

Abstract
Together with various hosts and environments, ubiquitous microbes interact closely with each other forming an intertwined system or community. Of interest, shifts of the relationships between microbes and their hosts or environments are associated with critical diseases and ecological changes. While advances in high-throughput Omics technologies offer a great opportunity for understanding the structures and functions of microbiome, it is still challenging to analyse and interpret the omics data. Specifically, the heterogeneity and diversity of microbial communities, compounded with the large size of the datasets, impose a tremendous challenge to mechanistically elucidate the complex communities. Fortunately, network analyses provide an efficient way to tackle this problem, and several network approaches have been proposed to improve this understanding recently. Here, we systemically illustrate these network theories that have been used in biological and biomedical research. Then, we review existing network modelling methods of microbial studies at multiple layers from metagenomics to metabolomics and further to multi-omics. Lastly, we discuss the limitations of present studies and provide a perspective for further directions in support of the understanding of microbial communities.
Keywords: microbiome, multi-omics, co-occurrence, network analysis, integrated analysis
Summary of microbiome studies
As complex biological systems, microbial communities are not merely a legion of microbe collections but are also related to other hosts or ecological communities [1]. Microbes participate in nearly all biogeochemical cycles on earth and dominate vital ecosystem functioning and productivity [2]. Human diseases [3, 4], climate change [5], crop yield [6], antibiotic resistance [7] and many other issues typically involve changes in microbial communities, which have thus been under intense scrutiny by researchers. Yet little is known about these complex communities, due to the heterogeneity and diversity of microbial compositions between individuals with different genetic information and under different exposures or environmental conditions [8, 9].
Traditional studies relied on culture techniques to explore the diversity and functions of microbial communities [10]. However, researchers have realized that more than 99% of microorganisms in nature could not be cultured alone in vitro [11]. Moreover, most biological functions cannot be attributed to just an independent individual [10], but rather to the complex microbial interactions among the whole community. Therefore, culture-independent methods are needed to fully describe uncultured microbes, as well as the microbiome and host–microbiome relationships within communities [10, 11].
The development of high-throughput Omics technologies opens a new era to microbiome studies with advantages of the high volume of data generation, high quality of data interpretation and fairly acceptable cost [12]. Such technologies boost the use of meta-omics approaches, including sequencing-based metagenomics and metatranscriptomics and mass spectrometry-based metaproteomics and metabolomics [13], to understand the microbial community with more advanced and accurate in situ methods that bypass the difficulties of species isolation [14]. Specifically, metagenomics provides the total genetic content contained in the community, enabling the elucidation of the compositions and functional potential of the whole community [11]. Metatranscriptomics has a focus on gene expression across the community [13] and characterizes functional changes across different contexts, in support of the inference of how microbiome interactions regulate community activities [15]. However, due to a lack of the finer-level details of actual function, metagenomic and metatranscriptomic analyses are not sufficient for drawing the actual biological mechanisms of a community. With this in mind, metaproteomics was proposed as a complementary approach to approximate the actual phenotypic traits by studying the protein content of microbial communities [16]. Additionally, metabolomics provides a detailed assessment of metabolites within a given biological sample [17], offering a more accurate snapshot of the global physiological state of the community. Together, these Omics technologies lay the foundation for a full description of microbial communities, including genes, RNA, protein and metabolites [18].
To our knowledge, a variety of international efforts have been directed at advancing microbial research. For example, the Human Microbiome Project gathers the microbiome profiles from different human body sites, such as the skin, oral cavity and gut, resulting in population-scale microbiome metagenomic data [19]. The Interactive Human Microbiome Project, focusing on the relationships between host and microbiome, provides multi-omics data from both the microbiome and human host [20, 21]. Additionally, Metagenomics of the Human Intestinal Tract provides genome information of the human intestinal microbiome, identifying previously unknown species and genes [22]. Collectively, these publicly available datasets provide a great opportunity to understand the complex microbiome. Given this landscape, the effective computational analysis techniques that keep up with vast amounts of data are necessary to uncover mechanistic insights into microbial communities.
Help along the way is provided by the great advances in complex network theories that, in the last few years, have made progress towards uncovering structures and functions of microbial communities [10]. Network models utilize experimental meta-omics data to evaluate complex communities from a global perspective and have been demonstrated powerful for studying microbiome and host–microbiome interactions [23, 24]. Throughout this review, we present the existing network analysis methods at multiple layers from metagenomics and metatranscriptomics to metaproteomics, metabolomics and multi-omics. In the following section, we overview network theories in biological research, which can be specifically applied to the microbial field.
Network theories in biological and biomedical research
The development of network theories provides a strong opportunity for applications in biological and biomedical research. A significant discovery of network theories is that biological systems share formation and evolution principles with many other complex systems in nature, such as the World Wide Web and social networks [25, 26]. This discovery lays the theoretical foundation for the application of network models to biological and biomedical research. Specifically, all biological systems can be viewed as networks wherein nodes represent the components in the systems (Box 1), such as metabolites in metabolic networks and genes or regulators in gene regulatory networks [27], and linking nodes according to their known or observed relationships [28].
Box 1. Definitions and clarifications of important terms in this manuscriptBasic definitions Node/Vertex——Nodes are basic discrete objects in networks. In a biological network, the node represents a member such as a taxon, a molecule, or a metabolite.
Edge/Arc——An edge refers to the line connecting nodes, which can be directed or not, weighted or not. In biological networks, they show specific relationships among the components, such as the correlation of abundance information or known biological reactions.
Neighbors of a node——Neighbors are nodes connected to a selected node via an edge.
Adjacency matrix——An adjacency matrix of a graph is a matrix 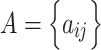 where
where 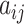 = 1 if and only if the node
= 1 if and only if the node 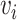 and node
and node 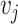 are neighbors.
are neighbors.
Operational taxonomic unit——Operational taxonomic units refer to clusters of organisms, generally grouped by sequence similarity or a specific taxonomic marker gene [29]. This concept has been extensively used to analyze the diversity of microbial communities, especially based on 16S ribosomal RNA sequencing data.
Network types
Random networks——A random network (i.e. the Erdős-Rényi network) consists of n nodes where each pair of nodes relates to probability p. The node degrees of this type of networks follow Poisson distribution [30].
Scale-free networks——In scale-free networks, most nodes have only a few edges, while a few nodes have a large number of edges [10]. This type of network is usually characterized by a power-law degree distribution (Table 1).
Microbial sequencing technologies
16S ribosomal RNA sequencing——16S ribosomal RNA is the component of the 30S subunit of a prokaryotic ribosome [44]. Due to the slow rates of evolution and mutation of the hyper variable region of genes coding 16S ribosomal RNA, such genes provide specific signature sequences of organisms [44]. 16S ribosomal RNA sequencing is widely used to identify the diversities in microbial communities thereby studying the phylogenetic relationships.
Whole metagenome shogun sequencing——Shotgun sequencing provides all genes present in a given biosample [45], which allows researchers to explore both taxonomic and functional information in a community.
Quantitative measurements of pairwise relationships
Bray-Curtis dissimilarity——Bray-Curtis index quantifies the dissimilarity between two different taxa over multiple samples. The calculation is as follows [46]:
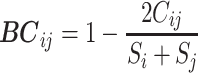 |
Where 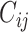 is the number of samples in which taxa i and taxa j are commonly occurring.
is the number of samples in which taxa i and taxa j are commonly occurring. 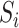 and
and 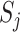 are the number of samples in which taxa i or taxa j occurred, respectively. This index is robust for compositional data and ranges from 0 and 1. 0 means two taxa are of strong relationships in these samples, while 1 means these two taxa are completely independent.
are the number of samples in which taxa i or taxa j occurred, respectively. This index is robust for compositional data and ranges from 0 and 1. 0 means two taxa are of strong relationships in these samples, while 1 means these two taxa are completely independent.
Kullback-Leibler dissimilarity——Kullback-Leibler index assesses the difference between two probability distributions. For any two taxa X and Y in microbial communities, the Kullback-Leibler dissimilarity 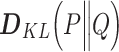 is calculated as follows [47]:
is calculated as follows [47]:
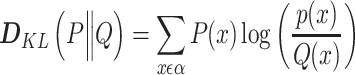 |
where P and Q represent the rank distributions of relative abundances of taxa X and Y, respectively. 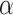 is a sample space of the ranks of relative abundances. The smaller
is a sample space of the ranks of relative abundances. The smaller 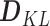 indicates the more significant relationships between the two taxa.
indicates the more significant relationships between the two taxa.
Pearson correlation coefficient—— Pearson correlation coefficient is a measure of the linear relationships between two taxa. For given n samples with pair abundance data of taxa X and taxa Y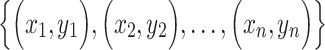 , the Pearson correlation coefficient
, the Pearson correlation coefficient 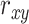 is defined as [48]:
is defined as [48]:
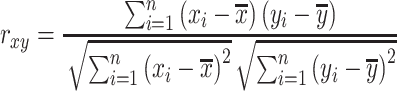 |
The correlation coefficient ranges from −1 to +1. The value of +1 or -1 means the relationship between two taxa is linearly dependent, where “+” means Y increases when X increases while “-” means Y decreases when X increases.
Spearman correlation coefficient——Spearman correlation coefficient, assessing the rank correlation of the relative abundances, is a measure of the monotonic relationships between two taxa (whether linear or not). For given n samples with pair abundance data of taxa X and taxa Y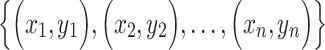 , the Spearman correlation coefficient
, the Spearman correlation coefficient 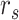 is defined as [49]:
is defined as [49]:
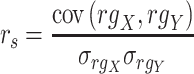 |
where 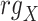 and
and 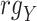 represent the ranks of abundance data of taxa X and taxa Y, respectively.
represent the ranks of abundance data of taxa X and taxa Y, respectively. 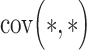 is the covariance of rank variables.
is the covariance of rank variables. 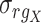 and
and 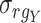 are the standard deviations of rank variables
are the standard deviations of rank variables 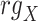 and
and 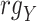 , respectively. The sign represents the relationships between taxa, which is the same as that in the Pearson correlation coefficient. A correlation value of +1 or −1 indicates X and Y are perfectly monotonically.
, respectively. The sign represents the relationships between taxa, which is the same as that in the Pearson correlation coefficient. A correlation value of +1 or −1 indicates X and Y are perfectly monotonically.
Researchers traditionally use random network models (Box 1) to describe real systems [30]. However, the expanded depth of network theories revealed that most organizational framework of biological networks is not randomly structured. These frameworks are better described as scale-free during evolution [50, 51], meaning that most nodes in the biological network have few edges but a few nodes have many. The scale-free architecture (Box 1) allows a given system to remain stable despite internal or external disturbances [10]. For example, the loss of many Escherichia coli genes has no effect on their growth rate [52], and the E. coli chemotaxis system can perform normal functions despite significant variations in ligand concentrations [53]. In addition, hierarchical architecture cannot be neglected in biological networks, which can be used to account for the relationships between relatively isolated modules in biological networks [10]. Specifically, a module is defined as a group of highly connected nodes that are relatively isolated from other nodes. However, the isolated groups are impossible in a scale-free network, and they tend to combine to form a hierarchical network [10]. Research has shown that the hierarchical structure arises in almost all cellular networks from metabolic, protein–protein interactions (PPIs) to gene regulatory networks [51, 54, 55]. Based on these general understandings, the biological networks can thus be studied in two ways: (1) top-down, from global structure to specific modules and nodes, or (2) bottom-up, from specific nodes and motifs to modules [56, 57]. Either way, all components within the biological system are considered as a whole, providing a comprehensive view of the system.
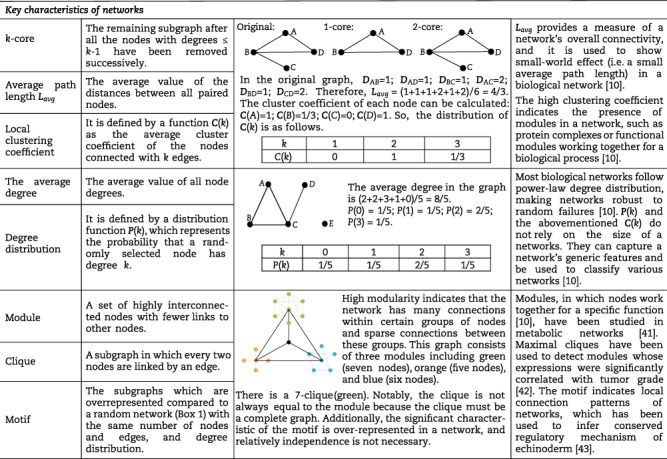
Based on the constructed networks, we can explore the underlying relationships among components using various network characteristics. These characteristics are described as quantified metrics by some concepts (e.g. degree and betweenness) in the network or graph theory. Table 1 summarizes some crucial concepts of network theories that have been used in biological and biomedical research [10, 36, 58]. For example, characteristics of the established network, including degree, betweenness and closeness centrality, have been used to identify hubs in phyllosphere communities [32]. Meanwhile, there are also valuable concepts whose applications in biology remain to be further explored, such as eigenvector centrality and coreness, which can measure the influence of specific nodes on the network and the robustness of networks, respectively. Understanding the biological networks by these measurements gives valuable insight into the underlying features and observable functions of the real microbial systems [10].
Table 1.
The crucial network theories in biological and biomedical research.
|
|
|

Network modelling methods and examples
The study of the microbiome, due to the species/strain diversity and highly complex interactions within communities or with the hosts or environments, is still in the exploration stage [8, 9]. In view of remarkable network theories in biology, network modelling methods are increasingly extended to microbial studies, and they offer a valuable representation of complex microbial relationships as a quantifiable method [10, 24]. We can portray the whole community based on meta-omics data and use network characteristics to identify the association between microbiome and hosts (Figure 1) [6, 31, 59–64]. Here, we discuss several network modelling methods of different types of microbial meta-omics data and summarize the common tools that contribute to network constructions [59].
Figure 1.
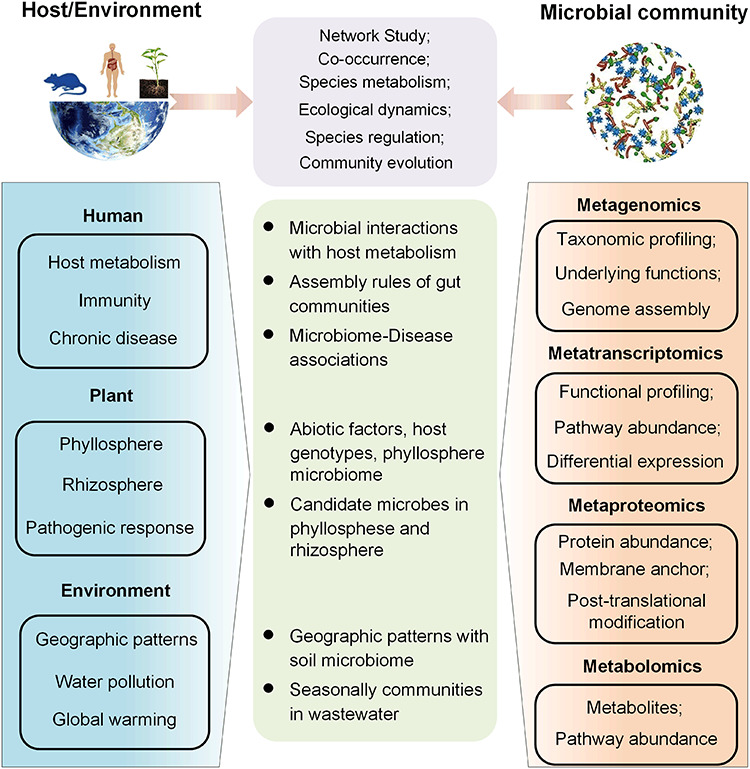
Overview of network studies on host–microbiome relationships. High-throughput Omics technologies produce a large amount of data, including metagenomic, metatranscriptomic, metaproteomic and metabolomic data, allowing the description of microbial communities at different layers. These molecular profiles of the microbial community can be viewed in the context of data describing the host and environment. Based on these different types of data, network analyses provide a viewpoint of host–environment–microbiome relationships.
Networks from metagenomics
Advanced sequencing technologies, including 16S ribosomal RNA (16S rRNA) sequencing and whole-metagenome shotgun sequencing (WMGS) (Box 1), yield metagenomic information that has been successfully applied to many fields, including human diseases, environmental research, etc. [65, 66]. These two technologies can characterize the taxonomic profiling of a given biospecimen [6], allowing the investigation of microbial associations, i.e. co-occurrence and co-exclusion. Furthermore, WMGS provides all genes in all organisms present in a given sample, enabling the understanding of underlying functional profiling within communities.
Networks at the taxonomic level
Based on sequencing data from either 16S rRNA or WMGS, various approaches have been proposed to construct microbial networks at the taxonomic level (i.e. co-occurrence networks) [31, 32, 67], offering insights into associations of microbes within the communities. Generally, the first step of such approaches is to determine the taxonomic profiling of the community, either by 16S rRNA-based taxa identification or mapping WMGS-based reads to reference catalogues [68]. Common analysis tools include QIIME2 [69], UPARSE [70] and MetaPhlAn2 [71] (Table 2). Then, the known microbial present–absent or abundance information is used to infer co-occurring and co-exclusive relationships between microbial taxa. The resulting co-occurrence network abstracts the biological taxa, such as species or optional taxonomic units (OTUs), into nodes [32, 67] and connects the nodes according to the relationships among taxa (Figure 2). Herein, we present such approaches as two groups according to the type of relationships, relationships between two nodes/taxa (pairwise relationships) and relationships among many different nodes/taxa (complex patterns). Specifically, we give two examples, corresponding to WMGS-based data (Example S1 in Supplementary Data) and 16S rRNA-based data (Example S2 in Supplementary Data), respectively, showing how to construct co-occurrence networks from different data types.
Table 2.
Meta-omics tools that contribute to network construction.
| Network | Tool | Yeara | Description | Output | Data |
|---|---|---|---|---|---|
| Co-occurrence network | UPARSE [70] | 2013 | An approach using UPARSE-OTU algorithms for producing clusters | OTUs in the community | Metagenomics |
| MetaPhlan2 [71] | 2015 | A tool for profiling composition of communities relying on unique clade-specific marker genes | Species in the community and their relative abundance | ||
| mOTUs2 [72] | 2019 | An approach based on marker gene OTUs that quantifies microbial species in metagenomic samples | Taxonomic assignment, relative abundance | ||
| MicroPro [73] | 2019 | A pipeline integrated reference-based methods with de novo assembly-based methods for microbial abundance from whole-genome sequencing | Reference-based known and assembly-binning-based unknown microbial abundance table | ||
| QIIME2 [69] | 2019 | An updated pipeline for demultiplexing and quality filtering, OTU picking, taxonomic assignment, phylogenetic reconstruction, visualizations | OTU table; taxonomy assignment based on reference | ||
| Genome-scale metabolic network | MetaQUBIC [74] | 2019 | An integrated biclustering-based pipeline for gene module detection that integrates both metagenomic and metatranscriptomic data | Functional enriched gene modules | Multi-omics |
| MetaTrans [75] | 2016 | A pipeline to analyse the structure and functions of active microbial communities from RNA-Seq data | Taxonomical abundance, functional assignment | Metatranscriptomics | |
| Co-occurrence network and genome-scale metabolic network | SAMSA [76] | 2016 | A pipeline for the analysis of gut microbiome data, focusing either on organism-specific activity or functional activity within sample | Organism abundance, RNA organism taxonomy list, function counts | |
| MG-RAST [77] | 2008 | A pipeline producing functional assignments of sequences in metagenome by comparing protein and nucleotide databases | Metabolic reconstruction and composition of the sample | Metagenomics/metatranscriptomics | |
| HUMAnN2 [78] | 2018 | A pipeline for profiling presence or absence and abundance of microbial pathways in a community from DNA/RNA reads | Species abundance and species-level metabolic pathway abundance in the community | ||
| PPI network | IdentiPy [79] | 2018 | A Python-based extensible engine peptide identification, postsearch validation, protein inference and quantification | Quantitative protein list | Metaproteomics |
| Trans-Proteomic Pipeline [80] | 2010 | A robust open-source standardized data processing pipeline for large-scale reproducible quantitative MS proteomics | Protein groups that sorted in descending order by probability | ||
| compleXView [81] | 2017 | A webserver that calculates measures of abundance, reproducibility and specificity to infer PPI | The PPI table | ||
| Metabolomics-driven metabolic network | Pathos [82] | 2011 | A webserver that analyses mass spectrometric data and displays metabolites and metabolic pathways | Metabolites and abundance changes, metabolic pathways | Metabolomics |
| MetaboAnalyst [83] | 2018 | A webserver for comprehensive metabolomic data analysis, interpretation and integration with other omics data | Biomarker identification, an MS peaks to pathways module | ||
| Netome [84] | 2018 | A tool for processing and analysing metabolomics data and exploring associations with other omics data and metadata | Metabolite abundances, testing association such as microbes and metabolites |
aPublication year.
Figure 2.
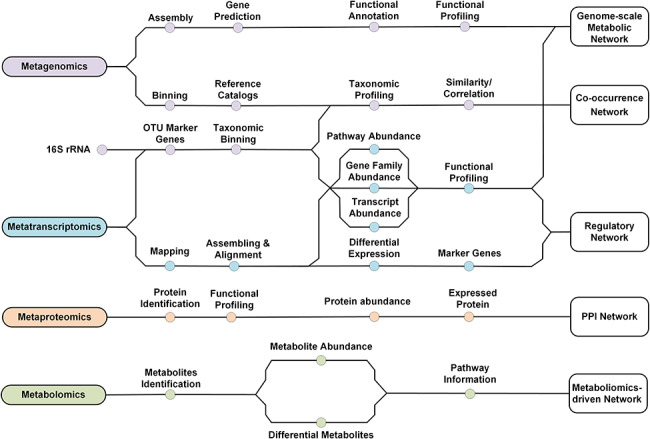
The network modelling methods applied to meta-omics data. Various approaches have been proposed to construct network models based on different types of microbial meta-omics data. From metagenomics, the taxonomic and underlying functional profiling can be inferred, and we can construct co-occurrence networks and genome-scale metabolic networks, respectively. From metatranscriptomics, not only can we construct co-occurrence and genome-scale metabolic networks, but also construct regulatory networks. Additionally, we can construct PPI networks and metabolomics-driven metabolic networks based on metaproteomics and metabolomics, respectively.
Pairwise relationships
The simplest and most prevalent method for constructing a co-occurrence network is to quantify the pairwise relationship between any two taxa within a community. Classically, there are two common quantitative measurements to estimate pairwise associations. The first one is (dis-)similarity indexes, such as Bray–Curtis and Kullback–Leibler indexes [67] (Box 1), which assess differences in two taxa over multiple samples. By calculating the (dis-)similarity scores and assessing the significance with a permutation test, all significant interactions could be regarded as links to construct the network. The other commonly used metric is the correlation methods, such as Pearson and Spearman correlation coefficients [31, 32] (Box 1). In such networks, a positive correlation coefficient may imply cross-feeding or co-aggregation between two taxa, while a negative coefficient may infer mutual exclusive interactions or niche differentiation [56].
However, constructing networks based on pairwise relationships is challenging. One such challenge comes from the use of relative compositional data, which introduces the problem of compositional bias. Specifically, a significant increase in one taxon’s abundance will result in a relative decrease in all others’ abundances. Additionally, given that correlation metrics cannot differentiate direct associations from the indirect ones, the faulty prediction of relationships between microbial taxa is a crucial challenge. For example, there will be inferred positive relationships between two taxa that share interaction partners (e.g. prey on a third taxon) while no direct relationships between them actually. Another major challenge is caused by data sparsity. In count or abundance data, zeros may imply that the taxon is absent, or it may imply that the taxon is present at levels below the detection limit. This ambiguity may cause particular issues with metrics based on presence or absence (e.g. Bray–Curtis) and may lead to spurious results.
Several computational approaches have been developed to address these concerns. Methods that attempt to minimize composition bias include SparCC [85], REBACCA [86] and CCLasso [87]. Given the large scale and sparsity of microbial networks, SparCC uses iterative approximation and the log transformation of composition data to estimate pairwise correlations [85]. While avoiding direct measurement of compositional data, SparCC is limited by high computational complexity [88]. By contrast, REBACCA and CCLasso are considerably faster due to the use of L1-norm shrinkage [87]. Furthermore, CCLasso used a loss function for data noise, thereby having a better performance in terms of compositional data. Additionally, the partial correlation approaches have been proposed to identify the direct relationships in a community. It has been successfully applied to identify the overall patterns of associations between viruses and bacteria [89]. However, this approach fails when the number of samples is far less than the number of taxa. Another approach to sparsity problems is the use of an ensemble approach, as proposed by Faust et al. [67], which considers the pros and cons of various similarity metrics and calculates a combined score from four individual metrics, including Bray–Curtis, Kullback–Leibler, Pearson and Spearman, to construct the co-occurrence network. Based on the network, the authors analysed the co-occurrence or co-exclusive patterns between pairwise microbes. Many of identified relationships are consistent with known cell-to-cell interactions (e.g. in the oral cavity, positive correlations were found between Fusobacterium and Capnocytophaga, Peptostreptococcus and Porphyromonas, where F. species have been reported as to be important organisms through physical contact with others) [67].
Complex patterns
Although the networks of pairwise relationships are relatively easy to construct, they are not able to capture the complex interactions among multiple taxa. One obvious alternative is to use regression-based analyses to infer relationships between a taxon and multiple other taxa (one-to-many relationships) [89]. In regression-based networks, relationship patterns are represented as directed hyperedges pointed from the independent taxa to dependent taxon [89]. Using this approach, van den Bergh identified independent associations of viruses in the upper respiratory tract of young children [89]. The results showed a positive correlation between enteroviruses and other poly-viruses, such as human bocavirus, parainfluenza viruses and human parechovirus [89], but a negative correlation between coronaviruses and human rhinoviruses, leading to disease discovery. However, it is important to recognize that overfitting is an inevitable problem in regression models, and thus, some measures (e.g. cross-validation, pruning and regularization) have to been taken to guard against this problem [90, 91]. Moreover, biological mechanisms underlying complex relationships extracted by regression-based methods are difficult to explain. For example, regression models imply a directionality, which may or may not reflect true biology (e.g. it is difficult to decipher whether a microbe affects or is affected by other microbes given relative abundance data). For these reasons, regression-based methods have not been extensively used.
Based on constructed networks, subsequent topological analysis can capture the underlying structures of the communities [6]. The basic network characteristics, from the degree to clustering coefficient and centrality, have been successfully applied to distinguish the different roles that microorganisms may play [6]. For example, combining node degree with betweenness centrality and closeness centrality, key nodes can be predicted as having an effect disproportionately crucial on the overall community [92, 93]. This approach was successfully applied to identify the mechanism of how abiotic factors and host genotype influence the phyllosphere microbial structure [32]. A more detailed case is given as Example S2 in Supplementary Data. Modularity is another common measure of local co-occurrence patterns of microbial networks. A module represents a group of physically linked microbes potentially working together to achieve a function [10]. Detecting modules sheds light on the complex assembly principle in communities [94]. A recent study has shown that modules in microbial communities are made up of various kinds of microbes rather than being dominated by a single taxon. From this observation, researchers deduced that the assembly patterns of the community were determined by environmental filters rather than species assortment [94], which has also been previously demonstrated [61].
Globally, co-occurrence network analyses have been extensively applied to study the microbiome in various contexts, including human diseases, plants and environments [6, 31]. Focusing on inflammatory bowel diseases (IBD), Bahtiyar et al. analysed conserved modules within co-occurrence networks from two different intestinal cohorts (with Crohn’s disease or with ulcerative colitis). The authors demonstrated that disturbances in these modules do have negative consequences for patients, such as poor responses to treatment or increased risk of relapse [95]. Further, Abbas et al. applied network-based features (node scoring integrated betweenness, closeness and average neighbour degree) within 973 samples (657 IBD and 316 healthy samples) to identify potential disease biomarkers [96], which significantly improves the predictive performance over other methods [96]. These studies suggest the significance of network studies to human health. Additionally, Poudel et al. proposed four types of frameworks to determine the roles of microbes in oak phyllosphere communities [6]. By constructing the co-occurrence network on OTUs, they first identified candidate taxa that contribute to maintaining the structure and function of the existing microbial community and determined whether these taxa had positive or negative relationships with host responses, pathogens as well as specific diseases under a given condition [6]. Such frameworks provide valuable insights into suppressing plant diseases or improving plant growth by the addition of appropriate microbes to a plant’s environment. To associate geographic patterns with symbiosis patterns of the microbial communities, Ma et al. collected soil samples from 110 continental-scale sites across 5 climate regions and constructed the co-occurrence networks based on OTUs [31]. Both the node-level and network-level topological analyses showed that soil microbiomes in the North seemed to have stronger phylogenetic relatedness but weaker ecological relatedness compared to those in the South [31].
Notably, the method chosen to measure relationships among taxa plays a key role in the resulting network. Therefore, several major factors should be seriously considered when choosing the network construction method, including the types of hypothetical relationships (e.g. the Pearson correlation assumes the relationships are linear, while regression approach assumes these are non-linear), the robustness to noise and outliers and the sensitivity to composition bias and data sparsity issues. Additionally, most studies focus on pairwise relationships within communities while lacking approaches that truly disentangle complex relationships among multiple taxa [56]. Additional developments will be required to solve this problem. More importantly, the networks at the taxonomic level fail to clarify potential mechanisms behind observed results due to the weak relationships between correlation and causality [97, 98]. Further experimental work is needed to validate relationships and specify possible causality.
Networks at the functional level
Given that communities could be functionally similar even if they differ at the taxonomic level [99], an alternative approach is proposed based on WMGS data to construct networks at the functional level (i.e. genome-scale metabolic networks, abbr. GMNs), which provides an important entry point for the mechanistic understanding of microbiome and host–microbiome relationships.
By quality filtering of raw data and gene prediction, we can predict present genes and infer their potential functions in microbial communities [100]. Then, we can utilize gene annotation information to find functional changes associated with host states at the level of a pathway or a module based on GMNs [60]. A common network construction approach is to identify functional profiling by genome annotation from current databases, such as the Kyoto Encyclopedia of Genes and Genomes [101], and then to identify all enzyme-coding genes. Based on prior reaction-level knowledge (biochemical reaction databases, e.g. MetaCyc [102]), each detected enzyme is annotated with one or more metabolic reactions in which it is involved. Hence, we can take the annotated enzymes or reactions as nodes and connect the enzymes that catalyse successive reactions as edges to construct metabolic networks (Figure 2) [60, 103]. In recent research, some tools have been proposed to determine the functional profiling from metagenomic data, such as HUMAnN2 and MG-RAST [77, 78], enabling more automatic reconstruction of GMNs (Table 2).
GMNs provide a global perspective to assess the overall metabolic capacity of a community. In this way, it is possible to associate special network topological features functionally with host states [60]. For example, Greenblum et al. constructed the phenotype-specific metabolic networks of the gut microbiome for different cohorts, including individuals suffering from obesity and IBD, along with healthy controls [60]. By analysing basic topological characteristics of the networks, they found that enzymes associated with obesity/IBD individuals were distributed at the periphery of the networks, and these enzymes were involved in different functions [60]. From these results, the authors inferred that the relationships of microbial metabolisms with the host differ in individuals with and without diseases [60]. Also, the network-level studies showed that the microbial communities of obesity/IBD individuals had lower modularity compared to healthy individuals [60], which could be a functional manifestation of the decrease of microbial diversity in diseased individuals [104]. Nevertheless, the analyses of overall communities cannot decipher functions to specific microbes, hindering the understanding of which microbes are associated with specific functions.
Another study gives a complementary viewpoint of understanding the functions of specific components based on the community-level network. Enzymes can be linked to the taxonomic groups according to the gene ID obtained from the read annotation [103]. Then, the Simpson index, analogized to the previous definition for identifying the dominating species in a sample, here is used to measure dominating taxonomic groups regarding a given function [103]. In this way, we can quantitatively evaluate the contribution of the key taxonomy groups to the whole community based on the constructed metabolic network [103]. Moreover, the pipeline developed by Franzosa et al. uses an annotated species-level pangenome database (i.e. ChocoPhlAn) to identify the functions of species within the community [78], allowing a deep understanding of species-level functional profiling.
Taken together, in contrast to taxonomic level networks, functional level networks not only focus on various taxa but also enable us to extract functional information (e.g. networks from HUMAnN2). Such approaches provide complementary views for taxonomic studies, bridging the gap between microbiome compositions and functions [60, 105]. However, networks at the functional level rely heavily on the complete and accurate genome annotation [106]. Therefore, frequent updating of biochemical information and efficient automatic annotation approach are indispensable, to avoid missing information in resulting metabolic networks and to get comprehensive conclusions.
Networks from metatranscriptomics
While metagenomics tries to explore the taxonomic and functional potential of the whole community [11], metatranscriptomic analyses produce a read-out of microbial genes that are transcribed as RNA [15]. By evaluating gene expression, we can understand the active functions of communities [13].
Currently, several efficient metatranscriptomic approaches have been developed to analyse the structure and active functions of microbial communities, such as HUMAnN2 [78], MetaTrans [75] and SAMSA [76]. These tools can identify taxonomic or functional profiling from metatranscriptomic data (Table 2). Consequently, we can construct the co-occurrence and metabolic network as described in metagenome section. The networks from metatranscriptomics provide more pertinent information on functional activity compared to the metagenomic description of the communities [63], as metatranscriptomics can reveal details of genes that are transcriptionally active under specific conditions and time [15]. Focusing on human gut microbiome, a seminal study compared the core metagenome and metatranscriptome in 372 men across different time points to evaluate gene and transcription pathways within individuals across time and between individuals [63]. Of particular interest, the authors identified a core set of metatranscripts that were universally transcribed across time and individuals [63]. This core set was enriched for genes essential for housekeeping functions and was often associated with different microbes, suggesting that different microbes may activate shared pathways [63]. These results perfectly demonstrate the complementary nature of analysing the metagenome and metatranscriptome together and how deviations in each contribute to a given state of an individual.
In addition, metatranscriptomic data could be applied to discover regulatory mechanisms and to construct global regulatory networks of the microbial communities (Figure 2) [15]. Such networks can represent gene-level interactions (e.g. activation and repression) between transcription factors and the corresponding target genes, supporting the mechanistic understanding of microbiome and host–microbiome relationships. However, transcriptional regulatory networks are still not as mature as metabolic networks [23], and relevant studies based on metatranscriptome are even rarer perhaps due to the incomplete transcriptional regulatory database and immature computational analysis techniques.
Globally, metatranscriptomics holds great potential to uncover the function and activity mechanisms of microbial communities. Nevertheless, it cannot be ignored that metatranscriptomic sequencing needs improvement, especially the isolation of high-quality RNA samples and the accurate detection of rapid responses to permutations [15]. In addition, mRNA is not always translated into the protein that carries out the actual function [15]. Therefore, the integration of metatranscriptomics with other omics can enable a comprehensive understanding of the microbial communities.
Networks from metaproteomics
Metaproteomics measures the proteins expressed by the entire microbial communities [107]. Using metaproteomic data, we can compare similarities and differences of protein expression in the communities and, more importantly, analyse PPI [107].
After the processing of proteomic samples and mass spectrometry, proteins in the microbial communities can be identified by IdentiPy or Trans-Proteomic Pipeline (Table 2) [79, 80]. From these measures, PPI networks are built where nodes represent proteins and edges denote physical interactions between proteins (Figure 2). Identification of protein interactions in bacterial species has been a powerful means to explore the role of proteins in pathways and pathogenesis [108]. A recent study explored the interactions of proteins in Streptococcus pneumoniae. Based on the topology of the PPI network, Wuchty et al. assigned putative functional roles to a large number of previously uncharacterized proteins, providing valuable hypotheses for future analysis of protein functions in S. pneumoniae [108].
However, current metaproteomic studies are still hampered due to sample complexity (e.g. sample heterogeneity and biomass amount) and redundant protein identification [109]. To take full advantage of the potential of metaproteomics, more efficient protein extraction, enhanced mass spectrometry and accurate protein identification are urgently needed [109]. Additionally, current PPI maps are far from complete, and the investigation of true PPIs from vast amounts of proteomic data is challenging [110, 111]. One common method integrates available genomic data (e.g. genome and transcriptomic data) to define likely interactions [110, 111]. Lv et al. predicted high confidence PPIs in cyanobacteria using seven different data types, including genome context and gene expression profiles [111]. Such an integrated approach provides a novel resource to subsequent functional analyses, e.g. the exploration of function-unknown proteins. Collectively, metaproteomics is not a completely stand-alone method, and its integration with other omics will provide a more comprehensive insight into microbial communities.
Networks from metabolomics
Metabolism, as the ultimate manifestation of an organism’s response to internal or external perturbation, is essential to maintaining the normal activities of organisms [112]. It thus plays a vital role in biological systems, and the microbiome plays a crucial role in maintaining a steady state of host metabolism [112, 113]. For example, microbes are crucial for breaking down larger molecules (e.g. proteins) into metabolites, and certain metabolites are only produced by the host in the presence of certain microbes [114]. Compared to other meta-omics information, the metabolites and metabolic pathways seem more conserved across species [115], thus providing a means for deciphering the functional mechanisms of the microbiome and host–microbiome relationships [116].
From raw metabolomics data, metabolic networks can be constructed by some automatic methods (Table 2), such as Pathos [82], MetaboAnalyst [83] and Netome [84]. Such networks (called metabolomics-driven networks to distinguish it from GMNs), consisting of nodes as metabolites and edges as metabolic reactions (Figure 2), provide a comprehensive description of a community’s metabolic processes.
Based on the constructed metabolic networks (both GMNs and metabolomics-driven networks), various topological-based approaches (as the section titled ‘Networks from Metagenomics’) have been used to associate network characteristics with functions of microbial communities. Furthermore, the mechanistic understanding of microbiome and host–microbiome relationships is the key focus of metabolic networks [61, 62]. Generally, the determination of relationships relies on metabolic interfaces (i.e. metabolites involved in metabolic exchanges) between species and communities, by which we can identify current microbiome and host–microbiome relationships specifically [61]. There are two common ways to identify metabolism interfaces. The first one is to predict the seed set according to the connectivity of the network, which has a complete theoretical framework. The seed set indicates the minimal set of exogenous compounds as the interface with other microbes or host [117]. This approach has been applied to assess interaction patterns between species [61]. A recent study detected the microorganisms’ seed set by the reverse-ecology framework [61]. Then, the study identified two interactions between species, including competition or synergism [61]. However, the results obtained by the seed set method depend solely on the metabolic interfaces computationally identified; thus the inferred interactions may be inaccurate. Another method is to identify metabolites that are exchanged between microbes and the host based upon the prior biological knowledge, mitigating the limitations of the computational method. Sung et al. manually collected an extensive data resource from a mass of published literature, including small-molecule transport and macromolecule degradation events of the gut microbiome. From these, they constructed a global metabolic transport network, called NJS16, providing a reference for determining microbiome or microbiome–host relationships [62]. Based on this global network, they measured the interactions between microbial taxa (e.g. a positive or negative effect on growth) and respectively built community-level metabolic influence networks for diseased and healthy individuals [62]. Such a study allows us to associate representative metabolic features with host states and infer the mechanisms of diseases. Despite its intuition in the biological sense, we have to realize that the prior knowledge may be incomplete or misleading due to gaps in experimental data collected to date. Given that the completeness and accuracy of this prior knowledge directly affect the downstream analysis, expanding experimental data is indispensable [62].
Globally, metabolic networks, including both metabolomics-driven networks and GMNs, focus on metabolic information pertaining to functions, providing a mechanistic understanding of behaviours of microbial communities. However, GMNs depend on the prediction of functions of communities from genome-scale data and emphasize the underlying functional profiling, lacking a true response to microbial phenotypes. By contrast, metabolomics-driven networks are sensitive to changing experimental and phenotype conditions due to the direct detection of metabolites, which seems more suitable for functional studies of communities. Nevertheless, the metabolomics-driven approaches rely on an accurate characterization of species- or strain-specific metabolites [116], making it difficult to scale well to study complex communities. Moreover, current experimental approaches to characterize the global metabolite profiling of microbial communities are costly and difficult [104]. Given that metagenomic and metatranscriptomic data are readily available, some computational approaches have been proposed to aid with these experimental challenges. A recent study used a model trained from known paired metabolomes and metagenomes within multiple samples to recover unobserved metabolites in new microbial communities that currently only have metagenomes available [116]. Such an idea provides useful insights into more reliable community metabolic trends. Together with many advances in metabolomics technologies and the integration with other omics [116], metabolomics would offer a wider application prospect to achieve accurate characterization of metabolic activities.
Networks from multi-omics
Analysis of each Omics independently does not yield a holistic view of a system. For example, due to the spatiotemporal specificity of gene expression and diversity of protein modifications, metagenomics analyses give no information as to which microbial characteristics are truly related to phenotype [18]. The networks from metatranscriptomics alone provide global expression information without a depiction of actual functions. The known transcriptomic regulatory information is extremely incomplete [23]. Additionally, metaproteomics and metabolomics analyses are limited in the number of identifiable proteins/metabolites and reactions [17]. Although each omics analysis has its limitations, the integration of these Omics approaches helps to maximize the interpretation of the taxonomies and functions of a microbial community.
The rapid advancement of experimental technologies allows the simultaneous extraction of samples representing the multi-omics information, including DNA, RNA, proteins and metabolites [118]. The integrated analysis of these Omics has got more attention in recent studies [18, 63, 64]. Such approaches provide a highly integrated landscape of environment-specific true expression and functions of the microbiome. For instance, Roume et al. integrated metagenomic, metatranscriptomic and metaproteomic data to construct a community-wide metabolic network [64]. Then, the authors evaluated the features of nodes and the levels of gene expression to extract genes encoding key functionalities, e.g. the subunits of ammonia and methane monooxygenase that are involved in nitrification and methane oxidation, respectively [64]. This study exemplifies the utility of combining different levels of evidence (copy number, metabolism, etc.) to link biological functions to microbial communities, deepening the understanding of communities’ metabolic capacity. Yet another example of multi-omics integration is the assessment of the shift of community structures linked to familial type 1 diabetes using the metagenome, metatranscriptome, metaproteome and metabolome [18]. The authors identified several populations of microbes that could contribute to functional differences (as measured by the metatranscriptome, metaproteome and metabolome) related to familial type 1 diabetes [18]. Collectively, these studies illustrate the value of multi-omics analysis for identifying relationships within microbial communities and their effect on the host.
The tools for multi-omics analysis are currently in progress. A recent study developed a computational pipeline, called MetaQUBIC, to predict condition-specific gene modules for microbial functional profiling based on metagenome and metatranscriptome [74], which provides a useful strategy to understand the functions of microbial communities and specific disease phenotypes. Furthermore, we can apply computational approaches, such as Bayesian networks, to elucidate the underlying molecular mechanisms of diseases by associating the microbial community, both at the level of taxonomy (metagenomics) and functionality (metatranscriptomics), with disease phenotypes and clinical measurements (Figure 3), providing mechanistic insights into host–microbiome relationships in the context of human health and disease [18].
Figure 3.
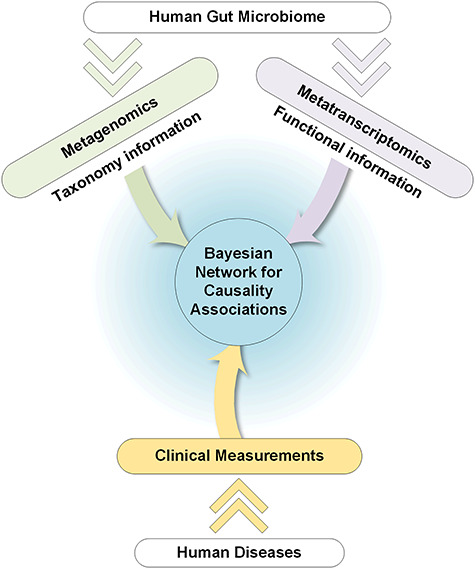
The exploration of host–microbiome relationships in the context of human health and disease. The gut microbiome plays an influential role in human diseases, including cancer, chronic diseases and inflammatory diseases. Integrated metagenomic and metatranscriptomic data of the microbial community with clinical measurements of the specific disease can unravel causal relationships between the microbiome and diseases, using the Bayesian network approaches for an example.
Multi-omics studies have been increasingly employed to decipher communities. Most of these studies focus on separate omics analysis first and then integrate different omics layers based on available data and extensive prior knowledge (e.g. enzymatic reactions to associate metagenomics and metabolomics), to provide a deeper understanding of complex microbiome [64]. However, such approaches may miss important associations among multiple omics layers. Much attention should be given to developing novel approaches for microbial multi-omics data integration. Given the current approaches used in other fields [119], one can try to extend those relevant to microbial studies. A promising example is the approach proposed by Tuncbag et al. [120], which uses a multi-weighted graph model to integrate multi-omics information measured in the same samples. This approach considers available properties of microbial communities, such as transcriptional profiling and proteomics, to explore structural and functional mechanisms. Another study applied the message-passing theory to fuse networks from multiple types of data, making use of the complementarity of different data types [121]. Tenenhaus et al. focused on a multivariate dimension reduction technique, sGCCA [122], to select common features from multi-omics datasets [123]. Based on such integrative data, both known and novel multi-omics biomarkers between multiple phenotypic groups were identified [123]. All these approaches represent a treasure trove that can be continually mined for in-depth insights into microbial communities. Notably, these approaches rely on accurate connections among different omics layers (i.e. corresponding relationships from gene and mRNA to protein and metabolomics), which is a well-known challenge of meta-omics data to be solved.
Conclusions and discussions
A significant challenge in microbiome research is understanding complex structures and functions and how perturbations of them impact the hosts. Making use of large amounts of meta-omics data, network-based analyses provide many significant insights into this issue. Here, we highlighted the merits and limitations of several common network-based approaches for microbial studies. Given that each approach has specific characteristics, the appropriate modelling methods should be selected for a specific objective of the study.
Globally, current approaches have some issues that need to be addressed before achieving a system-level understanding of microbiome. First, one of the major limitations is that meta-omics data lack direct associations of species/strains and specific functions. Although several tools (e.g. HUMAnN2) have been proposed to overcome this problem, they rely heavily on incomplete reference genomes [78], contributing to missing taxonomic and functional information of communities and influencing downstream network analyses. Thus, much attention should be given to solving the difficulties in metagenome assembly for a more comprehensive understanding of species without reference genomes. Currently, the major challenge for assembly is to bin assembled contigs from the whole community into species- or strain-level clusters [124]. Given that single-cell technologies can directly provide nucleotide frequency composition and gene content information of the target single cell and capture microbial diversity and heterogeneity at the species/strain level [124], the combination of metagenomics and single-cell genomics will improve the accuracy of metagenomic data binning [124]. Then, one could envision that a pseudo or draft genome, by scaffolding for contigs within each cluster based on the studies about organizational principles of the genome [125, 126], provides a reference for species/strains without a reference genome, giving a sufficient resolution of microbial communities on both taxonomic and functional layers. However, single-cell sequencing of microbial communities is not popular due to technical challenges. For example, isolating microbial cells from primary samples, especially from solid tissues, remains difficult [127]. Chimeric reads and uneven single-cell genome coverage are also inevitable due to the genome amplification process [127]. As such, technical improvements and refinements of single-cell workflows are promising for new insights into microbial studies. Superior whole-genome amplification chemistry and microfluidic droplet barcoding are beginning to help such advancements [128].
Second, network construction methods need to be further developed. Most studies focus on a specific layer of communities, e.g. gene regulation or metabolism. With the rapid development of experimental technologies, more integrated analyses of multi-omics, as referred to in ‘Network from Multi-omics’ section, should be introduced to form a network of networks and interpret all the properties of the communities as a whole. Moreover, the dynamics of microbial communities have not been well-described with most network methods. The majority of present microbial networks focus on the interactions in a selected time or environment. A promising research topic is the development of experimental methods and novel analysis pipelines to construct temporal networks, including at the gene level and spatiotemporal transcriptomic and metabolic levels [129]. The resulting networks, due to their dynamic nature, would make it possible to elucidate relationships over time within the microbiome or between the microbiome and its hosts. Notably, the resulting microbial networks are of large scale due to the high diversity and heterogeneity within communities, and to the intricate relationships between individuals, leading to the high complexity of subsequent analyses. For example, the shortest path length calculation of a selected node to all other nodes by Dijkstra algorithm with time complexity O(n2), where n is the number of nodes, is time-consuming for microbial networks containing a large number of nodes [130]. Another common example is the alignment between two networks to explore relationship variations between disease and healthy cohorts. Such alignment is extremely complex for networks with a large number of edges (e.g. the algorithm MAGNA++ with time complexity O(|E1| + |E2|) where |E1| and |E2| represent the number of edges of two networks, respectively [131]). Therefore, development of novel computational approaches is needed for more efficient analyses of microbial networks.
Last, additional experiments should be performed to effectively expand current knowledge. Increasing experimental data to more accurately annotate microbes and their functions are critical to improving the utility and accuracy of network models. To facilitate this process, active machine learning approaches could be applied to inform future experiments that would best improve the performance of current models and to build the most up-to-date models [132]. These two steps, including experimental selection and model update, are iteratively proceeded until the model can accurately adapt to current knowledge. These ideas will provide valuable insight into the construction of network models and, meanwhile, improve the generalization ability of the model in microbiome research.
In summary, the development of network models provides a powerful way to understand microbial communities. Combined with a large amount of data, a more widespread application of advanced network models is expected. Once such a pipeline is well developed, it will be possible to enhance the manipulation of the composition and function of microbial communities, giving rise to meaningful ideas for practical applications, especially disease prevention and treatment.
Authors’ Contributions
Q.M. and B.L. conceived the project. Z.L. and A.M. collected all the materials and prepared the figures and tables. All the authors wrote the manuscript. E.M. and M.M. helped revise the manuscript. The authors declare that they have no competing interests.
Supplementary Material
Zhaoqian Liu is a PhD student in School of Mathematics, Shandong University. Her research interests are graph theory and microbiome studies.
Anjun Ma is a PhD student at the Ohio State University. His primary research interest is metagenomics of the microbiome.
Ewy Mathé, PhD, is an assistant professor in the Department of Biomedical Informatics. Her primary research interests are epigenomics, genomics, nucleotide variants and metabolic patterns.
Marlena Merling is a PhD student at the Ohio State University, USA. Her research interests are network studies of microbiome.
Qin Ma, PhD, is an associate professor in the Department of Biomedical Informatics, the Ohio State University. Dr Ma has research experience over 10 years in omics data analysis and network modelling in microbial-related areas.
Bingqiang Liu, PhD, is a professor in School of Mathematics, Shandong University. His primary research strength is algorithm design in microbial regulatory network construction.
Key Points
The microbiome is a complex biological system that consists of a mass of microbes, including bacteria, archaea and viruses. These microbes closely interact with each other, as well as their hosts and environments, performing various biological functions.
Along with the development of complex network theories and increasing meta-omics data, network models have been widely applied to study microbial communities. For example, co-occurrence networks focus on co-occurrence or co-exclusion patterns between microbial taxa, while metabolic networks emphasize the functional level of microbial communities. In addition, regulatory and PPI networks based on meta-omics data should be further explored.
Networks that combine multi-omics data offer a unique multilayered viewpoint of the community and enable the assessment of how microbial communities affect and are affected by other factors (e.g. host genome and environment). These approaches have been used in various contexts, including human gut, plant and environment.
Despite much progress, there are still some challenges due to experimental and computational limitations, e.g. the detection of rare microbes, the incomplete genome annotation and the choice of network models. Therefore, future progress is expected in many directions.
The utility and predictive capacity of computational models are most directly affected by the amount and quality of experimental data. In turn, one could envision that computational methods, such as active machine learning, could effectively inform which experimental data are needed to subsequently enhance network models. These ideas will give valuable clues to obtain appropriate network models and, meanwhile, improve the generalizability of the model.
Supplementary Data
Supplementary data are available online at https://academic.oup.com/bib.
Funding
This work was supported by the National Natural Science Foundation of China (NSFC, 61772313), the Young Scholars Program of Shandong University (YSPSDU, 2015WLJH19) and the Innovation Method Fund of China (Ministry of Science and Technology of China, 2018IM020200). The project described was also supported by Award Number Grant UL1TR002733 from the National Center for Advancing Translational Sciences. This work used the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by the National Science Foundation #ACI-1548562. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Science Foundation and the National Institutes of Health.
References
- 1. Blaser MJ. The microbiome revolution. J Clin Invest 2014;124:4162–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Staley JT, Konopka A. Measurement of in situ activities of nonphotosynthetic microorganisms in aquatic and terrestrial habitats. Annu Rev Microbiol 1985;39:321–46. [DOI] [PubMed] [Google Scholar]
- 3. Blaser MJ. Who are we? Indigenous microbes and the ecology of human diseases. EMBO Rep 2006;7:956–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Huttenhower C, Gevers D, Knight R, et al. Structure, function and diversity of the healthy human microbiome. Nature 2012;486:207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bardgett RD, Freeman C, Ostle NJ. Microbial contributions to climate change through carbon cycle feedbacks. ISME J 2008;2:805. [DOI] [PubMed] [Google Scholar]
- 6. Poudel R, Jumpponen A, Schlatter DC, et al. Microbiome networks: a systems framework for identifying candidate microbial assemblages for disease management. Phytopathology 2016;106:1083–96. [DOI] [PubMed] [Google Scholar]
- 7. Mazel D, Davies J. Antibiotic resistance in microbes. Cell Mol Life Sci C 1999;56:742–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Arumugam M, Raes J, Pelletier E, et al. Enterotypes of the human gut microbiome. Nature 2011;473:174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sun Y, Li L, Xia Y, et al. The gut microbiota heterogeneity and assembly changes associated with the IBD. Sci Rep 2019;9:440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Barabasi A-L, Oltvai ZN. Network biology: understanding the cell’s functional organization. Nat Rev Genet 2004;5:101. [DOI] [PubMed] [Google Scholar]
- 11. Riesenfeld CS, Schloss PD, Handelsman J. Metagenomics: genomic analysis of microbial communities. Annu Rev Genet 2004;38:525–52. [DOI] [PubMed] [Google Scholar]
- 12. Di Bella JM, Bao Y, Gloor GB, et al. High throughput sequencing methods and analysis for microbiome research. J Microbiol Methods 2013;95:401–14. [DOI] [PubMed] [Google Scholar]
- 13. Helbling DE, Ackermann M, Fenner K, et al. The activity level of a microbial community function can be predicted from its metatranscriptome. ISME J 2012;6:902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhou J, He Z, Yang Y, et al. High-throughput metagenomic technologies for complex microbial community analysis: open and closed formats. MBio 2015;6:e02288–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bashiardes S, Zilberman-Schapira G, Elinav E. Use of metatranscriptomics in microbiome research. Bioinform Biol Insights 2016;10:BBI–S34610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wilmes P, Bond PL. Microbial community proteomics: elucidating the catalysts and metabolic mechanisms that drive the Earth’s biogeochemical cycles. Curr Opin Microbiol 2009;12:310–7. [DOI] [PubMed] [Google Scholar]
- 17. Tang J. Microbial metabolomics. Curr Genomics 2011;12:391–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Heintz-Buschart A, May P, Laczny CC, et al. Integrated multi-omics of the human gut microbiome in a case study of familial type 1 diabetes. Nat Microbiol 2017;2:16180. [DOI] [PubMed] [Google Scholar]
- 19. Methé BA, Nelson KE, Pop M, et al. A framework for human microbiome research. Nature 2012;486:215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Integrative HMP. The Integrative human microbiome project. Nature 2019;569:641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Integrative HMP. The Integrative human microbiome project: dynamic analysis of microbiome-host omics profiles during periods of human health and disease. Cell Host Microbe 2014;16:276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Qin J, Li R, Raes J, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010;464:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hao T, Wu D, Zhao L, et al. The genome-scale integrated networks in microorganisms. Front Microbiol 2018;9:296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Layeghifard M, Hwang DM, Guttman DS. Disentangling interactions in the microbiome: a network perspective. Trends Microbiol 2017;25:217–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dorogovtsev SN, Mendes JFF. Evolution of Networks: From Biological Nets to the Internet and WWW. New York: OUP Oxford, 2013. [Google Scholar]
- 26. Strogatz SH. Exploring complex networks. Nature 2001;410:268. [DOI] [PubMed] [Google Scholar]
- 27. Salgado H, Gama-Castro S, Peralta-Gil M, et al. RegulonDB (version 5.0): Escherichia coli K-12 transcriptional regulatory network, operon organization, and growth conditions. Nucleic Acids Res 2006;34:D394–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ma’ayan A. Introduction to network analysis in systems biology. Sci Signal 2011;4:tr5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Blaxter M, Mann J, Chapman T, et al. Defining operational taxonomic units using DNA barcode data. Philos Trans R Soc B Biol Sci 2005;360:1935–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Barabási A-L, Albert R. Emergence of scaling in random networks. Science (80-.). 1999;286:509–12. [DOI] [PubMed] [Google Scholar]
- 31. Ma B, Wang H, Dsouza M, et al. Geographic patterns of co-occurrence network topological features for soil microbiota at continental scale in eastern China. ISME J 2016;10:1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Agler MT, Ruhe J, Kroll S, et al. Microbial hub taxa link host and abiotic factors to plant microbiome variation. PLoS Biol 2016;14:e1002352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rio G, Koschützki D, Coello G. How to identify essential genes from molecular networks? BMC Syst Biol 2009;3:102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Feng Y, Wang Q, Wang T. Drug target protein-protein interaction networks: a systematic perspective. Biomed Res Int 2017;2017:1289259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Estrada E. Virtual identification of essential proteins within the protein interaction network of yeast. Proteomics 2006;6:35–40. [DOI] [PubMed] [Google Scholar]
- 36. Guimera R, Amaral LAN. Functional cartography of complex metabolic networks. Nature 2005;433:895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sharan R, Ulitsky I, Shamir R. Network-based prediction of protein function. Mol Syst Biol 2007;3:88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cheng F, Kovács IA, Barabási A-L. Network-based prediction of drug combinations. Nat Commun 2019;10:1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yoon J, Blumer A, Lee K. An algorithm for modularity analysis of directed and weighted biological networks based on edge-betweenness centrality. Bioinformatics 2006;22:3106–8. [DOI] [PubMed] [Google Scholar]
- 40. Newman MEJ, Girvan M. Finding and evaluating community structure in networks. Phys. Rev. E 2004;69:26113. [DOI] [PubMed] [Google Scholar]
- 41. Ma H-W, Zhao X-M, Yuan Y-J, et al. Decomposition of metabolic network into functional modules based on the global connectivity structure of reaction graph. Bioinformatics 2004;20:1870–6. [DOI] [PubMed] [Google Scholar]
- 42. Shi Z, Derow CK, Zhang B. Co-expression module analysis reveals biological processes, genomic gain, and regulatory mechanisms associated with breast cancer progression. BMC Syst Biol 2010;4:74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hinman VF, Nguyen AT, Cameron RA, et al. Developmental gene regulatory network architecture across 500 million years of echinoderm evolution. Proc Natl Acad Sci USA 2003;100:13356–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Woese CR, Fox GE. Phylogenetic structure of the prokaryotic domain: the primary kingdoms. Proc Natl Acad Sci USA 1977;74:5088–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hillmann B, Al-Ghalith GA, Shields-Cutler RR, et al. Evaluating the information content of shallow shotgun metagenomics. MSystems 2018;3:e00069–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. BRAy JR, CuRTIS JT. An ordination of upland forest communities of southern Wisconsin, ecological monographs. J Ecol Monogr 1957;27:325–49. [Google Scholar]
- 47. MacKay DJC, Mac Kay DJC. Information Theory, Inference and Learning Algorithms. Cambridge: Cambridge University Press, 2003. [Google Scholar]
- 48. Hotelling H. New light on the correlation coefficient and its transforms. J R Stat Soc Ser B 1953;15:193–232. [Google Scholar]
- 49. Myers JL, Well AD, LorchRF, Jr. Research Design and Statistical Analysis, New York: Routledge, 2013.
- 50. Ravasz E, Barabási A-L. Hierarchical organization in complex networks. Phys Rev E 2003;67:26112. [DOI] [PubMed] [Google Scholar]
- 51. Ravasz E, Somera AL, Mongru DA, et al. Hierarchical organization of modularity in metabolic networks. Science (80-) 2002;297:1551–5. [DOI] [PubMed] [Google Scholar]
- 52. Kolisnychenko V, Plunkett G, Herring CD, et al. Engineering a reduced Escherichia coli genome. Genome Res 2002;12:640–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Alon U, Surette MG, Barkai N, et al. Robustness in bacterial chemotaxis. Nature 1999;397:168. [DOI] [PubMed] [Google Scholar]
- 54. Yook S, Oltvai ZN, Barabási A. Functional and topological characterization of protein interaction networks. Proteomics 2004;4:928–42. [DOI] [PubMed] [Google Scholar]
- 55. Giot L, Bader JS, Brouwer C, et al. A protein interaction map of Drosophila melanogaster. Science (80-). 2003;302:1727–36. [DOI] [PubMed] [Google Scholar]
- 56. Faust K, Raes J. Microbial interactions: from networks to models. Nat Rev Microbiol 2012;10:538. [DOI] [PubMed] [Google Scholar]
- 57. Hartwell LH, Hopfield JJ, Leibler S, et al. From molecular to modular cell biology. Nature 1999;402:C47. [DOI] [PubMed] [Google Scholar]
- 58. Chen G, Wang X, Li X. Introduction to Complex Networks: Models Structures and Dynamics. Beijing: Higher Education Press, 2012. [Google Scholar]
- 59. Niu S-Y, Yang J, McDermaid A, et al. Bioinformatics tools for quantitative and functional metagenome and metatranscriptome data analysis in microbes. Brief Bioinform 2017;19:1415–29. [DOI] [PubMed] [Google Scholar]
- 60. Greenblum S, Turnbaugh PJ, Borenstein E. Metagenomic systems biology of the human gut microbiome reveals topological shifts associated with obesity and inflammatory bowel disease. Proc Natl Acad Sci USA 2012;109:594–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Levy R, Borenstein E. Metabolic modeling of species interaction in the human microbiome elucidates community-level assembly rules. Proc Natl Acad Sci USA 2013;110:12804–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sung J, Kim S, Cabatbat JJT, et al. Global metabolic interaction network of the human gut microbiota for context-specific community-scale analysis. Nat Commun 2017;8:15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Abu-Ali GS, Mehta RS, Lloyd-Price J, et al. Metatranscriptome of human faecal microbial communities in a cohort of adult men. Nat Microbiol 2018;3:356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Roume H, Heintz-Buschart A, Muller EEL, et al. Comparative integrated omics: identification of key functionalities in microbial community-wide metabolic networks. Biofilms and Microbiomes 2015;1:15007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Belda-Ferre P, Alcaraz LD, Cabrera-Rubio R, et al. The oral metagenome in health and disease. ISME J 2012;6:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Tan B, Ng CM, Nshimyimana JP, et al. Next-generation sequencing (NGS) for assessment of microbial water quality: current progress, challenges, and future opportunities. Front Microbiol 2015;6:1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Faust K, Sathirapongsasuti JF, Izard J, et al. Microbial co-occurrence relationships in the human microbiome. PLoS Comput Biol 2012;8:e1002606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Liu B, Gibbons T, Ghodsi M, et al. Accurate and fast estimation of taxonomic profiles from metagenomic shotgun sequences. Genome Biol 2011;12:P11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Bolyen E, Rideout JR, Dillon MR, et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol 2019;37:852–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Edgar RC. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods 2013;10:996. [DOI] [PubMed] [Google Scholar]
- 71. Truong DT, Franzosa EA, Tickle TL, et al. MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat Methods 2015;12:902. [DOI] [PubMed] [Google Scholar]
- 72. Milanese A, Mende DR, Paoli L, et al. Microbial abundance, activity and population genomic profiling with mOTUs2. Nat Commun 2019;10:1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Zhu Z, Ren J, Michail S, et al. MicroPro: using metagenomic unmapped reads to provide insights into human microbiota and disease associations. Genome Biol 2019;20:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Ma A, Sun M, McDermaid A, et al. MetaQUBIC: a computational pipeline for gene-level functional profiling of metagenome and metatranscriptome. Bioinformatics 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Martinez X, Pozuelo M, Pascal V, et al. MetaTrans: an open-source pipeline for metatranscriptomics. Sci Rep 2016;6:26447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Westreich ST, Korf I, Mills DA, et al. SAMSA: a comprehensive metatranscriptome analysis pipeline. BMC Bioinformatics 2016;17:399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Meyer F, Paarmann D, D’Souza M, et al. The metagenomics RAST server—a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics 2008;9:386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Franzosa EA, McIver LJ, Rahnavard G, et al. Species-level functional profiling of metagenomes and metatranscriptomes. Nat Methods 2018;15:962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Levitsky LI, Ivanov MV, Lobas AA, et al. IdentiPy: an extensible search engine for protein identification in shotgun proteomics. J Proteome Res 2018;17:2249–55. [DOI] [PubMed] [Google Scholar]
- 80. Deutsch EW, Mendoza L, Shteynberg D, et al. A guided tour of the trans-proteomic pipeline. Proteomics 2010;10:1150–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Solis-Mezarino V, Herzog F. compleXView: a server for the interpretation of protein abundance and connectivity information to identify protein complexes. Nucleic Acids Res 2017;45:W276–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Leader DP, Burgess K, Creek D, et al. Pathos: a web facility that uses metabolic maps to display experimental changes in metabolites identified by mass spectrometry. Rapid Commun Mass Spectrom 2011;25:3422–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Chong J, Soufan O, Li C, et al. MetaboAnalyst 4.0: towards more transparent and integrative metabolomics analysis. Nucleic Acids Res 2018;46:W486–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Rahnavard A, Hitchcock D, Pacheco JA, et al. Netome: a computational framework for metabolite profiling and omics network analysis. BioRxiv 2018;443903. [Google Scholar]
- 85. Friedman J, Alm EJ. Inferring correlation networks from genomic survey data. PLoS Comput Biol 2012;8:e1002687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Ban Y, An L, Jiang H. Investigating microbial co-occurrence patterns based on metagenomic compositional data. Bioinformatics 2015;31:3322–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Fang H, Huang C, Zhao H, et al. CCLasso: correlation inference for compositional data through Lasso. Bioinformatics 2015;31:3172–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Hirano H, Takemoto K. Difficulty in inferring microbial community structure based on co-occurrence network approaches. BMC Bioinformatics 2019;20:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Bergh MR, Biesbroek G, Rossen JWA, et al. Associations between pathogens in the upper respiratory tract of young children: interplay between viruses and bacteria. PLoS One 2012;7:e47711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Cawley GC, Talbot NLC. Preventing over-fitting during model selection via Bayesian regularisation of the hyper-parameters. J Mach Learn Res 2007;8:841–61. [Google Scholar]
- 91. Sarle WS. Stopped training and other remedies for overfitting. Comput Sci Stat 1996;352–60. [Google Scholar]
- 92. Berry D, Widder S. Deciphering microbial interactions and detecting keystone species with co-occurrence networks. Front Microbiol 2014;5:219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Daily G, Castilla JC, Lubchenco J, et al. Challenges in the quest for keystones. Sciences (New York) 1996;46:609–20. [Google Scholar]
- 94. Bakker MG, Schlatter DC, Otto-Hanson L, et al. Diffuse symbioses: roles of plant–plant, plant–microbe and microbe–microbe interactions in structuring the soil microbiome. Mol Ecol 2014;23:1571–83. [DOI] [PubMed] [Google Scholar]
- 95. Yilmaz B, Juillerat P, Øyås O, et al. Microbial network disturbances in relapsing refractory Crohn’s disease. Nat Med 2019;25:323. [DOI] [PubMed] [Google Scholar]
- 96. Abbas M, Matta J, Le T, et al. Biomarker discovery in inflammatory bowel diseases using network-based feature selection. PLoS One 2019;14:e0225382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Mainali K, Bewick S, Vecchio-Pagan B, et al. Detecting interaction networks in the human microbiome with conditional granger causality. PLoS Comput Biol 2019;15, e1007037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Bauer E, Thiele I. From network analysis to functional metabolic modeling of the human gut microbiota. MSystems 2018;3:e00209-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Taxis TM, Wolff S, Gregg SJ, et al. The players may change but the game remains: network analyses of ruminal microbiomes suggest taxonomic differences mask functional similarity. Nucleic Acids Res 2015;43:9600–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Qin J, Li Y, Cai Z, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012;490:55. [DOI] [PubMed] [Google Scholar]
- 101. Kanehisa M, Sato Y, Kawashima M, et al. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res 2015;44:D457–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Caspi R, Billington R, Fulcher CA, et al. The MetaCyc database of metabolic pathways and enzymes. Nucleic Acids Res 2017;46:D633–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Ofaim S, Ofek-Lalzar M, Sela N, et al. Analysis of microbial functions in the rhizosphere using a metabolic-network based framework for metagenomics interpretation. Front Microbiol 2017;8:1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Turnbaugh PJ, Hamady M, Yatsunenko T, et al. A core gut microbiome in obese and lean twins. Nature 2009;457:480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Jie Z, Xia H, Zhong S-L, et al. The gut microbiome in atherosclerotic cardiovascular disease. Nat Commun 2017;8:845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Danchin A, Ouzounis C, Tokuyasu T, et al. No wisdom in the crowd: genome annotation in the era of big data–current status and future prospects. J Microbial Biotechnol 2018;11:588–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Wilmes P, Bond PL. Metaproteomics: studying functional gene expression in microbial ecosystems. Trends Microbiol 2006;14:92–7. [DOI] [PubMed] [Google Scholar]
- 108. Wuchty S, Rajagopala SV, Blazie SM, et al. The protein interactome of Streptococcus pneumoniae and bacterial meta-interactomes improve function predictions. MSystems 2017;2:e00019-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Hettich RL, Pan C, Chourey K, et al. Metaproteomics: harnessing the power of high performance mass spectrometry to identify the suite of proteins that control metabolic activities in microbial communities. Anal Chem 2013;85:4203–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Skinnider MA, Stacey RG, Foster LJ. Genomic data integration systematically biases interactome mapping. PLoS Comput Biol 2018;14:e1006474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Lv Q, Ma W, Liu H, et al. Genome-wide protein-protein interactions and protein function exploration in cyanobacteria. Sci Rep 2015;5:15519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Fiehn O. Metabolomics—the link between genotypes and phenotypes. Funct Genom 2002;48:155–71. [PubMed] [Google Scholar]
- 113. Patti GJ, Yanes O, Siuzdak G. Innovation: metabolomics: the apogee of the omics trilogy. Nat Rev Mol Cell Biol 2012;13:263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Diether NE, Willing BP. Microbial fermentation of dietary protein: an important factor in diet–microbe–host interaction. Microorganisms 2019;7:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Chong J, Xia J. Computational approaches for integrative analysis of the metabolome and microbiome. Metabolites 2017;7:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Mallick H, Franzosa EA, Mclver LJ, et al. Predictive metabolomic profiling of microbial communities using amplicon or metagenomic sequences. Nat Commun 2019;10:3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Borenstein E, Kupiec M, Feldman MW, et al. Large-scale reconstruction and phylogenetic analysis of metabolic environments. Proc Natl Acad Sci USA 2008;105:14482–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Roume H, Heintz-Buschart A, Muller EEL, et al. Sequential isolation of metabolites, RNA, DNA, and proteins from the same unique sample. Methods Enzymol 2013;531:219–36. [DOI] [PubMed] [Google Scholar]
- 119. Noor E, Cherkaoui S, Sauer U. Biological insights through omics data integration. Curr Opin Syst Biol 2019;15:39–47. [Google Scholar]
- 120. Tuncbag N, McCallum S, Huang SC, et al. SteinerNet: a web server for integrating ‘omic’ data to discover hidden components of response pathways. Nucleic Acids Res 2012;40:W505–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Wang B, Mezlini AM, Demir F, et al. Similarity network fusion for aggregating data types on a genomic scale. Nat Methods 2014;11:333. [DOI] [PubMed] [Google Scholar]
- 122. Tenenhaus A, Philippe C, Guillemot V, et al. Variable selection for generalized canonical correlation analysis. Biostatistics 2014;15:569–83. [DOI] [PubMed] [Google Scholar]
- 123. Singh A, Shannon CP, Gautier B, et al. DIABLO: An integrative approach for identifying key molecular drivers from multi-omics assays. Bioinformatics 2019;35:3055–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Xu Y, Zhao F. Single-cell metagenomics: challenges and applications. Protein Cell 2018;9:501–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Ma Q, Xu Y. Global genomic arrangement of bacterial genes is closely tied with the total transcriptional efficiency. Genomics Proteomics Bioinformatics 2013;11:66–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Yin Y, Zhang H, Olman V, et al. Genomic arrangement of bacterial operons is constrained by biological pathways encoded in the genome. Proc Natl Acad Sci USA 2010;107:6310–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Tolonen AC, Xavier RJ. Dissecting the human microbiome with single-cell genomics. Genome Med 2017;9:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Woyke T, Doud DFR, Schulz F. The trajectory of microbial single-cell sequencing. Nat Methods 2017;14:1045–54. [DOI] [PubMed] [Google Scholar]
- 129. Buchweitz LF, Yurkovich JT, Blessing C, et al. Visualizing metabolic network dynamics through time-series metabolomics data. bioRxiv 2018;426106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Dijkstra EW. A note on two problems in connexion with graphs. Numer Math 1959;1:269–71. [Google Scholar]
- 131. Vijayan V, Saraph V, Milenković T. MAGNA++: maximizing accuracy in global network alignment via both node and edge conservation. Bioinformatics 2015;31:2409–11. [DOI] [PubMed] [Google Scholar]
- 132. Sverchkov Y, Craven M. A review of active learning approaches to experimental design for uncovering biological networks. PLoS Comput Biol 2017;13:e1005466. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.