

Abstract
Peripheral nerve section with subsequent disconnection of motor neuron (MN) cell bodies from their skeletal muscle targets leads to a rapid reactive response involving the recruitment and activation of microglia. In addition, the loss of afferent synapses on MNs occurs in concomitance with microglial reaction by a process described as synaptic stripping. However, the way in which postaxotomy‐activated microglia adjacent to MNs are involved in synaptic removal is less defined. Here, we used confocal and electron microscopy to examine interactions between recruited microglial cells and presynaptic terminals in axotomized MNs between 1 and 15 days after sciatic nerve transection in mice. We did not observe any bulk engulfment of synaptic boutons by microglia. Instead, microglial cells internalized small membranous‐vesicular fragments which originated from the acute disruption of synaptic terminals involving the activation of the necroptotic pathway. The presence of abundant extracellular vesicles in the perineuronal space after axotomy, together with the increased expression of phospho‐mixed lineage kinase domain‐like protein and, later, of extracellular vesicle markers, such as CD9, CD63, and flotillin, indicate that the vesicles mainly originated in synapses and were transferred to microglia. The upregulation of Rab7 and Rab10 in microglia interacting with injured MNs, indicated the activation of endocytosis. As activated microglia and synaptic boutons displayed positive C1q immunoreactivity, a complement‐mediated opsonization may also contribute to microglial‐mediated synaptic disruption. In addition to the relevance of our data in the context of neuroinflammation and MN disease, they should also be taken into account for understanding functional recovery after peripheral nerve injury.
Keywords: afferent synapses; extracellular vesicles, exosomes; microglia; motor neuron; nerve axotomy; necroptosis
Main Points
Early after axotomy, microglia recruited near injured motor neurons, emit processes that tend to contact their afferent synaptic terminals.
Extracellular vesicles resulting from necroptotic synaptic disruption are removed by microglia.
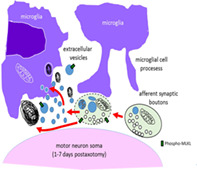
1. INTRODUCTION
The disconnection of motor neuron (MN) cell bodies from their skeletal muscle targets, as occurs after peripheral nerve injury, leads to rapid and profound reactive responses in the affected regions of the spinal cord. These reactive changes involve not only the severed neuronal cell bodies, but also their neighboring astroglial and microglial cells. In addition, the distal stump of injured peripheral nerve undergoes progressive disintegration in the context of Wallerian degeneration (Coleman & Freeman, 2010; Conforti, Gilley, & Coleman, 2014); conversely, the proximal stump regenerates as a consequence of the intrinsic growth capacity of the injured MN and of locally derived growth promoting factors (Chen, Yu, & Strickland, 2007). It seems that changes in retrograde signaling along with proximal axonal events inform the cell body of the distally located injury in order to activate a complex cellular response, which is eventually directed either to regeneration or, in certain cases, to cell death (Pollin, McHanwell, & Slater, 1991; Rishal & Fainzilber, 2014). The conspicuous structural changes that MN cell bodies suffer after axonal interruption are classically described within the concept of chromatolysis: a retrograde response mainly focused on alterations in the endoplasmic reticulum (ER) and in other organelles (Lieberman, 1971). Nevertheless, peripheral nerve transection also affects the stability of synaptic inputs on the corresponding MN cell bodies (Alvarez et al., 2020; Brannstrom & Kellerth, 1998; Sumner, 1975; Sumner & Sutherland, 1973). Pioneering studies by Blinzinger and Kreutzberg (1968) have shown that the loss of the afferent synaptic boutons on MNs following axotomy is mediated by recruited perineuronal microglial cells, leading to the introduction of the synaptic stripping concept. This notion is usually referred to as the glial‐mediated detachment of presynaptic terminals from the cell bodies or dendrites of axotomized MNs, and has been extended to other damaged CNS neurons (Kettenmann, Kirchhoff, & Verkhratsky, 2013). This process has been considered as favorable for the survival and functional recovery of injured neurons and, thereby, microglia should act in a protective manner (Cullheim & Thams, 2007; Kettenmann et al., 2013). In the seminal article on this subject (Blinzinger & Kreutzberg, 1968), it is stated that, in axotomized facial MNs, microglia‐mediated synaptic removal occurs in the absence of presynaptic bouton degeneration or phagocytosis. Conversely, microglia‐mediated phagocytosis of synaptic elements has been frequently reported within the context of both the postnatal remodeling of synapses and under a variety of pathologic conditions (Cullheim & Thams, 2007; Tremblay, Lowery, & Majewska, 2010). It has been suggested that, during development, microglia engulf and phagocyte synaptic boutons in order to eliminate supernumerary synapses by a mechanism involving complement (Paolicelli et al., 2011; Schafer et al., 2012; Sipe et al., 2016). However, the direct investigation of the way by which microglia ingest and destroy synaptic terminals has been barely addressed. This has been examined in detail by correlative advanced microscopical techniques in the hippocampus during developmental synaptic remodeling (Weinhard et al., 2018); although, interestingly, these authors have not reported any evidence for phagocytosis of dendritic spines. Instead, synaptic pruning occurs by selective partial phagocytosis of synaptic structures. The term trogocytosis, a name borrowed from the immune system process in which cells ingest small parts of their targets, was adopted to describe this process (Dopfer, Minguet, & Schamel, 2011).
Although the elimination of synaptic inputs in axotomized MNs and its association with microglial cell activation have been widely described for some time, the mechanism by which the afferent synaptic terminals disappear from the surface of injured MNs needs to be reexamined in depth. In a recent study (Salvany et al., 2019), we have reported that the degeneration of synaptic inputs on axotomized MNs occurs in a close relationship with processes of recruited microglia. This involves bouton terminal disruption and fragmentation into vesicular structures that seems to be “ingested” by microglia. It was suggested that this process may be comparable to that described as synaptic trogocytosis.
Here, by using ultrastructural and confocal microscope imaging, we revisited this issue by performing a more detailed examination of changes in perineuronal afferent inputs and glial cells in injured spinal cord MNs following sciatic nerve transection. We provide new data concerning the way in which synapses disintegrate during the local sterile neuroinflammation induced by a distal axonal lesion. It is known that peripheral nerve injury entails permanent changes within the spinal cord and brain circuitry that makes incomplete the functional restoration and clinical recovery (Alvarez et al., 2011; Delgado‐Garcia, Del Pozo, Spencer, & Baker, 1988; Lundborg, 2003; Navarro, Vivo, & Valero‐Cabre, 2007). For this reason, a more precise knowledge of the biological process involved in the loss and remodeling of afferent MN synaptic boutons after traumatic nerve lesions will provide new understanding and help to improve therapeutic interventions.
2. MATERIALS AND METHODS
2.1. Animals, surgical procedures, and tissue preparation
All the experiments were performed using adult CD1 mice obtained from Envigo (East Millstone, NJ). Mice were housed five to six per cage with ad libitum access to food and water under a 12 hr light/dark cycle. All animal experimentation procedures were performed according to the European Committee Council Directive and the norms established by the Generalitat de Catalunya (published as law in the Diari Oficial de la Generalitat de Catalunya 2073, 1995). All experiments were previously evaluated and approved by the Committee for Animal Care and Use of our university.
Adult (postnatal day [P] 60–90) mice were subjected to unilateral sciatic nerve transection. Animals were anaesthetized using a solution consisting of a combination of ketamine (100 mg/kg) and xylazine (10 mg/kg). The sciatic nerve was exposed at the femoral level and transected; a ligature was performed in the proximal segment in order to prevent spontaneous reinnervation. To minimize suffering, mice were subjected to postoperative analgesia with two subcutaneous injections of buprenorphine (0.05 mg/kg): one immediately after surgery and the other 24 hr after the intervention. Lumbar spinal cord samples were obtained 1, 3, 7, and 15 days after axotomy.
The colony stimulating factor 1 receptor (CSF‐1R) specific kinase inhibitor (PLX5622, Elmore et al., 2014) was generously provided by Plexxikon Inc. (Berkeley, CA), and formulated in AIN‐76A standard chow by Research Diets Inc. (New Brunswick, NJ). Two‐month‐old male CD1 mice, specific‐pathogen‐free housed, were treated with either vehicle or 1,200 mg/kg chow PLX5622 for 7 days. After that time, the mice had their sciatic nerves surgically intervened according to the procedure described above. The mice were then treated with the same dose of chow PLX5622 for a further 7 days. To verify PLX5622 consumption by the mice, their weight was monitored; no significant weight reduction was observed. To assess the effectiveness of PLX5622 in depleting microglial cells, the number of Iba1‐positive glial cells was assessed in serially sectioned brain striatal area. The treatment with PLX5622 resulted in a ~45% reduction in brain‐resident microglial cells evaluated after Iba1 immunostaining.
Mice were anesthetized and transcardially perfused with 4% PFA in 0.1 M phosphate buffer (PB) pH 7.4. Samples were postfixed for 24 hr in the same fixative, at 4°C, and then cryoprotected at 4°C with 30% sucrose in 0.1 M PB containing 0.02% sodium azide. Transverse cryostat sections (16 mm thick) were collected on gelatin‐coated glass slides.
2.2. Multiple fluorescent labeling and confocal microscopy
Cryostat sections were permeabilized with PBS containing 0.1% Triton X‐100 for 30 min, blocked with either 10% normal goat serum or normal horse serum in PBS for 1 hr at room temperature, and then incubated overnight at 4°C with an appropriate primary antibody mixture. The primary antibodies used are listed in Table 1.
TABLE 1.
Primary antibodies used for immunocytochemistry
| Target | Source | Host species | Used concentration |
|---|---|---|---|
| C1q | Abcam (ab182451) | Rabbit monoclonal | 1:1,000 |
| CD9 | Abcam (ab92726) | Rabbit monoclonal | 1:350 |
| CD11c | AbDserotec (MCA1369T) | Hamster monoclonal | 1:50 |
| CD63 | Abcam (ab217345) | Rabbit monoclonal | 1:100 |
| CD68 | AbDserotec (MCA1957T) | Rat monoclonal | 1:100 |
| Flotillin | Abcam (ab41927) | Rabbit polyclonal | 1:150 |
| Ionized calcium‐binding adaptor molecule 1 (IBA1) | Abcam (ab5076) | Goat polyclonal | 1:500 |
| Mac2 | Cerdalane (CL8942AP) | Rat monoclonal | 1:800 |
| MLKL (phospho S345) | Abcam (ab196436) | Rabbit monoclonal | 1:100 |
| PDC6IP‐Alix | Sigma‐Aldrich (HPA011905) | Rabbit polyclonal | 1:50 |
| Rab4 | Abcam (ab109009) | Rabbit monoclonal | 1:170 |
| Rab5 | Abcam (ab218624) | Rabbit monoclonal | 1:1,000 |
| Rab7 | Abcam (ab137029) | Rabbit monoclonal | 1:100 |
| Rab8A | Abcam (188574) | Rabbit monoclonal | 1:500 |
| Rab9 | Abcam (ab2810) | Mouse monoclonal | 1:200 |
| Rab10 | Abcam (ab237703) | Rabbit monoclonal | 1:500 |
| SV2 | Developmental Studies Hybridoma Bank | Mouse monoclonal | 1:1,000 |
| Synaptophysin | Synaptic Systems (101004) | Guinea pig polyclonal | 1/500 |
| TMEM119 | Abcam (ab209064) | Rabbit polyclonal | 1:300 |
| VAChT | Synaptic Systems (139105) | Guinea pig polyclonal | 1:500 |
Abbreviation: VAChT, vesicular acetylcholine transporter.
Once previously washed with PBS, sections were incubated for 1 hr with a combination of appropriate secondary antibodies labeled with one of the following fluorochromes (1:500): Alexa Fluor 488, Alexa Fluor 546 (ThermoFisher Scientific, Waltham, MA), cyanine 3, or cyanine 5 (Jackson Immuno‐Research Laboratories, West Grove, PA). Finally, the spinal cord sections were labeled with blue fluorescent NeuroTrace Nissl staining (1:150; Thermo Fisher Scientific) and mounted using an antifading medium containing 0.1 M Tris–HCl buffer (pH 8.5), 20% glycerol, 10% Mowiol, and 0.1% 1,4‐diazabicyclo[2.2.2]octane.
The slides were then examined under a FluoView FV‐500 or FluoView FV‐1000 Olympus laser‐scanning confocal microscope (Olympus, Tokyo, Japan). The MNs were imaged after obtaining optical sections (0.5 or 1 mm) of cell bodies. Digital images were analyzed using FV10‐ASW 3.1 Viewer (Olympus) and the ImageJ (National Institutes of Health [NIH], Bethesda, MD) software.
Immunolabeled profiles of the different protein markers examined were then manually counted on a screen for each MN soma. In axotomy experiments, we only analyzed cell bodies located in the pes 9 region of the lumbar 6 spinal cord segment, which corresponds to the sciatic motor column (Watson, Paxinos, Kayalioglu, & Heise, 2009). The pool of axotomized MNs was identified by their close interaction with recruited Iba1‐stained microglial cells. The intensity of microgliosis was evaluated by analyzing the percentage of either MN perimeter covered by microglia or the neuropile area adjacent to MNs occupied by microglia. The expression of CD68 in microglia was measured as the percentage of the area of CD68‐positive puncta with respect to Iba1‐positve profiles. The complexity of microglial branching was calculated as the number of triple‐point branches after analysis of the skeleton using ImageJ software (AnalyzeSkeleton [2D/3D] from http://imageJ.net/AnalyzeSkeleton) in 3D projected images of iba1‐immunostained cells. The number of afferent synaptic boutons contacting MNs was evaluated on synaptophysin‐immunolabeled sections after tracing a line along the periphery of the cell soma and manually counting the number of pixel profile picks. The colocalization of presynaptic markers with the other proteins that were examined was evaluated after pixel profiling around a line traced at the periphery of the MNs using the ImageJ plugin developed by Pierre Bourdoncle (bourdoncle@ijm.jussieu.fr).
2.3. Electron microscopy
Animals were perfused with either 2% PFA and 2% glutaraldehyde in PB (for conventional electron microscopy [EM]) or 4% PFA and 0.1% glutaraldehyde in PB (for ultrastructural immunolabeling). Dissected tissues were postfixed for 24 hr, at 4°C, in the same fixative solution. The samples were sectioned at 200 μm using a vibratome and postfixed with 1% OsO4 for 2 hr, and then contrasted with 0.5% uranyl acetate for 30 min; all these procedures were conducted at 4°C. After that, the samples were processed for Embed 812 (Electron Microscopy Sciences, Hatfield, PA) according to standard procedures. Ultrathin sections were counterstained with Reynold's lead citrate. For Iba1 ultrastructural immunolabeling with the preembedding procedure, 50‐μm‐thick vibratome sections were blocked in a solution containing: 10% BSA and 0.02% saponin in PBS, for 1 hr, prior to incubation with goat polyclonal anti‐Iba1 antibody (diluted 1:500 in 10% BSA/PBS containing 0.004% saponin), at 4°C, for 2 days, and washed five times in 1% BSA/TBS (3 × 1 min and 2 × 10 min). The sections were then incubated for 2 hr with a secondary anti‐goat biotinylated antibody (1:100, Vector Laboratories) and visualized using Vectastain Elite ABC (Vector Laboratories), followed by incubation in a 0.05% 3,3‐diaminobenzidine /0.01% H2O2 mixture. In some cases, peroxidase reactive sites were subjected to silver enhancement by incubating the sections in a solution containing 2.6% hexamethylenetetramine (Merck, Darmstadt, Germany), 0.2% silver nitrate (Merck), and 0.2% disodium tetraborate (Merck) for 10 min, at 60°C. Sections were rinsed in distilled water and treated with 0.05% gold chloride for 2 min. Finally, the sections were rinsed and incubated in 2.5% sodium thiosulfate (Merck) for 2 min. Tissue was postfixed in 2% OsO4 and flat embedded in Embed 812.
For postembedding immunogold analysis, lumbar spinal cords were sectioned at 200 μm using a McIlwain Tissue Chopper (Mickle Laboratory Engineering, Gomshall, UK). After glycerol cryoprotection, samples were plunged rapidly into liquid propane (−184°C) cooled by liquid nitrogen and processed for freeze substitution, using a Leica EM system (Leica Microsystems, Wetzlar, Germany). Tissues were low temperature embedded in Lowicryl HM20 resin (Electron Microscopy Sciences), following a protocol similar to that described elsewhere (Rubio & Wenthold, 1999). Ultrathin sections were collected using an Ultracut UC6 ultramicrotome (Leica) and picked up on formvar‐coated nickel grids. They were washed in PBS and in 50 mM glycine, and then blocked in 5 and 1% BSA. Sections were incubated for 1 hr with anti‐phospho‐mixed lineage kinase domain‐like protein (MLKL; 1:10) at room temperature. After being rinsed in 0.25 Tween 20, they were blocked in 1% BSA and incubated in 12 nm gold‐conjugated goat anti‐rabbit IgG (1:30, Sigma‐Aldrich, St. Louis, MO), for 30 min at room temperature. After being washed in PBS and distilled water, sections were counterstained with uranyl acetate and lead citrate. All observations were performed on a transmission electron microscope JEOL JEM 1010 (Akishima, Tokyo, Japan).
2.4. Statistical analysis
The data were expressed as the mean ± SEM. The statistical analysis was assessed by a Student's t test, or by one‐ or two‐way analysis of variance followed by the post hoc Bonferroni's test. The level of significance was established at p ≤ .05. GraphPad Prism 6 software was used for statistical analysis and graph presentation of data.
3. RESULTS
3.1. Microglial recruitment to axotomized MNs
Twenty four hours after nerve transection, Iba1, a pan‐marker for both resting and activated microglia (Ito et al., 1998), revealed a noticeable accumulation of these cells in areas of the spinal cord surrounding injured MNs. Recruited microglia extended large filopodia, which tended to contact MN surfaces, and increased in number and complexity from 1 to 7 days after axotomy (Figure 1a–e,o–q). Moreover, microglial morphology evolved from an amoeboid‐like pattern, seen 1 day after axotomy, to a highly ramified form 7 days postaxotomy (Figure 1q). Double labeling for Iba1 and the lysosomal‐associated protein CD68, which is present in phagocytic cells (da Silva & Gordon, 1999), demonstrated that once recruited, microglial cells displayed significant phagocytic activity in very close proximity to axotomized MN cell bodies (Figure 1f,j). The number of CD68‐positive particles within the microglial cytoplasm was observed to be already increased at 1day postaxotomy and reached their maximum number 3 days postlesion, before a subsequent decline (Figure 1p). The reduction in the number of CD68 particles found at 7 and 15 days after surgery did not entail a decrease in microgliosis during this period; in fact, the opposite occurred: microglial mobilization was observed to increase until 7 days after lesion (Figure 1o). Microglia enwrapping axotomized MNs displayed positive TMEM119 immunostaining (Figure 1k–n), a specific marker of resident adult microglia, which is absent in peripheral macrophages (Bennett et al., 2016). Whereas recruited microglial cells did not express activation markers such as CD11c or Mac‐2 (not shown), they exhibited other features indicative of an inflammatory response. For example, these cells were coated by endogenous IgGs, probably as a consequence of the local disruption of the blood brain barrier, and of the increased expression of Fc receptors at the microglial membrane (Supplementary Figure 1a–c). This finding is in agreement with previous observations (Liu, Aldskogius, & Svensson, 1998). In addition, the component of the classic complement pathway C1q was also notably upregulated in the recruited perineuronal microglia as early as 24 hr after axotomy, with a further and transient increase at 7 days postlesion (Figure 2a–i). C1q mediates synapse elimination during normal development and the aberrant activation of this mechanism may account for synaptic destruction in neurodegenerative diseases (Hong et al., 2016; Schafer et al., 2012; Stevens et al., 2007). We therefore wondered whether unwanted synaptic boutons on axotomized MNs could also be tagged by complement before loss. C1q immunoreactivity was analyzed for its colocalization with the general presynaptic protein marker synaptophysin. At the peak of C1q immunoreactivity, 7 days postaxotomy, there was partial colocalization (Pearson's correlation coefficient = .28) between C1q and the synaptophysin signal at the surface of the MN cell bodies (Figure 2h,i).
FIGURE 1.
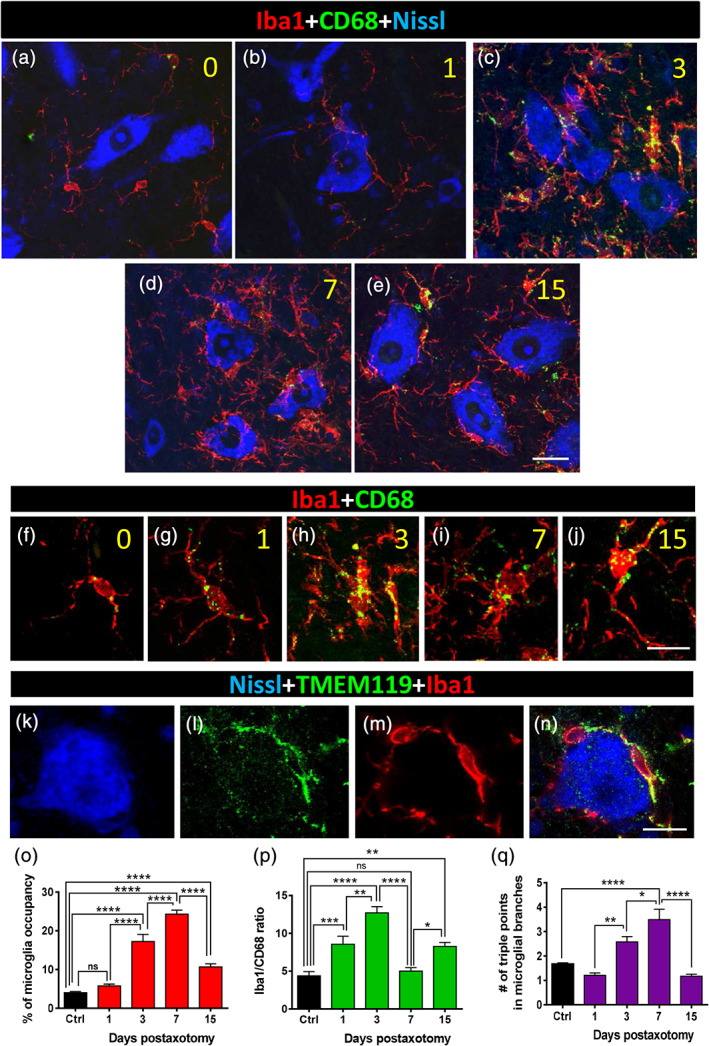
Microglial recruitment and activation around motor neuron (MN) somata after sciatic nerve axotomy. (a–e) Spinal cord sections displaying Nissl stained axotomized MN pools (blue) in combination with both Iba1 (red, as a general marker of microglia) and CD68 (green, for detection of lysosomes in activated microglia). Representative images taken on Days 0, 1, 3, 7, and 15 (numbers in yellow) after lesion are shown. (f–j) Details of size and distribution of CD68‐positive particles in individual Iba1‐positive microglial cells are shown after 0, 1, 3, 7, and 15 days postaxotomy (numbers in yellow). (k–n) In recruited microglial cells, Iba1‐immunolabeling (red) colocalizes with TMEM119 (green), as shown. (o–q) Quantification of MN surface occupancy by microglial cell processes, ratio of Iba1/CD68 occupied area (p), and complexity of microglial processes measured as a number of triple‐point branches (q) at different times after axotomy. Data in graph are shown as mean ± SEM, from 9 to 14 MNs (o,p), and 1,900–2,300 microglial branches (q), from nine mice, in projected Z‐stacks; *p < .05; **p < .01; ***p < .001; ****p < .0001, one‐way analysis of variance (ANOVA), Bonferroni's post hoc test. Scale bars: e = 20 μm (valid for a–d); j = 10 μm (valid for f–i); n = 10 μm (valid for k–m) [Color figure can be viewed at wileyonlinelibrary.com]
FIGURE 2.
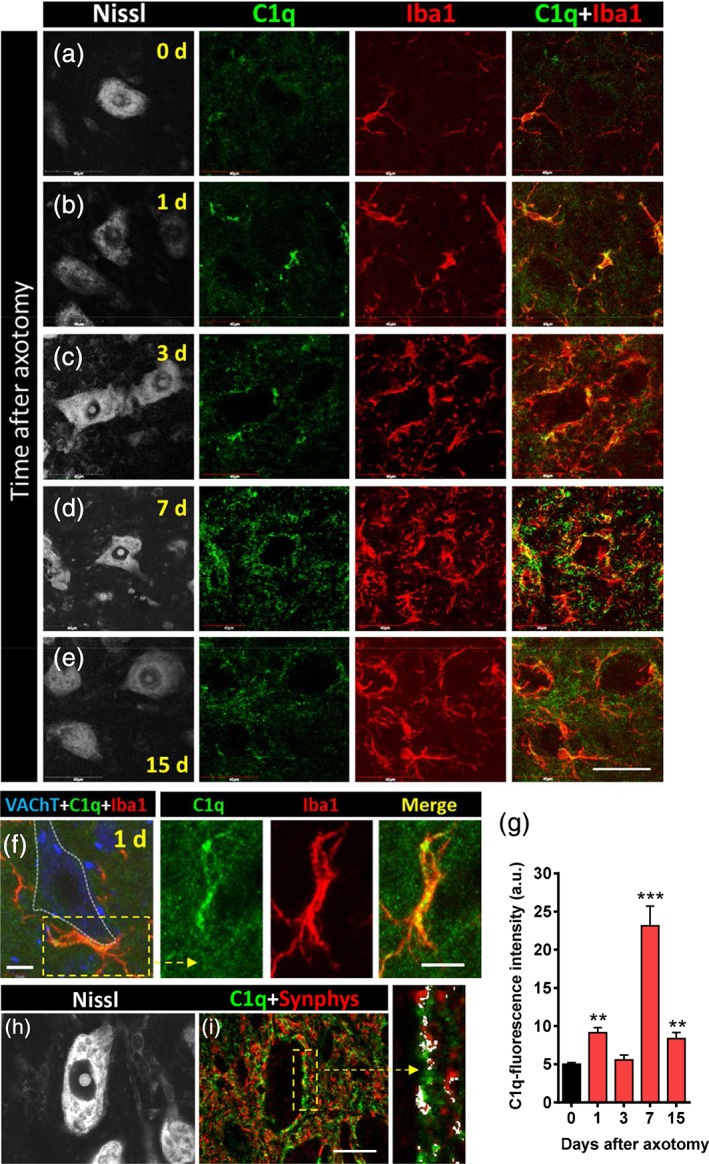
C1q expression is upregulated during microglial activation and recruitment that occurs in the vicinity of axotomized motor neurons (MNs). (a–e) Spinal cord MNs are visualized with fluorescent Nissl staining (gray) in conjunction with C1q‐ (green) and Iba1‐immunostaining (red) at different times after axotomy, as indicated (d = days). (f) Detail of recruited microglial cells adjacent to 1‐day‐axotomized MN soma (delimited by a dotted line); the boxed area is shown at a higher magnification in split and merged green and red channels (VAChT is shown in blue). (g) Quantification of the intensity of C1q fluorescence in Iba1‐positive profiles; data are shown as mean ± SEM; **p < .01, and ***p < .001 versus 0 days (control; Student's t test; n = 4–7 mice per condition). (h,i) C1q‐immunoreactivity (green) partially colocalizes with afferent synaptophysin‐immunostained synaptic boutons (red, Synphys) surrounding a 7‐day‐axotomized MN (visualized by Nissl staining, gray); the colocalized pixels are depicted in white in the enlarged panel. Scale bars: e = 40 μm (valid for a–d); (f) and enlarged boxed area = 10 μm; i = 20 μm (valid for h) [Color figure can be viewed at wileyonlinelibrary.com]
3.2. Loss of synaptic inputs on axotomized MNs
The loss of afferent synapses on MNs, induced by peripheral axotomy, was assessed after quantification of presynaptic synaptophysin puncta on the periphery of lesioned MN somas (Supplementary Figure 2a–d). The number of synaptophysin‐positive spots per 100 μm of MN perimeter was: control = 162.4 ± 2.74, and 7 days postaxotomy = 87.03 ± 2.2 (mean ± SEM, n = 85 and 109 measurements, respectively, from three animals; p < .0001). This agreed with the reported data which refer to the loss of about 50% of presynaptic terminals during the first week postaxotomy (Oliveira et al., 2004). We next evaluated the impact of the pharmacologically induced reduction of the microglial cell population on MN synaptic loss after nerve transection. To do this, we orally administered the selective CSF1R inhibitor PLX5622 (Elmore et al., 2014). The recruitment of microglia on the periphery of MN somata at 7 days postaxotomy was also substantially reduced by PLX5622 (percentage of microglial occupancy: axotomy = 25.98 ± 1.03 [n = 43], and axotomy + PLX5622 = 10.53 ± 0.61 [n = 22]; p < .0001; from six control and four PLX5622‐treated animals). In parallel, the effects of PLX5622 treatment on MN synapse loss were quantified after synaptophysin immunostaining, resulting in a moderate (~10%) increase in the number of synapses that remained after 7 days of peripheral nerve injury. The number of synaptophysin positive spots per 100 μm of MN perimeter was: axotomy = 87.03 ± 2.2 (n = 109), and axotomy + PLX5622 = 95.57 (n = 173), from three animals (p < .001).
We next analyzed the organization of synaptic terminals contacting the surface of normal and injured (axotomized) MN cell bodies of adult mouse by transmission EM. In intact MNs, almost the entire surface of the cell bodies and their proximal dendrites was covered by densely packed synaptic terminals and cellular processes separated by a very narrow cleft (~20 nm wide) of extracellular space (Figure 3a). Applying the classical morphological criteria previously established (Bodian, 1975; Conradi, 1969), it was possible to recognize the S, F, C, and M types of afferent bouton (Supplementary Figure 3a–e). This arrangement was noticeably altered as a consequence of peripheral nerve transection.
FIGURE 3.
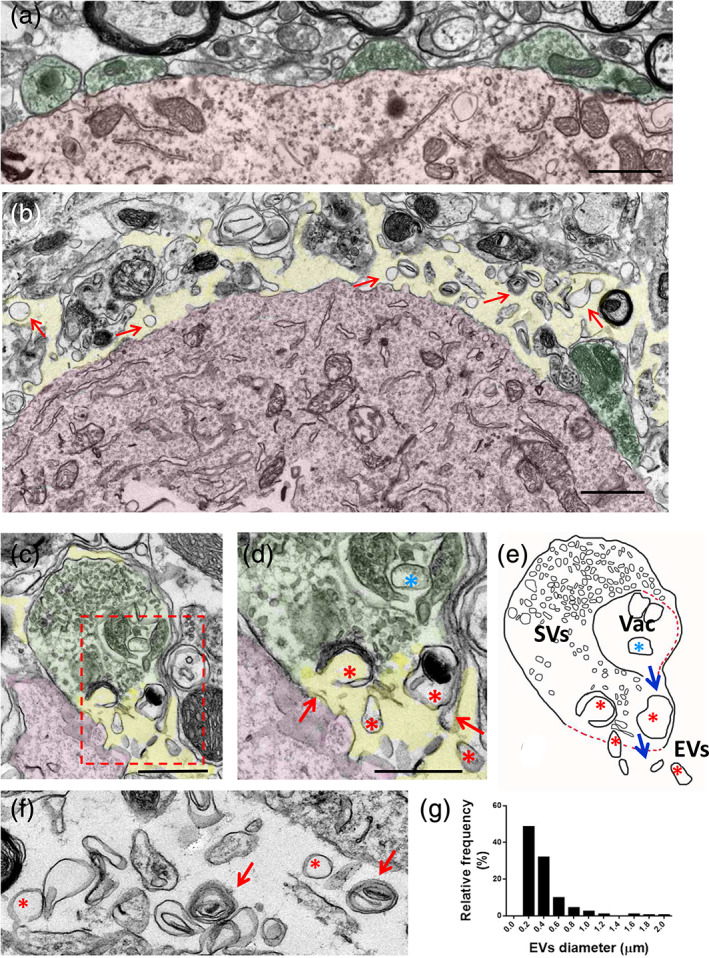
Ultrastructural analysis of the periphery of motor neuron (MN) cell bodies, adjacent synaptic afferents and cell structures in the spinal cord. (a) Under noninjured conditions, MN somata (shaded in red) are densely covered by terminal afferent synaptic boutons (shaded in green), with a narrow extracellular space. (b) Twenty‐four hours after axotomy, the extracellular space (shaded in yellow) is widened and synaptic terminals (green) are markedly detached from the MN surface (red); in addition, a number of extracellular membrane‐bound vesicles (arrows) can be seen “floating” in the extracellular space. (c) A nerve terminal afferent (green) contacting an MN dendrite (red) displaying a rupture of the presynaptic membrane and release of vesicular elements to the extracellular space (yellow), 24 hr after axotomy; the area delimited by the dashed rectangle is shown enlarged in (d), in which the points of rupture of presynaptic membrane are indicated with arrows; vesicles captured during their release to the extracellular space can be seen (red *); blue * indicates a large vesicle still remaining intracellularly. (e) A drawing based on image shown in (c), in which the process of rupture and release of extracellular vesicles (EVs) is more clearly depicted (SVs, synaptic vesicles; Vac, presynaptic vacuolar formation; dashed red lines indicate the sites of membrane disruption). (f) A detail of extracellular vesicles which are accumulated near MN surface 24 hr after axotomy, showing their pleomorphic appearance ranging from unilamellar (*) to multilamellar (arrow) organization. (g) Frequency distribution histogram of diameter of EVs that were accumulated at the perineuronal space adjacent to MN surface (n = 203 EVs), 24 hr after axotomy. Scale bars: (a) and (b) = 1 μm; (c), (d), and (f) = 500 nm [Color figure can be viewed at wileyonlinelibrary.com]
In contrast to the normal organization of synaptic afferents contacting MN surface, early (1–3 days) after lesion, a conspicuous detachment of the presynaptic membrane from its postsynaptic counterpart was observed in many nerve terminals (Figure 3a,b and Supplementary Figure 4a–f). Additionally, the narrow extracellular space seen in normal CNS was found to be highly enlarged. When the presynaptic compartment was partially detached, axosomatic terminals maintained some focal adherence to the MN surface at the puncta adherentia sites, which are considered to be purely adhesive structures (Peters, Palay, & Webster, 1976) (Supplementary Figure 4b). Many synaptic terminals, including C‐boutons, displayed noticeable changes in the distribution and size of their synaptic vesicles and related vacuolar structures. An increased accumulation of large endocytic‐like, or autophagosomal‐like, vesicles was found, irrespective of the synapse subtypes, at all the time‐points examined (1–15 days postlesion).
Due to the synapse detachment and loss, large areas of MN plasma membrane were devoid of presynaptic terminals. The widened extracellular space surrounding MN cell bodies contained a considerable amount of free vesicular‐like particles (Figure 3b and Supplementary Figure 4a–f). The accumulation of extracellular vesicles (EVs) was particularly prominent near MN surfaces denuded of synaptic terminals, suggesting that they resulted from the disruption of synaptic boutons. This was confirmed by examining the ultrastructural changes in synaptic boutons at early stages of the disruption (24 hr after nerve injury). At this time‐point, some afferent boutons undergoing cellular membrane rupture leading to the release of intracellular vesicular components to the extracellular space were observed (Figure 3c–e). The EVs displayed a marked variability in size, ranging from 100 to 2,000 nm (Figure 3g), and a great diversity of shapes. However, they were usually circular in appearance and exhibited either unilamellar or multilamellar organization (Figure 3f and Supplementary Figure 4d–f). As expected, recruited microglial cells were seen adjacent to injured MNs, and EVs were also abundant near the surface of some of these glial cells (Supplementary Figure 4c). Overall, these alterations affected S and F synaptic types at a similar degree, although C‐boutons appeared to be reluctant to the acute disruption.
Many remaining synaptic terminals displayed remarkable changes in the distribution and size of their vesicles and endocytic‐autophagosomal like vacuolar structures. This was found at all time‐points examined (1–15 days postlesion) and irrespective of the synapse subtypes. During microglial recruitment, some Iba1‐labeled microglial end‐foot profiles contacted still well preserved, but partially detached, presynaptic boutons (Figure 4a–i); in this case, the nerve terminals usually exhibited an abnormal accumulation of vacuoles regionally segregated from the areas in where the synaptic vesicles accumulated (Figure 4h,i). Other microglial profiles appeared adjacent to extracellular multilamellar bodies. Many structurally preserved nerve terminals, which withstand the acute (1–3 days) lytic disruption, exhibited an abnormal accumulation of multivesicular bodies (MVBs), and endocytic and double‐membrane bounded autophagosomal‐like structures. This suggests that an imbalance in the turnover of synaptic vesicles occurs prior to full synaptic degeneration seen at more advanced stages postlesion). Figure 5a–c, shows a striking example of Iba1‐labeled microglia, taken 7 days after axotomy, exhibiting a continuous sequence of early interaction, and posterior engulfment and internalization of EVs near the surface of an axotomized MN. The panels in Figure 5d–h show vacuoles of identical morphology in different locations, including inside synaptic terminals, in the extracellular space and in double‐membrane bounded intracellular microglial inclusions. This strongly suggests a sequential trafficking process from synapses to microglia by means of extracellular intermediaries. Several microglial processes, seen at 7 days postaxotomy, and extensive accumulations of this type of vesicle are depicted in Figure 5i.
FIGURE 4.
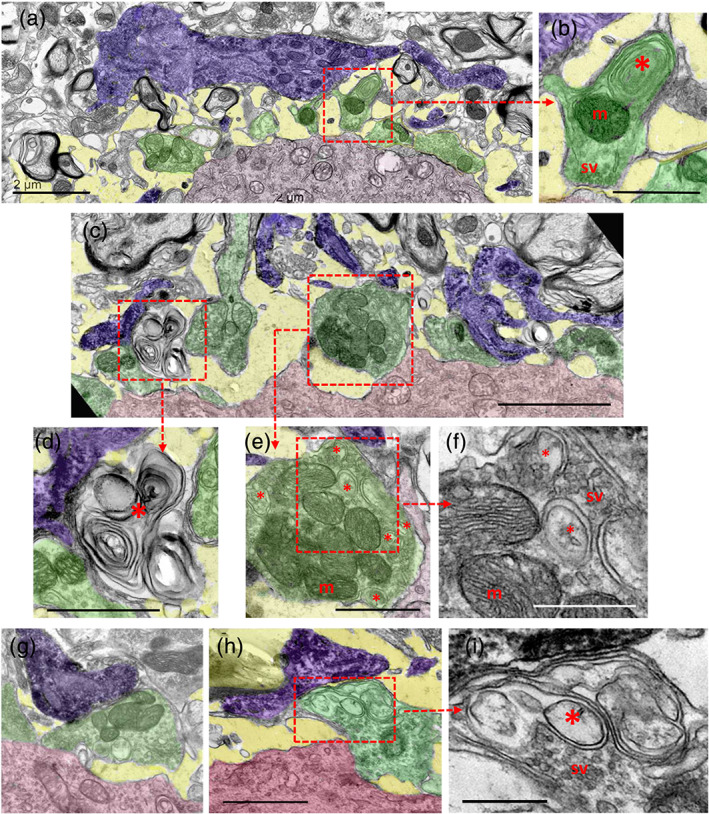
Ultrastructural analysis of the interactions of Iba1‐immunostained microglial profiles (shaded in blue) with synaptic afferents (shaded in green) on motor neuron (MN) cell bodies (shaded in red) 7 days postaxotomy. (a) Microglial processes in close proximity to altered synaptic boutons and a widened extracellular space (shaded in yellow) are depicted. (b) A higher magnification detail of a synaptic bouton (delimited by a dotted square) in (a) containing clustered synaptic vesicles in the active zones (sv), normal mitochondria (m) and abnormal concentrically arranged membranes of presumably endocytic origin (*). (c) Microglial cell processes (blue) interacting with altered and disrupted presynaptic boutons (green) on a 7‐day‐axotomized MN (red). (d) A higher magnification view of the corresponding area delimited in (c) showing a complex multilamellar body located near the MN surface and in close contact with a microglial process, presumably derived from a disrupted presynaptic bouton. (e) A detail of a partially detached presynaptic terminal contacted by microglial processes (which is delimited in (c)), showing abnormal accumulation of endocytic‐like vesicles (*), enlarged in (f); m = mitochondria. (g,h) Microglial processes (blue) contacting an altered afferent synaptic bouton (green) at the surface of MN cell bodies (red). (i) A detail of the area delimited in (h) showing an abnormal accumulation of membranous structures (*) in the region in which the synaptic terminal is closest to the microglial end‐foot process; note the normal clustering of synaptic vesicles in the zone contacting the postsynaptic MN. Scale bars: a = 2 μm, b = 1 μm, c = 2.5 μm, d and e = 1 μm, f = 500 μm, g = 1 μm (valid for h), and i = 500 nm [Color figure can be viewed at wileyonlinelibrary.com]
FIGURE 5.
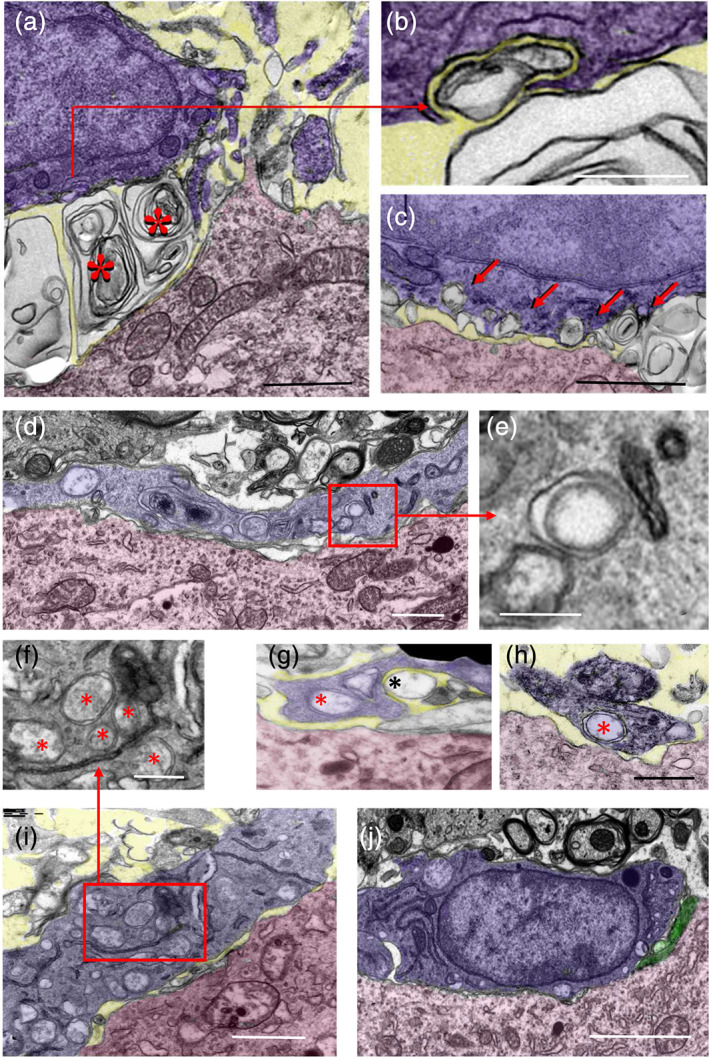
Ultrastructural analysis of the interactions of microglial cell bodies (shaded in blue) with motor neuron (MN) cell bodies (shaded in red) 7 days after axotomy. (a) Extracellular multilamellar bodies and vesicles (*) are seen interposed between an Iba1‐immunostained microglial cell and an axotomized MN cell body; note that extracellular membranous structures appear in sites that would normally be occupied by presynaptic boutons on an uninjured MN soma. This suggests that the extracellular material came from disrupted synaptic terminals. (b,c) Extracellular vesicles interact with microglial cell surfaces and undergo a process of enwrapping (b, [a high magnification detail of the indicated region in a]) and engulfment; in (c) there is a sequence of early contacts leading to a final engulfment (shown by the arrows). (d) A microglial cell profile (blue) covering the surface of an axotomized MN cell body (red), which appears to be completely denuded of synaptic terminals; note the presence of abundant double‐membrane endocytic/phagocytic inclusions inside the microglial cytoplasm (detailed in e). (g,h) An example of a microglial process (blue) close to an axotomized MN cell body (red); microglia interact with a single‐membrane‐bounded extracellular vesicle (black *) and contain a double‐membrane vesicular inclusion (red *), presumably resulting from extracellular vesicle endocytosis (g). The microglial nature of the processes containing a double‐membrane bounded inclusion (red *) is demonstrated by Iba1 ultrastructural immunolabeling (h). (i) A microglial cell process (blue) interacting with a surface of an axotomized MN cell body (red), which is depleted of afferent synaptic terminals; note the abundance of double membrane‐bounded inclusions (enlarged in (f)), presumably resulting from phagocytosis of EVs. (j) A microglial cell (blue) covering large areas of an axotomized MN surface (red) depleted of synaptic afferents (only a few of them remain, shaded in green); the microglial cell displays few phagocytic inclusions and a cytoplasmic organization suggestive of a less activated state. The extracellular space found in some panels is shaded in yellow. Scale bars: (a, c, d, and i = 1 μm; b) 250 nm; (e–h) = 500 nm; and (j) = 2.5 μm [Color figure can be viewed at wileyonlinelibrary.com]
The structural alterations in the postsynaptic regions facing damaged presynaptic terminals were more difficult to assess and to segregate from the broad cytoplasmatic organelle changes characteristic of chromatolytic neurons. However, in dendritic shafts, which are more restricted cytoplasmic regions adjacent to presynaptic terminals, an abnormal accumulation of vacuolar and phagosome‐like structures was observed. This was sometimes found in close relation to altered axodendritic boutons and microglial processes.
At 7 days after axotomy, the MN surface was notably depleted of synaptic boutons, and the surrounding extracellular space was largely expanded. Additionally, microglial cells recruited to the vicinity of MNs, emitted processes that contacted to some of the remaining axosomatic synaptic boutons or to the synaptically denuded MN surface (Figure 5j). EVs were also present in the perineuronal space, but at a lower density than that observed at 1–3 days postaxotomy. Many of the remaining axosomatic terminal boutons displayed a variety of involutive changes. These included: global darkening, the clumping of synaptic vesicles, and the accumulation of endosomal‐ or autophagosomal‐like vacuoles, as well as MVBs containing distinctive intraluminal vesicles.
Moreover, at 15 days after axotomy, recruited microglial cells were often seen covering extensive areas of the surface of axotomized MN cell bodies. These areas were completely devoid of synaptic terminals and displayed a narrow gap between the neuronal and microglial cell membranes, similarly to that occasionally seen at 7 days postaxotomy (Figure 5j).
It is known that axonal interruption in adult rats does not entail MN death, as occurs in young rats. In contrast, in adult mice, there is a slow loss of axotomized MNs in association with exacerbated microgliosis (Kiryu‐Seo, Gamo, Tachibana, Tanaka, & Kiyama, 2006; Yamada, Nakanishi, & Jinno, 2011). In concordance with these observations, we occasionally found some MNs that displayed a completely disrupted ultrastructural organization (Supplementary Figure 5a,b). These dying neurons were usually spatially related with altered microglial cells showing extensive vacuolation, and distended ER and perinuclear spaces, which are signs of cellular stress (Bisht et al., 2016).
At 15 days postlesion, axotomized cell bodies displayed hyperconvoluted nuclei (Figure 6a) and a reduced number of afferent synaptic terminals exhibiting a variety of structural alterations: detachment from the postsynaptic membrane, and a conspicuous accumulation of endosome and autophagosome‐lysosome‐like inclusions. Some terminals appeared to be clearly degenerated, appearing dark in color and with clumped synaptic vesicles (Figure 6b–i).
FIGURE 6.
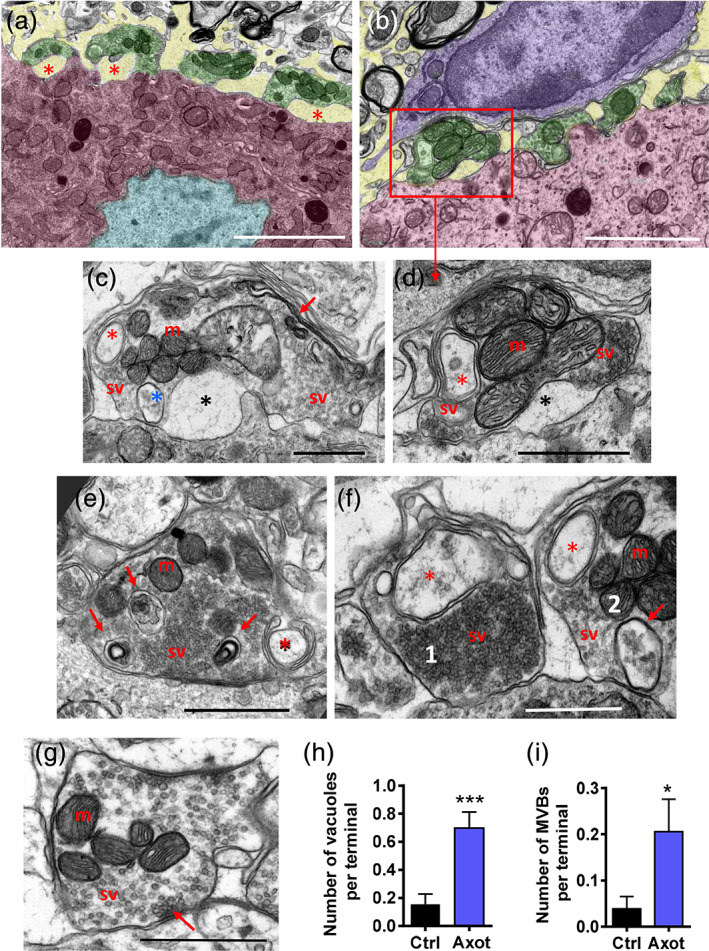
Changes in the organization of afferent synaptic terminals on motor neuron (MN) surfaces at 15 days postaxotomy. (a) An MN cell body displaying a wrinkled nucleus (light blue) and several detached synaptic terminals (green) with a widened extracellular space (yellow). (b) Another area of an axotomized MN (red) covered by altered synaptic boutons (green, detailed in d) interacting with a recruited microglial cell (blue); note that the microglial cell cytoplasm is devoid of endocytic/phagocytic inclusions, which is suggestive of a resting state. (c–f) Details of structural alterations observed in afferent synaptic terminals on a 15‐day‐axotomized MN; partial detachments of presynaptic and postsynaptic membranes (black asterisks), synaptic vesicles (sv) and mitochondria (m). Double‐membrane encircled‐endocytic‐like structures are indicated by red asterisks. A multivesicular body (blue, *) is shown in (c); the accumulation of highly folded intracellular membranes (c, arrows). (e) An accumulation of phagosome/lysosome inclusions (arrows). (f) An example of abnormally clustered and densely packed synaptic vesicles (sv) is depicted in terminal 1, in conjunction with an endocytic‐like vacuole; compare with the less clustered arrangement of synaptic vesicles (sv) in terminal 2, which also shows a multivesicular body (arrow), a double‐membrane containing endocytic‐like vesicles (*), and several mitochondria (m). (g) A control afferent synapse containing round synaptic vesicles (sv) clustered at the active zone (arrow); m = mitochondria. (h,i) Quantification of the number of endocytic/autophagosome‐like vacuoles (h) and multivesicular bodies (MVBs, i) in synaptic terminals contacting 15‐day‐axotomized MN bodies. Data are presented as mean ± SEM; *p < .05, and ***p < .001 versus Ctrl, Student's t test; n = 52–73 terminals from two animals per condition. Scale bars: a = 5 μm, b = 2 μm, and c–g = 1 μm [Color figure can be viewed at wileyonlinelibrary.com]
3.3. Activation of the necroptotic pathway is involved in synaptic disruption on axotomized MNs
Necroptosis is a regulated form of caspase‐independent cell death that is mediated by receptor‐interacting protein kinase 3 (RIPK3) and MLKL. In this type of programmed cell death, a rupture of cellular membranes occurs leading to the release of intracellular components; phosphorylation of MLKL by RIPK3 and its targeting to cell membranes is directly involved in the disruption of membrane integrity (Gong, Guy, Crawford, & Green, 2017; Grootjans, Vanden Berghe, & Vandenabeele, 2017; Wang et al., 2014). Broken plasma membrane may determine the formation of “bubbles” at the cell surface and the secretion of necroptotic EVs (Gong, Guy, Olauson, et al., 2017; Raden, Shlomovitz, & Gerlic, 2020). Interestingly, all these events fit very well with the ultrastructural observations we made during early stages (1–3 days) of afferent synapse acute disruption in axotomized MNs. For this reason, we explored, by immunolocalization of phospho‐MLKL (Figure 7a–l), whether the activation of the necroptotic pathway is involved in central synapse loss on injured MNs. In fact, a monoclonal antibody against phospho‐MLKL has been used as specific immunocytochemical marker for necroptosis in human diseased tissue (Wang et al., 2014). Using the same antibody, a positive phospho‐MLKL immunoreactivity was detected near the surface of axotomized MNs early (24 hr) after peripheral nerve injury; this was not seen in control noninjured MNs. The phospho‐MLKL signal was noticed in form of small puncta clustered at discrete regions of MN surface and closely related to processes of reactive microglia (Figure 7a–l). When individual sections of confocal z‐stacks were analyzed in detail, it was noticed that many phospho‐MLKL positive puncta were located in the narrow gap interposed between microglial and neuronal cells. Synaptophysin‐positive puncta, which normally surround MN cell bodies, were depleted at points in which phospho‐MLKL‐positive particles accumulated (Figure 7m–o). This is consistent with the assumption that phospho‐MLKL‐positive particles are derived from disrupted synaptic terminals. This was further corroborated by in situ localization of phospho‐MLKL by means of postembedding immunogold EM. A selective labeling of vacuoles in presynaptic bouton contacting MNs and in EVs located near MN surfaces was noticed (Figure 7p–q). Negligible labeling was observed in control sections after omitting the primary antibody. A positive immunofluorescence signal was also detected in the nuclei of neuronal cells both in control and axotomized conditions; although it is known that MLKL can translocate to the nucleus during necroptosis (Yoon, Bogdanov, Kovalenko, & Wallach, 2016, this aspect, was not further explored in our context.
FIGURE 7.
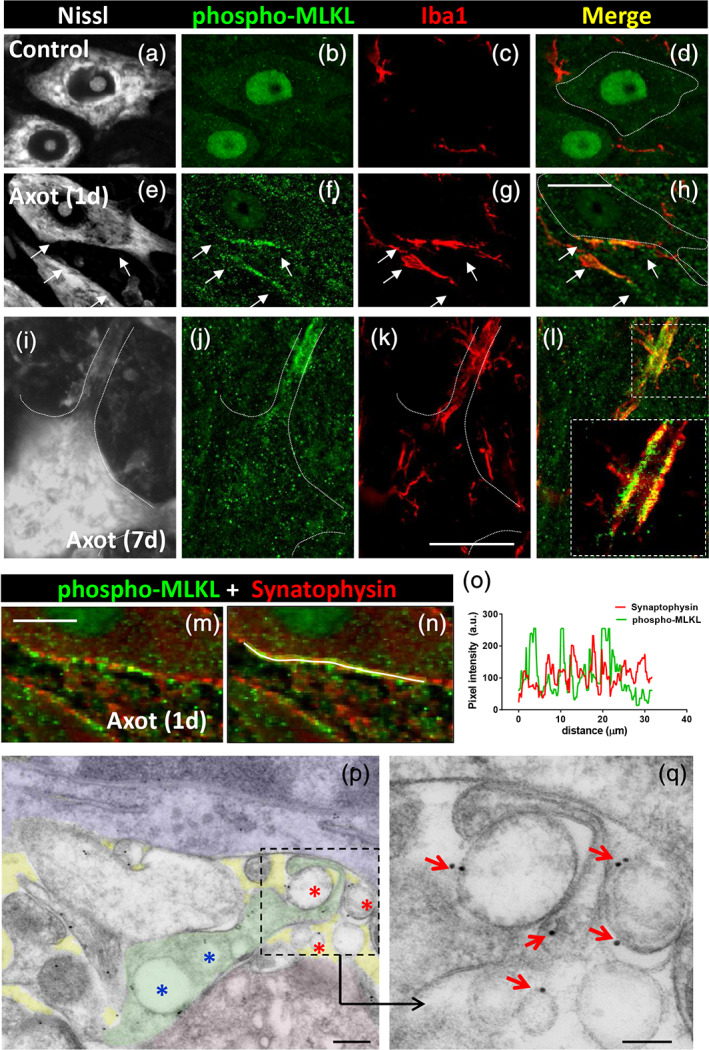
Activation of the necroptotic protein effector phospho‐MLKL near the surface of injured motor neuron (MN) cell bodies and dendrites, 24 hr after axotomy. (a–d) In noninjured MN cytoplasm, phospho‐MLKL‐immunoreactivity (green) is weak; only a positive signal is detected in nuclei; some Iba1‐positive microglial profiles (red) can be observed; MN somata were visualized by fluorescent Nissl staining (gray). (e–h) Twenty‐four hours after axotomy, clusters (delimited by arrows) of phospho‐MLKL‐positive particles (green) can be observed near the surface of injured MN cell bodies. Recruited Iba1‐positive microglial cell profiles (red) are seen in a close relationship with clustered phospho‐MLKL‐positive particles (green); MN somata are visualized with fluorescent Nissl staining (gray). (i–l) Clustered phospho‐MLKL‐positive particles (green) are seen at a site enwrapped by a recruited Iba1‐positive microglial cell (red), which is located on a dendrite of an MN (delimited by dashed line on a fluorescent Nissl stained image, gray) 24 hr postaxotomy; a detail of the distribution of Iba1 and phospho‐MLKL is shown in the enlarged inset in (l). (m–o) Clusters of MLKL positive particles (green), adjacent to the surface of a MN soma 24 hr postaxotomy, are interposed between the still remaining synaptophysin positive puncta (red), indicating that activated MLKL was associated to sites with disrupted synaptic boutons. The absence of colocalization of both phospho‐MLKL‐positive and synaptophysin‐positive puncta is shown in the pixel intensity profile (o) obtained along the line drawn in (n). (p) Postembedding immunogold localization of phospho‐MLKL at the membrane of extracellular vesicles (red *) interposed between an axotomized MN (red) and a microglial cell (blue); a synaptic terminal (green) displaying some labeled vacuoles (blue *) is also seen; extracellular space is shown shaded in yellow. The area delimited by dashed line in (p) is shown enlarged in (q), in which gold particles are more clearly visible in association with the membrane of EVs (arrows). Scale bars: (h) = 20 μm (valid for a–g), (k) = 20 μm (valid for i–l), (m) = 10 μm, (p) = 200 nm, and (q) = 100 nm [Color figure can be viewed at wileyonlinelibrary.com]
3.4. Identification of EV protein markers in association with an inflammatory response and synaptic disruption adjacent to axotomized MN cell bodies
EM analysis revealed that synaptic damage and local microglial recruitment, occurring near axotomized MNs, involved the generation and probable transcytosis of EVs. We therefore further explored whether any of the already identified proteins enriched in EVs, other than necroptotic EVs (Raden et al., 2020), were locally accumulated during this form of aseptic neuroinflammation. According to the ExoCarta database (http://www.exocarta.org), which catalogs molecular EVs components, we used antibodies against CD9, CD63, flotillin, and PDC6IP‐Alix in order to explore their presence in the ventral horn of the spinal cord during the neuroinflammatory response. Tissue samples were taken from mice at 7 days after sciatic nerve transection. Each EV marker was combined with Iba1 and VAChT immunolabeling to simultaneously visualize microglia and cholinergic synaptic profiles. In some cases, either the general synaptic bouton marker SV2 or synaptophysin was used instead of VAChT.
CD9, a member of tetraspanin family, is the protein most frequently identified in exosomes. Small CD9‐positive profiles were seen inside MN cytoplasm and also scattered in the adjacent neuropile (profile diameter = 0.53 ± 0.01 μm, n = 163) (Figure 8a). A slight CD9‐positive signal was also present in VAChT‐labeled cholinergic afferent terminals. However, after axotomy, there was a notable accumulation of CD9‐immunoreactive particles in association with cholinergic terminals, but not with any of the other afferent synaptic boutons (Figure 8b,e,f). In addition, CD9‐positive particles were noticed attached to the surface of processes of microglial cells recruited near axotomized MN cell bodies (Figure 8c,d). An ultrastructural examination of C boutons on 7‐day‐axotomized MNs showed increased pleomorphism in their synaptic vesicular content; this included an accumulation of large double‐membrane bounded vesicles (Figure 8g–j), which presumably corresponded to the CD9‐positive puncta observed under the confocal microscope.
FIGURE 8.
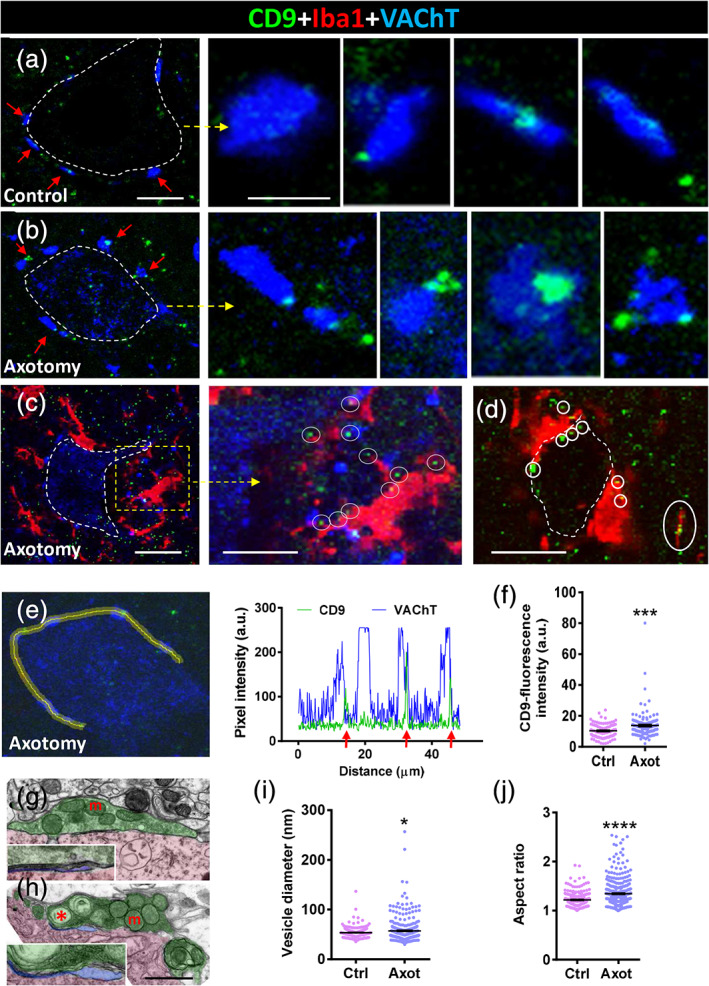
CD9‐immunoreactive puncta accumulate into VAChT‐positive synaptic afferents (C‐boutons) on motor neurons (MNs) at 7 days postaxotomy; CD9‐positive particles also interact with recruited microglia. (a) A control MN cell body (delimited by a dotted line) displaying VAChT‐positive synaptic terminals (blue, arrowed) and faint CD9‐positive puncta (green); arrowed synapses are shown at higher magnification in the neighboring panels (a). (b) A 7‐day‐axotomized MN (delimited by a dotted line) displaying VAChT‐positive synaptic terminals (blue, arrowed) and faint CD9‐positive puncta (green); arrowed synapses are shown at higher magnification in the right‐hand panels (b); note the increase in size and number of CD9 particles compared to the control. (c) Microglial cells (Iba1 immunostained, red) recruited in close proximity to a 7‐day‐axomized MN (delimited by a dotted line) interact with CD9‐containing particles (encircled), as shown at higher magnification in the right‐hand panel (c). (d) Another example of an axotomized MN showing recruited microglia (red) contacting CD9‐positive particles (green, encircled). (e) Pixel intensity (arbitrary units [a.u.]) profile along a line (yellow) traced on the VAChT‐positive synapses (blue) contacting MN surface, in order to simultaneously analyze CD9‐immunoreactivity (green) as depicted; arrows point out the association between the CD9‐signal and VAChT‐positive peaks. (f) Quantification of CD9 fluorescence intensity on VAChT‐positive C‐boutons on 7‐day‐axotomized MN. (g,h) Electron microscope micrographs showing C‐boutons on a control (g) and a 7‐day‐axotomized MN (h; synaptic terminals, shaded in green; MN cell body, shaded in red; and postsynaptic subsynaptic cistern, shaded in blue and enlarged in the insets); abnormally large vesicular structures (*) and dilated SSC are seen after axotomy in (h); m = mitochondria; (i,j) Quantification of the size (i) and shape (j, aspect ratio of vesicular membranous structures in C‐boutons; data are shown as mean ± SEM; *p < .05, ***p < .001, and ****p < .0001 versus Ctrl, Student's t test; n = 95–140 C‐boutons (f)), and 262–351 C‐bouton synaptic vesicles (i,j) (two animals per condition). Scale bars: a = 10 μm (valid for b); enlarged areas from a = 2 μm (valid for enlarged areas from b); c = 20 μm; enlarged area from c = 10 μm; d = 15 μm; and h = 1 μm (valid for g) [Color figure can be viewed at wileyonlinelibrary.com]
CD63 is another protein belonging to the tetraspanin family which is enriched in EVs and has been used in the characterization of EV subtypes and found in neuronal exosomes (Kowal et al., 2016; Men et al., 2019). In control MNs, CD63 immunostaining revealed a perinuclear punctate pattern that was reinforced at the MN periphery (Figure 9a). Overall, the CD63‐positive puncta were more abundant and larger than the CD9 positive particles (diameter: 0.71 ± 0.01 μm, n = 993). Axotomized MNs (7 days after nerve transection) showed an increased number of CD63 puncta surrounding their injured somata; they were otherwise covered by recruited microglial cells (Figure 9b–f). In sharp contrast to the CD9 immunostaining, no association of CD63‐positive particles and afferent cholinergic terminals was observed. However, when CD63 immunolabeling was combined with SV2 immunostaining, it was possible to observe a close relationship between CD63 puncta located at the MN surface and axosomatic synaptic terminals (Supplementary Figure 6a,b).
FIGURE 9.
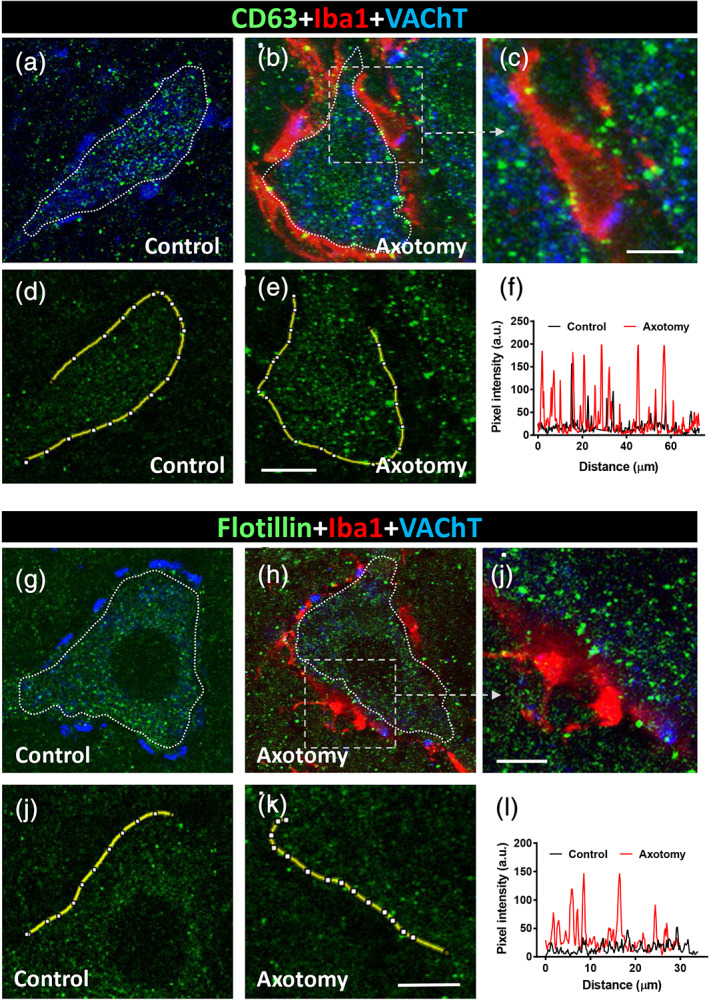
(a–f) CD63‐immunoreactivity in control and 7‐day‐axotomized motor neurons (MNs). (a) A control MN showing CD63‐immunolabeling in the form of abundant particles (green); no association is found with VAChT‐positive synaptic terminals (blue). (b) After axotomy, many CD63‐positive particles (green) are seen interacting with microglial cells (Iba1‐immnunolabeled, red) recruited at the MN surface; the boxed are is shown enlarged in (c). (d–f) Pixel intensity (arbitrary units [a.u.]) profile along a line (yellow) traced along the periphery of the control and axotomized MN cell bodies, showing the increase in CD63‐positive particles postaxotomy. (g–l) Flotillin‐immunoreactivity in control and 7‐day‐axotomized MNs. (g) A control MN showing flotillin‐immunolabeling in the form of abundant particles (green); no association is found with VAChT‐positive synaptic terminals (blue). (h) After axotomy, many flotillin‐positive profiles (green) are seen in close proximity to microglial cells (Iba1‐immnunolabeled, red) recruited at the MN surface; the boxed area is shown enlarged in (i). (j–l) Pixel intensity (arbitrary units [a.u.]) profile along a line (yellow) traced along the periphery of the control and axotomized MN cell bodies, showing the increase in flotillin‐positive particles postaxotomy. Scale bars: e = 10 μm (valid for a, b, d); c = 5 μm; k = 15 μm (valid for g, h, j); and i = 5 μm [Color figure can be viewed at wileyonlinelibrary.com]
Flotillin is another protein often used as an exosomal marker which has been detected in all types of EVs, baring or not tetraspanins (Baietti et al., 2012; Kowal et al., 2016). Flotillin is associated with “lipid rafts,” which are membrane subdomains that are enriched in cholesterol and sphingolipids and also localized in human neuronal cell bodies (Bickel et al., 1997; Girardot et al., 2003). It has also been shown that flotillin is involved in recycling the vesicle‐mediated trafficking of synaptic proteins and synaptogenesis (Bodrikov, Pauschert, Kochlamazashvili, & Stuermer, 2017). In spinal cord MNs, we noted that flotillin immunostaining displayed a particulate pattern (diameter profile: 0.59 ± 0.01 μm, n = 424). In axotomized cell bodies with extensive microglial covering, flotillin‐positive particles were seen to accumulate on the surface of neuronal cell bodies and, in particular, at the microglia‐neuronal interface (Figure 9g–l).
Another abundant component in the exosome proteome is PDC6IP‐Alix, a protein originally identified in relation to the apoptotic signaling pathway and later found to be connected to endocytic membrane trafficking and exosome formation (Odorizzi, 2006). On sections of spinal cord tissue, Alix‐immunoreactivity was detected in the form of small particles distributed within the MN cytoplasm; the particles delineated subdomains that probably represent intracellular membrane compartments such as ER or Golgi areas. In addition, the antibody used strongly detected intranuclear inclusions, which probably corresponded to nuclear speckles; we suggest that this localization could have been due to an unspecific cross reaction of the antibody used. This pattern of MN immunostaining did not change very much in the axotomized MNs that were covered by microglial cell processes (Supplementary Figure 7a,b).
3.5. Altered distribution of Rab GTPase proteins in MNs and microglia after axotomy
The Rab GTPase family of small‐molecular‐weight proteins comprises more than 60 gene products, with most of its members playing a role in vesicular trafficking in eukaryote cells (Zerial & McBride, 2001). They are also critically involved in the regulation of the synaptic function at presynaptic and postsynaptic sites (Mignogna & D'Adamo, 2018). Taking into account the dramatic changes that occur in the structural organization of the synaptic vesicular compartment observed in the synaptic terminals of axotomized MNs by EM, we used immunocytochemistry to analyze whether the localization of some of the Rab‐GTPase proteins in MNs and adjacent cells changed as a consequence of axotomy. Antibodies against Rab 4, 5, 7, 8A, 9, and 10 were used in sections of spinal cord taken from mice 3 or 7 days after sciatic nerve axotomy. Ventral horn MNs from both the ipsilateral (lesioned) and contralateral (control) sides were examined, after triple fluorescent immunostaining, for the simultaneous identification of each Rab protein. This was performed in combination with Iba1 and VAChT, which were, respectively, used as microglial and cholinergic synapse markers.
Rab4‐immunoreactivity revealed a pattern in the form of small particles scattered within the MN cell bodies, with no overt changes being found after axotomy. The Rab4 signal was, however, higher in the neuronal processes occupying the MN neuropile (including the MN axons), and this was substantially increased following axotomy. Similar results were found for Rab5 (Supplementary Figure 8a–d).
Rab7 is a factor which is recruited by nascent phagosomes and is essential for their fusion with late endosomes and/or lysosomes (Bucci, Thomsen, Nicoziani, McCarthy, & van Deurs, 2000; Harrison, Bucci, Vieira, Schroer, & Grinstein, 2003). In microglial cells, it has been shown that Rab7 is required for the fusion between pinosomes and lysosomes and for their size‐based sorting (Chen et al., 2015). In sections of spinal cord, Rab7 displayed a noticeable punctate pattern of immunoreactivity in the MN soma, under both control and axotomized conditions. The size of the Rab7‐positive particles was: 0.50 ± 0.01 μm (n = 220). Recruited microglia close to axotomized MNs displayed a notable accumulation of Rab7‐positive particles (Figure 10a–c). It should be noted that Rab7‐positive particles located inside microglial cells were significantly larger than those present in MN somata (1.13 ± 0.15 μm, n = 13, p < .0001).
FIGURE 10.
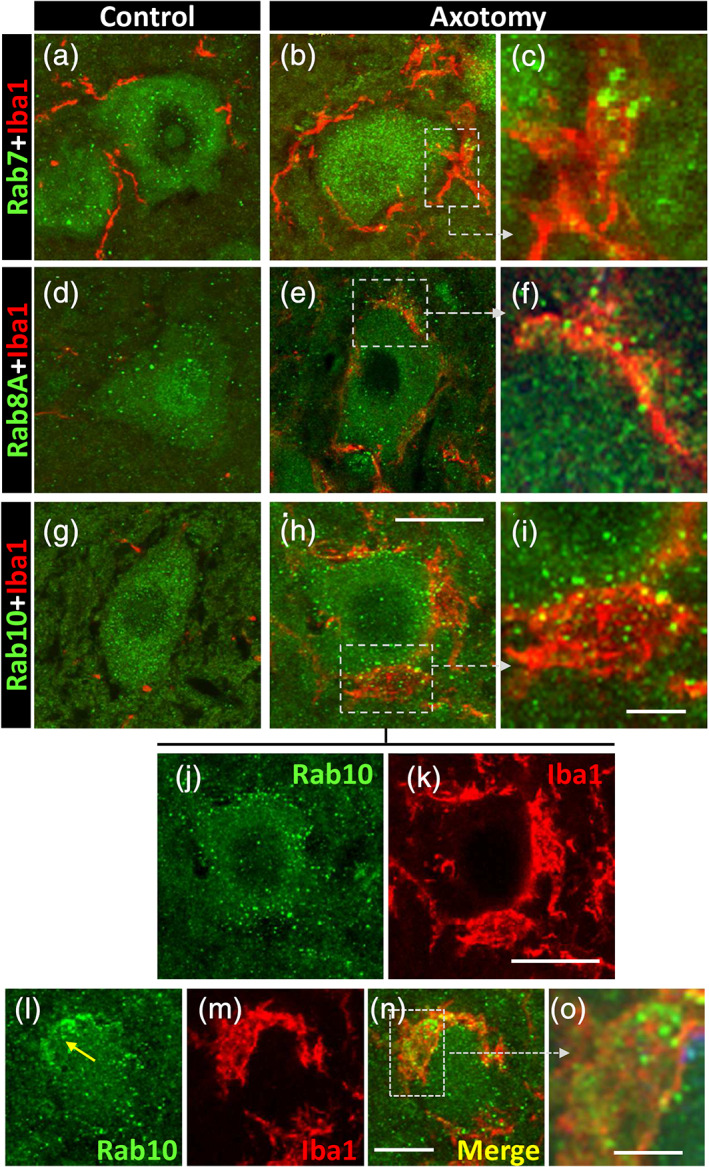
Expression of Rab GTPase proteins (green) in motor neurons (MNs) and microglia (red) in a basal condition and after axotomy, as indicated. (a–c) Compared to control, Rab7‐positive particles accumulate in recruited microglial cells adjacent to 3‐day‐axotomized MNs, as depicted in the enlarged boxed area in (c). (d–f) Abundant small Rab8A‐positive profiles are seen inside microglial cells covering a 7‐day‐axotomized MN; the area delimited by a square is shown at higher magnification in (f). (g–k) Numerous Rab10‐positive particles are mainly seen at the interface between microglia and the soma of a 7‐day‐axotomized MN; the area delimited by a square is shown enlarged in (i); (j,k) corresponds to spliced green and red channels from (h); the peripheral distribution of Rab10‐positive particles on MN soma is clearly visible in (j). (l–o) Illustrate the accumulation of Rab10‐positive particles inside microglia covering the surface of a 7‐day‐axotomized MN; an enlargement of the boxed area is shown in (o). Scale bars: h = 20 μm (valid for a, b, d, e, g, j, and k); i = 5 μm (valid for c and f); n = 20 μm (valid for l,m); and o = 5 μm [Color figure can be viewed at wileyonlinelibrary.com]
Rab8A has been shown to be enriched in macropinocytotic vesicles of activated macrophages (Wall et al., 2019). It has also been reported that Rab8A interacts with flotillin in the tubulovesicular recycling compartment (Solis et al., 2013). We found that Rab8A‐immunoreactiviry was present in MN somata in the form of small puncta (diameter: 0.55 ± 0.01 μm, n = 209), under both control and axotomized conditions. Some postaxotomy‐activated microglial cells, which interacted with MN somata, showed an accumulation of small Rab8A‐positive particles (Figure 10d–f). It is interesting to note that this location was highly comparable with the one that we found for flotillin in microglial cells after axotomy.
Rab10 has been identified as a novel protein which is predominantly localized in tubular endosomes (Etoh & Fukuda, 2019). As far as we know, no data have previously been published regarding Rab10 in microglia. Our findings relating to Rab10 after axotomy would seem to be of particular interest. Under control conditions, Rab10 was present in the form of small, homogeneously distributed, dots (diameter: 0.49 ± 0.008, n = 543) within the MN soma, while no particular association was observed with microglial cells. Seven days after axotomy, the Rab10 particles migrated to the periphery of the MN cell body and many particles were also observed in association with microglial cells covering lesioned MNs (Figure 10g–o).
4. DISCUSSION
During the developmental establishment of neuronal networks, microglia play a fundamental role via a process of activity‐dependent synaptic pruning and remodeling in order to achieve a stable neural function and establish robust behavior. In adults, however, synaptic loss is considered an early sign of various neurodegenerative conditions, including MN diseases (Fogarty, 2019; Vukojicic et al., 2019). It has been suggested that a reactivation of synaptic elimination mechanisms similar to those operating during development may occur in these disorders (Stephan, Barres, & Stevens, 2012). Accepting the putative relevance of the synaptic alteration within the context of MN pathology, we used a peripheral nerve lesion paradigm, which resulted in prominent microglial activation, to explore its impact on the fate of MN synaptic inputs. Microglial activation around axotomized MNs is an extensively documented process, but its effect on afferent synaptic bouton organization has been poorly addressed with rather confusing results. For a long time, it was assumed that activated microglia recruited close to MN cell bodies play a role as “synaptic strippers”; however, whether stripping represents a simple and reversible synaptic withdrawal or, conversely, leads to the degeneration and phagocytosis of synapses, has yet to be clearly established (Aldskogius, 2011; Alvarez et al., 2020; Linda et al., 2000; Möller et al., 1996; Moran & Graeber, 2004; Sumner, 1975). In axotomized facial MNs, Blinzinger and Kreutzberg (1968) have described how active microglia displaced intact synaptic terminals from neuronal perikarya; the absence of degenerating synaptic boutons and lack of apparent phagocytosis by microglia have also been reported. Our results in spinal cord, unambiguously demonstrate that a severe disruption of synaptic afferents to MNs occurs early after peripheral nerve transection in conjunction with prominent distant aseptic microglial mediated neuroinflammation. In agreement with the description Blinzinger and Kreutzberg (1968), we observed that in advanced stages of microglial migration toward axotomized MNs, large areas of the MN cell body surface appeared to be completely devoid of synaptic contacts and the vacant spaces were occupied by flattened microglial cells. In these areas, signs of synaptic degeneration were rarely visible. Nevertheless, this clearly contrasts with what occurs during the early steps of interaction between recruited microglial cells and axotomized MN cell bodies and their synapses. The partial discordance between observations could therefore have resulted from limited spatial and temporal sampling in different models of MN axotomy.
We observed that the way in which synaptic terminals are eliminated in the MN axotomy paradigm did not comprise their bulk engulfment by microglia. Instead of this, microglial cells internalize small membranous vesicular fragments arising from the disruption of synaptic terminals, which, presumably, were previously primed by their close proximity with microglial processes. It should be noted that processes from early recruited microglial cells postaxotomy preferentially contacted presynaptic boutons rather than the MN surface. Other microglial filopodia interacted with vesicular elements coming from the disassembly of previously altered synaptic boutons.
The ultrastructural organization of the area adjacent to the surface of MN somata abruptly changes as soon as 24 hr after peripheral nerve transection. During this early acute alteration, some synaptic terminals exhibit a rupture of presynaptic membranes with the concomitant release of intracellular vesicles that further accumulate at the extracellular space. This overall picture fits well with a process of necroptosis, and this was corroborated by the localization of the membrane‐disrupting effector MLKL at sites where presynaptic terminals degenerate and in neighboring EVs. Our negative results after western blot analysis could be because only a proportion of MNs in ventral horn belong to the axotomized sciatic nerve pool, and the corresponding synaptic disruption occurs in restricted subcompartments at a given time. Thus, the expression of necroptotic molecular markers such as phospho‐MLKL, would be hard to detect in the whole extracts. In any case, we think that, the accuracy of data from the in situ immunolocalization, both at the confocal and EM levels, are enough robust to show the involvement of the necroptotic pathway in the acute synaptic disruption occurring on axotomized MNs. As this probably occurs in the absence of the death of its parental neuron, we must assume that a necroptotic program may be locally activated in presynaptic axon terminals, as described for a form of apoptosis spatially confined to axons or synapses (Cusack, Swahari, Hampton Henley, Michael Ramsey, & Deshmukh, 2013; Mattson, Keller, & Begley, 1998).
Sites where phospho‐MLKL‐containing vesicles accumulate were usually closely associated with microglia that was attracted to axotomized MNs. One of the possible microglial‐derived factor that may initiate the activation of necroptotic pathway is tumor necrosis factor, as occurs in other neuropathological conditions (Chen et al., 2019; Ito et al., 2016; Pasparakis & Vandenabeele, 2015). However, one important aspect not resolved here is whether activated microglia induces a synaptic damage that precedes synaptic disruption or, conversely, an altered, neuronal‐autonomous, synaptic function, is as a primary event which promotes a chemotactic effect on microglia.
Experiments in which microglial proliferation has been prevented by means of antimitotic agents have not demonstrated any significant change in afferent synaptic loss on axotomized MNs (Graeber, Streit, & Kreutzberg, 1989; Svensson & Aldskogius, 1993). Our results obtained with the CSF1R blocking agent PLX5622 showed only ~10% of synaptic loss prevention, despite the ~46% reduction in microgliosis; this suggests a poor correlation between the strength of the microglial reaction and the number of synaptic boutons which are eliminated. A lack of correlation between synaptic stripping and microglial function has also been observed in MNs devoid of CSF1 (Akhter, Griffith, English, & Alvarez, 2019; Rotterman et al., 2019) and also in other conditions involving synaptic degeneration or plasticity (Perry & O'Connor, 2010; Tremblay & Majewska, 2011).
To a variable degree, nonacutely disrupted synaptic terminals contacting axotomized MNs showed an accumulation of membrane‐bound organelles, such as endosome‐like and phagosome‐like vacuoles, which are indicative of altered vesicle recycling and synaptic function. Early studies on synaptic structure and function have described how increased synaptic activity results in an accumulation of extensive presynaptic membrane infoldings which are generated as a consequence of the blocking of the vesicle recycling process (Haimann, Torri‐Tarelli, Fesce, & Ceccarelli, 1985; Solsona, Esquerda, & Marsal, 1981). Along these lines, it should also be noted that changes in the activity of MN afferent synapses occurs soon after peripheral nerve injury (Bichler et al., 2007). An accumulation of comparable vacuolar intermediates has been observed in presynaptic terminals when the conversion of bulk endosomes into synaptic vesicle is inhibited (Wu et al., 2014). We have also observed double‐membrane autophagosome‐like vesicles similar to those found after autophagy promotion by rapamycin (Hernandez et al., 2012), or by Sonic hedgehog (Petralia et al., 2013) or Basson depletion (Okerlund et al., 2017; Waites et al., 2013). In synaptic terminals, synaptic vesicles or proteins are eliminated by autophagy (Lüningschrör & Sendtner, 2018). Since the induction of reactive oxygen species (ROS) rapidly provokes a presynaptic promotion of autophagy (Hoffmann et al., 2019), it could be assumed that activated microglia in the proximity of synaptic terminals would release ROS (Block, Zecca, & Hong, 2007), resulting in local protein and organelle damage and in the activation of autophagic pathways. In addition, massive and spatially restricted microglial overactivation state may result in more extensive neurotoxic damage affecting MN survival as it is occasionally seen after axotomy.
Additionally, it should be taken into account that activation of MLKL does not definitively result in an irreversible commitment to cell death. In certain conditions, MLKL‐dependent membrane damage can be repaired to promote cell survival. In these circumstances, active MLKL induce the formation of bubbles at the surface of the cells that are released extracellularly (Gong, Guy, Olauson, et al., 2017). MLKL also associates to endosomes to facilitate the generation and release of EVs, independently of death induction (Yoon, Kovalenko, Bogdanov, & Wallach, 2017). These data are of interest because they may also account for the vacuolar changes we observed in synaptic terminals that escape from the acute “explosive” disruption. These terminals remain, at least several days, in contact with MNs, display abundant MVBs and endosome‐like vesicles, and are often in contact with microglial processes. Thus, in these terminals, the enhanced endosomal trafficking may be also a source of EVs other than “lytic EVs.” These EVs would correspond, based on their molecular composition, to exosomes. In these cases, phospho‐MLKL, would work antagonizing, instead of stimulating, the execution of lysis during necroptosis (see Yoon et al., 2017); this would result in the structural maintenance of synaptic terminals exhibiting a conspicuous alteration in their vacuolar system. Moreover, this scenario is in concordance with the increased density of particles displaying positive immunoreactivity for exosomal markers we found near the surface of axotomized MN somata 7 days postlesion. Thus, different populations of EVs such as necroptotic vesicles, plasma membrane‐derived vesicles, and vesicles derived from MVBs should be produced in a separate spatiotemporal sequence during the complex reactive cellular events occurring near the surface of axotomized MNs. However, we cannot exclude that a part of EVs may be secreted by microglia (Paolicelli, Bergamini, & Rajendran, 2019). Another point that should be considered here is the existing link between necroptosis and inflammation, by increasing the production and release of cytokines and chemokines (Orozco et al., 2019), as well as by activating the inflammosomal pathway, involving caspase 1 activation and cytokine IL‐1β secretion (Conos et al., 2017).
The degenerative signs we found in synaptic terminals 2 weeks after nerve transection are very similar to those described in other “synaptopathies” in the context of human prion diseases (Sikorska, Liberski, Giraud, Kopp, & Brown, 2004). Dark degenerating terminals have also been described in the initial stages of prion disease pathology (Jeffrey et al., 2000; Šišková, Reynolds, O'Connor, & Perry, 2013), even in the absence of the direct involvement of microglial cells (Šišková et al, 2009). This suggests that, at least in this case, synaptic degeneration and removal may be part of an autonomous neuronal process. Nevertheless, delayed microglia activation occurs in these models.
In the MN axotomy paradigm, the disconnection of cell bodies from the periphery is the stimulus that rapidly changes the MN status in a way that is coped with its immediate environment: that is, extracellular matrix, afferent synapses and glial cells. For example, it is conceivable that MNs or synapses rapidly switch from “do not eat me” to “eat me” signals, which are sensed by microglia. Mobilized microglia may release various factors, including matrix metalloproteinases (Kim et al., 2007; Konnecke & Bechmann, 2013), thrombospondin (Möller et al., 1996), and free radicals, which affect the extracellular matrix in the immediate MN environment (perineuronal net; Fawcett, Oohashi, & Pizzorusso, 2019). This may have consequences such as the widening of the extracellular space (i.e., local edema) or the detachment of presynaptic from postsynaptic structures. This would also contribute to weakening the blood‐spinal cord barrier, which has been shown to be disrupted in spinal cord after peripheral nerve lesions and to contribute to the influx of inflammatory mediators (Echeverry, Shi, Rivest, & Zhang, 2011); this is consistent with the endogenous IgGs that we have seen surrounding capillary vessels near axotomized MNs and also decorating the surface of recruited microglial cells. It is interesting to note that IgG extravasation exists during the initial stages of disease in ALS mouse models, in conjunction with muscle denervation and before MN death (Zhong et al., 2008). At the same time as escaped IgGs bind to the surface of microglial cells, presumably through Fc receptor upregulation, C1q also becomes overexpressed in microglia. It is conceivable that C1q produced by microglia binds vesicular debris coming from disrupted synaptic terminals in order to promote their opsonization and phagocytosis, in a comparable way to what happens during the clearance of apoptotic cells (Galvan, Greenlee‐Wacker, & Bohlson, 2012) or developmental synaptic remodeling and pathological synaptic loss (Hong et al., 2016; Schafer et al., 2012; Stevens et al., 2007). However, we must also point out that, in the absence of C1q, synapse elimination on MNs following axotomy is not reduced (Berg et al., 2012). This is probably due to a redundancy mechanism involved in the loss of afferent synapses on lesioned MNs.
We observed a type of double‐membrane‐bound vacuoles with an analogous morphology inside of synaptic terminals and internalized within microglial cells. This suggested that they could form part of a category of membrane vesicles that originated in nonacutely broken synapses and were subsequently transferred to microglia. In line with this interpretation, it was possible to detect free intercellular intermediates of this particular morphological type of vesicles. This observation is in concordance with the presynaptic secretion of EVs. The increased expression of EV markers such as CD9 and CD63, together with the presence of flotillin adjacent to neuronal surface and in close relation with microglial processes, would be concordant with this hypothesis. In an “in vitro” model of synaptic elimination, exosomes released by PC12 cells promoted microglial activation, with subsequent synaptic phagocytosis and pruning (Bahrini, Song, Diez, & Hanayama, 2015).
The increase in CD9‐containing particles restricted to the C‐boutons contacting axotomized MNs, is particularly intriguing. Several quite particular properties of vesicular trafficking and transcytotic transfer have been identified in this type of terminals (Caleo et al., 2018). We noticed that a pattern of vacuole accumulation in conjunction with dark degeneration predominated in C‐boutons of axotomized MNs; this phenotype is not usually seen in other types of synapses. C‐boutons are also preferential chemoattractive sites for microglial filopodia during their mobilization postaxotomy (Salvany et al., 2019). This indicates that the fate of afferent synaptic terminals on axotomized MNs is not entirely homogeneous and depends on their neurotransmitter‐specificity. It has also been reported that axotomy in MNs favors the predominant loss of excitatory inputs with respect to other input types (Alvarez et al., 2011; Spejo & Oliveira, 2015).
The ultrastructural morphology that we observed in the EVs “floating” in the perineuronal net of axotomized MNs was highly pleomorphic and exhibited a broad variation in size. These vesicles appear to belong to a heterogeneous population of EVs which includes exosomes and other EVs (in example, lytic or necroptotic vesicles). Used in a restrictive form, the term “exosome” is usually applied to small vesicles (50–100 nm) of endosomal or MVB origin that are released from cells. Other membrane vesicles released by the outward budding of cells include a heterogeneous population of elements without this size limitation that can even exceed 1 μm in length and that have sometimes been referred to as microvesicles or ectosomes. The more general term of EVs has been used for a collective denomination of these elements (Cocucci & Meldolesi, 2015; Colombo, Raposo, & Thery, 2014; Kowal et al., 2016). Exosomes can be released from neurons in an activity‐dependent manner (Fauré et al., 2006). In our system, we have seen synaptic boutons displaying a prominent concentration of MVBs; this suggests that they could be a source of exosome secretion in the perineuronal milieu. Other larger and pleomorphic vesicles observed in this enlarged extracellular space originate from a disruption of synaptic terminals and should be considered as cellular debris (Baxter et al., 2019). Similar alterations in membrane integrity may be induced by complement‐mediated cytolysis, a condition that should be further evaluated in MNs postaxotomy. We demonstrate that the fate of most of the EVs generated in the perineuronal space of axotomized MNs is their elimination by microglial cell phagocytosis. These data are in concordance with the progressive and transient accumulation of CD68‐positive puncta inside recruited and perisomatic microglia which, in turn, is indicative of lysosomal activity. However, the intimate relationship between exosomal markers such as CD9, CD63, and flotillin with the microglial cell surface indicates a previous process involving the endocytic trapping of EVs. The presence of Rab7 and Rab10 in microglia interacting with injured MNs is an indicator of the activation of endocytic activity by these cells.
Overall, our data reveal new mechanisms by which afferent synapses are removed from acutely injured MNs after peripheral nerve transection. Although microglial cells are actively involved in eliminating fragments of damaged presynaptic terminals, there is a lack of evidence to support any bulk engulfment of synaptic boutons. EVs are intermediate elements in this process of synaptic removal, which presents certain homologies with those described under the concept of synaptic trogocytosis during microglia‐mediated synaptic removal during development (Weinhard et al., 2018). Nevertheless, further studies are required for a more general validation of this phenomenon of synaptic piecemeal phagocytosis. Since the reduction of synaptic inputs in MNs is an early event in amyotrophic lateral sclerosis (ALS) (Chang & Martin, 2009; Jiang, Schuster, Fu, Siddique, & Heckman, 2009; Sasaki & Iwata, 1996; Schutz, 2005; Sunico et al., 2011; Vaughan, Kemp, Hatzipetros, Vieira, & Valdez, 2015) and spinal muscular atrophy (SMA) pathology (Cerveró et al., 2018; Mentis et al., 2011; Tarabal et al., 2014; Vukojicic et al., 2019), a more precise understanding of the fundamental mechanisms involved in the microglia‐synapse interaction in the injured MNs may help to define new therapeutic interventions. The impact of our observation on the pathogenetic mechanisms underlying ALS deserves further attention since the role of necroptosis in this disease remains controversial (Dermentzaki et al., 2019; Ito et al., 2016; Re et al., 2014). In any case, our results may prove significantly relevant in the context of functional recovery after peripheral nerve injury, in which the restoration of synaptic inputs to MNs is incomplete in spite of successful nerve regeneration and muscle reinnervation (Rotterman, Nardelli, Cope, & Alvarez, 2014).
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supporting information
Supplementary Figure 1 Endogenous IgGs decorate recruited microglial cells at the MN surface 7 days postaxotomy. (a‐c) Spinal cord section showing an axotomized MN (delimited by a dashed line) covered by Iba1‐immunostained microglia (red); note the association of endogenous IgGs (green) with microglial profiles. Scale bar: 20 μm
Supplementary Figure 2 Spinal cord section immunostained for synaptophysin (green) showing ventral horn MN cell bodies (visualized by fluorescent Nissl staining, blue) in control (a and b) and postaxotomy (c and d) conditions, as indicated. Note the reduction of synaptophisin positive puncta near the surface of MN cell bodies after axotomy. Scale bar: 40 μm (valid for a‐c)
Supplementary Figure 3 Ultrastructural morphology of normal synaptic boutons (shaded in green) contacting MN cell bodies (shaded in red). (a) Two presynaptic boutons containing synaptic vesicles clustered in the active zones (arrows); details of synaptic vesicle morphology are seen in the enlarged panels (b and c), showing flattened (b) and round (c) vesicles characteristic of F‐type and S‐type terminals, respectively. (d) A C‐type synaptic bouton is shown, displaying the characteristic subsynaptic cistern (arrows) that can be observed enlarged in (e); the intersynaptic extracellular gap is shaded in yellow. m = mitochondria. Scale bars: 0.5 μm in (a) and (d); 150 nm in (b), (c) and (e)
Supplementary Figure 4 Ultrastructural analysis of the periphery of MN cell bodies, and adjacent synaptic afferents and cell structures in the spinal cord. (a) Three days after axotomy, the extracellular space (shaded in yellow) is widened and synaptic terminals (green) are partially detached from the MN surface (red); in addition, a number of extracellular membrane‐bound vesicles (encircled in red) can be seen “floating” in the extracellular space. (b) A detail of two terminal synaptic boutons (green) contacting MN surface (red) are partially detached but maintain focal adherence at the puncta adherentia sites (red arrows); note the presence of abundant extracellular vesicles (*) in the extracellular space (yellow). (c) A recruited microglial cell (shaded in blue) in the proximity of the MN surface and interacting with abundant extracellular pleomorphic vesicles (indicated with red circles), 3 days after axotomy. (d‐f) A high magnification detail of the morphology of extracellular vesicles (arrows) near the MN surface, 3 days after axotomy. Scale bars: 1 μm (a‐c) and 500 nm (d‐f)
Supplementary Figure 5 Electron micrograph of a MN cell body (shaded in red) showing a marked disruption of intracellular organelle organization. (a) In the vicinity of the MNs, an altered microglial cell is seen displaying vacuolar changes in the endoplasmic reticulum and other organelles. Altered synaptic afferents are shaded in green, and the extracellular space at the MN‐microglia interface (shaded in yellow) can be seen expanded. A detailed view of MN cytoplasmic disorganization is shown at higher magnification in (b). Scale bars: (a) and (b) = 2.5 μm
Supplementary Figure 6 (a,b) An isolated MN cell body, 7 days postaxotomy, in a squash preparation immunostained for CD63 (green) and SV2 (red). Red spots, corresponding to afferent synaptic boutons, are seen covering the surface of the MN soma, in which abundant CD63 positive puncta can be seen. In the enlarged boxed area (a), the relationship between larger CD63‐positive spots and afferent boutons can be observed. Scale bars: a = 2 μm, b = 10 μm
Supplementary Figure 7 (a,b) Ventral horn MN cell bodes (delimited by dashed lines) double immunostained for PDC6IP‐Alix (green) and Iba1 (red) in control and 7 days postaxotomy. The axotomized MN can be identified by recruited Iba1‐positive microglial cells. However, the pattern of PDC6IP‐Alix does not show identifiable changes following axotomy. Scale bar: 20 μm (valid for a)
Supplementary Figure 8 Control and 7‐day axotomized MN cell bodies (delimited by dotted lines) immunostained for either Rab4 (a, b) or Rab5 (c, d) (green) combined with Iba1 (red). Note that the recruitment of Iba1‐positive microglial cells around axotomized MNs (b and d). Rab4 and Rab5‐immunoreactivity is increased in neuronal processes (arrows) adjacent to injured MNs. Scale bars: Scale bar: d = 20 μm (valid for a‐c)
ACKNOWLEDGMENTS
The authors thank Alaó Gatius, Alba Blasco, and Sílvia Gras for their help in some experiments, Anaïs Panosa from the SCT of Microscopy of the Universitat de Lleida for technical support with confocal microscopy, and the staff from the SCT Animal Facility of the Universitat de Lleida for mouse care and housing. The authors thank Lidia Delgado and M. Yolanda Muela, from the Unitat de Criomicroscòpia Electrònica (Centres Científics i Tecnològics de la Universitat de Barcelona), for technical support with ultrastructural immunolabeling, and Plexxikon Inc. for providing the CSFR1 inhibitor PLX5622. This work was supported by grants from the Ministerio de Ciencia, Innovación y Universidades cofinanced by Fondo Europeo de Desarrollo Regional (FEDER; RTI2018‐099278‐B‐I00 J. C. and J. E.), and from Jack Van den Hock a la Investigació de l'ELA – Fundació Miquel Valls. S. S. holds a grant from the Spanish Ministerio de Educación, Cultura y Deporte (FPU).
Salvany S, Casanovas A, Piedrafita L, et al. Microglial recruitment and mechanisms involved in the disruption of afferent synaptic terminals on spinal cord motor neurons after acute peripheral nerve injury. Glia. 2021;69:1216–1240. 10.1002/glia.23959
Sara Salvany and Anna Casanovas contributed equally to this work. Jordi Calderó and Josep E. Esquerda are senior co‐authors.
Funding information Ministerio de Ciencia, Innovación y Universidades (MICIU) and Fondo Europeo de Desarrollo Regional (FEDER), Grant/Award Number: RTI2018‐099278‐B‐I00; Spanish Ministerio de Educación, Cultura y Deporte; Jack Van den Hock a la Investigació de l'ELA ‐ Fundació Miquel Valls
DATA AVAILABILITY STATEMENT
All the data and original images of this article are available upon reasonable request by e‐mailing to the corresponding author.
REFERENCES
- Akhter, E. T. , Griffith, R. W. , English, A. W. , & Alvarez, F. J. (2019). Removal of the potassium chloride co‐transporter from the somatodendritic membrane of axotomized motoneurons is independent of BDNF/TrkB signaling but is controlled by neuromuscular innervation. eNeuro, 6(5), ENEURO.0172‐19.2019. 10.1523/ENEURO.0172-19.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldskogius, H. (2011). Mechanisms and consequences of microglial responses to peripheral axotomy. Frontiers in Bioscience (Scholar Edition), 3, 857–868. 10.2741/192 [DOI] [PubMed] [Google Scholar]
- Alvarez, F. J. , Rotterman, T. M. , Akhter, E. T. , Lane, A. R. , English, A. W. , & Cope, T. C. (2020). Synaptic plasticity on motoneurons after axotomy: A necessary change in paradigm. Frontiers in Molecular Neuroscience, 13, 68. 10.3389/fnmol.2020.00068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez, F. J. , Titus‐Mitchell, H. E. , Bullinger, K. L. , Kraszpulski, M. , Nardelli, P. , & Cope, T. C. (2011). Permanent central synaptic disconnection of proprioceptors after nerve injury and regeneration. I. Loss of VGLUT1/IA synapses on motoneurons. Journal of Neurophysiology, 106(5), 2450–2470. 10.1152/jn.01095.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahrini, I. , Song, J. H. , Diez, D. , & Hanayama, R. (2015). Neuronal exosomes facilitate synaptic pruning by up‐regulating complement factors in microglia. Scientific Reports, 5, 7989. 10.1038/srep07989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baietti, M. F. , Zhang, Z. , Mortier, E. , Melchior, A. , Degeest, G. , Geeraerts, A. , … David, G. (2012). Syndecan‐syntenin‐ALIX regulates the biogenesis of exosomes. Nature Cell Biology, 14(7), 677–685. 10.1038/ncb2502 [DOI] [PubMed] [Google Scholar]
- Baxter, A. A. , Phan, T. K. , Hanssen, E. , Liem, M. , Hulett, M. D. , Mathivanan, S. , & Poon, I. K. H. (2019). Analysis of extracellular vesicles generated from monocytes under conditions of lytic cell death. Scientific Reports, 9(1), 7538. 10.1038/s41598-019-44021-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett, M. L. , Bennett, F. C. , Liddelow, S. A. , Ajami, B. , Zamanian, J. L. , Fernhoff, N. B. , … Barres, B. A. (2016). New tools for studying microglia in the mouse and human CNS. Proceedings of the National Academy of Sciences of the United States of America, 113(12), E1738–E1746. 10.1073/pnas.1525528113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg, A. , Zelano, J. , Stephan, A. , Thams, S. , Barres, B. A. , Pekny, M. , … Cullheim, S. (2012). Reduced removal of synaptic terminals from axotomized spinal motoneurons in the absence of complement C3. Experimental Neurology, 237(1), 8–17. 10.1016/j.expneurol.2012.06.008 [DOI] [PubMed] [Google Scholar]
- Bichler, E. K. , Nakanishi, S. T. , Wang, Q. B. , Pinter, M. J. , Rich, M. M. , & Cope, T. C. (2007). Enhanced transmission at a spinal synapse triggered in vivo by an injury signal independent of altered synaptic activity. The Journal of Neuroscience, 27(47), 12851–12859. 10.1523/JNEUROSCI.1997-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bickel, P. E. , Scherer, P. E. , Schnitzer, J. E. , Oh, P. , Lisanti, M. P. , & Lodish, H. F. (1997). Flotillin and epidermal surface antigen define a new family of caveolae‐associated integral membrane proteins. Journal of Biological Chemistry, 272(21), 13793–13802. 10.1074/jbc.272.21.13793 [DOI] [PubMed] [Google Scholar]
- Bisht, K. , Sharma, K. P. , Lecours, C. , Sanchez, M. G. , El Hajj, H. , Milior, G. , … Tremblay, M. E. (2016). Dark microglia: A new phenotype predominantly associated with pathological states. Glia, 64(5), 826–839. 10.1002/glia.22966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blinzinger, K. , & Kreutzberg, G. (1968). Displacement of synaptic terminals from regenerating motoneurons by microglial cells. Zeitschrift für Zellforschung und Mikroskopische Anatomie, 85(2), 145–157. 10.1007/bf00325030 [DOI] [PubMed] [Google Scholar]
- Block, M. L. , Zecca, L. , & Hong, J. S. (2007). Microglia‐mediated neurotoxicity: Uncovering the molecular mechanisms. Nature Reviews. Neuroscience, 8(1), 57–69. 10.1038/nrn2038 [DOI] [PubMed] [Google Scholar]
- Bodian, D. (1975). Origin of specific synaptic types in the motoneuron neuropil of the monkey. The Journal of Comparative Neurology, 159(2), 225–243. 10.1002/cne.901590205 [DOI] [PubMed] [Google Scholar]
- Bodrikov, V. , Pauschert, A. , Kochlamazashvili, G. , & Stuermer, C. A. O. (2017). Corrigendum to "Reggie‐1 and reggie‐2 (flotillins) participate in Rab11a‐dependent cargo trafficking, spine synapse formation and LTP‐related AMPA receptor (GluA1) surface exposure in mouse hippocampal neurons" (Exp. Neurol. 289, pages 31‐45). Experimental Neurology, 293, 200. 10.1016/j.expneurol.2017.02.016 [DOI] [PubMed] [Google Scholar]
- Brannstrom, T. , & Kellerth, J. O. (1998). Changes in synaptology of adult cat spinal alpha‐motoneurons after axotomy. Experimental Brain Research, 118(1), 1–13. 10.1007/s002210050249 [DOI] [PubMed] [Google Scholar]
- Bucci, C. , Thomsen, P. , Nicoziani, P. , McCarthy, J. , & van Deurs, B. (2000). Rab7: A key to lysosome biogenesis. Molecular Biology of the Cell, 11(2), 467–480. 10.1091/mbc.11.2.467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caleo, M. , Spinelli, M. , Colosimo, F. , Matak, I. , Rossetto, O. , Lackovic, Z. , & Restani, L. (2018). Transynaptic action of botulinum neurotoxin type A at central cholinergic boutons. Journal of Neuroscience, 38(48), 10329–10337. 10.1523/Jneurosci.0294-18.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerveró, C. , Blasco, A. , Tarabal, O. , Casanovas, A. , Piedrafita, L. , Navarro, X. , … Calderó, J. (2018). Glial activation and central synapse loss, but not motoneuron degeneration, are prevented by the sigma‐1 receptor agonist PRE‐084 in the Smn2B/‐ mouse model of spinal muscular atrophy. Journal of Neuropathology and Experimental Neurology, 77(7), 577–597. 10.1093/jnen/nly033 [DOI] [PubMed] [Google Scholar]
- Chang, Q. , & Martin, L. J. (2009). Glycinergic innervation of motoneurons is deficient in amyotrophic lateral sclerosis mice A quantitative confocal analysis. American Journal of Pathology, 174(2), 574–585. 10.2353/ajpath.2009.080557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, A. Q. , Fang, Z. , Chen, X. L. , Yang, S. , Zhou, Y. F. , Mao, L. , … Hu, B. (2019). Microglia‐derived TNF‐alpha mediates endothelial necroptosis aggravating blood brain‐barrier disruption after ischemic stroke. Cell Death & Disease, 10(7), 487. 10.1038/s41419-019-1716-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, C. , Li, H. Q. , Liu, Y. J. , Guo, Z. F. , Wu, H. J. , Li, X. , … Duan, S. M. (2015). A novel size‐based sorting mechanism of pinocytic luminal cargoes in microglia. The Journal of Neuroscience, 35(6), 2674–2688. 10.1523/JNEUROSCI.4389-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Z. L. , Yu, W. M. , & Strickland, S. (2007). Peripheral regeneration. Annual Review of Neuroscience, 30, 209–233. 10.1146/annurev.neuro.30.051606.094337 [DOI] [PubMed] [Google Scholar]
- Cocucci, E. , & Meldolesi, J. (2015). Ectosomes and exosomes: Shedding the confusion between extracellular vesicles. Trends in Cell Biology, 25(6), 364–372. 10.1016/j.tcb.2015.01.004 [DOI] [PubMed] [Google Scholar]
- Coleman, M. P. , & Freeman, M. R. (2010). Wallerian degeneration, wld(s), and nmnat. Annual Review of Neuroscience, 33, 245–267. 10.1146/annurev-neuro-060909-153248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo, M. , Raposo, G. , & Thery, C. (2014). Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annual Review of Cell and Developmental Biology, 30, 255–289. 10.1146/annurev-cellbio-101512-122326 [DOI] [PubMed] [Google Scholar]
- Conforti, L. , Gilley, J. , & Coleman, M. P. (2014). Wallerian degeneration: An emerging axon death pathway linking injury and disease. Nature Reviews. Neuroscience, 15(6), 394–409. 10.1038/nrn3680 [DOI] [PubMed] [Google Scholar]
- Conos, S. A. , Chen, K. W. , de Nardo, D. , Hara, H. , Whitehead, L. , Nunez, G. , … Vince, J. E. (2017). Active MLKL triggers the NLRP3 inflammasome in a cell‐intrinsic manner. Proceedings of the National Academy of Sciences of the United States of America, 114(6), E961–E969. 10.1073/pnas.1613305114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conradi, S. (1969). Ultrastructure and distribution of neuronal and glial elements on the surface of the proximal part of a motoneuron dendrite, as analyzed by serial sections. ACTA Physiologica Scandinavica Supplement, 332, 49–64. [PubMed] [Google Scholar]
- Cullheim, S. , & Thams, S. (2007). The microglial networks of the brain and their role in neuronal network plasticity after lesion. Brain Research Reviews, 55(1), 89–96. 10.1016/j.brainresrev.2007.03.012 [DOI] [PubMed] [Google Scholar]
- Cusack, C. L. , Swahari, V. , Hampton Henley, W. , Michael Ramsey, J. , & Deshmukh, M. (2013). Distinct pathways mediate axon degeneration during apoptosis and axon‐specific pruning. Nature Communications, 4, 1876. 10.1038/ncomms2910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Silva, R. P. , & Gordon, S. (1999). Phagocytosis stimulates alternative glycosylation of macrosialin (mouse CD68), a macrophage‐specific endosomal protein. The Biochemical Journal, 338(Pt 3), 687–694. [PMC free article] [PubMed] [Google Scholar]
- Delgado‐Garcia, J. M. , del Pozo, F. , Spencer, R. F. , & Baker, R. (1988). Behavior of neurons in the abducens nucleus of the alert cat—III. Axotomized motoneurons. Neuroscience, 24(1), 143–160. 10.1016/0306-4522(88)90319-3 [DOI] [PubMed] [Google Scholar]
- Dermentzaki, G. , Politi, K. A. , Lu, L. , Mishra, V. , Perez‐Torres, E. J. , Sosunov, A. A. , … Przedborski, S. (2019). Deletion of Ripk3 prevents motor neuron death in vitro but not in Vivo. eNeuro, 6(1), ENEURO.0308‐18.2018. 10.1523/ENEURO.0308-18.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dopfer, E. P. , Minguet, S. , & Schamel, W. W. (2011). A new vampire saga: The molecular mechanism of T cell trogocytosis. Immunity, 35(2), 151–153. 10.1016/j.immuni.2011.08.004 [DOI] [PubMed] [Google Scholar]
- Echeverry, S. , Shi, X. Q. , Rivest, S. , & Zhang, J. (2011). Peripheral nerve injury alters blood‐spinal cord barrier functional and molecular integrity through a selective inflammatory pathway. Journal of Neuroscience, 31(30), 10819–10828. 10.1523/Jneurosci.1642-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmore, M. R. , Najafi, A. R. , Koike, M. A. , Dagher, N. N. , Spangenberg, E. E. , Rice, R. A. , … Green, K. N. (2014). Colony‐stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron, 82(2), 380–397. 10.1016/j.neuron.2014.02.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etoh, K. , & Fukuda, M. (2019). Rab10 regulates tubular endosome formation through KIF13A and KIF13B motors. Journal of Cell Science, 132(5), jcs226977. 10.1242/jcs.226977 [DOI] [PubMed] [Google Scholar]
- Fauré, J. , Lachenal, G. , Court, M. , Hirrlinger, J. , Chatellard‐Causse, C. , Blot, B. , … Sadoul, R. (2006). Exosomes are released by cultured cortical neurones. Molecular and Cellular Neurosciences, 31(4), 642–648. 10.1016/j.mcn.2005.12.003 [DOI] [PubMed] [Google Scholar]
- Fawcett, J. W. , Oohashi, T. , & Pizzorusso, T. (2019). The roles of perineuronal nets and the perinodal extracellular matrix in neuronal function. Nature Reviews. Neuroscience, 20(8), 451–465. 10.1038/s41583-019-0196-3 [DOI] [PubMed] [Google Scholar]
- Fogarty, M. J. (2019). Amyotrophic lateral sclerosis as a synaptopathy. Neural Regeneration Research, 14(2), 189–192. 10.4103/1673-5374.244782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvan, M. D. , Greenlee‐Wacker, M. C. , & Bohlson, S. S. (2012). C1q and phagocytosis: The perfect complement to a good meal. Journal of Leukocyte Biology, 92(3), 489–497. 10.1189/jlb.0212099 [DOI] [PubMed] [Google Scholar]
- Girardot, N. , Allinquant, B. , Langui, D. , Laquerriere, A. , Dubois, B. , Hauw, J. J. , & Duyckaerts, C. (2003). Accumulation of flotillin‐1 in tangle‐bearing neurones of Alzheimer's disease. Neuropathology and Applied Neurobiology, 29(5), 451–461. 10.1046/j.1365-2990.2003.00479.x [DOI] [PubMed] [Google Scholar]
- Gong, Y. N. , Guy, C. , Crawford, J. C. , & Green, D. R. (2017). Biological events and molecular signaling following MLKL activation during necroptosis. Cell Cycle, 16(19), 1748–1760. 10.1080/15384101.2017.1371889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong, Y. N. , Guy, C. , Olauson, H. , Becker, J. U. , Yang, M. , Fitzgerald, P. , … Green, D. R. (2017). ESCRT‐III acts downstream of MLKL to regulate necroptotic cell death and its consequences. Cell, 169(2), 286–300 e216. 10.1016/j.cell.2017.03.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graeber, M. B. , Streit, W. J. , & Kreutzberg, G. W. (1989). Formation of microglia‐derived brain macrophages is blocked by adriamycin. Acta Neuropathologica, 78(4), 348–358. 10.1007/BF00688171 [DOI] [PubMed] [Google Scholar]
- Grootjans, S. , Vanden Berghe, T. , & Vandenabeele, P. (2017). Initiation and execution mechanisms of necroptosis: An overview. Cell Death and Differentiation, 24(7), 1184–1195. 10.1038/cdd.2017.65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haimann, C. , Torri‐Tarelli, F. , Fesce, R. , & Ceccarelli, B. (1985). Measurement of quantal secretion induced by ouabain and its correlation with depletion of synaptic vesicles. The Journal of Cell Biology, 101(5 Pt 1), 1953–1965. 10.1083/jcb.101.5.1953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison, R. E. , Bucci, C. , Vieira, O. V. , Schroer, T. A. , & Grinstein, S. (2003). Phagosomes fuse with late endosomes and/or lysosomes by extension of membrane protrusions along microtubules: Role of Rab7 and RILP. Molecular and Cellular Biology, 23(18), 6494–6506. 10.1128/mcb.23.18.6494-6506.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez, D. , Torres, C. A. , Setlik, W. , Cebrian, C. , Mosharov, E. V. , Tang, G. , … Sulzer, D. (2012). Regulation of presynaptic neurotransmission by macroautophagy. Neuron, 74(2), 277–284. 10.1016/j.neuron.2012.02.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann, S. , Orlando, M. , Andrzejak, E. , Bruns, C. , Trimbuch, T. , Rosenmund, C. , … Ackermann, F. (2019). Light‐activated ROS production induces synaptic autophagy. The Journal of Neuroscience, 39(12), 2163–2183. 10.1523/JNEUROSCI.1317-18.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong, S. , Beja‐Glasser, V. F. , Nfonoyim, B. M. , Frouin, A. , Li, S. , Ramakrishnan, S. , … Stevens, B. (2016). Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science, 352(6286), 712–716. 10.1126/science.aad8373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito, D. , Imai, Y. , Ohsawa, K. , Nakajima, K. , Fukuuchi, Y. , & Kohsaka, S. (1998). Microglia‐specific localisation of a novel calcium binding protein, Iba1. Brain Research. Molecular Brain Research, 57(1), 1–9. 10.1016/s0169-328x(98)00040-0 [DOI] [PubMed] [Google Scholar]
- Ito, Y. , Ofengeim, D. , Najafov, A. , Das, S. , Saberi, S. , Li, Y. , … Yuan, J. (2016). RIPK1 mediates axonal degeneration by promoting inflammation and necroptosis in ALS. Science, 353(6299), 603–608. 10.1126/science.aaf6803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffrey, M. , Halliday, W. G. , Bell, J. , Johnston, A. R. , MacLeod, N. K. , Ingham, C. , … Fraser, J. R. (2000). Synapse loss associated with abnormal PrP precedes neuronal degeneration in the scrapie‐infected murine hippocampus. Neuropathology and Applied Neurobiology, 26(1), 41–54. 10.1046/j.1365-2990.2000.00216.x [DOI] [PubMed] [Google Scholar]
- Jiang, M. , Schuster, J. E. , Fu, R. , Siddique, T. , & Heckman, C. J. (2009). Progressive changes in synaptic inputs to motoneurons in adult sacral spinal cord of a mouse model of amyotrophic lateral sclerosis. The Journal of Neuroscience, 29(48), 15031–15038. 10.1523/JNEUROSCI.0574-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kettenmann, H. , Kirchhoff, F. , & Verkhratsky, A. (2013). Microglia: New roles for the synaptic stripper. Neuron, 77(1), 10–18. 10.1016/j.neuron.2012.12.023 [DOI] [PubMed] [Google Scholar]
- Kim, Y. S. , Choi, D. H. , Block, M. L. , Lorenzl, S. , Yang, L. , Kim, Y. J. , … Joh, T. H. (2007). A pivotal role of matrix metalloproteinase‐3 activity in dopaminergic neuronal degeneration via microglial activation. The FASEB Journal, 21(1), 179–187. 10.1096/fj.06-5865com [DOI] [PubMed] [Google Scholar]
- Kiryu‐Seo, S. , Gamo, K. , Tachibana, T. , Tanaka, K. , & Kiyama, H. (2006). Unique anti‐apoptotic activity of EAAC1 in injured motor neurons. The EMBO Journal, 25(14), 3411–3421. 10.1038/sj.emboj.7601225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konnecke, H. , & Bechmann, I. (2013). The role of microglia and matrix metalloproteinases involvement in neuroinflammation and gliomas. Clinical & Developmental Immunology, 2013, 914104–914115. 10.1155/2013/914104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowal, J. , Arras, G. , Colombo, M. , Jouve, M. , Morath, J. P. , Primdal‐Bengtson, B. , … Thery, C. (2016). Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proceedings of the National Academy of Sciences of the United States of America, 113(8), E968–E977. 10.1073/pnas.1521230113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman, A. R. (1971). The axon reaction: A review of the principal features of perikaryal responses to axon injury. International Review of Neurobiology, 14, 49–124. 10.1016/s0074-7742(08)60183-x [DOI] [PubMed] [Google Scholar]
- Linda, H. , Shupliakov, O. , Ornung, G. , Ottersen, O. P. , Storm‐Mathisen, J. , Risling, M. , & Cullheim, S. (2000). Ultrastructural evidence for a preferential elimination of glutamate‐immunoreactive synaptic terminals from spinal motoneurons after intramedullary axotomy. The Journal of Comparative Neurology, 425(1), 10–23. [PubMed] [Google Scholar]
- Liu, L. , Aldskogius, H. , & Svensson, M. (1998). Ultrastructural localization of immunoglobulin G and complement C9 in the brain stem and spinal cord following peripheral nerve injury: An immunoelectron microscopic study. Journal of Neurocytology, 27(10), 737–748. 10.1023/a:1006950917973 [DOI] [PubMed] [Google Scholar]
- Lundborg, G. (2003). Richard P. Bunge memorial lecture. Nerve injury and repair—A challenge to the plastic brain. Journal of the Peripheral Nervous System, 8(4), 209–226. 10.1111/j.1085-9489.2003.03027.x [DOI] [PubMed] [Google Scholar]
- Lüningschrör, P. , & Sendtner, M. (2018). Autophagy in the presynaptic compartment. Current Opinion in Neurobiology, 51, 80–85. 10.1016/j.conb.2018.02.023 [DOI] [PubMed] [Google Scholar]
- Mattson, M. P. , Keller, J. N. , & Begley, J. G. (1998). Evidence for synaptic apoptosis. Experimental Neurology, 153(1), 35–48. 10.1006/exnr.1998.6863 [DOI] [PubMed] [Google Scholar]
- Men, Y. , Yelick, J. , Jin, S. , Tian, Y. , Chiang, M. S. R. , Higashimori, H. , … Yang, Y. (2019). Exosome reporter mice reveal the involvement of exosomes in mediating neuron to astroglia communication in the CNS. Nature Communications, 10(1), 4136. 10.1038/s41467-019-11534-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mentis, G. Z. , Blivis, D. , Liu, W. , Drobac, E. , Crowder, M. E. , Kong, L. , … O'Donovan, M. J. (2011). Early functional impairment of sensory‐motor connectivity in a mouse model of spinal muscular atrophy. Neuron, 69(3), 453–467. 10.1016/j.neuron.2010.12.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mignogna, M. L. , & D'Adamo, P. (2018). Critical importance of RAB proteins for synaptic function. Small GTPases, 9(1–2), 145–157. 10.1080/21541248.2016.1277001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Möller, J. C. , Klein, M. A. , Haas, S. , Jones, L. L. , Kreutzberg, G. W. , & Raivich, G. (1996). Regulation of thrombospondin in the regenerating mouse facial motor nucleus. Glia, 17(2), 121–132. [DOI] [PubMed] [Google Scholar]
- Moran, L. B. , & Graeber, M. B. (2004). The facial nerve axotomy model. Brain Research. Brain Research Reviews, 44(2–3), 154–178. 10.1016/j.brainresrev.2003.11.004 [DOI] [PubMed] [Google Scholar]
- Navarro, X. , Vivo, M. , & Valero‐Cabre, A. (2007). Neural plasticity after peripheral nerve injury and regeneration. Progress in Neurobiology, 82(4), 163–201. 10.1016/j.pneurobio.2007.06.005 [DOI] [PubMed] [Google Scholar]
- Odorizzi, G. (2006). The multiple personalities of Alix. Journal of Cell Science, 119(Pt 15), 3025–3032. 10.1242/jcs.03072 [DOI] [PubMed] [Google Scholar]
- Okerlund, N. D. , Schneider, K. , Leal‐Ortiz, S. , Montenegro‐Venegas, C. , Kim, S. A. , Garner, L. C. , … Garner, C. C. (2017). Bassoon controls presynaptic autophagy through Atg5. Neuron, 93(4), 897–913 e897. 10.1016/j.neuron.2017.01.026 [DOI] [PubMed] [Google Scholar]
- Oliveira, A. L. , Thams, S. , Lidman, O. , Piehl, F. , Hokfelt, T. , Karre, K. , … Cullheim, S. (2004). A role for MHC class I molecules in synaptic plasticity and regeneration of neurons after axotomy. Proceedings of the National Academy of Sciences of the United States of America, 101(51), 17843–17848. 10.1073/pnas.0408154101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orozco, S. L. , Daniels, B. P. , Yatim, N. , Messmer, M. N. , Quarato, G. , Chen‐Harris, H. , … Oberst, A. (2019). RIPK3 activation leads to cytokine synthesis that continues after loss of cell membrane integrity. Cell Reports, 28(9), 2275–2287 e2275. 10.1016/j.celrep.2019.07.077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paolicelli, R. C. , Bergamini, G. , & Rajendran, L. (2019). Cell‐to‐cell communication by extracellular vesicles: Focus on microglia. Neuroscience, 405, 148–157. 10.1016/j.neuroscience.2018.04.003 [DOI] [PubMed] [Google Scholar]
- Paolicelli, R. C. , Bolasco, G. , Pagani, F. , Maggi, L. , Scianni, M. , Panzanelli, P. , … Gross, C. T. (2011). Synaptic pruning by microglia is necessary for normal brain development. Science, 333(6048), 1456–1458. 10.1126/science.1202529 [DOI] [PubMed] [Google Scholar]
- Pasparakis, M. , & Vandenabeele, P. (2015). Necroptosis and its role in inflammation. Nature, 517(7534), 311–320. 10.1038/nature14191 [DOI] [PubMed] [Google Scholar]
- Perry, V. H. , & O'Connor, V. (2010). The role of microglia in synaptic stripping and synaptic degeneration: A revised perspective. ASN Neuro, 2(5), e00047. 10.1042/AN20100024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters, A. , Palay, S. L. , & Webster, H. d. F. (1976). The fine structure of the nervous system: Neurons and their supporting cells. Philapelphia: Saunders Co. [Google Scholar]
- Petralia, R. S. , Schwartz, C. M. , Wang, Y. X. , Kawamoto, E. M. , Mattson, M. P. , & Yao, P. J. (2013). Sonic hedgehog promotes autophagy in hippocampal neurons. Biology Open, 2(5), 499–504. 10.1242/bio.20134275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollin, M. M. , McHanwell, S. , & Slater, C. R. (1991). The effect of age on motor neurone death following axotomy in the mouse. Development, 112(1), 83–89. [DOI] [PubMed] [Google Scholar]
- Raden, Y. , Shlomovitz, I. , & Gerlic, M. (2020). Necroptotic extracellular vesicles—Present and future. Seminars in Cell & Developmental Biology. (in press). 10.1016/j.semcdb.2020.08.011 [DOI] [PubMed] [Google Scholar]
- Re, D. B. , le Verche, V. , Yu, C. , Amoroso, M. W. , Politi, K. A. , Phani, S. , … Przedborski, S. (2014). Necroptosis drives motor neuron death in models of both sporadic and familial ALS. Neuron, 81(5), 1001–1008. 10.1016/j.neuron.2014.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rishal, I. , & Fainzilber, M. (2014). Axon‐soma communication in neuronal injury. Nature Reviews. Neuroscience, 15(1), 32–42. 10.1038/nrn3609 [DOI] [PubMed] [Google Scholar]
- Rotterman, T. M. , Akhter, E. T. , Lane, A. R. , MacPherson, K. P. , Garcia, V. V. , Tansey, M. G. , & Alvarez, F. J. (2019). Spinal motor circuit synaptic plasticity after peripheral nerve injury depends on microglia activation and a CCR2 mechanism. The Journal of Neuroscience, 39(18), 3412–3433. 10.1523/JNEUROSCI.2945-17.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotterman, T. M. , Nardelli, P. , Cope, T. C. , & Alvarez, F. J. (2014). Normal distribution of VGLUT1 synapses on spinal motoneuron dendrites and their reorganization after nerve injury. Journal of Neuroscience, 34(10), 3475–3492. 10.1523/Jneurosci.4768-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio, M. E. , & Wenthold, R. J. (1999). Differential distribution of intracellular glutamate receptors in dendrites. The Journal of Neuroscience, 19(13), 5549–5562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvany, S. , Casanovas, A. , Tarabal, O. , Piedrafita, L. , Hernandez, S. , Santafe, M. , … Esquerda, J. E. (2019). Localization and dynamic changes of neuregulin‐1 at C‐type synaptic boutons in association with motor neuron injury and repair. The FASEB Journal, 33(7), 7833–7851. 10.1096/fj.201802329R [DOI] [PubMed] [Google Scholar]
- Sasaki, S. , & Iwata, M. (1996). Synaptic loss in anterior horn neurons in lower motor neuron disease. Acta Neuropathologica, 91(4), 416–421. 10.1007/s004010050444 [DOI] [PubMed] [Google Scholar]
- Schafer, D. P. , Lehrman, E. K. , Kautzman, A. G. , Koyama, R. , Mardinly, A. R. , Yamasaki, R. , … Stevens, B. (2012). Microglia sculpt postnatal neural circuits in an activity and complement‐dependent manner. Neuron, 74(4), 691–705. 10.1016/j.neuron.2012.03.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schutz, B. (2005). Imbalanced excitatory to inhibitory synaptic input precedes motor neuron degeneration in an animal model of amyotrophic lateral sclerosis. Neurobiology of Disease, 20(1), 131–140. 10.1016/j.nbd.2005.02.006 [DOI] [PubMed] [Google Scholar]
- Sikorska, B. , Liberski, P. P. , Giraud, P. , Kopp, N. , & Brown, P. (2004). Autophagy is a part of ultrastructural synaptic pathology in Creutzfeldt‐Jakob disease: A brain biopsy study. International Journal of Biochemistry & Cell Biology, 36(12), 2563–2573. 10.1016/j.biocel.2004.04.014 [DOI] [PubMed] [Google Scholar]
- Sipe, G. O. , Lowery, R. L. , Tremblay, M. E. , Kelly, E. A. , Lamantia, C. E. , & Majewska, A. K. (2016). Microglial P2Y12 is necessary for synaptic plasticity in mouse visual cortex. Nature Communications, 7, 10905. 10.1038/ncomms10905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Šišková, Z. , Page, A. , O'Connor, v. , & Perry, V. H. (2009). Degenerating synaptic boutons in prion disease microglia activation without synaptic stripping. American Journal of Pathology, 175(4), 1610–1621. 10.2353/ajpath.2009.090372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Šišková, Z. , Reynolds, R. A. , O'Connor, V. , & Perry, V. H. (2013). Brain region specific pre‐synaptic and post‐synaptic degeneration are early components of neuropathology in prion disease. PLoS One, 8(1), e55004. 10.1371/journal.pone.0055004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solis, G. P. , Hulsbusch, N. , Radon, Y. , Katanaev, V. L. , Plattner, H. , & Stuermer, C. A. (2013). Reggies/flotillins interact with Rab11a and SNX4 at the tubulovesicular recycling compartment and function in transferrin receptor and E‐cadherin trafficking. Molecular Biology of the Cell, 24(17), 2689–2702. 10.1091/mbc.E12-12-0854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solsona, C. , Esquerda, J. E. , & Marsal, J. (1981). Effects of ouabain and electrical stimulation on the fine structure of nerve endings in the electric organ of Torpedo marmorata . Cell and Tissue Research, 220(4), 857–871. 10.1007/BF00210467 [DOI] [PubMed] [Google Scholar]
- Spejo, A. B. , & Oliveira, A. L. (2015). Synaptic rearrangement following axonal injury: Old and new players. Neuropharmacology, 96(Pt A), 113–123. 10.1016/j.neuropharm.2014.11.002 [DOI] [PubMed] [Google Scholar]
- Stephan, A. H. , Barres, B. A. , & Stevens, B. (2012). The complement system: An unexpected role in synaptic pruning during development and disease. Annual Review of Neuroscience, 35, 369–389. 10.1146/annurev-neuro-061010-113810 [DOI] [PubMed] [Google Scholar]
- Stevens, B. , Allen, N. J. , Vazquez, L. E. , Howell, G. R. , Christopherson, K. S. , Nouri, N. , … Barres, B. A. (2007). The classical complement cascade mediates CNS synapse elimination. Cell, 131(6), 1164–1178. 10.1016/j.cell.2007.10.036 [DOI] [PubMed] [Google Scholar]
- Sumner, B. E. (1975). A quantitative analysis of boutons with different types of synapse in normal and injured hypoglossal nuclei. Experimental Neurology, 49(2), 406–417. 10.1016/0014-4886(75)90097-7 [DOI] [PubMed] [Google Scholar]
- Sumner, B. E. , & Sutherland, F. I. (1973). Quantitative electron microscopy on the injured hypoglossal nucleus in the rat. Journal of Neurocytology, 2(3), 315–328. 10.1007/bf01104033 [DOI] [PubMed] [Google Scholar]
- Sunico, C. R. , Dominguez, G. , Garcia‐Verdugo, J. M. , Osta, R. , Montero, F. , & Moreno‐Lopez, B. (2011). Reduction in the motoneuron inhibitory/excitatory synaptic ratio in an early‐symptomatic mouse model of amyotrophic lateral sclerosis. Brain Pathology, 21(1), 1–15. 10.1111/j.1750-3639.2010.00417.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svensson, M. , & Aldskogius, H. (1993). Infusion of cytosine‐arabinoside into the cerebrospinal‐fluid of the rat‐brain inhibits the microglial cell‐proliferation after hypoglossal nerve injury. Glia, 7(4), 286–298. 10.1002/glia.440070404 [DOI] [PubMed] [Google Scholar]
- Tarabal, O. , Caraballo‐Miralles, V. , Cardona‐Rossinyol, A. , Correa, F. J. , Olmos, G. , Llado, J. , … Calderó, J. (2014). Mechanisms involved in spinal cord central synapse loss in a mouse model of spinal muscular atrophy. Journal of Neuropathology and Experimental Neurology, 73(6), 519–535. 10.1097/NEN.0000000000000074 [DOI] [PubMed] [Google Scholar]
- Tremblay, M. E. , Lowery, R. L. , & Majewska, A. K. (2010). Microglial interactions with synapses are modulated by visual experience. PLoS Biology, 8(11), e1000527. 10.1371/journal.pbio.1000527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay, M. E. , & Majewska, A. K. (2011). A role for microglia in synaptic plasticity? Communicative & Integrative Biology, 4(2), 220–222. 10.4161/cib.4.2.14506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan, S. K. , Kemp, Z. , Hatzipetros, T. , Vieira, F. , & Valdez, G. (2015). Degeneration of proprioceptive sensory nerve endings in mice harboring amyotrophic lateral sclerosis‐causing mutations. The Journal of Comparative Neurology, 523(17), 2477–2494. 10.1002/cne.23848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vukojicic, A. , Delestree, N. , Fletcher, E. V. , Pagiazitis, J. G. , Sankaranarayanan, S. , Yednock, T. A. , … Mentis, G. Z. (2019). The classical complement pathway mediates microglia‐dependent remodeling of spinal motor circuits during development and in SMA. Cell Reports, 29(10), 3087–3100 e3087. 10.1016/j.celrep.2019.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waites, C. L. , Leal‐Ortiz, S. A. , Okerlund, N. , Dalke, H. , Fejtova, A. , Altrock, W. D. , … Garner, C. C. (2013). Bassoon and piccolo maintain synapse integrity by regulating protein ubiquitination and degradation. The EMBO Journal, 32(7), 954–969. 10.1038/emboj.2013.27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall, A. A. , Condon, N. D. , Luo, L. , & Stow, J. L. (2019). Rab8a localisation and activation by Toll‐like receptors on macrophage macropinosomes. Philosophical Transactions of the Royal Society B: Biological Sciences, 374(1765), 20180151. 10.1098/rstb.2018.0151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, H. Y. , Sun, L. M. , Su, L. J. , Rizo, J. , Liu, L. , Wang, L. F. , … Wang, X. D. (2014). Mixed lineage kinase domain‐like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Molecular Cell, 54(1), 133–146. 10.1016/j.molcel.2014.03.003 [DOI] [PubMed] [Google Scholar]
- Watson, C. , Paxinos, G. , Kayalioglu, G. , & Heise, C. (2009). In Watson C., Paxinos G., & G K. (Eds.), The spinal cord (1st ed.). Amsterdam: Elsevier. [Google Scholar]
- Weinhard, L. , di Bartolomei, G. , Bolasco, G. , Machado, P. , Schieber, N. L. , Neniskyte, U. , … Gross, C. T. (2018). Microglia remodel synapses by presynaptic trogocytosis and spine head filopodia induction. Nature Communications, 9(1), 1228. 10.1038/s41467-018-03566-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, Y. , O'Toole, E. T. , Girard, M. , Ritter, B. , Messa, M. , Liu, X. , … de Camilli, P. (2014). A dynamin 1‐, dynamin 3‐ and clathrin‐independent pathway of synaptic vesicle recycling mediated by bulk endocytosis. eLife, 3, e01621. 10.7554/eLife.01621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada, J. , Nakanishi, H. , & Jinno, S. (2011). Differential involvement of perineuronal astrocytes and microglia in synaptic stripping after hypoglossal axotomy. Neuroscience, 182, 1–10. 10.1016/j.neuroscience.2011.03.030 [DOI] [PubMed] [Google Scholar]
- Yoon, S. , Bogdanov, K. , Kovalenko, A. , & Wallach, D. (2016). Necroptosis is preceded by nuclear translocation of the signaling proteins that induce it. Cell Death & Differentiation, 23(2), 253–260. 10.1038/cdd.2015.92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon, S. , Kovalenko, A. , Bogdanov, K. , & Wallach, D. (2017). MLKL, the protein that mediates necroptosis, also regulates endosomal trafficking and extracellular vesicle generation. Immunity, 47(1), 51–65 e57. 10.1016/j.immuni.2017.06.001 [DOI] [PubMed] [Google Scholar]
- Zerial, M. , & McBride, H. (2001). Rab proteins as membrane organizers. Nature Reviews. Molecular Cell Biology, 2(2), 107–117. 10.1038/35052055 [DOI] [PubMed] [Google Scholar]
- Zhong, Z. , Deane, R. , Ali, Z. , Parisi, M. , Shapovalov, Y. , O'Banion, M. K. , … Zlokovic, B. V. (2008). ALS‐causing SOD1 mutants generate vascular changes prior to motor neuron degeneration. Nature Neuroscience, 11(4), 420–422. 10.1038/nn2073 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1 Endogenous IgGs decorate recruited microglial cells at the MN surface 7 days postaxotomy. (a‐c) Spinal cord section showing an axotomized MN (delimited by a dashed line) covered by Iba1‐immunostained microglia (red); note the association of endogenous IgGs (green) with microglial profiles. Scale bar: 20 μm
Supplementary Figure 2 Spinal cord section immunostained for synaptophysin (green) showing ventral horn MN cell bodies (visualized by fluorescent Nissl staining, blue) in control (a and b) and postaxotomy (c and d) conditions, as indicated. Note the reduction of synaptophisin positive puncta near the surface of MN cell bodies after axotomy. Scale bar: 40 μm (valid for a‐c)
Supplementary Figure 3 Ultrastructural morphology of normal synaptic boutons (shaded in green) contacting MN cell bodies (shaded in red). (a) Two presynaptic boutons containing synaptic vesicles clustered in the active zones (arrows); details of synaptic vesicle morphology are seen in the enlarged panels (b and c), showing flattened (b) and round (c) vesicles characteristic of F‐type and S‐type terminals, respectively. (d) A C‐type synaptic bouton is shown, displaying the characteristic subsynaptic cistern (arrows) that can be observed enlarged in (e); the intersynaptic extracellular gap is shaded in yellow. m = mitochondria. Scale bars: 0.5 μm in (a) and (d); 150 nm in (b), (c) and (e)
Supplementary Figure 4 Ultrastructural analysis of the periphery of MN cell bodies, and adjacent synaptic afferents and cell structures in the spinal cord. (a) Three days after axotomy, the extracellular space (shaded in yellow) is widened and synaptic terminals (green) are partially detached from the MN surface (red); in addition, a number of extracellular membrane‐bound vesicles (encircled in red) can be seen “floating” in the extracellular space. (b) A detail of two terminal synaptic boutons (green) contacting MN surface (red) are partially detached but maintain focal adherence at the puncta adherentia sites (red arrows); note the presence of abundant extracellular vesicles (*) in the extracellular space (yellow). (c) A recruited microglial cell (shaded in blue) in the proximity of the MN surface and interacting with abundant extracellular pleomorphic vesicles (indicated with red circles), 3 days after axotomy. (d‐f) A high magnification detail of the morphology of extracellular vesicles (arrows) near the MN surface, 3 days after axotomy. Scale bars: 1 μm (a‐c) and 500 nm (d‐f)
Supplementary Figure 5 Electron micrograph of a MN cell body (shaded in red) showing a marked disruption of intracellular organelle organization. (a) In the vicinity of the MNs, an altered microglial cell is seen displaying vacuolar changes in the endoplasmic reticulum and other organelles. Altered synaptic afferents are shaded in green, and the extracellular space at the MN‐microglia interface (shaded in yellow) can be seen expanded. A detailed view of MN cytoplasmic disorganization is shown at higher magnification in (b). Scale bars: (a) and (b) = 2.5 μm
Supplementary Figure 6 (a,b) An isolated MN cell body, 7 days postaxotomy, in a squash preparation immunostained for CD63 (green) and SV2 (red). Red spots, corresponding to afferent synaptic boutons, are seen covering the surface of the MN soma, in which abundant CD63 positive puncta can be seen. In the enlarged boxed area (a), the relationship between larger CD63‐positive spots and afferent boutons can be observed. Scale bars: a = 2 μm, b = 10 μm
Supplementary Figure 7 (a,b) Ventral horn MN cell bodes (delimited by dashed lines) double immunostained for PDC6IP‐Alix (green) and Iba1 (red) in control and 7 days postaxotomy. The axotomized MN can be identified by recruited Iba1‐positive microglial cells. However, the pattern of PDC6IP‐Alix does not show identifiable changes following axotomy. Scale bar: 20 μm (valid for a)
Supplementary Figure 8 Control and 7‐day axotomized MN cell bodies (delimited by dotted lines) immunostained for either Rab4 (a, b) or Rab5 (c, d) (green) combined with Iba1 (red). Note that the recruitment of Iba1‐positive microglial cells around axotomized MNs (b and d). Rab4 and Rab5‐immunoreactivity is increased in neuronal processes (arrows) adjacent to injured MNs. Scale bars: Scale bar: d = 20 μm (valid for a‐c)
Data Availability Statement
All the data and original images of this article are available upon reasonable request by e‐mailing to the corresponding author.