

Abstract
An I(I)/(III) catalysis strategy to construct an enantioenriched fluorinated isostere of the iPr group is reported. The difluorination of readily accessible α‐CF3‐styrenes is enabled by the in situ generation of a chiral ArIF2 species to forge a stereocentre with the substituents F, CH2F and CF3 (up to 95 %, >20:1 vicinal:geminal difluorination). The replacement of the metabolically labile benzylic proton results in a highly preorganised scaffold as was determined by X‐ray crystallography (π→σ* and stereoelectronic gauche σ→σ* interactions). A process of catalyst editing is disclosed in which preliminary validation of enantioselectivity is placed on a structural foundation.
Keywords: agrochemistry, bioisostere, conformation, fluorine, organocatalysis
The iodine(I)/(III) catalysed construction of an enantioenriched fluorinated isostere of the iPr group is reported. The difluorination of readily accessible α‐CF3‐styrenes is enabled by the in situ generation of a chiral ArIF2 species to forge a stereocentre with the substituents F, CH2F and CF3 (up to 95 %, >20:1 vic:gem difluorination). Catalyst editing for preliminary validation of enantioselectivity is placed on a structural foundation.
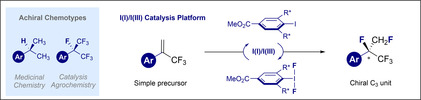
Short, unfunctionalised aliphatic groups (C1–C4) are ubiquitous structural features in the natural product repertoire, and are particularly conspicuous in polyketides and terpenes. [1] This is a logical consequence of iterative biosynthesis algorithms that process low molecular weight fragments into higher homologues. [2] Introduced under the auspices of acetyl‐ or propionyl‐CoA, [3] complemented by electrophilic paradigms involving methyltransferases (SAM), [4] these motifs appear to be vestigial in nature. However, they often encode for a highly specific function and thus delineating their biosynthetic origins has been intensively pursued. Indeed the value of harnessing small aliphatic groups to enhance the physicochemical profiles of drug candidates is exemplified by the “magic methyl effect”. [5] Interrogating the stereochemical course of enzymatic methylation has a venerable history, due to the achiral nature of this motif and the pre‐conditions associated with designing a chiral bioisostere to track the possible translation of stereochemical information.[ 6 , 7 ] Arigoni's celebrated synthesis of chiral acetic acid remains a tour de force in stereocontrolled synthesis, and a master class in orbital symmetry to craft an isotopically orthogonal motif (1H, 2H and 3H, Figure 1, top). [8] Whilst this isotope strategy remains expansive in the field of mechanistic enzymology, small fragment‐based bioisosterism in drug design relies on stable isotopes to enhance the pharmaco‐kinetics and ‐dynamics of drug candidate performance. [9] Molecular editing with fluorine (H→F) has proven to be particularly effective, [10] and is reflected in the increasing number of fluorinated small molecules reaching the market. [11] This is a consequence of fluorine's low steric demand, low polarisability and the stability of the C‐F bond. Given the success of achiral perfluoroalkyl groups in drug discovery, catalysis and agrochemistry, [12] routes to small, chiral, 3D fluoroalkane motifs would be advantageous to expand the available chemical space. This includes the C2 (BITE group) [13] which is a bioisosteric hybrid of the ethyl and trifluoromethyl groups. [14] Cognisant of the prevalence of (C3) isopropyl units in bioactive natural product leads and small molecule pharmaceuticals (Figure 1 centre), a catalysis‐based strategy to access a differentially fluorinated analogue of the iPr group was initiated. Harnessing I(I)/(III) catalysis, [15] it was envisaged that a formal 1,2‐addition of fluorine across the alkene moiety [16] of simple α‐trifluoromethyl styrenes would generate a stereogenic centre bearing F, CH2F and CF3 groups (Figure 1, bottom).
Figure 1.
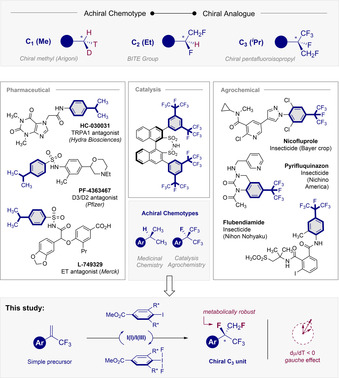
Top: Chiral bioisosteres of common aliphatic chemotypes. Centre: Selected functional small molecules containing the achiral iPr (pharmaceutical) and CF(CF3)2 (catalysis and agrochemical) units. [12] Bottom: Design of a main group catalysis approach to generate a chiral fluorinated analogue of iPr.
The success of this catalysis‐based approach would be contingent on the in situ oxidation of a chiral aryl iodide organocatalyst to generate an ArIF2 species [17] that would be sufficiently active to engage a sterically‐congested, electron‐deficient alkene. If successful, the resulting pentafluoroisopropyl surrogate would constitute a chiral C3 building block in which the lability of the methine proton is mitigated. [18] Moreover, the constituent hyperconjugative interactions intrinsic to the internal vicinal‐difluoro motif [19] would manifest themselves in conformation. To identify conditions that would enable the target fluorinated isopropyl motif to be generated from simple α‐trifluoromethyl styrenes, a process of reaction optimisation was conducted (Figure 2, 1 a→2 a). To that end, simple aryl iodides were investigated as inexpensive catalysts in conjunction with Selectfluor® as the terminal oxidant to generate the key ArI(III)F2 species. [17] Initial studies were performed in chloroform at ambient temperature using an amine:HF ratio of 1:7.5 and the reactions were examined by 19F NMR spectroscopy using an internal standard. Iodobenzene proved to be a perfectly effective catalyst for this transformation to generate (±)‐2 a and 3 a in a 2.5:1 ratio (86 % combined yield). The latter product arises from phenonium ion rearrangement and has been exploited in a range of catalysis‐based geminal difluorination processes. [20] Repeating the reaction with p‐iodotoluene (5) led to a notable improvement in yield (>95 %) with comparable regioselectivity in favour of the desired vicinal product 2 a (2.2:1). Electronic modulation was not well tolerated with the ester derivative 6 proving to be a less active catalyst under comparable conditions (26 %). Control experiments in the absence of catalyst led to <5 % yield and demonstrate the strongly deactivating nature of the trifluoromethyl group that inhibits background reactions such as those reported by Lal and co‐workers using HF sources and Selectfluor®. [21]
Figure 2.
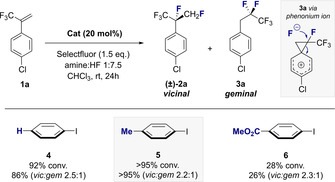
Catalyst identification. Standard reaction conditions: α‐CF3‐p‐chlorostyrene 1 a (0.2 mmol), catalyst (20 mol %), Selectfluor® (1.5 equiv), amine:HF 1:7.5 (0.5 mL), CHCl3 (0.5 mL), ambient temperature, 24 h. The yield is the sum of vicinal and geminal difluorination products. The regioselectivity ratio (vic:gem) and yield were determined by 19F NMR spectroscopy using α,α,α‐trifluorotoluene as internal standard. Control experiment without catalyst: yield <5 %.
To explore the scope and limitations of this catalysis‐based difluorination of α‐CF3‐styrenes, the effect of Brønsted acidity [22] was probed as a function of the amine:HF ratio. [16b] This led to the identification of methods A, B and C, reflecting amine:HF ratios of 1:4.5, 1:6 and 1:7.5 respectively (Figures 3 and 4). Method A proved to be highly effective in generating the electron rich products 2 b–2 d with high levels of regioselectivity favouring formation of the desired vicinal product (>20:1, up to 86 %). The presence of the CF3 group clearly distinguish this substrate class from the parent styrenes, which rearrange to generate the geminal product. [20] Control reactions again confirmed the necessity for the catalyst. The seemingly subtle change to Method B proved to be optimal for substrates 2 e–2 k, enabling the generation of alkyl derivatives (2 e–2 h, up to >20:1, vic:gem) as well as the electron deficient aniline derivative 2 i (74 %, 15.5:1).
Figure 3.
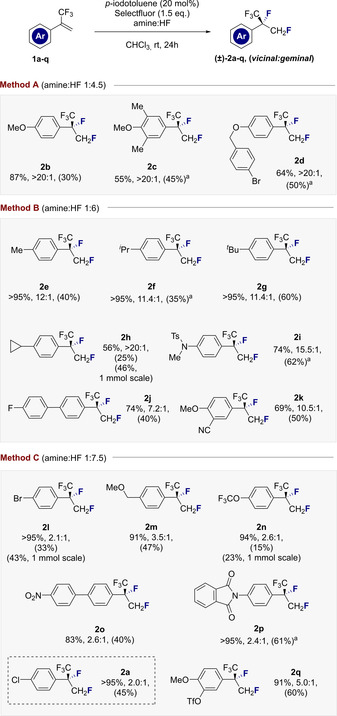
Establishing the scope of the vicinal difluorination of α‐CF3‐ styrenes to generate a highly fluorinated isopropyl group. The yield is the sum of vicinal and geminal difluorination products. The regioselectivity ratio (vic:gem) and yield were determined by 19F NMR spectroscopy using α,α,α‐trifluorotoluene as internal standard. Isolated yields of the vicinal products are given in parentheses. a Reaction time increased to 48 h. Arbitrary enantiomer of the product shown. N.B.: The products are often highly volatile and care must be taken in the isolation.
Figure 4.
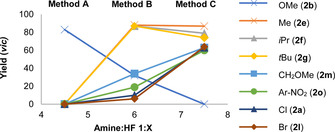
The effect of Brønsted acidity on catalysis.
Comparable efficiency and selectivity were also noted for the biphenyl system 2 j and adduct 2 k. Augmenting the amine:HF ratio further to Method C provided optimised conditions to access products 2 a,l–2 q. Whereas employing higher HF ratios/ Brønsted acidities under the conditions developed by this laboratory tend to favour 1,1‐difluorination, [20f] electron‐deficient α‐CF3‐styrenes proved to be notably more recalcitrant to rearrangement and the vicinal products predominated throughout. Given the importance of aryl bromides in contemporary medicinal chemistry, where the C(sp2)‐Br provides a handle for subsequent cross‐coupling, the synthesis of 2 l was conducted on a 1 mmol scale. Despite the volatility of the product, the vicinal product could be isolated in 43 % yield. Products 2 m, 2 n and 2 o behaved similarly and were generated in a vicinal:geminal ratio of ca. 3:1. Given the prominence of aniline fragments bearing isopropyl units in drug and agrochemical discovery (See Figure 1), the phthalimide 2 p was generated cleanly in 61 % yield. Finally, access to the disubstituted aryl 2 q was realised, this time with an improvement in regioselectivity (5.0:1). Having established conditions to enable the vicinal difluorination of α‐CF3‐styrenes via I(I)/(III) catalysis, attention was focussed on a preliminary validation of an enantioselective variant. Whilst catalyst p‐iodotoluene 5 is a highly competent catalyst, sites to append stereodirecting groups are conspicuously absent. The investigation was therefore repeated with resorcinol derivatives 7–9 in which proximal stereocentres might induce enantioinduction. Whereas catalysts 7 and 8 proved to be moderately effective, balancing the electronic effects of the resorcinol with a p‐CO2Me in catalyst 9 led to notably superior catalysis (87 % yield, vicinal:geminal 3:1). As the logical next step, C2‐symmetric resorcinol derivatives were investigated as summarised in Figure 5.[ 23 , 24 ] Reactions were performed under standard conditions with an amine:HF ratio of 1:7.5 in CHCl3 at ambient temperature. Initially, the effect of modifying the substituent X was assessed using the methyl esters 10–13. Counterintuitively, augmenting the steric footprint at site X had a detrimental effect on selectivity. Catalyst 10 (X=Me) proved to be most effective, generating compound 2 a with 86:14 e.r. (>95 % conversion, 88 % combined yield). Structural editing at site Y was not tolerated as exemplified by catalysts 14–16. As a control series, the C1‐symmetric catalysts 17–19 were examined (Figure 5, lower). Direct comparison of 17 with the most promising scaffold 10 confirmed the importance of C2 symmetry (72:28 versus 86:14 e.r.). Interestingly, substituting the methyl ester for benzyl (catalyst 18) did not erode selectivity, although efficiency was decreased. Moreover, the α‐Bn catalyst (19) proved to be less efficient than the C2‐symmetric derivative 12. Having identified catalyst 10 as the most promising scaffold to validate an enantioselective process (please see the ESI for additional details) a representative selection of α‐CF3‐styrenes were subjected to the general catalysis conditions using 10 (Figure 6).
Figure 5.
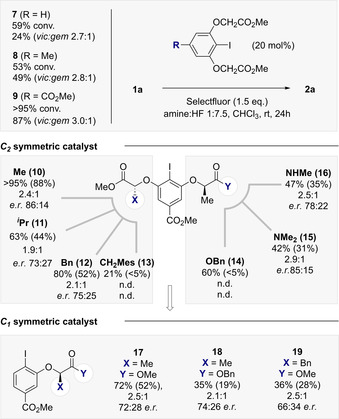
Catalyst optimisation to enable preliminary validation of enantioselection. The conversion and combined yield (in parentheses) was determined by 19F NMR spectroscopy of the crude reaction mixture using α,α,α‐trifluorotoluene as internal standard. Enantioselectivity determined by chiral HPLC. Standard reaction conditions: α‐CF3‐p‐chlorostyrene 1 a (0.2 mmol), catalyst (20 mol %), Selectfluor® (1.5 equiv), amine:HF 1:7.5 (0.5 mL), CHCl3 (0.5 mL), ambient temperature, 24 h.
Figure 6.
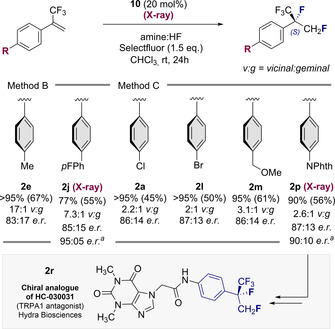
The yield is the sum of vicinal and geminal difluorination products. The regioselectivity ratio (vic:gem) and yield were determined by 19F NMR spectroscopy using α,α,α‐trifluorotoluene as internal standard. Isolated yield of the vicinal product is given in parentheses. Enantioselectivity determined by chiral HPLC. a After recrystallisation. N.B.: The products are often highly volatile and care must be taken in the isolation.
Gratifyingly, the methyl derivative underwent smooth difluorination to generate 2 e (>95 %, 17.1 vicinal:geminal, 83:17 e.r.). The fluorinated biaryl system 2 j was compatible with the conditions and could be prepared with a regioselectivity of 7:1 vicinal:geminal and 85:15 e.r (95:05 e.r. after recrystallisation). The p‐Cl and p‐Br derivatives 2 a and 2 l were prepared with 86:14 and 87:13 e.r., respectively, 2 m in 86:14 e.r. and the protected amine 2 p in 87:13 e.r. (90:10 e.r. after recrystallisation). Gratifyingly, compounds 2 j and 2 p were crystalline allowing the (S)‐configuration of the new stereocentre to be assigned (vide infra). [25] Finally, the phthalimide derivative 2 p was processed to an analogue of the TRPA1 antagonist HC‐030031 2 r in a short synthetic sequence (Figure 6, lower. Full details in the ESI).
To complement the plenum of methods available to construct short, unfunctionalised aliphatic groups for drug discovery, a catalysis‐based strategy to access chiral, fluorinated surrogates of the isopropyl group has been developed. This serves to expands the current portfolio of fluorine drug modules for drug discovery (Figure 7, centre). [26] Despite the intrinsic steric and electronic challenges associated with generating highly fluorinated stereocentres, this I(I)/I(III) catalysis platform enables α‐CF3‐styrenes to undergo smooth vicinal difluorination (up to >20:1 vicinal:geminal). Importantly, the CF3 group effectively inhibits the dominant phenonium ion rearrangement associated with electron rich styrenes, allowing products such as 2 b to be generated with excellent levels of regiocontrol (>20:1). Finally, preliminary validation of an enantioselective variant is disclosed. Whilst the sterically demanding phenyl and trifluoromethyl substituents (VvdW (CF3)=39.8 Å3)[ 10e , 27 ] render this intermolecular process challenging, it is gratifying to observe encouraging levels of enantioselectivity. A tentative induction model is proposed in which facial discrimination in the enantiodetermining fluorination is a precondition of selectivity. Since X‐ray analyses of 2 j and 2 p confirm that the major enantiomer is (S)‐configured (Figure 7), it is conceivable that stabilising electrostatic interactions (RCF2 δ−F⋅⋅⋅δ+H‐CH2R), [28] may bias catalyst‐substrate preorganisation. [29] Simple steric discrimination (CF3 vs. Ph) is not consistent with the selectivities observed the C1‐symmetric catalysts. The solid‐state analysis also reveals a stereoelectronic gauche effect (σ→σ*; φFCCF=69.9° and 51° for 2 j and 2 p, respectively) and that the CF3 group is orthogonal to the plane of the π system (π→σ*). [30] Exploring the physicochemical profile of this new motif in the context of drug discovery and contemporary agrochemistry is the focus of ongoing studies and will be reported in due course.
Figure 7.
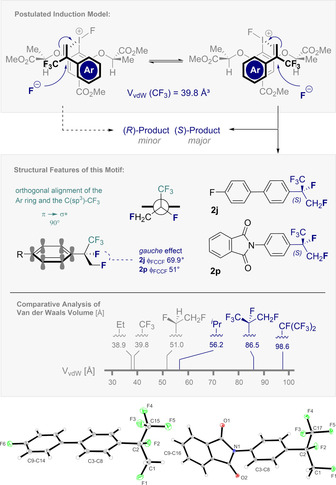
Postulated induction model and the X‐ray structure of compound 2 j and 2 p. Thermal ellipsoids are shown at 15 % probability. CCDC 2044630 (2 j) and 2044631 (2 p).
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We acknowledge generous financial support from the Westfälische Wilhelms‐Universität Münster, the European Research Council (ERC Consolidator Grant—Project Number 818949‐RECON, to RG), the DFG (Cluster of Excellence “Cells in Motion—CiM” (FF‐2013‐10) and SFB 858) and the Alexander von Humboldt Foundation (post‐doctoral research fellowship to JJM). We thank Mr. Tomáš Neveselý (WWU Münster) for helpful discussions. Open access funding enabled and organized by Projekt DEAL.
S. Meyer, J. Häfliger, M. Schäfer, J. J. Molloy, C. G. Daniliuc, R. Gilmour, Angew. Chem. Int. Ed. 2021, 60, 6430.
In memory of Prof. Dr. François Diederich (1952–2020)
Contributor Information
M. Sc. Stephanie Meyer, https://www.uni‐muenster.de/Chemie.oc/gilmour/
Prof. Dr. Ryan Gilmour, Email: ryan.gilmour@uni-muenster.de.
References
- 1.
- 1a. Eschenmoser A., Ruzicka L., Jeger O., Arigoni D., Helv. Chim. Acta 1955, 38, 1890; [Google Scholar]
- 1b. Eschenmoser A., Arigoni D., Helv. Chim. Acta 2005, 88, 3011. [Google Scholar]
- 2.
- 2a. Cane D. E., Chem. Rev. 1997, 97, 2463; [DOI] [PubMed] [Google Scholar]
- 2b. Staunton J., Weissman K. J., Nat. Prod. Rev. 2001, 18, 380. [DOI] [PubMed] [Google Scholar]
- 3. Nivina A., Yuet K. P., Khosla C., Chem. Rev. 2019, 119, 12524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Broderick J. B., Duffus B. R., Duschene K. S., Shepard E. M., Chem. Rev. 2014, 114, 4229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.
- 5a. Schönherr H., Cernak T., Angew. Chem. Int. Ed. 2013, 52, 12256; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 12480; [Google Scholar]
- 5b. Wencel-Delord J., Nat. Chem. 2020, 12, 505. [DOI] [PubMed] [Google Scholar]
- 6. Floss H. G., Lee S., Acc. Chem. Res. 1993, 26, 116. [Google Scholar]
- 7. Lüthy J., Rétey J., Arigoni D., Nature 1969, 221, 1213. [DOI] [PubMed] [Google Scholar]
- 8. Townsend C. A., Scholl T., Arigoni D., J. Chem. Soc. Chem. Commun. 1975, 921. [Google Scholar]
- 9. Patani G. A., LaVoie E. J., Chem. Rev. 1996, 96, 3147. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Müller K., Faeh C., Diederich F., Science 2007, 317, 1881; [DOI] [PubMed] [Google Scholar]
- 10b. O'Hagan D., Chem. Soc. Rev. 2008, 37, 308;18197347 [Google Scholar]
- 10c. Zimmer L. E., Sparr C., Gilmour R., Angew. Chem. Int. Ed. 2011, 50, 11860; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 12062; [Google Scholar]
- 10d. O'Hagan D., Deng H., Chem. Rev. 2015, 115, 634; [DOI] [PubMed] [Google Scholar]
- 10e. Meanwell N. A., J. Med. Chem. 2018, 61, 5822. [DOI] [PubMed] [Google Scholar]
- 11. Han J., Remete A. M., Dobson L. S., Kiss L., Izawa K., Moriwaki H., Soloshonok V. A., O'Hagan D., J. Fluorine Chem. 2020, 239, 109639. [Google Scholar]
- 12.
- 12a. Purser S., Moore P. R., Swallow S., Gouverneur V., Chem. Soc. Rev. 2008, 37, 320; [DOI] [PubMed] [Google Scholar]
- 12b. El. Qacemi M., Rendine S., Mainfisch P., in Fluorine in Life Sciences: Pharmaceuticals, Medicinal Diagnostics, and Agrochemicals, Elsevier, Amsterdam, 2009, pp. 607–623; [Google Scholar]
- 12c. Ogawa Y., Tokunaga E., Kobayashi O., Hirai K., Shibata N., iScience 2020, 23, 101467; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12d. Guin J., Rabalakos C., List B., Angew. Chem. Int. Ed. 2012, 51, 8859; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 8989. [Google Scholar]
- 13.
- 13a. Erdeljac N., Kehr G., Ahlqvist M., Knerr L., Gilmour R., Chem. Commun. 2018, 54, 12002; [DOI] [PubMed] [Google Scholar]
- 13b. Erdeljac N., Bussmann K., Schöller A., Hansen F., Gilmour R., ACS Med. Chem. Lett. 2019, 10, 1336; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13c. Erdeljac N., Thiehoff C., Jumde R., Daniliuc C., Hoeppner S., Faust A., Hirsch A., Gilmour R., J. Med. Chem. 2020, 63, 6225. [DOI] [PubMed] [Google Scholar]
- 14. Molnár I. G., Thiehoff C., Holland M. C., Gilmour R., ACS Catal. 2016, 6, 7167. [Google Scholar]
- 15.
- 15a. Kohlhepp S. V., Gulder T., Chem. Soc. Rev. 2016, 45, 6270; [DOI] [PubMed] [Google Scholar]
- 15b. Arnold A. M., Ulmer A., Gulder T., Chem. Eur. J. 2016, 22, 8728; [DOI] [PubMed] [Google Scholar]
- 15c. Doobary S., Lennox A. J. J., Synlett 2010, 31, 1333. [Google Scholar]
- 16.
- 16a. Molnár I. G., Gilmour R., J. Am. Chem. Soc. 2016, 138, 5004; [DOI] [PubMed] [Google Scholar]
- 16b. Scheidt F., Schäfer M., Sarie J. C., Daniliuc C. G., Molloy J. J., Gilmour R., Angew. Chem. Int. Ed. 2018, 57, 16431; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 16669; [Google Scholar]
- 16c. Banik S. M., Medley J. W., Jacobsen E. N., J. Am. Chem. Soc. 2016, 138, 5000; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16d. Haj M. K., Banik S. M., Jacobsen E. N., Org. Lett. 2019, 21, 4919; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16e. Doobary S., Sedikides A. T., Caldora H. P., Poole D. L., Lennox A. J. J., Angew. Chem. Int. Ed. 2020, 59, 1155; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 1171. [Google Scholar]
- 17.
- 17a. Weinland R. F., Stille W., Ber. Dtsch. Chem. Ges. 1901, 34, 2631; [Google Scholar]
- 17b. Edmunds J. J., Motherwell W. B., J. Chem. Soc. Chem. Commun. 1989, 881; [Google Scholar]
- 17c. Sarie J. C., Thiehoff C., Mudd R. J., Daniliuc C. G., Kehr G., Gilmour R., J. Org. Chem. 2017, 82, 11792; [DOI] [PubMed] [Google Scholar]
- 17d.For the analogous chlorinated system, see Sarie J. C., Neufeld J., Daniliuc C. G., Gilmour R., ACS Catal. 2019, 9, 7232. [Google Scholar]
- 18. Riley R. J., Curr. Opin. Drug Discovery Dev. 2001, 4, 45. [PubMed] [Google Scholar]
- 19.
- 19a. Thiehoff C., Rey Y. P., Gilmour R., Isr. J. Chem. 2017, 57, 92; [Google Scholar]
- 19b. Aufiero M., Gilmour R., Acc. Chem. Res. 2018, 51, 1701. [DOI] [PubMed] [Google Scholar]
- 20.For examples, see
- 20a. Hara S., Nakahigashi J., Ishi-I K., Fukuhura T., Yoneda N., Tetrahedron Lett. 1998, 39, 2589; [Google Scholar]
- 20b. Ilchenko N. O., Tasch B. O. A., Szabó K. J., Angew. Chem. Int. Ed. 2014, 53, 12897; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 13111; [Google Scholar]
- 20c. Kitamura T., Muta K., Oyamada J., J. Org. Chem. 2015, 80, 10431; [DOI] [PubMed] [Google Scholar]
- 20d. Banik S. M., Medley J. W., Jacobsen E. N., Science 2016, 353, 51; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20e. Ilchenko N. O., Szabó K. J., J. Fluorine Chem. 2017, 203, 104; [Google Scholar]
- 20f. Scheidt F., Neufeld J., Schäfer M., Thiehoff C., Gilmour R., Org. Lett. 2018, 20, 8073. [DOI] [PubMed] [Google Scholar]
- 21. Lal G. S., J. Org. Chem. 1993, 58, 2791. [Google Scholar]
- 22.For a study on the effect of Brønsted acidity on the activation of PhICl2 by TFA, see Cotter J. L., Andrews L. J., Keefer R. M., J. Am. Chem. Soc. 1962, 84, 793. [Google Scholar]
- 23.For selected examples, see
- 23a. Fujita M., Yoshida Y., Miyata K., Wakisaka A., Sugimura T., Angew. Chem. Int. Ed. 2010, 49, 7068; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 7222; [Google Scholar]
- 23b. Haubenreisser S., Wöste T. H., Martínez C., Ishihara K., Muñiz K., Angew. Chem. Int. Ed. 2016, 55, 413; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 422. [Google Scholar]
- 24.For a seminal report on the use of a catalytic chiral aryl iodide in enantioselective oxidative fluorination, see Suzuki S., Kamo T., Fukushi K., Hiramatsu T., Tokunaga E., Dohi T., Kita Y., Shibata N., Chem. Sci. 2014, 5, 2754. [Google Scholar]
- 25.Deposition numbers 2044630 (2j), 2044631 (2p) and 2047146 (10) contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.
- 26.The Van der Waals volumes of the groups shown in Figure 7 were calculated according to: Zhao Y. H., Abraham M. H., Zissimos A. M., J. Org. Chem. 2003, 68, 7368. Please see [DOI] [PubMed] [Google Scholar]; Bondi A., J. Phys. Chem. 1964, 68, 441. [Google Scholar]
- 27. Jagodzinska M., Huguenot F., Candiani G., Zanda M., ChemMedChem 2009, 4, 49. [DOI] [PubMed] [Google Scholar]
- 28.For an example of selectivity being influenced by electrostatic repulsion from a CF3 group, see: Katagiri T., Yamaji S., Handa M., Irie M., Uneyama K., Chem. Commun. 2001, 2054. [DOI] [PubMed] [Google Scholar]
- 29.
- 29a. Zhou B., Haj M. K., Jacobsen E. N., Houk K. N., Xue X.-S., J. Am. Chem. Soc. 2018, 140, 15206; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29b. Wang Q., Lübcke M., Biosca M., Hedberg M., Eriksson L., Himo F., Szabó K. J., J. Am. Chem. Soc. 2020, 142, 20048–20057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schaefer T., Schurko R. W., Sebastian R., Hruska F. E., Can. J. Chem. 1995, 73, 816. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary