

Abstract
The synthesis of calcium complexes ligated by three different chiral iminophosphonamide ligands, L‐H (L=[Ph2P{N(R)CH(CH3)Ph}2]), L′‐H (L′=[Ph2P{NDipp}{N(R)CH(CH3)Ph}]), (Dipp=2,6‐iPr2C6H3), and L′′‐H (L′′=[Ph2P{N(R)CH(CH3)naph}2]), (naph=naphthyl) is presented. The resulting structures [L 2Ca], [L′ 2Ca], and [L′′ 2Ca] represent the first examples of enantiopure homoleptic calcium complexes based on this type of ligands. The calcium complexes show blue–green photoluminescence (PL) in the solid state, which is especially bright at low temperatures. Whereas the emission of [L′′ 2Ca] is assigned to the fluorescence of naphthyl groups, the PL of [L 2Ca] and [L′ 2Ca] is contributed by long‐lived phosphorescence and thermally activated delayed fluorescence (TADF), with a strong variation of the PL lifetimes over the temperature range of 5–295 K. Furthermore, an excellent catalytic activity was found for these complexes in hydroboration of ketones at room temperature, although no enantioselectivity was achieved.
Keywords: calcium, homoleptic, hydroboration, iminophosphonamide, photoluminescence
Glowing bones: Enantiopure homoleptic iminophosphonamide calcium complexes have been prepared. These show rich photoluminescence properties and catalytic activity in the hydroboration of ketones.

Introduction
Over the last few decades, the rapid development in the field of photophysics and photochemistry is largely dominated by transition metal‐based complexes owing to their highly luminescent nature. [1] In particular expensive transition metals (i.e., Ir, Ru, Pt, and Au) have been studied extensively. [2] This circumstance may be a limiting factor for a mass production of economical organic light‐emitting diodes (OLED) devices for lighting applications. Accordingly, a search for alternatives based on earth‐abundant metals, also including main‐group metals, has recently become an active and challenging research area. [3] The well‐known aluminum compound Alq3 (q=8‐hydroxyquinolinate) and its derivatives may be viewed as proponents of practically important, highly luminescent complexes of the main‐group metals. [4] For group 1 and group 2 metals, however, only few molecular compounds with prominent emission have been described so far. [5]
Very recently, we have synthesized a chiral version of an iminophosphonamide ligand (L‐H) and found that it allows further derivatization into alkali and group 13 metal complexes.[ 5b , 6 ] The complexes [M2(L)2] (M=alkali metals, Figure 1) showed exciting photoluminescence (PL) properties, including bright long‐lived phosphorescence at low temperature and thermally activated delayed fluorescence (TADF) with a high quantum yield at ambient temperature. Inspired by the results obtained for the alkali metal complexes, we aimed to obtain the analogous complexes of L‐H and similar ligands also for group 2 metals as well as characterize their PL properties. Note that for the whole ligand class of iminophosphonamides only a few examples of alkaline earth metal complexes are known. [7] Moreover, only one heteroleptic calcium complex based on a chiral iminophosphonamide has been reported, with the chirality solely introduced at one nitrogen center. [8] None of these reported alkaline earth metal complexes based on iminophosphonamides showed PL.
Figure 1.

Enantiopure dimeric alkali metal complexes with iminophosphonamide ligands.
Furthermore, from the view point of green and sustainable chemistry, alkaline‐earth metal complexes are considered as benign catalysts for activation of synthetic intermediates due to their low electronegativity. [9] Thus, complexes based on alkaline‐earth metals are highly attractive catalysts for a wide range of chemical transformations including the Tishchenko reaction, [10] ring‐opening polymerization, [11] hydroboration, [12] hydroamination, [13] hydrophosphination of alkenes and alkynes, [14] coupling of alkynes with carbodiimides, [15] and cross dehydrocoupling. [16] The known catalysts based on alkaline‐earth‐metal complexes are mostly heteroleptic in nature.[ 12b , 12e , 12f , 17 ] However, catalysts based on homoleptic alkaline‐earth‐metal complexes are limited possibly due to saturation in the coordination sites.[ 10b , 12d , 14b ]
Herein, we describe an extended series of enantiopure iminophosphonamine ligands bearing different substituents at the nitrogen atom(s) of the NPN backbone (L‐H, L′‐H, and L′′‐H, Figure 2). These were used to prepare solvent‐free tetracoordinated calcium complexes 1–3, respectively. To the best of our knowledge, these complexes represent the first examples of enantiopure homoleptic alkaline‐earth‐metal complexes based on chiral iminophosphonamides. Furthermore, similar to the alkali metals ligated by L, [5b] calcium complexes with L and L′ ligands also show TADF. In addition, all calcium complexes were found to be very efficient catalysts for the hydroboration of ketones although no enantioselectivity was observed for the reaction products.
Figure 2.

Structural representation of the iminophosphonamine ligands under discussion.
Results and Discussion
Synthesis and characterization
Enantiopure iminophosphonamine ligands
The ligand L‐H was obtained using the previously reported procedure. [5b] In order to modify the steric factor of L‐H, a new ligand L′‐H was synthesized by replacing one chiral center (R−CH(CH3)Ph) with a Dipp (Dipp=2,6‐iPr2C6H3) group. Specifically, L′‐H was synthesized via the Staudinger reaction between the known compounds Dipp‐N3 [18] and HN[R−CH(CH3)Ph](PPh2) [19] (Scheme 1). The ligand was characterized by multinuclear NMR spectroscopy. The 31P{1H} NMR spectrum of L′‐H exhibits a single peak at δ=−10.9 ppm, significantly shifted upfield in comparison to δ=36.6 ppm observed for HN[R−CH(CH3)Ph](PPh2). In the 1H NMR spectrum, the NH proton appears as a doublet of doublets at δ=2.81 ppm (3 J HH=9.5 Hz, 2 J PH=9.5 Hz), indicating the absence of any E/Z isomerization in contrast to L‐H. [5b] A signal at δ=4.64 ppm (3 J HH=6.8 Hz, 3 J HH=10 Hz, 3 J PH=10 Hz) is assigned to [NHCH(CH3)Ph] whereas the methyl protons [HNCH(CH 3)Ph] appear as a doublet at δ=1.26 ppm (3 J HH=6.8 Hz). Finally, the IR spectrum of L′‐H shows the characteristic NH stretching frequency at 3347 cm−1 (Figure S34).
Scheme 1.

Synthesis of the ligand L′‐H.
Single crystals of L′‐H suitable for X‐ray analysis were grown from hot n‐heptane (Figure 3). The ligand L′‐H crystallized in the orthorhombic chiral space group P212 121 with one molecule in the asymmetric unit. The P−N1 (1.536(4) Å) bond is significantly shorter than the P−N2 (1.660(4) Å) bond, indicating a double bond P=N1. Therefore, the hydrogen atom could be assigned to N2. [5b] The N1−P−N2 angle of 116.7(2)° is smaller than in ligand L‐H and comparable to the angle in related achiral compounds.[ 5b , 20 ] No strand structure was observed in the crystalline state, suggesting no hydrogen bridging to the neighboring molecule.
Figure 3.

Molecular structure of L′‐H in the solid state. All hydrogen atoms except for the amine proton are omitted for clarity. Selected bond lengths [Å]: P−N1 1.536(4), P−N2 1.660(4); selected bond angles [°]: N1−P−N2 116.7(2).
Further on, to enhance bulk and the π‐character at the nitrogen substituents a modified ligand L′′‐H was synthesized by the incorporation of naphthyl substituents. Specifically, it was synthesized by conducting the Staudinger reaction between {R−CH(CH3)naph}N3 [19] and HN{R−CH(CH3)naph}(PPh2), which was synthesized from H2N{R−CH(CH3)naph}, and ClPPh2, as described earlier (Scheme 2). [21] Similar to L‐H, the 1H NMR spectrum of L′′‐H also shows E/Z tautomerization. Due to this tautomerization two signal sets for naph(CH)CH3 (δ=5.49, 5.17 ppm) and naph(CH)CH 3 (δ=1.81, 1.21 ppm) are observed. Appearance of two signal sets in pairs for naph(CH)CH3 (δ=29.9, 25.7 ppm) and naph(CH)CH3 (δ=51.2, 46.3 ppm) in the 13C{1H} spectrum further confirms the non‐equivalent nature of the methine and methyl carbon atoms of the ligand L′′‐H. The corresponding NH proton in the 1H NMR spectrum is observed as a broad signal at δ=2.88 ppm. Additionally, in the 31P{1H} NMR spectrum a singlet at δ=3.6 ppm is detected. Finally, in the IR spectrum of L′′‐H the NH stretching frequency is found at 3372 cm−1.
Scheme 2.

Synthesis of the ligand L′′‐H. The first step (a) was reported earlier. [21]
Colorless single crystals of the ligand L′′‐H, suitable for X‐ray analysis, were obtained from hot n‐heptane. Similar to the ligand L‐H, L′′‐H also crystallized in the chiral space group P21 with two molecules in the asymmetric unit (Figure 4). Similar to the ligand L′‐H, no strand structure was observed in the solid state. The phosphorous atom in the NPN moiety is tetrahedrally coordinated. The P−N1 bond length (1.675(4) Å) is within the range of a P−N single bond, whereas the P−N2 bond length of 1.544(4) Å is in the range of a P=N double bond. Therefore, the hydrogen atom could be clearly assigned to the nitrogen atom N1. The N1−P1−N2 bond angle (119.6(2)°) is wider than observed for L′‐H (116.7(2)°) but narrower than in L‐H (121.6(2)°).
Figure 4.

Molecular structure of L′′‐H in the solid state. All hydrogen atoms except for the amine proton are omitted for clarity. Selected bond lengths [Å]: P1−N1 1.675(4), P1−N2 1.544(4); selected bond angles [°]: N1−P1−N2 119.6(2).
Calcium complexes
As the starting point for calcium complexation, we chose our previously reported enantiopure ligand L‐H and reacted it with [Ca{N(SiMe3)2}2(THF)2] in a 2:1 ratio.
This afforded the corresponding homoleptic complex [L2Ca] (1) in good yield (Scheme 3). Complex 1 is solvent free and monomeric in nature.
Scheme 3.

Synthesis of complex 1.
Complex 1 was further characterized by multinuclear NMR spectroscopy, elemental analysis and IR spectroscopy. The absence of the NH stretching frequency in the IR spectrum and the absence of the NH resonance in the 1H NMR spectrum indicates complete deprotonation of the iminophosphonamide ligand in 1. Furthermore, the 31P{1H} NMR spectrum of 1 shows a significant downfield shift in comparison to the metal‐free ligand L‐H: δ=24.5 (1) versus 2.7 ppm (L‐H). In the 1H NMR spectra, the characteristic resonances that can be assigned to the methine protons (i.e., Ph(CH)CH3) and the methyl protons (i.e., Ph(CH)CH 3) are observed. In complex 1, the Ph(CH)CH3 resonance is well resolved and appears as a doublet of quartets at δ=4.04 ppm, whereas two sets of signals at δ=4.73 and 4.38 ppm are observed for the ligand due to E/Z isomerization and tautomerization.
Single crystals suitable for X‐ray crystallography were obtained from hot n‐hexane. Complex 1 crystallizes in the monoclinic chiral space group C2. The crystal structure shows two halves of the molecule in the asymmetric unit. Thus, a crystallographic C 2 axis goes along P1−Ca−P2. The central calcium metal ion in 1 is tetracoordinated by four N atoms without any coordinating solvent and adopts a distorted tetrahedral geometry (Figure 5).
Figure 5.

Molecular structure of complex 1 in the solid state. All hydrogen atoms are omitted for clarity. Selected bond lengths [Å]: Ca−N1 2.365(3), Ca−N2 2.344(2), Ca−P1 2.9884(14), Ca−P2 2.9801(14), P1−N1 1.606(3), P2−N2 1.605(2); selected bond angles [°]: P2−Ca‐P1 180.0, N2−Ca−N1 137.17(9), N2′−Ca−N1′ 137.18(9) N2−Ca−N2′ 64.76(11), N1−Ca−N1′ 64.64(13), N2−Ca−N1′ 133.94(9), N2′−Ca−N1 133.94(9), N1−P1−N1′ 103.9(2), N2−P2−N2′ 102.9(2).
The Ca−N bond distances (Ca−N1 2.365(3) Å and Ca−N2 2.344(2) Å) are consistent with literature reports on similar compounds. [22] The P1−Ca−P2 arrangement is clearly linear (180°), and the two CaN2P planes intersect at an angle of 86.04(3)°. The P−N bond distances in complex 1 are almost equal and in the range between single and double bonds. The NPN bond angle of ligand L‐H (121.6(2)°) significantly decreases upon coordination (N1−P1−N1′ 103.9(2)° and N2−P2−N2′ 102.9(2)°).
Similar to the synthesis of 1, the asymmetric ligand L′‐H was reacted with [Ca{N(SiMe3)2}2(THF)2] in a 2:1 ratio to obtain monomeric homoleptic calcium complex [L′ 2Ca] (2) (Scheme 4).
Scheme 4.
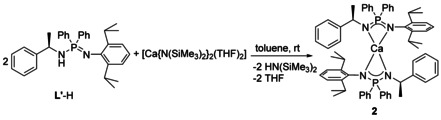
Synthesis of complex 2.
The absence of a NH signal in the 1H NMR spectrum of 2 suggests a full conversion of the ligand L′‐H. Furthermore, a doublet of quartets at δ=3.67 ppm and a doublet at δ=1.65 ppm are observed, which can be assigned to the Ph(CH)CH3 and the Ph(CH)CH 3 proton resonances, respectively. These well‐resolved resonances indicate a symmetric coordination of both ligands in solution. However, the signals corresponding to the Dipp groups show broad resonances, which may be the result of a hindered rotation. The 31P{1H} NMR spectrum of 2 appears to show a single peak at δ=22.6 ppm, which is significantly shifted downfield compared to the ligand L′‐H (δ=−10.9 ppm). Finally, the absence of any NH stretching band in the IR spectrum of 2 suggests complete deprotonation of the ligand by [Ca{N(SiMe3)2}2(THF)2].
Complex 2 crystallized in the orthorhombic chiral space group P212121 with one molecule in the asymmetric unit (Figure 6). The central calcium atom in complex 2 is tetracoordinated and adopts a distorted tetrahedral geometry. However, due to systematic twinning the quality of the single crystal X‐ray diffraction data was poor. This problem could not be so far solved by using different conditions for the crystallization. Nevertheless, the connectivity, which is shown as pictorial representation in Figure 6, could be deduced from the difference Fourier map.
Figure 6.

Pictorial representation of 2 in the solid state, based on the X‐ray diffraction connectivity data. All hydrogen atoms are omitted for clarity.
Ligand variation in the coordination sphere of the calcium center was further realized by the synthesis of the homoleptic tetracoordinated calcium complex [L′′ 2Ca] (3). Complex 3 was obtained by reaction of L′′‐H with [Ca{N(SiMe3)2}2(THF)2] in a 2:1 molar ratio (Scheme 5). Analogous to 1, the appearance of a single set of resonances for the methine and methyl protons in the 1H NMR spectrum of complex 3 suggests its symmetric nature. This is in contrast to L′′‐H, which showed two different sets of resonance corresponding to methine [i.e., naph(CH)CH3] and methyl [i.e., naph(CH)CH 3] due to E/Z tautomerization. In the 1H NMR spectrum of 3, the naph(CH)CH3 appears as a doublet of quartets at δ=4.86 ppm, whereas for naph(CH)CH 3 a doublet at δ=1.44 ppm is observed. The 31P{1H} NMR spectrum shows a single peak at δ=27.3 ppm, which is significantly shifted downfield compared to δ=3.6 ppm for the ligand.
Scheme 5.

Synthesis of complex 3.
Complex 3 crystallizes in the monoclinic chiral space group P21 with one molecule in the asymmetric unit (Figure 7). Unlike in complex 1, where the central calcium atom is exactly in the plane of the coordinating N2P ligand backbones, in complex 3, it is slightly above this plane with the angle P1−Ca−P2 equal to 170.35(2)°. The deviation of the central metal from the N2P backbone is reflected in the dihedral angles of 1.327(10)° and 6.887° between N1−P1−N2 and N1−Ca−N2 planes and N3−P2−N4 and N3−Ca−N4 planes, respectively. This slight displacement of the calcium atom is likely to minimize the repulsion between the naphthyl substituents. The Ca−N bond distances in 3 are in the range from 2.370(2) to 2.394(2) Å, similar to complexes 1 and 2.
Figure 7.

Molecular structure of 3 in the solid state. All hydrogen atoms are omitted for clarity. Selected bond lengths [Å] Ca−N1 2.390(2), Ca−N2 2.370(2), Ca−N3 2.394(2), Ca−N4 2.374(2), Ca−P1 3.0033(9), Ca−P2 3.0106(8), P1−N1 1.599(2), P1−N2 1.601(2), P2−N3 1.604(2), P2−N4 1.607(2); selected bond angles [°]: P1−Ca−P2 170.35(2), N1−Ca−N2 63.97(6), N3−Ca−N4 63.90(7), N1−Ca−N3 167.91(7), N2−Ca−N4 147.51(7), N1−Ca−N4 118.11(7), N1−P1−N2 104.01(10), N3−P2−N4 103.61(10).
Photoluminescence properties
We recently reported on the unusual PL properties of the alkali metal complexes [L 2M2] (M=Li, Na, K, Rb, Cs) with the ligand L‐H. [5b] In contrast to the fluorescent ligand, these complexes demonstrate bright blue–green phosphorescence at low temperatures (<100 K) in the solid state, which transforms upon raising the temperature above ≈150 K to TADF emission from the (relaxed) singlet state S1 thermally populated from the lower‐lying close triplet state T1. [23]
We were further able to show that the TADF results from the dimeric structure (i.e., the alkali metal participates in the TADF) and correlates with the mutual orientation of the two ligands. Accordingly, we were interested whether this mechanism is also observed for the novel monometallic Ca‐based complexes and how the nitrogen substituents and their respective orientation influence the PL properties.
Similar to the alkali metal complexes [L 2M2], all three calcium complexes 1–3 are colorless crystalline solids showing a broad blue–green photoluminescence upon UV excitation below ≈400 nm. The emission and excitation (PLE) spectra of 1–3 as well as the temperature dependencies of the integrated PL intensity are shown in Figure 8.
Figure 8.

Left: photoluminescence excitation (PLE) and emission (PL) spectra of compounds 1–3 at 7, 150, and 295 K. The spectra were recorded/excited at 480/330 nm, respectively. Right: integrated PL intensities plotted against the temperature in the range of 7–295 K.
In case of calcium complex 1, not only the PL spectra but also the temperature dependencies of the PL intensity (Figure 8, right panel) and decay lifetime (Figure S47) are rather similar to those observed for the dimeric alkali metal complexes of the same ligand. [5b] The latter dependence shows a characteristic TADF transition from a long‐lived phosphorescence with a lifetime of ca. 5.6 ms below 100 K to much faster PL kinetics with an effective lifetime of 24 μs at 295 K (Figure 9). This transition occurs over ≈130–180 K and correlates with the nonmonotonic dependence of the integrated PL intensity versus temperature (Figure 8). The latter also resembles the curves measured for [L 2Na2], [L 2K2] and [L 2Rb2] complexes. A simple TADF model of thermally equilibrated S1 and T1 states [Eq (S1) in the Supporting Information][ 23a , 23c ] can be applied to the data shown in Figure 9 to estimate the energy separation ΔE between these states as ca. 1200 cm−1 (148 meV). This value is notably larger in comparison with ΔE obtained for the alkali metal complexes (ca. 600–750 cm−1). A PL quantum yield of 22 % was determined for 1 using an integrating sphere under ambient conditions and excitation at 330 nm (Table S2).
Figure 9.

PL decay time of solid complex 1 versus temperature. The PL was excited with a ns‐pulsed laser at 337 nm, recorded at 480 nm, and fit with monoexponential curves. The red curve depicts the fit according to Equation (S1) (see text and the Supporting Information). The fit yielded the energy separation ΔE between S1 and T1 states of about 1200 cm−1. Below T≈100 K the emission is predominantly phosphorescence with a lifetime of ca. 5.6 ms. Increasing the temperature activates the TADF mechanism, resulting in an effective PL lifetime of 24 μs at room temperature.
Similar to 1, the PL of complex 2 shows the minor fast fluorescence and major long‐lived (τ=30 ms at 5 K) phosphorescence components below ≈20 K (Figure S49). By contrast, a TADF‐like transition to faster PL kinetics occurs already between ≈20 and ≈75 K, indicating a much smaller energy gap ΔE between S1 and T1 states, roughly estimated as ≈330 cm−1 (Figure S49). The PL decay above ≈20 K is non‐monoexponential. In addition, non‐radiative electronic relaxation significantly reduces the PL intensity upon raising the temperature (Figure 8), resulting in a low quantum yield of <1 % at 295 K (Table S2). Correspondingly, it is not surprising that the simple TADF model of equilibrated S1 and T1 states [Eq. (S1)] describes the PL decay versus temperature (Figure S48) only relatively poorly. By further increasing the temperature above ≈200 K, the fast (fluorescence) component becomes dominant in 2. The discrepancy in PL between 1 and 2 might be explained by different structural (ligand) arrangements in the solid state (Figures 5 and 6). This explanation would agree with our previous study on the alkali metal complexes, where pronounced correlations between the structural and photophysical properties were observed. [5b]
Finally, in contrast to 1 and 2, only a fast PL component with a lifetime <10 ns (time resolution of our apparatus) could be detected for complex 3 at low temperatures as well as under ambient conditions. This can be assigned to the fluorescence of the naphthyl moieties in 3. Its efficiency amounts to 7 % at room temperature. Similar to 1 and 2, a substantial decrease in the PL efficiency, that is, an increase in nonradiative excited‐state relaxation, was observed for complex 3 by raising the temperature over the range of 7–295 K (Figure 8).
Catalytic hydroboration of ketones
Hydroboration is the addition of B−H bond across an unsaturated system such as alkenes, alkynes, aldehydes, ketones, and imines to form compounds with C−B, C−O, and C−N bonds, respectively. In particular, hydroboration of ketones with boranes (R2BH) is an atom‐economical reaction to form borate esters (R2BOCHR2), which are useful synthetic intermediates for the formation of functionalized alcohols. For hydroboration of C=O‐containing compounds, pinacolborane (HBpin) or catecholborane (HBcat) are mostly considered as suitable mild reductants. [24]
The hydroboration catalysts based on alkaline earth metals reported so far are mostly heteroleptic in nature.[ 12b , 12e , 12f , 17 ] The homoleptic catalysts for this chemical transformation are extremely rare possibly due to saturation in coordination sites. [12d] With this background, one of the goals of our study was to probe a performance of tetra coordinated complexes 1–3 as catalysts for hydroboration of ketones. As a preliminary test, the reaction between acetophenone and HBpin was conducted in C6D6 and monitored continuously via 1H NMR measurements, with a catalyst loading of 1 mol % and ferrocene as an internal standard. Regardless of which complex was used, we observed almost complete conversion into the corresponding alkoxypinacolboronate ester within 5 min at room temperature. High catalytic activity of 1–3 was found for various acetophenones substituted at the para position with electron‐withdrawing (Br, NO2) or electron‐donating (NH2, CH3, and OCH3) groups (Table 1). Such catalytic efficiency of 1–3 and fast conversion of the substrates may be related to their coordinatively unsaturated nature. Perhaps due to this unsaturation in coordination sites the conversion requires less time in comparison to previously reported homoleptic alkaline earth metal catalysts. [12d] Since the catalysts 1–3 are enantiopure, one might expect enantiomeric excess in the product. However, practically no enantiomeric excess could be observed, also by conducting the reactions at lower temperature (down to −20 °C), likely due to the fast conversion of the ketones into their corresponding alkoxypinacolboronate esters.
Table 1.
Hydroboration of ketones using complexes 1–3 as catalysts.[a]
|
Entry |
Substrate |
Product |
Catalyst |
NMR yield[b] [%] |
|---|---|---|---|---|
|
1 |
|
|
1 |
>99 |
|
2 | ||||
|
3 | ||||
|
2 |
|
|
1 |
>99 |
|
2 | ||||
|
3 | ||||
|
3 |
|
|
1 |
>99 |
|
2 |
96 |
|||
|
3 |
>99 |
|||
|
4 |
|
|
1 |
>99 |
|
2 | ||||
|
3 | ||||
|
5 |
|
|
1 |
>99 |
|
2 | ||||
|
3 | ||||
|
6 |
|
|
1 |
>99 |
|
2 |
97 |
|||
|
3 |
>99 |












[a] Conditions: 1 mol % of catalyst 1–3, C6D6, rt. [b] Yields were calculated based on 1H NMR spectroscopy, using ferrocene as the internal standard.
The catalytic activity of catalysts 1–3 for acetophenone can be compared with the known catalysts based on alkaline earth metals. We used 1 mol % of catalyst and observed in most cases a quantitative conversion in less than 5 min resulting, in a turnover frequency (TOF) of >1200 h−1.
The heteroleptic Mg‐based catalysts reported by Fohlmeister and Stasch showed high catalytic activity with a TOF of >8486 h−1 towards a related ketone (i.e., 2‐adamantanone). [17b] Li et al. showed a TOF of 2000 h−1 for the reduction of acetophenone by using highly reactive asymmetric β‐diketiminate magnesium(I) complexes. [12g] Furthermore, related to our complexes, the amidinate stabilized calcium catalyst, [PhC(NiPr)2CaI], [12f] showed a TOF of only 9.5 h−1. In comparison, magnesium‐based catalysts such as {CH[C(Me)NAr]2MgnBu} (Ar=2,6‐iPr2C6H3) reported by Arrowsmith et al. [12b] and bulky amido magnesium methyl‐based catalysts from Ma et al. [17a] showed higher TOFs of 23.5 and 667 h−1, respectively. Additionally, Yadav et al. reported homoleptic calcium and magnesium complexes based on methylpyridinato β‐diketiminate ligands, which showed TOFs of about 16 h−1. [12d] The above examples evidence the highly active nature of catalysts 1–3.
These results prompted us to explore its ability to reduce alkenes under similar conditions. However, no conversion was observed in the reaction of styrene with HBpin using complexes 1–3 as catalysts, even at a relatively high temperature of 120 °C.
To gain some insight into the mechanism underlying the catalytic reaction, vibrational spectroscopic investigations were carried out accompanied by theoretical DFT calculations. In situ NMR studies were not conclusive. The complexation of acetophenone to calcium complex 1 in n‐heptane solution, which is a key step of the catalytic cycle, was investigated by Raman spectroscopy. On one hand, a red‐shift of the C=O valence mode of acetophenone from 1694 to 1651 cm−1 is seen upon complexation of the C=O group to the calcium center, on the other hand a rather negligible red‐shift of its valence phenyl band from 1600 to 1597 cm−1 is observed (Figure 10), which in total confirms an underlying weak calcium–oxygen contact.
Figure 10.

Extract of the experimental (a, b) and calculated (c, d) Raman spectra of acetophenone and its complex with 1 (bands associated to acetophenone and its complex are marked by # and *, respectively, those of the solvent n‐heptane are marked by §).
The theoretical frequency calculations confirm these experimental findings (Figure 10). The structure of the complexation between 1 and acetophenone indicates a weak calcium−oxygen bond (3.690 Å); its complexation energy was calculated to be about −13 kJ mol−1. The C=O distance of acetophenone (1.227 Å) is elongated to 1.235 Å upon complexation. However, this large Ca−O distance is expected for the borane to enable an attack on the acetophenone molecule during the catalytic cycle without any enantiomeric excess.
Conclusions
The reaction of the chiral iminophosphonamide ligands L‐H, L′‐H and L′′‐H with [Ca{N(SiMe3)2}2(THF)2] leads to the formation of the homoleptic calcium complexes [L 2Ca] (1), [L′ 2Ca] (2) and [L′′ 2Ca] (3), respectively. All these complexes are tetracoordinated without coordinating solvent molecules. In the solid state, complexes 1–3 show spectrally similar blue–green PL, which is relatively bright in the case of 1 and 3 also at ambient temperature. However, the PL of 3 is the fluorescence of the naphthyl moieties, whereas the emission of 1 and 2 is contributed by phosphorescence at cryogenic temperatures and thermally activated delayed fluorescence (TADF) at elevated temperatures. On the other hand, the characteristic parameters such as the TADF activation temperature and PL efficiency differ significantly in 1 and 2. These results suggest that the photophysical properties of iminophosphonamide complexes of both alkali and alkaline earth metals, including the TADF process, can be strongly varied by modification of the ligand. This suggests that the TADF quantum yield (22 % for 1 at room temperature) might be further increased by a ligand optimization. A high catalytic activity of complexes 1–3 was demonstrated in intermolecular hydroboration of various ketones at room temperature. Unfortunately, no enantioselectivity could be achieved, also when conducting the catalytic reactions at low temperature.
Experimental Section
All manipulations of air‐sensitive materials were performed under the rigorous exclusion of oxygen and moisture in flame‐dried Schlenk‐type glassware, either on a dual manifold Schlenk line, interfaced to a high vacuum (10−3 Torr) line, or in an argon‐filled MBraun glove box. Hydrocarbon solvents (toluene, n‐heptane, n‐pentane) were dried by using a MBraun solvent purification system (SPS‐800), degassed and stored under vacuo. n‐hexane was pre‐dried over CaCl2 before decantation and distillation over potassium and storage over 4 Å molecular sieves. Tetrahydrofuran (THF) was distilled under nitrogen over potassium benzophenone ketyl before storage over lithium aluminum hydride (LiAlH4). All substrates [ketones and 4,4,5,5‐tetramethyl‐1,3,2‐dioxaborolane (HBpin)] for hydroboration reactions were purchased from either ABCR or Sigma–Aldrich and used without any further purification.
NMR spectra were recorded on a Bruker Avance III 300 MHz or Avance III 400 MHz spectrometer. Chemical shifts were measured relative to the characteristic solvent resonances as internal standards [7.16 ppm (1H) and 128.06 ppm (13C)]. They are expressed in ppm and reported relative to tetramethylsilane. 85 % phosphoric acid was used as external reference for 31P‐ and 31P{1H}‐NMR. Elemental analyses were carried out with a Vario Micro Cube (Elementar Analysensysteme GmbH). IR spectra were obtained on a Bruker Tensor 37 FTIR spectrometer equipped with a room temperature DLaTGS detector, a diamond ATR (attenuated total reflection) unit, and a nitrogen‐flushed chamber. In terms of their intensity, the signals were classified into the categories vs=very strong, s=strong, m=medium, w=weak and vw=very weak. The Raman spectra were taken using a Bruker Senterra II Raman microscope using a laser with an excitation wavelength of 532 nm.
The theoretical calculations were performed by means of the program package TURBOMOLE 7.3 without constraints of symmetry using the RI‐DFT/BP‐86 method [25] and def2‐SV(P) basis sets for all atoms. [26] Vibrational frequencies and Raman intensities were calculated analytically using the modules aoforce [27] and egrad. [28]
For synthesis of (R)‐α‐methylnaphthyl azide the literature procedure was exactly followed. [29] Dipp‐azide (Dipp=2,6‐iPr2C6H3), [18] HN[R−CH(CH3)Ph](PPh2), [19] HN[R−CH(CH3)naph](PPh2), [21] P,P‐diphenyl‐N,N′‐bis((R)‐1‐phenylethyl)phosphinimidic amide, (R)‐HPEPIA (L‐H), [5b] and [Ca{N(SiMe3)2}2(THF)2] [30] were also prepared according to already described procedures.
Synthesis of P,P ‐diphenyl‐N‐[(R)‐1‐phenylethyl],N′‐(2′,6′‐diisopropylaniline)phosphinimidic amide, (R)‐HPEDippPIA (L′‐H): Ph2PN(R‐*CHMePh) (3.2 g, 10.61 mmol, 1.00 equiv) was dissolved in 30 mL of THF and cooled to −20 °C. A solution of Dipp‐azide (2.4 g, 11.67 mmol, 1.1 equiv) in 20 mL of THF was added slowly. After complete addition, the reaction mixture was warmed to ambient temperature whereupon liberation of N2 gas was observed. The reaction mixture was stirred at ambient temperature for 16 h. After evaporation of the solvent under reduced pressure, a viscous solid was obtained, which was washed with n‐pentane (4*20 mL) and dried in vacuum to obtain the desired product as white powder. Single crystals of the title compound suitable for X‐ray analysis were obtained by recrystallization from n‐heptane.
Yield (based on crystals): 3.57 g (70 %); 1H NMR (C6D6, 400 MHz): δ=7.78–7.66 (m, 4 H, o−Arphos−H), 7.25–7.22 (m, 2 H, Ar−H), 7.10–6.94 (m, 12 H, o,m,p‐Ar−H), 4.64 (ddq, 3 J HH=6.8 Hz, 3 J HH=10 Hz, 3 J PH=10 Hz, 1 H, HNCH), 3.67 (sept, 3 J HH=6.9 Hz, 2 H, HC(CH3)2), 2.81 (dd, 3 J HH=9.5 Hz, 2 J PH=9.5 Hz, 1 H, NH), 1.26 (d, 3 J HH=6.8 Hz, 3 H, HNCHCH 3), 1.183 (d, 3 J HH=6.9 Hz, 6 H, HC(CH 3)2), 1.180 ppm (d, 3 J HH=6.9 Hz, 6 H, HC(CH 3)2); 13C{1H} NMR (C6D6, 100 MHz): δ=146.2 (d, 3 J PC=4.8 Hz, PNHCHC q),144.6 (Ar−C Dipp),142.0 (d, 2 J PC=7.2 Hz, P=NC q), 134.8 (d, 1 J PC=121 Hz, Arphos C q), 134.2 (d, 1 J PC=127 Hz, Arphos C q), 132.3 (d, 2 J PC=9.8 Hz, o‐Arphos−CH), 132.2 (d, 2 J PC=9.8 Hz, o‐Arphos−CH), 131.1 (d, 3 J PC=2.8 Hz, Ar−CH), 130.9 (d, 3 J PC=2.9 Hz, Ar−CH) 128.7 (Ar−C), 128.5 (Ar−C), 128.2 (Ar−C), 127.0 (Ar−C), 126.2 (Ar−C), 123.3 (d, 4 J PC=1.3 Hz, o‐Ar−CH), 119.9 (d, J PC=2.5 Hz, Ar−C), 50.8 (P−NCHCH3), 29.1 (CH(CH3)2), 26.1 (d, 3 J PC=4.0 Hz, Ph(CH)CH3), 24.13 (CH(CH3)2), 24.06 ppm (CH(CH3)2); 31P{1H} NMR (C6D6, 162 MHz): δ=−10.9 ppm; IR (ATR): =3347 (w), 3054 (vw), 2971 (w), 2961 (vw), 2864 (w), 1585 (w), 1596 (w), 1450 (m), 1434 (vs), 1401 (vw), 1354 (m), 1295 (w), 1257 (w), 1202 (w), 1185 (w), 1117 (m), 1101 (m), 1087 (m), 1075 (m), 1064 (m), 1028 (w), 1017 (w), 950 (m), 833 (w), 794 (vw), 771 (m), 749 (vs), 695(vs), 625 (s), 649 (vw), 600 (m), 534 (s), 511 (vs), 500 (vs), 447 cm−1 (w); elemental analysis: calcd [%] for [C32H37N2P] (480.64): C 79.97, H 7.76, N 5.83; found: C 79.42, H 7.63, N 5.80.
Synthesis of P,P ‐diphenyl‐N,N ′‐bis[(R)‐1‐naphthylethyl]phosphinimidic amide, (R)‐HNEPIA (L′′‐H): Ph2PN(R−*CHMenaph) (7.20 g, 20.26 mmol, 1.00 equiv.) was dissolved in THF (40 mL) and cooled to −20 °C. A solution of 2.37 g (R)‐α‐methylnaphthyl azide (22.28 mmol, 1.10 equiv.) in THF (20 mL) was added slowly. After complete addition, the reaction mixture was warmed to ambient temperature whereupon liberation of N2 was observed. The reaction mixture was stirred for 12 h at ambient temperature. After evaporation of the solvent under reduced pressure, a viscous solid was obtained, which was washed with n‐pentane (4*20 mL) and dried in vacuum to obtain the desired product as white powder. Single crystals of the title compound suitable for X‐ray analysis were obtained by recrystallization from n‐heptane.
Yield (based on crystals): 6.7 g (63 %); 1H NMR (C6D6, 400 MHz): δ=8.44 (br, 2 H, Ar−H), 8.04 (br, 2 H, Ar−H), 7.77–7.69 (br, 3 H, Ar−H), 7.52–7.19 (m, 10 H, Ar−H), 7.09–6.95 (m, 7 H, Ar−H), 5.52–5.45 (m, 1 H, P=NCH), 5.17 (br, 1 H, HNCH), 2.88 (br, 1 H, NH), 1.81 (br, 3 H, P=NCHCH 3), 1.21 ppm (br, 3 H, HNCHCH 3); 13C{1H} NMR (C6D6, 100 MHz): δ=148.0 (d, J PC=11.3 Hz, Ar−C), 142.6 (Ar−C), 135.6 (d, J PC=23.5 Hz, Ar−C), 134.5 (Ar−C), 134.2 (Ar−CH), 132.9 (d, J PC=8.2 Hz, Ar−CH), 132.1 (d, J PC=8.6 Hz, Ar−CH), 131.4 (Ar−CH), 130.8 (Ar−CH), 129.0 (d, J PC=20.5 Hz, Ar−CH), 127.9 (Ar−CH), 127.6 (Ar−CH), 126.5 (Ar−CH), 126.1 (d, J PC=8.2 Hz, Ar−CH), 125.5 (d, J PC=13.1 Hz, Ar−CH), 125.0 (d, J PC=16.1 Hz, Ar−CH), 124.7 (Ar−CH), 124.3 (Ar−CH), 123.4 (Ar−CH), 122.4 (Ar−CH), 51.2 (P=NCH), 46.3 (HNCH), 29.9 (d, 3 J PC=11.8 Hz, P=NCHCH3), 25.7 ppm (HNCHCH3); 31P{1H} NMR (C6D6, 162 MHz): δ=3.6 ppm; IR (ATR): =3372 (m), 3074 (m),3051 (m) 2965 (m), 2921 (w), 2858 (vw), 2826 (vw), 2166 (vw), 2104 (vw), 1594 (m), 1508 (m), 1480 (vw), 1435 (m), 1392 (s), 1374 (m), 1329 (m), 1287 (s) 1261 (m), 1233 (vs), 1165 (s), 1111 (s), 1102 (m), 1080 (w), 1053 (w), 1024 (w),997 (w), 934 (m), 868 (w), 849 (w), 829 (s), 801 (m), 774 (s), 750 (s), 717(m), 694 (m), 639 (m), 612 (w), 545 (w), 524 (m), 483 (w), 434 cm−1 (w); elemental analysis calcd [%] for [C36H33N2P] (524.65): C 82.42; H 6.34, N 5.34; found: C 82.10, H 6.23, N 5.40.
Synthesis of complex 1: [Ca{N(SiMe3)2}2(THF)2] (182.0 mg, 0.36 mmol, 1.00 equiv.) and L‐H (300.0 mg, 0.71 mmol, 2.00 equiv.) were dissolved in toluene (30 mL) and stirred at room temperature for 16 h. After removal of the solvent under reduced pressure resulted into white solid. The resulting white solid was washed with n‐pentane (5 mL). Single crystals suitable for X‐ray analysis were obtained from hot n‐hexane. The solvent was decanted, and the product was washed with cold n‐pentane (5 mL).
Yield (based on crystals): 190 mg (60 %). 1H NMR (C6D6, 400 MHz): δ=7.71–7.66 (m, 8 H, o‐Arphos−CH), 7.21–7.19 (m, 16 H, o‐Ar−CH and m‐Arphos−CH), 7.18–7.17 (m, 4 H, Ar−CH), 7.15–7.13 (m, 6 H, Ar−CH), 7.11–7.06 (m, 6 H, Ar−CH), 4.04 (dq, 3 J HH=6.5 Hz, 3 J PH=19.6 Hz, 4 H, Ph(CH)CH3), 1.38 ppm (d, 3 J HH=6.5 Hz, 12 H, Ph(CH)CH 3); 13C{1H} NMR (C6D6, 100 MHz): δ=152.0 (d, 3 J PC=14.0 Hz, Ar−C q), 135.9 (d, 1 J PC=85.4 Hz, Arphos−C q), 132.7 (d, 2 J PC=8.8 Hz, o‐Arphos−CH), 130.3 (d, 4 J PC=2.7 Hz, Ar−CH), 129.3 (Ar−CH), 127.9 (Ar−CH), 126.1 (Ar−CH), 125.9 (Ar−CH), 54.1 (Ph(CH)CH3), 28.4 ppm (d, 3 J PC=6.7 Hz, Ph(CH)CH3); 31P{1H} NMR (C6D6, 162 MHz): δ=24.5 ppm; IR (ATR): =3058 (vw), 3022 (vw), 2957 (vw), 2913 (vw), 2835 (vw), 1598 (vw), 1489 (m), 1479 (m), 1452 (m), 1433 (m), 1401 (m), 1361 (m), 1346 (m), 1309 (m), 1273 (w), 1236 (s), 1204 (s), 1176 (m), 1167 (m), 1156 (m), 1149 (m), 1118 (m), 1102 (w), 1087 (m), 1060 (m), 1025 (m), 1000 (s), 984 (m), 973 (m), 965 (s), 945 (vs), 911 (m), 859 (m), 837 (m), 822 (m), 780 (s), 772 (s), 761 (m), 749 (m), 719 (s), 714 (s), 695 (s), 622 (m), 608 (m), 597 (m), 574 (m), 565 (m), 510 (s), 463 (s), 446 (m), 434 cm−1 (vw); elemental analysis calcd (%) for [C56H56CaN4P2] (887.09): C 75.82, H 6.36, N 6.32; found: C 75.56, H 6.20, N 6.46.
Synthesis of complex 2: Following the similar procedure described above for 1, the reaction of [Ca{N(SiMe3)2}2(THF)2] (157.0 mg, 0.31 mmol, 1.00 equiv.) and L′‐H (300 mg, 0.62 mmol, 2.00 equiv.) afforded single crystals from n‐pentane suitable for X‐ray analysis. The solvent was decanted and the product was washed with cold n‐pentane (5 mL).
Yield (based on crystals): 220.0 mg (71 %); 1H NMR (C6D6, 400 MHz): δ=7.50–7.45 (m, 4 H, o‐Arphos−CH), 7.27–7.25 (m, 4 H, Ar−CH), 7.14–7.09 (m, 6 H, Ar−CH), 6.99–6.94 (m, 10 H, Ar−CH), 6.83–6.74 (m, 12 H, Ar−CH), 3.67 (dq, 3 J HH=6.5 Hz, 3 J PH=24.87 Hz, 2 H, Ph(CH)CH 3), 3.07 (br, 4 H, CH(CH3)2), 1.65 (d, 3 J HH=6.5 Hz, 6 H, Ph(CH)CH 3), 1.06–0.38 ppm (m, 24 H, CH(CH 3)2); 13C{1H} NMR (C6D6, 100 MHz): δ=152.5 (d, 3 J PC=8.9 Hz, Ar−C q), 144.6 (Ar−C q), 143.0 (d, J PC=4.3 Hz, Ar−C q), 135.8 (Ar−C), 134.9 (d, 2 J PC=8.6 Hz, Ar−CH), 133.9 (Ar−CH), 133.5 (d, J PC=8.9 Hz, Ar−CH), 132.4 (d, J PC=8.1 Hz, Ar−CH), 130.4 (d, J PC=2.5 Hz, Ar−CH), 130.2 (d, J PC=2.6 Hz, Ar−CH), 129.5 (Ar−CH), 127.7 (Ar−CH), 127.5 (d, J PC=10.9 Hz, Ar−CH), 126.5 (Ar−CH), 123.3 (Ar−CH), 122.02 (d, J PC=4.8 Hz, Ar−CH), 55.8 (Ph(CH)CH3), 29.6 (d, 3 J PC=10.9 Hz, Ph(CH)CH3), 28.5 (CH(CH3)2), 24.1 ppm (br, CH(CH3)2); 31P{1H} NMR (C6D6, 162 MHz): δ=22.6 ppm; IR (ATR): =3055 (vw), 2964 (m), 2954 (w), 2919 (w), 2863 (w), 1588 (w), 1492 (m), 1452 (s), 1432 (w), 1380 (vw), 1364 (vw), 1327 (vw), 1308 (m), 1255 (m), 1204 (vs), 1179 (s), 1146 (w), 1111 (w), 1099 (w), 1067 (w), 1047 (m), 1026 (m), 1004 (vw), 992 (vw), 973 (vw), 950 (vw), 931 (vw), 837 (vs), 787 (m), 775 (m), 756 (m), 743 (m), 715 (m), 696 (vs), 662 (m), 609 (m), 583 (m), 564 (s), 524 (s), 514 (m), 490 (m), 477 (m), 451 cm−1 (m); elemental analysis calcd [%] for [C64H72CaN4P2] (999.33): C 76.92, H 7.26, N 5.61; found: C 77.13, H 7.69, N 5.82.
Synthesis of complex 3: Following the similar procedure described above for 1, the reaction of [Ca{N(SiMe3)2}2(THF)2] (96.0 mg, 0.19 mmol, 1.00 equiv.) and L′′‐H (200.0 mg, 0.38 mmol, 2.00 equiv.) afforded single crystals from n‐pentane suitable for X‐ray analysis. The solvent was decanted and the product was washed with cold n‐pentane (5 mL).
Yield (based on crystals): 120.0 mg (58.1 %); 1H NMR (C6D6, 400 MHz): δ=7.77–7.70 (m, 8 H, o‐Arphos−CH), 7.65–7.60 (m, 12 H, Arphos−CH), 7.48–7.46 (m, 4 H, Ar−CH), 7.14–7.10 (m, 4 H, Ar−CH), 7.06–6.99 (m, 20 H, Ar−CH), 4.86 (dq, 3 J HH=6.4 Hz, 3 J PH=19.02 Hz, 4 H, naph(CH)CH3), 1.44 ppm (d, 3 J HH=6.44 Hz, 12 H, naph(CH)CH 3); 13C{1H} NMR (C6D6, 100 MHz): δ=147.4 (d, 3 J PC=12.67 Hz, Ar−C q), 135.8 (Ar−C), 134.9 (Ar−C), 134.6 (Ar−CH), 132.7 (d, J PC=8.7 Hz, Ar−CH), 130.9 (Ar−CH), 130.2 (d, J PC=2.5 Hz, Ar−CH), 129.5 (Ar−CH), 126.7 (Ar−CH), 126.5 (Ar−C), 126.0 (Ar−C), 125.3 (Ar−C), 123.3 (Ar−C), 123.2 (Ar−C), 49.7 (naph(CH)CH3), 28.9 ppm (d, 3 J PC=8.3 Hz, naph(CH)CH3); 31P{1H} NMR (C6D6, 162 MHz): δ=27.3 ppm; IR (ATR): =3052 (vw), 2959 (vw), 2917 (w), 2859 (w), 1595 (w), 1508 (w), 1481 (w), 1456 (m), 1434 (w), 1394 (w), 1364 (w), 1323 (w), 1310 (w), 1300 (w), 1256 (vw), 1235 (vw), 1172 (w), 1144 (w), 1133 (w), 1082 (vs), 1027 (m), 1014 (m), 1001 (w), 970 (m), 926 (m), 908 (m), 849 (s), 835 (s), 821 (s), 791 (m), 775 (m), 748 (m), 730 (m), 712 (vs), 697 (w), 645 (m), 628 (vw), 617 (m), 575 (m), 560 (m), 543 (vs), 532 (vs), 517 (m), 499 (m), 488 (vw), 432 (m), 420 cm−1 (vw); elemental analysis calcd [%] for [C72H64CaN4P2] (1087.33): C 79.53, H 5.93, N 5.15; found: C 79.72, H 6.28, N 5.18.
Hydroboration reactions
The catalyst, ferrocene (internal standard) and solid substrates (0.25 mmol ketone, if any) were weighed into an NMR tube in the argon‐filled glove box. C6D6 (about 0.5 mL) was condensed into the NMR tube and the mixture was frozen at −196 °C. The reactants HBpin (0.3 mmol) and ketone (liquid, 0.25 mmol) were injected onto the solid mixture under nitrogen using an oven‐dried Pasteur pipette, and the whole sample was melted and mixed just before insertion into the core of the NMR machine (t 0). The completion of the reaction was indicated by conversion of singlet of the methyl protons of the ketone into doublet of the final product.
NMR data of catalysis: The NMR data (1H, 11B, 13C{1H}) for the hydroboration products was consistent with the corresponding literature.
Ph(Me)CHOBpin [31]
 1H NMR (C6D6, 400 MHz): δ=7.34 (d, 3
J
HH=6.47 Hz, 2 H, o‐Ar−H), 7.15–7.12 (m, 2 H, m‐Ar−H), 7.04 (t, 3
J
HH=7.34 Hz, 1 H, p‐Ar−H), 5.38 (q, 3
J
HH=6.46 Hz, 1 H, PhCHCH3), 1.43 (d, 3
J
HH=6.47 Hz, 3 H, PhCHCH3), 1.02 (s, 6 H, Bpin−CH
3), 0.99 ppm (s, 6 H, Bpin‐CH
3); 13C{1H} NMR (C6D6, 100 MHz): δ=145.4 (Ar−C
q), 128.5 (Ar−CH), 127.4 (Ar−CH), 125.7 (Ar−CH), 82.5 (Bpin−C), 72.9 (OCHCH3Ph), 25.8 (PhCHCH3), 24.7 ppm (Bpin−CH3); 11B NMR (C6D6, 128 MHz): δ=22.6 ppm.
1H NMR (C6D6, 400 MHz): δ=7.34 (d, 3
J
HH=6.47 Hz, 2 H, o‐Ar−H), 7.15–7.12 (m, 2 H, m‐Ar−H), 7.04 (t, 3
J
HH=7.34 Hz, 1 H, p‐Ar−H), 5.38 (q, 3
J
HH=6.46 Hz, 1 H, PhCHCH3), 1.43 (d, 3
J
HH=6.47 Hz, 3 H, PhCHCH3), 1.02 (s, 6 H, Bpin−CH
3), 0.99 ppm (s, 6 H, Bpin‐CH
3); 13C{1H} NMR (C6D6, 100 MHz): δ=145.4 (Ar−C
q), 128.5 (Ar−CH), 127.4 (Ar−CH), 125.7 (Ar−CH), 82.5 (Bpin−C), 72.9 (OCHCH3Ph), 25.8 (PhCHCH3), 24.7 ppm (Bpin−CH3); 11B NMR (C6D6, 128 MHz): δ=22.6 ppm.
(4‐NO2Ph)(Me)CHOBpin [31]
 1H NMR (C6D6, 400 MHz): δ=7.83 (d, 3
J
HH=8.78 Hz, 2 H, o‐Ar−H), 7.05 (d, 3
J
HH=8.52 Hz, 2 H, m‐Ar−H), 5.19 (q, 3
J
HH=6.48 Hz, 1 H, OCHCH3), 1.27 (d, 3
J
HH=6.51 Hz, 3 H, OCHCH
3), 1.05 (s, 6 H, Bpin‐CH
3), 1.02 ppm (s, 6 H, Bpin−CH
3); 13C{1H} NMR (C6D6, 100 MHz): δ=152.1 (Ar−C
q), 147.4 (Ar−CH), 126.2 (Ar−CH), 123.6 (Ar−CH), 83.0 (Bpin−C), 71.9 (OCHCH3), 25.3 (OCHCH3), 24.6 ppm (Bpin−CH3); 11B NMR (C6D6, 128 MHz): δ=22.4 ppm.
1H NMR (C6D6, 400 MHz): δ=7.83 (d, 3
J
HH=8.78 Hz, 2 H, o‐Ar−H), 7.05 (d, 3
J
HH=8.52 Hz, 2 H, m‐Ar−H), 5.19 (q, 3
J
HH=6.48 Hz, 1 H, OCHCH3), 1.27 (d, 3
J
HH=6.51 Hz, 3 H, OCHCH
3), 1.05 (s, 6 H, Bpin‐CH
3), 1.02 ppm (s, 6 H, Bpin−CH
3); 13C{1H} NMR (C6D6, 100 MHz): δ=152.1 (Ar−C
q), 147.4 (Ar−CH), 126.2 (Ar−CH), 123.6 (Ar−CH), 83.0 (Bpin−C), 71.9 (OCHCH3), 25.3 (OCHCH3), 24.6 ppm (Bpin−CH3); 11B NMR (C6D6, 128 MHz): δ=22.4 ppm.
(4‐BrPh)(Me)CHOBpin [32]
 1H NMR (C6D6, 400 MHz): δ=7.23 (d, 3
J
HH=8.46 Hz, 2 H, o‐Ar−H), 7.00 (d, 3
J
HH=8.32 Hz, 2 H, m‐Ar−H), 5.20 (q, 3
J
HH=6.44 Hz, 1 H, OCHCH3), 1.32 (d, 3
J
HH=6.47 Hz, 3 H, OCHCH
3), 1.02 (s, 6 H, Bpin−CH
3), 0.99 ppm (s, 6 H, Bpin−CH
3); 13C{1H} NMR (C6D6, 100 MHz): δ=144.3 (Ar−C
q), 131.6 (Ar−CH), 127.5 (Ar−CH), 121.2 (Ar−CH), 82.7 (Bpin−C), 72.2 (OCHCH3), 25.5 (OCHCH3), 24.7 ppm (Bpin−CH3); 11B NMR (C6D6, 128 MHz): δ=22.4 ppm.
1H NMR (C6D6, 400 MHz): δ=7.23 (d, 3
J
HH=8.46 Hz, 2 H, o‐Ar−H), 7.00 (d, 3
J
HH=8.32 Hz, 2 H, m‐Ar−H), 5.20 (q, 3
J
HH=6.44 Hz, 1 H, OCHCH3), 1.32 (d, 3
J
HH=6.47 Hz, 3 H, OCHCH
3), 1.02 (s, 6 H, Bpin−CH
3), 0.99 ppm (s, 6 H, Bpin−CH
3); 13C{1H} NMR (C6D6, 100 MHz): δ=144.3 (Ar−C
q), 131.6 (Ar−CH), 127.5 (Ar−CH), 121.2 (Ar−CH), 82.7 (Bpin−C), 72.2 (OCHCH3), 25.5 (OCHCH3), 24.7 ppm (Bpin−CH3); 11B NMR (C6D6, 128 MHz): δ=22.4 ppm.
(4‐MePh)(Me)CHOBpin [33]
 1H NMR (C6D6, 400 MHz): δ=7.29 (d, 3
J
HH=8.07 Hz, 2 H, o‐Ar−H), 6.97 (d, 3
J
HH=7.83 Hz, 2 H, m‐Ar−H), 5.41 (q, 3
J
HH=6.44 Hz, 1 H, OCHCH3), 2.09 (s, 3 H, PhCH
3), 1.47 (d, 3JHH=6.46 Hz, 3 H, OCHCH
3), 1.03 (s, 6 H, Bpin−CH
3), 1.01 ppm (s, 6 H, Bpin−CH
3); 13C{1H} NMR (C6D6, 100 MHz): δ=142.5 (Ar−C
q), 136.6 (Ar−CH), 129.2 (Ar−CH), 125.7 (Ar−CH), 82.5 (Bpin−C), 72.8 (OCHCH3), 25.8 (OCHCH3), 24.7 (Bpin−CH3), 21.1 ppm (PhCH3); 11B NMR (C6D6, 128 MHz): δ=22.5 ppm.
1H NMR (C6D6, 400 MHz): δ=7.29 (d, 3
J
HH=8.07 Hz, 2 H, o‐Ar−H), 6.97 (d, 3
J
HH=7.83 Hz, 2 H, m‐Ar−H), 5.41 (q, 3
J
HH=6.44 Hz, 1 H, OCHCH3), 2.09 (s, 3 H, PhCH
3), 1.47 (d, 3JHH=6.46 Hz, 3 H, OCHCH
3), 1.03 (s, 6 H, Bpin−CH
3), 1.01 ppm (s, 6 H, Bpin−CH
3); 13C{1H} NMR (C6D6, 100 MHz): δ=142.5 (Ar−C
q), 136.6 (Ar−CH), 129.2 (Ar−CH), 125.7 (Ar−CH), 82.5 (Bpin−C), 72.8 (OCHCH3), 25.8 (OCHCH3), 24.7 (Bpin−CH3), 21.1 ppm (PhCH3); 11B NMR (C6D6, 128 MHz): δ=22.5 ppm.
(4‐NH2Ph)(Me)CHOBpin [34]
 1H NMR (C6D6, 400 MHz): δ=7.19 (d, 3
J
HH=8.20 Hz, 2 H, o‐Ar−H), 6.36 (d, 3
J
HH=8.23 Hz, 2 H, m‐Ar−H), 5.35 (q, 3
J
HH=6.31 Hz, 1 H, PhCHCH3), 3.05 (s, 2 H, PhNH
2), 1.47 (d, 3JHH=6.40 Hz, 3 H, OCHCH3), 1.03 (s, 6 H, Bpin−CH
3), 1.01 ppm (s, 6 H, Bpin−CH
3); 13C{1H} NMR (C6D6, 100 MHz): δ=146.6 (Ar−C
q), 134.6 (Ar−CH), 126.9 (Ar−CH), 114.9 (Ar−CH), 82.4 (Bpin−C), 72.9 (OCHCH3), 25.6 (OCHCH3), 24.7 ppm (Bpin−CH3); 11B NMR (C6D6, 128 MHz): δ=22.6 ppm.
1H NMR (C6D6, 400 MHz): δ=7.19 (d, 3
J
HH=8.20 Hz, 2 H, o‐Ar−H), 6.36 (d, 3
J
HH=8.23 Hz, 2 H, m‐Ar−H), 5.35 (q, 3
J
HH=6.31 Hz, 1 H, PhCHCH3), 3.05 (s, 2 H, PhNH
2), 1.47 (d, 3JHH=6.40 Hz, 3 H, OCHCH3), 1.03 (s, 6 H, Bpin−CH
3), 1.01 ppm (s, 6 H, Bpin−CH
3); 13C{1H} NMR (C6D6, 100 MHz): δ=146.6 (Ar−C
q), 134.6 (Ar−CH), 126.9 (Ar−CH), 114.9 (Ar−CH), 82.4 (Bpin−C), 72.9 (OCHCH3), 25.6 (OCHCH3), 24.7 ppm (Bpin−CH3); 11B NMR (C6D6, 128 MHz): δ=22.6 ppm.
(4‐OCH3Ph)(Me)CHOBpin [32]
 1H NMR (C6D6, 400 MHz): δ=7.27 (d, 3
J
HH=8.38 Hz, 2 H, o‐Ar−H), 6.75 (d, 3
J
HH=8.77 Hz, 2 H, m‐Ar−H), 5.36 (q, 3
J
HH=6.42 Hz, 1 H, PhCHCH3), 3.33 (s, 3 H, PhOCH
3), 1.46 (d, 3JHH=6.44 Hz, 3 H, OCHCH3), 1.04 (s, 6 H, Bpin−CH
3), 1.02 ppm (s, 6 H, Bpin−CH
3); 13C{1H} NMR (C6D6, 100 MHz): δ=159.4 (Ar−C
q), 137.5 (Ar−CH), 127.0 (Ar−CH), 114.0 (Ar−CH), 82.5 (Bpin−C), 72.6 (OCHCH3Ph), 54.8 (PhOCH3), 25.7 (OCHCH3), 25.0 ppm (Bpin−CH3); 11B NMR (C6D6, 128 MHz): δ=22.5 ppm.
1H NMR (C6D6, 400 MHz): δ=7.27 (d, 3
J
HH=8.38 Hz, 2 H, o‐Ar−H), 6.75 (d, 3
J
HH=8.77 Hz, 2 H, m‐Ar−H), 5.36 (q, 3
J
HH=6.42 Hz, 1 H, PhCHCH3), 3.33 (s, 3 H, PhOCH
3), 1.46 (d, 3JHH=6.44 Hz, 3 H, OCHCH3), 1.04 (s, 6 H, Bpin−CH
3), 1.02 ppm (s, 6 H, Bpin−CH
3); 13C{1H} NMR (C6D6, 100 MHz): δ=159.4 (Ar−C
q), 137.5 (Ar−CH), 127.0 (Ar−CH), 114.0 (Ar−CH), 82.5 (Bpin−C), 72.6 (OCHCH3Ph), 54.8 (PhOCH3), 25.7 (OCHCH3), 25.0 ppm (Bpin−CH3); 11B NMR (C6D6, 128 MHz): δ=22.5 ppm.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Financial support from KIT is gratefully acknowledged. The authors acknowledge computational support by the state of Baden‐Württemberg through bwHPC and the German Research Foundation (DFG) through grant no INST 40/467‐1 FUGG. Open access funding enabled and organized by Projekt DEAL.
B. Goswami, T. J. Feuerstein, R. Yadav, R. Köppe, S. Lebedkin, M. M. Kappes, P. W. Roesky, Chem. Eur. J. 2021, 27, 4401.
Dedicated to Prof. Hansgeorg Schnöckel on the occasion of his 80th birthday.
References
- 1. Campagna S., Photochemistry and Photophysics of Coordination Compounds I, Vol. 280, Springer, Berlin Heidelberg, 2007. [Google Scholar]
- 2.
- 2a. Yam V. W.-W., Lo K. K.-W., Chem. Soc. Rev. 1999, 28, 323–334; [Google Scholar]
- 2b. Yersin H., Rausch A. F., Czerwieniec R., Hofbeck T., Fischer T., Coord. Chem. Rev. 2011, 255, 2622–2652. [Google Scholar]
- 3.
- 3a. Larsen C. B., Wenger O. S., Chem. Eur. J. 2018, 24, 2039–2058; [DOI] [PubMed] [Google Scholar]
- 3b. Bizzarri C., Spuling E., Knoll D. M., Volz D., Bräse S., Coord. Chem. Rev. 2018, 373, 49–82; [Google Scholar]
- 3c. Büldt L. A., Wenger O. S., Angew. Chem. Int. Ed. 2017, 56, 5676–5682; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 5770–5776; [Google Scholar]
- 3d. Büldt L. A., Wenger O. S., Dalton Trans. 2017, 46, 15175–15177. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Liao S.-H., Shiu J.-R., Liu S.-W., Yeh S.-J., Chen Y.-H., Chen C.-T., Chow T. J., Wu C.-I., J. Am. Chem. Soc. 2009, 131, 763–777; [DOI] [PubMed] [Google Scholar]
- 4b. Chen C. H., Shi J., Coord. Chem. Rev. 1998, 171, 161–174. [Google Scholar]
- 5.
- 5a. Bestgen S., Schoo C., Neumeier B. L., Feuerstein T. J., Zovko C., Köppe R., Feldmann C., Roesky P. W., Angew. Chem. Int. Ed. 2018, 57, 14265–14269; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 14461–14465; [Google Scholar]
- 5b. Feuerstein T. J., Goswami B., Rauthe P., Köppe R., Lebedkin S., Kappes M. M., Roesky P. W., Chem. Sci. 2019, 10, 4742–4749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Goswami B., Yadav R., Schoo C., Roesky P. W., Dalton Trans. 2020, 49, 675–681. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Fleischer R., Stalke D., Inorg. Chem. 1997, 36, 2413–2419; [DOI] [PubMed] [Google Scholar]
- 7b. Stasch A., Angew. Chem. Int. Ed. 2014, 53, 10200–10203; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 10364–10367; [Google Scholar]
- 7c. Wingerter S., Pfeiffer M., Murso A., Lustig C., Stey T., Chandrasekhar V., Stalke D., J. Am. Chem. Soc. 2001, 123, 1381–1388. [Google Scholar]
- 8. Li N., Guan B.-T., Adv. Synth. Catal. 2017, 359, 3526–3531. [Google Scholar]
- 9.
- 9a. Cotton F. A., Wilkinson G., Gaus P. L., Basic Inorganic Chemistry, 3rd Ed. ed., Wiley, 1995; [Google Scholar]
- 9b. Hill M. S., Liptrot D. J., Weetman C., Chem. Soc. Rev. 2016, 45, 972–988; [DOI] [PubMed] [Google Scholar]
- 9c. Arrowsmith M., Hill M. S., Comprehensive Inorganic Chemistry II, (Second Edition) (Eds.: Reedijk J., Poeppelmeier K.), Elsevier, Amsterdam, 2013, pp. 1189–1216; [Google Scholar]
- 9d. Anker M. D., Hill M. S. in Encyclopedia of Inorganic and Bioinorganic Chemistry, Wiley, Chichester, 2017, pp. 1–23. [Google Scholar]
- 10.
- 10a. Crimmin M. R., Barrett A. G. M., Hill M. S., Procopiou P. A., Org. Lett. 2007, 9, 331–333; [DOI] [PubMed] [Google Scholar]
- 10b. Barman M. K., Baishya A., Nembenna S., J. Organomet. Chem. 2015, 785, 52–60; [Google Scholar]
- 10c. Day B. M., Mansfield N. E., Coles M. P., Hitchcock P. B., Chem. Commun. 2011, 47, 4995–4997; [DOI] [PubMed] [Google Scholar]
- 10d. Day B. M., Knowelden W., Coles M. P., Dalton Trans. 2012, 41, 10930–10933; [DOI] [PubMed] [Google Scholar]
- 10e. Cronin L., Manoni F., O′Connor C. J., Connon S. J., Angew. Chem. Int. Ed. 2010, 49, 3045–3048; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 3109–3112. [Google Scholar]
- 11.
- 11a. Chamberlain B. M., Cheng M., Moore D. R., Ovitt T. M., Lobkovsky E. B., Coates G. W., J. Am. Chem. Soc. 2001, 123, 3229–3238; [DOI] [PubMed] [Google Scholar]
- 11b. Yi W., Ma H., Dalton Trans. 2014, 43, 5200–5210; [DOI] [PubMed] [Google Scholar]
- 11c. Wang Y., Zhao W., Liu X., Cui D., Chen E. Y. X., Macromolecules 2012, 45, 6957–6965; [Google Scholar]
- 11d. Chisholm M. H., Gallucci J. C., Phomphrai K., Inorg. Chem. 2004, 43, 6717–6725; [DOI] [PubMed] [Google Scholar]
- 11e. Shueh M.-L., Wang Y.-S., Huang B.-H., Kuo C.-Y., Lin C.-C., Macromolecules 2004, 37, 5155–5162; [Google Scholar]
- 11f. Tsai Y.-H., Lin C.-H., Lin C.-C., Ko B.-T., J. Polym. Sci. Part A 2009, 47, 4927–4936; [Google Scholar]
- 11g. Cushion M. G., Mountford P., Chem. Commun. 2011, 47, 2276–2278; [DOI] [PubMed] [Google Scholar]
- 11h. Sarazin Y., Poirier V., Roisnel T., Carpentier J.-F., Eur. J. Inorg. Chem. 2010, 3423–3428; [Google Scholar]
- 11i. Zhong Z., Dijkstra P. J., Birg C., Westerhausen M., Feijen J., Macromolecules 2001, 34, 3863–3868. [Google Scholar]
- 12.
- 12a. Arrowsmith M., Hill M. S., Kociok-Köhn G., Chem. Eur. J. 2013, 19, 2776–2783; [DOI] [PubMed] [Google Scholar]
- 12b. Arrowsmith M., Hadlington T. J., Hill M. S., Kociok-Köhn G., Chem. Commun. 2012, 48, 4567–4569; [DOI] [PubMed] [Google Scholar]
- 12c. Arrowsmith M., Hill M. S., Hadlington T., Kociok-Köhn G., Weetman C., Organometallics 2011, 30, 5556–5559; [Google Scholar]
- 12d. Yadav S., Dixit R., Bisai M. K., Vanka K., Sen S. S., Organometallics 2018, 37, 4576–4584; [Google Scholar]
- 12e. Mukherjee D., Ellern A., Sadow A. D., Chem. Sci. 2014, 5, 959–964; [Google Scholar]
- 12f. Yadav S., Pahar S., Sen S. S., Chem. Commun. 2017, 53, 4562–4564; [DOI] [PubMed] [Google Scholar]
- 12g. Li J., Luo M., Sheng X., Hua H., Yao W., Pullarkat S. A., Xu L., Ma M., Org. Chem. Front. 2018, 5, 3538–3547; [Google Scholar]
- 12h. Luo M., Li J., Xiao Q., Yang S., Su F., Ma M., J. Organomet. Chem. 2018, 868, 31–35. [Google Scholar]
- 13.
- 13a. Crimmin M. R., Casely I. J., Hill M. S., J. Am. Chem. Soc. 2005, 127, 2042–2043; [DOI] [PubMed] [Google Scholar]
- 13b. Crimmin M. R., Arrowsmith M., Barrett A. G. M., Casely I. J., Hill M. S., Procopiou P. A., J. Am. Chem. Soc. 2009, 131, 9670–9685; [DOI] [PubMed] [Google Scholar]
- 13c. Arrowsmith M., Crimmin M. R., Barrett A. G. M., Hill M. S., Kociok-Köhn G., Procopiou P. A., Organometallics 2011, 30, 1493–1506; [Google Scholar]
- 13d. Zhang X., Emge T. J., Hultzsch K. C., Angew. Chem. Int. Ed. 2012, 51, 394–398; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 406–410. [Google Scholar]
- 14.
- 14a. Hu H., Cui C., Organometallics 2012, 31, 1208–1211; [Google Scholar]
- 14b. He M., Gamer M. T., Roesky P. W., Organometallics 2016, 35, 2638–2644; [Google Scholar]
- 14c. Liu B., Roisnel T., Carpentier J.-F., Sarazin Y., Chem. Eur. J. 2013, 19, 13445–13462; [DOI] [PubMed] [Google Scholar]
- 14d. Selikhov A. N., Plankin G. S., Cherkasov A. V., Shavyrin A. S., Louyriac E., Maron L., Trifonov A. A., Inorg. Chem. 2019, 58, 5325–5334. [DOI] [PubMed] [Google Scholar]
- 15.
- 15a. Schwamm R. J., Coles M. P., Organometallics 2013, 32, 5277–5280; [Google Scholar]
- 15b. Barrett A. G. M., Crimmin M. R., Hill M. S., Hitchcock P. B., Lomas S. L., Mahon M. F., Procopiou P. A., Suntharalingam K., Organometallics 2008, 27, 6300–6306; [Google Scholar]
- 15c. Schwamm R. J., Day B. M., Mansfield N. E., Knowelden W., Hitchcock P. B., Coles M. P., Dalton Trans. 2014, 43, 14302–14314; [DOI] [PubMed] [Google Scholar]
- 15d. Arrowsmith M., Crimmin M. R., Hill M. S., Lomas S. L., Heng M. S., Hitchcock P. B., Kociok-Köhn G., Dalton Trans. 2014, 43, 14249–14256. [DOI] [PubMed] [Google Scholar]
- 16. Dunne J. F., Neal S. R., Engelkemier J., Ellern A., Sadow A. D., J. Am. Chem. Soc. 2011, 133, 16782–16785. [DOI] [PubMed] [Google Scholar]
- 17.
- 17a. Ma M., Li J., Shen X., Yu Z., Yao W., Pullarkat S. A., RSC Adv. 2017, 7, 45401–45407; [Google Scholar]
- 17b. Fohlmeister L., Stasch A., Chem. Eur. J. 2016, 22, 10235–10246; [DOI] [PubMed] [Google Scholar]
- 17c. Manna K., Ji P., Greene F. X., Lin W., J. Am. Chem. Soc. 2016, 138, 7488–7491. [DOI] [PubMed] [Google Scholar]
- 18. Spencer L. P., Altwer R., Wei P., Gelmini L., Gauld J., Stephan D. W., Organometallics 2003, 22, 3841–3854. [Google Scholar]
- 19. Kamalesh Babu R. P., Krishnamurthy S. S., Nethaji M., Tetrahedron: Asymmetry 1995, 6, 427–438. [Google Scholar]
- 20. Stasch A., Chem. Eur. J. 2012, 18, 15105–15112. [DOI] [PubMed] [Google Scholar]
- 21. Ceder R. M., García C., Grabulosa A., Karipcin F., Muller G., Rocamora M., Font-Bardía M., Solans X., J. Organomet. Chem. 2007, 692, 4005–4019. [Google Scholar]
- 22.
- 22a. Barrett A. G. M., Crimmin M. R., Hill M. S., Hitchcock P. B., Procopiou P. A., Dalton Trans. 2008, 4474–4481; [DOI] [PubMed] [Google Scholar]
- 22b. Causero A., Ballmann G., Pahl J., Färber C., Intemann J., Harder S., Dalton Trans. 2017, 46, 1822–1831. [DOI] [PubMed] [Google Scholar]
- 23.
- 23a. Czerwieniec R., Yu J., Yersin H., Inorg. Chem. 2011, 50, 8293–8301; [DOI] [PubMed] [Google Scholar]
- 23b. Deaton J. C., Switalski S. C., Kondakov D. Y., Young R. H., Pawlik T. D., Giesen D. J., Harkins S. B., Miller A. J. M., Mickenberg S. F., Peters J. C., J. Am. Chem. Soc. 2010, 132, 9499–9508; [DOI] [PubMed] [Google Scholar]
- 23c. Yersin H., Czerwieniec R., Shafikov M. Z., Suleymanova A. F., in Highly Efficient OLEDs: Materials Based on Thermally Activated Delayed Fluorescence (Ed.: Yersin H.), Wiley-VCH, Weinheim, 2019, Chapter 1, pp. 1–60.. [Google Scholar]
- 24.
- 24a. Chong C. C., Kinjo R., ACS Catal. 2015, 5, 3238–3259; [Google Scholar]
- 24b. Shegavi M. L., Bose S. K., Catal. Sci. Technol. 2019, 9, 3307–3336. [Google Scholar]
- 25.
- 25a. Ahlrichs R., Bär M., Häser M., Horn H., Kölmel C., Chem. Phys. Lett. 1989, 162, 165–169; [Google Scholar]
- 25b. Perdew J. P., Phys. Rev. B 1986, 34, 7406–7406; [PubMed] [Google Scholar]
- 25c. Becke A. D., Phys. Rev. A 1988, 38, 3098–3100; [DOI] [PubMed] [Google Scholar]
- 25d. Perdew J. P., Phys. Rev. B 1986, 33, 8822–8824; [DOI] [PubMed] [Google Scholar]
- 25e. Sierka M., Hogekamp A., Ahlrichs R., J. Chem. Phys. 2003, 118, 9136–9148. [Google Scholar]
- 26. Weigend F., Ahlrichs R., Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [DOI] [PubMed] [Google Scholar]
- 27. Deglmann P., May K., Furche F., Ahlrichs R., Chem. Phys. Lett. 2004, 384, 103–107. [Google Scholar]
- 28. Rappoport D., Furche F., J. Chem. Phys. 2007, 126, 201104. [DOI] [PubMed] [Google Scholar]
- 29. Troyer T. L., Muchalski H., Hong K. B., Johnston J. N., Org. Lett. 2011, 13, 1790–1792. [DOI] [PubMed] [Google Scholar]
- 30. Panda T. K., Hrib C. G., Jones P. G., Jenter J., Roesky P. W., Tamm M., Eur. J. Inorg. Chem. 2008, 4270–4279. [Google Scholar]
- 31. Eedugurala N., Wang Z., Chaudhary U., Nelson N., Kandel K., Kobayashi T., Slowing I. I., Pruski M., Sadow A. D., ACS Catal. 2015, 5, 7399–7414. [Google Scholar]
- 32. Bagherzadeh S., Mankad N. P., Chem. Commun. 2016, 52, 3844–3846. [DOI] [PubMed] [Google Scholar]
- 33. Zhu Z., Wu X., Xu X., Wu Z., Xue M., Yao Y., Shen Q., Bao X., J. Org. Chem. 2018, 83, 10677–10683. [DOI] [PubMed] [Google Scholar]
- 34. Kumar G. S., Harinath A., Narvariya R., Panda T. K., Eur. J. Inorg. Chem. 2020, 467–474. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary