐40/deleted in colorectal cancer; UNC‐40/DCC) (Runko & Kaprielian, 2004), and insulin receptors (Hampton et al., 2018); reduction of PGRMC1 levels via genetic knockdown results in internalization of these receptors. Direct binding of PGRMC1 to MAP1LC3B and UVRAG is required for normal autophagy (Mir et al., 2013). Both lipid vesicle formation/trafficking and autophagy place large demands on cellular lipid membrane synthesis and degradation, and both PGRMC1 and TMEM97 directly regulate cholesterol synthesis. Most of TMEM97’s sequence (aa 10‐158) is an Expera domain, likely to possess direct sterol isomerase catalytic activity (Sanchez‐Pulido & Ponting, 2014). TMEM97 itself binds directly to NPC1 (the protein that is mutated in the sphingolipid storage disorder Niemann–Pick disease); reduction of TMEM97 upregulates NPC1 and restores cholesterol synthesis and trafficking in Niemann–Pick cells (Ebrahimi‐Fakhari et al., 2015). PGRMC1 interacts with Nr4a1 (an immediate early gene required for the induction of several genes encoding steroidogenic enzymes) to regulate neurosteroid synthesis or signaling in neuronal cell lines (Intlekofer et al., 2019) a possible protective mechanism in response to neuroinflammatory signaling. In non‐neuronal cells, PGRMC1 regulates cholesterol synthesis via two direct binding interactions; to P450 proteins (Rohe et al., 2009) and to Insig and SCAP (Hughes et al., 2007; Suchanek et al., 2005); under low cholesterol conditions, Insig/SCAP dissociates from PGRMC1 and translocates to the nucleus where they mediate SRE‐related gene transcription). Finally, both PGRMC1 and TMEM97 bind directly to the low‐density lipoprotein receptor (LDL); this complex binds LDL, apoE, and monomeric and oligomeric Aβ and contributes to their internalization into neurons (Riad et al., 2018).
α‐Synuclein oligomers bind directly to PGRMC1 in brain synaptosomes (Betzer et al., 2015), and α‐synuclein binds directly to LAMP‐2A in the process of chaperone‐mediated autophagy (Schneider & Cuervo, 2014; Vogiatzi et al., 2008). α‐Synuclein oligomers interact with and disrupt cholesterol‐rich lipid membranes and disrupt related downstream signaling effects, including the process of autophagy (Galvagnion, 2017; Galvagnion et al., 2016; Plotegher et al., 2017; Stefanovic et al., 2014; Van Maarschalkerweerd et al., 2015). Thus, allosteric antagonists of the sigma‐2 receptor proteins may alleviate the negative impacts of α‐synuclein oligomers through a variety of mechanisms.
The present results are the first demonstration that PD patient brain‐derived α‐synuclein oligomers and recombinant α‐synuclein oligomers both inhibit intracellular lipid vesicle trafficking, providing strong support for the disease relevance of recombinant α‐synuclein oligomers made using the described methods and the translational applicability of using recombinant oligomers in screens to develop drug candidates. These results are also the first demonstration that sigma‐2 allosteric antagonists effectively reduced both the α‐synuclein oligomer‐induced lipid vesicle trafficking deficit and upregulation of LAMP‐2A. The sigma‐2 receptor complex proteins regulate multiple cellular damage response pathways in which α‐synuclein directly participates, and that are impacted in PD. These results suggest that the ability of sigma‐2 allosteric antagonists to block α‐synuclein oligomer toxicity may result from the role of sigma‐2 receptor proteins in regulating intracellular lipid vesicle trafficking, autophagy, and cholesterol metabolism. These data support the hypothesis that targeting the sigma‐2 receptor complex with brain‐penetrant small molecule antagonists represents a tractable therapeutic approach to alleviating α‐synuclein‐induced pathology in PD and other α‐synucleinopathies.
DECLARATION OF TRANSPARENCY
The authors, reviewers and editors affirm that in accordance to the policies set by the Journal of Neuroscience Research, this manuscript presents an accurate and transparent account of the study being reported and that all critical details describing the methods and results are present.
CONFLICT OF INTEREST
All authors were employees of Cognition Therapeutics, Inc at the time this work was performed. There is no other conflict of interest to report.
AUTHOR CONTRIBUTIONS
All authors had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. Conceptualization, S.M.C., N.J.I., and H.S.; Methodology, C.S.L., R.Y., N.J.I., C.R., and K.M.L.; Software, C.S.L. and N.J.I.; Investigation, C.S.L., K.M.L., C.R., and R.Y.; Formal Analysis, N.J.I. and C.S.L.; Writing – Original Draft, C.S.L.; Writing – Review & Editing, S.M.C. and N.J.I.; Visualization, C.S.L. and N.J.I.; Supervision, S.M.C.; Project Administration, S.M.C. and H.S.; Funding Acquisition, S.M.C., N.J.I., and H.S.
PEER REVIEW
The peer review history for this article is available at https://publons.com/publon/10.1002/jnr.24782.
Supporting information
FIGURE S1 Full gel of that shown in Figure 2 without splicing or changes in exposure
TABLE S1
Transparent Science Questionnaire for Authors
Transparent Peer Review Report
ACKNOWLEDGMENTS
Funding was provided by Michael J Fox Foundation Therapeutics Pipeline Grant #11709. We thank Dr. Jennifer Kahle (IHS, Intl) for providing writing assistance.
Limegrover CS, Yurko R, Izzo NJ, et al. Sigma‐2 receptor antagonists rescue neuronal dysfunction induced by Parkinson’s patient brain‐derived α‐synuclein. J Neurosci Res. 2021;99:1161–1176. 10.1002/jnr.24782
Edited by Cristina Ghiani and Barrington Burnett. Reviewed by Iryna Benilova and Joseph B. Watson.
DATA AVAILABILITY STATEMENT
The non‐proprietary data sets associated with this study are available as Supporting Information Files.
REFERENCES
- Aarsland, D. , Creese, B. , Politis, M. , Chaudhuri, K. R. , Ffytche, D. H. , Weintraub, D. , & Ballard, C. (2017). Cognitive decline in Parkinson disease. Nature Reviews Neurology, 13(4), 217–231. 10.1038/nrneurol.2017.27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed, I. S. , Rohe, H. J. , Twist, K. E. , & Craven, R. J. (2010). Pgrmc1 (progesterone receptor membrane component 1) associates with epidermal growth factor receptor and regulates erlotinib sensitivity. Journal of Biological Chemistry, 285(32), 24775–24782. 10.1074/jbc.M110.134585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alon, A. , Schmidt, H. R. , Wood, M. D. , Sahn, J. J. , Martin, S. F. , & Kruse, A. C. (2017). Identification of the gene that codes for the σ 2 receptor. Proceedings of the National Academy of Sciences of the United States of America, 114(27), 7160–7165. 10.1073/pnas.1705154114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez‐Erviti, L. , Seow, Y. , Schapira, A. H. , Gardiner, C. , Sargent, I. L. , Wood, M. J. A. , & Cooper, J. M. (2011). Lysosomal dysfunction increases exosome‐mediated alpha‐synuclein release and transmission. Neurobiology of Disease, 42(3), 360–367. 10.1016/j.nbd.2011.01.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ardah, M. T. , Paleologou, K. E. , Lv, G. , Khair, S. B. A. , Kendi, A. K. , Al Minhas, S. T. , Al‐Tel, T. H. , Al‐Hayani, A. A. , Haque, M. E. , Eliezer, D. , & El‐Agnaf, O. M. A. (2014). Structure activity relationship of phenolic acid inhibitors of α‐synuclein fibril formation and toxicity. Frontiers in Aging Neuroscience, 6, 1–17. 10.3389/fnagi.2014.00197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auluck, P. K. , Caraveo, G. , & Lindquist, S. (2010). α‐Synuclein: Membrane interactions and toxicity in Parkinson’s disease. Annual Review of Cell and Developmental Biology, 26, 211–233. 10.1146/annurev.cellbio.042308.113313 [DOI] [PubMed] [Google Scholar]
- Barrett, P. J. , & Greenamyre, J. T. (2015). Post‐translational modification of α‐synuclein in Parkinson׳s disease. Brain Research, 1628, 247–253. 10.1016/j.brainres.2015.06.002 [DOI] [PubMed] [Google Scholar]
- Behrends, C. , Sowa, M. E. , Gygi, S. P. , & Harper, J. W. (2010). Network organization of the human autophagy system. Nature, 466(7302), 68–76. 10.1038/nature09204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben Gedalya, T. , Loeb, V. , Israeli, E. , Altschuler, Y. , Selkoe, D. J. , & Sharon, R. (2009). Alpha‐synuclein and polyunsaturated fatty acids promote clathrin‐mediated endocytosis and synaptic vesicle recycling. Traffic, 10(2), 218–234. 10.1111/j.1600-0854.2008.00853.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benner, E. J. , Banerjee, R. , Reynolds, A. D. , Sherman, S. , Pisarev, V. M. , Tsiperson, V. , Nemachek, C. , Ciborowski, P. , Przedborski, S. , Mosley, R. L. , & Gendelman, H. E. (2008). Nitrated alpha‐synuclein immunity accelerates degeneration of nigral dopaminergic neurons. PLoS ONE, 3(1), e1376. 10.1371/journal.pone.0001376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernal‐Conde, L. D. , Ramos‐Acevedo, R. , Reyes‐Hernández, M. A. , Balbuena‐Olvera, A. J. , Morales‐Moreno, I. D. , Argüero‐Sánchez, R. , Schüle, B. , & Guerra‐Crespo, M. (2020). Alpha‐synuclein physiology and pathology: A perspective on cellular structures and organelles. Frontiers in Neuroscience, 13, 1399. 10.3389/fnins.2019.01399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betzer, C. , Movius, A. J. , Shi, M. , Gai, W.‐P. , Zhang, J. , & Jensen, P. H. (2015). Identification of synaptosomal proteins binding to monomeric and oligomeric α‐Synuclein. PLoS ONE, 10(2), e0116473. 10.1371/journal.pone.0116473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer, G. , Torricelli, J. , Evege, E. , & Price, P. (1993). Optimized survival of hippocampal neurons in B27‐supplemented neurobasal, a new serum‐free medium combination. Journal of Neuroscience Research, 35, 567–576. 10.1002/jnr.490350513 [DOI] [PubMed] [Google Scholar]
- Burré, J. , Sharma, M. , & Südhof, T. C. (2014). α‐Synuclein assembles into higher‐order multimers upon membrane binding to promote SNARE complex formation. Proceedings of the National Academy of Sciences of the United States of America, 111(40), E4274–E4283. 10.1073/pnas.1416598111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai, Y.‐J.‐ J. , Kim, D. , Park, J. , Zhao, H. , Lee, S.‐J.‐ J. , & Chang, S. (2013). The secreted oligomeric form of α‐synuclein affects multiple steps of membrane trafficking. FEBS Letters, 587(5), 452–459. 10.1016/j.febslet.2013.01.008 [DOI] [PubMed] [Google Scholar]
- Choi, B.‐K. , Choi, M.‐G. , Kim, J.‐Y. , Yang, Y. , Lai, Y. , Kweon, D.‐H. , Lee, N. K. , & Shin, Y.‐K. (2013). Large α‐synuclein oligomers inhibit neuronal SNARE‐mediated vesicle docking. Proceedings of the National Academy of Sciences of the United States of America, 110(10), 4087–4092. 10.1073/pnas.1218424110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi, B.‐K.‐ K. , Kim, J.‐Y.‐ Y. , Cha, M.‐Y.‐ Y. , Mook‐Jung, I. , Shin, Y.‐K.‐ K. , & Lee, N. K. (2015). β‐Amyloid and α‐Synuclein cooperate to block SNARE‐dependent vesicle fusion. Biochemistry, 54(9), 1831–1840. 10.1021/acs.biochem.5b00087 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Cuervo, A. M. , Stefanis, L. , Fredenburg, R. , Lansbury, P. T. , & Sulzer, D. (2004). Impaired degradation of mutant alpha‐synuclein by chaperone‐mediated autophagy. Science, 305(5688), 1292–1295. 10.1126/science.1101738 [DOI] [PubMed] [Google Scholar]
- Cuervo, A. M. , & Wong, E. (2014). Chaperone‐mediated autophagy: Roles in disease and aging. Cell Research, 24(1), 92–104. 10.1038/cr.2013.153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danzer, K. M. , Haasen, D. , Karow, A. R. , Moussaud, S. , Habeck, M. , Giese, A. , Kretzschmar, H. , Hengerer, B. , & Kostka, M. (2007). Different species of alpha‐synuclein oligomers induce calcium influx and seeding. Journal of Neuroscience, 27(34), 9220–9232. 10.1523/JNEUROSCI.2617-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deas, E. , Cremades, N. , Angelova, P. R. , Ludtmann, M. H. R. , Yao, Z. , Chen, S. , Horrocks, M. H. , Banushi, B. , Little, D. , Devine, M. J. , Gissen, P. , Klenerman, D. , Dobson, C. M. , Wood, N. W. , Gandhi, S. , & Abramov, A. Y. (2016). Alpha‐Synuclein oligomers interact with metal ions to induce oxidative stress and neuronal death in Parkinson’s disease. Antioxidants & Redox Signaling, 24(7), 376–391. 10.1089/ars.2015.6343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deramecourt, V. , Bombois, S. , Maurage, C.‐A. , Ghestem, A. , Drobecq, H. , Vanmechelen, E. , Lebert, F. , Pasquier, F. , & Delacourte, A. (2006). Biochemical staging of synucleinopathy and amyloid deposition in dementia with Lewy bodies. Journal of Neuropathology and Experimental Neurology, 65(3), 278–288. 10.1097/01.jnen.0000205145.54457.ea [DOI] [PubMed] [Google Scholar]
- Diao, J. , Burré, J. , Vivona, S. , Cipriano, D. J. , Sharma, M. , Kyoung, M. , Südhof, T. C. , & Brunger, A. T. (2013). Native α‐synuclein induces clustering of synaptic‐vesicle mimics via binding to phospholipids and synaptobrevin‐2/VAMP2. eLife, 2013(2), 1–17. 10.7554/eLife.00592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diógenes, M. J. , Dias, R. B. , Rombo, D. M. , Vicente Miranda, H. , Maiolino, F. , Guerreiro, P. , Näsström, T. , Franquelim, H. G. , Oliveira, L. M. A. , Castanho, M. A. R. B. , Lannfelt, L. , Bergström, J. , Ingelsson, M. , Quintas, A. , Sebastião, A. M. , Lopes, L. V. , & Outeiro, T. F. (2012). Extracellular alpha‐synuclein oligomers modulate synaptic transmission and impair LTP via NMDA‐receptor activation. Journal of Neuroscience, 32(34), 11750–11762. 10.1523/JNEUROSCI.0234-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domert, J. , Sackmann, C. , Severinsson, E. , Agholme, L. , Bergström, J. , Ingelsson, M. , & Hallbeck, M. (2016). Aggregated alpha‐Synuclein transfer efficiently between cultured human neuron‐like cells and localize to lysosomes. PLoS ONE, 11(12), e0168700. 10.1371/journal.pone.0168700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebrahimi‐Fakhari, D. , Wahlster, L. , Bartz, F. , Werenbeck‐Ueding, J. , Praggastis, M. , Zhang, J. , Joggerst‐Thomalla, B. , Theiss, S. , Grimm, D. , Ory, D. S. , & Runz, H. (2015). Reduction of TMEM97 increases NPC1 protein levels and restores cholesterol trafficking in Niemann‐pick type C1 disease cells. Human Molecular Genetics, 25(16), 3588–3599. 10.1093/hmg/ddw204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteves, A. R. , Arduíno, D. M. , Swerdlow, R. H. , Oliveira, C. R. , & Cardoso, S. M. (2009). Oxidative stress involvement in alpha‐synuclein oligomerization in Parkinson’s disease cybrids. Antioxidants & Redox Signaling, 11, 439–448. 10.1089/ARS.2008.2247 [DOI] [PubMed] [Google Scholar]
- Esteves, M. , Cristóvão, A. C. , Saraiva, T. , Rocha, S. M. , Baltazar, G. , Ferreira, L. , & Bernardino, L. (2015). Retinoic acid‐loaded polymeric nanoparticles induce neuroprotection in a mouse model for Parkinson’s disease. Frontiers in Aging Neuroscience, 7, 20. 10.3389/fnagi.2015.00020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faul, F. , Erdfelder, E. , Lang, A.‐G. , & Buchner, A. (2007). G*Power 3: A flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behavior Research Methods, 39(2), 175–191. 10.3758/BF03193146 [DOI] [PubMed] [Google Scholar]
- Fields, C. R. , Bengoa‐Vergniory, N. , & Wade‐Martins, R. (2019). Targeting alpha‐Synuclein as a therapy for Parkinson’s disease. Frontiers in Molecular Neuroscience, 12, 1–14. 10.3389/fnmol.2019.00299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonseca, T. , Villar‐Piqué, A. , & Outeiro, T. (2015). The interplay between alpha‐Synuclein clearance and spreading. Biomolecules, 5(2), 435–471. 10.3390/biom5020435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvagnion, C. (2017). The role of lipids interacting with α‐Synuclein in the pathogenesis of Parkinson’s disease. Journal of Parkinson’s Disease, 7(3), 433–450. 10.3233/JPD-171103 [DOI] [PubMed] [Google Scholar]
- Galvagnion, C. , Brown, J. W. P. , Ouberai, M. M. , Flagmeier, P. , Vendruscolo, M. , Buell, A. K. , Sparr, E. , & Dobson, C. M. (2016). Chemical properties of lipids strongly affect the kinetics of the membrane‐induced aggregation of α‐synuclein. Proceedings of the National Academy of Sciences of the United States of America, 31, 1–6. 10.1073/pnas.1601899113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganapathy, M. E. , Prasad, P. D. , Huang, W. , Seth, P. , Leibach, F. H. , & Ganapathy, V. (1999). Molecular and ligand‐binding characterization of the sigma‐receptor in the Jurkat human T lymphocyte cell line. Journal of Pharmacology and Experimental Therapeutics, 289(1), 251–260. http://www.ncbi.nlm.nih.gov/pubmed/10087012 [PubMed] [Google Scholar]
- Garcia‐Esparcia, P. , Hernández‐Ortega, K. , Koneti, A. , Gil, L. , Delgado‐Morales, R. , Castaño, E. , Carmona, M. , & Ferrer, I. (2015). Altered machinery of protein synthesis is region‐ and stage‐dependent and is associated with α‐synuclein oligomers in Parkinson’s disease. Acta Neuropathologica Communications, 3(1), 76. 10.1186/s40478-015-0257-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampton, K. K. , Anderson, K. , Frazier, H. , Thibault, O. , & Craven, R. J. (2018). Insulin receptor plasma membrane levels increased by the progesterone receptor membrane component 1. Molecular Pharmacology, 94(1), 665–673. 10.1124/mol.117.110510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen, C. , & Li, J.‐Y. (2012). Beyond α‐synuclein transfer: Pathology propagation in Parkinson’s disease. Trends in Molecular Medicine, 18(5), 248–255. 10.1016/j.molmed.2012.03.002 [DOI] [PubMed] [Google Scholar]
- Hassink, G. C. , Raiss, C. C. , Segers‐Nolten, I. M. J. , Van Wezel, R. J. A. , Subramaniam, V. , Le Feber, J. , & Claessens, M. M. A. E. (2018). Exogenous α‐synuclein hinders synaptic communication in cultured cortical primary rat neurons. PLoS ONE, 13(3), 1–21. 10.1371/journal.pone.0193763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson, M. X. , Trojanowski, J. Q. , & Lee, V. M. Y. (2019). α‐Synuclein pathology in Parkinson’s disease and related α‐synucleinopathies. Neuroscience Letters, 709, 134316. 10.1016/j.neulet.2019.134316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong, H.‐S. , Maezawa, I. , Yao, N. , Xu, B. , Diaz‐Avalos, R. , Rana, S. , Hua, D. H. , Cheng, R. H. , Lam, K. S. , & Jin, L.‐W. (2007). Combining the rapid MTT formazan exocytosis assay and the MC65 protection assay led to the discovery of carbazole analogs as small molecule inhibitors of Abeta oligomer‐induced cytotoxicity. Brain Research, 1130(1), 223–234. 10.1016/j.brainres.2006.10.093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes, A. L. , Powell, D. W. , Bard, M. , Eckstein, J. , Barbuch, R. , Link, A. J. , & Espenshade, P. J. (2007). Dap1/PGRMC1 binds and regulates cytochrome P450 enzymes. Cell Metabolism, 5(2), 143–149. 10.1016/j.cmet.2006.12.009 [DOI] [PubMed] [Google Scholar]
- Hunn, B. H. M. , Cragg, S. J. , Bolam, J. P. , Spillantini, M. , & Wade‐Martins, R. (2015). Impaired intracellular trafficking defines early Parkinson’s disease. Trends in Neurosciences, 38(3), 178–188. 10.1016/j.tins.2014.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Intlekofer, K. A. , Clements, K. , Woods, H. , Adams, H. , Suvorov, A. , & Petersen, S. L. (2019). Progesterone receptor membrane component 1 inhibits tumor necrosis factor alpha induction of gene expression in neural cells. PLoS ONE, 14(4), e0215389. 10.1371/journal.pone.0215389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izzo, N. J. , Staniszewski, A. , To, L. , Fa, M. , Teich, A. F. , Saeed, F. , Wostein, H. , Walko, T. , Vaswani, A. , Wardius, M. , Syed, Z. , Ravenscroft, J. , Mozzoni, K. , Silky, C. , Rehak, C. , Yurko, R. , Finn, P. , Look, G. , Rishton, G. , … Catalano, S. M. (2014). Alzheimer’s therapeutics targeting amyloid beta 1–42 oligomers I: Abeta 42 oligomer binding to specific neuronal receptors is displaced by drug candidates that improve cognitive deficits. PLoS ONE, 9(11), e111898. 10.1371/journal.pone.0111898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izzo, N. J. , Xu, J. , Zeng, C. , Kirk, M. J. , Mozzoni, K. , Silky, C. , Rehak, C. , Yurko, R. , Look, G. , Rishton, G. , Safferstein, H. , Cruchaga, C. , Goate, A. , Cahill, M. A. , Arancio, O. , Mach, R. H. , Craven, R. , Head, E. , LeVine, H. , … Catalano, S. M. (2014). Alzheimer’s therapeutics targeting amyloid beta 1–42 oligomers II: Sigma‐2/PGRMC1 receptors mediate Abeta 42 oligomer binding and synaptotoxicity. PLoS ONE, 9(11), e111899. 10.1371/journal.pone.0111899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang, A. , Lee, H.‐J. , Suk, J.‐E. , Jung, J.‐W. , Kim, K.‐P. , & Lee, S.‐J. (2010). Non‐classical exocytosis of alpha‐synuclein is sensitive to folding states and promoted under stress conditions. Journal of Neurochemistry, 113(5), 1263–1274. 10.1111/j.1471-4159.2010.06695.x [DOI] [PubMed] [Google Scholar]
- Jiang, C. , Feng, Y. , Huang, X. , Xu, Y. , Zhang, Y. , Zhou, N. , Shen, X. , Chen, K. , Jiang, H. , & Liu, D. (2010). An enzyme‐linked immunosorbent assay to compare the affinity of chemical compounds for β‐amyloid peptide as a monomer. Analytical and Bioanalytical Chemistry, 396(5), 1745–1754. 10.1007/s00216-009-3420-6 [DOI] [PubMed] [Google Scholar]
- Karpinar, D. P. , Balija, M. B. G. , Kügler, S. , Opazo, F. , Rezaei‐Ghaleh, N. , Wender, N. , Kim, H.‐Y. , Taschenberger, G. , Falkenburger, B. H. , Heise, H. , Kumar, A. , Riedel, D. , Fichtner, L. , Voigt, A. , Braus, G. H. , Giller, K. , Becker, S. , Herzig, A. , Baldus, M. , … Zweckstetter, M. (2009). Pre‐fibrillar α‐synuclein variants with impaired β‐structure increase neurotoxicity in Parkinson’s disease models. EMBO Journal, 28(20), 3256–3268. 10.1038/emboj.2009.257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karube, H. , Sakamoto, M. , Arawaka, S. , Hara, S. , Sato, H. , Ren, C.‐H. , Goto, S. , Koyama, S. , Wada, M. , Kawanami, T. , Kurita, K. , & Kato, T. (2008). N‐terminal region of α‐synuclein is essential for the fatty acid‐induced oligomerization of the molecules. FEBS Letters, 582(25–26), 3693–3700. 10.1016/j.febslet.2008.10.001 [DOI] [PubMed] [Google Scholar]
- Kellie, J. F. , Higgs, R. E. , Ryder, J. W. , Major, A. , Beach, T. G. , Adler, C. H. , Merchant, K. , & Knierman, M. D. (2014). Quantitative measurement of intact alpha‐synuclein proteoforms from post‐mortem control and Parkinson’s disease brain tissue by intact protein mass spectrometry. Scientific Reports, 4, 5797. 10.1038/srep05797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreutzmann, P. , Wolf, G. , & Kupsch, K. (2010). Minocycline recovers MTT‐formazan exocytosis impaired by amyloid beta peptide. Cellular and Molecular Neurobiology, 30(7), 979–984. 10.1007/s10571-010-9528-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, H.‐J. , Patel, S. , & Lee, S.‐J. (2005). Intravesicular localization and exocytosis of α‐Synuclein and its aggregates. Journal of Neuroscience, 25(25), 6016–6024. 10.1523/JNEUROSCI.0692-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Y. , Qiang, M. , Wei, Y. , & He, R. (2011). A novel molecular mechanism for nitrated {alpha}‐synuclein‐induced cell death. Journal of Molecular Cell Biology, 3(4), 239–249. 10.1093/jmcb/mjr011 [DOI] [PubMed] [Google Scholar]
- Liu, Y. , & Schubert, D. (1997). Cytotoxic amyloid peptides inhibit cellular 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide (MTT) reduction by enhancing MTT formazan exocytosis. Journal of Neurochemistry, 69(6), 2285–2293. http://www.ncbi.nlm.nih.gov/pubmed/9375659 10.1046/j.1471-4159.1997.69062285.x [DOI] [PubMed] [Google Scholar]
- Luk, K. C. , Song, C. , O’Brien, P. , Stieber, A. , Branch, J. R. , Brunden, K. R. , Trojanowski, J. Q. , & Lee, V.‐ M.‐Y. (2009). Exogenous ‐synuclein fibrils seed the formation of Lewy body‐like intracellular inclusions in cultured cells. Proceedings of the National Academy of Sciences of the United States of America, 106(47), 20051–20056. 10.1073/pnas.0908005106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luth, E. S. , Bartels, T. , Dettmer, U. , Kim, N. C. , & Selkoe, D. J. (2015). Purification of α‐Synuclein from human brain reveals an instability of endogenous multimers as the protein approaches purity. Biochemistry, 54, 279–292. 10.1021/bi501188a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak, S. K. , McCormack, A. L. , Manning‐Bog, A. B. , Cuervo, A. M. , & Di Monte, D. A. (2010). Lysosomal degradation of α‐synuclein in vivo. Journal of Biological Chemistry, 285(18), 13621–13629. 10.1074/jbc.M109.074617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal, P. K. , Pettegrew, J. W. , Masliah, E. , Hamilton, R. L. , & Mandal, R. (2006). Interaction between Abeta peptide and alpha synuclein: Molecular mechanisms in overlapping pathology of Alzheimer’s and Parkinson’s in dementia with Lewy body disease. Neurochemical Research, 31(9), 1153–1162. 10.1007/s11064-006-9140-9 [DOI] [PubMed] [Google Scholar]
- Martin, Z. S. , Neugebauer, V. , Dineley, K. T. , Kayed, R. , Zhang, W. , Reese, L. C. , & Taglialatela, G. (2012). α‐Synuclein oligomers oppose long‐term potentiation and impair memory through a calcineurin‐dependent mechanism: Relevance to human synucleopathic diseases. Journal of Neurochemistry, 120(3), 440–452. 10.1111/j.1471-4159.2011.07576.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez‐Vicente, M. , & Cuervo, A. M. (2007). Autophagy and neurodegeneration: When the cleaning crew goes on strike. Lancet Neurology, 6(4), 352–361. 10.1016/S1474-4422(07)70076-5 [DOI] [PubMed] [Google Scholar]
- Martinez‐Vicente, M. , Talloczy, Z. , Kaushik, S. , Massey, A. C. , Mazzulli, J. , Mosharov, E. V. , Hodara, R. , Fredenburg, R. , Wu, D. C. , Follenzi, A. , Dauer, W. , Przedborski, S. , Ischiropoulos, H. , Lansbury, P. T. , Sulzer, D. , Cuervo, A. M. , Npejgjfe, P. , Martinez‐vicente, M. , Talloczy, Z. , … Cuervo, A. M. (2008). Dopamine‐modified α‐synuclein blocks chaperone‐mediated autophagy. Journal of Clinical Investigation, 118(2), 777–778. 10.1172/JCI32806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah, E. , Rockenstein, E. , Veinbergs, I. , Sagara, Y. , Mallory, M. , Hashimoto, M. , & Mucke, L. (2001). Beta‐amyloid peptides enhance alpha‐synuclein accumulation and neuronal deficits in a transgenic mouse model linking Alzheimer’s disease and Parkinson’s disease. Proceedings of the National Academy of Sciences of the United States of America, 98(21), 12245–12250. 10.1073/pnas.211412398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meade, R. M. , Fairlie, D. P. , & Mason, J. M. (2019). Alpha‐synuclein structure and Parkinson’s disease—Lessons and emerging principles. Molecular Neurodegeneration, 14(1), 29. 10.1186/s13024-019-0329-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medeiros, A. T. , Soll, L. G. , Tessari, I. , Bubacco, L. , & Morgan, J. R. (2017). α‐Synuclein dimers impair vesicle fission during clathrin‐mediated synaptic vesicle recycling. Frontiers in Cellular Neuroscience, 11, 1–15. 10.3389/fncel.2017.00388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mhyre, T. R. , Boyd, J. T. , Hamill, R. W. , & Maguire‐Zeiss, K. A. (2012). Parkinson’s disease. Sub‐Cellular Biochemistry, 65, 389–455. 10.1007/978-94-007-5416-4_16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mir, S. U. R. R. , Schwarze, S. R. , Jin, L. , Zhang, J. , Friend, W. , Miriyala, S. , Clair, D. S. , Craven, R. J. , St Clair, D. , & Craven, R. J. (2013). Progesterone receptor membrane component 1/Sigma‐2 receptor associates with MAP1LC3B and promotes autophagy. Autophagy, 9(10), 1566–1578. 10.4161/auto.25889 [DOI] [PubMed] [Google Scholar]
- Narayanan, V. , & Scarlata, S. (2001). Membrane binding and self‐association of alpha‐synucleins. Biochemistry, 40(33), 9927–9934. 10.1021/Bi002952n [DOI] [PubMed] [Google Scholar]
- Orenstein, S. J. , Kuo, S.‐H. , Tasset, I. , Arias, E. , Koga, H. , Fernandez‐Carasa, I. , Cortes, E. , Honig, L. S. , Dauer, W. , Consiglio, A. , Raya, A. , Sulzer, D. , & Cuervo, A. M. (2013). Interplay of LRRK2 with chaperone‐mediated autophagy. Nature Neuroscience, 16(4), 394–406. 10.1038/nn.3350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Outeiro, T. F. , Klucken, J. , Bercury, K. , Tetzlaff, J. , Putcha, P. , Oliveira, L. M. A. , Quintas, A. , McLean, P. J. , & Hyman, B. T. (2009). Dopamine‐induced conformational changes in alpha‐synuclein. PLoS ONE, 4(9), 1–11. 10.1371/journal.pone.0006906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paleologou, K. E. , Kragh, C. L. , Mann, D. M. , Salem, S. , Al‐Shami, R. , Allsop, D. , Hassan, A. H. , Jensen, P. H. , & El‐Agnaf, O. M. (2009). Detection of elevated levels of soluble alpha‐synuclein oligomers in post‐mortem brain extracts from patients with dementia with Lewy bodies. Brain, 132(Pt 4), 1093–1101. 10.1093/brain/awn349 [DOI] [PubMed] [Google Scholar]
- Papagiannakis, N. , Koros, C. , Stamelou, M. , Simitsi, A. M. , Maniati, M. , Antonelou, R. , Papadimitriou, D. , Dermentzaki, G. , Moraitou, M. , Michelakakis, H. , & Stefanis, L. (2018). Alpha‐synuclein dimerization in erythrocytes of patients with genetic and non‐genetic forms of Parkinson’s disease. Neuroscience Letters, 672, 145–149. 10.1016/j.neulet.2017.11.012 [DOI] [PubMed] [Google Scholar]
- Perrin, R. J. , Woods, W. S. , Clayton, D. F. , & George, J. M. (2001). Exposure to long chain polyunsaturated fatty acids triggers rapid multimerization of synucleins. Journal of Biological Chemistry, 276(45), 41958–41962. 10.1074/jbc.M105022200 [DOI] [PubMed] [Google Scholar]
- Plotegher, N. , Berti, G. , Ferrari, E. , Tessari, I. , Zanetti, M. , Lunelli, L. , Greggio, E. , Bisaglia, M. , Veronesi, M. , Girotto, S. , Dalla Serra, M. , Perego, C. , Casella, L. , & Bubacco, L. (2017). DOPAL derived alpha‐synuclein oligomers impair synaptic vesicles physiological function. Scientific Reports, 7, 1–16. 10.1038/srep40699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recasens, A. , Dehay, B. , Bové, J. , Carballo‐Carbajal, I. , Dovero, S. , Pérez‐Villalba, A. , Fernagut, P.‐O. , Blesa, J. , Parent, A. , Perier, C. , Fariñas, I. , Obeso, J. A. , Bezard, E. , & Vila, M. (2014). Lewy body extracts from Parkinson disease brains trigger α‐synuclein pathology and neurodegeneration in mice and monkeys. Annals of Neurology, 75(3), 351–362. 10.1002/ana.24066 [DOI] [PubMed] [Google Scholar]
- Riad, A. , Lengyel‐Zhand, Z. , Zeng, C. , Weng, C.‐C.‐ C. , Lee, V.‐ M.‐Y.‐ M.‐ Y. , Trojanowski, J. Q. , & Mach, R. H. (2020). The sigma‐2 receptor/TMEM97, PGRMC1, and LDL receptor complex are responsible for the cellular uptake of Aβ42 and its protein aggregates. Molecular Neurobiology, 57(9), 3803–3813. 10.1007/s12035-020-01988-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riad, A. , Zeng, C. , Weng, C.‐C.‐ C. , Winters, H. , Xu, K. , Makvandi, M. , Metz, T. , Carlin, S. , & Mach, R. H. (2018). Sigma‐2 receptor/TMEM97 and PGRMC‐1 increase the rate of internalization of LDL by LDL receptor through the formation of a ternary complex. Scientific Reports, 8(1), 16845. 10.1038/s41598-018-35430-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts, H. , & Brown, D. (2015). Seeking a mechanism for the toxicity of oligomeric α‐Synuclein. Biomolecules, 5(2), 282–305. 10.3390/biom5020282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts, R. F. , Wade‐Martins, R. , & Alegre‐Abarrategui, J. (2015). Direct visualization of alpha‐synuclein oligomers reveals previously undetected pathology in Parkinson’s disease brain. Brain, 138(Pt 6), 1642–1657. 10.1093/brain/awv040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohe, H. J. , Ahmed, I. S. , Twist, K. E. , & Craven, R. J. (2009). PGRMC1 (progesterone receptor membrane component 1): A targetable protein with multiple functions in steroid signaling, P450 activation and drug binding. Pharmacology & Therapeutics, 121(1), 14–19. 10.1016/j.pharmthera.2008.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roostaee, A. , Beaudoin, S. , Staskevicius, A. , & Roucou, X. (2013). Aggregation and neurotoxicity of recombinant α‐synuclein aggregates initiated by dimerization. Molecular Neurodegeneration, 8(1), 1–12. 10.1186/1750-1326-8-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Runko, E. , & Kaprielian, Z. (2004). Caenorhabditis elegans VEM‐1, a novel membrane protein, regulates the guidance of ventral nerve cord‐associated axons. Journal of Neuroscience, 24(41), 9015–9026. 10.1523/JNEUROSCI.2385-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sala, G. , Marinig, D. , Arosio, A. , & Ferrarese, C. (2016). Role of chaperone‐mediated autophagy dysfunctions in the pathogenesis of Parkinson’s disease. Frontiers in Molecular Neuroscience, 9, 1–7. 10.3389/fnmol.2016.00157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez‐Pulido, L. , & Ponting, C. P. (2014). TM6SF2 and MAC30, new enzyme homologs in sterol metabolism and common metabolic disease. Frontiers in Genetics, 5, 1–9. 10.3389/fgene.2014.00439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheper, W. , & Hoozemans, J. J. M. (2015). The unfolded protein response in neurodegenerative diseases: A neuropathological perspective. Acta Neuropathologica, 130(3), 315–331. 10.1007/s00401-015-1462-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider, J. L. , & Cuervo, A. M. (2014). Autophagy and human disease: Emerging themes. Current Opinion in Genetics and Development, 26, 16–23. 10.1016/j.gde.2014.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott, D. A. , Tabarean, I. , Tang, Y. , Cartier, A. , Masliah, E. , & Roy, S. (2010). A pathologic cascade leading to synaptic dysfunction in alpha‐synuclein‐induced neurodegeneration. Journal of Neuroscience, 30(24), 8083–8095. 10.1523/JNEUROSCI.1091-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shihabuddin, L. S. , Brundin, P. , Greenamyre, J. T. , Stephenson, D. , & Sardi, S. P. (2018). New frontiers in Parkinson’s disease: From genetics to the clinic. Journal of Neuroscience, 38(44), 9375–9382. 10.1523/JNEUROSCI.1666-18.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silky, C. , Rehak, C. , Mozzoni, K. , Yurko, R. , Izzo, N. , Rishton, G. , Look, G. , Safferstein, H. , & Catalano, S. M. (2016). Characterization of alpha synuclein oligomers from Parkinson’s patient tissue. Program No. 42.12. 2016. Neuroscience Meeting Planner, San Diego, CA, Society for Neuroscience 2016 Online. [Google Scholar]
- Spillantini, M. G. , Schmidt, M. L. , Lee, V. M. , Trojanowski, J. Q. , Jakes, R. , & Goedert, M. (1997). Alpha‐synuclein in Lewy bodies. Nature, 388(6645), 839–840. 10.1038/42166 [DOI] [PubMed] [Google Scholar]
- Stefanovic, A. N. D. , Stöckl, M. T. , Claessens, M. M. A. E. , & Subramaniam, V. (2014). α‐Synuclein oligomers distinctively permeabilize complex model membranes. FEBS Journal, 281(12), 2838–2850. 10.1111/febs.12824 [DOI] [PubMed] [Google Scholar]
- Suchanek, M. , Radzikowska, A. , & Thiele, C. (2005). Photo‐leucine and photo‐methionine allow identification of protein‐protein interactions in living cells. Nature Methods, 2(4), 261–267. 10.1038/nmeth752 [DOI] [PubMed] [Google Scholar]
- Takeda, A. , Nyssen, O. P. , Syed, A. , Jansen, E. , Bueno‐de‐Mesquita, B. , & Gallo, V. (2014). Vitamin A and carotenoids and the risk of Parkinson’s disease: A systematic review and meta‐analysis. Neuroepidemiology, 42(1), 25–38. 10.1159/000355849 [DOI] [PubMed] [Google Scholar]
- Thomas, P. , Pang, Y. , & Dong, J. (2014). Enhancement of cell surface expression and receptor functions of membrane progestin receptor α (mPRα) by progesterone receptor membrane component 1 (PGRMC1): Evidence for a role of PGRMC1 as an adaptor protein for steroid receptors. Endocrinology, 155(3), 1107–1119. 10.1210/en.2013-1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong, J. , Wong, H. , Guttman, M. , Ang, L. C. , Forno, L. S. , Shimadzu, M. , Rajput, A. H. , Muenter, M. D. , Kish, S. J. , Hornykiewicz, O. , & Furukawa, Y. (2010). Brain α‐synuclein accumulation in multiple system atrophy, Parkinson’s disease and progressive supranuclear palsy: A comparative investigation. Brain, 133(1), 172–188. 10.1093/brain/awp282 [DOI] [PubMed] [Google Scholar]
- Tong, L. , Balazs, R. , Thornton, P. L. , & Cotman, C. W. (2004). β‐amyloid peptide at sublethal concentrations downregulates brain‐derived neurotrophic factor functions in cultured cortical neurons. Journal of Neuroscience, 24(30), 6799–6809. 10.1523/JNEUROSCI.5463-03.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torre, V. , & Nicholls, J. (Eds.). (1998). Neural circuits and networks. In Proceedings of the NATO advanced study Institute on Neuronal Circuits and Network. Springer. 10.1007/978-3-642-58955-3 [DOI] [Google Scholar]
- Tsigelny, I. F. , Crews, L. , Desplats, P. , Shaked, G. M. , Sharikov, Y. , Mizuno, H. , Spencer, B. , Rockenstein, E. , Trejo, M. , Platoshyn, O. , Yuan, J. X. J. , & Masliah, E. (2008). Mechanisms of hybrid oligomer formation in the pathogenesis of combined Alzheimer’s and Parkinson’s diseases. PLoS One, 3(9), e3135. 10.1371/journal.pone.0003135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsika, E. , Moysidou, M. , Guo, J. , Cushman, M. , Gannon, P. , Sandaltzopoulos, R. , Giasson, B. I. , Krainc, D. , Ischiropoulos, H. , & Mazzulli, J. R. (2010). Distinct region‐specific α‐Synuclein oligomers in A53T transgenic mice: Implications for neurodegeneration. Journal of Neuroscience, 30(9), 3409–3418. 10.1523/JNEUROSCI.4977-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Laar, V. S. , Chen, J. , Zharikov, A. D. , Bai, Q. , Di Maio, R. , Dukes, A. A. , Hastings, T. G. , Watkins, S. C. , Greenamyre, J. T. , St Croix, C. M. , & Burton, E. A. (2020). α‐Synuclein amplifies cytoplasmic peroxide flux and oxidative stress provoked by mitochondrial inhibitors in CNS dopaminergic neurons in vivo. Redox Biology, 37, 101695. 10.1016/j.redox.2020.101695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Maarschalkerweerd, A. , Vetri, V. , & Vestergaard, B. (2015). Cholesterol facilitates interactions between α‐synuclein oligomers and charge‐neutral membranes. FEBS Letters, 589(19), 2661–2667. 10.1016/j.febslet.2015.08.013 [DOI] [PubMed] [Google Scholar]
- Vogiatzi, T. , Xilouri, M. , Vekrellis, K. , & Stefanis, L. (2008). Wild type α‐synuclein is degraded by chaperone‐mediated autophagy and macroautophagy in neuronal cells. Journal of Biological Chemistry, 283(35), 23542–23556. 10.1074/jbc.M801992200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, B. , Abraham, N. , Gao, G. , & Yang, Q. (2016). Dysregulation of autophagy and mitochondrial function in Parkinson’s disease. Translational Neurodegeneration, 5(1), 19. 10.1186/s40035-016-0065-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winner, B. , Jappelli, R. , Maji, S. K. , Desplats, P. A. , Boyer, L. , Aigner, S. , Hetzer, C. , Loher, T. , Vilar, M. , Campioni, S. , Tzitzilonis, C. , Soragni, A. , Jessberger, S. , Mira, H. , Consiglio, A. , Pham, E. , Masliah, E. , Gage, F. H. , & Riek, R. (2011). In vivo demonstration that alpha‐synuclein oligomers are toxic. Proceedings of the National Academy of Sciences of the United States of America, 108(10), 4194–4199. 10.1073/pnas.1100976108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong, Y. C. , & Krainc, D. (2017). α‐Synuclein toxicity in neurodegeneration: Mechanism and therapeutic strategies. Nature Medicine, 23(2), 1–13. 10.1038/nm.4269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xilouri, M. , Brekk, O. R. , & Stefanis, L. (2016). Autophagy and alpha‐synuclein: Relevance to Parkinson’s disease and related synucleopathies. Movement Disorders, 31(2), 178–192. 10.1002/mds.26477 [DOI] [PubMed] [Google Scholar]
- Xilouri, M. , Vogiatzi, T. , Vekrellis, K. , Park, D. , & Stefanis, L. (2009). Abberant alpha‐synuclein confers toxicity to neurons in part through inhibition of chaperone‐mediated autophagy. PLoS One, 4(5), e5515. 10.1371/journal.pone.0005515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, J. , Zeng, C. , Chu, W. , Pan, F. , Rothfuss, J. M. , Zhang, F. , Tu, Z. , Zhou, D. , Zeng, D. , Vangveravong, S. , Johnston, F. , Spitzer, D. , Chang, K. C. , Hotchkiss, R. S. , Hawkins, W. G. , Wheeler, K. T. , & Mach, R. H. (2011). Identification of the PGRMC1 protein complex as the putative sigma‐2 receptor binding site. Nature Communications, 2(2), 380. 10.1038/ncomms1386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, Z. , Xu, X. , Xiang, Z. , Zhou, J. , Zhang, Z. , Hu, C. , & He, C. (2010). Nitrated alpha‐synuclein induces the loss of dopaminergic neurons in the substantia nigra of rats. PLoS One, 5(4), e9956. 10.1371/journal.pone.0009956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue, Z. , Friedman, L. , Komatsu, M. , & Tanaka, K. (2009). The cellular pathways of neuronal autophagy and their implication in neurodegenerative diseases. Biochimica et Biophysica Acta ‐ Molecular Cell Research, 1793(9), 1496–1507. 10.1016/j.bbamcr.2009.01.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, J.‐H. , Chung, T. , & Oldenburg, K. (1999). A simple statistical parameter for use in evaluation and validation of high throughput screening assays. Journal of Biomolecular Screening, 4(2), 67–73. 10.1016/j.ajo.2013.06.011 [DOI] [PubMed] [Google Scholar]
- Zhang, M. , Robitaille, M. , Showalter, A. D. , Huang, X. , Liu, Y. , Bhattacharjee, A. , Willard, F. S. , Han, J. , Froese, S. , Wei, L. , Gaisano, H. Y. , Angers, S. , Sloop, K. W. , Dai, F. F. , & Wheeler, M. B. (2014). Progesterone receptor membrane component 1 is a functional part of the GLP‐1 receptor complex in pancreatic beta cells. Molecular & Cellular Proteomics, 1(416), 3049–3062. 10.1074/mcp.M114.040196 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
FIGURE S1 Full gel of that shown in Figure 2 without splicing or changes in exposure
TABLE S1
Transparent Science Questionnaire for Authors
Transparent Peer Review Report
Data Availability Statement
The non‐proprietary data sets associated with this study are available as Supporting Information Files.