

Abstract
The PtII linker [ethylenediamineplatinum(II)]2+, coined Lx, has emerged as a novel non‐conventional approach to antibody–drug conjugates (ADCs) and has shown its potential in preclinical in vitro and in vivo benchmark studies. A crucial improvement of the Lx conjugation reaction from initially <15 % to ca. 75–90 % conjugation efficiency is described, resulting from a systematic screening of all relevant reaction parameters. NaI, a strikingly simple inorganic salt additive, greatly improves the conjugation efficiency as well as the conjugation selectivity simply by exchanging the leaving chloride ligand on Cl‐Lx‐drug complexes (which are direct precursors for Lx‐ADCs) for iodide, thus generating I‐Lx‐drug complexes as more reactive species. Using this iodide effect, we developed a general and highly practical conjugation procedure that is scalable: our lead Lx‐ADC was produced on a 5 g scale with an outstanding conjugation efficiency of 89 %.
Keywords: antibody–drug conjugates, conjugation, linkers, Lx, platinum
PtII complexes containing a payload, a bidentate ligand, and a halido leaving ligand (Cl−, Br− or I−) can be conjugated to native monoclonal antibodies in a very efficient way by using iodide salts, such as NaI, as simple inorganic additives to the aqueous conjugation mixtures. This method works reliably for various mAbs, consistently giving conjugation efficiencies in the range of 75–90 %.
Introduction
Antibody–drug conjugates (ADCs) are Trojan horses of medicinal chemistry: they are a targeted approach to treat cancer allowing selective delivery of effector molecules (payloads), such as cytotoxic or immune‐modulating therapeutics, to malignant cells, thereby sparing healthy cells. The advancements in the ADC field and the role of organic chemistry in ADC development were recently highlighted in a review. [1] Currently, nine ADCs have been approved by the Food and Drug Administration (FDA): Adcetris, Kadcyla, Mylotarg, Besponsa, Polivy, Padcev, Enhertu, Trodelvy, and Blenrep (the last two approved in 2020), with >80 ADCs currently in active clinical evaluation. [2a] ADCs are utmost complex to develop[ 2b , 2c , 2d ] and comprise three components: a disease‐selective monoclonal antibody (mAb), a small‐molecule therapeutic payload (drug), and a linker which connects the two parts. Linkers are the most modifiable part of an ADC and often form a critical factor in the efficiency and costs of the production process, as well as in the therapeutic efficacy and tolerability of an ADC.[ 3a , 3b , 3c ]
To attach a drug‐linker moiety to a mAb, a bioconjugation procedure is applied. Generally, the conjugation step is key for every ADC technology, since it not only dictates the nature and characteristics of the bioconjugate, but also the efficiency of the conjugation, that is, the percentage of the offered drug‐linker moiety that is effectively coupled to the antibody. An efficient conjugation reaction will strongly contribute to the ultimate success of any ADC.
Typical conjugation approaches are the stochastic (random) conjugation to lysine (Lys) residues and the conjugation to reduced cysteine (Cys) residues in the hinge region of the mAb, along with a variety of more recently developed techniques. [4] The site‐specific conjugation [5] was clearly a major trend during the last years; nevertheless, a recent publication showed that heterogeneous stochastic conjugation can be beneficial over homogeneous site‐specific conjugation. [5a] However, even modern site‐specific conjugation procedures using, for example, genetically engineered amino acids or enzymatic approaches [6] mainly focus on the efficacy of the resulting ADCs or improvement of their pharmacokinetics/pharmacodynamics. Surprisingly, little attention is paid to the overall conjugation efficiency and manufacturability of the developed procedures. Therefore, desired conversions are typically achieved by addition of a large excess of a drug‐linker reagent, just to ensure a complete conjugation. Even a recent paper describing manufacturing of 1.7 g of an ADC under good manufacturing practice (GMP) using a site‐specific technology still uses as much as 10 equiv of the payload‐spacer moiety to obtain an average drug‐to‐antibody ratio (DAR) of 1.6, which corresponds to a 16 % conjugation efficiency only. [7] Thus, currently there are clear unmet needs in the development and manufacturing of ADCs.
Some years ago, we introduced a novel approach to an improved ADC linker design by developing a cationic metal‐organic PtII‐based linker, [ethylenediamineplatinum(II)]2+, which we coined Lx.[ 8a , 8b , 8c , 8d ] The Lx conjugation is a stochastic conjugation technology, which (uniquely among ADC conjugation methods) addresses histidine (His) residues of native unmodified antibodies for a stable drug‐linker attachment. This straightforward ADC linker technology might be a valuable addition to the field of next‐generation ADCs based on its in vitro and in vivo performance, as summarized in our recent technology review. [8c]
The Lx‐technology is based on the remarkable stability of PtII–N coordination compounds [9] and a high affinity of the PtII center towards N‐ and S‐donor biomolecules, [10] including proteins. [11]
The concept consists of two key steps: “complexation” and conjugation (Scheme 1 depicts details of the initial conjugation conditions and Scheme 4, left, shows the general Lx‐approach, as it was initially described [8a] ).
Scheme 1.
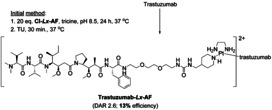
The initial conditions of the Lx conjugation. The drug–Lx part of the ADC is taken into square brackets, showing its corresponding pH independent charge on PtII.
Scheme 4.
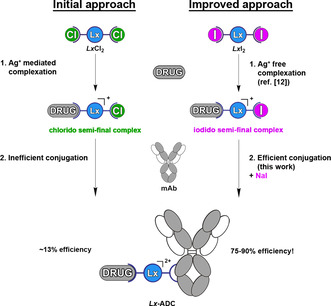
The initial Lx conjugation approach to ADCs with its two crucial steps: complexation and conjugation (left) and the advanced Lx technology, with both key steps greatly improved by use of iodide as a ligand and as a salt additive (right).
In the first step (complexation), the commercially available precursor complex LxCl2 bearing two cis‐oriented chlorido leaving ligands can be coordinated to payloads/drugs bearing a suitable coordination group (CG). Typically, CG is an N‐heterocycle, such as piperidine, which provides a stable bond to Lx; all constructs used in this work utilize piperidine as a CG. As a result of the complexation step, stable intermediate products, which we coined semi‐final complexes, are obtained comprising the payload and one chlorido leaving ligand. These semi‐final products contain a positively charged PtII center, which increases the water solubility of drug‐linker moieties compared to analogous non‐Lx containing constructs. Recently, we described an improvement of the complexation reaction and the development of a highly efficient, versatile, and green approach to iodido semi‐final complexes, [12] which unexpectedly turned out to be beneficial for the Lx conjugation, as will be outlined below. Both chlorido and iodido semi‐final compounds can be stored at low temperatures for >1 year without decomposition.
In the second step (conjugation), the obtained semi‐final complexes are conjugated to the imidazole moiety of His residues of unmodified mAbs, taking advantage of a high intrinsic preference of the PtII center to such N‐donor ligands. [13] In principle, besides His, PtII is able to coordinate to reduced Cys (which are not present in native mAbs) and to methionine (Met) residues of proteins.[ 10b , 14 , 28 ] Therefore, we introduced a thiourea (TU) quenching step [15] to ensure stability and reproducibility of the obtained Lx conjugates. Owing to a high affinity of TU (which was even suggested as a rescue agent in cisplatin therapy) [10b] to PtII, it removes the kinetically formed but thermodynamically labile Met‐bound Lx‐payload complexes, leaving only the thermodynamically more stable and kinetically inert His‐bound Lx‐payload complexes intact. [16]
The bottleneck of our initial approach (Scheme 1; Supporting Information, Section 2) was the low conjugation efficiency of ca. 13 %. To obtain an average DAR of about 2.5, which is the desired DAR for our proposed clinical lead candidate ADC trastuzumab‐Lx‐AF, [8a] as much as 20 equiv of the linker‐payload semi‐final complex Cl‐Lx‐AF were required, meaning that ca. 87 % of this precious compound was wasted. Such a poor conjugation efficiency is clearly unacceptable for any clinical application.
We therefore set the goal to develop a general and efficient conjugation procedure which could be easily and reproducibly applied on a large scale.
Results and Discussion
Herein we describe a comprehensive optimization study of the Lx conjugation procedure and important insights and findings that were made. The validation of the newly developed method, the multigram scale synthesis of a lead Lx‐ADC, and the advanced Lx‐technology are described below. The full description of all experiments is presented in the Supporting Information, Sections 3–7.
To substantially improve the Lx conjugation procedure, we performed a broad and detailed optimization of all relevant reaction parameters. For this optimization work, we choose a Desferal (DFO) based model semi‐final complex 1 a (Figure 1) and investigated its conjugation reaction to trastuzumab (Herceptin).
Figure 1.
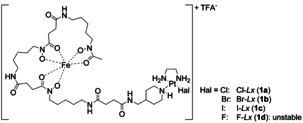
Structures of the Hal‐Lx‐DFO‐FeIII (Hal‐Lx) semi‐final complexes 1 a–c used as substrates for the current optimization study. Hal=halide.
The model compound 1 a contains a DFO–FeIII complex, coordinated to Lx via a piperidine coordination group, the same CG as used in our clinical lead candidate. [8a] It has several important advantages that make it a valuable model compound for the intended optimization study (Supporting Information, Section 3.1). For the method of determination of the conjugation efficiency used for this optimization study, see the Supporting Information, Section 3.2 and Figure S1.
First, a broad screening of different buffers in the pH range of 2.7–10.4 (Supporting Information, Section 3.3) revealed the optimal buffer, 20 mM 2‐[4‐(2‐hydroxyethyl)piperazin‐1‐yl] ethanesulfonic acid (HEPES) of pH 8.1, which provided a minor increase in the conjugation efficiency (from ca. 15 % to ca. 21 %; Supporting Information, Figure S3) compared to the previously used tricine buffer of pH 8.5. [8a]
In the next step, we investigated the influence of inorganic salts on the Lx conjugation efficiency, since they can interact both with a protein and with a semi‐final complex 1. Assuming that anions would have a stronger effect than cations, we performed a broad screening of sodium salts (Figure 2; Supporting Information, Section 3.5.1).
Figure 2.
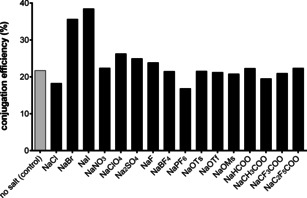
Conjugation efficiency in the presence of various sodium salt additives (30 mM). Screening of anions. Ts=tosyl, Tf=triflyl, Ms=mesyl.
Confirming an early observation, [16] it was found that NaClO4 and Na2SO4 showed a minor increase in the conjugation efficiency, likely explainable by their interaction with the mAb. [17] However, most spectacular results were observed when NaI and (to a lesser extent) NaBr were added. Addition of NaI resulted in a conjugation efficiency of ca. 38 % (ca. 36 % when NaBr was used), which was significantly higher compared to the conjugation without addition of a salt (ca. 21 %) or using the negative control salt NaIO3 (ca. 22 %) that also contains iodine, but cannot release the iodide anion. In this case, the interaction of the salt with the protein is of minor importance and this outcome can be rationalized by an interaction of the salt (NaI or NaBr) with the PtII complex 1 a. Most likely, the enhancement happened through an intermediate formation of the bromido and iodido semi‐final complexes 1 b and 1 c (Figure 1), respectively, from the parent chlorido semi‐final complex 1 a, which was later shown to be a rapid process (Supporting Information, Section 3.7). Interestingly, a diiodido PtII carbohydrate complex was described as less reactive towards a model N‐nucleophile compared to its dichlorido counterpart. [18] Later, slower binding of the diiodido analog of cisplatin, cis‐PtI2(NH3)2, to DNA was confirmed and was explained by its slower hydrolysis. [19] Thus, previous reports suggested that an I–Lx complex should be less reactive towards N‐nucleophiles than a Cl–Lx complex, but we observed exactly the opposite. Therefore, our current finding of a faster reaction of I–PtII complexes with biomolecules compared to their chlorido counterparts is non‐trivial and counterintuitive and remains unprecedented.
Generally, after an initial finding that cis‐PtI2(NH3)2 was less anticancer‐active compared with cisplatin, [20] the I–PtII complexes, which were since then stigmatized to be inert and unreactive, [21] remained in the sleeping beauty slumber for successive four decades, being totally neglected and overlooked by medicinal chemists. Nevertheless, they remained to be extensively utilized as very useful synthetic precursors (by Ag+ mediated ligand exchange) for numerous cisplatin analogues, for example, in the well‐known Dhara's method for the synthesis of cisplatin [22a] and its modified procedures.[ 22b , 22c ] However, I‐PtII complexes are now experiencing their renaissance [23] following the recognition of their intriguing properties, so that we will probably see a lot of interesting novel anti‐cancer compounds of this class in the near future.
After successful screening of anions, a large variety of iodide and bromide salts has been screened (Supporting Information, Section 3.5.2). It was found that the nature of the cation did not have a profound effect on the Lx conjugation efficiency (Supporting Information, Figure S8). Therefore, we decided to implement NaI routinely as a simple inorganic salt additive for the Lx conjugation reactions, at a concentration of 10 mM (Supporting Information, Figure S7), which corresponds to an I−:PtII ratio of about 6:1.
Besides inorganic salts, also other additives that may have effects on the Lx conjugation reaction, such as amino acids, were tested (Supporting Information, Section 3.6). Namely, if these compounds are able to coordinate to PtII, they can compete with coordination sites of the antibody for binding to Lx. This can have an effect on the Lx conjugation efficiency depending on the reactivity of the intermediate PtII additive thus formed. For example, L‐His is present in the clinical formulation buffer of Herceptin (trastuzumab) and can therefore affect the Lx conjugation (see below).
Previous reports suggest that formation of stable PtII‐His complexes can be more efficient owing to transplatination after formation of kinetically favored but thermodynamically less stable PtII–Met complexes.[ 10b , 24 ] Based on this knowledge, our working hypothesis for the Lx conjugation was that initially the coordination of an Lx semi‐final complex 1 to Met residues of an antibody takes place, followed by a shift of a payload‐Lx moiety to His residues. We assumed therefore that the addition of Met derivatives or other soft nucleophiles could increase the Lx conjugation efficiency. [25] Moreover, these experiments would provide further insight into the binding sites of Lx on antibodies (a particular attention was paid to His, Met, Cys, Lys, and arginine (Arg) as potential binding spots for Lx) and into the influence of NaI on the affinity of Lx to those amino acids.
It was found that none of the tested additives significantly increased the Lx conjugation efficiency and that additives capable of coordination to Lx diminished the conjugation efficiency (Supporting Information, Figure S9), thus disproving the hypothesis that Met‐adducts are productive intermediates on the way towards the His‐adducts. [26] Met and Cys almost completely inhibited the Lx conjugation, His (and imidazole as its heterocyclic core) considerably diminished it, whereas Lys and Arg were found much less inhibiting (Figure 3). It confirmed our previous conclusion and literature knowledge that Cys (in case the mAb is reduced before conjugation), Met, and His are preferable Lx binding sites. Most interestingly, it appeared that Met inhibits the Lx conjugation stronger in the absence of NaI, whereas for His the opposite is the case (Figure 3). This unexpected finding suggests that addition of NaI to the conjugation mixture improves the selectivity/affinity of the Lx conjugation towards the desired His‐binding over the undesired Met‐binding (see also below).
Figure 3.
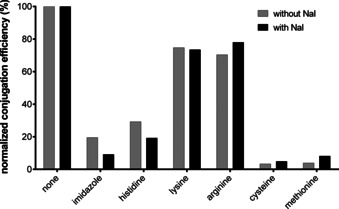
Normalized conjugation efficiency in the presence of selected additives (30 mM), in the absence or presence of NaI (10 mM).
Having achieved a conjugation efficiency of about 38 %, at this stage we decided to fine‐tune the Lx stoichiometry and lowered the excess of Cl‐Lx (1 a) to 5 equiv instead of 20 equiv (Supporting Information, Section 3.7). This led to an increase in the conjugation efficiency to about 48 %, corresponding to the desired average DAR of about 2.5 (Supporting Information, Figure S11). Thus, the optimization achieved to this point allowed use of less payload‐Lx to obtain the desired average DAR, resulting in an increased conjugation efficiency due to a more efficient conjugation to accessible His residues.
In a next stage, we investigated the effect of the TU quenching/washing step (Supporting Information, Section 3.10) that was introduced to ensure stability of Lx conjugates by stripping off weakly bound Lx‐complexes (see above). While having a profound effect on the conjugation efficiency in the absence of NaI (indicating profound formation of weakly Met‐bound Lx‐payload complexes) for conjugation times <24 h, the effect of the TU quenching step became hardly detectable when NaI had been present during the conjugation (Supporting Information, Figure S15).
This observation confirms that addition of NaI does not only make the Lx‐complex more reactive towards proteins (resulting in higher efficiency of the conjugation), but it also makes it more selective for the desired stable binding to His at the cost of the undesired weak binding to Met, in line with results above. This improved selectivity could be explained by a more rapid equilibrium between starting I‐Lx complexes and labile dead‐end Met‐Lx complexes in the presence of an excess of NaI.
Based on conclusions from earlier experiments, [16] no removal of L‐His, which can compete with His binding sites of trastuzumab for conjugation, from the Lx conjugation mixtures was undertaken yet. However, after it was found that L‐His was indeed detrimental for the Lx conjugation reaction (ca. 80 % decrease in the presence of NaI; Figure 3), a buffer exchange step of the antibody before the Lx conjugation had to be introduced (Supporting Information, Section 3.11). Consequently, removal of L‐His from the native trastuzumab formulation buffer greatly improved the conjugation efficiency (from ca. 45 % to ca. 80 %) after 24 h conjugation time (Supporting Information, Figure S17).
To demonstrate the generality of the Lx conjugation, it was applied to five representative mAbs (Supporting Information, Section 3.12). We found similar conjugation efficiencies in the range of 74–78 % after 24 h conjugation time, indicating that our newly developed conjugation procedure is not restricted to trastuzumab but can be applied to a broad range of mAbs (Supporting Information, Figure S19).
Finally, the effect of the halido leaving ligand on the conjugation efficiency was investigated in detail (Supporting Information, Section 3.13). For that purpose, we compared all three halido semi‐final complexes 1 a–c (Figure 1) that were synthesized and isolated for this experiment (Supporting Information, Scheme S15). As first, their hydrolytic stability was assessed. We found that stabilizing concentrations of the corresponding halide salts were necessary to preserve the semi‐final complexes 1 a–c from hydrolysis (that is, formation of an unreactive HO‐Lx species), when they were incubated under Lx conjugation conditions, but without the mAb. Noteworthy, the concentration of the corresponding stabilizing salt reflected the hydrolytic stability of the semi‐final complexes. For Cl‐Lx (1 c), a high concentration of 200 mM NaCl was required to keep >90 % of the semi‐final complex intact for 4 h under the conjugation conditions (Supporting Information, Figure S25), whereas for Br‐Lx (1 b) and I‐Lx (1 c), concentrations of 50 mM NaBr (Supporting Information, Figure S26) and 10 mM NaI (Supporting Information, Figure S27), respectively, were found to be sufficient. Therefore, the hydrolytic stability of semi‐final complexes follows the row: 1 c>1 b>1 a, which is fully in line with the described higher hydrolytic stability of for example, cis‐PtI2(NH3)2 vs. cisplatin. [19] Therefore, to explore the net effect of the halido leaving ligand without interference of hydrolysis, conjugations of complexes 1 a–c were performed in the presence of the corresponding halide salts, in concentrations sufficient to preserve the aforementioned semi‐final complexes from hydrolysis (Figure 4 A). We found that the conjugation efficiency was highest for I‐Lx (1 c)/10 mM NaI (90 %), followed by Br‐Lx (1 b)/50 mM NaBr (74 %), and was lowest for Cl‐Lx (1 a)/200 mM NaCl (34 %). An attempt to prepare the F‐Lx complex 1 d (Figure 1) failed: it turned out to be too unstable towards hydrolysis to be isolated.
Figure 4.
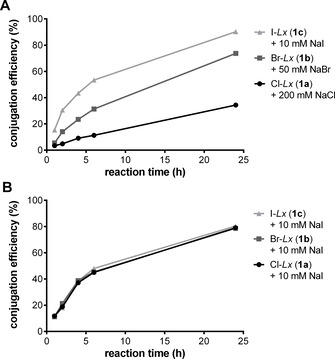
Conjugation efficiency obtained in the presence of corresponding sodium halide salts for stabilization of the Hal‐Lx complexes 1 a–c (A) and in the presence of NaI (10 mM) for the Hal‐Lx complexes 1 a–c (B).
Another important finding is that the halido leaving ligand of the starting Hal‐Lx complexes 1 can be freely chosen from iodido, bromido, and chlorido ligands: in case an external iodide source will be added into the Lx conjugation mixture, all semi‐final complexes 1 will generate the desired Lx‐ADC at the same rate and with the same efficiency (ca. 80 % after 24 h; Figure 4 B) because of a rapid exchange of a halido to the iodido ligand, thus resulting in a common semi‐final compound 1 c. Hence, regardless of which semi‐final complex is used (be it Cl–Lx‐drug, Br–Lx‐drug, or I–Lx‐drug complex) the addition of a certain concentration of an iodide salt to the aqueous conjugation mixture ensures an outstanding efficiency of the Lx conjugation reaction. The iodide effect was already noticeable at an I−:PtII ratio of about 0.6:1 (thus, at catalytic concentrations; Supporting Information, Figure S7), however, a ratio of about 30:1 is standardly used in current optimized procedures.
As yet we do not fully understand why the iodido species perform so much better in the Lx conjugation reaction compared with the chlorido species and considerably better than the bromido species, both in terms of conjugation efficiency and conjugation selectivity. A main reason might be that I–PtII complexes are thermodynamically more stable (which, for example, can be seen in their higher hydrolytic stability described above), but at the same time they are kinetically more labile compared with Cl–PtII complexes (Br–PtII complexes being in between [27] ), allowing a very efficient step towards conjugation. Interestingly, an X‐ray crystallographic structural analysis of an adduct of cis‐PtI2(NH3)2 with lysozyme revealed that, although the complex also preferred a His residue for binding, it was not the iodide but rather the ammonia ligand that was released. [28a] Recently, the same authors described a complicated binding behavior of cis‐PtI2(NH3)2 to cytochrome c, indicating that loss of both iodide and ammonia ligands is possible. [28b] Having a stabilizing bidentate ligand ethylenediamine as a structural part of Lx, we never observed its release, despite the notoriously strong trans effect of iodide.
The Lx conjugation has been optimized successfully, as presented in detail above. However, the biological properties of the conjugates obtained by the newly developed method required validation. In particular, in case that the optimized method would deliver inferior conjugates the whole optimization endeavor would become useless.
Therefore, first the Cl‐Lx‐DFO‐FeIII model semi‐final complex 1 a was used to compare the optimized and the initial methods and to verify that the properties of the thus obtained Lx conjugates 2 a and 2 b remained unaltered (Supporting Information, Section 4). FeIII can be easily replaced by the positron emitting isotope 89ZrIV, a widely used nuclide in the positron emission tomography (PET) imaging of mAbs.[ 8b , 29 ] Thus, by means of 89Zr labeling, the comparison of the human epidermal growth factor receptor 2 (HER2) binding properties (Supporting Information, Figure S30), serum stability (Supporting Information, Figure S31), biodistribution (Figure 5 A), and pharmacokinetics (Figure 5 B) revealed that the properties of the two conjugates 2 a/2 b were identical, while the Lx conjugation efficiency increased from ca. 13 % to about 76 % (Scheme 2).
Figure 5.
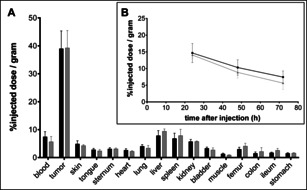
Biodistribution 72 h after injection (A) and blood kinetics (B) of 4 mg kg−1 dose of radiolabeled Lx conjugates 89 Zr‐2 a (black bars and black line; initial method) and 89 Zr‐2 b (gray bars and gray line; optimized method) in NCI‐N87 xenograft bearing mice (N=6/group), both assessed by 89Zr counting. The error bars represent the standard deviation (SD).
Scheme 2.
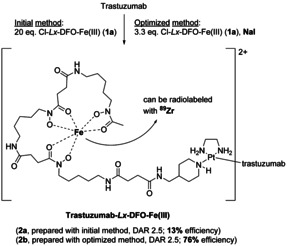
Synthesis of the Lx conjugates trastuzumab‐Lx‐DFO‐FeIII (2 a and 2 b) from the chlorido semi‐final complex Cl‐Lx‐DFO‐FeIII (1 a), comparing the initial and the optimized Lx conjugation methods.
Next, we applied the Lx conjugation procedure using iodide (optimized method) to the semi‐final complex Cl‐Lx‐AF (1 f) that bears the supertoxic payload auristatin F, yielding AF‐based Lx conjugate 3 b with ca. 85 % efficiency (Scheme 3; Supporting Information, Section 5). Its properties were compared to the properties of the conjugate 3 a obtained via the initial Lx conjugation method with ca. 13 % efficiency.
Scheme 3.
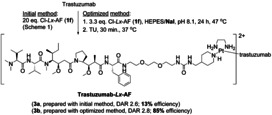
Synthesis of the Lx conjugates trastuzumab‐Lx‐AF (3 a and 3 b) from the chlorido semi‐final complex Cl‐Lx‐AF (1 f), comparing the initial and the optimized Lx conjugation methods.
It was confirmed that the DAR as well as the payload distribution on the antibody were similar for both Lx conjugates (Supporting Information, Section 5.2). These results also confirmed our hypothesis that the optimized Lx conjugation method provides an impressive improvement of the conjugation efficiency without changing/affecting the properties of the final Lx‐ADC product (thus, 3 a=3 b). Expectedly, the cell titer blue (CTB) based cytotoxicity bioassay of conjugates 3 a and 3 b provided very similar picomolar half‐maximal inhibitory concentration (IC50) values for both conjugates in HER2‐positive cell lines NCI‐N87, SK‐OV‐3, and JIMT‐1 (results for JIMT‐1 are shown in Figure 6; for all cell lines in the Supporting Information, Figure S34).
Figure 6.
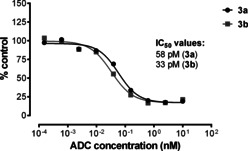
The IC50 values of the Lx conjugates 3 a and 3 b in JIMT‐1, determined by a CTB assay.
Thus, we successfully confirmed the validity of the optimized method for two payloads: DFO and AF.
Additionally, stability of the lead product Lx‐ADC was confirmed by a formulation stability study of a batch 3 bstab (Supporting Information, Section 6).
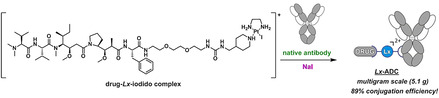
To test the scalability of the newly developed Lx conjugation method, we performed a multigram (5.1 g) scale synthesis of our lead Lx‐ADC 3 c (Supporting Information, Section 7) from a trastuzumab biosimilar and 2.5 equiv of I‐Lx‐AF (1 g), which was produced previously on a 16 g scale using the recently published procedure for the synthesis of iodido semi‐final complexes. [12]
After the conjugation using the optimized method, the obtained product 3 c was quality controlled against five products of milligram scale conjugation reactions (3 cref) that were run in parallel to the multigram conjugation using the same conjugation mixture; the results of the quality control of the obtained multigram scale batch are summarized in the Supporting Information, Table S29.
The obtained multigram scale conjugate 3 c showed properties, such as protein content (Supporting Information, Table S30), monomeric purity (Supporting Information, Figure S36 and Table S31), average DAR (Supporting Information, Figure S38 and Table S32), and potency (Supporting Information, Figure S39 and Table S33), very similar to the milligram scale reference conjugates 3 cref (thus, 3 c=3 cref). This confirmed that the upscaling went successfully without any complications. With the determined DAR of about 2.2 (by size‐exclusion chromatography‐mass spectrometry (SEC‐MS)), which is in the anticipated range of 2.1–2.8, the conjugation efficiency of about 89 % was found to be high and fully met the expectations.
The results obtained in this work convincingly show that the Lx‐linker technology has become a mature bioconjugation method and the Lx‐platform advanced from a promising proof of concept (Scheme 4, left) to a straightforward, robust, and general approach to ADCs (Scheme 4, right).
Both key steps of the greatly improved Lx‐technology, complexation and conjugation, are aligned and work highly synergistically. Namely, the recently developed direct Ag‐free complexation [12] method of preparing the iodido semi‐final complexes delivers perfect precursors for the subsequent Lx conjugation, as was discovered in the course of the optimization work described above. They are conveniently obtained from a simple PtII precursor LxI2[ 12 , 30 ] and subsequently they can be directly used in the herein developed iodide‐promoted/‐catalyzed Lx conjugation procedure, having already the optimal leaving ligand (iodide instead of chloride) installed. Logically enough, the iodido semi‐final complexes are currently used as first‐choice next‐generation precursors for the Lx conjugations.
However, the Lx conjugation does not depend on the nature of the halido leaving ligand of the semi‐final complex (see above): it can be produced as an iodido (currently our routine and preferred synthesis route), [12] bromido or chlorido complex depending on the particular synthetic need. This convincingly demonstrates that the Lx‐technology in its current advanced state is highly modular and versatile, offering (based on its technical aspects and favorable properties of the obtained ADCs)[ 8a , 8c ] a valuable addition to the repertoire of the currently existing bioconjugation approaches.
Conclusion
The exhaustive optimization described above has resulted in a successful and rather surprising finding [31] that the addition of iodide salts to the conjugation mixture greatly improves the efficiency of the Lx conjugations.
The key mechanistic step of the discovered iodide effect is a rapid halide exchange of the initially used chlorido semi‐final complexes, [8a] resulting in the formation of the corresponding iodido semi‐final complexes, which are hydrolytically more stable but at the same time more reactive and more selective for His.
The current Lx conjugation technology making use of the discovered iodide effect has matured to become a general, robust, and preparatively simple approach to ADCs. The demonstrated conjugation efficiency of 89 % during the synthesis of our lead Lx‐ADC on a multigram scale was remarkably high. The method allows a straightforward coupling of various payloads to native mAbs without any chemical pre‐modification or genetical engineering of the antibodies. The new Lx‐technology utilizes NaI, a harmless salt that is used industrially as a nutritional supplement to treat and prevent iodine deficiency, as a simple additive.
Further studies of new opportunities provided by the discovered iodide effect are currently ongoing. Future work will also focus on the GMP production of our lead Lx‐ADCs, toxicological studies, and first‐in‐human trials.
Conflict of interest
E. Merkul, J. A. Muns, N. J. Sijbrandi, and B. Nijmeijer are employed by LinXis B.V., H.‐J. Houthoff and G. van Rheenen are Executive Advisors of LinXis B.V., H.‐J. Houthoff has ownership (including patents) in LinXis B.V., G.A.M.S. van Dongen is a non‐profit scientific advisory board member of LinXis B.V., J. Reedijk is an advisor of LinXis B.V.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The authors thank Prof. Dr. Dario Neri (ETH Zürich) for critical reading of the manuscript.
E. Merkul, J. A. Muns, N. J. Sijbrandi, H.-J. Houthoff, B. Nijmeijer, G. van Rheenen, J. Reedijk, G. A. M. S. van Dongen, Angew. Chem. Int. Ed. 2021, 60, 3008.
Dedicated to Alisa Lucretia Merkul on the occasion of her 4th birthday
References
- 1. Nicolaou K. C., Rigol S., Angew. Chem. Int. Ed. 2019, 58, 11206–11241; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 11326–11363. [Google Scholar]
- 2.
- 2a. Joubert N., Beck A., Dumontet C., Denevault-Sabourin C., Pharmaceuticals 2020, 13, 245; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2b. Ponziani S., Di Vittorio G., Pitari G., Cimini A. M., Ardini M., Gentile R., Iacobelli S., Sala G., Capone E., Flavell D. J., Ippoliti R., Giansanti F., Int. J. Mol. Sci. 2020, 21, 5510; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2c. Coats S., Williams M., Kebble B., Dixit R., Tseng L., Yao N.-S., Tice D. A., Soria J.-C., Clin. Cancer Res. 2019, 25, 5441–5448; [DOI] [PubMed] [Google Scholar]
- 2d. Tang H., Liu Y., Yu Z., Sun M., Lin L., Liu W., Han Q., Wei M., Jin Y., Front. Pharmacol. 2019, 10, 373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.
- 3a. Saber H., Leighton J. K., Regul. Toxicol. Pharmacol. 2015, 71, 444–452; [DOI] [PubMed] [Google Scholar]
- 3b. Acchione M., Kwon H., Jochheim C. M., Atkins W. M., mAbs 2012, 4, 362–372; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3c. Alley S. C., Benjamin D. R., Jeffrey S. C., Okeley N. M., Meyer D. L., Sanderson R. J., Senter P. D., Bioconjugate Chem. 2008, 19, 759–765. [DOI] [PubMed] [Google Scholar]
- 4. Tsuchikama K., An Z., Protein Cell 2018, 9, 33–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.
- 5a. Yoder N. C., Bai C., Tavares D., Widdison W. C., Whiteman K. R., Wilhelm A., Wilhelm S. D., McShea M. A., Maloney E. K., Ab O., Wang L., Jin S., Erickson H. K., Keating T. A., Lambert J. M., Mol. Pharm. 2019, 16, 3926–3937; [DOI] [PubMed] [Google Scholar]
- 5b. Zhou Q., Biomedicines 2017, 5, 64. [Google Scholar]
- 6. Ebenig A., Juettner N. E., Deweid L., Avrutina O., Fuchsbauer H.-L., Kolmar H., ChemBioChem 2019, 20, 2411–2419. [DOI] [PubMed] [Google Scholar]
- 7. Matsuda Y., Clancy C., Tawfiq Z., Robles V., Mendelsohn B. A., ACS Omega 2019, 4, 20564–20570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.
- 8a. Sijbrandi N. J., Merkul E., Muns J. A., Waalboer D. C. J., Adamzek K., Bolijn M., Montserrat V., Somsen G. W., Haselberg R., Steverink P. J. G. M., Houthoff H.-J., van Dongen G. A. M. S., Cancer Res. 2017, 77, 257–267; [DOI] [PubMed] [Google Scholar]
- 8b. Muns J. A., Montserrat V., Houthoff H.-J., Codée-van der Schilden K., Zwaagstra O., Sijbrandi N. J., Merkul E., van Dongen G. A. M. S., J. Nucl. Med. 2018, 59, 1146–1151; [DOI] [PubMed] [Google Scholar]
- 8c. Merkul E., Sijbrandi N. J., Muns J. A., Aydin I., Adamzek K., Houthoff H.-J., Nijmeijer B., van Dongen G. A. M. S., Expert Opin. Drug Delivery 2019, 16, 783–793; [DOI] [PubMed] [Google Scholar]
- 8d. del Solar V., Contel M., J. Inorg. Biochem. 2019, 199, 110780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Reedijk J., Platinum Met. Rev. 2008, 52, 2–11. [Google Scholar]
- 10.
- 10a. Bugarčić Ž. D., Bogojeski J., Petrović B., Hochreuther S., van Eldik R., Dalton Trans. 2012, 41, 12329–12345; [DOI] [PubMed] [Google Scholar]
- 10b. Reedijk J., Eur. J. Inorg. Chem. 2009, 1303–1312; [Google Scholar]
- 10c. Reedijk J., Chem. Rev. 1999, 99, 2499–2510. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Casini A., Reedijk J., Chem. Sci. 2012, 3, 3135–3144; [Google Scholar]
- 11b. Timerbaev A. R., Hartinger C. G., Aleksenko S. S., Keppler B. K., Chem. Rev. 2006, 106, 2224–2248. [DOI] [PubMed] [Google Scholar]
- 12. Merkul E., Sijbrandi N. J., Aydin I., Muns J. A., Peters R. J. R. W., Laarhoven P., Houthoff H.-J., van Dongen G. A. M. S., Green Chem. 2020, 22, 2203–2212. [Google Scholar]
- 13. Deubel D. V., J. Am. Chem. Soc. 2002, 124, 5834–5842. [DOI] [PubMed] [Google Scholar]
- 14. Ivanov A. I., Christodoulou J., Parkinson J. A., Barnham K. J., Tucker A., Woodrow J., Sadler P. J., J. Biol. Chem. 1998, 273, 14721–14730. [DOI] [PubMed] [Google Scholar]
- 15.
- 15a. van Dongen G. A. M. S., Sijbrandi N. J., Waalboer D. C. J., Houthoff H.-J., WO 2016/144171 A1, September 15, 2016;
- 15b. Lempers E. L. M., Reedijk J., Inorg. Chem. 1990, 29, 217–222. [Google Scholar]
- 16. Waalboer D. C. J., Muns J. A., Sijbrandi N. J., Schasfoort R. B. M., Haselberg R., Somsen G. W., Houthoff H.-J., van Dongen G. A. M. S., ChemMedChem 2015, 10, 797–803. [DOI] [PubMed] [Google Scholar]
- 17.
- 17a. Majumdar R., Manikwar P., Hickey J. M., Samra H. S., Sathish H. A., Bishop S. M., Middaugh C. R., Volkin D. B., Weis D. D., Biochemistry 2013, 52, 3376–3389; [DOI] [PubMed] [Google Scholar]
- 17b. Zhang-van Enk J., Mason B. D., Yu L., Zhang L., Hamouda W., Huang G., Liu D., Remmele R. L., Zhang J., Mol. Pharm. 2013, 10, 619–630. [DOI] [PubMed] [Google Scholar]
- 18. Berger I., Nazarov A. A., Hartinger C. G., Groessl M., Valiahdi S.-M., Jakupec M. A., Keppler B. K., ChemMedChem 2007, 2, 505–514. [DOI] [PubMed] [Google Scholar]
- 19. Marzo T., Pillozzi S., Hrabina O., Kasparkova J., Brabec V., Arcangeli A., Bartoli G., Severi M., Lunghi A., Totti F., Gabbiani C., Quiroga A. G., Messori L., Dalton Trans. 2015, 44, 14896–14905. [DOI] [PubMed] [Google Scholar]
- 20. Cleare M. J., Hoeschele J. D., Bioinorg. Chem. 1973, 2, 187–210. [Google Scholar]
- 21. Basolo F., Gray H. B., Pearson R. G., J. Am. Chem. Soc. 1960, 82, 4200–4203. [Google Scholar]
- 22.
- 22a. Dhara S. C., Indian J. Chem. 1970, 8, 193–194; [Google Scholar]
- 22b. Talman E. G., Brüning W., Reedijk J., Spek A. L., Veldman N., Inorg. Chem. 1997, 36, 854–861; [Google Scholar]
- 22c. Wilson J. J., Lippard S. J., Chem. Rev. 2014, 114, 4470–4495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.
- 23a. Štarha P., Vančo J., Trávníček Z., Coord. Chem. Rev. 2019, 380, 103–135; [Google Scholar]
- 23b. Musumeci D., Platella C., Riccardi C., Merlino A., Marzo T., Massai L., Messori L., Montesarchio D., Dalton Trans. 2016, 45, 8587–8600; [DOI] [PubMed] [Google Scholar]
- 23c. Messori L., Cubo L., Gabbiani C., Álvarez-Valdés A., Michelucci E., Pieraccini G., Ríos-Luci C., León L. G., Padrón J. M., Navarro-Ranninger C., Casini A., Quiroga A. G., Inorg. Chem. 2012, 51, 1717–1726. [DOI] [PubMed] [Google Scholar]
- 24.
- 24a. Van Boom S. S. G. E., Chen B. W., Teuben J.-M., Reedijk J., Inorg. Chem. 1999, 38, 1450–1455; [Google Scholar]
- 24b. Hahn M., Wolters D., Sheldrick W. S., Hulsbergen F. B., Reedijk J., J. Biol. Inorg. Chem. 1999, 4, 412–420; [DOI] [PubMed] [Google Scholar]
- 24c. Chen Y., Guo Z., del Socorro Murdoch P., Zang E., Sadler P. J., J. Chem. Soc. Dalton Trans. 1998, 1503–1508; [Google Scholar]
- 24d. Fröhling C. D. W., Sheldrick W. S., Chem. Commun. 1997, 1737–1738; [Google Scholar]
- 24e. Lempers E. L. M., Reedijk J., Inorg. Chem. 1990, 29, 1880–1884. [Google Scholar]
- 25. Barnham K. J., Djuran M. I., del Socorro Murdoch P., Ranford J. D., Sadler P. J., J. Chem. Soc. Dalton Trans. 1995, 3721–3726. [Google Scholar]
- 26. Teuben J.-M., Reedijk J., J. Biol. Inorg. Chem. 2000, 5, 463–468. [DOI] [PubMed] [Google Scholar]
- 27. Marzo T., Bartoli G., Gabbiani C., Pescitelli G., Severi M., Pillozzi S., Michelucci E., Fiorini B., Arcangeli A., Quiroga A. G., Messori L., Biometals 2016, 29, 535–542. [DOI] [PubMed] [Google Scholar]
- 28.
- 28a. Messori L., Marzo T., Gabbiani C., Valdes A. A., Quiroga A. G., Merlino A., Inorg. Chem. 2013, 52, 13827–13829; [DOI] [PubMed] [Google Scholar]
- 28b. Tolbatov I., Marzo T., Cirri D., Gabbiani C., Coletti C., Marrone A., Paciotti R., Messori L., Re N., J. Inorg. Biochem. 2020, 209, 111096. [DOI] [PubMed] [Google Scholar]
- 29. Verel I., Visser G. W., Boellaard R., Stigter-van Walsum M., Snow G. B., van Dongen G. A. M. S., J. Nucl. Med. 2003, 44, 1271–1281. [PubMed] [Google Scholar]
- 30. Merkul E., Sijbrandi N. J., Muns J. A., van Dongen G. A. M. S., Steverink P. J. G. M., Houthoff H.-J., WO 2019/125153 A1, June 27, 2019.
- 31. Merkul E., Sijbrandi N. J., Muns J. A., van Dongen G. A. M. S., Steverink P. J. G. M., Houthoff H.-J., WO 2019/125154 A2, June 27, 2019.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary