

Abstract
Key points
Mouse chromaffin cells in acute adrenal slices exhibit two distinct spiking patterns, a repetitive mode and a bursting mode.
A sodium background conductance operates at rest as demonstrated by the membrane hyperpolarization evoked by a low Na+‐containing extracellular saline.
This sodium background current is insensitive to TTX, is not blocked by Cs+ ions and displays a linear I‐V relationship at potentials close to chromaffin cell resting potential. Its properties are reminiscent of those of the sodium leak channel NALCN.
In the adrenal gland, Nalcn mRNA is selectively expressed in chromaffin cells.
The study fosters our understanding of how the spiking pattern of chromaffin cells is regulated and adds a sodium background conductance to the list of players involved in the stimulus‐secretion coupling of the adrenomedullary tissue.
Abstract
Chromaffin cells (CCs) are the master neuroendocrine units for the secretory function of the adrenal medulla and a finely‐tuned regulation of their electrical activity is required for appropriate catecholamine secretion in response to the organismal demand. Here, we aim at deciphering how the spiking pattern of mouse CCs is regulated by the ion conductances operating near the resting membrane potential (RMP). At RMP, mouse CCs display a composite firing pattern, alternating between active periods composed of action potentials spiking with a regular or a bursting mode, and silent periods. RMP is sensitive to changes in extracellular sodium concentration, and a low Na+‐containing saline hyperpolarizes the membrane, regardless of the discharge pattern. This RMP drive reflects the contribution of a depolarizing conductance, which is (i) not blocked by tetrodotoxin or caesium, (ii) displays a linear I‐V relationship between −110 and −40 mV, and (iii) is carried by cations with a conductance sequence g Na > g K > g Cs. These biophysical attributes, together with the expression of the sodium‐leak channel Nalcn transcript in CCs, state credible the contribution of NALCN. This inaugural report opens new research routes in the field of CC stimulus‐secretion coupling, and extends the inventory of tissues in which NALCN is expressed to neuroendocrine glands.
Keywords: acute mouse adrenal slice, chromaffin cell excitability, resting membrane potential, sodium background conductance, sodium leak channel NALCN, spiking pattern
Key points
Mouse chromaffin cells in acute adrenal slices exhibit two distinct spiking patterns, a repetitive mode and a bursting mode.
A sodium background conductance operates at rest as demonstrated by the membrane hyperpolarization evoked by a low Na+‐containing extracellular saline.
This sodium background current is insensitive to TTX, is not blocked by Cs+ ions and displays a linear I‐V relationship at potentials close to chromaffin cell resting potential. Its properties are reminiscent of those of the sodium leak channel NALCN.
In the adrenal gland, Nalcn mRNA is selectively expressed in chromaffin cells.
The study fosters our understanding of how the spiking pattern of chromaffin cells is regulated and adds a sodium background conductance to the list of players involved in the stimulus‐secretion coupling of the adrenomedullary tissue.
Introduction
Regulation of cellular excitability is a crucial process whereby cells ensure their physiological function (neurotransmission, muscle contraction, hormone secretion …). In the adrenal medullary tissue, the excitability of neuroendocrine chromaffin cells (CCs) is critical for the well‐described process of ‘stimulus‐secretion coupling’, leading to catecholamine secretion. In response to a stressful episode, catecholamines (mainly adrenaline (epinephrine)), which are the first hormones to be released into the blood circulation, actively contribute to the adaptive response of the organism to stress by their peripheral actions on various organs and tissues.
CC excitability relies on intricate mechanisms, supported by the interplay between cholinergic and peptidergic innervation (Guerineau, 2020), a gap junctional electrical coupling between CCs (Colomer et al. 2012; Hodson et al. 2015; Guerineau, 2018) and the palette of ion channels expressed at the plasma membrane (Lingle et al. 2018). The major stimulus for catecholamine secretion comes from the preganglionic command (neurogenic control) ensured by the electrical activity of the splanchnic nerve whose cholinergic nerve terminals synapse onto CCs. The synaptic release of acetylcholine activates CC nicotinic receptors leading to depolarization and activation of Ca2+ influx. This in turn depolarizes cells and facilitates action potential (AP) firing. Increasing splanchnic nerve discharge enhances catecholamine secretion, indicating how significant is the contribution of the neurogenic control to CC excitability and ensuing catecholamine secretion. However, the regulation of CC excitability is much more puzzling when non‐neurogenic components of electrical activity are added to the scenario. First, a local intercellular electrical communication mediated by gap junctions between CCs also contributes to cell excitability and catecholamine secretion (Martin et al. 2001; Colomer et al. 2008b ; Desarmenien et al. 2013; Guerineau, 2018). Second, the repertoire of ion channels expressed in CCs robustly patterns cell excitability, not only through voltage‐gated channels involved in AP generation, but also through leak channels, which operate near the resting membrane potential (RMP). While ion channels contributing to AP generation are well described, as recently reviewed (Lingle et al. 2018), CC leak channels are less well known. Among leak channel families, two pore‐domain potassium channels and transient receptor potential channels are present in CCs (Tesfai et al. 2001; Hu et al. 2009; Kim & Kang, 2015; Guarina et al. 2017). Extending these studies in acute slices of mouse CCs, we describe here the presence of a Na+‐permeant background conductance. By controlling the RMP value, this conductance exerts a substantial role in CC excitability. Taking into account common electrophysiological and pharmacological attributes, we propose the sodium leak channel NALCN as a likely candidate to support this Na+ background conductance.
Methods
Ethical approval
Ethical approval for all experimental protocols was obtained from the Institute of Functional Genomics Animal Care and Use Committee, Montpellier, France. Housing and experimental procedures were approved by the French Agriculture and Forestry Ministry (A34‐ 172‐41). All procedures in this study conformed to the animal welfare guidelines of the European Community and were approved by the French Ministry of Higher Education, Research and Innovation (authorizations no. 49‐2011‐18 and 49–247).
Adrenal slice preparation
Acute slices were prepared from 8‐ to 12‐week‐old adult C57BL/6J male mice (Janvier, Le Genest‐St‐Isle, France), as described in the rat (Martin et al. 2001). Briefly, after removal, the glands were kept in ice‐cold saline for 2 min. Before slicing, a gland was unsheathed from the surrounding fat tissue and was then transferred to the stage of a vibratome (DTK‐1000, D.S.K, Dosaka EM CO. LTD, Kyoto, Japan). Slices (150 μm thickness) were cut with a razor blade and transferred to a storage chamber maintained at 37°C, containing Ringer saline (in mM): 125 NaCl, 2.5 KCl, 2 CaCl2, 1 MgCl2, 1.25 NaH2PO4, 26 NaHCO3, 12 glucose. The saline was continuously bubbled with carbogen (95% O2/5% CO2) and buffered to pH 7.4.
Electrophysiology
Adrenal slices were transferred to a recording chamber attached to the stage of an upright microscope (Axioskop FS2, Zeiss, Le Pecq, France) and continuously superfused with Ringer saline at 34°C. All experiments were performed using the patch‐clamp technique and electrophysiological signals were acquired using an EPC‐9 patch‐clamp amplifier (HEKA Electronik, Lambrecht/Pfalz, Germany) and Pulse software. Signals were sampled at 10 kHz and analysed with Igor Pro 7 (version 7.02, WaveMetrics Inc., Lake Oswego, OR, USA). An Ag/AgCl pellet was used as a reference ground electrode.
Whole‐cell recordings
Patch pipettes were pulled from borosilicate glass and filled with the following internal solution (in mM): 140 potassium gluconate, 2 MgCl2, 1.1 EGTA, 5 HEPES, 3 Mg‐ATP, 0.5 Na‐GTP, titrated to pH 7.2 with KOH. Osmolarity was adjusted to 295 mOsm with potassium gluconate and pipette resistance was 5–7 MΩ. Pipette and cell capacitances were fully compensated and the series resistance was compensated at 75%–80%. Membrane potential was recorded in the current‐clamp mode and filtered at 3 kHz. In some experiments, potassium gluconate was replaced by an equimolar concentration of CsCl.
Loose‐patch recordings
For extracellular recordings of spontaneous currents in the loose‐patch configuration, pipettes were filled with the following saline (in mM): 125 NaCl, 2.5 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES, 10 glucose, buffered to pH 7.4 with NaOH. Osmolarity was adjusted to 295 mOsm with NaCl and pipette resistance was 5–10 MΩ. Once the tip of the pipette was positioned at the surface of a CC, a minimal suction pressure was applied (seal resistance <500 MΩ) and the electrical activity was recorded in the voltage‐clamp mode (0 mV) of the loose cell‐attached configuration (Almers et al. 1983). This method allows monitoring of membrane excitability under physiological conditions and stable recordings of firing rate can therefore be obtained (Perkins, 2006; Alcami et al. 2012).
Ionic selectivity of I Nab
This issue was addressed by changing intracellular K+ and Na+ concentrations without variation in intracellular chloride concentration. The other ionic species were maintained constant in both intracellular and extracellular media. The ionic composition of media is reported in Table 1. A voltage ramp protocol (−130 to −50 mV, 3 s duration) was applied first in the control condition and then in low Na+ conditions. To achieve low Na+ conditions, 110 mM extracellular NaCl was replaced by an equimolar concentration of NMDG‐Cl or Tris‐Cl. The Na+ background current was obtained by subtracting the current trace during low Na+ application from the control current. The reversal potential E rev of the ‘low Na+‐sensitive current’ was calculated from a linear extrapolation applied to the subtracted current. A calculated E rev was determined for each recording condition ([K+]i = 25, 50, 130 and 140 mM). The g Na/g K conductance ratio was then calculated, as follows:
| (1) |
| (2) |
Table 1.
Ionic concentrations of intracellular and extracellular media used to determine I Nab ionic selectivity
| Intracellular media | d‐Gluconic acid (sodium salt) (mM) | d‐Gluconic acid (potassium salt) (mM) | NaCl (mM) | KCl (mM) |
|---|---|---|---|---|
| [K+]i = 25 mM | 75 | 0 | 50 | 25 |
| [K+]i = 50 mM | 75 | 0 | 25 | 50 |
| [K+]i = 130 mM | 10 | 75 | 10 | 55 |
| [K+]i = 140 mM, with regular Na+ | 0 | 140 | 10 | 0 |
| [K+]i = 140 mM, with low Na+ | 0 | 140 | 0.5 | 0 |
| Extracellular media | d‐Gluconic acid (sodium salt) (mM) | NaCl (mM) | KCl (mM) | NMDG (mM) |
|---|---|---|---|---|
| control | 47.9 | 72.5 | 2.5 | 0 |
| low [Na+] | 0 | 15 | 2.5 | 105.4 |
Note that NaHCO3 and NaH NaH2PO4 concentrations remained constant at 26 mM and 1.1 mM, respectively.
Assuming that at RMP, g K and g Na >>> g Cl and g Ca, the same eqn (2) is used to calculate α for cells exhibiting a regular or a bursting firing pattern and for the silent cells.
In situ hybridization
In situ hybridizations were performed as previously described (Venteo et al. 2016). Mice were killed by CO2 inhalation followed by cervical dislocation. Adrenal glands were dissected in PBS and fixed for 1 h at room temperature (RT) by immersion in 4% paraformaldehyde (PFA). Tissues were rinsed twice in PBS before overnight immersion in 30% sucrose/PBS at 4°C and frozen in OCT compound. Transverse 14 μM thick sections were cut on a cryostat, collected on Superfrost Plus slides Fisher Scientific (Illkirch, France) and stored at −80°C until use. An 1162 bp mouse Nalcn cDNA fragment covering nucleotides 998–2159 (reference sequence no. NM_177393) was amplified from adult mouse brain and cloned in the pBluescript II SK(+) plasmid (Agilent Technologies Cat. No. 212205) using standard molecular biology techniques. Sense and antisense Digoxigenin‐labelled RNA probes were generated in a 20 μl reaction containing 1 μg of linearized plasmid (linearized with XbaI and KpnI, respectively) using the DIG RNA labelling mix (Roche) and T3 and T7 RNA polymerase respectively (Promega) following the manufacturer's instructions. The probe for Dbh (Pattyn et al. 1999) was kindly provided by Dr J.‐F. Brunet (Institut de Biologie de l'Ecole Normale Supérieure, Paris) and synthesized using the Fluorescein‐labelling kit (Roche Diagnostics) according to the manufacturer's instructions. Probes were purified on MicroSpin G50 columns (GE Healthcare). Double in situ hybridizations were performed on 14 μM thick frozen sections prepared as above. DIG‐ and Fluorescein‐labelled RNA probes were mixed in hybridization buffer and applied to sections. After hybridization at 70°C overnight, sections were washed twice in 50% formamide, 1× saline sodium citrate buffer, 0.1% Tween‐20 for 1 h at 70°C, twice in 1× maleic acid buffer, 0.1% Tween‐20 (MABT) for 30 min before blocking in blocking buffer (MABT, 2% blocking reagent from Roche, 20% inactivated sheep serum) for 2 h at room temperature. Sections were then exposed to a 1:2000 dilution of anti‐Fluo‐alkaline phosphatase‐conjugate antibody (Roche Diagnostics) in blocking buffer overnight at 4°C. After washing for 30 min in MABT, the bound Fluo‐probe was visualized by an alkaline phosphatase‐catalysed colour reaction using Fast Red tablets (Sigma‐Aldrich) according to the manufacturer's instructions. The colour reaction was stopped in 0.1 M Tris (pH 8.2), the slides were mounted temporarily in 90% glycerol and 0.1 M Tris, pH 8.2 and the images were acquired on AxioImager D1 microscope (Zeiss). After washing for 15 min in 0.1 M Tris (pH 8.2), the alkaline phosphatase activity was then inactivated by incubating with 100 mM glycine and 0.1% Tween‐20 (pH 2.2) for 30 min. The sections were washed twice in PBS, 0.1% Tween‐20 for 15 min, post‐fixed in 4% PFA in PBS for 10 min at RT, washed in PBS, 0.1% Tween‐20 for 20 min, twice in MABT buffer for 30 min, blocked again in blocking buffer for 2 h, and incubated overnight with an anti‐Digoxigenin antibody, conjugated with alkaline phosphatase (1:2000, Roche Diagnostics) at 4°C. After washing as above, slides were incubated with NBT‐BCIP (Roche Diagnostics) staining solution according to the manufacturer's instructions and the reaction stopped by washing in water. Fast Red precipitates were then removed by incubating the slides in increasing concentrations of ethanol, culminating in two final incubations in 100% ethanol for 10 min. Photographs of the NBT/BCIP results were then taken for comparison with those showing the Fast Red results on the same sections. The Fast Red signals were converted into pseudo‐red and the NBT/BCIP signals into pseudo‐green fluorescent colours. The pseudo‐red fluorescent images were then carefully overlaid with pseudo‐green fluorescent images. This sequential approach permits unequivocal identification of co‐expression at the single cell level.
Quantification of mRNA expression levels by real‐time PCR
Total RNA was extracted from macrodissected adrenal medulla using the RNeasy Microextraction kit (Qiagen, Courtaboeuf, France). RNA (500 ng) was reverse transcribed using the QuantiTect Reverse Transcription kit (Qiagen) in a final volume of 10 μl. Real‐time PCR analyses of the target genes and the reference genes Gusb, Hprt and Gapdh were performed using Sybr Green PCR master mix (Applied Biosystems, Foster City, CA, USA) with 1:100 of the reverse‐transcription reaction, and were carried out on an ABI 7500 SDS Real‐Time PCR system (Applied Biosystems). Primer sequences of target genes are given in Table 2 and the concentration used was 300 nM for all genes. After an initial denaturation step for 10 min at 95°C, the thermal cycling conditions were 40 cycles at 95°C for 15 s and 60°C for 1 min (LightCycler 480, Roche Diagnostic). The amplification specificity was checked by melting curve analysis. Each sample value was determined from triplicate measurements. Expression of the target transcripts was normalized to the mean of the expression level of the three reference genes according to the formula E = 2−(Ctmean[Target] − Ctmean[Reference]), where Ctmean is the mean threshold cycle.
Table 2.
Primer sequences used for quantitative RT‐PCR
| Genename | GenBank accession number | Protein name | Forward primer (5′−3′) | Reverse primer (5′−3′) |
|---|---|---|---|---|
| Nalcn | NM_177393.4 | NALCN | agatggagaggctgcacaat | tctacagagcgataggagagca |
| Unc79 | NM_001081017.2 | UNC79 | gccaattcgggatatggtt | tctctgtgggcgtgttctc |
| Unc80 | NM_175510.3 | UNC80 | catcaaatcagacgcaggtg | ggagcagtcgagaacctcct |
|
Nlf1/ Fam155A |
NM_173446.2 | Nlf1 | accaggactacgaccaccac | gccaggctttgtagacaatct |
| Hprt | NM_012583.2 | Hprt | gaccggttctgtcatgtcg | acctggttcatcatcactaatcac |
| B2m | NM_009735.3 | B2M | tatgctatccagaaaacccctcaa | gtatgttcggcttcccattctc |
Solutions and chemicals
Tetrodotoxin (TTX) was purchased from Latoxan (Cat. No. L8503) and ouabain octahydrate from Sigma‐Aldrich (Cat. No. O3125).
Statistical analysis
Statistics were performed with Prism 8 (version 8.0.2, GraphPad, San Diego, CA, USA). Numerical data are expressed as the means ± standard deviation. Student's unpaired t test was used to compare means when appropriate. The non‐parametrical Wilcoxon matched pairs test was used to compare two related samples. One‐way ANOVA followed, if significant interaction, by a Dunnett or a Tukey post hoc test was used when appropriate. Percentages were compared using a contingency table and the Fisher's exact test. The Spearman's rank correlation coefficient ρ was used to measure the strength and direction of association between two ranked parameters. Differences with P < 0.05 were considered significant, with asterisks indicating significance levels: * P < 0.05, ** P < 0.01, *** P < 0.001 and **** P < 0.0001.
Results
Spontaneous electrical activity of mouse CCs in situ: two distinct firing patterns
The passive biophysical membrane properties of mouse adrenal CCs in acute slices are summarized in Table 3. The data were collected from both spontaneously firing and silent cells, and our results are in agreement with those reported by other groups (Nassar‐Gentina et al. 1988; Moser, 1998; Marcantoni et al. 2009; Martinez‐Espinosa et al. 2014; Vandael et al. 2015 b; Guarina et al. 2017).
Table 3.
Passive electrical membrane properties of mouse CCs in situ
| Membrane capacitance (pF) | Input resistance (GΩ) | Resting membrane potential (mV) |
|---|---|---|
|
7.14 ± 2.55 (n = 323 cells) |
1.13 ± 0.72 (n = 323 cells) |
−43.4 ± 7.5 (n = 225 cells) |
Sixty‐two per cent of mouse CCs in situ exhibit spontaneous AP discharges (n = 225 cells). The analysis of the discharge pattern was performed on 88 cells (54 spiking cells and 34 silent cells). Interestingly, cells fire with two distinct patterns, a regular firing mode (Fig. 1A ) and an irregular firing mode indicative of bursts (Fig. 1B ). To discriminate between the two discharge patterns, the regularity of the firing discharge was investigated by calculating the coefficient of variation (Cv) of inter‐spike interval (ISI) distribution. As expected, a regular firing mode is associated with a Cv < 1 and a bursting mode with a Cv > 1 (Fig. 1A and 1B , right panels). Note that the percentage of cells exhibiting a regular electrical activity or a bursting pattern does not significantly differ (48% versus 52% for the regular and bursting mode, respectively, P > 0.05 ( = 0.8274), Fisher's exact test). We next analysed the AP parameters in consecutive spikes in single bursts (Fig. 2). Changes in AP peak amplitude, AHP (anti‐peak) amplitude, half‐width duration, interspike interval (ISI) and intraburst instantaneous firing frequency were plotted as a function of the AP position in the burst. Whatever the number of APs in a burst (<10, Fig. 2Aa ) or >10 (Fig. 2Ba ), the evolution of AP parameters is the same (Fig. 2Ab and 2Bb ). As illustrated by the pooled data in Fig. 2C (15 bursts, 10 cells, AP number in a burst ranging from 5 to 92), the peak amplitude and the AHP amplitude gradually decrease (with a mean reduction between the first and the last AP of 73.8% ± 30.6%, n = 15 bursts and 18.4% ± 10.1%, n = 15 bursts, for peak amplitude and AHP amplitude, respectively), the half‐width duration increases (153.1% ± 125%, n = 15 bursts), the ISI decreases (48.8% ± 33.1%, n = 15 bursts) and the instantaneous intra‐burst firing frequency increases (183.6% ± 171.9%, n = 15 bursts).
Figure 1. Presence of two distinct firing patterns in mouse adrenal CCs in situ .
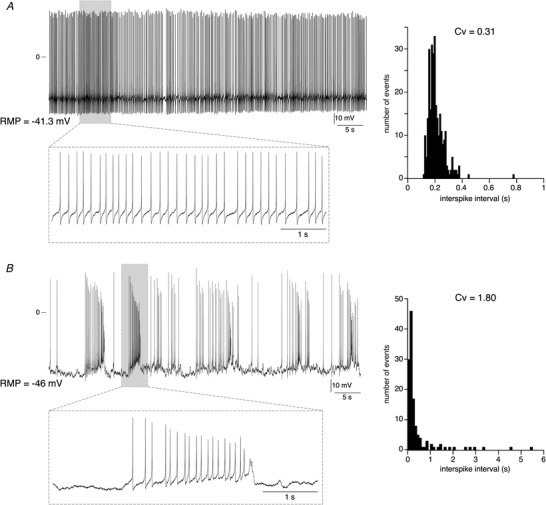
Representative chart recordings of two CCs exhibiting a regular firing (A) or an irregular firing with burst (B). Insets: expanded time scale illustrating a 5 s firing in the regular cell and in the bursting cell. The histograms on the right illustrate the distribution of the inter‐spike intervals (10 ms bin for the cell in A and 100 ms bin for the cell in B), from which the coefficients of variation (Cv) were calculated.
Figure 2. Analysis of AP parameters in bursts.
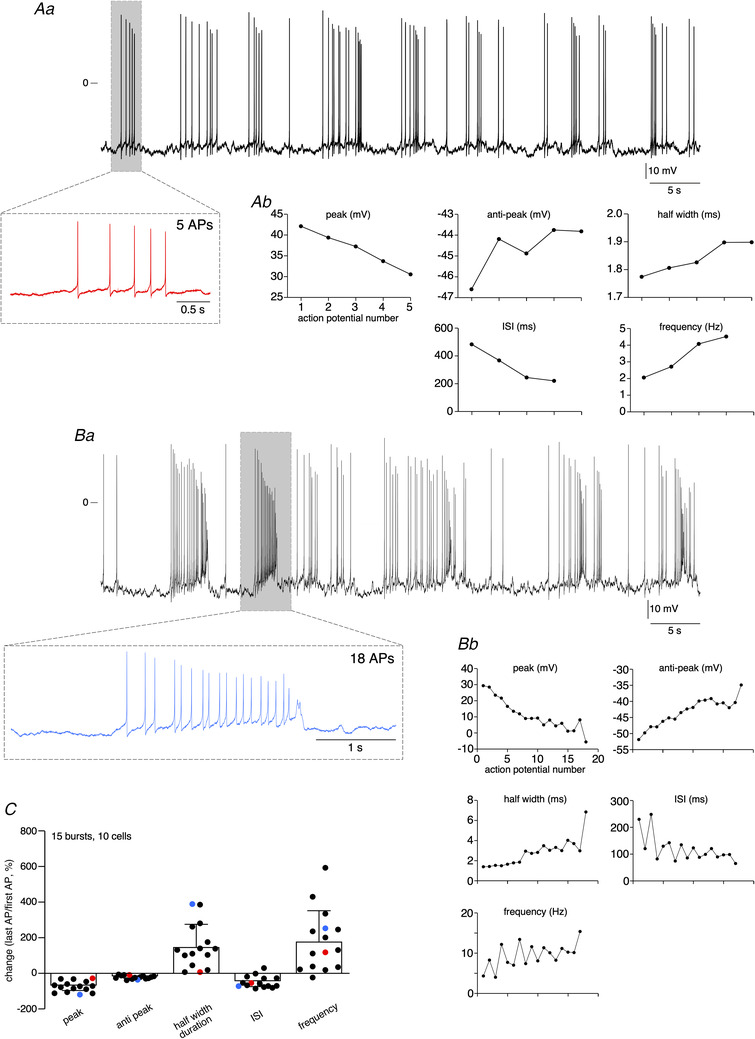
Evolution of AP peak amplitude, AHP (anti‐peak) amplitude, half‐width duration, interspike interval (ISI) and intra‐burst instantaneous firing frequency measured in consecutive spikes within a burst (Ab and Bb). Example of two mouse CCs, with an AP number in a burst <10 (Aa) and >10 (Ba). C, pooled data of changes between the first AP and the last AP in a burst (data collected from 15 bursts, 10 cells). AP parameters corresponding to the cell illustrated in Aa are plotted in red and those corresponding to the cell illustrated in Ba are plotted in blue.
To further characterize the regular and bursting firing patterns, we compared parameters of APs engaged in one or the other discharge mode (Fig. 3). We first performed a phase‐ plane plot analysis of AP discharges (Fig. 3Aa ). It shows that AP threshold is significantly more hyperpolarized in bursting cells. This result is confirmed by the pooled data coming from the analysis of the first AP in a burst versus the first AP in a regular pattern (data collected from 22 cells randomly selected out of the 54 spiking cells recorded (11 cells illustrative of the regular pattern and 11 cells illustrative of the bursting pattern, Fig. 3B ). The mean value of the AP threshold is −32.4 ± 2.1 mV (n = 11 cells) for the ‘bursting’ AP versus −28.8 ± 2.8 mV (n = 11 cells) for the ‘regular’ AP, P < 0.05 ( = 0.0026), unpaired t test). Similarly, AP peak amplitude notably differs between a regular and a bursting firing, as such the distribution of events is significantly broader in bursting cells and the associated variance enhanced (65 ± 55.9, n = 25 cells versus 16 ± 10.2 in regular cells, n = 28 cells, P < 0.0001, unpaired t test, Fig. 3Ab ). This reflects the gradual diminution of AP amplitude within bursts (Fig. 1B , inset), likely to be due to the inactivation of Na+ channels activated during AP. Contrasting with bursting cells, AP amplitude is uniform throughout the discharge in cells with a regular firing pattern (Fig. 1A , inset). Interestingly, the significant difference in AP peak amplitude observed between APs engaged in a regular pattern and APs engaged in a bursting spiking mode is also found when only the first AP was taking into analysis (37.9 ± 9.7 mV, n = 11 cells for the ‘bursting’ AP versus 29.4. ± 7.3 mV, n = 11 cells for the ‘regular’ AP, P < 0.05 ( = 0.0322), unpaired t test, Fig. 3B ). The other AP parameters (AHP amplitude and half‐width duration) did not significantly differ between the ‘bursting’ AP and the ‘regular’ AP (−52.6 ± 6.1 mV, n = 11 cells versus −53.0 ± 2.5 mV, n = 11 cells for AHP amplitude, P > 0.05 ( = 0.8276), unpaired t test and 1.8 ± 0.4 ms, n = 11 cells versus 2.0 ± 0.4 ms, n = 11 cells for half‐width duration, P > 0.05 ( = 0.2491), unpaired t test, Fig. 3B ).
Figure 3. Analysis of action potential parameters in regular and bursting spiking cells.
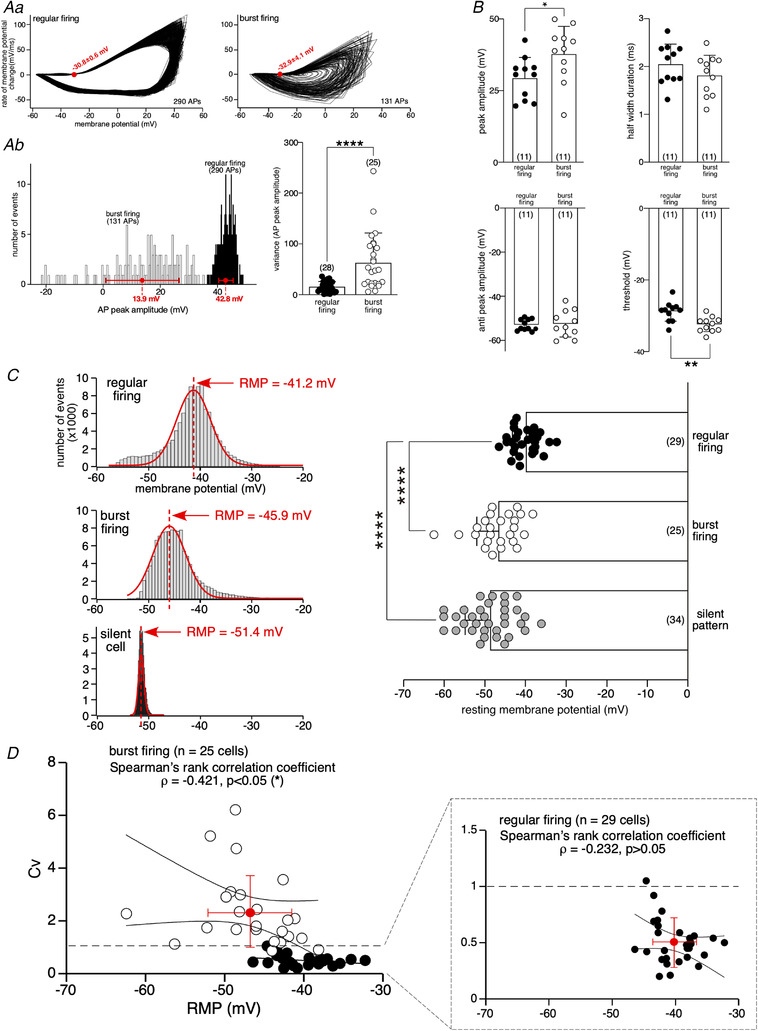
Aa, phase‐plane plot analysis of APs of the two cells illustrated in Fig. 1. 299 APs were analysed in the regular cell and 131 in the bursting cell. The AP threshold (values plotted in red on the phase‐plane plots) is significantly more polarized in bursting cells (right histogram for pooled data, ** P < 0.01, unpaired t test. Ab, analysis of action potential peak amplitude showing a broader distribution in the bursting spiking cell (right histogram for pooled data, *** P < 0.001, unpaired t test). B, analysis of the first AP in a burst versus the first AP in a regular pattern. Significant differences were found for the peak amplitude (* P < 0.05, unpaired t test) and for the threshold (** P < 0.01, unpaired t test). Data were collected from 22 cells randomly selected (11 cells with a regular pattern and 11 cells with a bursting pattern. The AHP amplitude and the half‐width duration do not significantly differ between regular and bursting firing modes. C, determination of RMP from the sample (fraction of time) distribution of membrane potential values from the two cells recorded in Fig. 1 and from one silent cell. Pooled data (plotted in the right panel) show a significant difference in RMP between regular and bursting cells (**** P < 0.0001, one‐way ANOVA, Tukey's post hoc test), with a more polarized RMP in bursting CCs. Silent cells also displayed a significantly more polarized RMP compared to regular cells (**** P < 0.0001, one‐way ANOVA, Tukey's post hoc test). No difference in RMP was found between bursting and silent cells (P > 0.05, one‐way ANOVA, Tukey's post hoc test). D, graphs plotting the RMP value of 29 regular cells and 25 bursting cells over the corresponding Cv. The Cv threshold allowing to discriminate between regular cells and bursting cells was positioned at Cv = 1 (dashed lines). The Spearman's rank correlation coefficient ρ, combined with the confidence band at 95%, shows a significant correlation between RMP and Cv values for bursting cells (ρ = −0.421, P < 0.05, but not for regular cells (ρ = −0.232, P > 0.05). The data for regular cells are expanded into the inset.
Another crucial parameter that might differ between the regular and bursting spiking modes is the RMP. Because the determination of RMP cannot be reliably assessed by direct measurement on chart recordings of spontaneously firing cells, we calculated its mean value from the distribution of the membrane potential values all along the recording. As illustrated in Fig. 3C , membrane potential values yielded a unimodal distribution that can be fitted by a single Gaussian curve. Contrasting with membrane potentials rapidly scanned during APs, the RMP corresponds to a stable potential state towards which the cell tends to move and remain. Accordingly, the RMP value is defined by the peak of the Gaussian curve. The RMP analysis was applied to cells exhibiting regular discharges, to cells exhibiting a bursting activity and to silent cells. As illustrated in Fig. 3C (left panel), a significant difference in RMP values is found between a regular (−40.0 ± 3.4 mV, n = 29 cells, corresponding to g Na/g K = 0.34, see eqn (2) with RMP = E rev) and a bursting (−46.8 ± 5.4 mV, n = 25 cells, corresponding to g Na/g K = 0.30) spiking pattern (P < 0.0001, one‐way ANOVA, Tukey's post hoc test), Fig. 3C , right panel). The RMP value of silent cells (−48.8 ± 6.1 mV, n = 34 cells, corresponding to g Na/g K = 0.29) does not significantly differ from the RMP value of bursting cells (P > 0.05 ( = 0.3048), one‐way ANOVA, Tukey's post hoc test), but differs from the RMP value of regular cells (P < 0.0001, one‐way ANOVA, Tukey's post hoc test). This raises the issue of whether the spiking pattern (regular versus bursting) correlates with the RMP value. Correlation analyses between the Cv (indicative of a regular or a bursting firing mode) and the RMP values reveal a significant correlation for bursting activity only (Spearman's rank correlation coefficient ρ = −0.421 for bursting cells (n = 25 cells), P < 0.05 ( = 0.036) versus −0.232, for regular cells (n = 29 cells), P > 0.05 ( = 0.226), Fig. 3D ). When RMP value is plotted over the AP threshold, a significant correlation is found for both a regular and a bursting spiking pattern (Spearman's rank correlation coefficient ρ = 0.651 for bursting cells (n = 25 cells), P < 0.001 ( = 0.0002) versus 0.504, for regular cells (n = 29 cells), P < 0.01 ( = 0.0027); data not shown). Interestingly, the slope of the linear regression used to fit the data is similar between the two firing modes (0.62 and 0.61 for regular and bursting cells, respectively; data not shown). This suggests that the cellular mechanisms contributing to depolarize cells from RMP to the spiking threshold might be similar during a regular or a bursting activity.
The presence of two distinct firing patterns raises the question of whether those two spiking modes can occur in the same cell or in two distinct CC populations. To address this issue, the spontaneous electrical activity of individual CCs was recorded for a longer period of time (2—5 min). As illustrated in Fig. 4A , the discharge mode can alternate during a 5 min recording, switching from a regular (at the beginning, highlighted section a) to a bursting mode (at the end, highlighted section b). Reciprocally, the firing pattern can switch from a bursting to a regular spiking mode, as shown in another cell recorded during two successive 1 min recordings (Fig. 4B ). These findings indicate that these firing patterns do not reflect two different cell populations. To rule out a possible contribution of the ‘whole‐cell’ configuration mode in generating the switch between the two firing patterns, we next carried out long‐lasting recordings in the loose‐patch configuration (Fig. 5). Spontaneous AP currents were observed in 43% of cells (n = 9/21 cells). Consistent with the ‘whole‐cell’ recordings, both bursting (Fig. 5A , left traces) and regular (Fig. 5A , right traces) patterns were observed. Over an 8 min period, the electrical activity of one individual cell alternated between regular, bursting and silent modes (Fig. 5B and highlighted sections a, b and c for expanded chart recordings and associated Cv calculation). Collectively, our results demonstrate that (i) the two firing modes are not assigned to distinct CC populations and (ii) within the same CC, the electrical activity is a dynamically regulated process, with the expression of various discharge patterns.
Figure 4. Dynamic switch between a regular and a bursting spiking pattern in a same CC.
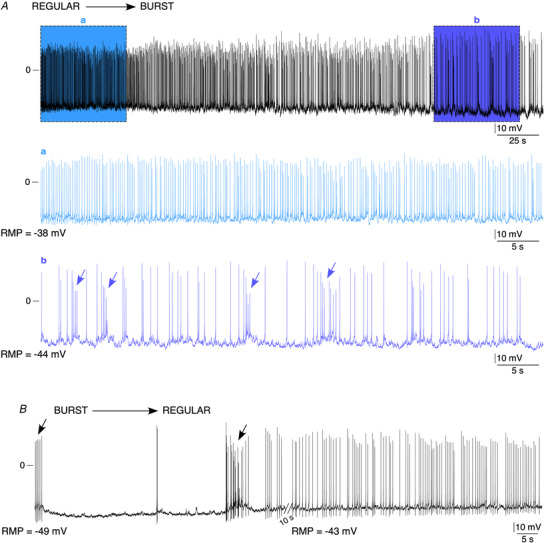
A, example of a cell whose spontaneous electrical activity was recorded for 5 min and in which the spiking pattern switched from a regular to a bursting mode. Highlighted a and b sections: expanded time scale showing the regular mode at the beginning of the recording (section a) and the bursting mode at the end of the recording (section b). The arrows point to bursts. B, example of a cell recorded for two successive 1 min period (10 s interval between recordings), in which the bursting mode observed during the first 1 min recording switched to a regular mode during the second 1 min recording.
Figure 5. Loose‐patch recordings of a spontaneously firing mouse CCs.
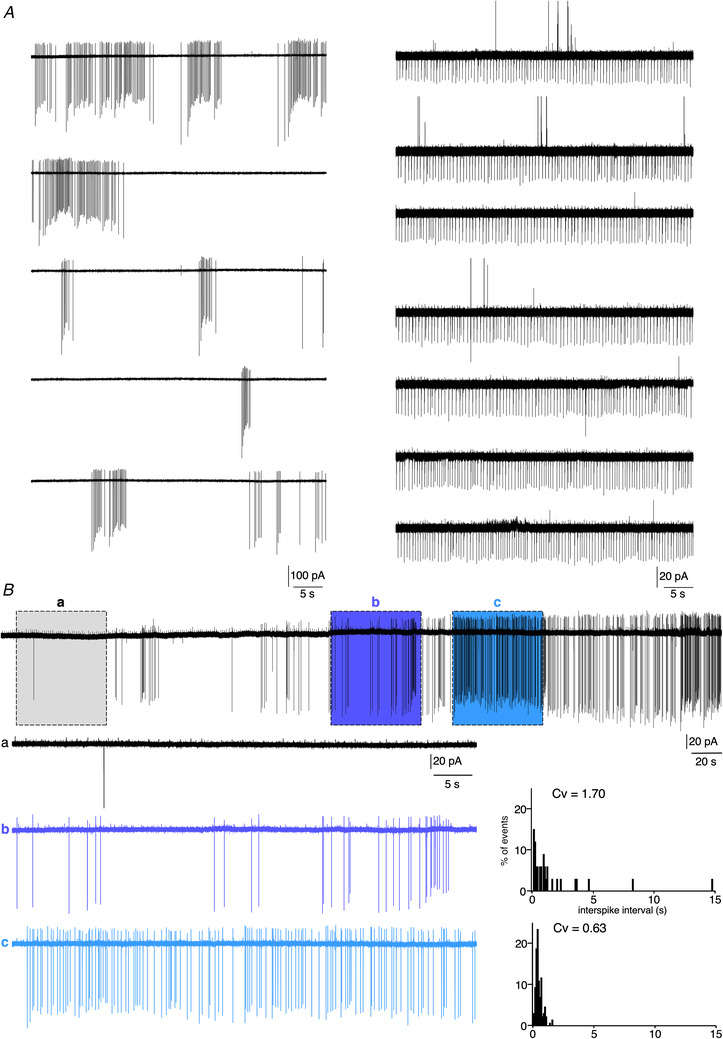
A, spontaneous action potential currents recorded in two individual CCs. Left traces (5 successive 1 min recordings) illustrate an arrangement of the currents as bursts and right traces (7 successive 1 min recordings) exemplify a cell with a regular action potential current activity. B, chart recording (8 min duration) showing that a same cell can alternatively exhibit periods of regular (highlighted section c), irregular bursting (highlighted section b) and no firing (highlighted section a). The corresponding distributions of inter‐spike intervals and calculated Cv are plotted in the right histograms.
Could the switch in electrical activity mode be related to RMP? To address this issue, we investigated the effect of prolonged depolarization or hyperpolarization, from the spontaneous RMP recorded over a 1 min period, on the firing pattern and Cv value. Depolarizations and hyperpolarizations were induced by injecting positive and negative currents, respectively. A representative example is shown in Fig. 6A and pooled data were plotted in Fig. 6B . At RMP (Fig. 6A , black trace), the cell exhibits a regular pattern (Cv = 0.64). Upon hyperpolarization (−8.6 mV from RMP, Fig. 6A , blue trace) or depolarization (+8.6 mV from RMP, Fig. 6A , red trace), the spiking pattern switches to a bursting mode (Cv = 1.13 and 1.11, respectively). The mean Cv value was 0.64 ± 0.24 at RMP and increased to 1.72 ± 0.93 under hyperpolarized conditions (n = 8 cells, P < 0.01 ( = 0.0078), Wilcoxon matched‐pairs signed‐rank test, Fig. 6B ). Under depolarized conditions, the mean Cv value increased from 0.66 ± 0.43 to 1.23 ± 0.55 (n = 4 cells, P > 0.05 ( = 0.125), Wilcoxon matched‐pairs signed‐rank test, Fig. 6B ). The transition towards a bursting pattern in response to membrane hyperpolarization and, although to a lesser extent, to membrane depolarization, argues for a crucial role of membrane potential near the resting value in controlling electrical cell activity and therefore cell function. Note that the highest AP frequency occurred at RMP (Fig. 6C for data obtained in 25 cells in which membrane potential was alternatively hyperpolarized and/or depolarized from RMP). A consistent observation is the firing frequency peaking at RMP compared to hyperpolarized or depolarized potentials (one‐way ANOVA, Dunnett as post hoc test, P < 0.005 ( = 0.0033). The mean firing frequencies were 2.02 ± 1.77 Hz, 0.71 ± 0.88 Hz and 1.04 ± 1.15 Hz, at RMP (n = 25 cells), in hyperpolarized (n = 25 cells) and depolarized (n = 16 cells) conditions, respectively. The frequency was significantly higher at RMP (P < 0.005 ( = 0.0021) when compared to the hyperpolarized condition, one‐way ANOVA, Dunnett's post hoc test, and P < 0.05 ( = 0.049) when compared to the depolarized condition, one‐way ANOVA, Dunnett's post hoc test). Taking this further, hyperpolarizing currents have been injected in silent cells (n = 12 cells; data not shown). In 10 cells (83.4%), no spiking activity was evoked during the 1 min recording period. In the two remaining cells, one cell exhibited one AP and the other cell displayed a series of APs. Overall, these results reveal a tight regulation of the electrical activity pattern of mouse CCs in situ by small fluctuations of the membrane potential, indicating a decisive role of the ionic conductances operating in this window of potentials in governing CC excitability. Following this line, our observation of a relationship between the ratio g Na/g K and the mode of discharge, (0.34 in regular, 0.30 in bursting and 0.29 in silent cells), led us to hypothesize that a background g Na might contribute to the mode of discharge.
Figure 6. Membrane potential‐driven spiking pattern.
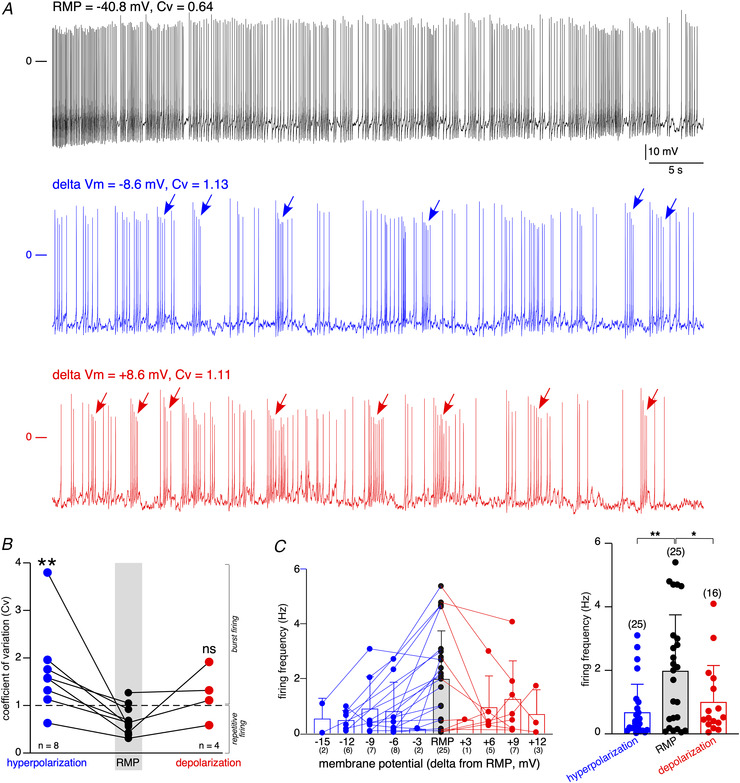
A, representative AP discharges recorded in one individual CC current‐clamped at RMP (black trace), hyperpolarized (blue trace, 8.6 mV as negative delta V m) or depolarized (red trace, 8.6 mV as positive delta V m). For each recording conditions, the Cv was calculated, given a value of 0.64 at RMP, 1.13 for hyperpolarized and 1.11 for depolarized conditions. Setting a threshold value of 1 for Cv to discriminate between regular (Cv < 1) and bursting (Cv > 1) cells, Cv values argue for a switch from a regular to a bursting mode. B, pooled data showing a significant increase in Cv in response to membrane hyperpolarization (** P = 0.0078, Wilcoxon matched‐pairs signed‐rank test), but not to membrane depolarization (P = 0.125, Wilcoxon matched‐pairs signed‐rank test). C, pooled data illustrating the firing frequency as a function of membrane potential (RMP and hyperpolarization/depolarization from RMP). The number of recorded cells is indicated below the value of membrane potential change. In panels B and C, the grey region of interest underlies the results obtained at RMP. [Color figure can be viewed at wileyonlinelibrary.com]
A sodium background conductance operates near RMP and contributes to RMP
Among the background conductances reported to contribute to RMP, Na+‐permeant conductances are eligible candidates (Ren, 2011). To investigate whether such conductances contribute to RMP in mouse CCs, spontaneous electrical activity was recorded at RMP upon an extracellular Na+ challenge. Decreasing external [Na+] from 152.2 to 42.2 mM (110 mM NaCl substituted with 110 mM NMDG‐Cl) elicited a membrane hyperpolarization (−9.6 ± 11.8 mV, n = 30 cells), abrogating APs (Fig. 7). Similar results were obtained upon Tris substitution, which led to a mean membrane hyperpolarization of −7.1 ± 7.1 mV (n = 17 cells, not shown). On return to standard bath solution (i.e. 152.2 mM Na+), CCs recovered their initial RMP and, in some cells, the spiking behaviour also resumed (Fig. 7A ). Interestingly, the hyperpolarizing effect of low Na+‐containing saline did not depend on the pattern of electrical activity. Indeed, it was observed both in regular (−14.5 ± 13.6 mV, n = 10 cells, P < 0.05 ( = 0.0137), Wilcoxon matched‐pairs signed‐rank test, Fig. 7B , left panel) and in bursting cells (−6.7 ± 8.5 mV, n = 10 cells, P < 0.05 ( = 0.0273), Wilcoxon matched‐pairs signed‐rank test, Fig. 7B , middle panel). In silent cells perfused with a low Na+‐containing saline, a membrane hyperpolarization also occurred (−7.6 ± 12.0 mV, n = 10 cells, P > 0.05 ( = 0.1934), Wilcoxon matched‐pairs signed‐rank test, Fig. 7B , right panel), although the mean hyperpolarization value did not reach the significance threshold. In addition, pooled data from the 30 recorded cells indicate that the amplitude of membrane hyperpolarization did not depend on the spiking pattern, regular, bursting or silent (P = 0.3712, Kruskal‐Wallis test). Interestingly, considering a g Na/g K of 0.34 for a regular pattern, 0.30 for a bursting pattern, and 0.29 in silent cells, the theoretical hyperpolarization induced by changing E Na from +151 mV to + 117 mV (NMDG replacement, see eqn (1) with E rev = RMP) is −9.4 mV for a regular pattern, ‐ 7.7 mV for a bursting one and −7.6 mV in silent cells. Those theoretical values are close to the experimental values (−14 mV, −6.7 mV and −7.6 mV for regular, bursting and silent patterns, respectively). Thus, our results reveal that a Na+ conductance contributes to the RMP of mouse CCs, leading to a membrane hyperpolarization away from RMP upon lowering the extracellular Na+ concentration. Accordingly, switching to voltage‐clamp at −50 mV (a physiologically relevant membrane potential close to CC RMP), we observed that low Na+‐containing saline induced an outward current in all the 21 cells tested (NMDG replacement, Fig. 8Aa , change in current amplitude 19.0 ± 16.8 pA, n = 9 cells and Tris replacement, Fig. 8Ab , change in current amplitude 7.6 ± 9.7 pA, n = 12 cells). Note the smaller current amplitude change in Tris‐containing saline compared with NMDG‐containing saline, reflecting a better permeability of Tris over NMDG. Considering a mean value of 0.3 for g Na/g K and a mean resistance of 1.13 GΩ (see Table 3) and assuming that the channel is impermeant to NMDG, the expected amplitude of a current induced by a 110 mV change in Na+ driving force would be 22 pA, a theoretical value close to the measured value of 19 pA.
Figure 7. Extracellular Na+‐dependency of resting membrane potential.
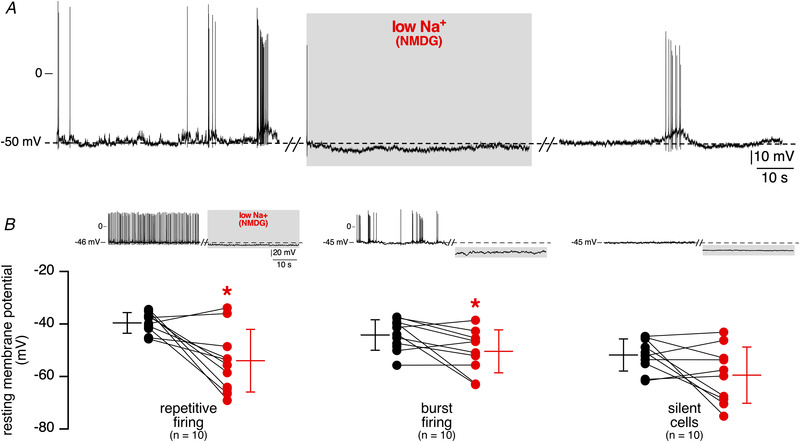
A, membrane hyperpolarization in response to bath‐applied low Na+‐containing saline (NaCl substituted by an equimolar concentration of NMDG‐Cl) in a spontaneously spiking (bursting) CC at RMP. Note the reversibility of the effect with AP resuming upon re‐administration of a standard extracellular Na+ concentration. B, pooled data of low extracellular Na+‐induced RMP changes as a function of the firing discharge pattern. Representative chart recordings of low Na+ (NMDG‐replaced)‐elicited membrane potential changes are plotted above each group of cells (left panel for regular spiking cells, middle panel for bursting cells and right panel for silent cells). The mean value of resting membrane potential was −39.8 ± 3.9 mV in control saline versus −54.5 ± 11.9 mV in low Na+‐containing saline for cells with regular firing (n = 10 cells, P < 0.05 ( = 0.0137), Wilcoxon matched‐pairs signed‐rank test), −44.2 ± 5.9 mV in control saline versus −50.8 ± 8.2 mV in low Na+‐containing saline for cells with burst firing pattern (n = 10 cells, P < 0.05 ( = 0.0273), Wilcoxon matched‐pairs signed‐rank test) and −52.2 ± 6.1 mV in control saline versus −59.9 ± 10.7 mV in low Na+‐containing saline for silent cells (n = 10 cells, P > 0.05 ( = 0.1934), Wilcoxon matched‐pairs signed‐rank test). [Color figure can be viewed at wileyonlinelibrary.com]
Figure 8. Changes in holding and steady‐state currents in response to bath‐applied low Na+ saline.
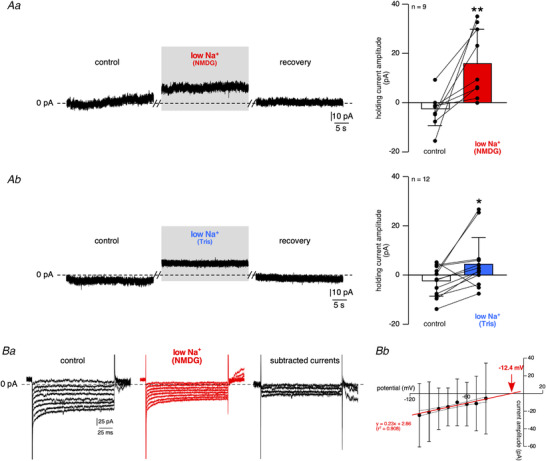
A, holding current recording. CCs were voltage‐clamped at −50 mV (a value near RMP). Substitution of NaCl by NMDG (Aa) or Tris (Ab) decreases the holding current, with the appearance of an outward current (mean amplitude: −2.9 ± 6.6 pA versus 16.1 ± 14.1 pA, in control and NMDG‐containing saline, respectively, n = 9 cells, P < 0.01 ( = 0.0078), Wilcoxon matched‐pairs signed‐rank test and −2.8 ± 6.1 pA versus 4.8 ± 10.8 pA, in control and Tris‐containing saline, respectively, n = 12 cells, P < 0.05 ( = 0.0161), Wilcoxon matched‐pairs signed‐rank test). B, steady‐state membrane currents evoked by voltage steps (150 ms duration) made from an initial holding potential of −30 mV in 10 mV decrements. Note the substantial decrease of the current amplitude in response to bath‐applied low Na+ (NMDG replacement, Ba). Bb, I‐V relationship of the subtracted currents (n = 26), combined with the confidence band at 95%. Current amplitudes were calculated from the average of a 10 ms window at the end of each 150 ms step. The ‘low Na+‐sensitive current’ exhibits a linear I‐V relationship, indicative of its voltage independency. The current reverses at −12.4 mV, arguing for a mixed ionic current. [Color figure can be viewed at wileyonlinelibrary.com]
To further explore the electrophysiological properties of the outward current observed upon low Na+ application, voltage‐clamped CCs were submitted to voltage step commands in control and low Na+ (NMDG replacement)‐containing saline (Fig. 8B ). The sustained Na+ current was obtained by subtracting the current during low Na+ application from the control current. Cells were held at −30 mV and stepped from −40 to −110 mV (150 ms duration, 10 mV decrements, Fig. 8Ba ). In response to bath‐applied low Na+, the current amplitude was reduced for each voltage step. As shown by pooled data collected from 26 cells (Fig. 8Bb ), the sustained Na+ current displayed a linear I‐V relationship and exhibited a mean E rev value of −12.4 mV, which suggests the contribution of a non‐selective cationic conductance (see also Fig. 13).
Figure 13. I Nab is more permeant to Na+ than to K+ .
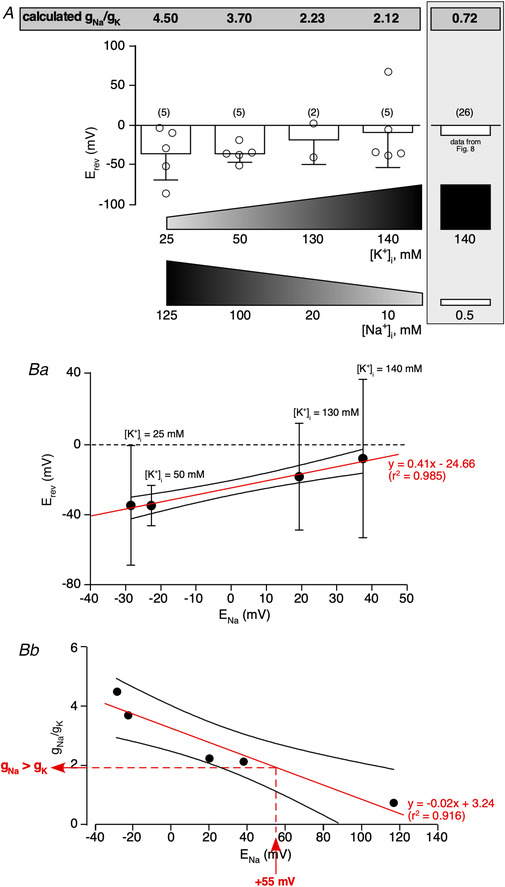
Ionic selectivity of I Nab was assessed by changing intracellular K+ and Na+ concentrations (K+ concentrations 25, 50, 130 and 140 mM and Na+ concentrations 125, 100, 20, 10). The ionic concentrations highlighted in a grey filled rectangle correspond to the condition of a low (0.5 mM) intracellular Na+ concentration, which was used in experiments described in Fig. 6. Under each ionic condition, I Nab was recorded through a voltage ramp command (−110 to −50 mV, 3 s duration). A, the E rev value of I Nab was calculated, for each recording condition, from a linear extrapolation applied to the subtracted currents. g Na/g K ratios were then calculated as described in Methods. B, robust correlation of E rev over E Na (plotted with the confidence band at 95%), with the restriction of [Na+]I ≥ 10 mM (Ba) and of g Na/g K over E Na (also plotted with the confidence band at 95%, Bb). Data were fitted with a linear regression (r 2 = 0.985 and 0.916, respectively). Note that under physiological conditions (i.e. E Na = +55 mV), g Na/g K is close to 2, indicating that I Nab is about twice as permeant to Na+ as to K+. [Color figure can be viewed at wileyonlinelibrary.com]
Altogether, the results argue for the presence of a Na+‐permeant background conductance operating near RMP. Under physiological conditions (i.e. in the presence of extracellular Na+), this conductance would exert a depolarizing effect, probably contributing to maintaining the RMP value above the equilibrium potential for K+. In the rest of the paper, this Na+‐sensitive conductance will be designated as I Nab, which stands for sodium background current.
Pharmacological features of I Nab: effects of TTX, Cs+ and ouabain
Because (i) our results support a significant contribution of extracellular Na+ to electrical activity and (ii) voltage‐gated Na+ channels (Nav) are engaged in AP generation in excitable cells, including mouse CCs (Vandael et al. 2015 b), we examined the sensitivity of the low Na+‐induced hyperpolarization to TTX (Fig. 9A ). Bath‐applied 0.5 μM TTX almost completely abolished APs, as expected, but (i) without the hyperpolarizing effect reminiscent of lowering extracellular Na+ (mean TTX‐induced membrane potential change +2.0 ± 11.8 mV, n = 12 cells) and (ii) without blocking the hyperpolarizing effect of low Na+‐containing saline (NMDG substitution, mean hyperpolarization of −6.0 ± 6.6 mV, n = 12 cells). In agreement with this finding is the result obtained in cells voltage‐clamped at −50 mV, in which TTX per se did not change the baseline of the holding current and did not inhibit the outward current induced by bath‐applied low Na+ (Fig. 9B , NMDG replacement). This demonstrates that I Nab is not carried by TTX‐sensitive Nav channels. Supporting this is the finding that, in many cells (n = 16/23 cells, 69.6%), APs could be electrically evoked in low Na+ (NMDG replacement)‐containing saline (data not shown).
Figure 9. I Nab is insensitive to TTX.
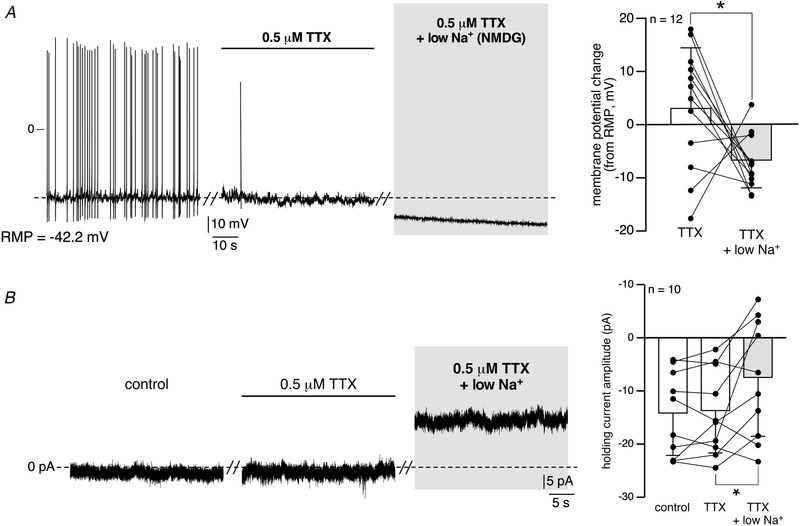
A, recording of the electrical activity of a mouse CC current‐clamped at RMP. The cell was sequentially challenged by a bath‐application of TTX (0.5 μM), followed by a low Na+ (NMDG substitution) + TTX‐containing saline. As expected, TTX in control saline drastically reduced the discharge of action potentials. In the presence of TTX, application of low Na+‐containing saline still induced a significant membrane hyperpolarization (* P = 0.042, Wilcoxon matched‐pairs signed‐rank test), thus indicating that I Nab is TTX independent. Pooled data are plotted in the right histogram. B, chart recordings of the holding current in a cell voltage‐clamped at −50 mV, before, during TTX application and in response to bath‐applied TTX + low Na+. Pooled data in the right histogram (* P = 0.027, Wilcoxon matched‐pairs signed‐rank test) show that low Na+‐induced holding current decrease persists in the presence of TTX.
To pursue the pharmacological characterization of the outward current, we performed additional experiments in the presence of intracellular Cs+ (140 mM in the patch pipette) to block most of the voltage‐gated K+ currents. As illustrated in Fig. 10A , the outward current induced by bath‐applied low Na+ (NMDG‐ (Fig. 10Aa ) or Tris‐substitution (Fig. 10Ab )) persisted. Besides, intracellular Cs+ did not inhibit the decrease in steady‐state current amplitude evoked by low Na+‐containing saline (Fig. 10B ). Under this experimental condition of high intracellular Cs+, however, the calculated mean E rev of I Nab displayed a shift towards a positive value (+29 mV) compared to the value calculated in high intracellular K+ (−12.4 mV). This indicates that I Nab is permeant to Cs+, but less so to K+.
Figure 10. I Nab is not blocked by Cs+ .
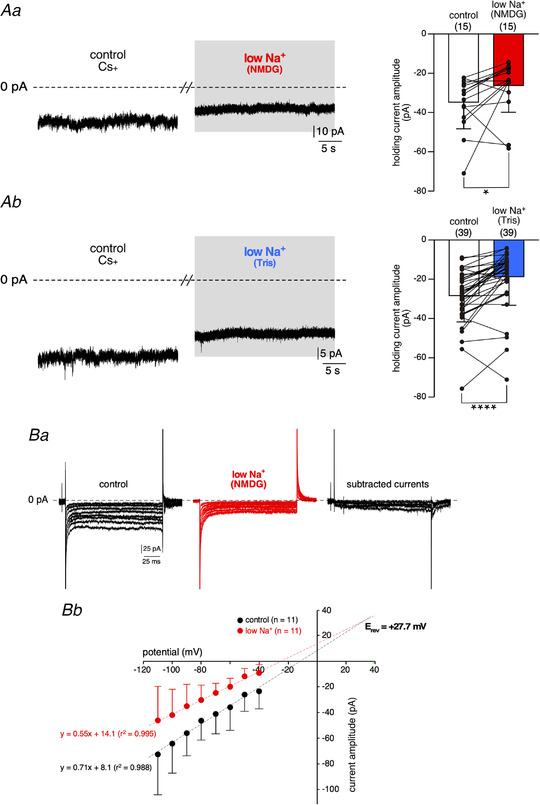
A, effects of low Na+ saline on the holding current of CCs patched with a pipette solution containing 140 mM CsCl and clamped at −50 mV. Bath‐applied low Na+ salines (NMDG‐ (Aa) or a Tris‐ (Ab) replaced) decreased the holding current value, indicating that I Nab is not inhibited by Cs+. Mean holding current amplitude: −35.3 ± 13.4 pA versus −26.8 ± 13.5 pA, in control and NMDG‐containing saline, respectively, n = 15 cells, P < 0.05 ( = 0.0256), Wilcoxon matched‐pairs signed‐rank test and −28.9 ± 13.5 pA versus −19.3 ± 14.6 pA, in control and Tris‐containing saline, respectively, n = 39 cells, P < 0.0001, Wilcoxon matched‐pairs signed‐rank test). B, recordings of steady‐state currents in CCs filled with Cs+ and clamped at −30 mV. Ba, bath application of low Na+ (NMDG substitution) reduced the amplitude of membrane currents evoked by voltage step commands (150 ms duration, 10 mV decrements). The I‐V relationship plotted in Bb shows that I Nab is linear in the voltage range from −110 to −40 mV. E rev was determined as the x‐axis‐intercept value calculated from extrapolated linear regression of currents recorded in control and low Na+ conditions. [Color figure can be viewed at wileyonlinelibrary.com]
Another possible candidate for supporting I Nab could be the Na+/K+ ATPase pump, the putative contribution of which was determined by using ouabain (Fig. 11). By blocking the Na+/K+ pump, ouabain prevents Na+ efflux from the cell and therefore increases the intracellular Na+ concentration. Ouabain per se (300 μM, bath application) did have a hyperpolarizing effect (Fig. 11Ab and B for pooled data), but did not prevent the low Na+‐evoked membrane hyperpolarization (mean amplitude −10.2 ± 12.2 mV, n = 4 cells), in both spontaneously firing and silent CCs (Fig. 11Aa , Ab and B for pooled data). This result disqualifies the Na+/K+ pump as a major contributor to I Nab.
Figure 11. The Na+/K+ ATPase pump blocker ouabain does not block I Nab .
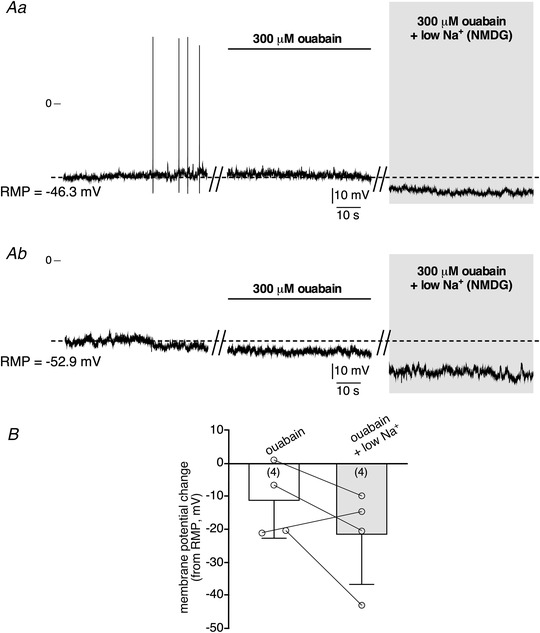
A, recording of the electrical activity of mouse CCs current‐clamped at RMP. The cells were sequentially challenged by a bath‐application of ouabain (300 μM), followed by a low Na+ (NMDG substitution) + ouabain‐containing saline. Ouabain per se induced a membrane hyperpolarization, both in spontaneously firing (Aa) and silent (Ab) cells. B, pooled data illustrating the membrane potential changes in response to low Na+ in the presence of ouabain. A further application of low Na+ saline in presence of ouabain increased hyperpolarization in both cells.
Sodium leak channel NALCN: a plausible contributor to I Nab
Consistent with the pharmacological (TTX insensitivity and no blockade by Cs+) and electrophysiological attributes (linear I‐V relationship, E rev corresponding to a non‐selective cationic conductance) reported above, a credible candidate for I Nab would be the sodium‐leak channel NALCN (Lu et al. 2007), also known to contribute to RMP in neuronal cells (Lu et al. 2007; Lu & Feng, 2011; Ren, 2011; Xie et al. 2013; Yeh et al. 2017). The absence of antibodies raised against mouse NALCN precluded an identification at the protein level. Therefore, to determine whether NALCN could participate in I Nab, we investigated the expression of Nalcn transcript in the mouse adrenal tissue (Fig. 12). Real‐time PCR revealed that Nalcn mRNA is present in the adrenal glands (Fig. 12A , n = 4 mice). As expected, Nalcn mRNA was detected in the brain, but not in the liver (Swayne et al. 2009). Note also the expression in the adrenal glands and brain of Unc79, Unc80 and Nlf‐1 transcripts, three members of the NALCN channelosome (Cochet‐Bissuel et al. 2014). Because whole adrenal glands were used in qPCR experiments, we could not discriminate between Nalcn expression in the cortex, in the medulla, or in both tissues. We therefore carried out in situ hybridization experiments (Fig. 12B ). As illustrated in Fig. 12Ba , the antisense Nalcn mRNA probe stained the adrenomedullary tissue (labelled with a Dbh probe), but not the cortex. Further, a double in situ hybridization (Fig. 12Bb ) allowed us to identify CCs as the cells expressing Nalcn mRNA in the mouse adrenal gland. This first report of Nalcn mRNA expression in adrenal CCs contributes to identifying NALCN as a highly plausible candidate channel supporting I Nab.
Figure 12. Expression of Nalcn transcript in the mouse adrenal gland, with a specific localisation in CCs.
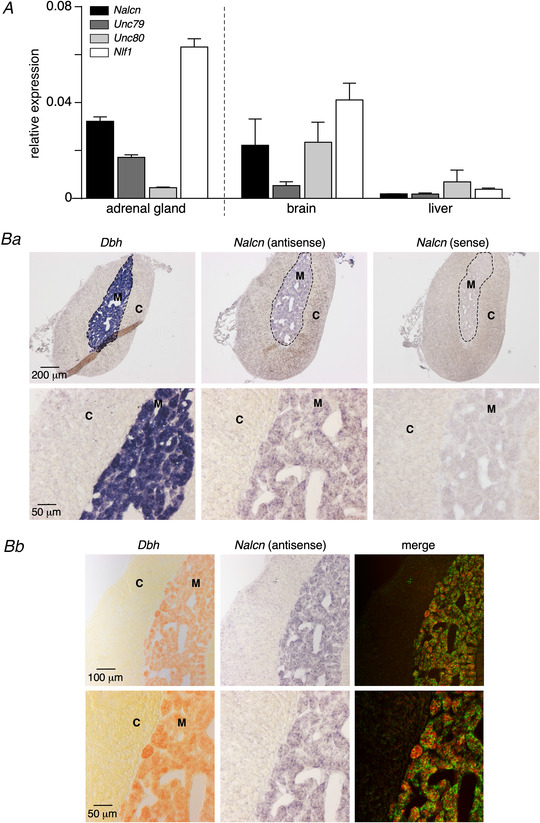
A, real‐time PCR determination of transcripts encoding NALCN and the ancillary proteins Unc79, Unc80 and NLF‐1 in the mouse adrenal tissue (n = 4 mice). Mouse brain was used as a positive tissue expressing NALCN and the liver as a negative tissue. Note that whole adrenal glands (including both cortex and medulla) were used. B, double in situ hybridization used for the co‐detection of Nalcn and dopamine beta hydroxylase (Dbh) transcripts in the mouse adrenal gland. Ba, specific detection of Nalcn mRNA with the antisense probe showing that Nalcn expression is restricted to the adrenomedullary tissue (M, specifically labelled by Dbh mRNA, which encodes a biosynthetic catecholamine enzyme) and excluded from the cortex (C). Specific sense probe showed no labelling. Bb, co‐localisation of Nalcn (pseudo green fluorescent labelling) and Dbh (pseudo red fluorescent labelling) arguing for a specific expression of NALCN in CCs.
NALCN channels generate a mixed ion current with the permeability sequence P Na>P K>P Cs (Lu et al. 2007). We therefore performed experiments aiming at characterizing the ionic selectivity of I Nab to K+ and Na+. To address this issue, CCs were patched with pipette solutions containing modified concentrations of K+ and Na+ (see Methods and Table 1). A voltage ramp protocol (−130 to −50 mV, 3 s duration) was applied in a control extracellular saline and then in low Na+‐containing saline. For each intracellular solution, the reversal potential of I Nab was determined and the associated ratio g Na/g K was calculated (see equations in Methods, Table 4 and Fig. 13A ). Regarding this analysis, g Na/g K appears to be concentration dependent, since when [Na+]i is lower, g Na/g K is also lower (0.72 in 0.5 mM [Na+]i). Supporting a significant permeability of I Nab to Na+ is the robust correlation between calculated E rev values for each experimental condition and the corresponding E Na values (r 2 of the linear regression = 0.985, Fig. 13Ba ), with the restriction of [Na+]i ≥10 mM. Similarly, calculated g Na/g K values robustly correlate with E Na (r 2 = 0.916, Fig. 13Bb ). Interestingly, this analysis also indicates that under physiological conditions (i.e. E Na = +55 mV), I Nab is about twice as permeant to Na+ ions as to K+ ions.
Table 4.
Determination of the g Na/g K ratio for I Nab in different ionic conditions
| [K+]i = 25 mM (n = 5 cells) | [K+]i = 50 mM (n = 5 cells) | [K+]i = 130 mM (n = 2 cells) | [K+]i = 140 mM (n = 5 cells) | [K+]i = 140 mM (n = 26 cells) | |
|---|---|---|---|---|---|
| [Na+]i (mM) | 125 | 100 | 20 | 10 | 0.5 |
| [Na+]o (mM) | 42.1 | 42.1 | 42.1 | 42.1 | 42.1 |
| [K+]i (mM) | 25 | 50 | 130 | 140 | 140 |
| [K+]o (mM) | 2.5 | 2.5 | 2.5 | 2.5 | 2.5 |
| E Na (mV) | −29 | −23 | +20 | +38 | +117 |
| E K (mV) | −61 | −79 | −104 | −106 | −106 |
|
E rev for I Nab (mV) |
−34.8 | −34.9 | −18.4 | −8.1 | −12.4 |
| g Na/g K | 4.50 | 3.70 | 2.23 | 2.12 | 0.72 |
The calculation of gNa/gK ratio indicates that INab is more permeant to Na+ than to K+ (g Na/g K > 1), except when the intracellular Na+ concentration is low (0.5 mM, last column, highlighted in grey). Under this experimental condition, INab becomes more permeant to K+ rather than to Na+ (g Na/g K < 1).
Collectively, our findings point to the presence of a cation‐permeant conductance in mouse adrenal CCs, mainly carried by Na+ ions and most probably supported by NALCN.
Discussion
This study reports the first description of a Na+‐permeant conductance operating near the resting membrane potential in mouse adrenal CCs in situ. In addition, we show that mouse CCs in acute slices exhibit a complex firing discharge pattern, consisting of intermittent periods of regular firing and irregular bursting spiking.
Regular and bursting spontaneous spiking modes in CCS: a peculiarity of mouse CCs?
Of all species in which the CC spontaneous electrical activity has been recorded, mouse CCs appear to exhibit a unique firing pattern alternating between regular and bursting modes. Indeed, contrasting with the mouse, rat CCs appear to spontaneously fire with a regular pattern, both in control and stress conditions (Colomer et al. 2008 a). In our study, bursts of APs were recorded in more than 50% of CCs firing at RMP. Although to a lesser extent, a spontaneous bursting pattern has been found in mouse CCs, both in cultured dissociated cells (Marcantoni et al. 2010; Vandael et al. 2015 b) and in vivo in anaesthetized animals (Desarmenien et al. 2013). In humans, less than 10% of adrenal CCs exhibit spontaneous APs (Hernandez‐Vivanco et al. 2017) and in the spiking cells, although not investigated in detail, the AP pattern seems to be irregular, alternating between firing and silence periods, but without typical bursts. The data resulting from our analysis of APs engaged in a burst is consistent with previous results in dissociated mouse CCs (Vandael et al. 2015 b), as such consecutive APs display a gradual reduction in the peak amplitude, AHP amplitude and ISI associated with an increase in the half‐width duration and in intra‐burst instantaneous firing frequency. Interestingly, we noted that the peak amplitude of the first AP in a burst is larger compared to the peak amplitude of APs coming from a regular firing pattern. This may be explained by the more polarized RMP found in cells exhibiting burst discharges, leading to increased Nav channel availability for further generation of APs.
It is noteworthy that a bursting electrical activity can be elicited by neurotransmitters (reviewed in Carbone et al. 2019) or by manipulating CC ion channel expression and availability (Nassar‐Gentina et al. 1988; Marcantoni et al. 2010; Martinez‐Espinosa et al. 2014; Vandael et al. 2015 a, b ; Lingle et al. 2018) or extracellular pH (Guarina et al. 2017). Which mechanism(s) contribute(s) to those two firing patterns in the mouse, and how does a CC switch from a regular to a bursting firing mode? A modification of the permeability ratio between Na+ and K+ conductances may take place. Indeed, the g Na/g K ratios are 0.34 and 0.30 for regular and bursting patterns, respectively. This indicates that the membrane is more permeable to Na+ and/or less permeable to K+ ions during a regular pattern than during bursts. If we assume a role of I Nab in this process, we can therefore propose that the open probability of the channels supporting I Nab would vary along recording time, switching from an active mode to a silent mode, as described for K+ channels (Bossu & Gahwiler, 1996). How could I Nab contribute to firing? This should be seen in a general excitability scheme in which the regulation of RMP by ion channels (including I Nab) and the ion channels expressed by the cell play a central role. Above RMP (i.e. upon I Nab activation), subsequent activation of Nav and Cav channels would contribute to trigger a spiking activity. Below RMP (i.e. upon I Nab closure), the resulting hyperpolarization would, in turn, de‐inactivate voltage‐sensitive channels operating close to RMP (such as the M‐type K+ channels or low threshold Ca2+ channels) leading thus to cell depolarization and subsequent triggering of bursts of APs (Wallace et al. 2002; Crunelli et al. 2018). The plateau current observed during the burst could result from an equilibrium between Na+, Ca2+ and K+ currents, and subsequent activation of SK and/or BK channels driven by a Ca2+ entry through voltage‐gated Ca2+ channels would end the burst. From a less biophysical and more physiological point of view, it is tempting to pair the regular firing pattern to basal splanchnic nerve sympathetic tone and the bursting firing pattern to the increased sympathetic tone as found during stress response. This could be verified if CC electrical activity resulted solely from the synaptic inputs. But it is unlikely for at least four reasons. First, CCs fire APs even in the absence of synaptic neurotransmission (Marcantoni et al. 2010; Vandael et al. 2015 b). Second, in acute slices, only few CCs (<20%) display spontaneous excitatory synaptic events due to the elimination of numerous nerve fibres during the slicing procedure (Barbara et al. 1998). Third, at least in the rat, CCs fire spontaneously with a regular pattern, both in control and stress conditions (Colomer et al. 2008 a). Fourth, the two firing modes are found in hexamethonium‐containing saline, a blocker of postsynaptic α3‐containing nicotinic acetylcholine receptors (A. Milman and N. C. Guerineau, unpublished data). Another explanation, more conceivable, would be changes in ion channel availability, as observed for voltage‐gated Na+ channels (Vandael et al. 2015 b) or in external milieu parameters such as pH (Guarina et al. 2017). Additional experiments would be required to ascertain this latter hypothesis.
Sodium dependency of resting membrane potential of mouse CCs: a critical contribution to CC excitability
By challenging cells with a low Na+‐containing saline (NMDG or Tris replacement), we evidenced here in mouse CCs the presence of a Na+‐permeant conductance operating near RMP, indicating a Na+ dependency of RMP. This finding is consistent with membrane hyperpolarization reported in freshly dissociated gerbil CCs (Douglas et al. 1967) and thin bovine adrenal medulla slices (Baker & Rink, 1975), in response to extracellular Na+ replacement. In two other studies, an AP blockade without membrane hyperpolarization (Brandt et al. 1976) or no change in membrane potential (Nassar‐Gentina et al. 1988) has been observed in response to extracellular Na+ deprivation. Taking anterior pituitary cells and pancreatic beta cells as other representative examples ofendocrine/neuroendocrine tissues, the replacement of extracellular Na+ collectively converges toward a robust membrane hyperpolarization (>10–20 mV) and subsequent abrogation of APs for anterior pituitary (Simasko, 1994; Sankaranarayanan & Simasko, 1996; Simasko & Sankaranarayanan, 1997; Kwiecien et al. 1998; Tsaneva‐Atanasova et al. 2007; Kucka et al. 2010, 2012; Liang et al. 2011; Tomic et al. 2011; Zemkova et al. 2016; Kayano et al. 2019) and for pancreatic insulin‐secreting cells (Ribalet & Beigelman, 1982; de Miguel et al. 1988; Worley et al. 1994; Garcia‐Barrado et al. 1996). Also supporting a critical contribution of I Nab to cell excitability, the implementation of a persistent Na+ conductance in computational modelling is a pre‐requisite to simulating cell excitability in a reliable manner (Fridlyand et al. 2009). I Nab would behave as a regulator of bursting. As exemplified in modelled pancreatic beta cells, decreasing I Nab conductance reduces the ability of cells to burst (Fridlyand et al. 2009). Also of note, blocking I Nab only is sufficient to abolish the electrical activity and to hyperpolarize cell membrane, as observed during experimental conditions (Tsaneva‐Atanasova et al. 2007; Liang et al. 2011).
Resting membrane potential of CCs: an intricate contribution of K+ and Na+ permeabilities
Compared to RMP of neuronal cells (∼ −80 mV), the RMP value of CCs, which share a common embryonic origin with neurons, is much more depolarized, both in the mouse (mean values of −43 mV in the present study, −49 mV in Marcantoni et al. 2009, −54 mV in Nassar‐Gentina et al. 1988)) and in the rat (mean values of −52 mV in Sanz‐Lazaro et al. 2019, −61 mV in Barbara & Takeda, 1996, −64 mV in Colomer et al. 2008 a and −68 mV in Kajiwara et al. 1997; Martin et al. 2001). A RMP value quite distant from the equilibrium potential for K+ suggests the involvement of ions with positive equilibrium potentials, such as Na+. Two hypotheses, not mutually exclusive, are currently proposed to account for a notable Na+ contribution to RMP, which are (i) the stochastic activation of voltage‐gated Na+ channels able to push cells to initiate firing (Schmich & Miller, 1997; Schneidman et al. 1998; Dorval, 2006) and (ii) the presence of a persistent depolarizing resting cation flux (Lingle et al. 2018). In excitable cells, RMP is determined by background depolarizing (sodium‐permeant) conductances and hyperpolarizing (potassium‐permeant) conductances (such as SK channels, Kir‐ or K2P‐mediated currents) (Hille, 2001; Vandael et al. 2012). With a mean g Na/g K ratio of 0.3, our data indicate that RMP in mouse CCs is mainly driven by K+ permeabilities, regardless of the firing pattern (regular, bursting or silent). However, the external Na+ substitution demonstrates an additional contribution of Na+ ions in setting the RMP. At RMP, this Na+‐permeant background conductance is almost 2 times more permeant to Na+ than to K+.
A persistent Na+‐permeant conductance (alternatively named I Nab, I BNC, I b,Na, I NS,Na or I NaP according to the studies) involved in setting‐up the RMP value and the ability of cells to fire (i) is not restricted to endocrine/neuroendocrine tissues, but is also present in other excitable cells (Jones, 1989; Noble et al. 1992; Spindler et al. 1998; Raman et al. 2000; Eggermann et al. 2003; Pena & Ramirez, 2004; Atherton & Bevan, 2005; Khaliq & Bean, 2010; LeSauter et al. 2011; Tremblay et al. 2011; Reinl et al. 2015) and (ii) is not found only in vertebrates (Lear et al. 2005; Gouwens & Wilson, 2009; Xie et al. 2013; Flourakis et al. 2015; Gao et al. 2015) pointing towards a crucial role of this conductance in regulating cell and tissue function.
Possible contributors to I Nab
NALCN as a candidate
The finding that I Nab is mainly carried by Na+ ions directs the search for the molecular identity of the channels toward Na‐permeant ionic contributors. In this regard, the Na+‐leak channel NALCN appears as a plausible candidate, sharing at least four attributes with I Nab. First, I NALCN is TTX insensitive, is not blocked by Cs+ (Lu et al. 2007; Bouasse et al. 2019) and displays a linear I‐V relationship (Lu et al. 2007). Second, the ionic selectivity for Na+ and K+ reported in our study (g Na>g K) is in agreement with the permeability sequence reported for I NALCN (Lu et al. 2007). In addition, our data indicate that I Nab is also permeant to Cs+ with the sequence g Cs<g K<g Na). Third, in neuronal cells, NALCN supports a resting Na+‐permeant depolarizing conductance, which contributes to maintaining RMP and thus the ability of cells to fire (Lu et al. 2007; Ren, 2011; Xie et al. 2013; Gao et al. 2015; Ford et al. 2018). Fourth, our finding that the Nalcn transcript is expressed in mouse adrenal CCs supports a possible contribution of NALCN to I Nab. Although not investigated in CCs in particular, the detection in the mouse adrenal gland of mRNAs encoding NALCN‐associated proteins (Unc79, Unc80 and NLF‐1), which seem to be mandatory for the generation of NALCN currents (Bouasse et al. 2019; Chua et al. 2020), also argues for a possible role of NALCN‐mediated current in the adrenal tissue. Note that Nalcn mRNA has been found in pancreatic insulin‐secreting cells (Swayne et al. 2009), neuroendocrine cells sharing common stimulation‐secretion coupling attributes with adrenal CCs. However, despite all these arguments robustly converging on a NALCN‐mediated current, the contribution of NALCN channels to I Nab remains to be ascertained. At present, the electrophysiological study of NALCN‐mediated currents in native tissues is severely hampered by the lack of drugs able to selectively block the channel, rendering laborious the establishment of a clear‐cut electrophysiological signature of the current carried by NALCN. The non‐specific blocker gadolinium (Gd3+) is frequently used to block I NALCN. However, its use was not appropriate here considering its ability to inhibit other ion channels, including voltage‐gated Ca2+ channels (Biagi & Enyeart, 1990), major components of the adrenal stimulus‐secretion coupling (Guerineau et al. 2012; Lingle et al. 2018). Recently, Bouasse and colleagues (Bouasse et al. 2019) deciphered new electrophysiological features of the NALCN current via the heterologous expression of recombinant human NALCN and associated channelosome in a neuronal cell line. Under these experimental conditions, NALCN current exhibits a time‐dependent inactivation and a non‐linear I‐V relationship indicative of voltage sensitivity (Bouasse et al. 2019). This differs from the biophysical properties described here for I Nab in mouse CCs. Several explanations, not mutually exclusive, can account for this discrepancy. First, I Nab and I NALCN are two distinct currents. Second, I NALCN displays separate biophysical features when endogenously expressed in situ and when it is heterologously expressed in vitro in a recipient cell line (Bouasse et al. 2019). Third, the electrophysiological properties of I NALCN differ between mouse (our study) and human orthologues (Bouasse et al. 2019).
Contribution of other channels?
Although NALCN is a convincing candidate for supporting I Nab in CCs, other ‘leak’ ion channels may also contribute to a background conductance. First, the hyperpolarization‐activated cyclic nucleotide‐gated (HCN) channels are blocked by Cs+ while I Nab is not blocked by Cs+. In addition, I h, the HCN channel‐related current, is about three times more permeant to K+ than to Na+ (Solomon & Nerbonne, 1993; Budde et al. 1994; Simasko & Sankaranarayanan, 1997). Also consistent with the fact that HCN channels do not support I Nab, at −50 mV, a value close to CC RMP, I h is unlikely to be active (half‐maximal activation voltage found around −100 mV; Simasko & Sankaranarayanan, 1997). Spontaneously firing CCs do not hyperpolarize enough to reach the voltages required to activate I h. It is also interesting to note that an I h current has not yet been deciphered in CCs. Second, the two‐pore‐domain potassium (K2P) channels which are known as ‘leak’ channels are responsible for leak background potassium‐permeant conductances. Although K2P channels can change their ionic selectivity under changes in extracellular milieu (such as pH), and, as found for TWIK‐1, can become Na+ permeant (Chatelain et al. 2012), these channels are unlikely to contribute to I Nab. Indeed, even when displaying an ionic selectivity to Na+, the P Na/P K ratio for TWIK‐1 channels remains <1 (Chatelain et al. 2012), a value far from the ratio of 2.88 we found for I Nab. Also, only TREK and TASK channels have been reported to be expressed in the rodent adrenal medullary tissue (Inoue et al. 2008; Kim & Kang, 2015; Guarina et al. 2017). Third, the family of transient receptor potential (TRP) channels deserves consideration. Mouse CCs express TRPM4 channels (Mathar et al. 2010). In addition, the TRP canonical (TRPC) channels constitute the largest family of cation‐conducting channels. Interestingly, some TRP canonical (TRPC) channels are constitutively active (Trebak et al. 2003; Nichols et al. 2007) and may therefore represent putative candidates for supporting I Nab. Hence, a TTX‐resistant background‐depolarizing Na+ current, presumably supported by the TRPC channel family, has been described in anterior pituitary cells, and appears involved in AP firing and for hormone secretion (Kucka et al. 2012).
Physiological relevance of I Nab in CCs: a role in catecholamine secretion?
Our data show that I Nab contributes to RMP in mouse adrenal CCs and that membrane potential changes occurring near the resting value remodel the spiking pattern (regular/bursting modes). The combination of these two observations raises the stimulating hypothesis of a role for I Nab in regulating the firing mode. Depending on I Nab current density expressed in a given CC, at a given time, one would expect the cell to fire either with a regular or a bursting mode. This might be of particular physiological interest because the bursting mode has been reported to efficiently stimulate catecholamine secretion in rodent CCs (Duan et al. 2003; Vandael et al. 2015 b). This would link I Nab to catecholamine secretion but whether and how I Nab contributes to catecholamine secretion remains an open question. We think that this issue requires identification of the ion channel(s) supporting I Nab. NALCN is certainly a candidate, but the absence of pharmacological tools to selectively block NALCN current prevents further electrophysiological characterization. The use of Nalcn‐deficient animals could represent an alternative strategy to pursue physiological investigations.
From a physiological point of view, whether I Nab does require receptor activation is an interesting issue. The recording of I Nab in the presence of hexamethonium reveals that I Nab does not require activation of α3‐containing nicotinic acetylcholine receptors (A. Milman and N. C. Guérineau, unpublished data). However, additional experiments will be necessary since, at the splanchnic nerve‐CC synapse, not only acetylcholine is released but also various neuropeptides, such as the pituitary adenylate cyclase activating peptide PACAP (reviewed in Guerineau, 2020). Of particular interest are the findings that Na+‐permeant voltage‐independent inward conductances can be activated by neurotransmitters and neuropeptides. In this context, PACAP is a remarkable example, inasmuch it induces a TTX‐insensitive sub‐threshold membrane depolarization that depends on extracellular Na+ in bovine CCs (Tanaka et al. 1996). Such an alternate route of Na+ influx, independent of APs but resulting possibly in depolarization, is also found in insulinoma cells, in which the neuropeptide PACAP stimulates a voltage‐independent Na+‐permeant conductance (E rev of −28 mV), convincingly resembling I Nab (Leech et al. 1995). Along the same line, it is of note that NALCN channels can be activated by peptides and neurotransmitters (Lu et al. 2009), such as substance P (Kim et al. 2012) and acetylcholine (Swayne et al. 2009). Assuming that NALCN channels might be activated by PACAP, and because PACAP is a major neurotransmitter for stress transduction in the adrenomedullary tissue (Smith & Eiden, 2012; Eiden et al. 2018), I NALCN and/or I Nab may then have a decisive and unanticipated role in the adrenal stimulus‐secretion coupling. Again, the identification of channel(s) supporting I Nab would greatly assist in the study of regulatory mechanisms.
More generally, could I Nab be involved in hormone release in endocrine/neuroendocrine tissues? Regarding this, extensive works have investigated the effect of a low Na+‐ containing saline on hormone secretion. Although with a certain degree of variability (cell type, hormone studied, basal or stimulated secretion … ), lowering extracellular Na+ collectively converges toward a decrease in hormone release in the anterior pituitary (Collu et al. 1984; Saith et al. 1984; Kato et al. 1988; Sankaranarayanan & Simasko, 1996; Takano et al. 1996; Yang et al. 2008; Kucka et al. 2012), the insulin‐secreting pancreas (Hales & Milner, 1968; Malaisse et al. 1971; Hellman et al. 1974; Lambert et al. 1974; Herchuelz et al. 1980) and the adrenal medullary tissue (Banks et al. 1969; Kilpatrick et al. 1981; Lemaire et al. 1981; Role et al. 1981; Knight & Baker, 1983; Wada et al. 1984). These data are consistent with a regulatory action of the background Na+ conductance on RMP and cell excitability. Indeed, under physiological conditions, the depolarizing action of I Nab contributes to bringing the secretory cells into an electrical status favouring the firing, and subsequent [Ca2+]i increases, which are required for the secretory process. Catecholamine secretion by adrenal CCs is a key step in the adaptive mechanisms triggered by an organism to cope with stress. Assuming a substantial role of I Nab in catecholamine secretion, it would be of interest to investigate whether and how the biophysical properties of I Nab remodel in a ‘stressed’ adrenal medullary tissue.
Additional information
Competing interests
The authors declare no conflict of interest.
Author contributions
N.C.G., P.L. and J.L.B. designed research; A.M, S.V., J.L.B. and N.C.G. analysed data; A.M. and S.V. performed research; A.M. designed molecular tools; P.F. developed software to analyse AP parameters and N.C.G. wrote the paper. All authors approved the final version of the manuscript and all those that qualify for authorship are listed.
Funding
This work was supported by grants from Centre National de la Recherche Scientifique, Institut National de la Santé et de la Recherche Médicale, the Laboratory of Excellence ‘Ion Channel Science and Therapeutics’ (LabEx ICST, grant ANR‐11‐LABX‐0015‐01) and Fondation pour la Recherche Médicale. A. Milman was supported by a fellowship from the LabEx ICST.
Supporting information
Statistical Summary Document
Acknowledgements
We thank Drs Michel G. Desarménien, Sophie Nicole and Jean Chemin for critical reading of the manuscript.
Biography
Alexandre Milman obtained his Masters degree at Pierre and Marie Curie University in Paris (France). He then moved to the University of Montpellier (France) to complete his PhD thesis in neuroscience (2018) under the supervision of Dr Nathalie C. Guérineau in Philippe Lory's team at the Institute of Functional Genomics. During his PhD, Alexandre Milman's research focused on the role of a sodium background conductance in the regulation of mouse chromaffin cell excitability. He was supported by a fellowship from the Laboratory of Excellence (LabEx) ‘Ion Channel Science and Therapeutics’.

Edited by: Peying Fong & Florian Lesage
Linked articles: This article is highlighted in a Perspectives article by Tuluc. To read this article, visit https://doi.org/10.1113/JP281353.
Data availability statement
The datasets generated and analysed during the present study are available from the corresponding author upon reasonable request.
References
- Alcami P, Franconville R, Llano I & Marty A (2012). Measuring the firing rate of high‐resistance neurons with cell‐attached recording. J Neurosci 32, 3118–3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almers W, Stanfield PR & Stuhmer W (1983). Lateral distribution of sodium and potassium channels in frog skeletal muscle: measurements with a patch‐clamp technique. J Physiol 336, 261–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atherton JF & Bevan MD (2005). Ionic mechanisms underlying autonomous action potential generation in the somata and dendrites of GABAergic substantia nigra pars reticulata neurons in vitro. J Neurosci 25, 8272–8281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker PF & Rink TJ (1975). Catecholamine release from bovine adrenal medulla in response to maintained depolarization. J Physiol 253, 593–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks P, Biggins R, Bishop R, Christian B & Currie N (1969). Sodium ions and the secretion of catecholamines. J Physiol 200, 797–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbara JG, Poncer JC, McKinney RA & Takeda K (1998). An adrenal slice preparation for the study of chromaffin cells and their cholinergic innervation. J Neurosci Methods 80, 181–189. [DOI] [PubMed] [Google Scholar]
- Barbara JG & Takeda K (1996). Quantal release at a neuronal nicotinic synapse from rat adrenal gland. Proc Natl Acad Sci U S A 93, 9905–9909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biagi BA & Enyeart JJ (1990). Gadolinium blocks low‐ and high‐threshold calcium currents in pituitary cells. Am J Physiol Cell Physiol 259, C515–C520. [DOI] [PubMed] [Google Scholar]
- Bossu JL & Gahwiler BH (1996). Distinct modes of channel gating underlie inactivation of somatic K+ current in rat hippocampal pyramidal cells in vitro. J Physiol 495 383–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouasse M, Impheng H, Servant Z, Lory P & Monteil A (2019). Functional expression of CLIFAHDD and IHPRF pathogenic variants of the NALCN channel in neuronal cells reveals both gain‐ and loss‐of‐function properties. Sci Rep 9, 11791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt BL, Hagiwara S, Kidokoro Y & Miyazaki S (1976). Action potentials in the rat chromaffin cell and effects of acetylcholine. J Physiol 263, 417–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budde T, White JA & Kay AR (1994). Hyperpolarization‐activated Na+‐K+ current (Ih) in neocortical neurons is blocked by external proteolysis and internal TEA. J Neurophysiol 72, 2737–2742. [DOI] [PubMed] [Google Scholar]
- Carbone E, Borges R, Eiden LE, Garcia AG & Hernandez‐Cruz A (2019). Chromaffin cells of the adrenal medulla: physiology, pharmacology, and disease. Compr Physiol 9, 1443–1502. [DOI] [PubMed] [Google Scholar]
- Chatelain FC, Bichet D, Douguet D, Feliciangeli S, Bendahhou S, Reichold M, Warth R, Barhanin J & Lesage F (2012). TWIK1, a unique background channel with variable ion selectivity. Proc Natl Acad Sci U S A 109, 5499–5504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua HC, Wulf M, Weidling C, Rasmussen LP & Pless SA (2020). The NALCN channel complex is voltage sensitive and directly modulated by extracellular calcium. Sci Adv 6, eaaz3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cochet‐Bissuel M, Lory P & Monteil A (2014). The sodium leak channel, NALCN, in health and disease. Front Cell Neurosci 8, 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collu R, Lafond J, Marchisio AM, Eljarmak D & Ducharme JR (1984). Sodium ions: their role and mechanism of action in the control of prolactin release. Endocrinology 114, 1302–1307. [DOI] [PubMed] [Google Scholar]
- Colomer C, Lafont C & Guerineau NC (2008a). Stress‐induced intercellular communication remodeling in the rat adrenal medulla. Ann N Y Acad Sci 1148, 106–111. [DOI] [PubMed] [Google Scholar]
- Colomer C, Martin AO, Desarmenien MG & Guerineau NC (2012). Gap junction‐mediated intercellular communication in the adrenal medulla: An additional ingredient of stimulus‐secretion coupling regulation. Biochim Biophys Acta 1818, 1937–1951. [DOI] [PubMed] [Google Scholar]
- Colomer C, Olivos Ore LA, Coutry N, Mathieu MN, Arthaud S, Fontanaud P, Iankova I, Macari F, Thouennon E, Yon L, Anouar Y & Guerineau NC (2008b). Functional remodeling of gap junction‐mediated electrical communication between adrenal chromaffin cells in stressed rats. J Neurosci 28, 6616–6626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crunelli V, Lorincz ML, Connelly WM, David F, Hughes SW, Lambert RC, Leresche N & Errington AC (2018). Dual function of thalamic low‐vigilance state oscillations: rhythm‐regulation and plasticity. Nat Rev Neurosci 19, 107–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Miguel R, Tamagawa T, Schmeer W, Nenquin M & Henquin JC (1988). Effects of acute sodium omission on insulin release, ionic flux and membrane potential in mouse pancreatic B‐cells. Biochim Biophys Acta 969, 198–207. [DOI] [PubMed] [Google Scholar]
- Desarmenien MG, Jourdan C, Toutain B, Vessieres E, Hormuzdi SG & Guerineau NC (2013). Gap junction signalling is a stress‐regulated component of adrenal neuroendocrine stimulus‐secretion coupling in vivo. Nat Commun 4, 2938. [DOI] [PubMed] [Google Scholar]
- Dorval AD (2006). The rhythmic consequences of ion channel stochasticity. Neuroscientist 12, 442–448. [DOI] [PubMed] [Google Scholar]
- Douglas WW, Kanno T & Sampson SR (1967). Influence of the ionic environment on the membrane potential of adrenal chromaffin cells and on the depolarizing effect of acetylcholine. J Physiol 191, 107–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan K, Yu X, Zhang C & Zhou Z (2003). Control of secretion by temporal patterns of action potentials in adrenal chromaffin cells. J Neurosci 23, 11235–11243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggermann E, Bayer L, Serafin M, Saint‐Mleux B, Bernheim L, Machard D, Jones BE & Muhlethaler M (2003). The wake‐promoting hypocretin‐orexin neurons are in an intrinsic state of membrane depolarization. J Neurosci 23, 1557–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eiden LE, Emery AC, Zhang L & Smith CB (2018). PACAP signaling in stress: insights from the chromaffin cell. Pflugers Arch 470, 79–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flourakis M, Kula‐Eversole E, Hutchison AL, Han TH, Aranda K, Moose DL, White KP, Dinner AR, Lear BC, Ren D, Diekman CO, Raman IM & Allada R (2015). A conserved bicycle model for circadian clock control of membrane excitability. Cell 162, 836–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford NC, Ren D & Baccei ML (2018). NALCN channels enhance the intrinsic excitability of spinal projection neurons. Pain 159, 1719–1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fridlyand LE, Jacobson DA, Kuznetsov A & Philipson LH (2009). A model of action potentials and fast Ca2+ dynamics in pancreatic beta‐cells. Biophys J 96, 3126–3139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao S, Xie L, Kawano T, Po MD, Guan S, Zhen M, Pirri JK & Alkema MJ (2015). The NCA sodium leak channel is required for persistent motor circuit activity that sustains locomotion. Nat Commun 6, 6323. [DOI] [PubMed] [Google Scholar]
- Garcia‐Barrado MJ, Gilon P, Sato Y, Nenquin M & Henquin JC (1996). No evidence for a role of reverse Na+‐Ca2+ exchange in insulin release from mouse pancreatic islets. Am J Physiol Endocrinol Metab 271, E426‐433. [DOI] [PubMed] [Google Scholar]
- Gouwens NW & Wilson RI (2009). Signal propagation in Drosophila central neurons. J Neurosci 29, 6239–6249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarina L, Vandael DH, Carabelli V & Carbone E (2017). Low pHo boosts burst firing and catecholamine release by blocking TASK‐1 and BK channels while preserving Cav1 channels in mouse chromaffin cells. J Physiol 595, 2587–2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerineau NC (2018). Gap junction communication between chromaffin cells: the hidden face of adrenal stimulus‐secretion coupling. Pflugers Arch 470, 89–96. [DOI] [PubMed] [Google Scholar]
- Guerineau NC (2020). Cholinergic and peptidergic neurotransmission in the adrenal medulla: A dynamic control of stimulus‐secretion coupling. IUBMB Life 72, 553–567. [DOI] [PubMed] [Google Scholar]
- Guerineau NC, Desarmenien MG, Carabelli V & Carbone E (2012). Functional chromaffin cell plasticity in response to stress: focus on nicotinic, gap junction, and voltage‐gated Ca2+ channels. J Mol Neurosci 48, 368–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hales CN & Milner RD (1968). The role of sodium and potassium in insulin secretion from rabbit pancreas. J Physiol 194, 725–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellman B, Idahl LA, Lernmark A, Sehlin J & Taljedal IB (1974). The pancreatic beta‐cell recognition of insulin secretagogues. Effects of calcium and sodium on glucose metabolism and insulin release. Biochem J 138, 33–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herchuelz A, Sener A & Malaisse WJ (1980). Regulation of calcium fluxes in rat pancreatic islets: calcium extrusion by sodium‐calcium countertransport. J Membr Biol 57, 1–12. [DOI] [PubMed] [Google Scholar]
- Hernandez‐Vivanco A, Sanz‐Lazaro S, Jimenez‐Pompa A, Garcia‐Magro N, Carmona‐Hidalgo B, Perez‐Alvarez A, Caba‐Gonzalez JC, Tabernero A, Alonso YGS, Passas J, Blazquez J, Gonzalez‐Enguita C, de Castro‐Guerin C & Albillos A (2017). Human native Cav1 channels in chromaffin cells: contribution to exocytosis and firing of spontaneous action potentials. Eur J Pharmacol 796, 115–121. [DOI] [PubMed] [Google Scholar]
- Hille B (2001). Ion channels of excitable membranes. Sinauer Associates, Sunderland, MA, USA. [Google Scholar]
- Hodson DJ, Legros C, Desarmenien MG & Guerineau NC (2015). Roles of connexins and pannexins in (neuro)endocrine physiology. Cell Mol Life Sci 72, 2911–2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu G, Oboukhova EA, Kumar S, Sturek M & Obukhov AG (2009). Canonical transient receptor potential channels expression is elevated in a porcine model of metabolic syndrome. Mol Endocrinol 23, 689–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue M, Harada K, Matsuoka H, Sata T & Warashina A (2008). Inhibition of TASK1‐like channels by muscarinic receptor stimulation in rat adrenal medullary cells. J Neurochem 106, 1804–1814. [DOI] [PubMed] [Google Scholar]
- Jones SW (1989). On the resting potential of isolated frog sympathetic neurons. Neuron 3, 153–161. [DOI] [PubMed] [Google Scholar]
- Kajiwara R, Sand O, Kidokoro Y, Barish ME & Iijima T (1997). Functional organization of chromaffin cells and cholinergic synaptic transmission in rat adrenal medulla. Jpn J Physiol 47, 449–464. [DOI] [PubMed] [Google Scholar]
- Kato M, Hattori MA & Suzuki M (1988). Inhibition by extracellular Na+ replacement of GRF‐induced GH secretion from rat pituitary cells. Am J Physiol Endocrinol Metab 254, E476–E481. [DOI] [PubMed] [Google Scholar]
- Kayano T, Sasaki Y, Kitamura N, Harayama N, Moriya T, Dayanithi G, Verkhratsky A & Shibuya I (2019). Persistent Na+ influx drives L‐type channel resting Ca2+ entry in rat melanotrophs. Cell Calcium 79, 11–19. [DOI] [PubMed] [Google Scholar]
- Khaliq ZM & Bean BP (2010). Pacemaking in dopaminergic ventral tegmental area neurons: depolarizing drive from background and voltage‐dependent sodium conductances. J Neurosci 30, 7401–7413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilpatrick DL, Slepetis R & Kirshner N (1981). Ion channels and membrane potential in stimulus‐secretion coupling in adrenal medulla cells. J Neurochem 36, 1245–1255. [DOI] [PubMed] [Google Scholar]
- Kim BJ, Chang IY, Choi S, Jun JY, Jeon JH, Xu WX, Kwon YK, Ren D & So I (2012). Involvement of Na+‐leak channel in substance P‐induced depolarization of pacemaking activity in interstitial cells of Cajal. Cell Physiol Biochem 29, 501–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D & Kang D (2015). Role of K(2)p channels in stimulus‐secretion coupling. Pflugers Arch 467, 1001–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight DE & Baker PF (1983). Stimulus‐secretion coupling in isolated bovine adrenal medullary cells. Q J Exp Physiol 68, 123–143 [DOI] [PubMed] [Google Scholar]
- Kucka M, Kretschmannova K, Murano T, Wu CP, Zemkova H, Ambudkar SV & Stojilkovic SS (2010). Dependence of multidrug resistance protein‐mediated cyclic nucleotide efflux on the background sodium conductance. Mol Pharmacol 77, 270–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucka M, Kretschmannova K, Stojilkovic SS, Zemkova H & Tomic M (2012). Dependence of spontaneous electrical activity and basal prolactin release on nonselective cation channels in pituitary lactotrophs. Physiol Res 61, 267–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiecien R, Robert C, Cannon R, Vigues S, Arnoux A, Kordon C & Hammond C (1998). Endogenous pacemaker activity of rat tumour somatotrophs. J Physiol 508, 883–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert AE, Henquin JC & Malvaux P (1974). Cationic environment and dynamics of insulin secretion. I. Effect of low concentrations of sodium. Endocrinology 95, 1069–1077. [DOI] [PubMed] [Google Scholar]
- Lear BC, Lin JM, Keath JR, McGill JJ, Raman IM & Allada R (2005). The ion channel narrow abdomen is critical for neural output of the Drosophila circadian pacemaker. Neuron 48, 965–976. [DOI] [PubMed] [Google Scholar]
- Leech CA, Holz GG & Habener JF (1995). Pituitary adenylate cyclase‐activating polypeptide induces the voltage‐independent activation of inward membrane currents and elevation of intracellular calcium in HIT‐T15 insulinoma cells. Endocrinology 136, 1530–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemaire S, Derome G, Tseng R, Mercier P & Lemaire I (1981). Distinct regulations by calcium of cyclic GMP levels and catecholamine secretion in isolated bovine adrenal chromaffin cells. Metabolism 30, 462–468. [DOI] [PubMed] [Google Scholar]
- LeSauter J, Silver R, Cloues R & Witkovsky P (2011). Light exposure induces short‐ and long‐term changes in the excitability of retinorecipient neurons in suprachiasmatic nucleus. J Neurophysiol 106, 576–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Z, Chen L, McClafferty H, Lukowski R, MacGregor D, King JT, Rizzi S, Sausbier M, McCobb DP, Knaus HG, Ruth P & Shipston MJ (2011). Control of hypothalamic‐pituitary‐adrenal stress axis activity by the intermediate conductance calcium‐activated potassium channel, SK4. J Physiol 589, 5965–5986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingle CJ, Martinez‐Espinosa PL, Guarina L & Carbone E (2018). Roles of Na+, Ca2+, and K+ channels in the generation of repetitive firing and rhythmic bursting in adrenal chromaffin cells. Pflugers Arch 470, 39–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu B, Su Y, Das S, Liu J, Xia J & Ren D (2007). The neuronal channel NALCN contributes resting sodium permeability and is required for normal respiratory rhythm. Cell 129, 371–383. [DOI] [PubMed] [Google Scholar]
- Lu B, Su Y, Das S, Wang H, Wang Y, Liu J & Ren D (2009). Peptide neurotransmitters activate a cation channel complex of NALCN and UNC‐80. Nature 457, 741–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu TZ & Feng ZP (2011). A sodium leak current regulates pacemaker activity of adult central pattern generator neurons in Lymnaea stagnalis. PLoS One 6, e18745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malaisse WJ, Malaisse‐Lagae F & Brisson G (1971). The stimulus‐secretion coupling of glucose‐induced insulin release. II. Interaction of alkali and alkaline earth cations. Horm Metab Res 3, 65–70. [DOI] [PubMed] [Google Scholar]
- Marcantoni A, Carabelli V, Vandael DH, Comunanza V & Carbone E (2009). PDE type‐4 inhibition increases L‐type Ca2+ currents, action potential firing, and quantal size of exocytosis in mouse chromaffin cells. Pflugers Arch 457, 1093–1110. [DOI] [PubMed] [Google Scholar]
- Marcantoni A, Vandael DH, Mahapatra S, Carabelli V, Sinnegger‐Brauns MJ, Striessnig J & Carbone E (2010). Loss of Cav1.3 channels reveals the critical role of L‐type and BK channel coupling in pacemaking mouse adrenal chromaffin cells. J Neurosci 30, 491–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin AO, Mathieu MN, Chevillard C & Guerineau NC (2001). Gap junctions mediate electrical signaling and ensuing cytosolic Ca2+ increases between chromaffin cells in adrenal slices: A role in catecholamine release. J Neurosci 21, 5397–5405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez‐Espinosa PL, Yang C, Gonzalez‐Perez V, Xia XM & Lingle CJ (2014). Knockout of the BK beta2 subunit abolishes inactivation of BK currents in mouse adrenal chromaffin cells and results in slow‐wave burst activity. J Gen Physiol 144, 275–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathar I, Vennekens R, Meissner M, Kees F, Van der Mieren G, Camacho Londono JE, Uhl S, Voets T, Hummel B, van den Bergh A, Herijgers P, Nilius B, Flockerzi V, Schweda F & Freichel M (2010). Increased catecholamine secretion contributes to hypertension in TRPM4‐deficient mice. J Clin Invest 120, 3267–3279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser T (1998). Low‐conductance intercellular coupling between mouse chromaffin cells in situ. J Physiol 506 195–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nassar‐Gentina V, Pollard HB & Rojas E (1988). Electrical activity in chromaffin cells of intact mouse adrenal gland. Am J Physiol Cell Physiol 254, C675–C683. [DOI] [PubMed] [Google Scholar]
- Nichols RA, Dengler AF, Nakagawa EM, Bashkin M, paul BT, Wu J & Khan GM (2007). A constitutive, transient receptor potential‐like Ca2+ influx pathway in presynaptic nerve endings independent of voltage‐gated Ca2+ channels and Na+/Ca2+ exchange. J Biol Chem 282, 36102–36111. [DOI] [PubMed] [Google Scholar]
- Noble D, Denyer JC, Brown HF & DiFrancesco D (1992). Reciprocal role of the inward currents ib, Na and i(f) in controlling and stabilizing pacemaker frequency of rabbit sino‐atrial node cells. Proc Biol Sci 250, 199–207. [DOI] [PubMed] [Google Scholar]
- Pattyn A, Morin X, Cremer H, Goridis C & Brunet JF (1999). The homeobox gene Phox2b is essential for the development of autonomic neural crest derivatives. Nature 399, 366–370. [DOI] [PubMed] [Google Scholar]
- Pena F & Ramirez JM (2004). Substance P‐mediated modulation of pacemaker properties in the mammalian respiratory network. J Neurosci 24, 7549–7556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins KL (2006). Cell‐attached voltage‐clamp and current‐clamp recording and stimulation techniques in brain slices. J Neurosci Methods 154, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman IM, Gustafson AE & padgett D (2000). Ionic currents and spontaneous firing in neurons isolated from the cerebellar nuclei. J Neurosci 20, 9004–9016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinl EL, Cabeza R, Gregory IA, Cahill AG & England SK (2015). Sodium leak channel, non‐selective contributes to the leak current in human myometrial smooth muscle cells from pregnant women. Mol Hum Reprod 21, 816–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren D (2011). Sodium leak channels in neuronal excitability and rhythmic behaviors. Neuron 72, 899–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribalet B & Beigelman PM (1982). Effects of sodium on beta‐cell electrical activity. Am J Physiol Cell Physiol 242, C296–C303. [DOI] [PubMed] [Google Scholar]
- Role LW, Leeman SE & Perlman RL (1981). Somatostatin and substance P inhibit catecholamine secretion from isolated cells of guinea‐pig adrenal medulla. Neuroscience 6, 1813–1821. [DOI] [PubMed] [Google Scholar]
- Saith S, Bicknell RJ & Schofield JG (1984). Different sodium requirements for 86Rb efflux and for growth hormone and prolactin secretion from bovine anterior pituitary cells. Mol Cell Endocrinol 35, 47–54. [DOI] [PubMed] [Google Scholar]
- Sankaranarayanan S & Simasko SM (1996). A role for a background sodium current in spontaneous action potentials and secretion from rat lactotrophs. Am J Physiol Cell Physiol 271, C1927–C1934. [DOI] [PubMed] [Google Scholar]
- Sanz‐Lazaro S, Jimenez‐Pompa A, Carmona‐Hidalgo B, Ubeda M, Munoz L, Caba‐Gonzalez JC, Hernandez‐Vivanco A, Lopez‐Garcia S, Albillos A & Albillos A (2019). The firing frequency of spontaneous action potentials and their corresponding evoked exocytosis are increased in chromaffin cells of CCl4 ‐induced cirrhotic rats with respect to control rats. J Neurochem 148, 359–372. [DOI] [PubMed] [Google Scholar]
- Schmich RM & Miller MI (1997). Stochastic threshold characterization of the intensity of active channel dynamical action potential generation. J Neurophysiol 78, 2616–2630. [DOI] [PubMed] [Google Scholar]
- Schneidman E, Freedman B & Segev I (1998). Ion channel stochasticity may be critical in determining the reliability and precision of spike timing. Neural Comput 10, 1679–1703. [DOI] [PubMed] [Google Scholar]
- Simasko SM (1994). A background sodium conductance is necessary for spontaneous depolarizations in rat pituitary cell line GH3. Am J Physiol Cell Physiol 266, C709–C719. [DOI] [PubMed] [Google Scholar]
- Simasko SM & Sankaranarayanan S (1997). Characterization of a hyperpolarization‐activated cation current in rat pituitary cells. Am J Physiol Endocrin Metab 272, E405–E414. [DOI] [PubMed] [Google Scholar]
- Smith CB & Eiden LE (2012). Is paCAP the major neurotransmitter for stress transduction at the adrenomedullary synapse? J Mol Neurosci 48, 403–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon JS & Nerbonne JM (1993). Hyperpolarization‐activated currents in isolated superior colliculus‐projecting neurons from rat visual cortex. J Physiol 462, 393–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spindler AJ, Noble SJ, Noble D & LeGuennec JY (1998). The effects of sodium substitution on currents determining the resting potential in guinea‐pig ventricular cells. Exp Physiol 83, 121–136. [DOI] [PubMed] [Google Scholar]
- Swayne LA, Mezghrani A, Varrault A, Chemin J, Bertrand G, Dalle S, Bourinet E, Lory P, Miller RJ, Nargeot J & Monteil A (2009). The NALCN ion channel is activated by M3 muscarinic receptors in a pancreatic beta‐cell line. EMBO Rep 10, 873–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takano K, Takei T, Teramoto A & Yamashita N (1996). GHRH activates a nonselective cation current in human GH‐secreting adenoma cells. Am J Physiol Endocrinol Metab 270, E1050–E1057. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Shibuya I, Nagamoto T, Yamashita H & Kanno T (1996). Pituitary adenylate cyclase‐activating polypeptide causes rapid Ca2+ release from intracellular stores and long lasting Ca2+ influx mediated by Na+ influx‐dependent membrane depolarization in bovine adrenal chromaffin cells. Endocrinology 137, 956–966. [DOI] [PubMed] [Google Scholar]
- Tesfai Y, Brereton HM & Barritt GJ (2001). A diacylglycerol‐activated Ca2+ channel in PC12 cells (an adrenal chromaffin cell line) correlates with expression of the TRP‐6 (transient receptor potential) protein. Biochem J 358, 717–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomic M, Kucka M, Kretschmannova K, Li S, Nesterova M, Stratakis CA & Stojilkovic SS (2011). Role of nonselective cation channels in spontaneous and protein kinase A‐stimulated calcium signaling in pituitary cells. Am J Physiol Endocrinol Metab 301, E370–E379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trebak M, Vazquez G, Bird GS & Putney JW Jr (2003). The TRPC3/6/7 subfamily of cation channels. Cell Calcium 33, 451–461. [DOI] [PubMed] [Google Scholar]
- Tremblay C, Berret E, Henry M, Nehme B, Nadeau L & Mouginot D (2011). Neuronal sodium leak channel is responsible for the detection of sodium in the rat median preoptic nucleus. J Neurophysiol 105, 650–660. [DOI] [PubMed] [Google Scholar]
- Tsaneva‐Atanasova K, Sherman A, van Goor F & Stojilkovic SS (2007). Mechanism of spontaneous and receptor‐controlled electrical activity in pituitary somatotrophs: experiments and theory. J Neurophysiol 98, 131–144. [DOI] [PubMed] [Google Scholar]
- Vandael DH, Marcantoni A & Carbone E (2015a). Cav1.3 channels as key regulators of neuron‐like firings and catecholamine release in chromaffin cells. Curr Mol Pharmacol 8, 149–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandael DH, Ottaviani MM, Legros C, Lefort C, Guerineau NC, Allio A, Carabelli V & Carbone E (2015b). Reduced availability of voltage‐gated sodium channels by depolarization or blockade by tetrodotoxin boosts burst firing and catecholamine release in mouse chromaffin cells. J Physiol 593, 905–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandael DH, Zuccotti A, Striessnig J & Carbone E (2012). Cav1.3‐driven SK channel activation regulates pacemaking and spike frequency adaptation in mouse chromaffin cells. J Neurosci 32, 16345–16359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venteo S, Laffray S, Wetzel C, Rivat C, Scamps F, Mechaly I, Bauchet L, Raoul C, Bourinet E, Lewin GR, Carroll P & Pattyn A (2016). Fxyd2 regulates Adelta‐ and C‐fiber mechanosensitivity and is required for the maintenance of neuropathic pain. Sci Rep 6, 36407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada A, Yashima N, Izumi F, Kobayashi H & Yanagihara N (1984). Involvement of Na influx in acetylcholine receptor mediated secretion of catecholamines from cultured bovine adrenal medulla cells. Neurosci Lett 47, 75–80. [DOI] [PubMed] [Google Scholar]
- Wallace DJ, Chen C & Marley PD (2002). Histamine promotes excitability in bovine adrenal chromaffin cells by inhibiting an M‐current. J Physiol 540, 921–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worley JF 3rd, McIntyre MS, Spencer B & Dukes ID (1994). Depletion of intracellular Ca2+ stores activates a maitotoxin‐sensitive nonselective cationic current in beta‐cells. J Biol Chem 269, 32055–32058. [PubMed] [Google Scholar]
- Xie L, Gao S, Alcaire SM, Aoyagi K, Wang Y, Griffin JK, Stagljar I, Nagamatsu S & Zhen M (2013). NLF‐1 delivers a sodium leak channel to regulate neuronal excitability and modulate rhythmic locomotion. Neuron 77, 1069–1082. [DOI] [PubMed] [Google Scholar]
- Yang SK, Wang K, Parkington H & Chen C (2008). Involvement of tetrodotoxin‐resistant Na+ current and protein kinase C in the action of growth hormone (GH)‐releasing hormone on primary cultured somatotropes from GH‐green fluorescent protein transgenic mice. Endocrinology 149, 4726–4735. [DOI] [PubMed] [Google Scholar]
- Yeh SY, Huang WH, Wang W, Ward CS, Chao ES, Wu Z, Tang B, Tang J, Sun JJ, Esther van der Heijden M, Gray PA, Xue M, Ray RS, Ren D & Zoghbi HY (2017). Respiratory network stability and modulatory response to substance P require Nalcn. Neuron 94, 294–303.e4 e294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zemkova H, Tomic M, Kucka M, Aguilera G & Stojilkovic SS (2016). Spontaneous and CRH‐induced excitability and calcium signaling in mice corticotrophs involves sodium, calcium, and cation‐conducting channels. Endocrinology 157, 1576–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Statistical Summary Document
Data Availability Statement
The datasets generated and analysed during the present study are available from the corresponding author upon reasonable request.