

Abstract
Circular RNAs (circRNAs) are a unique class of RNA molecule identified more than 40 years ago which are produced by a covalent linkage via back-splicing of linear RNA. Recent advances in sequencing technologies and bioinformatics tools have led directly to an ever-expanding field of types and biological functions of circRNAs. In parallel with technological developments, practical applications of circRNAs have arisen including their utilization as biomarkers of human disease. Currently, circRNA-associated bioinformatics tools can support projects including circRNA annotation, circRNA identification and network analysis of competing endogenous RNA (ceRNA). In this review, we collected about 100 circRNA-associated bioinformatics tools and summarized their current attributes and capabilities. We also performed network analysis and text mining on circRNA tool publications in order to reveal trends in their ongoing development.
Keywords: circRNA, bioinformatics tools, text mining, next generation sequencing, non-coding RNA, disease biomarker
Introduction
Circular RNAs (circRNAs) are biologically active nucleic acid molecules that exist in closed loop RNA forms and lack polyadenylated tails in contrast to messenger RNAs [1]. CircRNAs are generally categorized as non-coding RNA (ncRNA), although some circRNAs have protein-coding potential [2, 3]. CircRNAs were first discovered and identified in the 1970s in plant viroids [4] and subsequently in the cytoplasm of eukaryotic cells [5]. Initial progress in this field was likely slow due to the predominance of linear RNAs [6], and circRNAs were considered a byproduct of RNA splicing [7]. However, recent advances in next generation sequencing and related bioinformatics tools have accelerated research. These molecules have been identified in many species including humans [8], mouse [9], nematode [10], plant [11] and archaea [12]. A timeline describing development of the circRNA field is shown in Figure 1.
Figure 1.
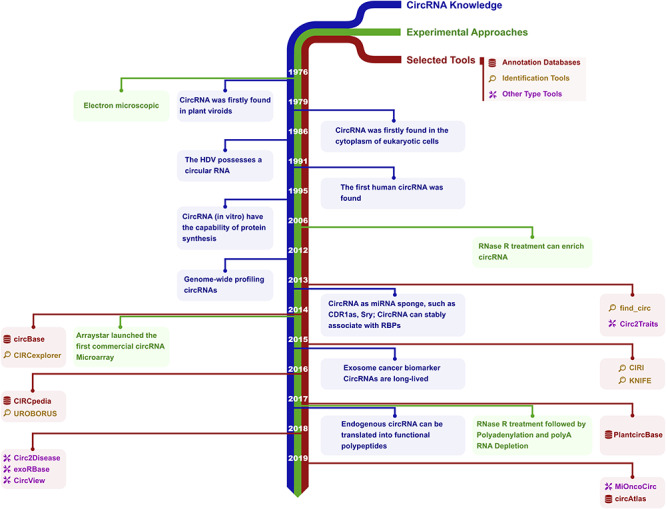
Historical timeline of circRNA research. The development of knowledge, experimental and computational tools of circRNA is illustrated. Blue, green and red marks are the circRNA discoveries related to biology, experimental approaches and representative bioinformatics tools, respectively. In 1976, circRNAs were identified as circular RNA genomes in plant viroids by electron microscopy [4]. With similar methods, circRNA was found in the cytoplasm of eukaryotic cells in 1979 [5] and in the hepatitis delta virus (HDV) in 1986 [170]. In 1991, the first human circRNA was identified [171]. In 1995, circRNA was shown to have the capability of protein synthesis in vitro [2]. In 2006, RNase R treatment was found to enrich circRNA [33]. By 2012, genome-wide profiling of circRNAs by RNA-Seq [8] was demonstrated. In 2013, circRNA was shown to act as a miRNA sponge with examples such as CDR1as [9] and Sry [172]. CircRNAs could stably associate with RNA binding proteins (RBPs), such as Argonaute protein (AGO) and RNA polymerase II [9, 31]. In 2014, Arraystar launched the first commercial circRNA microarray, and its expression was profiled by array [173]. In 2015, circRNAs were proposed as cancer biomarkers, and could be detected in the exosome [174]. CircRNAs also were shown to be long-lived [32]. In 2017, endogenous circRNA was shown to be translated into functional polypeptides [16, 166, 175]. New methods were developed for isolating highly pure circRNA using RNase R treatment followed by polyadenylation and polyA+ RNA depletion (RPAD) in 2017 [176].
Figure 2.
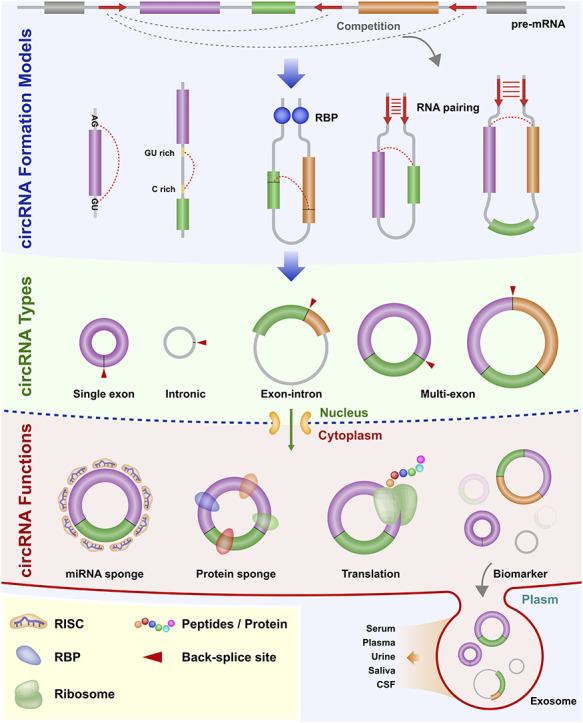
Biogenesis of circRNA in animal. CircRNA formation models: the back-splicing circularization requires the help of the both-end motif (Flanking AG-GU), complementary sequences (ALU elements) or RBPs. circRNA types: single exon, intronic, exon-intron and multi-exon. circRNA functions: miRNA sponge, protein sponge, translation and biomarker. Abbreviations: cerebrospinal fluid, CSF; RNA-induced silencing complex, RISC; RNA-binding protein, RBP.
While this review focuses on circRNA-related bioinformatics tools, other recent reviews provide perspectives on understanding the functions of circRNA with evidence gathered from regulation in tissues and subcellular locations, biogenesis, degradation, translation, transport, interactions and evolutionary conservation [1, 6]. Experimental approaches for discovery and profiling of circRNAs have also been recently reviewed elsewhere [3, 13, 14]. A set of studies have reported on N6-methyladenine (m6A)-associated degradation [15], translation of circRNAs [16], expression in cancer, fusion circRNAs [17–20] and expression in various body fluids or exosomes that point towards their potential as biomarkers [21, 22].
The biogenesis of circRNA is shown in Figure 2. Similar to linear mRNAs, circRNAs are derived from linear precursor mRNAs (pre-mRNAs) transcribed by RNA polymerase II [23]. CircRNA can be generated via back-splicing (Figure 2). For example, intronic complementary sequence (ICS) pairing or RNA binding protein (RBP) action across introns can facilitate circRNA biogenesis by bringing the distal flank back-spliced exons into close proximity and thus facilitate back-splicing [1, 24]. Notably, one gene can generate different circRNAs, which can be affected by the competition of RNA pairing across the flanking introns [24, 25]. Based on different combinations of exons and introns in the final circRNA sequence, circRNA can be divided into three categories that include exonic circRNA [24], circular intronic [26] and exon-intron circRNA [27]. Notably, exonic circRNA can be formed as a single- or multi-exon molecule.
Figure 3.
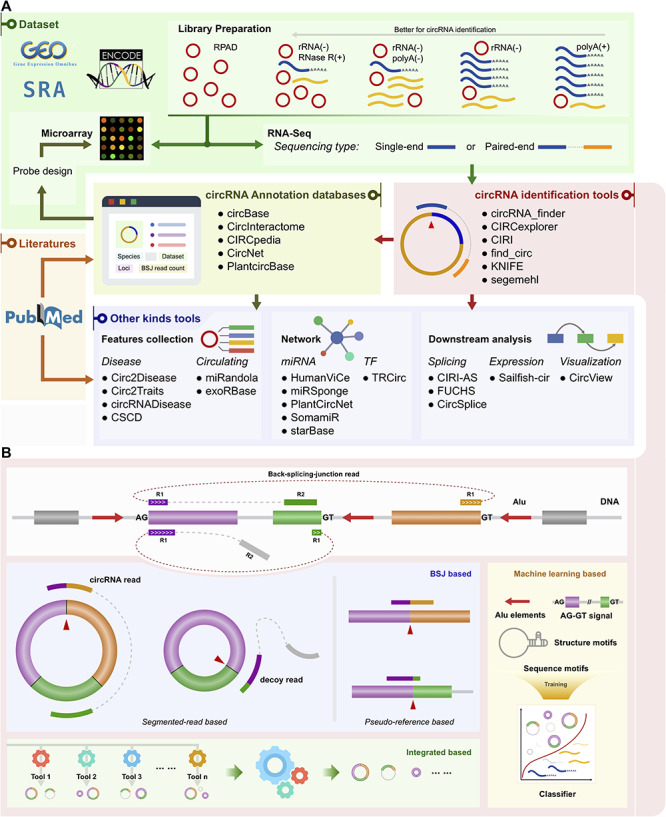
Schematic of classified circRNA bioinformatics tools. (A) Schematic of classified circRNA bioinformatics tools. Examples of tools are listed for each category. polyA(+): polyA selected; polyA(−): polyA depleted; rRNA(−): rRNA depleted; RNase R(+): treated with RNase R; RPAD: RNase R treatment followed by polyadenylation and polyA(+) RNA depletion. (B) Categories of circRNA identification tools. The top is a model of BSJ read mapping to the genome. R1 and R2 represent read1 and read2, respectively, in a paired-end RNA-Seq dataset. Three illustrations of categories are shown below the model. The categories are BSJ-based, integrated-based and machine learning-based. BSJ-based can be more finely divided into two sub-classes: segmented-read based and pseudo-reference based.
CircRNAs have been suggested to have multiple functions. CircRNA can act as a miRNA sponge or competing endogenous RNA (ceRNA) which competes for miRNA pairing with other RNAs [9, 28, 29]. CircRNAs can also regulate transcription in the nucleus [1] as well as bind to protein factors [23]. CircRNAs are enriched in the brain in comparison to other tissues [30]. Compared to linear RNAs, circRNAs are more stable in tissues, have a longer lifetime and are more resistant to RNase R [31–33]. CircRNA has been shown to not only be involved in a variety of human diseases [22] including cancers [34], neurodegenerative conditions [35] and cardiovascular illnesses [36] but also participate in plant stress responses [37].
Advances in high throughput sequencing and associated bioinformatics tools and databases have provided new opportunities to facilitate an understanding of circRNAs. The first genome-wide circRNA profiling was described in 2012 [8]. In 2013, find_circ became the first publicly available pipeline for circRNA identification [9]. In this review, we collected and analyzed ~100 tools including web services, databases, stand-alone programs and pipeline tools. We present an overview and discuss trends in the development of these circRNA bioinformatics tools. Based upon our findings, we also discuss the current challenges of circRNA analysis and the clinical implications of circRNA function.
Bioinformatics tools used in circRNA research
Since 2012, a large number of bioinformatics tools have been developed for circRNA study. Although the functions of circRNA tools are diverse, they can be classified into three main categories that include circRNA identification tools, circRNA annotation databases and other tools (Figure 3A). A few tools such as disease-associated circRNA databases are technically databases but do not contain primary data. These are listed in Supplementary Table 1. We group tools with similar functions together. Tools that primarily recognize circRNAs are classified as circRNA identification tools. Databases that provide basic annotation information are classified as circRNA annotation databases. Finally, stand-alone tools or online services are classified as other tools.
Figure 4.
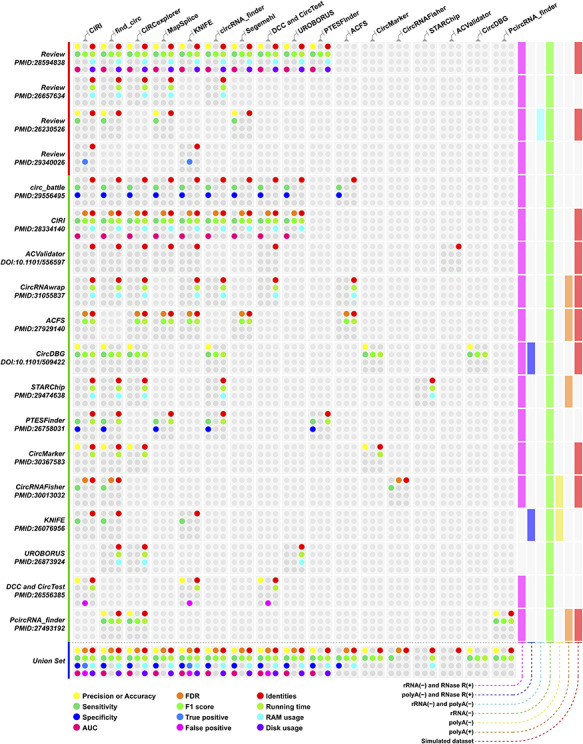
Performance index of circRNA identification methods evaluated by different studies. The colored dot represents different performance indices, and the corresponding performance index for each dot is provided in the legend of the figure. Rows are different studies and classified by publication types, and the last row is the union set of each column. The columns represent circRNA identification tools, and the order is based on mentioned occurrences in those studies. Details regarding the dataset used in these studies are available in Supplementary Table 2.
CircRNA identification tools
All circRNA identification tools are listed in Table 1. Prior to identification, RNA library construction and sequencing provide the raw reads needed. As Figure 3A shows, several library preparation methods can be used for RNA sequencing and subsequent circRNA identification. In theory, polyA+ selected RNAs should not contain circRNAs. Nonetheless, libraries sequenced from this selection of RNA may still contain a tiny proportion of circRNA, because selection is not absolutely accurate (see Figure 3A) [14]. Despite this minor imperfection, polyA+ selected datasets can be used as negative controls [38]. Another factor in identification is specificity. To improve this aspect of identification, the false-positive rate of back spliced junction (BSJ) reads can be reduced in various ways. For example, BSJ reads can be supported by its presence in different samples, by a strict cut-off for read counts or by determining whether the circRNA enrichment step is adopted in the library preparation [14]. Most identification tools prefer a circRNA-enriched RNA-Seq dataset as an input. Another strategy is to use paired-end read sequencing to improve the identification of decoy reads that are typically discarded as alignment artifacts. This can be helpful for filtering out false-positive circRNAs [8, 39]. The support of paired-end or single-end read datasets for each tool is listed in Table 1. Notably, most tools have an essential step termed remapping for discovering BSJ reads [14]. Except for segemehl [40, 41], k-mer based tools and machine-learning-based tools, all identification tools depend on external aligners, with Bowtie, BWA and STAR as the common choices.
Table 1.
Selected circRNA identification tools
| Tool name | ST | TT | IT | QS | ATMR | AC | PL | LU | CV | Platform | Link | Ref |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CIRCexplorer | PE, SE | De novo; annotation | pip, Conda, Docker | YES | STAR, BWA | BSJ | Python | 2019 | v2.3.8 | Unix/Linux | https://github.com/YangLab/CIRCexplorer2 | [24, 46] |
| CircPro | PE, SE | De novo; annotation | MID | YES | BWA (CIRI2) | BSJ | Perl | 2017 | — | Unix/Linux | http://bis.zju.edu.cn/CircPro | [73] |
| MapSplice | PE, SE | De novo; annotation | Conda | YES | Bowtie | BSJ | Python | 2016 | v2.2.1 | Unix/Linux | http://www.netlab.uky.edu/p/bioinfo/MapSplice2 | [48] |
| circRNA_finder | PE, SE | De novo | MID | YES | STAR | BSJ | Perl, AWK | 2019 | v1.2 | Unix/Linux | https://github.com/orzechoj/circRNA_finder | [51] |
| CircRNAFisher | PE | De novo | MID | YES | Bowtie2 | BSJ | Perl | 2019 | v0.1 | Unix/Linux | https://github.com/duolinwang/CircRNAFisher | [65] |
| miARma | PE, SE | De novo | Docker, Virtualbox image | YES | BWA (CIRI) | BSJ | Perl, Python, R | 2018 | v1.7.5 | Unix/Linux, Windows | https://sourceforge.net/projects/miarma/ | [67] |
| CIRI | PE, SE | De novo | MID | YES | BWA | BSJ | Perl | 2017 | v2.0.6 | Unix/Linux | https://sourceforge.net/projects/ciri/ | [43–45] |
| ACFS | PE, SE | De novo | MID | YES | BWA, BLAT | BSJ | Perl | 2016 | v2 | Unix/Linux | https://github.com/arthuryxt/acfs | [59] |
| find_circ | SE | De novo | MID | YES | Bowtie2 | BSJ | Python | 2015 | v1.2 | Unix/Linux | https://github.com/marvin-jens/find_circ | [9] |
| ACValidator | PE, SE | Annotation | pip, MID | NULL | BWA,Bowtie2 | BSJ | Python, Shell | 2019 | v1.0 | Unix/Linux | https://github.com/tgen/ACValidator | [56] |
| ANNOgesic | PE, SE | Annotation | pip3, Docker | NULL | segemehl | BSJ | Python | 2019 | v1.0.8 | Unix/Linux | https://github.com/Sung-Huan/ANNOgesic | [63] |
| BIQ | PE, SE | Annotation | nmp | NULL | k-mer (no need aligner) | BSJ | C++, Perl, JavaScript | 2019 | v0.2.0 | Unix/Linux, Windows | https://github.com/pmenzel/biq | [58] |
| CircDBG | PE | Annotation | CR | NULL | k-mer (no need aligner) | BSJ | C++ | 2019 | — | Unix/Linux | https://github.com/lxwgcool/CircDBG | [54] |
| CircMarker | PE, SE | Annotation | CR, MID | NULL | k-mer (no need aligner) | BSJ | C++ | 2019 | — | Unix/Linux | https://github.com/lxwgcool/CircMarker | [53] |
| circtools | PE, SE | Annotation | pip3, SS, Bioconductor | YES | DCC, FUCHS | BSJ | Python, R | 2019 | Release 1.1.0.8 | Unix/Linux | https://github.com/dieterich-lab/circtools | [69] |
| DCC and CircTest | PE, SE | Annotation | SS | YES | STAR | BSJ | Python, R | 2019 | — | Unix/Linux | https://github.com/dieterich-lab/ | [47] |
| NCLcomparator | PE, SE | Annotation | MID | YES | STAR, BLAT | BSJ | Shell | 2019 | v1.0.0 | Unix/Linux | https://github.com/TreesLab/NCLcomparator | [55] |
| PRAPI | PE, SE | Annotation | MID, pip | YES | CIRIexplorer | BSJ | Python, R | 2019 | V6.2.0 | Unix/Linux | http://www.bioinfor.org/bioinfor/tool/PRAPI/ | [72] |
| PTESFinder | PE, SE | Annotation | MID | YES | Bowtie | BSJ | Java, Shell | 2019 | v1 | Unix/Linux | https://sourceforge.net/projects/ptesfinder-v1/ | [52] |
| Ularcirc | PE, SE | Annotation | devtools, Conda, Bioconductor | YES | STAR | BSJ | R | 2019 | v1.3.24 | Unix/Linux, Windows | https://github.com/VCCRI/Ularcirc | [49] |
| AutoCirc | PE, SE | Annotation | MID | YES | Bowtie2 | BSJ | C++, Perl | 2018 | v1.3 | Unix/Linux | https://github.com/chanzhou/AutoCirc | [64] |
| hppRNA | PE, SE | Annotation | SS | YES | STAR (DCC and CircTest) | BSJ | Perl, R | 2018 | v1.3.7 | Unix/Linux, Windows | https://sourceforge.net/projects/hpprna/ | [66] |
| Identify circularRNA reads | PE | Annotation | MID | YES | STAR | BSJ | Perl | 2018 | - | Unix/Linux, Windows | https://bitbucket.org/snippets/MSmid/Le949d/identify-circularrna-reads | [57] |
| ROP | PE, SE | Annotation | SS | YES | TopHat-Fusion, CIRCexplorer2 | BSJ | Shell | 2018 | v1.1.2 | Unix/Linux | https://github.com/smangul1/rop | [61] |
| segemehl | PE, SE | Annotation | CR, Conda | NULL | segemehl | BSJ | C++ | 2018 | Release 0.3.4 | Unix/Linux | https://www.bioinf.uni-leipzig.de/Software/segemehl/ | [40, 41] |
| STARChip | PE, SE | Annotation | MID | YES | STAR | BSJ | Perl, R, Shell | 2018 | v1.3e | Unix/Linux | https://github.com/LosicLab/STARChip | [62] |
| UROBORUS | PE, SE | Annotation | MID | YES | Bowtie | BSJ | Perl | 2018 | v2.0.6 | Unix/Linux | https://github.com/WGLab/UROBORUS | [50] |
| circScan | PE, SE | Annotation | CR | YES | Bowtie2 | BSJ | C++, Perl | 2017 | v0.1 | Unix/Linux | https://github.com/sysu-software/circscan | [70] |
| circTools (starBase) | PE, SE | Annotation | CR, MID | NULL | Bowtie2 | BSJ | C++ | 2017 | v0.1 | Unix/Linux | http://starbase.sysu.edu.cn/starbase2/circTools.php | [71] |
| circseq-cup | PE, SE | Annotation | MID | NULL | TopHat, STAR, segemehl | BSJ | Perl, Python | 2016 | v1.0 | Unix/Linux | https://github.com/bioinplant/circseq-cup/ | [68] |
| exceRpt | PE, SE | Annotation | Docker | YES | STAR, Bowtie2 | BSJ | R, Sell, Perl | 2016 | v4.3.2 | Unix/Linux, Windows | http://github.gersteinlab.org/exceRpt/ | [97] |
| KNIFE | PE, SE | Annotation | MID | YES | Bowtie2, Bowtie | BSJ | Python, R, Perl, Shell | 2016 | v1.5 | Unix/Linux | https://github.com/lindaszabo/KNIFE | [39] |
| NCLscan | PE | Annotation | CR | YES | BWA, BLAT | BSJ | Python, C++ | 2016 | v1.6 | Unix/Linux | https://github.com/TreesLab/NCLscan | [60] |
| CircularRNAPipeline | PE, SE | Annotation | MID | YES | CIRCExplorer, TopHat-Fusion | BSJ | Python | 2015 | — | Unix/Linux | https://github.com/huboqiang/TanglabCircularRNAPipeline | [75, 76] |
| circ_battle | PE, SE | De novo, annotation | — | YES | — | IPA | — | 2018 | — | Web-based | http://174.138.53.214:3838/circ_battle/ | [77] |
| CirComPara | PE, SE | Annotation | SS, Docker | YES | — | IPA | Python, R | 2019 | v1.0 | Unix/Linux | http://github.com/egaffo/CirComPara | [79] |
| CircRNAwrap | PE, SE | Annotation | MID | YES | — | IPA | Perl, Shell, R | 2019 | — | Unix/Linux | https://github.com/liaoscience/circRNAwrap | [82] |
| RAISE | PE, SE | Annotation | MID | YES | — | IPA | Shell | 2017 | — | Unix/Linux | https://github.com/liaoscience/RAISE | [80] |
| PcircRNA_finder | PE, SE | Annotation | MID | YES | — | IPA | Perl, Python | 2016 | — | Unix/Linux | http://ibi.zju.edu.cn/bioinplant/tools/manual.htm | [81] |
| DeepCirCode | — | De novo; annotation | devtools | NULL | — | ML | R | 2019 | v1.0.0 | Unix/Linux, Windows | https://github.com/BioDataLearning/DeepCirCode | [86] |
| WebCircRNA | — | De novo; annotation | — | NULL | — | ML | Python | 2018 | — | Web-based, Unix/Linux | https://rth.dk/resources/webcircrna/ | [84] |
| PredcircRNA | — | De novo; annotation | MID | NULL | — | ML | Python | 2017 | — | Unix/Linux | https://github.com/xypan1232/PredcircRNA | [83] |
| PredicircRNATool | — | annotation | MID | NULL | — | ML | MATLAB | 2016 | — | Unix/Linux, Windows | https://sourceforge.net/projects/predicircrnatool/files/ | [85] |
| CPSS | PE, SE | annotation | — | YES | — | NULL | Perl, PHP, R | 2017 | v2.0 | Web-based | http://114.214.166.79/cpss2.0/ | [74] |
Header abbreviations: AC, algorithm category; ST, sequencing type; TT, tools type; IT, installation type; LU, last update; CV, current version; Ref, reference; QS, quantification support; ATMR, aligner or tools or method required; PL, programming language.
Column descriptions: ‘Tool Name’ is the name of related tools. ‘ST’ column describes the tool supported sequencing type. ‘TT’ column describes the type of the tool. If the tool needs a gene annotation file, the tool is labeled with ‘annotation’, otherwise, it is labeled with ‘De novo’. ‘IT’ column describes the tool supported installation methods, such as Docker image installation and Conda installation. ‘QS’ column describes whether the tool can be used to measure the quantification of circRNAs. ‘ATMR’ column describes the aligner or method of the tool. ‘AC’ column describes the algorithm of the tool. ‘PL’ column describes which programming language is used to develop the tool. ‘LU’ column describes the latest update time of the tool. ‘CV’ column describes the current version of the tool. ‘Platform’ column describes the tool supported platforms. ‘Link’ column provides the homepage of the tool or the link which gives more detailed information about the tool. ‘Ref’ column provides the related publications.
Other abbreviations: BSJ, back-splice-junction-based method; IPA, integrated prediction algorithms; ML, machine learning-based method; PE, paired-end; SE, single-end; MID, manually install dependencies; CR, compile required (make command); SS, setup script is available.
Based on Table 1, most circRNA identification tools belong to the stand-alone category. Nearly all tools release their source code via GitHub and the most popular program language is Python, with R and Perl also being popular languages and Shell as a common choice for building a pipeline. Most pipelines run under Linux or a UNIX-like system. Some tools need to be compiled from source code and some tools need to be manually installed with the dependency environment. Currently, it is more convenient to install tools in Linux, with many tools supporting easy installation methods, such as Conda (bioconda), Docker, Python package index (PyPI) and BiocManager (Bioconductor). Most have well-written documents and tutorials to help users install and run their tools. Although installation can be straightforward, some computer science skills may be needed (e.g. programming, Linux system knowledge) to perform an analysis. Therefore, a well-designed, free, online interface or stand-alone tools with a user-friendly interface is generally required for users without advanced computational training.
Based on implementation form, circRNA identification tools can be divided into three categories (see Table 1 column AC and Figure 3B) that include BSJ-based, integrated-based and machine learning-based.
BSJ-based circRNA identification tools
The BSJ read represents a molecular signature to identify a circRNA. Many tools recognize circRNA by identifying the BSJ read. Different strategies for mapping BSJs have been discussed in previous studies [42]. Most algorithms embedded in tools are based on splitting the reads (called segmented-read-based), while several other tools are based on a pre-defined BSJ and flanking sequence of a circRNA. These tools then map the read directly to that pseudo-reference for discovering a BSJ (called pseudo-reference based) (see Figure 3B). Below, we describe some representative BSJ-based circRNA identification tools.
The first RNA-Seq-based circRNA prediction tool utilizing the identification of back-spliced sequencing reads was Find_circ [9]. CIRI [43–45], a more robust tool that came later, scans through sequence data first to identify junction reads and then implements multiple filtration strategies to remove false positives. CIRCexplorer [24, 46] also identifies junction reads out of back-spliced exons and intron lariats; however, the latest version supports circRNA alternative splicing analysis and de novo assembly for circular RNA transcripts. In contrast to tools that support raw RNA-Seq reads, DCC and CircTest [47] use the output of STAR read mapper to identify BSJ reads. Further improvements in sensitivity and specificity of circRNA identification can be found in KNIFE [39], which uses a logistic generalized linear model (GLM). A splicing-focused tool can be found in MapSplice [48], while segemehl [40, 41] uses read alignment to detect circRNAs. MapSplice [48] and segemehl [40, 41] have been developed as split alignment tools, and the circRNA detection function is a new feature supported in the latest version. BSJ-based tools with narrower applications include Ularcirc [49] and UROBORUS [50], which can detect circRNAs with low expression levels in RNA-Seq datasets without RNase R treatment. Finally, circRNA_finder [51] is a de novo circRNA forecasting tool without the need of gene annotations or exon-intron structure. This is useful for the prediction of circRNA with proximal splice sites.
While identification of the BSJ is essential for circRNA identification, algorithm implementations may differ between tools. The following offer a few examples. PTESFinder [52] is based on post-transcriptional exon shuffling structure identification and subsequent mapping to a sequence model, while CircMarker [53] is k-mers-based and CircDBG [54] is De Bruijn graph-based. NCLcomparator [55] is a nonco-linear (circular, trans-spliced or fusion RNAs) transcript-detecting tool based on post-screening the nonco-linear transcript detected by other tools. Additional details concerning different strategies of searching for donor and acceptor anchors have already been discussed in previous studies [38, 42]. ACValidator [56] is a circRNA validation tool in silico which is based on assembly. Taking different approaches, ‘Identify circularRNA reads’ [57] does not rely on unmapped reads or known splice junctions, while BIQ [58] builds an index of BSJ-spanning k-mers based on the genome annotation and provides a search function for querying the BSJ reads.
Some specific challenges to detecting circular RNAs remain such as the presence of fusion events. One such tool that can overcome this challenge is ACFS [59], which uses single-end and paired-end RNA-Seq data to identify fusion circRNAs from chromosome translocation events. Another tool to detect nonco-linear transcripts such as fusions, trans-splicing and circRNA is embodied in NCLscan [60]. Similar to this, ROP [61] can profile repeats, circRNAs and gene fusions, while STARChip [62] supports circRNA abundance quantification, annotation and fusion prediction. Species differences in splicing detection represent another challenge. Species-specific tools include ANNOgesic [63], which is a bacterial and archaeal genome annotation tool and can be used to detect circRNAs and other genomic features. AutoCirc [64] is another tool that can be applied to all species with an available genome sequence. A different approach to solving challenges is to provide a statistical score for predicted circRNAs. This has been implemented in CircRNAFisher [65], which can perform systematic de novo circRNA prediction with p-values based on BSJ overlapping reads and discordant BSJ spanning reads. Another challenge is identifying important molecules and variations within the circRNA discovery data set, which requires some multifunctionalism. A multifunctional tool hppRNA [66] can perform read mapping, sequence variation and fusion gene detection and identify circRNAs. Another multifunctional tool, miARma [67], can identify mRNAs, miRNAs and circRNAs in any sequenced organism. CIRCexplorer [24] could also be included as a multifunctional tool since it contains a module for analysis of the alternative splicing of circRNA. Circseq-cup [68] can also be used to assemble the full-length sequence of circRNA based on BSJ reads and their corresponding paired-end reads. Circtools [69] is capable of detecting and reconstructing circRNAs, circRNA primer design, RBP enrichment screenings and differential exon usage analysis.
CircRNAs can also be identified from CLIP-Seq, ISO-Seq, Ribo-Seq and miRNA-Seq data sets and many tools exist to support this activity. A basic CLIP-Seq-based circRNA identification tool based on the recognition of BSJ reads is CircScan [70]. A more complicated tool is CircTools (starBase) [71] that is a pipeline consisting of three separate software tools (circSeeker, circAnno and clipSearch) to detect and annotate circRNAs and their interactions with miRNAs by using CLIP-Seq. A more comprehensive pipeline for ISO-Seq data analysis, including alternative transcription initiation, alternative splicing and circRNA identification, is PRAPI [72]. A specialized tool that can identify circRNA that has protein-coding potential and junction reads is Ribo-Seq CircPro [73]. Another comprehensive pipeline is CPSS [74] that can perform ncRNA detection and abundance quantification, as well as detect circRNA from a miRNA-Seq dataset. A pipeline tool that can detect circRNA from raw fastq data is CircularRNAPipeline [75, 76]. With CIRCexplorer in the pipeline, it has been able to detect circRNAs from single cell RNA-Seq datasets. With these pipelines, researchers can study circRNA using a larger dataset in addition to reusing previous datasets analyzed for other purposes.
Integrated circRNA identification tools
Some identification tools can be classified as ensemble tools, as they integrate current stable tools and merge the results. It has been demonstrated that integration of different circRNA identification tools can reduce the false-positive rate [13, 77, 78]. There are some RNase R sensitive circRNAs (e.g. circ_CDR1as), which can be significantly depleted by RNase R treatment. Therefore, it is problematic to only use RNase R-treated library preparation-based tools to identify these RNase R-sensitive circRNAs [59]. Therefore, the identification or confirmation of the existence of a circRNA needs to be supported by different identification tools, as well as identified from multiple datasets with different treatments. Undoubtedly, different methods and samples can enhance the reliability of circRNA candidates.
Towards this goal, integrated pipelines for circRNA identification are now available. For example, CirComPara [79] is a circRNA identification, abundance quantification and annotation pipeline that integrates CIRCexplorer, CIRI, find_circ and segemehl. Users can also compare or combine different circRNA prediction algorithms (circRNA_finder, CIRCexplorer, CIRI, find_circ, MapSplice, ACSF, DCC, KNIFE and UROBORUS) to improve the sensitivity and specificity of circRNA identification using circ_battle [77]. Another example is RAISE [80], which can also measure circRNA abundance and predict circRNA structure, with the help of MapSplice, find_circ, ACFS and circRNA_finder. In the plant field, PcircRNA_finder [81] can be used to identify exonic circRNAs based on BSJ read identification and available gene annotation, using find_circ, MapSplice and segemehl. An even more comprehensive pipeline tool is CircRNAwrap [82], which can identify (by using KNIFE, find_circ, CIRI, CIRCexplorer, MapSplice, ACFS, circRNA_finder and DCC), predict transcripts (by including RAISE and CIRI-as) and estimate abundance (by using sailfish-cir).
Machine learning-based circRNA identification tools
Although most circRNA identification tools need RNA-Seq datasets as input, there remain some tools that apply machine learning techniques to predict circRNA using circRNA knowledge and features of known circRNA to train a classification model. Machine learning algorithms allow the model to learn knowledge from real circRNAs. As more knowledge about circRNA is gained, more features can be used as machine learning input to build more reliable classification models and more rules can be used as filter steps in most tools. Common features are ALU repeats, structure motifs and sequence motifs [31, 83]. Examples of this approach include PredcircRNA [83], which aims to distinguish circRNA from other long noncoding RNAs (lncRNAs) based on a multiple kernel learning algorithm. WebCircRNA [84] is trained based on a stem cell dataset and can further predict stem cell-specific circRNAs. PredicircRNATool [85] uses conformational and thermodynamic properties in flanking regions to predict circRNAs, while DeepCirCode [86] deploys a convolutional neural network (CNN) with flanking 50 nt sequence of the back-splicing site as input to predict human circRNA. DeepCirCode is the first deep learning model for predicting circRNA.
Comparisons across circRNA identification tools
Recently, several reviews have evaluated previously published or commonly used identification tools. Meanwhile, some newly designed tools tend to compare their own tools with the most commonly used or cited in the field as part of their original publication. We collected comparison data of identification tools that were found in reviews and in original tool publications. Figure 4 shows the comparison criteria and datasets. Additional information extracted from the publication is listed in Supplementary Table 2.
Figure 6.
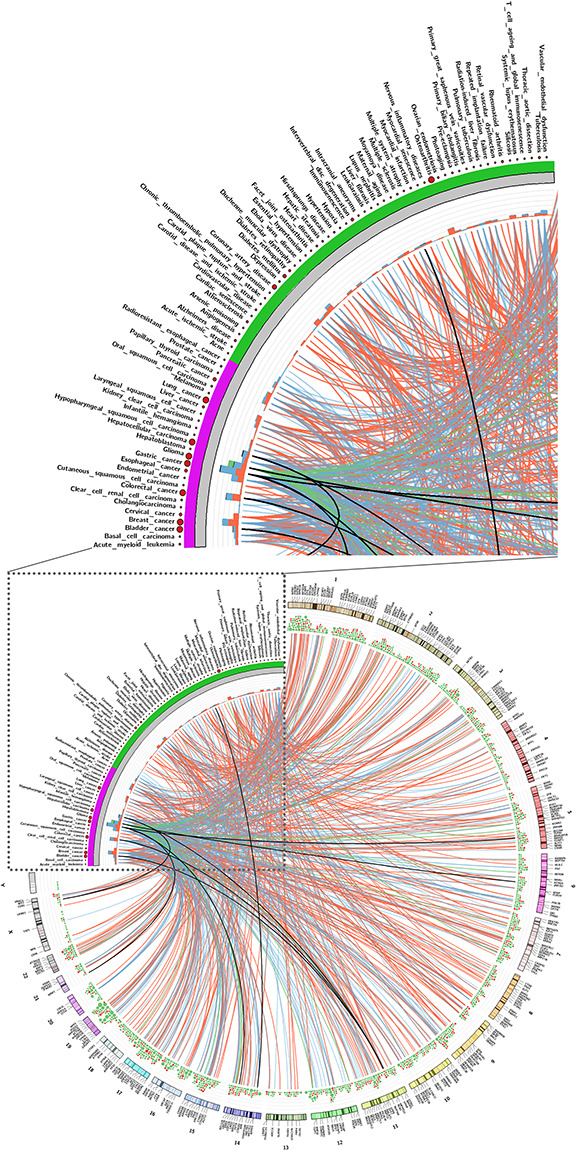
CircRNA-disease association. The top figure is an enlargement of a corner of the bottom figure. Association with circRNAs and diseases, recorded in Circ2Disease, CircR2Disease and circRNADisease. All associations were manually curated from the literature. Purple marks are cancers and other diseases are in green. The circRNA host gene symbols and chromosome label are shown in the outer layer of the genome ideogram. The scatter plot represents the number of publications for each disease, and the stacked bar plot the number of circRNAs related with each disease, and the color represents the regulation direction. The links connect the diseases and circRNAs. The different regulation directions are marked by color: red indicates circRNA up-regulated in a disease, blue indicates down-regulated, green indicates unclear regulation direction, and black, circRNA that can be detected up and downregulated in different publications. In the gene density layer, red represents the disease-related circRNAs and green represents the functional circRNA records in the CircFunBase. A high-resolution figure (the gene symbol is visible) and the association details are available in Supplementary Figure 3 and Supplementary Table 3, respectively.
Most tools listed in Figure 4 were tested using different real and simulated datasets with variable criteria. CIRI (16 times), find_circ (15 times), CIRCexplorer (12 times), KNIFE (9 times), MapSplice (8 times) and circRNA_finder (8 times) were the top five tools chosen as benchmark methods or counterpart tools. All tools in all publications were compared on the basis of circRNA identification with the putative circRNAs overlap between tools in each study. There were ~40 identification tools, but less than half of the tools have been carefully compared. Based on comparison results, MapSplice is more reliable but time-consuming [78, 87]. KNIFE, segemehl, CIRI, PTESFinder and CIRCexplorer achieved the best sensitivity [87]. UROBORUS is an efficient tool that can detect circRNAs with low expression levels in rRNA(−) samples without RNase R treatment [50].
We observed that ‘rRNA(−) and RNase R(+)’ and rRNA(−) were common comparison datasets used in these studies. The read count fold change for a candidate circRNA between ‘rRNA(−) and RNase R(+)’ and rRNA(−) samples was used to assess the level of false positive circRNAs, but this may have potential statistical problems [14, 78]. Based on Supplementary Table 2, most RNA-Seq datasets were performed at 100 or 101 bp sequencing mode. This was consistent with a study [13] that recommended at least 100-bp sequencing to obtain sufficient read length to align to the BSJ site.
Simulated RNA-Seq datasets are necessary for benchmarking and can be generated in multiple ways. In CIRI package, a Perl script called ‘CIRI_simulator.pl’ can generate circular transcript and read length simulation datasets with various coverages. CircRNAFisher creates a generated dataset by down-sampling from the real dataset. In a previous work [87], a read simulator named ART [88] was used to generate a simulated RNA-Seq dataset and indel and substitution variants were introduced into the reads. Chen and coworkers [89] applied benchmarking [90] to generate paired-end reads with different transcript expression levels. While some studies used simulated data to test their discovery and identification algorithm, it remains challenging to determine their usefulness because simulated data may not recapitulate the actual state or background of the linear and circular transcriptome [14, 44].
CircRNA quantification
In addition to discovery and annotation of circRNA, it is imperative to study the expression levels of circRNAs using different techniques to gain further insight into their function. In addition to RNA-Seq, microarrays represent a platform for circRNA profiling [91]. To our knowledge, there are at least three commercial microarrays designed for circRNA. They are Agilent circRNA Array (Santa Clara, USA), CapitalBio technology circRNA array (Beijing, China) and Arraystar circRNA microarray (Rockville, USA). To print a microarray, the probe design may require a circRNA annotation database or curation of sequence data from scientific literature. The probes present on the microarray can be based on the annotation of the BSJ read in the database, such as in circBase. In this case, the user does not need to design the probes (see Figure 3). An interesting microarray tool is ReCirc [92] that re-annotates noncircRNA microarrays’ probes to BSJ reads. The array analysis tools are not different from those for noncircRNA, such as Agilent feature extraction software [91], which can analyze hybrid features. By querying GEO (Query date: 2019/09/16) microarray platform for circRNA, the three species available are Homo sapiens, Mus musculus and Rattus norvegicus. Due to the limited data available, this review will only cover identification or profiling tools based on RNA-Seq datasets (see Table 1).
Some circRNAs identification tools can also include the ability to calculate circRNA expression values. The methods of profiling circRNA vary widely. One study applied BSJ read and normal splicing junction read ratio (named circular-to-linear ratio, CLR) to represent the ratio of circRNA and linear RNA to obtain an overall expression value [93]. Using a different strategy, Sailfish-cir [94] quantified circRNA abundance by transforming circRNA to a pseudo-linear transcript. Another study applied the UROBORUS pipeline to the unmapped read and used normalized BSJ read count to represent the expression of circRNA [95]. In DCC and CircExplorer, junction read counts were normalized to reads per million mapped reads (RPM) [96]. STARChip quantifies circRNA via counting reads aligned to the BSJ. Intended for low input miRNA-Seq samples, exceRpt [97] also calculates circRNA expression. Similar to quantitation of other RNA-Seq molecules, there appears to be no consensus on the appropriate unit of measurement for expression.
CircRNA annotation databases
Many current circRNA database portals collect putative circRNAs from the literature based on various identification tools and specific NGS datasets, while others apply a unified pipeline to process RNA-Seq datasets and store circRNA prediction results. As development of these databases in ongoing, the expansion and improvement of circRNA database content and quality remains an important task [3, 14, 78, 87].
Some databases are specifically designed for circRNA. These databases store circRNA species, loci and BSJ read count data. A good example of this is circBase [98], which hosts animal circRNAs, sequences and genomic coordinates. The latest version of circBase annotates circRNAs based on data from nine publications. Alternatively, manually curated circRNAs can be found in CircFunBase [99]. CIRCpedia [46, 100] is another example but is more comprehensive because it contains circRNA annotations and expression profiles from six species with data from different cell types or tissues. CircRNADb [101] is also a comprehensive database, but features human circRNA with annotated exonic circRNAs and circRNA with protein-coding potential. The circRNA annotation information in CircRNADb was extracted from the literature. Other databases of note include CircBank [102], which contains human circRNA from different sources and proposes a new standard nomenclature, PigcirNet [103], which contains pig circRNA genome annotation, circRNA catalog and tissue-specific expression level data, and AtCircDB [104], which contains tissue-specific Arabidopsis circRNAs. The circRNA identification tools used in AtCircDB are CIRCexplorer and CIRI. Plant databases include PlantcircBase [105], which collects publicly available BSJ positions of circRNAs across 16 different plant organisms and provides a tool, BLASTcirc, for prediction of circRNAs from query sequences, PlantCircNet [106], which contains circRNA-miRNA-mRNA interactions, and CropCircDB [107], which stores predicted and validated crop circRNAs associated with abiotic stress. Figure 5A displays the circRNA gene density recorded in each database, and Figure 5B provides the species distribution of each database.
Figure 5.
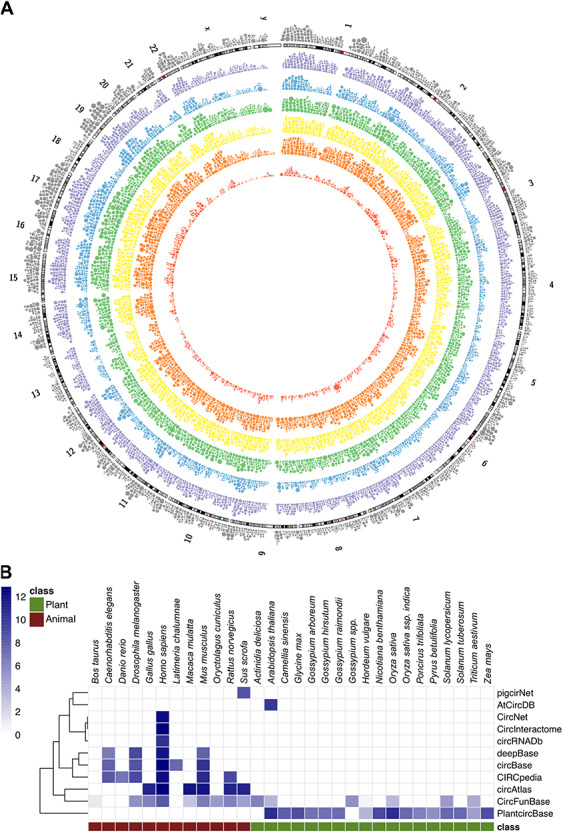
CircRNA density illustration and species support in each database. (A) The human circRNA gene’s density in each database. The outermost layer is the linear gene density of human (UCSC hg19). Outside-in oriented, the different colored layers are circRNA gene density based on circAtlas, circBase, circNet, CIRCpedia, circRNAdb and circFunBase, respectively (coordinates based on hg19 version). The size of the dot represents the count of circRNA in every 1 million bases. (B) The number of different species circRNA records in each database. The count is log transformed. The literature source for each database is shown in Supplementary Figure 1.
Some databases aim to collect ncRNA information with circRNA as a part of the database. circRNA data provided include their interaction with other ncRNAs as well as expression data. An example of this can be found in CircInteractome [108, 109], which provides functions for retrieving RBP-binding and miRNA-binding sites on human circRNAs and siRNA design tools for circRNA silencing. For CircInteractome, the coordinates of a circRNA is based on circBase. A further example is CircNet [110], which provides circRNA integrated miRNA-target networks, expression profiles, genomic annotations and sequences of circRNA. Similarly, DeepBase [111–113] can annotate and discover small RNAs, lncRNAs and circRNAs from next generation sequencing data. The circRNAs provided in DeepBase were extracted from circBase and literature. Wide coverage can be found in circAtlas [114], which contains millions of circRNAs and supports seven species and a variety of tissues.
Nomenclature across databases is a current challenge. Presently, there is no unified nomenclature for circRNA (see Table 2), and IDs used between different databases are not universal. Unified nomenclature would greatly facilitate data integration from different circRNA databases. CircBase uses an ID defined by species_circ_number, circ+host gene symbol or convention name. Some databases also use the host gene symbol with accession numbers or BSJ loci as ID. CircBank made an attempt to unify nomenclature for circRNA. The scheme proposed was ‘hsa-circHUGO-#’ (HUGO is the HUGO symbol of the host gene, ‘#’ represents a number based on the position of cirRNAs in the host gene). Because of the complexity of circRNA, such as alternative splicing, a standard nomenclature for circRNA may be most stable if it is based on sequence. Nonetheless, unified nomenclature remains a significant challenge for the field.
Table 2.
Selected circRNA databases
| Tool name | Organisms | ID nomenclature example | Annotation tools | LU | CV | Download | Link | Ref |
|---|---|---|---|---|---|---|---|---|
| circBase | hsa, mmu, cel, lch, lme | hsa_circ_0001571 | Manually curated | 2017 | — | FASTA, BED (hg19) | http://www.circbase.org/ | [98] |
| CIRCpedia | hsa, mmu, cel, dme, dre, rno | HSA_CIRCpedia_77727 | CIRCexplorer2 | 2018 | v2 | CSV, FASTA (hg19, hg38) | http://www.picb.ac.cn/rnomics/circpedia | [100] |
| deepBase | hsa, mmu, cel, dme | hsa-circRNA11326-1 | Manually curated, circBase | 2016 | v2.0 | BED | http://rna.sysu.edu.cn/deepBase/ | [111–113] |
| CircFunBase | hsa, gga, mml, mmu, rno, ssc, hvu, osa, ath, tae, sly, gsp, ade, bta, ssc, ocu, dme | circCAX2 or hsa_circ_0001946 | Manually curated | 2019 | — | API (hg19) | http://bis.zju.edu.cn/CircFunBase/index.php | [99] |
| circAtlas | hsa, gga, mml, mmu, rno, ssc | hsa-ABCC2_000058 | CIRI2, DCC, CIRCexplorer2, MapSplice | 2019 | v2.0 | BED (hg19) | http://circatlas.biols.ac.cn/ | [114] |
| CircBank | hsa | hsa_circA1CF_001 | circBase | 2019 | — | FASTA, BED | http://www.circbank.cn/ | [102] |
| CircInteractome | hsa | hsa_circ_0041946 | circBase | 2018 | — | XLS | https://circinteractome.nia.nih.gov/ | [108, 109] |
| CircNet | hsa | circ-ZEB1.33 | manually curated | 2016 | — | FASTA, GTF (hg19) | http://syslab5.nchu.edu.tw/CircNet/ | [110] |
| circRNADb | hsa | hsa_circ_11016 | manually curated | 2016 | v1.0 | FASTA, GTF (hg19) | http://202.195.183.4:8000/circrnadb/circRNADb.php | [101] |
| pigcirNet | ssc | AEMK02000123.1:10975|24427 | find_circ | 2019 | v2 | TXT | http://lnc.rnanet.org/circ | [103] |
| CropCircDB | osa, zma | osa-circ41-OS10T0167300 | CIRCexplorer2, CIRI2 | 2018 | — | CSV | http://deepbiology.cn/crop/ | [107] |
| PlantcircBase | ath, gma, hvu, osa, sly, tae, zma, gar, ghi, gra, ptr, stu, csi, nbe, pbe, osi | Gh_A01G0075_circ_g.1 or ghi_circ_000001, A01:626066|628727 | manually curated, modified CIRI2, BLASTcirc | 2019 | Release 4.0 | TXT, FASTA | http://ibi.zju.edu.cn/plantcircbase/ | [105] |
| PlantCircNet | ath, gma, hvu, osa, sly, tae, zma, bdi | ath-circ1-AT1G01490 | find_circ, CIRI, MapSplice, CircRNAFinder, UROBORUS | 2017 | — | CSV, FASTA | http://bis.zju.edu.cn/plantcircnet/index.php | [106] |
| AtCircDB | ath | ath-circ20893-AT1G76930 | CIRCexplorer2, CIRI2 | 2017 | — | — | http://genome.sdau.edu.cn/circRNA | [104] |
Column descriptions: ‘Tool Name’ is the name of related databases. ‘Organisms’ column provides the database supported organisms. ‘ID Nomenclature Example’ gives an example of circRNA from the database. ‘Annotation Tools’ column provides the database used annotation tools. ‘LU’ column provides the latest update time of the database. ‘CV’ column describes the current version of the database. ‘Download’ column provides the data format of available data from the database. ‘Link’ column provides the homepage link of the database. ‘Ref’ column provides the related publications.
Species abbreviations: bta, Bos taurus; cel, Caenorhabditis elegans; dme, Drosophila melanogaster; dre, Danio rerio; gga, Gallus gallus; hsa, Homo sapiens; lch, Latimeria chalumnae; lme, Latimeria menadoensis; mml, Macaca mulatta; mmu, Mus musculus; ocu, Oryctolagus cuniculus; rno, Rattus norvegicus; ssc, Sus scrofa; ade, Actinidia deliciosa; ath, Arabidopsis thaliana; bdi, Brachypodium distachyon; csi, Camellia sinensis; gar, Gossypium arboreum; ghi, Gossypium hirsutum; gma, Glycine max; gra, Gossypium raimondii; gsp, Gossypium spp.; hvu, Hordeum vulgare; nbe, Nicotiana benthamiana; osa, Oryza sativa; osi, Oryza sativa ssp. indica; pbe, Pyrus betulifolia; ptr, Poncirus trifoliata; sly, Solanum lycopersicum; stu, Solanum tuberosum; tae, Triticum aestivum; zma, Zea mays. Header abbreviations: Ref, reference; LU, last update; CV, current version.
Linking circRNA and features
Many databases collect various properties of circRNA rather than being a primary source of circRNA sequence information. For example, rSNPBase [115] is a database that collects SNP-associated regulatory elements including circRNA region elements. Other features may be associated with a disease. A listing of these databases is in Supplementary Table 1. The clinical implications of circRNA is a topic of great interest because circRNAs are promising biomarkers for human diseases [22]. CircRNAs are amenable as biomarkers because of their long life cycle, enrichment in specific cells and detection in various body fluids [22]. To exploit these advantages, some databases have been built to store the relationship between circRNA and human disease. Meanwhile, for plants, CropCircDB is the only database that contains crop circRNAs during response to abiotic stress.
Circ2Disease [116], circRNADisease [117] and CircR2Disease [118] are manually curated, experimentally validated circRNA-disease association databases. Similarly, Circ2Traits [119] stores potential associations between circRNA and diseases in human beings. CSCD [120] and MiOncoCirc [17] are cancer-specific circRNA databases aiming to facilitate the functional study of cancer-specific circRNAs, while HDncRNA [121] and LncRNADisease [122] store disease-associated ncRNAs including circRNAs. Specific databases useful for biomarker discovery include exoRBase [123], which contains human blood exosome-associated RNA (mRNA, lncRNA and circRNA) (Supplementary Figure 2), miRandola [124], which stores extracellular ncRNAs including circRNAs and BBBomics [125], which stores omics data of human blood-brain barrier including miRNA, lncRNA and circRNA.
Figure 6 illustrates the data collected from these representative databases. As depicted, circRNAs were associated with many diseases, in particular, cancer. However, it was observed that some circRNA-disease association evidence collected from different databases was conflicting. This may reflect different criteria for disease association or differences in disease population samples. Circ2Disease, CircR2Disease and circRNADisease appear to be the important databases to record and search disease-related circRNAs. The associations covering a variety of diseases are based on manual curation of the literature.
CircRNA network identification
Abundant circRNAs can function as miRNA sponges [1] and RBP sponges [126]. The construction of a circRNA network can be based on the interaction of circRNAs with miRNAs, lncRNAs and RBPs. Our previous study found that the circRNA tools and miRNA tools have a strong connection [127]. As Supplementary Table 1 shows, there are many databases collecting the interactions of circRNAs and miRNAs, as well as lncRNAs and transcription factors (TFs). The importance of these interactions is highlighted by the support of many databases for circRNA interactions in their latest releases.
CircRNA can act as a competing endogenous RNA (ceRNA), which can regulate other RNA transcripts by competing for shared miRNAs. Decoding the ceRNA network is a new field for cancer biomarker discovery for which circRNAs may play an important role [128]. For example, circRNA-MYLK can compete with VEGFA for miR-29a in bladder cancer [129], and circDOCK1 can compete with BIRC3 for miR-196a-5p in oral squamous cell carcinoma [130]. Many databases collect the circRNA ceRNA network such as starBase [131, 132], SomamiR [133, 134], LncACTdb [135], AFCMEasyModel [136] and HumanViCe [137]. Some databases only store experimentally supported or putative miRNA sponge circRNA candidates such as miRNAsponge [138] and miRSponge [139]. In contrast, StarScan [140] and ACT [141] are web services for predicting small RNA target sites on circRNAs. Other databases record the interaction network in which circRNA participates. RAID [142] and Arena-Idb [143] store human ncRNA interaction network information and include circRNA as one of the major ncRNA classes in the database. TRCirc [144] stores TF-circRNA regulatory network data from approximately 100 cell types, while NetMiner [145] and circlncRNAnet [146] are tools that can reveal the biological roles of circRNA by network analysis.
Other types of circRNA-associated tools
Although most tools are circRNA identification tools, circRNA research is not limited to circRNA discovery. A variety of tools with different functions have been developed and are listed in Table 3.
Table 3.
Selected other circRNA tools
| Name | Main features | LU | CV | Platform | PL | Link | Ref |
|---|---|---|---|---|---|---|---|
| CircSplice | Alternative splicing | 2019 | — | Unix/Linux | Perl | https://github.com/GeneFeng/CircSplice | [149] |
| CIRI-AS | Alternative splicing | 2018 | v1.2 | Unix/Linux | Perl | https://sourceforge.net/projects/ciri/files/CIRI-AS/ | [147] |
| FUCHS | Alternative splicing | 2018 | Release 0.2.0 | Unix/Linux | Python, R | https://github.com/dieterich-lab/FUCHS/tree/master/GCB_testset | [148] |
| Equivalent-Junctions | Ambiguous splice sites distinguish circRNA and linear splicing | 2019 | — | Unix/Linux | Python | https://github.com/salzmanlab/Equivalent-Junctions | [164] |
| P_RNA_scaffolder | circRNAs assemble | 2019 | — | Unix/Linux | Perl, Shell | https://github.com/CAFS-bioinformatics/P_RNA_scaffolder | [155] |
| CIRI-full | circRNAs assemble | 2018 | v2.0 | Unix/Linux | Java | https://sourceforge.net/projects/ciri-full/ | [45, 151] |
| Sailfish-cir | Expression | 2016 | v0.11 | Unix/Linux | Python | https://github.com/zerodel/Sailfish-cir | [94] |
| IntronPicker | Flanking sequence | 2017 | — | Unix/Linux | Shell | https://github.com/alexandruioanvoda/IntronPicker | [161] |
| ReCirc | Microarrays probe re-annotating program | 2019 | — | Unix/Linux, Windows | R | http://licpathway.net:8080/ReCirc/ | [92] |
| NetMiner | Network (co-expression) | 2017 | v1.0.0 | Unix/Linux | Perl, R | https://github.com/czllab/NetMiner | [145] |
| CircPrimer | Primer Design | 2018 | v1.2 | Windows | Delphi | http://www.bioinf.com.cn/ | [162] |
| CIRCpseudo | Pseudo-gene, circRNA-derived pseudogenes detection | 2017 | — | Unix/Linux | Perl | https://github.com/YangLab/CIRCpseudo | [163] |
| ViennaRNA | Structure | 2019 | v2.4.14 | Unix/Linux, Windows | C++ | https://www.tbi.univie.ac.at/RNA/ | [159, 160] |
| Supernmotifs | Structure | 2017 | v1.2 | Unix/Linux | C++ | http://jpsglouzon.github.io/supernmotifs/ | [156] |
| SpliceV | Visualization | 2019 | — | Unix/Linux | Python | https://github.com/flemingtonlab/SpliceV | [153] |
| CircView | Visualization | 2018 | v1.0 | Unix/Linux, Windows | Java | https://github.com/GeneFeng/CircView | [152] |
| TERate | 4sUDRB-Seq | 2016 | — | Unix/Linux | Shell | https://github.com/YangLab/TERate | [165] |
Header abbreviations: LU, last update; CV, current version; Ref, reference; PL, programming language.
Column descriptions: ‘Name’ is the name of the tool. ‘Main Features’ column describes the key feature of the tool. ‘LU’ column provides the latest update time of the tool. ‘CV’ column describes the current version of the tool. ‘Platform’ column describes the tool supported platforms. ‘PL’ column describes the programming language of the tool. ‘Link’ column provides the homepage link or the link which gives more detailed information about the tool. ‘Ref’ column provides the related publications.
Downstream tools
Downstream tools for circRNA analysis are capable of alternative splicing detection, circRNA assembly, primer design, structure prediction and visualization. Among these, CIRI-AS [147], FUCHS [148] and CircSplice [149] are tools used for circRNA alternative splicing analysis. ASmiR [150] stores miRNA target data for alternatively spliced linear and circRNAs in 11 plant species. Sailfish-cir [94] is a circRNA quantification tool based on the output of circRNA identification tools (CIRI, KNIFE, circRNA_finder) and circRNA transformed pseudo-linear transcripts. CIRI-full [45, 151] is a tool aiming to assemble the full-length sequence of circRNA based on transcriptome data, which relies on the outputs of CIRI and CIRI-AS.
There are also some tools that are difficult to categorize or that are not dedicated circRNA tools. However, these tools may be significant and useful. One example is CircView [152], a standalone software for visualizing and exploring circRNA based on the output of different circRNA identification tools. Another example is SpliceV [153], an analysis and visualization tool to plot all relevant forward- and back-splice data, with exon and single nucleotide level coverage information from RNA-Seq experiments. Another useful tool more oriented towards miRNA is miRToolsGallery [127, 154]. This miRNA tool gathering database includes some tools that can be used to analyze circRNAs. Other tools include P_RNA_scaffolder [155], for genome scaffolding using paired-end RNA-Seq data, the outputs of which may recover the structure of both protein-coding genes and circRNAs; Supernmotifs [156], an alignment-free method to visualize and compare the secondary structures of linear or circular RNAs; TSCD [157], which gathers tissue-specific circRNA data from humans and mice; and circ_rna_human_brain [158], which records specific circRNAs in the human brain. ViennaRNA [159, 160] is a highly regarded and heavily used RNA tool that implements algorithms for circRNA secondary structure prediction. Although ViennaRNA supports the circular genome, it can also be applied to circRNA analysis. ACFS [59] has applied ViennaRNA to estimate the energy of circRNA secondary structure, while IntronPicker [161] can be used to extract intron sequences flanking the circRNAs. AutoBLAST can use the output of IntronPicker to perform reverse complementary match analysis. Other tools include CircPrimer [162], which provides an algorithm for circRNA annotation and can determine the specificity of circRNA primers. CIRCpseudo [163] can identify circRNA-derived pseudo-genes, while Equivalent-Junctions [164] can identify equivalent junctions prevalent in circRNA and linear junctions. TERate [165] calculates transcription elongation rates (TER), which can be used to calculate TER of nascent circRNA-producing genes based on 4sUDRB-Seq. Clearly, there exists a large range of tools which the circRNA field can easily repurpose.
Beyond the functions of tools themselves, analysis of circRNA publications may provide some insights into the direction and main capabilities of the field. We applied text mining and network analysis on publications of circRNA tools and display these results in Figure 7.
Figure 7.
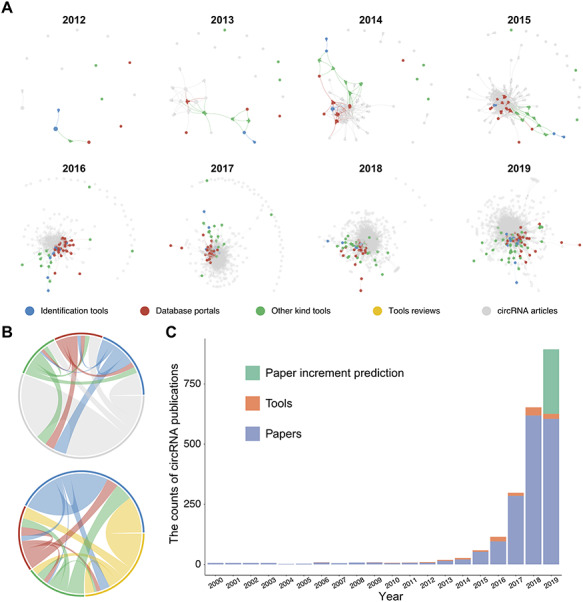
Illustration of interactions of circRNA publications and publication count by year. (A) circRNA article citation network by years. Each node refers to a publication, the edge represents the citation relationship and the size of the node is scaled by PageRank score. The network was drawn with a force-directed layout. Different types of publications or reviews are labeled with a different color. (B) Chord diagram represents the interaction strength of each different type of circRNA publication. (C) Number of publications related to circRNAs from 2000 to 2019. The publication list was searched on 17 July 2019 in PubMed with keywords (‘circRNA’ AND ‘circular RNA’). Red represents the publication of a circRNA tool, and blue represents a regular circRNA research article. Green represents the prediction increment of the publication, based on the number from 2016, 2017 and 2018 with a simple regression model.
Figure 7A shows the circRNA article citation network from 2012 to 2019. The tools were ranked based on each network with the PageRank algorithm and are listed in Supplementary Table 4. From Figure 7A, we observed that these bioinformatics tools have played a significant role in the rise and development of the circRNA field. The community of bioinformatics tools is concentrated in one area of the entire network. As Figure 7B shows, identification tools are more likely to be cited by circRNA studies. Both circRNA database tools and other kinds of tools heavily rely on identification tools, while identification tools do not necessarily require circRNA databases or other kinds of tools. This result is consistent with the data shown in Tables 1–3. In Figure 7C, the histogram clearly illustrates that the study of circRNA has experienced explosive growth since 2012. Some key studies have played a role in promoting this process [38]. The circRNA bioinformatics toolbox is growing, with the exception of a slight decline in 2017. As the field grows in maturity, additional tools are in development. Taken together, this analysis shows a field that is growing and deeply integrated with the development of bioinformatics tools. Identification tools are still dominant suggesting that additional functional tools may be needed.
Future perspectives
As a new and increasingly popular research area, circRNA is still in its infancy, but its complexity has already begun to emerge. For example, a subset of circRNAs has shown unexpected properties and functions, such as protein-coding potential and miRNA sponge actions. Regardless, the precise molecular and biochemical function of the majority of circRNAs remains unknown. Moreover, when new findings are made, they need to be more rapidly incorporated into new circRNA databases or used to update current ones. This would help to facilitate further investigations as well as the comparison of orthologs across species and comparison between closely related circRNA molecules within species. Currently, this requires tedious, low throughput and resource draining manual checks.
Although protein-coding circRNAs have been found in eukaryotes [166], to our best knowledge, the experimental evidence for circRNA translation in plants is still missing and new and specific tools for predicting the protein-coding potential of plant circRNAs are needed. However, assembling full length circRNA is a prerequisite for protein-coding prediction. Some tools are already able to assemble circRNAs but more may be necessary. Emerging purification technologies of circRNA like RPAD may lead to an increase in circRNA assembly accuracy. After assembly, tools for protein-coding prediction or other functions may be based on Ribo-Seq or only dependent on the sequence features. Follow-up experimental validation for assembled circRNAs may then be applied to further strengthen the case for existence of many circRNAs.
Some circRNAs are sensitive to RNase R, such as circ_CDR1as [59], and are expressed at a low level [1]. For the study of RNase R-sensitive circRNAs, the RNase R(+) library preparation method will lead to a relatively low abundance of the circRNA. Therefore, the type of RNA-Seq library preparation method may greatly impact downstream results. Additionally, different kinds of NGS datasets and IP-based datasets (e.g. CLIP-Seq, ISO-Seq and Ribo-Seq) can be a source of BSJ and this may enrich the range of circRNAs identified. Within annotation databases, the maximum human circRNA number is 380 827 as indicated in circAtlas, and the minimum number is 3799 in CircFunBase. This is a nearly 100 times difference. In 2017, only 282 circRNAs were validated by other experimental techniques [87]. Although this number may have increased substantially in the past 2 years, this is still a noticeably small proportion. Substantial conservation of circRNA expression in mammals and plants [3, 167] suggests that circRNAs have a functional role and also provides evidence for the existence of putative circRNAs. Thus, to identify or confirm a circRNA, it would be necessary to have not only different identification tools, but also multiple datasets with different treatments, sequencing types and species. These different datasets from a diversity of sources can enhance the reliability of circRNA candidates and their function. With the increasing number of tools, there remain many requirements for confidence in a circRNA dataset. Simulated datasets can complement the RNase R(+) dataset that are used to validate a tool, but a better statistical model is needed to simulate the circRNA dataset. In the future, there will be a great need for this type of tool.
Technical challenges are often solved by applying existing tools for use in problems that were not foreseen or intended. This solution also applies to circRNA identification. For example, ACValidator is an in silico circRNA validator that has a dependency on the Trinity assembler [168]. Similarly, sailfish-cir takes circRNA data from different outputs and quantifies them based upon the Sailfish framework [169]. MapSplice also directly borrows the concept of a fusion gene to accommodate new problems. CircDBG [54] applies the De Bruijn graph, widely used in genome assembly, to find potential donor/acceptor sites [168]. CircRNA identification problems can also be translated into classification problems. Although only a limited number of circRNA formation features have been found, we can use this data to train a classifier that helps to predict novel circRNAs. Currently, machine learning-based tools are beginning to emerge, but are few in number.
As early as 2014, the nomenclature for circRNA was discussed [3], although each database tends to create a unique system to represent circRNA genes. For example, CircBank developed a standard nomenclature of circRNA. However, 5 years later, we observed that there remains no uniform or dominant standard. Databases have not yet formed a uniform accession/ID annotation, and this will lead to many difficulties and inefficient work for future experimental as well as computational research on circRNA. Bioinformaticians have been encouraged to develop additional tools for ID conversion, such as an ID converter app embedded in circAtlas [114]. We suggest referring to the existing well-known database (RefSeq, GenBank and Ensembl) practices for annotating circRNAs.
As the research on circRNA progresses, more features, knowledge and functions of this molecule are being revealed. Many circRNA databases associated with human diseases exist, while few databases concerning plant diseases or stress exists. The increasingly important role of circRNA in disease processes and the detection of circRNAs in human body fluids and exosomes make circRNA a class of molecules likely to remain in the spotlight. The increasing role of circRNA in health and disease suggests not only more applications in the future, but also points towards the development of more specialized circRNA tools to meet these needs.
Supplementary Material
{kind=link}
Acknowledgments
The authors thank all members of the Garry Wong Laboratory who provided critical comments and feedback. We are grateful to Catherine Teebay for improving the language of the manuscript. This work was performed in part at the high performance computing cluster (HPCC), which is supported by the Information and Communication Technology Office (ICTO) of the University of Macau. We thank all authors who developed tools included in this survey and apologize to any authors whose tools were omitted unintentionally.
Liang Chen is an associate professor of the Department of Computer Science, Key Laboratory of Intelligent Manufacturing Technology of Ministry of Education, Shantou University. His research interest is focused on machine learning and non-coding RNA bioinformatics.
Changliang Wang is a PhD student of the Faculty of Health Sciences, University of Macau. His research interests include data integration analysis, database construction, aging and cancer-associated research.
Huiyan Sun is an assistant professor of the School of Artificial Intelligence, Jilin University. She is interested in developing and applying statistical and computational methods for mining cancer omic data.
Juexin Wang is a research scientist in the Department of Electrical Engineering and Computer Science and Bond Life Science Center, University of Missouri. His research interest is focused on machine learning with applications in bioinformatics.
Yanchun Liang is a professor at the College of Computer Science and Technology, Jilin University. He is also a professor at the School of Computer Science, Zhuhai College of Jilin University. His research interests include computational intelligence, machine learning methods, text mining and bioinformatics.
Yan Wang is a professor at the College of Computer Science and Technology, Jilin University. His research interest is focused on data mining, machine learning and community network analysis.
Garry Wong is a professor at the Faculty of Health Sciences, University of Macau. His laboratory focuses on Parkinson’s disease, aging and environmental stress and development of bioinformatics tools to investigate these processes.
Funding
This work was partially supported by the STU Scientific Research Foundation for Talents (no. 35941918), National Natural Science Foundation of China (no. 61972174, 61902144, 61602207, 61572227, 61572228), Guangdong Key-Project for Applied Fundamental Research (2018KZDXM076), Guangdong Premier Key-Discipline Enhancement Scheme (2016GDYSZDXK036) and University of Macau Faculty of Health Sciences (MYRG2016-00101-FHS).
References
- 1. Li X, Yang L, Chen LL. The biogenesis, functions, and challenges of circular RNAs. Mol Cell 2018;71:428–42. [DOI] [PubMed] [Google Scholar]
- 2. Chen CY, Sarnow P. Initiation of protein synthesis by the eukaryotic translational apparatus on circular RNAs. Science 1995;268:415–7. [DOI] [PubMed] [Google Scholar]
- 3. Jeck WR, Sharpless NE. Detecting and characterizing circular RNAs. Nat Biotechnol 2014;32:453–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sanger HL, Klotz G, Riesner D, et al. . Viroids are single-stranded covalently closed circular RNA molecules existing as highly base-paired rod-like structures. Proc Natl Acad Sci U S A 1976;73:3852–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hsu MT, Coca-Prados M. Electron microscopic evidence for the circular form of RNA in the cytoplasm of eukaryotic cells. Nature 1979;280:339–40. [DOI] [PubMed] [Google Scholar]
- 6. Patop IL, Wust S, Kadener S. Past, present, and future of circRNAs. EMBO J 2019;38:e100836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cocquerelle C, Mascrez B, Hetuin D, et al. . Mis-splicing yields circular RNA molecules. FASEB J 1993;7:155–60. [DOI] [PubMed] [Google Scholar]
- 8. Salzman J, Gawad C, Wang PL, et al. . Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PLoS One 2012;7:e30733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Memczak S, Jens M, Elefsinioti A, et al. . Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013;495:333–8. [DOI] [PubMed] [Google Scholar]
- 10. Ivanov A, Memczak S, Wyler E, et al. . Analysis of intron sequences reveals hallmarks of circular RNA biogenesis in animals. Cell Rep 2015;10:170–7. [DOI] [PubMed] [Google Scholar]
- 11. Ye CY, Chen L, Liu C, et al. . Widespread noncoding circular RNAs in plants. New Phytol 2015;208:88–95. [DOI] [PubMed] [Google Scholar]
- 12. Danan M, Schwartz S, Edelheit S, et al. . Transcriptome-wide discovery of circular RNAs in Archaea. Nucleic Acids Res 2012;40:3131–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kristensen LS, Andersen MS, Stagsted LVW, et al. . The biogenesis, biology and characterization of circular RNAs. Nat Rev Genet 2019;20:675–91. [DOI] [PubMed] [Google Scholar]
- 14. Szabo L, Salzman J. Detecting circular RNAs: bioinformatic and experimental challenges. Nat Rev Genet 2016;17:679–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Park OH, Ha H, Lee Y, et al. . Endoribonucleolytic cleavage of m(6)A-containing RNAs by RNase P/MRP complex. Mol Cell 2019;74:494–507.e498. [DOI] [PubMed] [Google Scholar]
- 16. Yang Y, Fan X, Mao M, et al. . Extensive translation of circular RNAs driven by N(6)-methyladenosine. Cell Res 2017;27:626–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Vo JN, Cieslik M, Zhang Y, et al. . The landscape of circular RNA in cancer. Cell 2019;176:869–81.e813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Guarnerio J, Bezzi M, Jeong JC, et al. . Oncogenic role of fusion-circRNAs derived from cancer-associated chromosomal translocations. Cell 2016;165:289–302. [DOI] [PubMed] [Google Scholar]
- 19. Bach DH, Lee SK, Sood AK. Circular RNAs in cancer. Mol Ther Nucleic Acids 2019;16:118–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhao X, Cai Y, Xu J. Circular RNAs: biogenesis, mechanism, and function in human cancers. Int J Mol Sci 2019;20:pii: E3926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang Y, Liu J, Ma J, et al. . Exosomal circRNAs: biogenesis, effect and application in human diseases. Mol Cancer 2019;18:116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang Z, Yang T, Xiao J. Circular RNAs: promising biomarkers for human diseases. EBioMedicine 2018;34:267–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ashwal-Fluss R, Meyer M, Pamudurti NR, et al. . circRNA biogenesis competes with pre-mRNA splicing. Mol Cell 2014;56:55–66. [DOI] [PubMed] [Google Scholar]
- 24. Zhang XO, Wang HB, Zhang Y, et al. . Complementary sequence-mediated exon circularization. Cell 2014;159:134–47. [DOI] [PubMed] [Google Scholar]
- 25. Chen LL. The biogenesis and emerging roles of circular RNAs. Nat Rev Mol Cell Biol 2016;17:205–11. [DOI] [PubMed] [Google Scholar]
- 26. Zhang Y, Zhang XO, Chen T, et al. . Circular intronic long noncoding RNAs. Mol Cell 2013;51:792–806. [DOI] [PubMed] [Google Scholar]
- 27. Li Z, Huang C, Bao C, et al. . Exon-intron circular RNAs regulate transcription in the nucleus. Nat Struct Mol Biol 2015;22:256–64. [DOI] [PubMed] [Google Scholar]
- 28. Tay Y, Rinn J, Pandolfi PP. The multilayered complexity of ceRNA crosstalk and competition. Nature 2014;505:344–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chen L, Wong G. Transcriptome informatics. In: Encyclopedia of Bioinformatics and Computational Biology. Amsterdam, Elsevier, 2019, 324–40.
- 30. You X, Vlatkovic I, Babic A, et al. . Neural circular RNAs are derived from synaptic genes and regulated by development and plasticity. Nat Neurosci 2015;18:603–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jeck WR, Sorrentino JA, Wang K, et al. . Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA 2013;19:141–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Enuka Y, Lauriola M, Feldman ME, et al. . Circular RNAs are long-lived and display only minimal early alterations in response to a growth factor. Nucleic Acids Res 2016;44:1370–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Suzuki H, Zuo Y, Wang J, et al. . Characterization of RNase R-digested cellular RNA source that consists of lariat and circular RNAs from pre-mRNA splicing. Nucleic Acids Res 2006;34:e63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kristensen LS, Hansen TB, Veno MT, et al. . Circular RNAs in cancer: opportunities and challenges in the field. Oncogene 2018;37:555–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kumar L, Shamsuzzama, Haque R, et al. . Circular RNAs: the emerging class of non-coding RNAs and their potential role in human neurodegenerative diseases. Mol Neurobiol 2017;54:7224–34. [DOI] [PubMed] [Google Scholar]
- 36. Aufiero S, Reckman YJ, Pinto YM, et al. . Circular RNAs open a new chapter in cardiovascular biology. Nat Rev Cardiol 2019;16(8):503–14. [DOI] [PubMed] [Google Scholar]
- 37. Zhao W, Chu S, Jiao Y. Present scenario of circular RNAs (circRNAs) in plants. Front Plant Sci 2019;10:379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jakobi T, Dieterich C. Computational approaches for circular RNA analysis. Wiley Interdiscip Rev RNA 2019;10(3):e1528. [DOI] [PubMed] [Google Scholar]
- 39. Szabo L, Morey R, Palpant NJ, et al. . Statistically based splicing detection reveals neural enrichment and tissue-specific induction of circular RNA during human fetal development. Genome Biol 2015;16:126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hoffmann S, Otto C, Kurtz S, et al. . Fast mapping of short sequences with mismatches, insertions and deletions using index structures. PLoS Comput Biol 2009;5:e1000502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hoffmann S, Otto C, Doose G, et al. . A multi-split mapping algorithm for circular RNA, splicing, trans-splicing and fusion detection. Genome Biol 2014;15:R34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gao Y, Zhao F. Computational strategies for exploring circular RNAs. Trends Genet 2018;34:389–400. [DOI] [PubMed] [Google Scholar]
- 43. Gao Y, Wang J, Zhao F. CIRI: an efficient and unbiased algorithm for de novo circular RNA identification. Genome Biol 2015;16:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gao Y, Zhang J, Zhao F. Circular RNA identification based on multiple seed matching. Brief Bioinform 2018;19:803–10. [DOI] [PubMed] [Google Scholar]
- 45. Zheng Y, Ji P, Chen S, et al. . Reconstruction of full-length circular RNAs enables isoform-level quantification. Genome Med 2019;11:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhang XO, Dong R, Zhang Y, et al. . Diverse alternative back-splicing and alternative splicing landscape of circular RNAs. Genome Res 2016;26:1277–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cheng J, Metge F, Dieterich C. Specific identification and quantification of circular RNAs from sequencing data. Bioinformatics 2016;32:1094–6. [DOI] [PubMed] [Google Scholar]
- 48. Wang K, Singh D, Zeng Z, et al. . MapSplice: accurate mapping of RNA-seq reads for splice junction discovery. Nucleic Acids Res 2010;38:e178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Humphreys DT, Fossat N, Tam PP, et al. . Ularcirc: visualisation and enhanced analysis of circular RNAs via back and canonical forward splicing, bioRxiv, 2018, 318436. [DOI] [PMC free article] [PubMed]
- 50. Song X, Zhang N, Han P, et al. . Circular RNA profile in gliomas revealed by identification tool UROBORUS. Nucleic Acids Res 2016;44:e87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Westholm JO, Miura P, Olson S, et al. . Genome-wide analysis of drosophila circular RNAs reveals their structural and sequence properties and age-dependent neural accumulation. Cell Rep 2014;9:1966–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Izuogu OG, Alhasan AA, Alafghani HM, et al. . PTESFinder: a computational method to identify post-transcriptional exon shuffling (PTES) events. BMC Bioinformatics 2016;17:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Li X, Chu C, Pei J, et al. . CircMarker: a fast and accurate algorithm for circular RNA detection. BMC Genomics 2018;19:572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Li X, Wu Y. Detecting circular RNA from high-throughput sequence data with de Bruijn graph, bioRxiv, 2019, 509422. [DOI] [PMC free article] [PubMed]
- 55. Chen CY, Chuang TJ. NCLcomparator: systematically post-screening non-co-linear transcripts (circular, trans-spliced, or fusion RNAs) identified from various detectors. BMC Bioinformatics 2019;20:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sekar S, Geiger P, Adkins J, et al. . ACValidator: a novel assembly-based approach for in silico validation of circular RNAs, bioRxiv, 2019, 556597. [DOI] [PMC free article] [PubMed]
- 57. Smid M, Wilting SM, Uhr K, et al. . The circular RNome of primary breast cancer. Genome Res 2019;29:356–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Menzel P, Meyer IM. BIQ: a method for searching circular RNAs in transcriptome databases by indexing backsplice junctions, bioRxiv, 2019, 556993.
- 59. You X, Conrad TO. Acfs: accurate circRNA identification and quantification from RNA-Seq data. Sci Rep 2016;6:38820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Chuang TJ, Wu CS, Chen CY, et al. . NCLscan: accurate identification of non-co-linear transcripts (fusion, trans-splicing and circular RNA) with a good balance between sensitivity and precision. Nucleic Acids Res 2016;44:e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Mangul S, Yang HT, Strauli N, et al. . ROP: dumpster diving in RNA-sequencing to find the source of 1 trillion reads across diverse adult human tissues. Genome Biol 2018;19:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Akers NK, Schadt EE, Losic B. STAR chimeric post for rapid detection of circular RNA and fusion transcripts. Bioinformatics 2018;34:2364–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Yu SH, Vogel J, Forstner KU. ANNOgesic: a Swiss army knife for the RNA-seq based annotation of bacterial/archaeal genomes. GIGASCIENCE 2018;7:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Zhou C, Molinie B, Daneshvar K, et al. . Genome-wide maps of m6A circRNAs identify widespread and cell-type-specific methylation patterns that are distinct from mRNAs. Cell Rep 2017;20:2262–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Jia GY, Wang DL, Xue MZ, et al. . CircRNAFisher: a systematic computational approach for de novo circular RNA identification. Acta Pharmacol Sin 2019;40:55–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Wang D. hppRNA-a Snakemake-based handy parameter-free pipeline for RNA-Seq analysis of numerous samples. Brief Bioinform 2018;19:622–6. [DOI] [PubMed] [Google Scholar]
- 67. Andres-Leon E, Nunez-Torres R, Rojas AM. miARma-Seq: a comprehensive tool for miRNA, mRNA and circRNA analysis. Sci Rep 2016;6:25749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Ye CY, Zhang X, Chu Q, et al. . Full-length sequence assembly reveals circular RNAs with diverse non-GT/AG splicing signals in rice. RNA Biol 2017;14:1055–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Jakobi T, Uvarovskii A, Dieterich C. Circtools—a one-stop software solution for circular RNA research. Bioinformatics 2018;35(13):2326–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Li B, Zhang X-Q, Liu S-R, et al. . Discovering the interactions between circular RNAs and RNA-binding proteins from CLIP-seq data using circScan, bioRxiv, 2017, 115980.
- 71. Zhang XQ, Yang JH. Discovering circRNA-microRNA interactions from CLIP-Seq data. Methods Mol Biol 2018;1724:193–207. [DOI] [PubMed] [Google Scholar]
- 72. Gao Y, Wang H, Zhang H, et al. . PRAPI: post-transcriptional regulation analysis pipeline for Iso-Seq. Bioinformatics 2018;34:1580–2. [DOI] [PubMed] [Google Scholar]
- 73. Meng X, Chen Q, Zhang P, et al. . CircPro: an integrated tool for the identification of circRNAs with protein-coding potential. Bioinformatics 2017;33:3314–6. [DOI] [PubMed] [Google Scholar]
- 74. Wan C, Gao J, Zhang H, et al. . CPSS 2.0: a computational platform update for the analysis of small RNA sequencing data. Bioinformatics 2017;33:3289–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Fan X, Zhang X, Wu X, et al. . Single-cell RNA-seq transcriptome analysis of linear and circular RNAs in mouse preimplantation embryos. Genome Biol 2015;16:148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Dang Y, Yan L, Hu B, et al. . Tracing the expression of circular RNAs in human pre-implantation embryos. Genome Biol 2016;17:130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Hansen TB. Improved circRNA identification by combining prediction algorithms. Front Cell Dev Biol 2018;6:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Hansen TB, Veno MT, Damgaard CK, et al. . Comparison of circular RNA prediction tools. Nucleic Acids Res 2016;44:e58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Gaffo E, Bonizzato A, Kronnie GT, et al. . CirComPara: a multi-method comparative bioinformatics pipeline to detect and study circRNAs from RNA-seq data. Non-Coding RNA 2017;3:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Li L, Zheng YC, Kayani MUR, et al. . Comprehensive analysis of circRNA expression profiles in humans by RAISE. Int J Oncol 2017;51:1625–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Chen L, Yu Y, Zhang X, et al. . PcircRNA_finder: a software for circRNA prediction in plants. Bioinformatics 2016;32:3528–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Li L, Bu D, Zhao Y. CircRNAwrap—a flexible pipeline for circRNA identification, transcript prediction, and abundance estimation. FEBS Lett 2019;593(11):1179–89. [DOI] [PubMed] [Google Scholar]
- 83. Pan X, Xiong K. PredcircRNA: computational classification of circular RNA from other long non-coding RNA using hybrid features. Mol BioSyst 2015;11:2219–26. [DOI] [PubMed] [Google Scholar]
- 84. Pan X, Xiong K, Anthon C, et al. . WebCircRNA: classifying the circular RNA potential of coding and noncoding RNA. Genes (Basel) 2018;9:536–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Liu Z, Han J, Lv H, et al. . Computational identification of circular RNAs based on conformational and thermodynamic properties in the flanking introns. Comput Biol Chem 2016;61:221–5. [DOI] [PubMed] [Google Scholar]
- 86. Wang J, Wang L. Deep learning of the back-splicing code for circular RNA formation. Bioinformatics 2019;35(24):5235–42. [DOI] [PubMed] [Google Scholar]
- 87. Zeng X, Lin W, Guo M, et al. . A comprehensive overview and evaluation of circular RNA detection tools. PLoS Comput Biol 2017;13:e1005420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Huang W, Li L, Myers JR, et al. . ART: a next-generation sequencing read simulator. Bioinformatics 2012;28:593–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Chen I, Chen CY, Chuang TJ. Biogenesis, identification, and function of exonic circular RNAs. Wiley Interdiscip Rev RNA 2015;6:563–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Holtgrewe M, Emde AK, Weese D, et al. . A novel and well-defined benchmarking method for second generation read mapping. BMC Bioinformatics 2011;12:210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Li S, Teng S, Xu J, et al. . Microarray is an efficient tool for circRNA profiling. Brief Bioinform 2018;20:1420–33. [DOI] [PubMed] [Google Scholar]
- 92. Zhao J, Li X, Guo J, et al. . ReCirc: prediction of circRNA expression and function through probe reannotation of non-circRNA microarrays. Mol Omics 2019;15:150–63. [DOI] [PubMed] [Google Scholar]
- 93. Rybak-Wolf A, Stottmeister C, Glazar P, et al. . Circular RNAs in the mammalian brain are highly abundant, conserved, and dynamically expressed. Mol Cell 2015;58:870–85. [DOI] [PubMed] [Google Scholar]
- 94. Li M, Xie X, Zhou J, et al. . Quantifying circular RNA expression from RNA-seq data using model-based framework. Bioinformatics 2017;33:2131–9. [DOI] [PubMed] [Google Scholar]
- 95. Xu T, Wu J, Han P, et al. . Circular RNA expression profiles and features in human tissues: a study using RNA-seq data. BMC Genomics 2017;18:680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Nicolet BP, Engels S, Aglialoro F, et al. . Circular RNA expression in human hematopoietic cells is widespread and cell-type specific. Nucleic Acids Res 2018;46:8168–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Rozowsky J, Kitchen RR, Park JJ, et al. . exceRpt: a comprehensive analytic platform for extracellular RNA profiling. Cell Syst 2019;8:352–357.e353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Glazar P, Papavasileiou P, Rajewsky N. circBase: a database for circular RNAs. RNA 2014;20:1666–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Meng X, Hu D, Zhang P, et al. . CircFunBase: a database for functional circular RNAs. Database (Oxford) 2019;2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Dong R, Ma XK, Li GW, et al. . CIRCpedia v2: an updated database for comprehensive circular RNA annotation and expression comparison. Genomics Proteomics Bioinformatics 2018;16:226–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Chen X, Han P, Zhou T, et al. . circRNADb: a comprehensive database for human circular RNAs with protein-coding annotations. Sci Rep 2016;6:34985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Liu M, Wang Q, Shen J, et al. . Circbank: a comprehensive database for circRNA with standard nomenclature. RNA Biol 2019;16:899–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Liang G, Yang Y, Niu G, et al. . Genome-wide profiling of sus scrofa circular RNAs across nine organs and three developmental stages. DNA Res 2017;24:523–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Ye J, Wang L, Li S, et al. . AtCircDB: a tissue-specific database for Arabidopsis circular RNAs. Brief Bioinform 2019;20:58–65. [DOI] [PubMed] [Google Scholar]
- 105. Chu Q, Zhang X, Zhu X, et al. . PlantcircBase: a database for plant circular RNAs. Mol Plant 2017;10:1126–8. [DOI] [PubMed] [Google Scholar]
- 106. Zhang P, Meng X, Chen H, et al. . PlantCircNet: a database for plant circRNA–miRNA–mRNA regulatory networks. Database 2017;2017:bax089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Wang K, Wang C, Guo B, et al. . CropCircDB: a comprehensive circular RNA resource for crops in response to abiotic stress. Database (Oxford) 2019;2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Dudekula DB, Panda AC, Grammatikakis I, et al. . CircInteractome: a web tool for exploring circular RNAs and their interacting proteins and microRNAs. RNA Biol 2016;13:34–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Panda AC, Dudekula DB, Abdelmohsen K, et al. . Analysis of circular RNAs using the web tool circinteractome. Methods Mol Biol 2018;1724:43–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Liu YC, Li JR, Sun CH, et al. . CircNet: a database of circular RNAs derived from transcriptome sequencing data. Nucleic Acids Res 2016;44:D209–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Yang JH, Shao P, Zhou H, et al. . deepBase: a database for deeply annotating and mining deep sequencing data. Nucleic Acids Res 2010;38:D123–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Yang JH QLH. DeepBase: annotation and discovery of microRNAs and other noncoding RNAs from deep-sequencing data. Methods Mol Biol 2012;822:233–48. [DOI] [PubMed] [Google Scholar]
- 113. Zheng LL, Li JH, Wu J, et al. . DeepBase v2.0: identification, expression, evolution and function of small RNAs, LncRNAs and circular RNAs from deep-sequencing data. Nucleic Acids Res 2016;44:D196–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Ji P, Wu W, Chen S, et al. . Expanded expression landscape and prioritization of circular RNAs in mammals. Cell Rep 2019;26:3444–60.e3445. [DOI] [PubMed] [Google Scholar]
- 115. Guo L, Wang J. rSNPBase 3.0: an updated database of SNP-related regulatory elements, element-gene pairs and SNP-based gene regulatory networks. Nucleic Acids Res 2018;46:D1111–d1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Yao D, Zhang L, Zheng M, et al. . Circ2Disease: a manually curated database of experimentally validated circRNAs in human disease. Sci Rep 2018;8:11018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Zhao Z, Wang K, Wu F, et al. . circRNA disease: a manually curated database of experimentally supported circRNA-disease associations. Cell Death Dis 2018;9:475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Fan C, Lei X, Fang Z, et al. . CircR2Disease: a manually curated database for experimentally supported circular RNAs associated with various diseases. Database (Oxford) 2018;2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Ghosal S, Das S, Sen R, et al. . Circ2Traits: a comprehensive database for circular RNA potentially associated with disease and traits. Front Genet 2013;4:283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Xia S, Feng J, Chen K, et al. . CSCD: a database for cancer-specific circular RNAs. Nucleic Acids Res 2018;46:D925–d929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Wang WJ, Wang YM, Hu Y, et al. . HDncRNA: a comprehensive database of non-coding RNAs associated with heart diseases. Database (Oxford) 2018;2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Bao Z, Yang Z, Huang Z, et al. . LncRNADisease 2.0: an updated database of long non-coding RNA-associated diseases. Nucleic Acids Res 2019;47:D1034–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Li S, Li Y, Chen B, et al. . exoRBase: a database of circRNA, lncRNA and mRNA in human blood exosomes. Nucleic Acids Res 2018;46:D106–d112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Russo F, Di Bella S, Vannini F, et al. . miRandola 2017: a curated knowledge base of non-invasive biomarkers. Nucleic Acids Res 2018;46:D354–d359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Kalari KR, Thompson KJ, Nair AA, et al. . BBBomics-human blood brain barrier Transcriptomics hub. Front Neurosci 2016;10:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Hentze MW, Preiss T. Circular RNAs: splicing's enigma variations. EMBO J 2013;32:923–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Chen L, Heikkinen L, Wang C, et al. . Trends in the development of miRNA bioinformatics tools. Brief Bioinform 2019;20:1836–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Qi X, Lin Y, Chen J, et al. . Decoding competing endogenous RNA networks for cancer biomarker discovery. Brief Bioinform 2019;20:1–17. [DOI] [PubMed] [Google Scholar]
- 129. Zhong Z, Huang M, Lv M, et al. . Circular RNA MYLK as a competing endogenous RNA promotes bladder cancer progression through modulating VEGFA/VEGFR2 signaling pathway. Cancer Lett 2017;403:305–17. [DOI] [PubMed] [Google Scholar]
- 130. Wang L, Wei Y, Yan Y, et al. . CircDOCK1 suppresses cell apoptosis via inhibition of miR196a5p by targeting BIRC3 in OSCC. Oncol Rep 2018;39:951–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Yang JH, Li JH, Shao P, et al. . starBase: a database for exploring microRNA-mRNA interaction maps from Argonaute CLIP-Seq and Degradome-Seq data. Nucleic Acids Res 2011;39:D202–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Li JH, Liu S, Zhou H, et al. . starBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res 2014;42:D92–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Bhattacharya A, Ziebarth JD, Cui Y. SomamiR: a database for somatic mutations impacting microRNA function in cancer. Nucleic Acids Res 2013;41:D977–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Bhattacharya A, Cui Y. SomamiR 2.0: a database of cancer somatic mutations altering microRNA-ceRNA interactions. Nucleic Acids Res 2016;44:D1005–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Wang P, Li X, Gao Y, et al. . LncACTdb 2.0: an updated database of experimentally supported ceRNA interactions curated from low- and high-throughput experiments. Nucleic Acids Res 2019;47:D121–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Tarek MM. AFCMEasyModel: an easy interface for modeling competing endogenous RNA networks using ODEs, bioRxiv, 2017, 241026.
- 137. Ghosal S, Das S, Sen R, et al. . HumanViCe: host ceRNA network in virus infected cells in human. Front Genet 2014;5:249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Pan X, Wenzel A, Jensen LJ, et al. . Genome-wide identification of clusters of predicted microRNA binding sites as microRNA sponge candidates. PLoS One 2018;13:e0202369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Wang P, Zhi H, Zhang Y, et al. . miRSponge: a manually curated database for experimentally supported miRNA sponges and ceRNAs. Database (Oxford) 2015;2015:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Liu S, Li JH, Wu J, et al. . StarScan: a web server for scanning small RNA targets from degradome sequencing data. Nucleic Acids Res 2015;43:W480–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Lin YC, Lee YC, Chang KL, et al. . Analysis of common targets for circular RNAs. BMC Bioinformatics 2019;20:372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Yi Y, Zhao Y, Li C, et al. . RAID v2.0: an updated resource of RNA-associated interactions across organisms. Nucleic Acids Res 2017;45:D115–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Bonnici V, Caro G, Constantino G, et al. . Arena-Idb: a platform to build human non-coding RNA interaction networks. BMC Bioinformatics 2018;19:350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Tang Z, Li X, Zhao J, et al. . TRCirc: a resource for transcriptional regulation information of circRNAs. Brief Bioinform 2018;20:2327–33. [DOI] [PubMed] [Google Scholar]
- 145. Yu H, Jiao B, Lu L, et al. . NetMiner-an ensemble pipeline for building genome-wide and high-quality gene co-expression network using massive-scale RNA-seq samples. PLoS One 2018;13:e0192613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Wu SM, Liu H, Huang PJ, et al. . circlncRNAnet: an integrated web-based resource for mapping functional networks of long or circular forms of noncoding RNAs. GIGASCIENCE 2018;7:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Gao Y, Wang J, Zheng Y, et al. . Comprehensive identification of internal structure and alternative splicing events in circular RNAs. Nat Commun 2016;7:12060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Metge F, Czaja-Hasse LF, Reinhardt R, et al. . FUCHS-towards full circular RNA characterization using RNAseq. PeerJ 2017;5:e2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Feng J, Chen K, Dong X, et al. . Genome-wide identification of cancer-specific alternative splicing in circRNA. Mol Cancer 2019;18:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Wang H, Wang H, Zhang H, et al. . The interplay between microRNA and alternative splicing of linear and circular RNAs in eleven plant species. Bioinformatics 2019;35(17):3119–26. [DOI] [PubMed] [Google Scholar]
- 151. Zheng Y, Detection ZF. Reconstruction of circular RNAs from transcriptomic data. Methods Mol Biol 2018;1724:1–8. [DOI] [PubMed] [Google Scholar]
- 152. Feng J, Xiang Y, Xia S, et al. . CircView: a visualization and exploration tool for circular RNAs. Brief Bioinform 2018;19:1310–6. [DOI] [PubMed] [Google Scholar]
- 153. Ungerleider NA, Flemington EK. SpliceV: analysis and publication quality printing of linear and circular RNA splicing, expression and regulation, bioRxiv, 2019, 509661. [DOI] [PMC free article] [PubMed]
- 154. Chen L, Heikkinen L, Wang C, et al. . miRToolsGallery: a tag-based and rankable microRNA bioinformatics resources database portal. Database (Oxford) 2018;2018:bay004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Zhu BH, Xiao J, Xue W, et al. . P_RNA_scaffolder: a fast and accurate genome scaffolder using paired-end RNA-sequencing reads. BMC Genomics 2018;19:175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Glouzon JS, Perreault JP, Wang S. The super-n-motifs model: a novel alignment-free approach for representing and comparing RNA secondary structures. Bioinformatics 2017;33:1169–78. [DOI] [PubMed] [Google Scholar]
- 157. Xia S, Feng J, Lei L, et al. . Comprehensive characterization of tissue-specific circular RNAs in the human and mouse genomes. Brief Bioinform 2017;18:984–92. [DOI] [PubMed] [Google Scholar]
- 158. Gokoolparsadh A, Anwar F. Voineagu I. The landscape of circular RNA expression in the human brain, bioRxiv. 2018, 500991. [DOI] [PubMed]
- 159. Hofacker IL, Fontana W, Stadler PF, et al. . Fast folding and comparison of RNA secondary structures. Monatsh Chem 1994;125:167–88. [Google Scholar]
- 160. Hofacker IL, Stadler PF. Memory efficient folding algorithms for circular RNA secondary structures. Bioinformatics 2006;22:1172–6. [DOI] [PubMed] [Google Scholar]
- 161. Cortes-Lopez M, Gruner MR, Cooper DA, et al. . Global accumulation of circRNAs during aging in Caenorhabditis elegans. BMC Genomics 2018;19:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Zhong S, Wang J, Zhang Q, et al. . CircPrimer: a software for annotating circRNAs and determining the specificity of circRNA primers. BMC Bioinformatics 2018;19:292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Dong R, Zhang XO, Zhang Y, et al. . CircRNA-derived pseudogenes. Cell Res 2016;26:747–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Dehghannasiri R, Szabo L, Salzman J. Ambiguous splice sites distinguish circRNA and linear splicing in the human genome. Bioinformatics 2018;35:1263–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Zhang Y, Xue W, Li X, et al. . The biogenesis of nascent circular RNAs. Cell Rep 2016;15:611–24. [DOI] [PubMed] [Google Scholar]
- 166. Pamudurti NR, Bartok O, Jens M, et al. . Translation of CircRNAs. Mol Cell 2017;66:9–21.e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Chu Q, Bai P, Zhu X, et al. . Characteristics of plant circular RNAs. Brief Bioinform 2018;00:1–9. [DOI] [PubMed] [Google Scholar]
- 168. Grabherr MG, Haas BJ, Yassour M, et al. . Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 2011;29:644–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Patro R, Mount SM, Kingsford C. Sailfish enables alignment-free isoform quantification from RNA-seq reads using lightweight algorithms. Nat Biotechnol 2014;32:462–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Kos A, Dijkema R, Arnberg AC, et al. . The hepatitis delta (delta) virus possesses a circular RNA. Nature 1986;323:558–60. [DOI] [PubMed] [Google Scholar]
- 171. Nigro JM, Cho KR, Fearon ER, et al. . Scrambled exons. Cell 1991;64:607–13. [DOI] [PubMed] [Google Scholar]
- 172. Hansen TB, Jensen TI, Clausen BH, et al. . Natural RNA circles function as efficient microRNA sponges. Nature 2013;495:384–8. [DOI] [PubMed] [Google Scholar]
- 173. Qu S, Song W, Yang X, et al. . Microarray expression profile of circular RNAs in human pancreatic ductal adenocarcinoma. Genom Data 2015;5:385–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Li Y, Zheng Q, Bao C, et al. . Circular RNA is enriched and stable in exosomes: a promising biomarker for cancer diagnosis. Cell Res 2015;25:981–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Legnini I, Di Timoteo G, Rossi F, et al. . Circ-ZNF609 is a circular RNA that can be translated and functions in myogenesis. Mol Cell 2017;66:22–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Panda AC, De S, Grammatikakis I, et al. . High-purity circular RNA isolation method (RPAD) reveals vast collection of intronic circRNAs. Nucleic Acids Res 2017;45:e116. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.