

Abstract
Herein, a conceptually distinct approach was developed that allowed for the dicarbofunctionalization of alkynes at room temperature using simple, bench‐stable alkyl iodides and a second molecule of alkyne as coupling partner. Specifically, the photochemical activation of palladium complexes enabled this strategic dicarbofunctionalization via addition of alkyl radicals from secondary and tertiary alkyl iodides and formation of an intermediate palladium vinyl complex that could undergo subsequent Sonogashira reaction with a second alkyne molecule. This alkylation–alkynylation sequence allowed the one‐step synthesis of 1,3‐enynes including heteroarenes and biologically active compounds with high efficiency without exogenous photosensitizers or oxidants and now opens up pathways towards cascade reactions via photochemical palladium catalysis.
Keywords: alkynes, dicarbofunctionalization, enynes, palladium, radical coupling
Dicarbofunctionalization of alkynes at room temperature is achieved using simple, bench‐stable alkyl iodides and a second molecule of alkyne as coupling partner. This alkylation–alkynylation sequence allows the one‐step synthesis of 1,3‐enynes including heteroarenes and biologically active compounds with high efficiency without exogenous photosensitizers or oxidants.

Palladium‐catalyzed cross‐coupling reactions constitute one of the pillars of chemistry and have significantly shaped modern organic synthesis methodology and applications, for example, in materials’ sciences or drug development. [1] Since the discovery of Pd‐catalyzed cross‐coupling reactions, [2] advances in catalyst and ligand design have opened up a broad array of synthetic transformations to push the boundaries of the classic oxidative addition–transmetalation–reductive elimination mechanism. Only recently, the discovery of photochemical Pd‐catalyzed cross‐coupling reactions opened up a new paradigm. [3] This strategy harnesses the photochemical properties of palladium complexes that often feature a strong absorption in the visible light region.[ 3 , 4 , 5 , 6 , 7 , 8 , 9 ] Hence, palladium complexes can take up a dual function as both photosensitizers and cross‐coupling catalysts and thus contrast the developments in dual photoredox/ transition metal‐catalyzed transformations that require two different catalysts for the sensitization and coupling event. [10] Moreover, it has recently been demonstrated that photoexcitation of Pd‐complexes facilitates the formation of an intermediate radical and PdI complex, which does not proceed via single‐electron transfer processes (Scheme 1). [5]
Scheme 1.
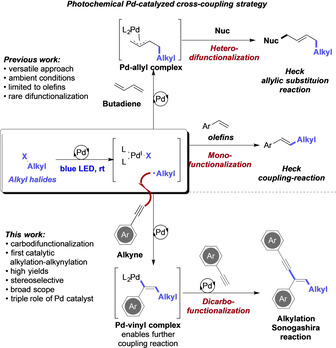
Photochemical Pd‐catalyzed cross‐coupling reactions.
Today, the application of photochemical Pd‐catalysis is rapidly advancing as an important strategy to unveil the reactivity of alkyl halides in Pd‐catalyzed cross‐coupling reactions, which are typically challenging substrates owing to the ease of β‐hydride elimination. In this context, the photochemical, palladium‐catalyzed Heck‐reaction of unactivated alkyl halides witnessed significant advances as reported by the groups of Fu, [6] Gevorgyan,[ 3 , 7 ] Glorius, [8] and Rueping. [5] Currently available approaches focus on a singular catalytic bond‐forming event, while difunctionalization reactions of hydrocarbon frameworks remain rare. In this context, Glorius and co‐workers and Gevorgyan and co‐workers recently reported on the reactivity of buta‐1,3‐dienes under photochemical palladium catalyzed conditions to allow for the 1,4‐difunctionalization via a Heck–allylic substitution reaction cascade (Scheme 1).[ 7f , 8b ]
Against this background and our interest in metal‐catalyzed C−C bond‐forming reactions, [11] we became intrigued in studying difunctionalization reactions of alkynes via multiple Pd‐catalyzed bond forming events. [12] Specifically, we hypothesized that photochemical activation of a Pd‐complex would allow the formation of alkyl radicals that undergo addition reaction with terminal alkynes to furnish an intermediate Pd‐vinyl complex, which in turn could be applied to subsequent Pd‐catalyzed cross‐coupling reactions to realize photocatalytic dicarbofunctionalization reactions. In this context, Nevado and co‐workers [13] and Li et al. [14] recently reported on their developments in dicarbofunctionalization reactions to allow for formal alkylation–arylation or dialkylation reactions of alkynes, yet requiring forcing thermal reaction conditions [13a] or the use of reactive organometallic reagents. [14]
We initiated these studies by examining the reaction of phenylacetylene 1 a with cyclohexyl iodide 2 a under photoinduced palladium‐catalyzed reaction conditions. Indeed, we obtained the 1,3‐enyne product 3 a in 55 % yield and high stereoselectivity [diastereomeric ratio (d.r.) >20:1] without formation of by‐products from alkyne dimerization (4). [15] This transformation now showcases one of the first examples of a catalytic alkylation‐alkynylation reaction and now provides a one‐step access towards important 1,3‐enynes. [16] We thus examined different reaction parameters, such as stoichiometry, solvent, base, palladium precursor, or ligand to further improve the yield of 3 a (for the full details of our optimization studies, please see Tables S1–S7 in the Supporting Information). [17] Of all bases and solvents tested, the combination of Cs2CO3 and THF and an excess of phenylacetylene 1 a gave the best yield of the 1,3‐enyne product (Table 1, entry 5). Monodentate phosphine ligands, such as RuPhos (Table 1, entry 6) failed to provide the desired alkylation‐alkynylation product 3 a, instead the homo‐coupling product 4 was obtained exclusively. Bidentate phosphine ligands gave the dicarbofunctionalization without formation of the product of homo‐coupling, albeit in lower yield (Table S1 in the Supporting Information). [17] When using 10 mol % of the palladium catalyst, the product 3 a was obtained in 81 % isolated yield (Table 1, entry 7). A control experiment showed the necessity of the photochemical reaction conditions as no reaction occurred in the dark (Table 1, entry 9). Similarly, alkyl bromides proved incompatible and only trace amounts of the 1,3‐enyne product were observed (Table 1, entry 8). To probe the potential of thermally induced processes, we examined the reaction under thermal conditions in the dark, yet, no desired 1,3‐enyne product was found.
Table 1.
Reaction optimization.[a]
|
| ||
|---|---|---|
|
Entry |
Deviations from above |
Yield[b] (3 a) [%] |
|
1 |
– |
55 |
|
2 |
1 equiv. 1 a, 3 equiv. 2 a |
40 |
|
3 |
Pd source: PdCl2/Pd(TFA)2/Pd2(dba)3 |
trace/24/16 |
|
4 |
K2CO3 instead of Cs2CO3 |
trace |
|
5 |
THF instead of PhH solvent |
62 |
|
6 |
10 mol % RuPhos instead of Xantphos |
3 a: n.d. 4: 63 % |
|
7 |
10 mol % Pd(OAc)2, 20 mol % Xantphos, THF solvent |
83 (81)[c] |
|
8 |
cyclohexyl bromide instead of 2 a |
trace |
|
9 |
reaction in the dark |
no reaction |

[a] Reaction conditions: 0.6 mmol 1 a (3 equiv.), 0.2 mmol 2 a (1 equiv.), 0.4 mmol (2 equiv.) Cs2CO3, 5 mol % Pd(OAc)2, 10 mol % Xantphos were dissolved in 1 mL benzene under argon atmosphere, irradiated with 3 W blue led (470 nm) at room temperature overnight. [b] The yield was determined by 1H NMR spectroscopy of the crude reaction. The internal standard was CHBr3. [c] Isolated yield.
With the optimized reaction condition in hand, we next examined the application of this enyne synthesis (Scheme 2). Electron‐donating substituents, such as methyl (3 d, 3 l), methoxy (3 e, 3 m), and dimethylamino (3 f) were found compatible, and the corresponding dicarbofunctionalization products were obtained in moderate yield and excellent stereoselectivity.
Scheme 2.
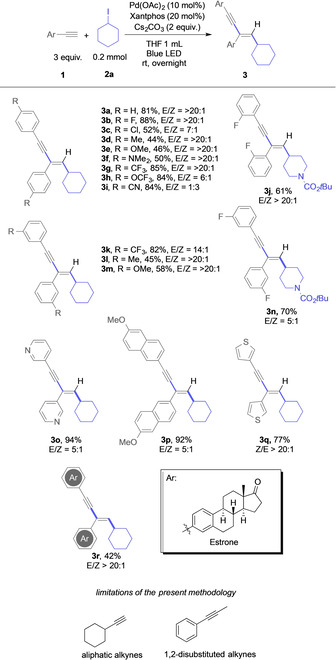
Evaluation of different alkynes.
Electron‐deficient alkynes gave an increased yield, which might be related to a more facile radical addition, albeit with lower stereoselectivity (3 g, 3 h, 3 i, 3 k). A notable limitation was observed for halogen substituents. Fluorine‐substituted arylalkynes gave the desired 1,3‐enyne product (3 b, 3 j) in good yield with excellent stereoselectivity. In the case of 4‐chlorophenyl acetylene, the yield of the enyne 3 c dropped significantly due to background Sonogashira coupling reaction. 4‐Bromophenyl acetylene, however, gave only trace amounts of the desired product. Further studies involved the use of heterocyclic substrates. To our delight, heterocycle‐ or carbocycle‐substituted alkynes were also compatible under the present conditions, and the heterocycle‐substituted 1,3‐enyne products were isolated in excellent yield (3 o–q). An Estrone‐derived alkyne was also found compatible in this transformation, and the corresponding product 3 r was obtained in 42 % yield, thus demonstrating the application in late‐stage functionalization.
The limitations of the present methodology lie within the use of aliphatic, or 1,2‐disubstituted alkynes, which both did not provide the enyne coupling product under the present reaction conditions; instead, decomposition of cyclohexyl iodide took place, and the formation of complex reaction mixtures was observed. The missing reactivity of 1,2‐disubstituted alkynes can be explained by the missing C(sp)−H proton that is important for the final coupling step to access the enyne.
Next, we examined the compatibility of different alkyl iodides. Primary alkyl iodides gave only trace amounts of the coupling product and decomposition of the alkyl iodide as observed by 1H NMR spectroscopy of the crude reaction mixture (for more details, please see the Supporting Information), which might be reasoned by the instability of primary radical. Secondary, acyclic alkyl iodides gave the corresponding 1,3‐enyne products 5 a and 5 b in moderate yield and high stereoselectivity (Scheme 3). We then studied different ring sizes of cyclic alkyl iodides, ranging from 5‐ to 8‐membered ring size, which gave the 1,3‐enyne products 5 c–e in good yield and excellent stereoselectivity for small ring sizes. The stereoselectivity was slightly reduced in the case of cycloheptyl and cyclooctyl iodide. Heterocyclic iodides, including tetrahydrofuran 2 f, tetrahydropyran 2 g, and piperidine 2 h, reacted smoothly to the heterocycle‐substituted 1,3‐enyne products 5 f–h in good yield and stereoselectivity. In a scale‐up experiment of 5 g to 1 mmol, the desired 1,3‐enyne product was obtained in slightly reduced yield, which might be related to the low efficiency of the 3 W blue LED lamps and thus missing penetration of the light into the reaction mixture. Tertiary alkyl iodides, such as tert‐butyl‐iodide 2 i and adamantyl iodide 2 j similarly gave the 1,3‐enyne product with 63 and 84 % yield, respectively (5 i,j). The epiandosterone‐derived iodide derivative 2 k gave the desired 1,3‐enyne product 5 k in moderate yield.
Scheme 3.
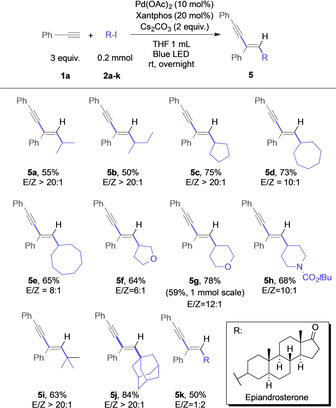
Evaluation of alkyl iodides.
To gain insight into the reaction mechanism, several control experiments were studied. In a first experiment, we studied the on/off experiment of the reaction of phenylacetylene with cyclohexyl iodide (Figure 1). When the light was switched off, the reaction did not proceed further; after turning the light back on, the reaction resumed, which indicates that the reaction only proceeds under constant irradiation.
Figure 1.
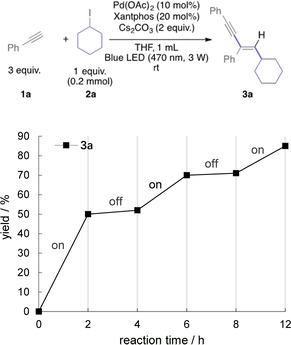
On/off experiment.
In a second set of experiments, we probed the participation of radical intermediates. When 0.5 equiv. of the radical quencher (2,2,6,6‐tetramethylpiperidin‐1‐yl)oxyl (TEMPO) was added to the reaction mixture, the pathway towards 1,3‐enyne formation was completely inhibited, and a 46 % yield of Cy‐TEMPO 6 was obtained as determined by 1H NMR spectroscopy of the crude reaction mixture (Scheme 4 a). We next probed the reaction with (cyclopropyliodomethyl)‐benzene 2 l, which can readily undergo ring‐opening reactions via a radical clock mechanism to give a primary alkyl radical (Scheme 4 b). Consistently with the above findings, no reaction product was formed due to the missing reactivity of primary alkyl radicals, and decomposition of iodo alkane 2 l was observed. To further prove the radical mechanism, a radical clock experiment was studied with 5‐iodohex‐1‐ene 2 m. We could indeed observe an inseparable mixture of the direct radical coupling product 5 m and product 5 m’, which arises from radical formation, followed by intramolecular radical cyclization with the pendant olefin (Scheme 4 b). This observation further supports the radical mechanism and moreover demonstrates that the intramolecular radical cyclization is disfavored over the radical coupling process. To further elucidate the role of radical intermediates in the reaction mechanism, different alkyl‐substituted alkynes were studied. When tert‐butyl acetylene 1 s was applied as substrate, no desired 1,3‐enyne was founded in the reaction mixture. Contrarily, trimethylsilane (TMS)‐acetylene 1 t gave the corresponding 1,3‐enyne in moderate yield (Scheme 4 c). For this phenomenon, we rationalize that addition of alkyl radicals to the terminal alkyne generates a highly reactive vinyl radical. While a neighboring aromatic ring or TMS substitution can stabilize this vinyl radical, alkyl groups only poorly stabilize this intermediate, resulting in missing reaction to the 1,3‐enyne product.
Scheme 4.
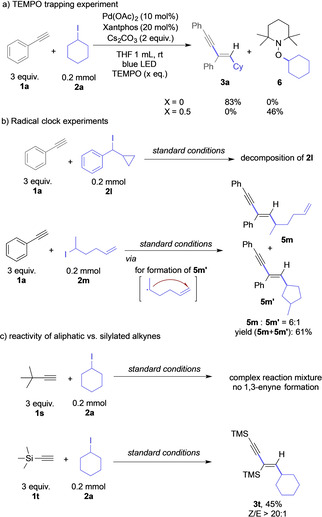
Control experiments on the participation of radical intermediates.
Next, we aimed at an understanding of the second C−C bond‐forming step. We examined different viable intermediates that could rationalize the observed reactivity. First, we proposed that a radical mechanism of alkyl iodide and phenylacetylene could result in the formation of the corresponding vinyl iodide. The latter could further react with another molecule of phenylacetylene 1 a via Sonogashira coupling reaction. We thus studied the reaction of vinyl iodide 7 and phenylacetylene 1 a under the optimized conditions (Scheme 5 a). The corresponding 1,3‐enyne 3 a was obtained with moderate yield under the irradiation of blue light. Interestingly, the 1,3‐enyne 3 a was also observed in the dark, yet significantly higher reaction temperatures are required to obtain a comparable yield, which shows that the second C−C bond forming reaction occurs at room temperature only in the presence of visible light with high efficiency. Moreover, NMR spectroscopy and GC–MS analysis of the crude reaction mixture of cyclohexyl iodide 2 a with phenylacetylene 1 a under optimized conditions did not reveal the presence of vinyl iodide 7, which shows that vinyl iodide 7 is either a short‐lived intermediate or might not even be formed at all. Next, we examined the potential of a formal Sonogashira reaction–hydroalkynylation reaction mechanism (Scheme 5 b). Such a mechanism would involve the formation of a 1,2‐disubstitued alkyne 8, which could undergo a hydroalkynylation reaction under the present reaction conditions. Yet, a control experiment with 8 showed no reaction and both reactants remained untouched under the present reaction conditions, thus excluding a reaction mechanism via a hydroalkynylation reaction. Finally, we examined the potential of formation of higher homologues of 3 a that stem from multiple consecutive reactions with phenylacetylene (Scheme 5 c). For this purpose, we studied the reaction of cyclohexyl iodide 2 a under optimized conditions, yet with a large excess of phenylacetylene 1 a. However, no higher homologues of 3 a were identified via GC–MS or NMR spectroscopy, which rules out a radical addition pathway in the second bond‐forming event and further supports the Sonogashira coupling mechanism. At this point it is important to note that attempts at the coupling of two different terminal alkynes did not lead to the formation of any crossover reaction products (for details, please see the Supporting Information).
Scheme 5.
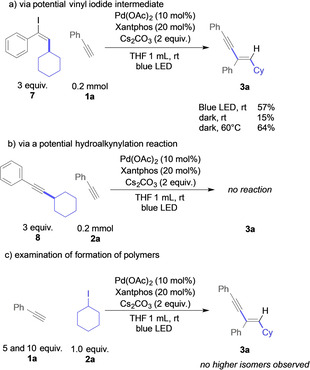
Control experiments on the role of the alkyne reaction partner.
Based on the above control experiments and previous reports,[ 5 , 17 ] we hypothesize the following mechanism (Scheme 6): Photoexcitation of a Pd0 complex A provides a photoexcited state A* that reacts with alkyl iodide compound 2 a to give a PdI complex and alkyl radical B. Radical addition between B and one molecule of phenylacetylene 1 a generates vinyl radical Int‐I, which is stabilized by the pendant aromatic ring. Contrarily, a corresponding alkyl‐substituted vinyl radical would lack this stabilization and thus undergo decomposition. The linear geometry of vinyl radical Int‐I results in coordination to PdI from the sterically less hindered face resulting in the cis geometry of aryl and alkyl group. The vinyl PdII complex Int‐II can further undergo Sonogashira coupling reaction with another molecule of phenylacetylene 1 a by deprotonation of Cs2CO3 via Int‐III to afford the 1,3‐enyne product 3 a and regenerate the Pd0 catalyst. In the case of 1,2‐disubstituted alkynes this Sonogashira reaction is impossible and, resulting in no formation of the 1,3‐enyne product.
Scheme 6.
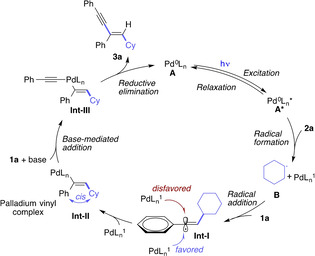
Proposed reaction mechanism.
In the proposed mechanism, the photoinduced palladium catalysis takes up different functions. First, the Pd0 complex acts as a photosensitizer that is also key to access a PdI complex and an alkyl radical and thus initiates this dicarbofunctionalization reaction. Second, the palladium catalyst plays a key role in enabling the subsequent Sonogashira coupling process at room temperature via excitation with visible light to afford the final 1,3‐enyne product.
In summary, we have developed a visible‐light‐induced palladium‐catalyzed alkylation‐alkynylation reaction to construct C(sp2)−C(sp3)/C(sp2)−C(sp) bonds in one step from aromatic alkynes and alkyl iodides under mild reaction conditions without the need of exogenous photosensitizers or oxidants. Various alkynes and iodide compounds including bioactive derivatives are compatible to afford densely functionalized 1,3‐enynes with good yield and stereoselectivity. A possible reaction mechanism was proposed and involves a palladium vinyl intermediate, which now opens up new a pathway in photochemical palladium catalysis towards cascade reactions and multi‐functionalization reactions.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
R.M.K. thanks the German Science Foundation for financial support. Open access funding enabled and organized by Projekt DEAL.
Z. Yang, R. M. Koenigs, Chem. Eur. J. 2021, 27, 3694.
References
- 1.Selected reviews:
- 1a. Biffis A., Centomo P., Del Zotto A., Zecca M., Chem. Rev. 2018, 118, 2249–2295; [DOI] [PubMed] [Google Scholar]
- 1b. Ruiz-Castillo P., Buchwald S. L., Chem. Rev. 2016, 116, 12564–12649; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1c. Zhu C., Liu J., Li M. B., Bäckvall J.-E., Chem. Soc. Rev. 2020, 49, 341–353; [DOI] [PubMed] [Google Scholar]
- 1d. Fricke C., Sperger T., Mendel M., Schoenebeck F., Angew. Chem. Int. Ed. 2021, 10.1002/anie.202011825; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1e. Liu Q., Dong X., Li J., Xiao J., Dong Y., Liu H., ACS Catal. 2015, 5, 6111–6137; [Google Scholar]
- 1f. Roughley S. D., Jordan A. M., J. Med. Chem. 2011, 54, 3451–3479; [DOI] [PubMed] [Google Scholar]
- 1g. Brown D. G., Boström J., J. Med. Chem. 2016, 59, 4443–4458. [DOI] [PubMed] [Google Scholar]
- 2.Selected review: Johansson Seechurn C. C. C., Kitching M. O., Colacot T. J., Snieckus V., Angew. Chem. Int. Ed. 2012, 51, 5062–5085; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 5150–5174. [Google Scholar]
- 3. Parasram M., Chuentragool P., Sarkar D., Gevorgyan V., J. Am. Chem. Soc. 2016, 138, 6340–6343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.
- 4a. Chuentragool P., Kurandina D., Gevorgyan V., Angew. Chem. Int. Ed. 2019, 58, 11586–11598; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 11710–11722; [Google Scholar]
- 4b. Sumino S., Fasano A., Fukuyama T., Ryu I., Acc. Chem. Res. 2014, 47, 1563–1574; [DOI] [PubMed] [Google Scholar]
- 4c. Kancherla R., Muralirajan K., Sagadevan A., Rueping M., Trends Chem. 2019, 1, 510–523; [Google Scholar]
- 4d. Ye S., Xiang T., Li X., Wu J., Org. Chem. Front. 2019, 6, 2183–2199; [Google Scholar]
- 4e. Zhou W.-J., Cao G.-M., Zhang Z.-P., Yu D.-G., Chem. Lett. 2019, 48, 181–191. [Google Scholar]
- 5. Kancherla R., Muralirajan K., Maity B., Zhu C., Krach P. E., Cavallo L., Rueping M., Angew. Chem. Int. Ed. 2019, 58, 3412–3416; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 3450–3454. [Google Scholar]
- 6.
- 6a. Wang G.-Z., Shang R., Cheng W.-M., Fu Y., J. Am. Chem. Soc. 2017, 139, 18307–18312; [DOI] [PubMed] [Google Scholar]
- 6b. Wang G.-Z., Shang R., Fu Y., Synthesis 2018, 50, 2908–2914; [Google Scholar]
- 6c. Wang G.-Z., Shang R., Fu Y., Org. Lett. 2018, 20, 888–891; [DOI] [PubMed] [Google Scholar]
- 6d. Cheng W.-M., Shang R., Fu Y., Nat. Commun. 2018, 9, 5215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.
- 7a. Chuentragool P., Parasram M., Shi Y., Gevorgyan V., J. Am. Chem. Soc. 2018, 140, 2465–2468; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7b. Ratushnyy M., Parasram M., Wang Y., Gevorgyan V., Angew. Chem. Int. Ed. 2018, 57, 2712–2715; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 2742–2745; [Google Scholar]
- 7c. Kurandina D., Parasram M., Gevorgyan V., Angew. Chem. Int. Ed. 2017, 56, 14212–14216; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 14400–14404; [Google Scholar]
- 7d. Parasram M., Chuentragool P., Wang Y., Shi Y., Gevorgyan V., J. Am. Chem. Soc. 2017, 139, 14857–14860; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7e. Chuentragool P., Yadagiri D., Morita T., Sarkar S., Chuentragool P., Wang Y., Gevorgyan V., Angew. Chem. Int. Ed. 2019, 58, 1794–1798; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 1808–1812; [Google Scholar]
- 7f. Cheung K. P. S., Kurandina D., Yata T., Gevorgyan V., J. Am. Chem. Soc. 2020, 142, 9932–9937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.
- 8a. Koy M., Sandfort F., Tlahuext-Aca A., Quach L., Daniliuc C. G., Glorius F., Chem. Eur. J. 2018, 24, 4552–4555; [DOI] [PubMed] [Google Scholar]
- 8b. Huang H.-M., Bellotti P., Pflueger P. M., Schwarz J. L., Heidrich B., Glorius F., J. Am. Chem. Soc. 2020, 142, 10173–10183. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Zhou W.-J., Cao G.-M., Shen G., Zhu X.-Y., Gui Y.-Y., Ye J.-H., Sun L., Liao L.-L., Li J., Yu D.-G., Angew. Chem. Int. Ed. 2017, 56, 15683–15687; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 15889–15893; [Google Scholar]
- 9b. Sun L., Ye J.-H., Zhou W.-J., Yu D.-G., Org. Lett. 2018, 20, 3049–3052; [DOI] [PubMed] [Google Scholar]
- 9c. Czyz M. L., Weragoda G. K., Horngren T. H., Connell T. U., Gomez D., O'Hair R. A. J., Polyzos A., Chem. Sci. 2020, 11, 2455–2463; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9d. Adamik R., Földesi T., Novak Z., Org. Lett. 2020, 22, 8091–8095; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9e. Kim D., Lee G. S., Kim D., Hong S. H., Nature Commun. 2020, 11, 5266; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9f. Chen L., Guo L.-N., Liu S., Liu L., Duan X.-H., Chem. Sci. 2021, 10.1039/D0SC04399K. [DOI] [Google Scholar]
- 10.Selected articles:
- 10a. Prier C. K., Rankic D. A., MacMillan D. W. C., Chem. Rev. 2013, 113, 5322–5363; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10b. Huang X., Meggers E., Acc. Chem. Res. 2019, 52, 833–847; [DOI] [PubMed] [Google Scholar]
- 10c. Fabry D. C., Rueping M., Acc. Chem. Res. 2016, 49, 1969–1979; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10d. Reiser O., Acc. Chem. Res. 2016, 49, 1990–1996; [DOI] [PubMed] [Google Scholar]
- 10e. Tellis J. C., Kelly C. B., Primer D. N., Jouffroy M., Patel N. R., Molander G. A., Acc. Chem. Res. 2016, 49, 1429–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.
- 11a. Empel C., Koenigs R. M., Synlett 2019, 30, 1929–1934; [Google Scholar]
- 11b. Yang Z., Möller M., Koenigs R. M., Angew. Chem. Int. Ed. 2020, 59, 5572–5576; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 5620–5624; [Google Scholar]
- 11c. Yang Z., Pei C., Koenigs R. M., Org. Lett. 2020, 22, 7234–7238; [DOI] [PubMed] [Google Scholar]
- 11d. Jana S., Empel C., Pei C., Aseeva P., Nguyen T. V., Koenigs R. M., ACS Catal. 2020, 10, 9925–9931; [Google Scholar]
- 11e. Hock K. J., Knorrscheidt A., Hommelsheim R., Ho J., Weissenborn M. J., Koenigs R. M., Angew. Chem. Int. Ed. 2019, 58, 3630–3634; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 3669–3673. [Google Scholar]
- 12.For selected reviews:
- 12a. Yoshida H., ACS Catal. 2016, 6, 1799–1811; [Google Scholar]
- 12b. Chinchilla R., Nájera C., Chem. Rev. 2014, 114, 1783–1826; [DOI] [PubMed] [Google Scholar]
- 12c. Shimizu Y., Kanai M., Tetrahedron Lett. 2014, 55, 3727–3737; [Google Scholar]
- 12d. Besset T., Poisson T., Pannecoucke X., Eur. J. Org. Chem. 2015, 2765–2789. [Google Scholar]
- 13.
- 13a. Li Z., Garcia-Dominguez A., Nevado C., Angew. Chem. Int. Ed. 2016, 55, 6938–6941; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 7052–7055; [Google Scholar]
- 13b. Li Z., Garcia-Dominguez A., Nevado C., J. Am. Chem. Soc. 2015, 137, 11610–11613. [DOI] [PubMed] [Google Scholar]
- 14. Li W., Yu S., Cai J., Zhao Y., Angew. Chem. Int. Ed. 2020, 59, 14404–14408; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 14510–14514. [Google Scholar]
- 15. Morozov O. S., Asachenko A. F., Antonov D. V., Kochurov V. S., Paraschuk D. Y., Nechaev M. S., Adv. Synth. Catal. 2014, 356, 2671–2678. [Google Scholar]
- 16.
- 16a. Nussbaumer P., Leitner I., Marz K., Stuetz A., J. Med. Chem. 1995, 38, 1831–1836; [DOI] [PubMed] [Google Scholar]
- 16b. Saito S., Yamamoto Y., Chem. Rev. 2000, 100, 2901–2916; [DOI] [PubMed] [Google Scholar]
- 16c. Liu Y., Nishiura M., Wang Y., Hou Z., J. Am. Chem. Soc. 2006, 128, 5592–5593; [DOI] [PubMed] [Google Scholar]
- 16d. Chen G., Wang L., Thompson D. W., Zhao Y., Org. Lett. 2008, 10, 657–660. [DOI] [PubMed] [Google Scholar]
- 17. Cheung C. W., Zhurkin F. E., Hu X., J. Am. Chem. Soc. 2015, 137, 4932–4935. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary