

Abstract
The Hepatitis C virus nonstructural protein 5A (NS5A) is a membrane‐associated protein involved in multiple steps of the viral life cycle. Direct‐acting antivirals (DAAs) targeting NS5A are a cornerstone of antiviral therapy, but the mode‐of‐action of these drugs is poorly understood. This is due to the lack of information on the membrane‐bound NS5A structure. Herein, we present the structural model of an NS5A AH‐linker‐D1 protein reconstituted as proteoliposomes. We use highly sensitive proton‐detected solid‐state NMR methods suitable to study samples generated through synthetic biology approaches. Spectra analyses disclose that both the AH membrane anchor and the linker are highly flexible. Paramagnetic relaxation enhancements (PRE) reveal that the dimer organization in lipids requires a new type of NS5A self‐interaction not reflected in previous crystal structures. In conclusion, we provide the first characterization of NS5A AH‐linker‐D1 in a lipidic environment shedding light onto the mode‐of‐action of clinically used NS5A inhibitors.
Keywords: cell-free synthesis, hepatitis C virus, NS5A, paramagnetic relaxation enhancement, solid-state NMR
The membrane orientation of the hepatitis C virus NS5A protein was assessed by combining a cell‐free protein synthesis approach with highly sensitive 1H‐detected solid‐state NMR. Insertion of lipids chelated with a paramagnetic Gd3+ ion allowed to orient the protein with respect to its membrane anchor using PRE. This information allowed to propose a model for the interaction of NS5A with a direct acting antiviral.

Introduction
Hepatitis C virus (HCV) infection is a major cause of chronic hepatitis, with approximately 70 million infected individuals worldwide, which frequently further develops into liver cirrhosis and hepatocellular carcinoma. [1] HCV is an enveloped virus of the Flaviviridae family with a single‐stranded RNA genome. The open reading frame is translated into a single polyprotein which is further processed into ten individual membrane‐bound proteins (reviewed in [2, 3, 4]). Structural proteins encoded in the N‐terminal part of the HCV polyprotein serve as building blocks of viral particles, whereas nonstructural proteins in the C‐terminal part contribute to the formation of membrane‐bound replication organelles and viral genome amplification (reviewed in [5, 6, 7, 8, 9, 10]). NS5A is a RNA‐binding protein exerting multiple functions in the viral life cycle.[ 11 , 12 ] It is involved in viral genome replication, the assembly of infectious virus particles and the counteraction of cellular antiviral defense. Viral replication complexes contain nonstructural proteins (NS) 3/4A, 4B, 5A and 5B, and consist of double‐membrane vesicles (DMV) that are derived from the ER membrane, a process orchestrated mainly by NS5A, and more precisely AHD1.[ 13 , 14 ] NS5A is a 447 amino acids long monotopic membrane phosphoprotein lacking enzymatic activity and exerting its multiple functions by acting as central interaction hub with viral and cellular proteins.[ 15 , 16 ] For a long time NS5A was considered as non‐druggable, but high‐content screens with cell‐based HCV systems identified direct‐acting antivirals (DAAs) targeting NS5A and blocking virus replication and assembly with exceptionally high potency. [17] For that reason, NS5A inhibitors became a cornerstone of antiviral therapy, allowing virus elimination in more than 95 % of treated individuals. While the exact mode‐of‐action of NS5A inhibitors remains largely unknown, it is assumed that membrane association of NS5A [18] is required for its interaction with members of this drug class such as Daclatasvir. [19] Binding of Daclatasvir for example also forms, besides evidence from structural biology,[ 20 , 21 , 22 ] biochemistry[ 23 , 24 ] and reverse genetics, [25] one of the strongest arguments in favor of a dimeric state of NS5A in cells. Indeed, its palindromic topology strongly supports binding across the dimer interface of the NS5A protein. [17] Notably, the majority of resistance mutations cluster in the membrane‐proximal region, often in the linker connecting the N‐terminal amphipathic α‐helix (AH) with domain 1(D1).[ 17 , 26 , 27 ] However, the precise mode‐of‐action is unknown because of the lack of structural information of NS5A in association with membranes.
The 3D structure of full length NS5A has not been solved because every (sub)domain requires a distinct structural approach. Therefore, only structures of individual domains are known. First, the helical structure of the AH, representing the membrane anchor, was determined in detergent micelles by solution‐state NMR. [28] Second, X‐ray crystallography revealed for D1 several structures with a similar monomer, but different dimer interfaces.[ 21 , 29 , 30 ] Finally, D2 and D3 were determined to form, in isolation[ 31 , 32 ] and together with D1, [33] ensemble (un)structures, characterizing them as being intrinsically disordered according to solution‐state NMR.[ 31 , 32 , 34 ] The signals of D2/D3 and D1D2D3 mostly overlay, which indicates that there is only limited interactions between the domains 2/3 and D1. [33] Despite the central role membrane lipids play in NS5A function, they were absent in all these structural studies. Notably, the orientation of the protein with respect to the lipids remained obscure. So far, structural studies in presence of the membrane anchor and lipids have been prevented by the fact that both sample preparation and structural analyses are particularly demanding for NS5A. This is indeed often the case for integral membrane proteins, but also for peripheral membrane proteins anchored to the membrane through an independently interacting domain. As a consequence, the position of membrane‐anchored proteins with respect to the lipid bilayer could in some cases be approximated for instance using solution NMR and micelles,[ 35 , 36 , 37 ] but is still mainly predicted using computational methods that account for the hydrophobic, hydrogen bonding and electrostatic interactions with the anisotropic water‐lipid environment.[ 29 , 38 ]
Only recently solid‐state NMR, one of the most adequate methods to study membrane‐bound proteins,[ 39 , 40 , 41 , 42 , 43 , 44 ] increased its mass sensitivity by a spectacular factor of >100 through the more sensitive proton‐detection possible under fast magic‐angle spinning (MAS) NMR.[ 45 , 46 , 47 , 48 , 49 ] This technical advance makes the method now compatible with the analysis of an important class of proteins, which are eukaryotic membrane proteins from viruses[ 50 , 51 ] and humans.[ 52 , 53 ] As they can often only be produced in relatively small quantities (e.g. <1 mg), these new approaches are central for their structural investigation. In this context, the measurement of paramagnetic relaxation enhancements which allow to probe intermolecular distances,[ 54 , 55 , 56 , 57 , 58 , 59 , 60 ] surface accessibility, and ligand binding,[ 61 , 62 , 63 , 64 , 65 , 66 ] and the orientation of a protein with respect to the lipid layer[ 67 , 68 , 69 , 70 , 71 , 72 ] opens now also to this type of proteins.
We here show that sub‐milligram amounts of NS5A AH‐linker‐D1 (which for simplicity in the following we will refer to as NS5A‐AHD1) can be produced, directly in detergent‐solubilized form, using eukaryotic wheat‐germ cell‐free protein synthesis in presence of mild detergents, and that it can be reconstituted for structural studies in a phospholipid environment. Using these samples, we could record high‐quality proton‐detected NMR spectra of NS5A‐AHD1 allowing for de novo sequential assignments and thus for structural and dynamic analysis with single amino‐acid‐residue resolution. Based on this, we find that the AH domain exhibits dynamics very different from the D1 domain and determine the orientation of D1 relative to the membrane plane using NMR paramagnetic relaxation enhancements. Our results suggest that D1 monomers undergo a so far unreported type of self‐interaction upon membrane association.
Results
Cell‐free‐synthesized and lipid‐reconstituted NS5A‐AHD1 yields high‐quality NMR spectra
We prepared the NS5A‐AHD1 protein, comprising the membrane‐anchoring AH, a linker region and the globular D1 domain (HCV strain JFH1), using wheat‐germ cell‐free protein synthesis (WG‐CFPS). This produced NS5A‐AHD1 in a solubilized form through direct addition of mild detergents to the reaction, at protein yields compatible with structural studies. The sample preparation process is summarized in Figures 1 and S1.
Figure 1.
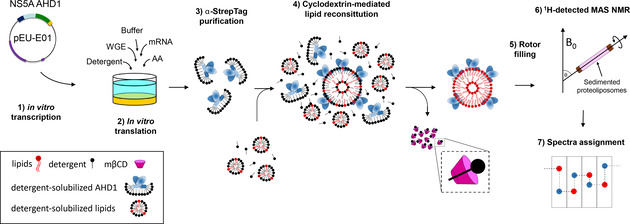
NMR sample preparation of NS5A‐AHD1 protein. In vitro transcribed mRNA (1) was mixed with wheat‐germ extract (WGE), labelled amino acids (AA) and detergent for protein synthesis using a bilayer reaction (2). AHD1 protein was purified on a Strep‐Tactin column in one step using the Strep‐tag (3). NS5A‐AHD1 was reconstituted by cyclodextrin (mβCD)‐mediated removal of detergent molecules (4).[ 51 , 73 ] NS5A‐AHD1 proteoliposomes were sedimented into a 0.7 mm rotor (5) for NMR spectra acquisition (6) followed by protein backbone assignment (7).
In order to compensate for the small sample amount, we systematically used detection of the sensitive 1H spins at around 100 kHz MAS.[ 47 , 48 , 49 , 74 ] We first recorded a fingerprint hNH spectrum of NS5A‐AHD1 (Figure 2 a), which revealed narrow proton lines (100±20 Hz (full width at half maximum) at 100 kHz MAS and 850 MHz proton frequency) and showed good spectral dispersion. To further explore the protein's fingerprint, we recorded a 3D hCANH spectrum of which the different planes are overlaid with the hNH 2D (Figure 2 b). One can see that most signals are observed in both spectra, thus showing sufficient signal‐to‐noise ratio (SNR). 131 resonances out of the 204 expected ones (one per residue, except prolines) are detected. Thus, while the majority of amino acids are seen with a good SNR of 3–10 in these spectra using dipolar couplings for polarization transfers, a non‐negligible part of the protein shows disorder or dynamic behavior on the μs‐ms time scale, which explains the absence of the remaining resonances.
Figure 2.
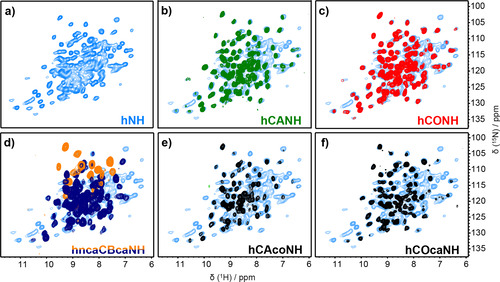
2D and all planes from 3D spectra of uniformly 2H/13C/15N labelled NS5A‐AHD1. a) 2D hNH spectrum in light blue. b–f) Overlay of all signal‐containing NH planes along the carbon dimension of hCANH, hCONH, hCAcoNH, hCOcaNH and hncaCBcaNH 3D spectra, on the 2D hNH of panel (a) as background.
To summarize, we produced a NS5A‐AHD1 NMR sample harnessing synthetic biology approaches, which resulted in high‐quality spectra of the membrane‐bound protein allowing sequential assignments (vide infra). While parts of the protein are not visible due to dynamics, no signs for aggregation are observed.
Resonance assignments by combining 3D spectroscopy and selective labeling
We recorded 3D spectra for sequential assignment and Figure 2 c–f shows the 2D HN planes of the 3D NS5A‐AHD1 hCONH, hncaCBcaNH, hCAcoNH, and hCOcaNH[ 46 , 48 , 75 , 76 ] spectra, overlaid onto the 2D hNH spectrum (experimental parameters are given in Tables S1 and S2). In the hCONH (Figure 2 c), nearly as many peaks as in the hCANH are present, indicating that inter‐residue connections can be obtained for most signals. Additional inter‐residue information can be obtained from the hCAcoNH, hCOcaNH and hncaCBcaNH 3D spectra, which show however a smaller number of resonances. [75]
The identification of starting and anchor points is key in the de novo sequential assignment process, and amino‐acid selective labeling, as can be obtained from cell‐free synthesis,[ 77 , 78 , 79 , 80 , 81 ] was in this context central for success. We prepared three different selectively labelled samples (Figure S2, Table S3 and Table S4) on which both 3D hCANH and hCONH experiments were recorded (Figure 3). The hCONH should show only signals for labelled pairs, and indeed for the ten G/T/Y pairs in the sequence, nine signals are observed in the hCONH spectrum (Figure 3 g), meaning that only one signal remained unobserved.
Figure 3.
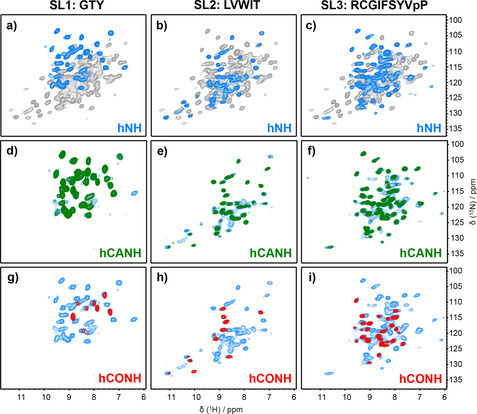
2D and 3D spectra of selectively labelled NS5A‐AHD1 samples. a–c) Two‐dimensional hNH spectra of selectively labelled samples in blue are overlaid on the hNH spectra of uniformly labelled sample in gray. d–i) Overlay of all signal‐containing NH planes along the carbon dimension of hCANH (green) and hCONH (red) 3D spectra, on the 2D hNH spectra (blue).
Using all spectra, we could assign, in a sequential manner, 87 residues (for representative strip plots and connectivities see Figures S3a,b). We also verified assignments using FLYA [82] (Figure S4). Resulting assignments are indicated in Figures S5 and 4a, and are deposited at the BMRB (50380). As seen in Figure 4 a, only few strong peaks remain unassigned, and completeness is 66 %. Unassigned residues concentrate in the N‐ and C‐terminal parts, as well as in three loop regions (Figure 4 c).
Figure 4.

NS5A‐AHD1 resonance assignments. a) Planes from three sections of the hCANH. b) NS5A‐AHD1 secondary chemical shifts (δΔCα‐δΔCβ). Three negative values in a row are indicative for β strands. TALOS+ [85] secondary structure plotted above the sequence in green, and from the X‐ray structure [29] (PDB 1zh1) of NS5A D1 in white. The N‐terminal helix [28] (PDB 1r7E) is colored in white. Grey boxes the indicate the length of protein constructs used. Grey bars show glycine residues. c) Residues sequentially assigned are colored in green, residues without assignment are mapped in grey on the monomer of a homology model based on the x‐ray structure (PDB 1zh1 [29] ).
We calculated secondary chemical shifts [83] (δΔCα‐δΔCβ) (after correcting for 2H isotope shift [84] ) and predicted secondary structure with TALOS+. [85] The β‐strands from secondary chemical shifts and those predicted by TALOS+ [85] (Figure 4 b, grey) are very similar to those observed in the X‐ray structure 1zh1 [29] of the isolated NS5A D1 domain (Figure 4 b).
The NMR resonance assignments obtained are fully de novo, meaning that no previous knowledge was available from solution or 13C‐detected chemical shifts, which has to our knowledge not been achieved before for a membrane‐bound protein using such small quantities. A highly homogenous sample, versatile selective labeling through cell‐free protein synthesis, high magnetic fields, and most importantly proton‐detected fast MAS were enabling here.
The membrane anchor is either flexible or disordered
Interestingly, no assignments could be obtained for the N‐terminal 40 residues, including the N‐terminal amphipathic AH, as well as for the C‐terminal 39 residues (including the 11 amino‐acid tag). Furthermore, the three longer loops are largely missing in the spectra.
We therefore designed selective‐labeling scheme 2 (SL2, Tables S3, S4) to clarify whether resonances of AH could not be assigned due to overlap, or because they are simply absent from the spectra. In the hCONH spectrum of this sample, 21 resonances for pairs were expected (Figure S2), with 8 in AH, 10 in D1 and 3 in the C‐terminal. However, only 12 resonances were visible, out of which 9 in D1, and 3 from backfolded tryptophan side chains. This shows that the AH domain is indeed not visible in the spectra at the given signal to noise. This suggests AH internal dynamics which compromise the polarization transfer or lead to line broadening beyond detection. Alternatively, it could be significantly statically disordered; this is however unlikely, since no featureless broad peaks are observed.
We thus used the SL2 sample to further investigate whether the dynamic behavior of the AH domain is impacted by the lipid environment, and tested different lipid compositions, lipid‐to‐protein ratios, as well as temperatures above and below the lipid phase transition, using a 2D hcoNH spectrum as read out. Figure 5 a shows the hNH (blue), all planes of 3D hCONH (red), and 2D hcoNH (black) spectra recorded under standard conditions. Panels Figure 5 b–f show for reference the hNH from Figure 5 a, peaks from 3D hCONH as red crosses, and in black 2D hcoNH spectra recorded under the different conditions detailed in the panels. One can see that, apart from some variations in intensity (Figure S6, Table S5), spectra appeared very similar, and no additional strong peaks belonging to the AH domain can be seen. Still, some weak signals already present at higher temperatures in the 2D hcoNH grow slightly more intense at lower temperatures (highlighted by red arrows in Figure 5), which could hint at a slowdown of molecular motions. It is however unlikely that the dynamics of the AH domain can become sufficiently slow in the studied temperature range, nor do the data support that the domain is aggregated due to a lack of lipids in the LPR 0.5 condition. No additional signals were observed when replacing the HN CP steps by INEPT transfers neither, as shown in Figure S7. This points to μs–ms dynamics of the AH domain, which stayed dynamic under all conditions investigated.
Figure 5.
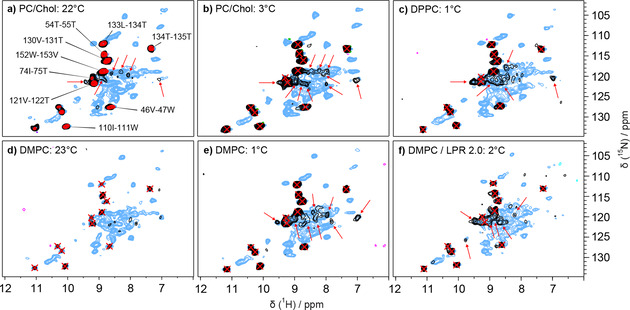
Impact of lipid environment and temperature on NS5A‐AHD1. SL2 NS5A‐AHD1 protein was reconstituted into PC/Chol (a, b), DPPC (c) or DMPC (d–f) lipids at LPR 0.5 (a–e) or at LPR 2 (f). Spectra in PC/Chol are recorded above the lipid phase transition temperature (T m) (T m=−6 °C), while those in DPPC are recorded below (T m=41 °C). Spectra in DMPC are recorded around and below (T m=24 °C). 2D hcoNH (black) and hNH spectra (blue) were acquired at 60 kHz in 0.7 mm rotors at conditions close to room temperature (a, d) or close to 0 °C (b, c, e, f). All planes from hCONH 3D spectra (red) of a standard SL2 sample were overlaid on the 2D hcoNH and hNH in (a) used as reference. Red arrows in (a), (b) and (c) point out newly detected resonances in the hcoNH.
We thus conclude that the AH domain, as well as several loops, remain flexible in the lipid‐bound NS5A‐AHD1 protein. Dynamic behavior on the μs–ms time scale is a common observation in multidomain proteins, and NS5A‐AHD1 is no exception here. This behavior is often at the origin of poor crystal growth in X‐ray studies, and also of signal loss in cryo‐EM due to frozen‐in disorder at cryogenic temperatures. Attempts to slow down the motion sufficiently to observe sizeable NMR signals failed. Lower temperatures were not investigated, since below the freezing point, spectral resolution is degraded in NMR spectra. Direct observation of the detailed conformation thus remains hidden; but in contrast to other techniques, NMR can indirectly investigate the orientation of the D1 part of the protein with respect to the lipid‐bound anchor through paramagnetic relaxation enhancements. The results from this type of experiments for NS5A‐AHD1 will be described below.
PREs reveal the interaction interface of NS5A‐AHD1 with lipids
To determine the orientation of the D1 domain with respect to the lipid membrane, we measured the protein paramagnetic relaxation enhancement (PRE)[ 80 , 81 ] as induced by Gd3+ paramagnetic centers[ 61 , 86 ] located within molecular cages chelated to the lipid headgroups (Gd3+‐DPPE[ 69 , 70 , 71 ]). PREs thus strongest affect membrane‐proximal regions of the protein. We added Gd3+‐DPPE (and diamagnetic Lu3+‐DPPE as reference) at a ratio of 1:20 to the lipid mixture during reconstitution, and measured relative peak intensities of NS5A‐AHD1 in 2D hNH (Figure 6 a), as well as 3D hCANH spectra (Figure 6 b) in the presence of Gd3+ or Lu3+, respectively (Figure 6 a). We then measured peak attenuations in both 2D and 3D spectra, and used, where resolved, information from the 2D because of the better SNR. Peaks overlapped in 2D were measured in the 3D hCANH spectra. Figure 6 c (and Table S6) report the resulting attenuations, with three classes (more than 0.4; 0.4–0.6; less than 0.6) color coded. Figure 6 d shows the measured attenuations color coded on a NS5A‐AHD1 homology model (PDB 1zh1 (D1) [29] and 1r7e (AH) [28] ). One can see that the different classes clearly separate into distinct regions on the 3D structure (in the orientation shown strong attenuation at the bottom, and weak attenuations on the top). This allows to establish the orientation of the D1 monomer relative to the AH, which is in interaction with the lipids containing the paramagnetic tags. The larger the PRE, the closer the residues are to the lipids, as shown in Figure 6 d, where the amphipathic helix anchor, as well as its localization with respect to the membrane, are sketched. Y93, the main site for NS5A DAA resistance mutations, is highlighted in yellow and points towards the lipid plane in this model.
Figure 6.
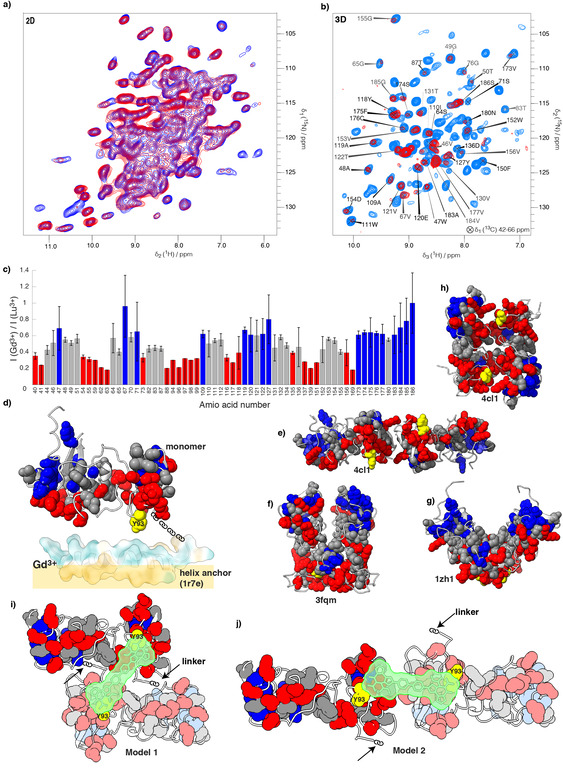
Membrane orientation of NS5A‐AHD1. a) Overlaid 2D hNH spectra of the NS5A‐AHD1 protein in the presence of Lu3+‐DPPE (blue) or Gd3+‐DPPE lipids (red) mixed with PC/Chol lipids. b) Overlaid 3D hCANH spectra of the NS5A‐AHD1 protein in the presence of Lu3+‐DPPE (light blue) or Gd3+‐DPPE lipids (red) mixed with PC/Chol lipids. c) Ratios of peak heights in the presence of Gd3+‐DPPE lipids (I (Gd3+)) or of Lu3+‐DPPE lipids (I (Lu3+)) extracted from the 2D and 3D correlation spectra. Individual values are shown in Table S6. The PREs of signals that vanished in the Gd3+ spectra were estimated to be equal or smaller than the ratio of noise in the Gd3+ spectra to the signal intensity in the Lu3+ spectra. These PREs can be recognized by the asymmetric error bars highlighting them as being the lower limit of attenuation. Values were normalized relative to residue S186. Attenuations are color coded according to 1–0.6 (blue); 0.6–0.4 grey; and 0.4–0 red. d) PREs color coded on the homology models produced by Swiss‐Model [87] based on X‐ray structure of D1 (one monomer) of 1zh1 [29] as a monomer and 1r7e [28] of the HA. The amphipathic helix is color coded according to hydrophobicity using ChimeraX, [88] and shown with its lipophilic part pointing towards the lipid membrane (yellow rectangle). The blue‐grey‐red qualification shows increasing PRE. The chain between the monomer and AH represents the linker. The main site for NS5A DAA resistance mutations, Y93, is shown in yellow. e) to h) represent dimers from the homology models based on the indicated crystal structures (1zh1, [29] 3fqm, [21] 4cl1 [30] ). None of them yields a consistent pattern of the PRE towards a fictitious lipid membrane. i, j) Sketches of two possible dimer models compatible with the PRE data. The monomers were positioned (by hand) such that Daclatasvir (in green) can contact in a symmetric manner both Y93 (yellow) side chains. All images of the molecular structures have been produced by ChimeraX. [88]
The three crystal structures of D1 available[ 21 , 29 , 30 ] show very similar monomer structures but quite different dimer arrangements, as shown in panels e–h in Figure 6. From the PREs reported on these structures, one can deduce that none of the reported dimers yields a PRE pattern that would be consistent with the membrane associated D1 dimer. This leads us to suspect that the dimer structures in the X‐ray crystal structures are in all cases determined by the crystal packing, and are at variance to the D1 structure of NS5A associated with a membrane. Instead, our data would be compatible for example, with a flat dimer arrangement comprising a twofold symmetry around the vertical axis (see Discussion).
Discussion
Solid‐state NMR has recently emerged as a versatile approach to investigate membrane‐associated proteins, and we show herein that it can be used to successfully study membrane‐bound NS5A‐AHD1, which has escaped structural characterization so far. Our NMR analysis is based on a de novo sequential assignment of NS5A‐AHD1, made possible by protein samples synthesized using a cell‐free approach, and fast MAS 1H‐detected solid‐state NMR experiments. Sequential assignments were obtained, without any prior knowledge from solution or solid‐state NMR chemical shifts. Amino‐acid‐selective labeling, facilitated in cell‐free synthesis, was important in this context.
Several parts of NS5A‐AHD1 are not observed in any of the NMR spectra recorded here. Missing signals from several residues are common in protein spectra, also for proton‐detected ones as in the recent example of Rpo4/7.[ 89 , 90 ] Prominently, resonances from the AH segment remained unobserved, indicating that it shows dynamic behavior. This feature may be related to the multi‐functional roles of the membrane‐associated NS5A, which forms complexes with several host proteins of relevance in various steps of the viral replication cycle.[ 18 , 19 , 91 , 92 ]
The PRE measurements allowed us to discriminate between different possible orientations of the protein with respect to the lipid plane. While intermolecular distance restraints would be highly precious to support these data, their measurement would need dissociation of the dimers, and reassembly thereof from differentially labelled NS5A monomers into mixed labeled dimers.[ 93 , 94 ] No conditions providing native reassembly of NS5A dimers have been described, and their development is arduous with the small sample amounts produced from cell‐free synthesis. Also, reassembly could impact the dimer structure produced directly on exit from the ribosome, and thus probably close‐to‐native. The monomer, as extracted from the crystal structure [29] can clearly be positioned with respect to the lipid membrane, and this results in positioning Y93 towards the membrane interface (Figure 6 d). However, none of the dimers from the different crystal structures can explain the orientation we observed here. Thus, a NS5A dimer[ 21 , 29 , 30 ] must form differently in the presence of a lipid bilayer than in crystals. In order to establish a model of the membrane‐associated NS5A‐AHD1 dimer, we took several considerations into account. First, as the spectra do not show multiple resonances for a single atom, the dimer must be symmetric. Second, because of that and based on the PREs, the second monomer in the dimer must be in the same plane and oriented towards the membrane with the same interface. Third, Daclatasvir binding should be possible in a symmetric manner to both monomers across the NS5A dimer interface.[ 17 , 19 , 24 ] Forth, inhibitor binding should occur in proximity to the class‐defining resistance mutation, amino‐acid residue Y93, of both monomers. Two models compatible with these restraints were established, and are sketched in Figures 6 i, j. Of note, the observation that several NS5A DAA resistance mutations reside in the linker region, [27] favors model 1 over 2 for D1 dimerization, as it positions the drug in closer proximity to the linkers (indicated by the black arrows). These observations suggest a mode of action where the NS5A inhibitor localizes at the interface between D1 and lipids, in a position where it has the potential to interfere with, or disrupt, NS5A membrane interactions.
Conclusion
We have analyzed an NMR sample of the membrane‐anchored NS5A‐AHD1, and found exceptional spectral resolution with a sample amount of only 200 μg of protein which allowed us to assign the NMR spectrum to 66 %. The data revealed that D1 is rigid and well‐ordered, while the amphipathic AH is dynamic, and remains unobserved. Inference from PRE effects allowed us to deduce the orientation of D1 with respect to the membrane lipids layer. At variance to D1 dimer structures in crystals, in the presence of lipids D1 most likely forms a flat dimer creating a membrane proximal surface that includes the predominant site of NS5A DAA resistance mutations. These results do not only provide models for the mode‐of‐action of HCV direct‐acting antivirals, but also demonstrate the power of recently developed highly sensitive proton‐detection solid‐state NMR techniques suitable to analyze structures of eukaryotic membrane bound proteins often available only in small quantities.
Methods
Methods are described in detail in the SI Appendix, including plasmids used, cell‐free synthesis and lipid reconstitution of NS5A‐AHD1 proteoliposomes, and solid‐state NMR experiments with parameter tables.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was supported by the French Agence Nationale de Recherches sur le Sida et les Hépatites Virales (ANRS), the French Agence Nationale de Recherche (A.B., ANR‐14‐CE09‐0024B) and the LABEX ECOFECT (A.B., ANR‐11‐LABX‐0048) within the Université de Lyon program Investissements d'Avenir (A.B., ANR‐11‐IDEX‐0007), an ERC Advanced Grant (B.H.M., grant number 741863, FASTER), by the Swiss National Science Foundation (B.H.M., grant number 200020_188711). R.B. is supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation)—Project Number 112927078—TRR 83. We greatly acknowledge technical support by Riccardo Cadalbert, Andreas Hunkeler and Alexander Däpp (solid‐state NMR group, ETH Zurich). We thank François Penin for having initiated and supported the project.
V. Jirasko, A. Lends, N.-A. Lakomek, M.-L. Fogeron, M. E. Weber, A. A. Malär, S. Penzel, R. Bartenschlager, B. H. Meier, A. Böckmann, Angew. Chem. Int. Ed. 2021, 60, 5339.
Contributor Information
Ralf Bartenschlager, Email: Ralf.Bartenschlager@med.uni-heidelberg.de.
Beat H. Meier, Email: beme@ethz.ch.
Anja Böckmann, Email: a.bockmann@ibcp.fr.
References
- 1. Razavi H., Gastroenterology Clinics of NA 2020, 49, 179–189. [DOI] [PubMed] [Google Scholar]
- 2. Hoffman B., Liu Q., Liver Int. 2011, 31, 1449–1467. [DOI] [PubMed] [Google Scholar]
- 3. Niepmann M., Curr. Top. Microbiol. Immunol. 2013, 369, 143–166. [DOI] [PubMed] [Google Scholar]
- 4. Hellen C. U., Pestova T. V., J. Viral Hepatitis 1999, 6, 79–87. [DOI] [PubMed] [Google Scholar]
- 5. Penin F. O., Dubuisson J., Rey F. A., Moradpour D., Pawlotsky J.-M., Hepatology 2004, 39, 5–19. [DOI] [PubMed] [Google Scholar]
- 6. Bartenschlager R., Penin F., Lohmann V., André P., Trends Microbiol. 2011, 19, 95–103. [DOI] [PubMed] [Google Scholar]
- 7. Shulla A., Randall G., Curr. Opin. Virol. 2012, 2, 725–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lindenbach B. D., Rice C. M., Nat. Rev. Microbiol. 2013, 11, 688–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Suzuki T., Ishii K., Aizaki H., Wakita T., Adv. Drug Delivery Rev. 2007, 59, 1200–1212. [DOI] [PubMed] [Google Scholar]
- 10. Neufeldt C. J., Cortese M., Acosta E. G., Bartenschlager R., Nat. Rev. Microbiol. 2018, 16, 125–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tellinghuisen T. L., Evans M. J., von Hahn T., You S., Rice C. M., J. Virol. 2007, 81, 8853–8867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ross-Thriepland D., Harris M., J. Gen. Virol. 2015, 96, 727–738. [DOI] [PubMed] [Google Scholar]
- 13. Romero-Brey I., Berger C., Kallis S., Kolovou A., Paul D., Lohmann V., Bartenschlager R., MBio 2015, 6, e00759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Romero-Brey I., Merz A., Chiramel A., Lee J. Y., Chlanda P., Haselman U., Santarella-Mellwig R., Habermann A., Hoppe S., Kallis S., et al., PLoS Pathog. 2012, 8, e1003056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. de Chassey B., Navratil V., Tafforeau L., Hiet M. S., Aublin-Gex A., Agaugué S., Meiffren G., Pradezynski F., Faria B. F., Chantier T., et al., Mol. Syst. Biol. 2008, 4, 230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Reiss S., Rebhan I., Backes P., Romero-Brey I., Erfle H., Matula P., Kaderali L., Poenisch M., Blankenburg H., Hiet M.-S., et al., Cell Host Microbe 2011, 9, 32–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gao M., Nettles R. E., Belema M., Snyder L. B., Nguyen V. N., Fridell R. A., Serrano-Wu M. H., Langley D. R., Sun J.-H., O'Boyle D. R., et al., Nature 2010, 465, 96–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Elazar M., Cheong K. H., Liu P., Greenberg H. B., Rice C. M., Glenn J. S., J. Virol. 2003, 77, 6055–6061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Berger C., Romero-Brey I., Radujkovic D., Terreux R., Zayas M., Paul D., Harak C., Hoppe S., Gao M., Penin F., et al., Gastroenterology 2014, 147, 1094—1095, e25. [DOI] [PubMed] [Google Scholar]
- 20. Tellinghuisen T. L., Foss K. L., Treadaway J. C., Rice C. M., J. Virol. 2008, 82, 1073–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Love R. A., Brodsky O., Hickey M. J., Wells P. A., Cronin C. N., J. Virol. 2009, 83, 4395–4403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lambert S. M., Langley D. R., Garnett J. A., Angell R., Hedgethorne K., Meanwell N. A., Matthews S. J., Protein Sci. 2014, 23, 723–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hwang J., Huang L., Cordek D. G., Vaughan R., Reynolds S. L., Kihara G., Raney K. D., Kao C. C., Cameron C. E., J. Virol. 2010, 84, 12480–12491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lim P. J., Chatterji U., Cordek D., Sharma S. D., Garcia-Rivera J. A., Cameron C. E., Lin K., Targett-Adams P., Gallay P. A., J. Biol. Chem. 2012, 287, 30861–30873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shanmugam S., Nichols A. K., Saravanabalaji D., Welsch C., Yi M., PLoS Pathog. 2018, 14, e1007177-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fridell R. A., Qiu D., Valera L., Wang C., Rose R. E., Gao M., J. Virol. 2011, 85, 7312–7320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fridell R. A., Qiu D., Wang C., Valera L., Gao M., Antimicrob. Agents Chemother. 2010, 54, 3641–3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Penin F., Brass V., Appel N., Ramboarina S., Montserret R., Ficheux D., Blum H. E., Bartenschlager R., Moradpour D., J. Biol. Chem. 2004, 279, 40835–40843. [DOI] [PubMed] [Google Scholar]
- 29. Tellinghuisen T. L., Marcotrigiano J., Rice C. M., Nature 2005, 435, 374–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lambert S. M., Langley D. R., Garnett J. A., Angell R., Hedgethorne K., Meanwell N. A., Matthews S. J., Protein Sci. 2014, 23, 723–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Verdegem D., Badillo A., Wieruszeski J.-M., Landrieu I., Leroy A., Bartenschlager R., Penin F., Lippens G., Hanoulle X., J. Biol. Chem. 2011, 286, 20441–20454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hanoulle X., Badillo A., Verdegem D., Penin F., Lippens G., Protein Pept. Lett. 2010, 17, 1012–1018. [DOI] [PubMed] [Google Scholar]
- 33. Badillo A., Receveur-Brechot V., Sarrazin S., Cantrelle F.-X., Delolme F., Fogeron M.-L., Molle J., Montserret R., Böckmann A., Bartenschlager R., et al., Biochemistry 2017, 56, 3029–3048. [DOI] [PubMed] [Google Scholar]
- 34. Hanoulle X., Badillo A., Wieruszeski J.-M., Verdegem D., Landrieu I., Bartenschlager R., Penin F., Lippens G., J. Biol. Chem. 2009, 284, 13589–13601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Valentine K. G., Peterson R. W., Saad J. S., Summers M. F., Xu X., Ames J. B., Wand A. J., Structure 2010, 18, 9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liang B., Tamm L. K., Nat. Struct. Mol. Biol. 2016, 23, 468–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mercredi P. Y., Bucca N., Loeliger B., Gaines C. R., Mehta M., Bhargava P., Tedbury P. R., Charlier L., Floquet N., Muriaux D., et al., J. Mol. Biol. 2016, 428, 1637–1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lomize M. A., Pogozheva I. D., Joo H., Mosberg H. I., Lomize A. L., Nucleic Acids Res. 2012, 40, D370–D376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. McDermott A., Annu. Rev. Biophys. 2009, 38, 385–403. [DOI] [PubMed] [Google Scholar]
- 40. Franks W. T., Linden A. H., Kunert B., van Rossum B.-J., Oschkinat H., Eur. J. Cell Biol. 2012, 91, 340–348. [DOI] [PubMed] [Google Scholar]
- 41. Ulrich S. J., Glaubitz C., Acc. Chem. Res. 2013, 46, 2164–2171. [DOI] [PubMed] [Google Scholar]
- 42. Retel J. S., Nieuwkoop A. J., Hiller M., Higman V. A., Barbet-Massin E., Stanek J., Andreas L. B., Franks W. T., van Rossum B.-J., Vinothkumar K. R., et al., Nat. Commun. 2017, 8, 2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Schubeis T., Le Marchand T., Andreas L. B., Pintacuda G., J. Biomol. NMR 2018, 287, 140–152. [DOI] [PubMed] [Google Scholar]
- 44. Opella S. J., Marassi F. M., Arch. Biochem. Biophys. 2017, 628, 92–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Struppe J., Quinn C. M., Lu M., Wang M., Hou G., Lu X., Kraus J., Andreas L. B., Stanek J., Lalli D., et al., Solid State NMR 2017, 87, 117–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Barbet-Massin E., Pell A. J., Retel J. S., Andreas L. B., Jaudzems K., Franks W. T., Nieuwkoop A. J., Hiller M., Higman V., Guerry P., et al., J. Am. Chem. Soc. 2014, 136, 12489–12497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Agarwal V., Penzel S., Szekely K., Cadalbert R., Testori E., Oss A., Past J., Samoson A., Ernst M., Böckmann A., et al., Angew. Chem. Int. Ed. 2014, 53, 12253–12256; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 12450–12453. [Google Scholar]
- 48. Böckmann A., Ernst M., Meier B. H., J. Magn. Reson. 2015, 253, 71–79. [DOI] [PubMed] [Google Scholar]
- 49. Lecoq L., Schledorn M., Wang S., Smith-Penzel S., Malär A. A., Callon M., Nassal M., Meier B. H., Böckmann A., Front. Mol. Biosci. 2019, 6, 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. David G., Fogeron M.-L., Schledorn M., Montserret R., Haselmann U., Penzel S., Badillo A., Lecoq L., André P., Nassal M., et al., Angew. Chem. Int. Ed. 2018, 57, 4787–4791; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 4877–4882. [Google Scholar]
- 51. Jirasko V., Lakomek N.-A., Penzel S., Fogeron M.-L., Bartenschlager R., Meier B. H., Böckmann A., ChemBioChem 2020, 21, 1453–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Joedicke L., Mao J., Kuenze G., Reinhart C., Kalavacherla T., Jonker H. R. A., Richter C., Schwalbe H., Meiler J., Preu J., et al., Nat. Chem. Biol. 2018, 14, 284–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lehnert E., Mao J., Mehdipour A. R., Hummer G., Abele R., Glaubitz C., Tampé R., J. Am. Chem. Soc. 2016, 138, 13967—13974. [DOI] [PubMed] [Google Scholar]
- 54. Sengupta I., Nadaud P. S., Jaroniec C. P., Acc. Chem. Res. 2013, 46, 2117–2126. [DOI] [PubMed] [Google Scholar]
- 55. Pell A. J., Pintacuda G., Grey C. P., Prog. Nucl. Magn. Reson. Spectrosc. 2019, 111, 1–271. [DOI] [PubMed] [Google Scholar]
- 56. Wiegand T., Lacabanne D., Keller K., Cadalbert R., Lecoq L., Yulikov M., Terradot L., Jeschke G., Meier B. H., Böckmann A., Angew. Chem. Int. Ed. 2017, 56, 3369–3373; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 3418–3422. [Google Scholar]
- 57. Perez A., Gaalswyk K., Jaroniec C. P., MacCallum J. L., Angew. Chem. Int. Ed. 2019, 58, 6564–6568; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 6636–6640. [Google Scholar]
- 58. Öster C., Kosol S., Hartlmüller C., Lamley J. M., Iuga D., Oss A., Org M.-L., Vanatalu K., Samoson A., Madl T., et al., J. Am. Chem. Soc. 2017, 139, 12165–12174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Linser R., Fink U., Reif B., J. Am. Chem. Soc. 2009, 131, 13703–13708. [DOI] [PubMed] [Google Scholar]
- 60. Aucoin D., Xia Y., Theint T., Nadaud P. S., Surewicz K., Surewicz W. K., Jaroniec C. P., J. Struct. Biol. 2019, 206, 36–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ullrich S. J., Hölper S., Glaubitz C., J. Biomol. NMR 2014, 58, 27–35. [DOI] [PubMed] [Google Scholar]
- 62. Pintacuda G., Giraud N., Pierattelli R., Böckmann A., Bertini I., Emsley L., Angew. Chem. Int. Ed. 2007, 46, 1079–1082; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 1097–1100. [Google Scholar]
- 63. Pintacuda G., Otting G., J. Am. Chem. Soc. 2002, 124, 372–373. [DOI] [PubMed] [Google Scholar]
- 64. Clore G. M., Iwahara J., Chem. Rev. 2009, 109, 4108–4139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Keizers P. H. J., Ubbink M., Prog. Nucl. Magn. Reson. Spectrosc. 2011, 58, 88–96. [DOI] [PubMed] [Google Scholar]
- 66. Nitsche C., Otting G., Curr. Opin. Struct. Biol. 2018, 48, 16–22. [DOI] [PubMed] [Google Scholar]
- 67. Wang S., Munro R. A., Kim S.-Y., Jung K.-H., Brown L. S., Ladizhansky V., J. Am. Chem. Soc. 2012, 134, 16995–16998. [DOI] [PubMed] [Google Scholar]
- 68. Su Y., Mani R., Hong M., J. Am. Chem. Soc. 2008, 130, 8856–8864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Mazhab-Jafari M. T., Marshall C. B., Stathopulos P. B., Kobashigawa Y., Stambolic V., Kay L. E., Inagaki F., Ikura M., J. Am. Chem. Soc. 2013, 135, 3367–3370. [DOI] [PubMed] [Google Scholar]
- 70. Liu C., J. Biomol. NMR 2017, 68, 203–214. [DOI] [PubMed] [Google Scholar]
- 71. Raltchev K., Pipercevic J., Hagn F., Chem. Eur. J. 2018, 24, 5493–5499. [DOI] [PubMed] [Google Scholar]
- 72. Jaipuria G., Giller K., Leonov A., Becker S., Zweckstetter M., Chem. Eur. J. 2018, 24, 17606–17611. [DOI] [PubMed] [Google Scholar]
- 73. DeGrip W. J., Vanoostrum J., Bovee-Geurts P., Biochem. J. 1998, 330, 667–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Mandala V. S., Hong M., Curr. Opin. Struct. Biol. 2019, 183–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Penzel S., Smith A. A., Agarwal V., Hunkeler A., Org M.-L., Samoson A., Böckmann A., Ernst M., Meier B. H., J. Biomol. NMR 2015, 63, 165–186. [DOI] [PubMed] [Google Scholar]
- 76. Linser R., Dasari M., Hiller M., Higman V., Fink U., Lopez del Amo J. M., Markovic S., Handel L., Kessler B., Schmieder P., et al., Angew. Chem. Int. Ed. 2011, 50, 4508–4512; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 4601–4605. [Google Scholar]
- 77. Vinarov D. A., Lytle B. L., Peterson F. C., Tyler E. M., Volkman B. F., Markley J. L., Nat. Methods 2004, 1, 149–153. [DOI] [PubMed] [Google Scholar]
- 78. Tonelli M., Singarapu K. K., Makino S.-I., Sahu S. C., Matsubara Y., Endo Y., Kainosho M., Markley J. L., J. Biomol. NMR 2011, 51, 467–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Myshkin M. Y., Dubinnyi M. A., Kulbatskii D. S., Lyukmanova E. N., Kirpichnikov M. P., Shenkarev Z. O., J. Biomol. NMR 2019, 73, 531–544. [DOI] [PubMed] [Google Scholar]
- 80. Löhr F., Gebel J., Henrich E., Hein C., Dötsch V., J. Biomol. NMR 2019, 302, 50–63. [DOI] [PubMed] [Google Scholar]
- 81. Gregorio N. E., Levine M. Z., Oza J. P., Methods Protoc. 2019, 2, 24–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Schmidt E., Gath J., Habenstein B., Ravotti F., Szekely K., Huber M., Buchner L., Böckmann A., Meier B. H., Güntert P., J. Biomol. NMR 2013, 56, 243–254. [DOI] [PubMed] [Google Scholar]
- 83. Wishart D. S., Sykes B. D., Richards F. M., Biochemistry 1992, 31, 1647–1651. [DOI] [PubMed] [Google Scholar]
- 84. Smith A. A., Ravotti F., Testori E., Cadalbert R., Ernst M., Böckmann A., Meier B. H., J. Biomol. NMR 2017, 67, 109–119. [DOI] [PubMed] [Google Scholar]
- 85. Shen Y., Delaglio F., Cornilescu G., Bax A., J. Biomol. NMR 2009, 44, 213–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Teissier E., Zandomeneghi G., Loquet A., Lavillette D., Lavergne J.-P., Montserret R., Cosset F.-L., Böckmann A., Meier B. H., Penin F., Pécheur E.-I. PLoS ONE. 2011, 6, e15874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Waterhouse A., Bertoni M., Bienert S., Studer G., Tauriello G., Gumienny R., Heer F. T., de Beer T. A. P., Rempfer C., Bordoli L., et al., Nucleic Acids Res. 2018, 46, W296–W303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Goddard T. D., Huang C. C., Meng E. C., Pettersen E. F., Couch G. S., Morris J. H., Ferrin T. E., Protein Sci. 2018, 27, 14–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Torosyan A., Wiegand T., Schledorn M., Klose D., Güntert P., Böckmann A., Meier B. H., Front. Mol. Biosci. 2019, 6, 36–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Penzel S., Oss A., Org M.-L., Samoson A., Böckmann A., Ernst M., Meier B. H., J. Biomol. NMR 2019, 128, 12620–12611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Polyak S. J., Paschal D. M., McArdle S., Gale M. J., Moradpour D., Gretch D. R., Hepatology 1999, 29, 1262–1271. [DOI] [PubMed] [Google Scholar]
- 92. Brass V., Bieck E., Montserret R., Wölk B., Hellings J. A., Blum H. E., Penin F., Moradpour D., J. Biol. Chem. 2002, 277, 8130–8139. [DOI] [PubMed] [Google Scholar]
- 93. Zinke M., Fricke P., Lange S., Zinn-Justin S., Lange A., ChemPhysChem 2018, 19, 2457–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Etzkorn M., Böckmann A., Lange A., Baldus M., J. Am. Chem. Soc. 2004, 126, 14746–14751. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary