

Abstract
We report the supramolecular assembly of artificial metalloenzymes (ArMs), based on the Lactococcal multidrug resistance regulator (LmrR) and an exogeneous copper(II)–phenanthroline complex, in the cytoplasm of E. coli cells. A combination of catalysis, cell‐fractionation, and inhibitor experiments, supplemented with in‐cell solid‐state NMR spectroscopy, confirmed the in‐cell assembly. The ArM‐containing whole cells were active in the catalysis of the enantioselective Friedel–Crafts alkylation of indoles and the Diels–Alder reaction of azachalcone with cyclopentadiene. Directed evolution resulted in two different improved mutants for both reactions, LmrR_A92E_M8D and LmrR_A92E_V15A, respectively. The whole‐cell ArM system required no engineering of the microbial host, the protein scaffold, or the cofactor to achieve ArM assembly and catalysis. We consider this a key step towards integrating abiological catalysis with biosynthesis to generate a hybrid metabolism.
Keywords: artificial metalloenzymes, biocatalysis, copper, in cell NMR spectroscopy, in vivo catalysis
Artificial metalloenzymes were created by supramolecular assembly in the cytoplasm of E. coli cells. The cells were then applied to the enantioselective biocatalysis of a Friedel–Crafts alkylation of indoles (see picture) and a Diels–Alder reaction. Directed evolution of the artificial metalloenzymes inside the cells provided improved variants for both whole‐cell biocatalytic reactions.
Introduction
Artificial metalloenzymes (ArM), which are hybrids of catalytically active transition metal complexes embedded in protein scaffolds, have emerged as a promising approach for biocatalysis of reactions that have no equivalent in nature.[ 1 , 2 ] This approach gives rise to rudimentary enzymes that can subsequently be tailored for the reaction of interest by employing the power of site‐directed mutagenesis and/or directed evolution.[ 3 , 4 , 5 ] For further developments, it is highly desirable to achieve the assembly and application of ArMs in bacterial cells. This will allow for whole cell biocatalysis, which is attractive from an economical perspective, is convenient for directed evolution and ultimately will be important towards achieving the goal of creating a hybrid metabolism, that is, a biosynthetic pathway augmented with new‐to‐nature chemistry. [6] Yet, application in whole cells present some major challenges, which include assembly of the artificial enzyme from a heterologously expressed protein and an exogenously added metal complex and the mutual incompatibility and inactivation of transition metal complexes and biological components, in particular glutathione.[ 7 , 8 , 9 ] Recently, the first reports of application of ArMs in cells appeared. In these studies, the above‐mentioned challenges were circumvented by creating the ArM in the periplasm or on the cell surface, where the GSH concentration is minimal and there are less barriers to achieving incorporation of the metal cofactor.[ 10 , 11 , 12 , 13 , 14 , 15 ] Cytoplasmic assembly has been achieved for artificial metallo‐heme enzymes, using bacteria containing co‐expressed natural and engineered heme transporters.[ 16 , 17 , 18 , 19 ]
Previously, we have reported on ArMs based on the Lactococcal multidrug resistance regulator (LmrR) protein from Lactococcus lactis. [20] LmrR is a transcription factor that forms a homodimer with a large hydrophobic pocket, known to promiscuously bind many small planar compounds. [21] We have shown that ArMs can be formed from LmrR by self‐assembly upon addition of a copper(II) 1,10‐phenanthroline complex (CuII‐Phen).[ 22 , 23 ] The CuII‐Phen complex binds to LmrR with low micromolar affinity and is primarily located between the two central tryptophans, one from each monomer (W96/W96′). This ArM was found to be an excellent catalyst for the enantioselective vinylogous Friedel–Crafts alkylation of indoles. Moreover, mutagenesis of residues in the hydrophobic pocket showed significant effect on catalysis, with a few mutations, notably M8A and A92E giving rise to significant improvement of activity and selectivity of the catalyzed reaction. [24]
Herein, we report the spontaneous self‐assembly of functional LmrR‐based ArMs in the cytoplasm of Escherichia coli and subsequent application in whole cell biocatalysis and directed evolution.
Results and Discussion
Catalysis in Whole Cells
To establish the tolerance of LmrR/CuII‐Phen against cellular components, we performed the catalysis of the reaction of α,β‐unsaturated 2‐acyl‐1‐methylimidazole (1) with 2‐methylindole (2 a) in cell‐free extracts. For this purpose, LmrR was expressed heterologously in E. coli C43(DE3) cells, the cells were lysed and the cell debris pelleted to yield cell free extracts, to which CuII‐Phen was added. Significant catalysis and enantioselective product formation was observed (Figure 1 b). In contrast, expressing Small Ubiquitin‐like MOdifier (SUMO) as a control protein in combination with CuII‐Phen only gave rise to low yield of product and low enantioselectivity in cell‐free extract.
Figure 1.
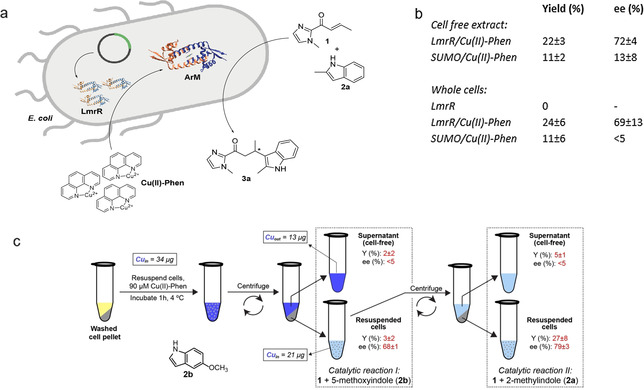
a) Self‐assembly of CuII‐Phen/LmrR artificial metalloenzyme in E. coli cells and catalyzed enantioselective Friedel–Crafts alkylation reaction. b) Results of catalysis of the enantioselective Friedel–Crafts alkylation reaction of 1 with 2 a by CuII‐Phen/LmrR and CuII‐Phen/SUMO in cell‐free extract and whole cells. In vitro catalysis: 120 μM LmrR, 90 μM CuII‐Phen, 1 mM 1 and 2 a in 20 mM MOPS, 150 mM NaCl, pH 7 at 4 °C for 30 min; in vivo catalysis: E. coli C43(DE3) cells over‐expressing LmrR (from a 0.5 mL overnight culture; OD600nm=4), 90 μM CuII‐Phen, 1 mM 1 and 2 a in catalysis buffer at 4 °C. Values are given as the average of independent duplicate experiments, each performed in duplo. Errors margins are standard deviations. c) Experimental design of the catalysis experiments with whole cells and supernatant. Values of copper content as determined by ICP‐OES are given in solid boxes. In dashed boxes are the samples that were used for catalytic reaction I: the Friedel–Crafts alkylation of 2 b with 1, and catalytic reaction II: the Friedel–Crafts alkylation of 2 a with 1.
Next, we took the whole E. coli cells expressing LmrR and incubated them with CuII‐Phen. The cells were then washed to remove excess, unbound CuII‐Phen and incubated with substrates (Figure 1 a). Remarkably, we still observed accelerated catalysis and enantioselective product formation. In the absence of CuII‐Phen, or using SUMO instead of LmrR did not give rise to catalysis. Thus, it was confirmed that both LmrR protein and exogenously added CuII‐Phen are essential for catalysis.
To confirm that the catalysis occurs in the cells, we incubated E. coli cells expressing LmrR with CuII‐Phen for 1 h and then pelleted the cells and washed with buffer to remove unbound CuII‐Phen. The copper content of the cells was quantified using inductively coupled plasma optical emission spectrometry (ICP‐OES; Figure 1 c). The data shows that a significant portion of the copper added is retained by the cell fraction suggesting the cells did indeed take up the CuII‐Phen complex. Comparison of the relative amounts of copper found in the cell fraction and supernatant fraction suggest that the overall concentration of CuII‐Phen in the cell fraction sample is around 50 μM. Notably, this does not necessarily mean that all this CuII‐Phen has been internalized; it also includes the possibility of copper complex that is bound on the surface of the cells. Based on the protein expression yields, the overall concentration of LmrR in the sample is in the low μM range and, hence, the CuII‐Phen is present in excess. It should be noted that the local concentration of the ArM in the cells will be significantly higher. The excess CuII‐Phen will contribute to the background reaction, causing a lowering of the ee value as compared to the experiments with isolated protein. Yet, the control experiments with SUMO (Figure 1 a) and the combined enantioselective catalysis results (vide infra) show that the overall outcome of catalysis is dominated by the ArM, due to the rate acceleration it provides.
The cell and the supernatant fractions were both incubated with substrates 1 and 2 b for the Friedel–Crafts alkylation reaction. Low activity was observed in both cases, which is in part related to the fact that 2 b is a less preferred substrate for LmrR/CuII‐Phen (see below), but only in the cell fraction significant enantioselectivity was observed in the product (Figure 1 b). The cell fraction was then again subjected to the same procedure and then a fresh batch of substrates, in this case 1 and 2 a, which is a preferred indole, [22] was added. Again, enantioselective product formation was only observed from the cell sample. These results confirm that the ArM assembled in the cell is responsible for this catalysis. Moreover, it also shows that the ArM has not leaked out into the buffer/supernatant, indicating that the structural integrity of cells is preserved.
Next, a series of LmrR mutants, for which the activity and enantioselectivity is known were tested in the cell experiments. [24] A good correlation was observed between activity and selectivity of the isolated ArM and that of the ArM in whole cells, even though the enantioselectivities in whole cells were generally lower than those found with isolated enzymes, most likely due to the background reaction (Figure 2 a, Table S1). For example, the mutant A92E, which we reported as the most active and selective ArM to date for this reaction [24] also gives significantly higher yield and enantioselectivity than the wild type in the cell experiments. Similarly, mutations that had a detrimental effect on catalysis in isolated proteins also showed this effect in cells.
Figure 2.
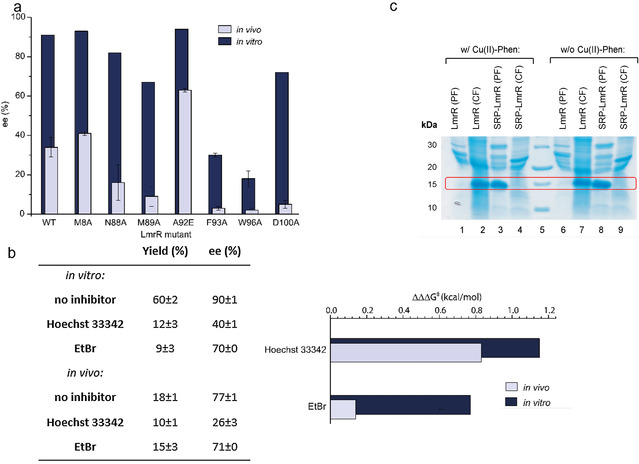
a) Enantioselectivity of the Friedel–Crafts alkylation reaction of 1 with 2 a, catalyzed by isolated CuII‐Phen/LmrR artificial enzyme mutants (dark blue) and CuII‐Phen/LmrR artificial enzyme mutants in whole E. coli cells (light blue). b) Left: Effect of addition of Hoechst 33342 and ethidium bromide (EtBr; 4 equivalents compared to CuII‐phen) on the yield and enantioselectivity of the reaction of 1 with 2 b, catalyzed by isolated CuII‐Phen/LmrR artificial metalloenyzmes and in whole cells. Values are given as the average of independent duplicate experiments, each performed in duplo. Errors are given as standard deviations. Right: Difference in enantioselectivity in the reaction of 1 with 2 b catalyzed by isolated CuII‐Phen/LmrR artificial metalloenzymes (dark blue bars) and in whole cells (light blue bars upon addition of inhibitors, compared to w/o inhibitor). A larger bar signifies a larger effect of the inhibitor on the enantioselectivity. Enantioselectivity differences are represented as ΔΔΔG ǂ, which is calculated using ΔΔΔG ǂ=ΔΔG ǂ (w/o inhibitor)−ΔΔG ǂ (w inhibitor) and ΔΔG ǂ=RTln(er), in which er is the enantiomeric ratio: % major enantiomer/% minor enantiomer. c) SDS PAGE of a E. coli cell fractionation experiment to determine protein localization for LmrR and SRP‐LmrR with and without CuII‐Phen. PF=periplasmic fraction; CF=cytoplasmic fraction.
From previous work on the LmrR protein we know that it binds Hoechst 33342 and ethidium bromide with high affinity in its hydrophobic pocket and that they can inhibit catalysis by competing for binding with CuII‐Phen and the substrates. [24] Hoechst 33342 is cell permeable, whereas ethidium has difficulties crossing the double membrane barrier of E. coli and is normally used to stain dead E. coli cells, which have permeable cell membranes. Fluorescence microscopy confirmed that Hoechst 33342 was readily taken up by the LmrR expressing cells, whereas ethidium bromide was not (Figures S4–S6). Indeed, using these dyes in combination with isolated LmrR/CuII‐Phen gave rise to a strongly reduced yield and enantioselectivity in catalysis in the reaction of 1 with 2 b (Figure 2 b). Incubating E. coli cells expressing LmrR with Hoechst 33342 prior to addition of CuII‐Phen, we found that in catalysis the enantioselectivity of the product was significantly decreased. In contrast, no significant effect on the enantioselectivity was observed for cells incubated with ethidium bromide. This becomes especially apparent when expressing the enantioselectivity differences w/ and w/o inhibitor in ΔΔΔG ǂ (Figure 2 b, inset).
We further investigated the cellular localization of CuII‐Phen/LmrR in E. coli by performing cell fractionation experiments. These experiments showed that LmrR and CuII‐Phen/LmrR are localized exclusively in the cytoplasm of E. coli (Figure 2 c). This strongly suggests that the ArM assembles in the cytoplasm of E. coli. For comparison, a variant of LmrR (called SRP‐LmrR) containing a N‐terminal periplasmic localization signal known to transport proteins to the periplasm through the SRP pathway, was constructed. [25] As expected, SRP‐LmrR and CuII‐Phen/SRP‐LmrR were found to exclusively localize in the periplasm of E. coli.
Combined these experiments support that the LmrR/CuII‐Phen ArM is assembled in the cytoplasm of E. coli and that the structural integrity of the cellular membrane is maintained during the experiments.
In‐Cell NMR Studies of ArM Assembly
While the reactivity data is fully in agreement with in vivo assembly of the ArM, it does not provide direct evidence. For this reason, the assembly of the ArM in E. coli Lemo21 (DE3) cells was studied by in‐cell NMR spectroscopy. We focused on the LmrR_A92E mutant, since this has a higher binding affinity for CuII‐Phen. [24] Due to high molecular crowding in E. coli cells, [26] in‐cell solution‐state NMR spectroscopy was not feasible. [27] Instead, we employed dynamic nuclear polarization (DNP)‐supported solid‐state NMR (DNP‐ssNMR) spectroscopy, which can probe protein structure inside cells irrespective of protein size and molecular crowding at greatly enhanced sensitivity.[ 28 , 29 ] For reference, we first obtained solution‐ and solid‐state NMR evidence for in vitro assembly of the ArM using near‐complete resonance assignments (Figure 3 a, see also Tables S9, S10) and by probing paramagnetic quenching effects exerted by CuII. 2D experiments in both soluble (Figures S10, S11) and microcrystalline (Figure S12) samples revealed site‐selective paramagnetic quenching, along with faster longitudinal relaxation rates (R1) in the ssNMR spectra (Figure S13). Additionally, small chemical shift perturbations (CSPs) occurred in the residues away from the CuII‐Phen binding site (Figure S14), consistent with the structural plasticity exhibited by LmrR when bound to different compounds.[ 21 , 30 , 31 , 32 ]
Figure 3.
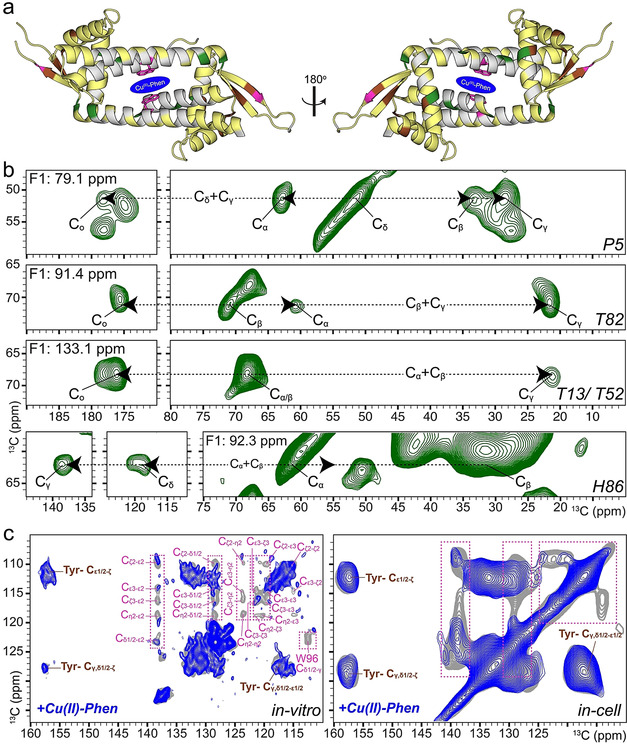
In vitro and in‐cell NMR studies of the LmrR_A92E/CuII‐Phen artificial metalloenzyme. a) Summary of NMR analysis: Residues used for both solution‐ and solid‐state NMR analysis are plotted on the crystal structure of LmrR (PDB ID: 3F8F) in yellow, green, pink and brown. Green residues were identified in the 3D 13C (DQ‐SQ‐SQ) DNP‐ssNMR spectrum (b). Tryptophan (pink) and tyrosine (brown) residues are used for analysis in (c), in which the selective reduction of NMR signal intensities for Trp aromatic signals confirms proper in‐cell assembly of the ArM. In‐cell experiments were performed in E. coli Lemo21 (DE3).
For the in‐cell DNP‐ssNMR experiments, LmrR‐specific isotope labeling was achieved using the antibiotic rifampicin to suppress native E. coli polymerases during T7 RNA polymerase mediated expression.[ 33 , 34 , 35 , 36 ] The signal contributions from the cellular background were reduced further by eliminating the non‐protein cellular background labeling using a specialized algal amino‐acid mixture (devoid of Trp, Cys, Asn and Gln) for isotope labeling. [37] Secondly, the cellular background was deuterated, which also leads to high DNP enhancements on the molecule of interest. [38] Lastly signal contributions from unincorporated, isotope labeled amino acids were removed by expressing in unlabeled medium during the last quarter of the expression time. The resulting in‐cell DNP‐ssNMR samples exhibited an approximately 100‐fold increase in LmrR signals (Figure S15a) and allowed us to rapidly record different multidimensional ssNMR experiments. The 2D DNP‐ssNMR spectra (Figure S16, S17) were in very good agreement with the in vitro spectrum of LmrR. In line with our 2D data sets, analysis of the 3D data sets suggested qualitative agreement between our in vitro assignments and the backbone correlations observed in cells. In spite of the increased NMR line width at low‐temperature 400 MHz DNP conditions, [39] we could obtain several spectral strips in the 3D experiment (see materials and methods) which unambiguously matched with the backbone assignments determined in vitro (Figure 3 b, Figures S18–S20). We correlated the Cα, Cβ assignments to the side chains and the carbonyl chemical shifts to confirm the respective amino acid type (Figure 3 b). The solution‐state NMR spectrum of the lysates further confirmed that well folded LmrR is the only labeled molecule in the sample, and additionally showed no visible signs of protein degradation (Figure S21).
As a final test for assembly of the ArM in cells, we tracked the paramagnetic effect of adding CuII‐Phen upon Trp NMR resonances of LmrR. For this purpose, a modified expression protocol was used (SI Material and Methods NMR studies) that led to the labeling of all amino acids, including Trp. In the presence of CuII‐Phen, we observed a selective reduction of the Trp aromatic side‐chain signal intensity, while Tyr residues, which are present far away from the CuII‐Phen binding region, do not display paramagnetic quenching effects (Figure 3 a,c and Figure S22). Additionally, a clear increase in the longitudinal relaxation rate (R1) was also observed (Figure S23) in full accordance with the spectral changes observed in vitro (Figure 3 c, Figure S12, S13). To validate our DNP‐ssNMR analysis, we finally analyzed the clear cell lysates of replicate cells used for in vivo DNP‐ssNMR experiments using solution NMR (Figure S24). The CSPs due to CuII‐Phen binding largely followed a similar trend to in vitro CSPs (Figure S25), and we observed a complete quenching of Trp96 side chain resonance (Figure S24, S26) suggesting tight binding to the protein, indicating the presence of stably assembled ArM in the lysate.
Taken together, these results provide direct spectroscopic evidence for the in vivo assembly of the ArM. Moreover, the copper complex is predominantly present in the CuII state. This is consistent with the observed activity in catalysis, further confirming that the reduction of CuII to CuI, [40] due to cellular reductants, does not play a significant role.
Directed Evolution
Having established that these LmrR based ArMs are assembled in vivo and can be employed in whole cell biocatalysis, we aimed to exploit this for directed evolution of the ArMs. The reaction of enone 1 with 5‐methoxyindole (2 b), which is a less good substrate than 2‐methylindole (2 a) for enantioselective vinylogous Friedel–Crafts alkylation, was selected for this purpose. An Alanine scan of residues in the hydrophobic pocket of LmrR was performed to identify positions of interest (Table S2). The trend of enantioselectivity from in vivo catalysis and activity from in vitro catalysis matched well for the Alanine mutants (Figure 2 a).
LmrR_A92E was chosen as the starting point for directed evolution because it was shown to be significantly more active than LmrR. [24] Site‐saturation libraries with NDT codon degeneracy were designed for positions 8, 14, 88, 89, 93 and 100, which were identified in the alanine scan as positions of interest (Figure 4 a). From one round of screening (in total ca. 250 clones) the mutant LmR_A92E_M8D was found to show the highest activity and enantioselectivity (Table S14). Enzyme kinetics studies for the reaction of 1 and 2 b confirmed that the catalytic efficiency (k cat/K M) of LmrR_A92E_M8D was 185.9 M−1 min−1, almost three times higher than that of the starting point, LmrR_A92E (73.3 M−1 min−1; Figure S29). While M8 in LmrR was known from previous studies to be a privileged residue for ArM optimization,[ 24 , 41 , 42 ] this always entailed mutation to another hydrophobic residue and the mutation M8D was not found before as beneficial for catalysis.
Figure 4.
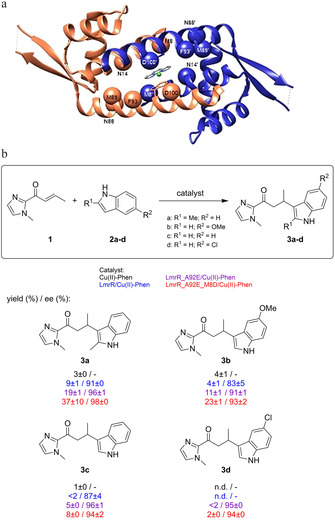
a) Structure of LmrR/CuII‐Phen with residues that were randomized during the directed evolution study indicated as spheres. b) Scope of the enantioselective Friedel–Crafts alkylation catalyzed by CuII‐Phen and CuII‐Phen/LmrR artificial metalloenzyme mutants. Conditions: 12 μM LmrR mutant, 9 μM CuII‐Phen, 1 mM 1 and 2 a in 20 mM MOPS, 150 mM NaCl, pH 7 at 4 °C. Reaction times: 30 min (3 a), 6 h (3 b, 3 c) or 24 h (3 d). Values are given as the average of independent duplicate experiments, each performed in duplo. Errors are given as standard deviations.
The scope of indoles was compared for ArMs based on three LmrR variants: LmrR, LmrR_A92E and LmrR_A92E_M8D) (Figure 4 b). A marked improvement in activity from LmrR to LmrR_A92E for the different indoles was found. This was further improved for the double mutant LmrR_A92E_M8D for all the indoles tested. It is remarkable to see that almost 40 % of 2 a is converted to product in thirty minutes by LmrR_A92E_M8D. This demonstrates that directed evolution is a powerful tool for improving the catalytic properties of ArM.
In Vivo ArM Catalysis of Diels–Alder Reactions
Having established the in vivo ArM‐catalyzed Friedel–Crafts alkylation, we sought to expand the scope to a different catalytic reaction. For this purpose, we selected the CuII catalyzed Diels–Alder reaction of azachalcone (4) with cyclopentadiene (5) (Table 1). [43] This is a reaction that we have reported on before using ArMs based on LmrR with covalently attached phenanthroline and bipyridine ligands, [44] but it was so far not investigated with supramolecular assembled ArMs. Performing the reaction with isolated wtLmrR/CuII‐Phen after 2 days of reaction provided the endo isomer of the Diels–Alder product 6 with 58 % ee (Table 1). Using whole cells containing the ArMs, assembled as described above, the Diels–Alder product was obtained in about 14 % yield with 8 % ee after 3 days, whereas control experiments with SUMO instead of LmrR resulted in racemic product. LmrR_A92E gave rise to a higher ee value, both in vitro and in vivo, confirming that also for this reaction the effect of mutations is detectable in the whole cell experiments (Table 1). Therefore, an alanine scan of various positions in LmrR and LmrR_A92E was performed to identify residues that could potentially be mutated to improve the ArM. Whole cell experiments with the various mutant ArMs showed detectable differences in both yield and enantioselectivity (Table S5). The results were verified independently with isolated ArMs and, even though the yields and ee values in the whole cell catalysis experiments were generally lower, a good correlation of the trends in yields and ee values was observed. From the whole cell experiments one variant, LmrR_A92E_V15A, was identified that gave rise to both higher yield and ee, that is, 20 and 45 %, respectively. This was confirmed in vitro using the isolated protein, giving the Diels–Alder product in 52 % yield and 84 % ee (Table 1). Control experiments with this mutant confirmed that the catalysis indeed occurred inside cells (Table S6).
Table 1.
Results of the in vivo and in vitro ArM‐catalyzed Diels–Alder reaction.
|
Protein |
In vivo[a] |
In vitro[b] |
||
|---|---|---|---|---|
|
|
yield [%][c] |
ee [%][c] |
yield [%][c] |
ee [%][c] |
|
SUMO |
14±1 |
0 |
|
|
|
LmrR |
14±4 |
8±2 |
22±5 |
58±6 |
|
LmrR_A92E |
15±6 |
17±1 |
33±10 |
68±2 |
|
LmrR_A92E_V15A |
20±3 |
45±0 |
52±7 |
84±3 |
[a] CuII‐Phen (180 μM), E. coli cell suspension (1 mL), 4 (1 mM) and 5 (33 mM) in 20 mM MOPS, 150 mM NaCl at pH 7, reaction time 48 h at 4 °C. [b] CuII‐Phen (90 μM), LmrR variant (120 μM), 4 (1 mM) and 5 (33 mM) in 20 mM MOPS, 150 mM NaCl at pH 7, reaction time 48 h at 4 °C. [c] Values for the endo isomer. Values represent the average of independent duplicate experiments, each performed in duplo. Errors are given as standard deviations.
Attempted directed evolution by randomization of 7 positions that were deemed potentially interesting from the alanine scanning did not result in further improved variants. Nevertheless, LmrR_A92E_V15A is a new variant: it was not found before as improved mutant in other reactions. Interestingly, this mutant performed poorly in the Friedel–Crafts reaction (Table S1). Conversely, the variant LmrR_A92E_M8D, which gave the best results in the Friedel–Crafts reaction, did not give rise to improved activity and selectivity in the Diels–Alder reaction (Table S6). This shows that the active site of the LmrR‐based ArM can be tailored specifically for catalysis of mechanistically different reactions. Understanding of the structural effect of these different mutations and their role in catalysis is not easily rationalized and will require molecular dynamics studies, as we performed before for the A92E mutation. [24]
Discussion
The remarkable and unexpected aspects of the current study are the straightforward in vivo assembly and apparent stability of the LmrR/CuII‐Phen artificial metalloenzymes in the cell's cytoplasm, as evident from the catalysis data, inhibition studies, cell fractionation experiments and the in‐cell NMR studies. E. coli cells over‐expressing the protein LmrR are simply incubated with CuII‐Phen abiological cofactor, resulting in uptake of the abiological cofactor and in vivo ArM assembly. For almost all ArM catalysis in vivo, the ArM and/or the bacterial cell had to be engineered to transport and localize the protein in the periplasm or on the cell surface, to reduce exposure to glutathione that is detrimental to many transition‐metal complexes.[ 7 , 10 , 15 ]
Copper complexes are also expected to be readily reduced to CuI and then be toxic to the cell due to the formation of reactive oxygen species (ROS). Yet, the combined results demonstrate unequivocally that the assembled LmrR/CuII artificial metalloenzymes are stable and catalytically active in the cytoplasm of the cell. This was further verified by performing the Friedel–Crafts reaction with isolated LmrR_A92E_M8D/ CuII‐Phen ArM in the presence of increasing amounts of glutathione, which is usually the main culprit for deactivation and instability of metal complexes and ArMs in cellular environments. The results indeed show that the ArM is still active in the presence of moderate concentrations of GSH (1 mM) and only at higher concentrations (10 mM) a significant detrimental effect on catalytic activity is observed, albeit that activity and enantioselectivity is even then still observed (Table S3). We hypothesize that the CuII‐Phen complex bound to the front entrance of the binding pocket of LmrR is protected from GSH, and other cellular components. The front entrance where the CuII ion is located has an overall negative charge due to the presence of multiple carboxylate rich residues. This may cause charge repulsion with glutathione and in this way protect the metal complex, as was recently reported also for a glycosylated albumin artificial metalloenzyme. [45] Intriguingly, this is reminiscent of the biological role of the protein LmrR, which has evolved to rapidly bind a plethora of different toxic compounds entering the cell, as start of the cellular drug resistance response in Lactococcus lactis.[ 20 , 31 ]
Conclusion
In conclusion, we have demonstrated herein that catalytically active artificial metalloenzymes can be self‐assembled in the cytoplasm of E. coli from heterologously produced LmrR and an exogenously added CuII‐Phen complex. The unique aspect of our system is that no extensive engineering of the microbial host, the protein scaffold or the cofactor is required, which makes it attractive for applications in whole cell biocatalysis and directed evolution, as demonstrated here. Further evolution is envisioned to give rise to mutants with catalytic efficiencies that can become competitive with natural enzymes. Moreover, in view of the catalytic versatility of LmrR‐based artificial metalloenzymes,[ 23 , 41 ] this work represents an important step forward towards achieving a hybrid metabolism by integrating artificial metalloenzymes in biosynthetic pathways.[ 6 , 16 ]
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank L. Villarino for assistance with synthetic chemistry, Dr. A. Iyer for assistance with live cell confocal microscopy, R. Leveson‐Gower for assistance with the MS measurements, Prof. P. Tordo, Dr. O. Ouari (Aix‐Marseille Université) for providing AMUPol for the DNP experiments and Dr. C. Mayer for advice and discussions. We thank Prof. I. Shimada (U. of Tokyo) for the NMR assignments of the wild‐type LmrR and Dr. H. van Ingen for providing access to the solution‐state NMR instrument along with J. van der Zwan and Dr. S. Xiang for technical support and discussions. This work was supported by the Netherlands Organisation for Scientific Research (NWO, projects 700.26.121 and 700.10.443 to M.B. and 724.013.003 to G.R.). In addition, S.N. was supported by the Netherlands’ Magnetic Resonance Research School (NMARRS, project number 022.005.029). G.R. acknowledges support from the Netherlands Ministry of Education, Culture and Science (Gravitation program no. 024.001.035).
S. Chordia, S. Narasimhan, A. Lucini Paioni, M. Baldus, G. Roelfes, Angew. Chem. Int. Ed. 2021, 60, 5913.
A previous version of this manuscript has been deposited on a preprint server (https://doi.org/10.26434/chemrxiv.12485993.v1).
References
- 1. Schwizer F., Okamoto Y., Heinisch T., Gu Y., Pellizzoni M. M., Lebrun V., Reuter R., Kohler V., Lewis J. C., Ward T. R., Chem. Rev. 2018, 118, 142–231. [DOI] [PubMed] [Google Scholar]
- 2. Wilson M., Whitesides G., J. Am. Chem. Soc. 1978, 100, 306–307. [Google Scholar]
- 3. Hyster T. K., Ward T. R., Angew. Chem. Int. Ed. 2016, 55, 7344–7357; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 7468–7482. [Google Scholar]
- 4. Markel U., Sauer D. F., Schiffels J., Okuda J., Schwaneberg U., Angew. Chem. Int. Ed. 2019, 58, 4454–4464; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 4500–4511. [Google Scholar]
- 5. Leveson-Gower R. B., Mayer C., Roelfes G., Nat. Rev. Chem. 2019, 3, 687–705. [Google Scholar]
- 6. Jeschek M., Panke S., Ward T. R., Trends Biotechnol. 2018, 36, 60–72. [DOI] [PubMed] [Google Scholar]
- 7. Wilson Y. M., Duerrenberger M., Nogueira E. S., Ward T. R., J. Am. Chem. Soc. 2014, 136, 8928–8932. [DOI] [PubMed] [Google Scholar]
- 8. Martínez-Calvo M., Mascareñas J. L., Coord. Chem. Rev. 2018, 359, 57–79. [Google Scholar]
- 9. Destito P., Vidal C., Lopéz F., Mascarenas J. L., Chem. Eur. J. 2020, 10.1002/chem.202003927. [DOI] [Google Scholar]
- 10. Jeschek M., Reuter R., Heinisch T., Trindler C., Klehr J., Panke S., Ward T. R., Nature 2016, 537, 661–665. [DOI] [PubMed] [Google Scholar]
- 11. Heinisch T., Schwizer F., Garabedian B., Csibra E., Jeschek M., Vallapurackal J., Pinheiro V. B., Marliere P., Panke S., Ward T. R., Chem. Sci. 2018, 9, 5383–5388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Grimm A. R., Sauer D. F., Polen T., Zhu L., Hayashi T., Okuda J., Schwaneberg U., ACS Catal. 2018, 8, 2611–2614. [Google Scholar]
- 13. Ghattas W., Dubosclard V., Wick A., Bendelac A., Guillot R., Ricoux R., Mahy J., J. Am. Chem. Soc. 2018, 140, 8756–8762. [DOI] [PubMed] [Google Scholar]
- 14. Zhao J., Rebelein J. G., Mallin H., Trindler C., Pellizzoni M. M., Ward T. R., J. Am. Chem. Soc. 2018, 140, 13171–13175. [DOI] [PubMed] [Google Scholar]
- 15. Liang A. D., Serrano-Plana J., Peterson R. L., Ward T. R., Acc. Chem. Res. 2019, 52, 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.J. Huang, Z. Liu, B. Bloomer, D. Clark, A. Mukhopadhyay, J. Keasling, J. Hartwig, ChemRxiv 2020, 10.26434/chemrxiv.11955174. [DOI]
- 17. Reynolds E. W., Schwochert T. D., McHenry M. W., Watters J. W., Brustad E. M., ChemBioChem 2017, 18, 2380–2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bordeaux M., Singh R., Fasan R., Bioorg. Med. Chem. 2014, 22, 5697–5704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sreenilayam G., Fasan R., Chem. Commun. 2015, 51, 1532–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Agustiandari H., Lubelski J., van Saparoea H., van den Berg B., Kuipers O. P., Driessen A. J. M., J. Bacteriol. 2008, 190, 759–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Madoori P. K., Agustiandari H., Driessen A. J. M., Thunnissen A. W. H., EMBO J. 2009, 28, 156–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bos J., Browne W. R., Driessen A. J. M., Roelfes G., J. Am. Chem. Soc. 2015, 137, 9796–9799. [DOI] [PubMed] [Google Scholar]
- 23. Roelfes G., Acc. Chem. Res. 2019, 52, 545–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Villarino L., Chordia S., Alonso-Cotchico L., Reddem E., Zhou Z., Thunnissen A. M. T., Maréchal J.-D., Roelfes G., ACS Catal. 2020, 10, 11783–11790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. van Bloois E., Winter R. T., Janssen D. B., Fraaije M. W., Appl. Microbiol. Biotechnol. 2009, 83, 679–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tabaka M., Sun L., Kalwarczyk T., Holyst R., Soft Matter 2013, 9, 4386–4389. [Google Scholar]
- 27. Serber Z., Keatinge-Clay A., Ledwidge R., Kelly A., Miller S., Dotsch V., J. Am. Chem. Soc. 2001, 123, 2446–2447. [DOI] [PubMed] [Google Scholar]
- 28. Narasimhan S., Scherpe S., Paioni A. L., van der Zwan J., Folkers G. E., Ovaa H., Baldus M., Angew. Chem. Int. Ed. 2019, 58, 12969–12973; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 13103–13107. [Google Scholar]
- 29. Narasimhan S., Pinto C., Paioni A. L., van der Zwan J., Folkers G. E., Baldus M., Nat. Protoc. 2021, 10.1038/s41596-020-00439-4. [DOI] [PubMed] [Google Scholar]
- 30. Takeuchi K., Tokunaga Y., Imai M., Takahashi H., Shimada I., Sci. Rep. 2014, 4, 6922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. van der Berg J. P., Madoori P. K., Komarudin A. G., Thunnissen A., Driessen A. J. M., PLoS One 2015, 10, e0135467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Takeuchi K., Imai M., Shimada I., Sci. Rep. 2017, 7, 267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Almeida F., Amorim G., Moreau V., Sousa V., Creazola A., Americo T., Pais A., Leite A., Netto L., Giordano R., Valente A., J. Magn. Reson. 2001, 148, 142–146. [DOI] [PubMed] [Google Scholar]
- 34. Galvão-Botton L., Katsuyama A., Guzzo C., Almeida F., Farah C., Valente A., FEBS Lett. 2003, 552, 207–213. [DOI] [PubMed] [Google Scholar]
- 35. Baker L. A., Daniels M., van der Cruijsen E. A. W., Folkers G. E., Baldus M., J. Biomol. NMR 2015, 62, 199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Baker L. A., Sinnige T., Schellenberger P., de Keyzer J., Siebert C. A., Driessen A. J. M., Baldus M., Grunewald K., Structure 2018, 26, 161–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kaplan M., Narasimhan S., de Heus C., Mance D., van Doorn S., Houben K., Popov-Čeleketić D., Damman R., Katrukha E. A., Jain P., Geerts W. J. C., Heck A. J. R., Folkers G. E., Kapitein L. C., Lemeer S., van Bergen en Henegouwen P. M. P., Baldus M., Cell 2016, 167, 1241–1251. [DOI] [PubMed] [Google Scholar]
- 38. Viennet T., Viegas A., Kuepper A., Arens S., Gelev V., Petrov O., Grossmann T. N., Heise H., Etzkorn M., Angew. Chem. Int. Ed. 2016, 55, 10746–10750; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 10904–10908. [Google Scholar]
- 39. Koers E. J., van der Cruijsen E. A. W., Rosay M., Weingarth M., Prokofyev A., Sauvee C., Ouari O., van der Zwan J., Pongs O., Tordo P., Maas W. E., Baldus M., J. Biomol. NMR 2014, 60, 157–168. [DOI] [PubMed] [Google Scholar]
- 40. Volentini S. I., Farias R. N., Rodriguez-Montelongo L., Rapisarda V. A., Biometals 2011, 24, 827–835. [DOI] [PubMed] [Google Scholar]
- 41. Villarino L., Splan K. E., Reddem E., Alonso-Cotchico L., Guttierez de Souza C., Lledos A., Maréchal J., Thunnissen A. W. H., Roelfes G., Angew. Chem. Int. Ed. 2018, 57, 7785–7789; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 7911–7915. [Google Scholar]
- 42. Zhou Z., Roelfes G., Nat. Catal. 2020, 3, 289–294. [Google Scholar]
- 43. Otto S., Bertoncin F., Engberts J., J. Am. Chem. Soc. 1996, 118, 7702–7707. [Google Scholar]
- 44. Bos J., Fusetti F., Driessen A. J. M., Roelfes G., Angew. Chem. Int. Ed. 2012, 51, 7472–7475; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 7590–7593. [Google Scholar]
- 45. Eda S., Nasibullin I., Vong K., Kudo N., Yoshida M., Kurbangalieva A., Tanaka K., Nat. Catal. 2019, 2, 780–792. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary