

Summary
White coat patterning is a feature of many dog breeds and is known to be coded primarily by the gene micropthalmia‐associated transcription factor (MITF). This patterning in the coat can be modified by other factors to produce the attractive phenotypes termed ‘ticked’ and ‘roan’ that describe the presence of flecks of color that vary in distribution and intensity within otherwise ‘clear’ white markings. The appearance of the pigment in the white patterning caused by ticking and roaning intensifies in the weeks after birth. We applied genome‐wide association to compare English Cocker Spaniels of roan phenotype (N = 34) with parti‐color (non‐roan) English Cocker Spaniels (N = 9) and identified an associated locus on CFA 38, CFA38:11 057 040 (P raw = 8.9 × 10−10, P genome = 2.7 × 10−5). A local case–control association in English Springer Spaniels comparing 11 ticked and six clear dogs identified indicative association with a different haplotype, CFA38:11 122 467G>T (P raw = 1.7 × 10−5) and CFA38:11 124 294A>C (P raw = 1.7 × 10−5). We characterize three haplotypes in Spaniels according to their putative functional variant profiles at CFA38:11 111 286C>T (missense), CFA38:11 131 841–11 143 239DUP.insTTAA (using strongly linked marker CFA38:11 143 243C>T) and CFA38:11 156 425T>C (splice site). In Spaniels, the haplotypes work as an allelic series including alleles (t, recessive clear; T, dominant ticked/parti‐color; and T R, incomplete dominant roan) to control the appearance of pigmented spots or flecks in otherwise white areas of the canine coat. In Spaniels the associated haplotypes are t (CCT), T (TCC) and T R (TTT) for SNP markers on CFA38 at 11 111 286C>T, 11 143 243C>T and 11 156 425T>C respectively. It is likely that other alleles exist in this series and together the haplotypes result in a complex range of patterning that is only visible when dogs have white patterning resulting from the epistatic gene Micropthalmia‐associated transcription factor (the S‐locus).
Keywords: coat‐color, pigmentation, USH2A, usherin
Introduction
Domestic dogs exhibit a wide range of coat colors and coat patterns, including white patterning that may occur with or without pigmented spots or interspersed hairs. The broader absence of pigmentation in defined regions of the coat (white patterning) is primarily affected by the activity of the micropthalmia‐associated transcription factor (MITF) gene (Karlsson et al. 2007). White patterning on dogs may range from as little as a white chest spot on an otherwise self‐colored (or solid‐colored) dog to an entirely white dog (known as extreme white). The extent of the white patterning is increased by the presence of a small interspersed nuclear element and affected by a length polymorphism, both in the vicinity of the promoter region of the MITF gene. The longer the length polymorphism, the more extensive is the white patterning on the dog (Körberg et al. 2014).
Two well‐described variations of white patterning are roan (Fig. 1a) and ticked (Fig. 1b), which are both characterized by the presence of pigmented hairs in otherwise clear white patterned areas of the coat. Pups that will exhibit the pattern variants as adults are born with clear white patterning and pigmentation develops in the first weeks of life (Fig. 1c). Roan and ticked are described by various names across dog breeds, including roan in the English Cocker Spaniel (ECS) and German Short‐haired Pointer, blue in the Australian Cattle Dog and Belton in the English Setter. Colloquially, ticked is regarded as the appearance of pigmented spots in white areas of the body. Pigmented spots from ticking range in size from only a few hairs to coin‐sized, and are most dense on the muzzle and legs (Fig. 1b). In contrast, pigmentation in the white patterning associated with roan is a mixture of pigmented and white hairs throughout the areas of the coat that would otherwise be white (Little 1953), with the phenotype dispersed relatively evenly across the body. ECSs that have visible white markings lacking roan are known as parti‐color. The white‐patterned areas in parti‐color ECSs frequently contain pigmented spots (are ticked), but may be clear.
Figure 1.

Coat patterning in Spaniel breeds: (a) roan; (b) ticked; and (c) appearance of pigmentation in white‐patterned area of coat is not present in pups before loss of birth coat. The image demonstrates a disparity in ticked phenotype between pups and their dam (images under license from iStockPhoto.com).
Interestingly, the spots that are characteristic of the Dalmatian dog breed share two features with roan: the appearance of pigmented spots in white‐patterned areas in the first weeks after birth and the dispersal of the pigmentation evenly across the body. Unlike roan, however, Dalmatians have well‐defined pigmented spots in the white‐patterned areas of the coat rather than interspersed pigmented and white hairs. The size and definition of the spots in the Dalmatian breed are predicted to be controlled by a locus that is in LD with the hyperuricosurea locus on CFA3 (Bannasch et al. 2008).
In classic literature, the two phenotypes of ticked and roan have been presumed to be controlled by two separate loci (Little 1953); however, even in the early characterization of the trait it was accepted that the phenotypic observations might equally have been explained by a multiallelic single locus. The accepted phenotypic dominance hierarchy places roan as dominant over ticked and both dominant over ‘clear’ (no pigmented spots or few spots). However, the range of the phenotypes displayed by individuals suggests a complex inheritance.
The hue of the pigmented hair associated with ticked or roan pigmentation patterns is controlled by many separate coat color loci (Little 1953). When individuals have red/yellow pigmented base color, it is sometimes difficult to discern light roan from lightly ticked individuals, particularly when the dogs are young. Interestingly, pigmented skin spots in the absence of corresponding pigmented fur appear in non‐ticked dog breeds, although the inheritance of this characteristic is as yet unexplored.
In the current study, we apply within‐breed genome‐wide association mapping to identify a locus that is strongly associated with the appearance of pigmentation in white patches of the coat in the ECS and English Springer Spaniel (ESS). Spaniels are a group of dogs in the sporting group that includes dogs bred for indicating, flushing and retrieving small game (American Kennel Club 2020). As the name implies, Spaniel breeds are derived from dogs that were taken from Spain to Britain in the Middle Ages, where the modern breeds were developed (Australian National Kennel Council 2006). Alongside other Spaniel breeds such as the American Cocker Spaniel and the Cavalier King Charles Spaniel, the ECS and ESS are broadly described as small to medium‐sized short‐legged dogs with large, drooping ears. Although they are closely related, the ECS can be distinguished from the ESS by its smaller stature.
Materials and methods
Samples
Information relating to dogs used in the various analyses follows.
Samples used in genome‐wide association
Forty‐three dogs of the ECS breed – 34 roan (19 male and 15 female) and nine parti‐color individuals (seven female, two male) – were analyzed by genome‐wide association. All were privately owned dogs that were sampled on presentation at Norwegian veterinary clinics for health checks, behavioral consultation or medical procedures unrelated to their coat color. All samples were collected as part of diagnostic procedures and remains were stored with the owner’s consent. Coat colors were visually phenotyped at the time of sample collection. Self‐colored ECSs were excluded from the association analysis because the segregation of roan and parti‐color cannot be observed on this genetic background – although the patterns are expected to segregate freely in HWE in the absence of white overlay.
Samples used for haplotype refinement
Limited genotyping with a denser group of markers was used to identify haplotypes segregating throughout the region of association with the roan coat pattern. The analysis used nine of the roan and nine parti‐color ECSs from the same group as used in the genome‐wide association. To test whether regional association separated ticked and clear individuals, a cohort of privately owned coat‐phenotyped ESS dogs (six clear and 11 ticked with ticking observed by photograph ranging from light to heavy) was assayed using the same markers.
Samples used for representative haplotype variant calling
Electronic data comprising a variant call file were available from a 590 dog resource published by the Dog Biomedical Variant Consortium (DBVC) (Jagannathan et al. 2019). The DBVC data were used for identifying putative functional variants on haplotypes observed in the homozygous state among the phenotyped ECSs and ESSs with the denser marker set.
Samples used to validate roan‐associated variants
Blood samples for all dogs were collected in ethylenediaminetetraacetic acid‐coated tubes and genomic DNA was isolated using DNeasy Tissue kit (Qiagen GmbH). All nucleic acid measurements and quality assessments were performed with a NanoDrop 1000 spectrophotometer (Thermo Fisher Scientific Inc.). Samples were collected under institutional ethics guidance (approval numbers N00/9‐2009/3/5109, 2015/902 and 2018‐1449).
Samples for the determination of ancestral alleles
Electronic data from wolf samples (N = 3) from the DBVC data (Jagannathan et al. 2019) and from Plassais et al. (2019) (N = 48) were assessed at loci represented in Table S2 to determine allelic history.
Genotyping and analysis
Genome‐wide association analysis
DNA samples for within‐breed genome‐wide association analysis were purified according to Affymetrix (Affymetrix) sample preparation guidelines and genotyped using the Affymetrix Version 2 (49 663 markers) Custom Canine SNP array (Affymetrix). They were called using Affymetrix’s snp5‐geno‐qc software.
The within‐breed (ECS) genome‐wide association analysis was conducted using standard case–control association analysis in plink (Purcell et al. 2007). SNPs with a genotyping rate of <10%, with strong deviation from Hardy–Weinberg expectation (P < 0.0005) and MAF of lower than 0.05 were removed. Genome‐wide significance was ascertained by Bonferroni correction using the adjusted option in plink (Purcell et al. 2007).
Haplotype refinement analysis
To define haplotypes, the primary regions of the association were assayed by 53 SNPs, including nine markers from the array and 44 new variants in the region. The samples were genotyped using the iPLEX Gold Mass ARRAY (Sequenom; Gabriel et al. 2009). The genotype data were analyzed using the typer 4.0 analyzer software (Sequenom) for cluster analysis. SNPs with a call rate of greater than 75% (‐‐geno 0.25) and a MAF of at least 5% were included in each analysis. Samples with a call rate of less than or equal to 75% (‐‐mind 0.25) were excluded from further analysis.
Genotypes from two dog breeds (ECS and ESS) were visually phased to identify haplotypes in the region of the most associated markers. To prevent ambiguity of haplotype calling, phasing was restricted to animals that were homozygous throughout the region of association. Tagging variants were identified to enable clear definition of observed homozygous haplotypes (Table S2).
Representative haplotype variant calling analysis
Coat patterns related to the mapped locus were not expected to affect animal genetic fitness (reproduction and longevity), thus the causal mutations for observed major phenotypes were expected to segregate in many breeds. This assumption enabled us to use haplotype representatives from general canine whole‐genome sequencing panels as proxies for homozygous ECSs and ESSs to identify potential functional variant sites in strong LD with the ECS/ESS observed haplotypes (Table S3).
The tabix tool (Li 2011) was used to extract called variants from the relevant region of the DBVC variant call file (vcf) (Jagannathan et al. 2019). From the ECS/ESS haplotype tag genotypes, individual DBVC dogs representing the relevant observed homozygous haplotypes throughout the region of association were identified. plink (Purcell et al. 2007) was used to convert the DBVC region to a standard tped file and to reduce the data to include only the homozygous haplotype representative dogs. Data were reconverted to vcf format to enable phasing of functional variants to individual haplotypes.
Variant call files from the representative individuals from across the region of association were uploaded as custom tracks onto the UCSC genome browser (genome.ucsc.edu). The variant annotation integrator (VAI) function in the UCSC genome browser (Hinrichs et al. 2016) was used to annotate potential functional SNP and small insertion‐deletion variants within the associated region for each individual.
Structural variants were visualized by plotting nucleotide mean variant cover from the DBVC data. Dogs were encoded and grouped according to their homozygosity for representative haplotypes shown in Table S3. Other dogs and canids were coded as unknown.
Validation analysis
Seven putative functional variants in the critical region were genotyped in 10 ESSs (various ticking density) and six ECSs [parti‐color (4), roan (2)] by Sanger sequencing (Australian Genome Research Facility, Sydney, Australia) employing capillary separation on an AB 3730xl machine. Primer sequences are provided in Table S4.
The presence of an observed structural variant was further investigated by PCR in 72 individuals [ECS roan (23), ECS parti‐color (5), ECS self‐color (black) (7), ESS ticked (6), ESS clear (4), Dalmatian (23), Australian Cattle Dog (2) and German Short‐haired Pointer (2)]. Primers were designed using Primer3 (Untergasser et al. 2012) to amplify the intervening product between the tandem duplicates. The PCR reaction was performed in a total volume of 20 µl using AmpliTaq Gold 360 Master Mix (Applied Biosystems). Each reaction contained 25 ng of template DNA and 1 μm of each primer (forward TCAACATCCATACTTCTCATGCTT; reverse TGCTCAACCTAAATGTGCTAA) at an annealing temperature of 55°C. PCR conditions were heat activation for 15 min at 95°C followed by 35 cycles of 45 s at 95°C, 45 s at 55°C and 1 min 30 s at 72°C and terminated with a final elongation step at 72 °C for 10 min. PCR products were visualized on a 1% agarose gel.
Results
Genome‐wide association analysis
In the ECS genome‐wide association analysis 26 014 SNPs were included after filtering. A total of 10 695 SNPs failed missingness, 16 067 failed the frequency test and 132 were excluded owing to deviation from HWE.
The whole‐genome association, Q–Q distribution of probabilities and multidimensional scaling plots are shown in Fig. 2. An interval of genome‐wide significant association with roan coat color spans CFA38:10 268 280–11 341,635 (canfam3.1). The 10 most strongly associated variants from genome‐wide association are shown in Table S1.
Figure 2.
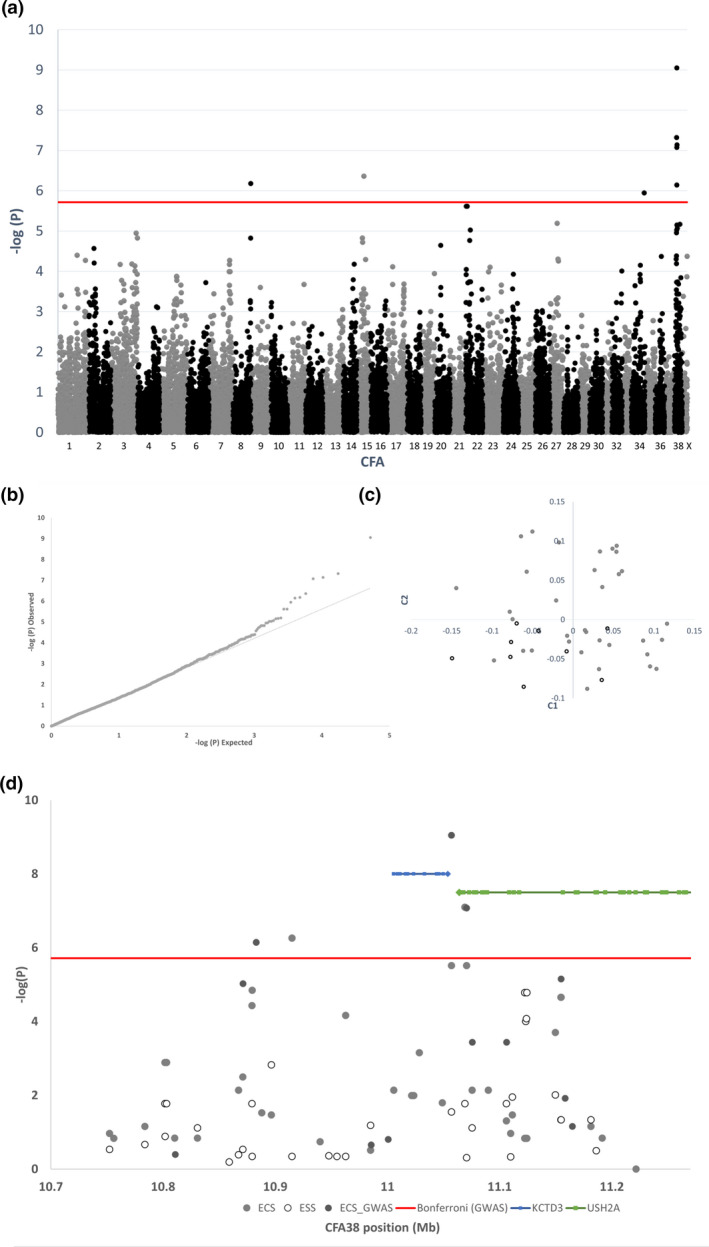
(a) Negative log of probability of whole‐genome association of roan vs. non‐roan in the English Cocker Spaniel (nine non‐roan and 34 roan) (b) Q–Q plot of association. (c) Multidimensional scaling plot of first two components of variation in samples. (d) Negative log of probability of local association of roan vs. non‐roan in English Cocker Spaniels and ticked vs. clear in English Springer Spaniels.
Haplotype refinement
Association over the denser marker set revealed the strongest association at CFA38:11 069 051G>A (P raw = 8.0 × 10−8) for roan vs. parti‐color in ECSs. Among ESSs, two markers demonstrated indicative association with ticked vs. clear: CFA38:11 122 467G>T (P raw = 1.7 × 10−5) and CFA38 11 124 294A>C (P raw = 1.7 × 10−5).
Haplotype refinement analysis of both ECS and ESS data revealed the presence of three major haplotypes (t, clear; T, ticked; and T R, roan). Other minor haplotypes exist in heterozygous animals (Table S2). ECSs genotyped as homozygous ticked but phenotyped as parti‐color exhibited the same haplotype at associated markers as some ESSs determined as ticked. Animals homozygous for the parti‐color haplotype did exhibit pigmented spotting in their white‐marked coat areas. Typically, these spots were more common at the muzzle and paws.
The defined area of investigation was restricted to a region where markers for ESSs with the clear haplotype agreed with the reference genome (canfam3.1 – Boxer breed) markers (known to be clear phenotype). This encompassed markers between and exclusive of CFA38:11 089 995–11 176 811 (Table S2). Two markers in this region (CFA38:11 163 111 and CFA38:11 163 250) did not differentiate the observed haplotypes and were unrepresented in the public data, suggesting that they were monomorphic.
ESSs that had been determined as clear had few (if any) spots. Homozygosity for the reference genome haplotype was not observed in the ECS samples analyzed. Roan ECSs were homozygous for a haplotype that diverged from the reference. One heavily marked ESS was homozygous for a fourth haplotype that was unobserved in the DBVC data but that was most similar to the roan haplotype (marked as TR* in Table S2). Genotyped markers did not clearly differentiate heavy and light ticking in the ESS haplotype refinement analysis, but did clearly differentiate the ticked from clear phenotypes in homozygous individuals.
Representative haplotype functional variant calling
Based on the variant calls available for DBVC haplotype‐representative animals, six putative functional variants were identified using the VAI (Hinrichs et al. 2016) that segregated among the three representative haplotypes in the DBVC data. DBVC homozygous representatives of haplotypes observed in the ECSs and ESSs are identified by their allelic haplotype in Table S3. Across all dogs, many additional haplotypes and putative functional variants exist that were not observed to segregate in either the ECS or ESS. Heterozygous dogs or dogs with haplotypes unobserved in the ECS or ESS are shown as unknown in Table S3 (N = 558).
Below we describe the putative functional variant profiles for three alleles within an allelic series based on proxy haplotype. The characterized alleles are described in phenotype order from least to most pigmented within the white‐patterned areas of the coat (Fig. 3). Information relating to the functions of putative functional variants in the region as determined by Ensembl (Yates et al. 2020) is shown in Table S5. Other neutral variants segregate with the haplotypes and these are shown in Table S3.
Figure 3.
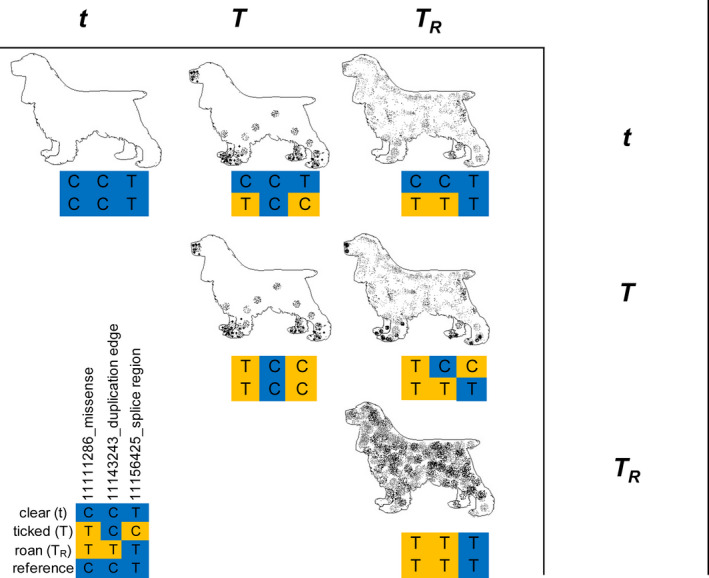
Illustration of allelic series hypothesis in the English Cocker Spaniel (ECS) and English Springer Spaniel (ESS). The roan allele is presumed to be codominant, whereas the ticked allele is codominant with roan but dominant in relation to clear. The following genotypes were observed in our haplotype refinement analysis: t/t (ESS = 2), T/t (ECS = 3, ESS = 1), T/T (ECS = 2, ESS = 1), T/T R (ECS = 1, ESS = 1), T R/T R (ECS = 8). No dogs were identified with the t/T R genotype.
In Spaniels, the reference genome haplotype clear (t) is differs from both the ticked and roan haplotypes by one missense SNP in usherin (USH2A; CFA38:g.11 111 286C>T) where the clear allele is C (Table 1). All observed ticked and roan haplotype representative animals have at least one copy of the T allele. Four of nine parti‐color ECSs used in haplotype characterization were heterozygous for the t haplotype; the remainder had no copies of the t haplotype. Two of six clear ESSs were homozygous for the t haplotype and four were heterozygous.
Table 1.
Description of variants on canine CFA38 segregating with haplotypes that represent observed coat phenotypes with putative functional variants within the region segregating among three homozygous haplotypes in bold type.
| Position | Gene | Variant source | Putative function | Coding change | SIFT | Reference | Alternate |
|---|---|---|---|---|---|---|---|
| 11 057 040 | hr 1 /wga 2 /DBVC 3 | Neutral | None | C | T | ||
| 11 106 067 | hr/wga/DBVC | Neutral | None | A | G | ||
| 11 085 443 | USH2A | DBVC | Missense | P/S | 0.00 | G | A |
| 11 111 286 | USH2A | hr/wga/DBVC | Missense | E/K | 0.95 | C | T |
| 11 122 467 | hr/DBVC | Neutral | None | G | T | ||
| 11 123 271 | hr/DBVC | Neutral | None | T | G | ||
| 11 123 896 | hr/DBVC | Neutral | None | A | G | ||
| 11 124 294 | hr | Neutral | None | C | A | ||
| 11 143 243 | USH2A | DBVC | Duplication edge | None | C | T | |
| 11 149 787 | hr/DBVC | Neutral | None | G | A | ||
| 11 154 628 | hr/DBVC | Neutral | None | G | A | ||
| 11 154 832 | hr/wga/DBVC | Neutral | None | A | C | ||
| 11 156 276 | USH2A | DBVC | Missense | Q/L | 0.33 | A | T |
| 11 156 425 | USH2A | DBVC | Splice‐region | Unknown | T | C | |
| 11 167 847 | USH2A | DBVC | Splice‐region | Unknown | GA | GAA/DEL | |
| 11 169 445 | USH2A | DBVC | Missense | P/S | 0.45 | G | A |
| 11 176 811 | hr | Neutral | None | A | C |
A further putative functional variant in strong linkage with the roan haplotype but outside of the main analysis region (at 11 085 443) is shown also.
Haplotype refinement.
Whole‐genome association.
Dog Biomedical Variant Consortium.
Spaniels with the ticked (T) haplotype exhibit the alleles A and C at missense variant CFA38:g.11 156 276T>A and splice‐site variant CFA38:g.11 156 425T>C respectively, whereas both roan and clear Spaniels have the T allele at both sites (Table 1). In addition, ticked Spaniels differ from the clear haplotype at CFA38:g.11 111 286C>T. One parti‐color ECS was homozygous for haplotype T; two ticked ESSs were homozygous for haplotype T.
The roan (T R) haplotype is diverged from the reference/clear haplotype at four putative functional sites. Two are missense variants CFA38:g.11 085 443G>A and CFA38:g.11 111 286C>T (Table S3). The variant at CFA38:g.11 085 443G>A lies proximal to the genomic region that clearly distinguishes the three haplotypes based on concordance of the clear and reference haplotypes. The T R haplotype has two additional variants that affect the secondary structure of the DNA in the region (see the next section).
Structural variants
By analysis of regional coverage in representative individuals, we identified a duplicated DNA segment (11 398 bp) lying within the 67th intron of USH2A and spanning CFA38:11 131 841–11 143 239 (canfam3.1), which is unique to the T R haplotype (Fig. 2; Table S6). Subsequent testing for the presence of the PCR product capturing the junction between the duplicated segments identified the product in roan ECSs, all Dalmatians (typically described as evenly spotted), German Short‐haired Pointer (typically roan) and Australian Cattle Dogs (typically roan). The PCR product was not captured in parti‐color ECSs (ticked) nor Australian ESS (clear or ticked). All animals identified with the PCR product had pigmented spots or interspersed pigmented hairs in their white markings. One of 23 coat‐phenotyped roan ECSs tested for the product reported a negative result based on owner‐reported color. All other of 72 results were concordant with expectation based on coat phenotype (Table S6). Roan is a common phenotype in ECSs and is not observable when dogs do not have white patterning from the activity of MITF. In such dogs, the duplication is expected to freely segregate in accordance with Hardy–Weinberg expectation. In accordance with this expectation, black ECSs (where ticked and roan are unobserved) segregated the product.
The duplication allele is in strong LD with a variant at CFA38:11 143 243C>T (reference allele is C) that lies within 5 bp of the distal duplication edge but outside of the duplication. The variant acts as a proxy for the presence of the segmental duplication on the roan haplotype and enables the assessment of heterozygosity for the duplication. The dosage of the T‐allele at the marker SNP is completely concordant with the expected copy number as ascertained by sequence coverage of the region of the duplication in the T R dogs with the WGS (DBVC animals showing the T R haplotype in Table S3). For DBVC breeds with observable ticking (i.e. breeds that commonly display white markings), the variant co‐occurs with roan patterning in the affected breeds (i.e. Australian Cattle Dog, Dalmatian, German Wire‐haired Pointer). Animals assessed as homozygous for the duplication according to this proxy have even dispersion of pigment throughout the white markings, i.e. the pigmentation favors neither the feet nor the muzzle.
DBVC coverage data (Fig. S1) show the presence of a VNTR mutation at CFA38:11 112 907–11 113 501. This variant lies close to an intron–exon boundary (ENSCAFG00000010731.4 exon 62/71) and it may affect the phenotypic expression of the USH2A gene. Ambiguity in the reference assembly at this site precludes effective assay of the variant at this time.
Validation analysis
Two variants of seven sequenced putatively functional variants (Table S8) segregated perfectly with roan phenotype (duplication bridge product and missense CFA38:g.11 169 445G>A) in the Australian genotyped Spaniels. Missense variant CFA38:g.11 169 445G>A segregates more broadly in the wider population and the roan‐associated A allele is present as a homozygote in observable non‐roan breed representatives, e.g. SID00101, Cavalier King Charles Spaniel, and SRR2094385, Beagle (Table S3). Sequencing shows that the Spaniel roan differs from the roan proxy haplotype at variants CFA38:11 085 443 (deleterious missense) and CFA38:11 156 276 (tolerated missense), where Spaniels have the reference and derived alleles respectively. Further, deleterious missense variant CFA38:11 085 443G>A that seemed strongly associated with roan in homozygous dogs from Table S3 did not segregate in Spaniels, showing that these variants are not causative for the roan phenotype. Together the results suggest that the segmental duplication variant (CFA38:11 131 841–11 143 239DUP.insTTAA) is the most likely causative variant for roan. The duplication associated marker CFA38:11 143 243C>T is homozygous in one ESS without the duplication, showing that linkage with the duplication is imperfect in Spaniels. Ticking density among dogs without the roan haplotype is not clearly associated with any of the genotyped variants. However, no animals homozygous for the clear haplotype were sampled in this analysis.
Determination of ancestral alleles
Wider data for the Plassais cohort (Plassais et al. 2019) are represented in Table S9. All loci shown in Table S3 were polymorphic in the wolf samples other than CFA38:g.11 085 443G>A and CFA38:g.11 143 243C>T, which describe the wider roan haplotype (T R, wolf monomorphic for reference alleles G and C respectively; Table S10; Jagannathan et al. 2019; Plassais et al. 2019).
Discussion
The purpose of the initial study was to identify the genetic basis of the coat‐patterning phenotype roan. Mapping of this phenotype without regard to the presence or absence of white patterning in ECSs misidentifies the causative locus as the white‐patterning locus (MITF; data not shown) because frequently ECSs with white patterning are also roan. Restricting the analysis to only animals that have white patterning from the activity of MITF correctly identifies the causative locus as being in LD with USH2A on CFA38. Parti‐color in the ECS is relatively rare and only five animals of 35 sampled in Australia had this phenotype (Table S7).
In the course of exploring the genomic association with roan phenotype, we asked whether ticked might be controlled by a related mechanism using ESSs which segregate both ticked and non‐ticked phenotypes. The analysis including ESSs revealed that the most common non‐roan haplotype in the ECSs was shared with the ticked ESSs and that homozygous non‐ticked ESSs had a third haplotype in the same region that was concordant with the reference genome sequence (from a clear Boxer).
We have identified a region on canine CFA38 that segregates the roan and parti‐color phenotypes in ECSs with genome‐wide significance. Hair length, which is controlled by the gene fibroblast growth‐factor (FGF5; Cadieu et al. 2009), can also affect our perception of color dispersion in the coat with shorter hair resulting in spots with precise edges whereas spots in long hair appear as diffuse. This may explain the discrepancy of one ECS in validation that is probably ticked based on inspection of a photograph.
On CFA38, the Affymetrix array marker most associated with roan in the ECS was CFA38:11 057 040 (P raw = 8.9 × 10−10). Further examination of genetic markers in the same region distinguished clear from ticked ESSs with the most associated markers at CFA38:11 122 467 and CFA38:11 124 294 (both P raw = 1.7 × 10−5) based on a denser marker set with a limited number of phenotyped dogs. Observation of variant call files from dogs of various breeds with representative haplotypes in public‐domain WGS data across the region of association identified further putative functional variants that segregate in LD with the three common haplotypes identified in coat‐phenotyped ECSs and ESSs. We have labelled these haplotypes as t (recessive clear and reference genome haplotype), T (ticked) and T R (roan). The phene has been allocated in OMIA as: coat color, ticked in Canis lupus familiaris (002264‐9615).
Although our evidence strongly implicates the involvement of one gene (USH2A), the authors do not suggest that there is classic monogenic inheritance of these phenotypes. Rather, the three haplotypes of this allelic series together with others yet to be characterized result in a complex range of clear, ticked and roan patterns in the two phenotyped breeds.
The haplotypes that were characterized in our phenotyped Spaniels ignore other haplotypes present in domestic dogs, including rarer haplotypes in ECSs and ESSs. An illustration representing our current understanding of the most simplistic interaction of genotype and phenotype in ECSs and ESSs at this locus is shown in Fig. 3. Results from validation (Tables S2 and S8) suggest that in Spaniels the ticked phenotype can result from a large number of haplotypes in the region, whereas roan and clear phenotypes each seem to be clearly concordant with single haplotypes.
Sampled wolves did not segregate the T R (roan) haplotype, suggesting that this haplotype evolved in domestic dogs. Other variants associated with the clear and ticked haplotypes did segregate in wolves.
A number of putative functional variants occur throughout the region of association in proxy animals; however, those with the strongest LD on the characterized haplotypes consist of four missense variants in the gene USH2A (CFA38:g.11 085 443G>A, CFA38:g.11 111 286C>T, CFA38:g.11 156 276A>T and CFA38:g.11 169 445G>A), two splicing variants in USH2A (CFA38:g11 156 425T>C and CFA38:g.11 167 847GA>GAA/DEL) and two structural variants (CFA38:11 112 907–11 113 501VNTR and CFA38:g11 131 841–11 143 239DUP.insTTAA).
The roan haplotype (T R) segregates strongly with the duplication of 11 kb located within the 67th intron of USH2A (CFA38:g11 131 841–11 143 239DUP.insTTAA). Evidence of the duplication is reported elsewhere (Ensembl), although not in relation to any particular phenotype (Decker et al. 2015). Puzzlingly, homozygous dogs from two breeds with disparate phenotypes (Dalmatian and Australian Cattle Dog) share a common haplotype with other roan breeds throughout the wider region of association. Their phenotypic divergence may be affected by the VNTR structural variant (CFA38:11 112 907–11 113 501VNTR) or variants elsewhere in the genome. Coverage data (Table S6) suggest polymorphism for copy number between breeds at the VNTR locus. Australian Cattle Dogs in both public data resources (which are most always roan) demonstrated either homozygosity or heterozygosity for the T R haplotype, supporting a proposed (incomplete) dominant mode of inheritance of the roan phenotype. One identified variant (CFA38:g.11 156 425T>C) differentiating the T (ticked) allele from roan and clear alleles in the cohort used for haplotype characterization affects gene splicing and this may alter somatic expression of the USH2A transcript.
The USH2A gene encodes the Usherin‐2A protein and is found in the basement membrane of the extracellular matrix (ECM). Usherin‐2A comprises a pentaxin domain with multiple laminin epidermal growth factor and many fibronectin type III motifs (Stelzer et al. 2016). In turn, laminin and fibronectin are proteins of the ECM and interact with other ECM proteins such as collagen. All variants that are associated with the coat patterning phenotypes examined in this research occur in the fibronectin region of the gene. Public data from the canine (Hoeppner et al. 2014) suggest that there are two alternatively spliced isoforms of USH2A that differ according to the presence or absence of an exon between exons 69 and 70 (ENSCAFG00000010731.4). The missing exon is represented in canine EST DN750099 (Genbank).
In humans, variants within USH2A are associated with Usher Syndrome type IIA (OMIM entry no. 276901), an autosomal recessive disorder characterized by varying severities of aural, visual and neurological impairment (Dreyer et al. 2008). The skin, eye and inner ear originate from the migration of neural crest cells. USH2A‐associated hearing impairment is believed to occur as a result of disruptions to hair bundle formation and stereocilia differentiation within cochlear hair cells of the inner ear, whereas retinitis pigmentosa, also commonly associated with Usher Syndrome type IIA, is caused by defects in the retinal pigment epithelium (made from melanin and lipofuscin). To the authors’ knowledge, the prevalence of ocular and/or aural disorders in roan or ticked Spaniel dogs has not yet been investigated but may be of interest given the role of USH2A in aural and visual phenotypes. Further, the mechanisms by which the USH2A variants functionally influence ticked or roan canine coat are at this time unclear.
Two breeds with the T R haplotype (Dalmatian and Australian Cattle Dog) have some individuals that exhibit congenital sensorineural deafness (Stritzel et al. 2009; Sommerlad et al. 2014). The two breeds additionally have high frequency of the extreme white coat color at the white markings locus (MITF). The interaction of variants between these two loci in deafness‐affected breeds remains under investigation.
Conflict of interest
The authors have no conflicts to declare.
Supporting information
Figure S1. Regional coverage by representative haplotype clearly showing the presence of the segmental duplication and VNTR expansion on the T R haplotype: (a) haplotype t (no structural variant, gray) vs. haplotype T R (dark blue); (b) haplotype T (no structural variant, light blue) vs. haplotype T R (dark blue).
Table S1. Segregation of the 10 most associated variants from within‐breed whole‐genome‐wide association analysis for parti‐color (non‐roan) and roan coat patterns in the English Cocker Spaniel.
Table S2. Haplotype defining genotypes for English Cocker Spaniels (ECSs) and English Springer Spaniels (ESSs) with coat phenotypes and showing homozygous haplotypes.
Table S3. Dog Biomedical Variant Consortium Dogs showing breed and haplotype representation (homozygous individuals only).
Table S4. Primers for capture of putative functional variants segregating among identified haplotypes.
Table S5. Putative functional variants in usherin (USH2A) as predicted by the variant table of Ensembl (Yates et al. 2020) for canfam3.1.
Table S6. Summary of variant calls and cover by representative haplotype (DBVC data: t, clear; T, ticked; TR, roan, unknown?).
Table S7. Positive or negative result for structural variant bridge segment in coat phenotyped Australian dogs defined by positive capture of fragment in PCR using primers (forward, TCAACATCCATACTTCTCATGCTT; reverse, TGCTCAACCTAAATGTGCTAA) and an annealing temperature of 55°C.
Table S8. Genotyping of identified putative functional variants in five ECSs and 11 ESSs.
Table S9. Genotypes for 722 dogs and canids derived from Plassais et al. (2019) for variants segregating with three coat phenotype haplotypes: t (clear), T (ticked) and T R (roan) in ESSs and ECSs.
Table S10. Genotypes for wolves derived from Plassais et al. (2019) for variants segregating with three coat phenotype alleles t (clear), T (ticked) and T R (roan) in ESSs and ECSs.
Acknowledgements
We gratefully acknowledge the assistance of the Canine Health Foundation (02157‐MOU) and the Dalmatian Club of NSW who have provided financial or in‐kind support to this work. In addition, we thank the owners of all animals participating in this project. KLT is a Distinguished Professor of the Swedish Research Council. The authors acknowledge the Sydney Informatics Hub, a Core Research Facility, of the University of Sydney for access to the High‐Performance Computing cluster ‘Artemis’.
References
- American Kennel Club (2020) Spaniels , viewed 17 September 2020. https://www.akc.org/sports/spaniels/.
- Australian National Kennel Council (2006) Extended breed standard of The Cocker Spaniel , viewed 17 September 2020. http://ankc.org.au/media/pdf/Cocker_Spaniel_BSE_amen12.pdf.
- Bannasch D., Safra N., Young A., Karmi N., Schaible R.S. & Ling G.V. (2008) Mutations in the SLC2A9 gene cause hyperuricosuria and hyperuricemia in the dog. PLoS Genetics 4, e1000246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadieu E., Neff M.W., Quignon P. et al. (2009) Coat variation in the domestic dog is governed by variants in three genes. Science 326, 150–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decker B., Davis B.W., Rimbault M. et al. (2015) Comparison against 186 canid whole‐genome sequences reveals survival strategies of an ancient clonally transmissible canine tumor. Genome Research 25, 1646–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreyer B., Brox V., Tranebjaerg L., Rosenberg T., Sadeghi A.M., Moller C. & Nilssen O. (2008) Spectrum of USH2A mutations in Scandinavian patients with Usher syndrome type II. Human Mutation 29, 451. [DOI] [PubMed] [Google Scholar]
- Gabriel S., Ziaugra L. & Tabbaa D. (2009) SNP genotyping using the Sequenom MassARRAY iPLEX platform. Current Protocols in Human Genetics 60, 2–12. [DOI] [PubMed] [Google Scholar]
- Hinrichs A.S., Raney B.J., Speir M.L. et al. (2016) UCSC data integrator and variant annotation integrator. Bioinformatics 32, 1430–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeppner M.P., Lundquist A., Pirun M. et al. (2014) An improved canine genome and a comprehensive catalogue of coding genes and non‐coding transcripts. PLoS One 9, e91172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagannathan V., Drogemuller C., Leeb T. & Dog Biomedical Variant Database C (2019) A comprehensive biomedical variant catalogue based on whole genome sequences of 582 dogs and eight wolves. Animal Genetics 50, 695–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson E.K., Baranowska I., Wade C.M. et al. (2007) Efficient mapping of mendelian traits in dogs through genome‐wide association. Nature Genetics 39, 1321–8. [DOI] [PubMed] [Google Scholar]
- Körberg I.B., Sundström E., Meadows JRS et al. (2014) A simple repeat polymorphism in the MITF‐M promoter is a key regulator of white spotting in dogs. PLoS One 9, e104363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. (2011) Tabix: fast retrieval of sequence features from generic TAB‐delimited files. Bioinformatics 27, 718–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little C.C. (1953) An analysis of coat‐color inheritance in the dog. Science 118, 572. [Google Scholar]
- Plassais J., Kim J., Davis B.W., Karyadi D.M., Hogan A.N., Harris A.C., Decker B., Parker H.G. & Ostrander E.A. (2019) Whole genome sequencing of canids reveals genomic regions under selection and variants influencing morphology. Nature Communications 10, 1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell S., Neale B., Todd‐Brown K. et al. (2007) PLINK: a tool set for whole‐genome association and population‐based linkage analyses. American Journal of Human Genetics 81, 559–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommerlad S.F., Morton J.M., Johnstone I., O'Leary C.A. & Seddon J.M. (2014) Consequences of a screening programme on the prevalence of congenital hereditary sensorineural deafness in the Australian Cattle Dog. Animal Genetics 45, 855–62. [DOI] [PubMed] [Google Scholar]
- Stelzer G., Rosen R., Plaschkes I. et al. (2016) The GeneCards suite: from gene data mining to disease genome sequence analysis. Current Protocols in Bioinformatics 54, 1.30.1–33. [DOI] [PubMed] [Google Scholar]
- Stritzel S., Wohlke A. & Distl O. (2009) A role of the microphthalmia‐associated transcription factor in congenital sensorineural deafness and eye pigmentation in Dalmatian dogs. Journal of Animal Breeding and Genetics 126, 59–62. [DOI] [PubMed] [Google Scholar]
- Untergasser A., Cutcutache I., Koressaar T., Ye J., Faircloth B.C., Remm M. & Rozen S.G. (2012) Primer3—new capabilities and interfaces. Nucleic Acids Research 40, e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates A.D., Achuthan P., Akanni W. et al. (2020) Ensembl 2020. Nucleic Acids Research 48, D682–D688. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Regional coverage by representative haplotype clearly showing the presence of the segmental duplication and VNTR expansion on the T R haplotype: (a) haplotype t (no structural variant, gray) vs. haplotype T R (dark blue); (b) haplotype T (no structural variant, light blue) vs. haplotype T R (dark blue).
Table S1. Segregation of the 10 most associated variants from within‐breed whole‐genome‐wide association analysis for parti‐color (non‐roan) and roan coat patterns in the English Cocker Spaniel.
Table S2. Haplotype defining genotypes for English Cocker Spaniels (ECSs) and English Springer Spaniels (ESSs) with coat phenotypes and showing homozygous haplotypes.
Table S3. Dog Biomedical Variant Consortium Dogs showing breed and haplotype representation (homozygous individuals only).
Table S4. Primers for capture of putative functional variants segregating among identified haplotypes.
Table S5. Putative functional variants in usherin (USH2A) as predicted by the variant table of Ensembl (Yates et al. 2020) for canfam3.1.
Table S6. Summary of variant calls and cover by representative haplotype (DBVC data: t, clear; T, ticked; TR, roan, unknown?).
Table S7. Positive or negative result for structural variant bridge segment in coat phenotyped Australian dogs defined by positive capture of fragment in PCR using primers (forward, TCAACATCCATACTTCTCATGCTT; reverse, TGCTCAACCTAAATGTGCTAA) and an annealing temperature of 55°C.
Table S8. Genotyping of identified putative functional variants in five ECSs and 11 ESSs.
Table S9. Genotypes for 722 dogs and canids derived from Plassais et al. (2019) for variants segregating with three coat phenotype haplotypes: t (clear), T (ticked) and T R (roan) in ESSs and ECSs.
Table S10. Genotypes for wolves derived from Plassais et al. (2019) for variants segregating with three coat phenotype alleles t (clear), T (ticked) and T R (roan) in ESSs and ECSs.