

Abstract
Brain involvement in myotonic dystrophy type 1 (DM1) is characterized by heterogeneous cognitive, behavioral, and affective symptoms and imaging alterations indicative of widespread grey and white matter involvement. The aim of the present study was to systematically review the literature on brain pathology in DM1. We conducted a structured search in EMBASE (index period 1974–2017) and MEDLINE (index period 1887–2017) on December 11, 2017, using free text and index search terms related to myotonic dystrophy type 1 and brain structures or regions. Eligible studies were full‐text studies reporting on microscopic brain pathology of DM1 patients without potentially interfering comorbidity. We discussed the findings based on the anatomical region and the nature of the anomaly. Neuropathological findings in DM1 can be classified as follows: (1) protein and nucleotide deposits; (2) changes in neurons and glial cells; and (3) white matter alterations. Most findings are unspecific to DM1 and may occur with physiological aging, albeit to a lesser degree. There are similarities and contrasts with Alzheimer's disease; both show the appearance of neurofibrillary tangles in the limbic system without plaque occurrence. Likewise, there is myelin loss and gliosis, and there are dilated perivascular spaces in the white matter resemblant of cerebral small vessel disease. However, we did not find evidence of lacunar infarction or microbleeding. The various neuropathological findings in DM1 are reflective of the heterogeneous clinical and neuroimaging features of the disease. The strength of conclusions from this study's findings is bounded by limited numbers of participants in studies, methodological constraints, and lack of assessed associations between histopathology and clinical or neuroimaging findings.
Keywords: brain, microscopic pathology, myotonic dystrophy, systematic review
INTRODUCTION
Myotonic dystrophy type 1 (DM1) is a complex multisystem disease, caused by a cytosine–thymine–guanine (CTG) microsatellite repeat expansion in the 3′‐untranslated region of the dystrophia myotonica protein kinase (DMPK) gene, that has autosomal dominant inheritance. 1 DM1 is a chronic progressive disease characterized by reduced survival, high burden of disease, and loss of quality of life. 2 , 3 Brain involvement in DM1 is phenotypically heterogeneous. 4 Features include variable combination of developmental disorders, cognitive deficits in various domains, personality and behavioral disturbances, affective symptoms, and sleep disorders. 5 , 6 , 7 These clinical features of brain involvement in DM1 are accompanied by alterations in structural and functional brain imaging, as summarized in several recent reviews. 8 , 9 , 10
Despite progress in the understanding of brain correlates of these non‐motor manifestations of DM1, partly due to increasingly sophisticated imaging techniques, less is known about pathological alterations that underly these now well‐recognized clinical and imaging phenotypes in DM1. Various histopathological features have been observed in postmortem brain samples of patients with DM1. 4 In the current study, we systematically reviewed the published literature on microscopic brain alterations in the human DM1 brain. A thorough and comprehensive literature review was conducted to identify gaps in the present knowledge and to inform future studies evaluating microscopic brain alterations in DM1. These goals comport with suggestions and advice recently formulated by the scientific community in DM1. 11 , 12 Therefore, the aim of this study was to provide a systematic overview of the literature on microscopic pathology in the human DM1 brain.
METHODS
Search strategy
We conducted a systematic review of the literature and, where possible, aggregated homologous data. Prior to the commencement of data analysis, we registered the protocol of this study in PROSPERO, an international prospective register of systematic reviews, under ID 42018085626. It can be accessed at: http://www.crd.york.ac.uk/PROSPERO/display_record.php?ID=CRD42018085626.
A single author (RW) conducted a search in EMBASE (index period 1974–2017) and MEDLINE (index period 1887–2017) on December 11, 2017. AC, JR, and BK supervised the search, which was conducted in collaboration with an information specialist of the university library. We used free text and index search terms related to myotonic dystrophy type 1 and brain structures or regions (Appendix I). Additional studies were identified through cross‐referencing. Because we were unaware of the number, sizes, and types of studies to expect, we modified the eligibility criteria during the course of the study, as we felt the initial criteria to be too restrictive or too loose (see Appendix II).
Eligibility criteria and study selection
Studies were eligible if they reported on at least one microscopic pathological brain feature in ≥ 1 patient with confirmed clinical or genetic DM1 diagnosis according to the authors and no evidence of potentially relevant co‐morbidity (i.e. brain disease other than DM1) that could interfere with outcomes of interest. We excluded studies for reasons as follows: (1) unavailability of full text; (2) language of publication other than English, Dutch, French, or German; (3) double reporting (i.e. dual publication of same research results); and (4) conference abstracts. After deduplication, study selection comprised an initial round of title and abstract screening, followed by a second round of full‐text screening. Both screening rounds were independently performed by two authors (RW and KO). Differences in the selection of studies were discussed to meet consensus.
Data extraction
Data extraction was carried out independently by RW and KO. We extracted the following data from each selected study: study characteristics (first author, year of publication, brain areas, and pathological alterations under study); patient characteristics (number, sex, age at death, DM1 disease class, and clinical or genetic DM1 diagnosis). DM1 disease class was defined as either congenital, infantile, juvenile, adult, or late‐onset DM1. Where applicable, we also collected these variables for healthy or disease control subjects that were presented in the included studies.
Data synthesis
To summarize our findings, we classified the pathological alterations reported in the included studies and counted the number of studies reporting these pathological alterations. Accordingly, we subdivided our results on the basis of the most frequently reported alterations.
RESULTS
Search and selection results
Our primary search resulted in a total of 2099 records (Fig. 1). After deduplication and title and abstract screening, a total of 102 records remained for full‐text screening. We excluded 63 studies, mostly for reasons of absence of outcome of interest (n = 40), unsuitable publication type/conference abstract (n = 17), and language restriction (n = 5). We included 39 studies from our primary search and included an additional two studies after cross‐referencing, making a total of 41 studies (Fig. 2). 13 , 14 , 15 , 16 , 17 , 18 , 19 , 20 , 21 , 22 , 23 , 24 , 25 , 26 , 27 , 28 , 29 , 30 , 31 , 32 , 33 , 34 , 35 , 36 , 37 , 38 , 39 , 40 , 41 , 42 , 43 , 44 , 45 , 46 , 47 , 48 , 49 , 50 , 51 , 52 , 53
Fig 1.
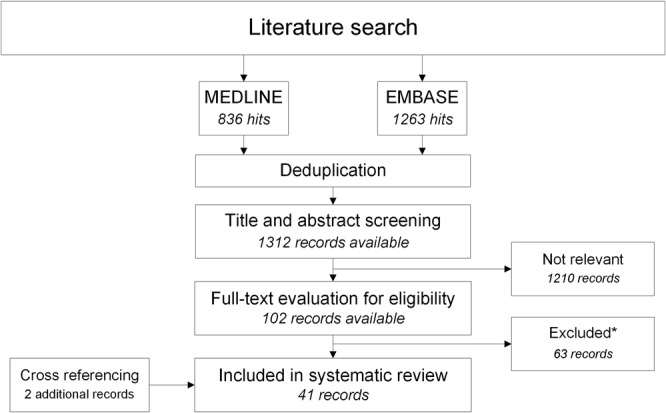
Literature search and study selection with a flow chart of literature search and study selection. *See text for reasons for exclusion. This figure has been created using Microsoft Visio 2007, saved in .jpg image format.
Fig 2.
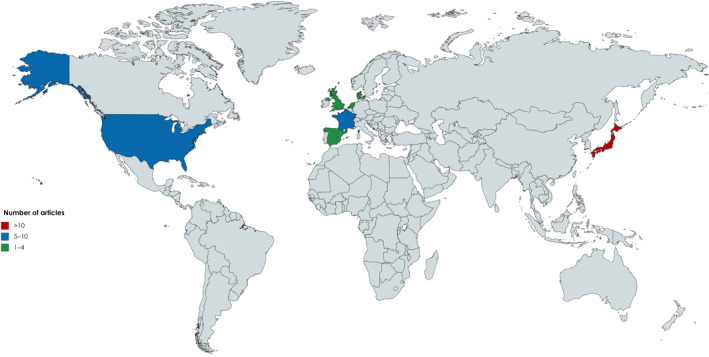
Country of origin of studies. The world map shows country of origin of brain pathology studies in DM1. All studies originate from a total of seven countries. The map has been created with Mapchart.net (https://mapchart.net/) under a CC BY‐SA 4.0 license.
Study characteristics
A total of 130 DM1 patients were included in 41 studies (Table 1). However, as some research groups assessed different pathological aspects in studies on identical subjects (e.g. the group of Ono and colleagues), the number of unique subjects is likely considerably lower. Most studies included a limited number of participants; the largest series investigated the brain of 17 patients and the median number of patients was five. Healthy (n = 292) and disease controls (n = 598) were present in 23 and 16 studies, respectively. The most frequently included disease controls were patients with Duchenne muscular dystrophy (n = 253, two studies), Alzheimer's disease (n = 56, 11 studies), amyotrophic lateral sclerosis (n = 28, six studies) and progressive supranuclear palsy (n = 30, five studies). The most common pathologic features were: (1) protein or nucleotide deposits; (2) cellular alterations in neurons or glial cells; and (3) white matter alterations. For each feature, the relevant study and brain structures in which they were present are shown in Table 2.
Table 1.
Overview of included studies
| Author, year, reference | ID | No. P (M:F) | Age at death (mean ± SD in years) | DM1 disease class ‡ (c:i:j:a:lo) | No. HC | No. DC | Brain areas | Histopathological changes evaluated |
|---|---|---|---|---|---|---|---|---|
| Rosman, 1967 13 | 1 | 4 (2:2) | 47 ± 7 | ? | 0 | 0 | CC | Neuronal heterotopia; disordered lamination |
| Culebras, 1973 14 | 2 | 6 (4:2) | 48 ± 9 | ‐:‐:+:+:‐ | 0 | 3 | CC | Neuronal heterotopia; disordered lamination; astrocytes |
| Thalamus | IIB | |||||||
| Wisniewski, 1975 15 | 3 | 1 (0:1) | 48 | ‐:‐:‐:‐:+ | 0 | 0 | Thalamus | IIB |
| Sarnat, 1976 16 | 4 | 4 (1:3) | 0 † | +:‐:‐:‐:‐ | 0 | 0 | CC | Neuronal size |
| WM | Gliosis | |||||||
| Pena, 1980 17 | 5 | 2 (2:0) | 0; 62 | +:?:?:?:? | 40 | 0 | Thalamus | IIB |
| Young, 1981 18 | 6 | 4 (3:1) | 0 | +:‐:‐:‐:‐ | 0 | 0 | WM | Gliosis |
| BS: medullary pyramids | Gliosis | |||||||
| Ono § , 1989 19 | 7 | 4 (2:2) | 62 ± 4 | ? | 8 | 8 | Thalamus | IIB |
| SN | IIB; MB | |||||||
| HPC | NFT; GVD; plaques | |||||||
| Yoshimura, 1990 20 | 8 | 1 (1:0) | 61 | ‐:‐:‐:+:‐ | 3 | 0 | CC; hypothalamus; NBM; BG; HPC; amygdala; BS; CB; olfactory bulb | Neuronal loss; neuronal size; neuronal degeneration; gliosis; NFT; plaques |
| Thalamus | IIB; neuronal size; neuronal degeneration; gliosis | |||||||
| Garcia‐Alix, 1991 21 | 9 | 4 (4:0) | 0 † | +:‐:‐:‐:‐ | 0 | 0 | WM | Neuronal heterotopia; necrosis; hemorrhage; astrocytes |
| CB | Neuronal heterotopia | |||||||
| Kiuchi, 1991 22 | 10 | 7 (4:3) | 49 ± 8 | ? | 18 | 0 | CC: limbic cortex | NFT; plaques |
| HPC | Neuronal loss | |||||||
| BS: LC, raphe nuclei; WM | NFT | |||||||
| Hageman, 1993 23 | 11 | 5 (3:2) | 0; 4 | +:‐:‐:‐:‐ | 0 | 0 | WM | Periventricular leukomalacia |
| Thalamus | IIB | |||||||
| Moriuchi, 1993 24 | 12 | 17 (?:?) | 50 ±? | ? | 0 | 312 | CC | Neuronal loss |
| ? | NFT | |||||||
| Abe, 1994 25 | 13 | 1 (0:1) | 54 | ? | 0 | 0 | CC; BS | Neuronal loss |
| WM | Myelin loss | |||||||
| Oyanagi, 1994 26 | 14 | 5 (2:3) | 53 ± 9 | ? | 24 | 0 | CC: temporal | NFT; GVD; Hirano; plaques |
| Thalamus | IIB | |||||||
| BG | IIB; NFT | |||||||
| SN | IIB; MB | |||||||
| Lidang Jensen, 1995 27 | 15 | 2 (1:1) | 0 † | +:‐:‐:‐:‐ | 0 | 0 | CC | Neuronal size; neuronal heterotopia |
| BS: hypoglossal nucleus | Neuronal size | |||||||
| Ono § , 1995 28 | 16 | 7 (4:3) | 62 ± 5 | ? | 8 | 0 | BS: raphe nuclei | Neuronal size; gliosis |
| Ono § , 1995 29 | 17 | 1 (0:1) | 53 | ‐:‐:‐:+:‐ | 0 | 0 | BS: raphe nuclei, MRF | Neuronal size; gliosis |
| Ono § , 1996 30 | 18 | 7 (4:3) | 62 ± 5 | ? | 8 | 0 | BS: MRF | Neuronal loss |
| Ono § , 1996 31 | 19 | 8 (5:3) | 62 ± 4 | ? | 0 | 9 | thalamus | IIB |
| Vermersch, 1996 32 | 20 | 2 (2:0) | 53; 61 | ‐:+:‐:+:‐ | 20 | ? | CC: entorhinal, inferior temporal | NFT; plaques |
| thalamus; BG | IIB | |||||||
| HPC; BS; amygdala | NFT | |||||||
| Ono, 1997 33 | 21 | 8 (5:3) | 62 ± 4 | ? | 8 | 9 | SN | IIB |
| Watanabe, 1997 34 | 22 | 1 (1:0) | 48 | +:‐:‐:‐:‐ | 0 | 9 | CC | Neuronal loss; neuronal size; neuronal heterotopia; gliosis; NFT; plaques |
| Thalamus | IIB | |||||||
| Amygdala | NFT | |||||||
| HPC, NBM | Neuronal size | |||||||
| BS: SN | NFT; gliosis; MB | |||||||
| WM | Neuronal heterotopia | |||||||
| Ono § , 1998 35 | 23 | 8 (4:4) | 63 ±? | ? | 12 | 0 | BS: raphe nuclei | Neuronal size |
| Ono § , 1998 36 | 24 | 8 (4:4) | 63 ±? | ? | 10 | 0 | BS: MRF | Neuronal size |
| Mizukami, 1999 37 | 25 | 1 (1:0) | 58 | ? | 0 | 0 | CC: entorhinal cortex | Neuronal loss; neuronal size; astrocytes; NFT |
| Thalamus | IIB | |||||||
| BS: SN, pons: LC, medulla | Neuronal size; Lewy bodies | |||||||
| CB: ventral hemisphere | Neuronal loss; gliosis | |||||||
| NBM | Neuronal loss; gliosis; NFT | |||||||
| WM | Myelin loss; axonal loss; neuronal neuronal heterotopia | |||||||
| Spillantini, 1999 38 | 26 | 1 (0:1) | 54 | ? | 2 | 10 | HPC | NFT; plaques |
| Endo, 2000 39 | 27 | 11 (7:4) | +:‐:‐:+:‐ | 30 | 0 | CC | Neuronal heterotopia; disordered lamination; NFT | |
| Thalamus | NFT; IIB | |||||||
| BG; BS | NFT; neuronal heterotopia | |||||||
| CB | Neuronal heterotopia | |||||||
| WM | Periventricular leukomalacia | |||||||
| Kawashima, 2000 40 | 28 | 6 (3:3) | 59 ± 12 | ? | 40 | 59 | BG | SLI |
| Ono § , 2001 41 | 29 | 8 (4:4) | 63 ± 5 | ? | 8 | 10 | CC; HPC; CB; BS: medullary arcuate nucleus | Neuronal size; neuronal loss |
| Kumada, 2002 42 | 30 | 7 (4:3) | 53 ± 10 | ? | 10 | 0 | thalamus; CB; CC | Intranuclear inclusions |
| SN | Neuronal size; gliosis; MB | |||||||
| Maurage, 2003 43 | 31 | 3 (3:0) | 64 ± 2 | ? | 15 | 32 | CC | NFT |
| WM | NFT | |||||||
| Jiang, 2004 44 | 32 | 10 (7:3) | 56 ±? | ‐:‐:‐:+:+ | 6 | 7 | CC; CB; HPC; thalamus; WM: subcortical, corpus callosum; BS: SN, tegmentum | RNA foci |
| Maurage, 2005 45 | 33 | 3 (3:0) | 59 ± 6 | +:‐:‐:+:‐ | 3 | 2 | CC: entorhinal; HPC; BS – LC, SN; CB | NFT |
| SN | MB | |||||||
| Yamazaki, 2005 46 | 34 | 5 (3:2) | 54 ± 20 | ? | 0 | 101 | WM: frontal | Tau‐positive fine granules |
| Oyamada, 2006 47 | 35 | 12 (6:6) | 55 ± 8 | ? | 4 | 0 | CC; HPC: subiculum | NFT, plaques |
| BS: SN, LC, central grey matter | NFT, plaques, Lewy bodies | |||||||
| Itoh, 2010 48 | 36 | 11 (5:6) | 60 ± 5 | ? | 0 | 0 | CC; CB; BG; HPC; BS | Neuronal size |
| CC: enthorhinal cortex; HPC; amygdala; BS | NFT, plaques | |||||||
| Thalamus | IIB | |||||||
| SN | MB | |||||||
| SN, LC | Gliosis, Lewy bodies | |||||||
| Deep WM | Gliosis | |||||||
| Dhaenens, 2011 49 | 37 | 5 (4:1) | 60 ± 4 | +:‐:‐:+:‐ | 3 | 0 | CC: entorhinal cortex; HPC; amygdala | NFT |
| Yamazaki, 2011 50 | 38 | 5 (4:1) | 57 ± 6 | ? | 9 | 23 | HPC | NFT; GVD |
| Nakamori, 2012 51 | 39 | 3 (2:1) | 55 ± 6 | ? | 3 | 4 | HPC | NFT; GVD |
| Jinnai, 2013 52 | 40 | 9 (5:4) | 60 ± 8 | ? | 0 | 0 | CC: entorhinal cortex; CB | Neuronal loss |
| WM; cerebellar WM | Myelin loss; dilate perivascular spaces; axonal loss; gliosis; capillary hyalinization | |||||||
| Entorhinal cortex; HPC | NFT; plaques | |||||||
| Michel, 2015 53 | 41 | ? | ? | +:‐:‐:‐:‐? | 0 | 0 | ? | RNA foci |
Study included deceased newborns.
Disease class: congenital‐onset DM1; infantile‐onset DM1; juvenile‐onset DM1, adult‐onset DM1 and late‐onset DM1, 54 according to the authors; + denotes presence, − denotes absence. BG, basal ganglia; BS, brain stem; CB, cerebellum; CC, cerebral cortex; GVD, granulovacuolar degeneration; HPC, hippocampus; IIB, intracytoplasmic inclusion body; LC, locus coeruleus; MB, Marinesco body; MRF, medullary reticular formation; NBM, nucleus basalis of Meynert; NFT, neurofibrillary tangle; SLI, skein‐like inclusions; SN, substantia nigra; WM, white matter.
Not all patients in these studies were unique.
DC, disease controls; F, female; HC, healthy controls; M, male; No., number; P, patients.
Table 2.
Summary of histopathological changes, brain regions, and studies
| Histopathological changes | Number of studies | Study IDs | Brain regions |
|---|---|---|---|
| 1. Accumulations of material | |||
| Intracytoplasmic eosinophilic inclusion bodies | 14 | 2; 3; 5; 7; 8; 11; 14; 19; 20; 21; 22; 25; 27; 36 |
Thalamus BG; BS: SN |
| Marinesco bodies | 6 | 7; 14; 22; 30; 33; 36 | SN |
| Neurofibrillary degeneration | 19 | 7; 8; 10; 12; 14; 20; 22; 25; 26; 27; 31; 33; 34; 35; 36; 37; 38; 39; 40 |
Limbic system: amygdala; HPC; CC; limbic Cortex; olfactory bulb CB; BG; NBM; BS: raphe; hypothalamus WM |
| Granulovacuolar degeneration | 4 | 7; 14; 38; 39 | HPC; CC: temporal |
| Plaques | 10 | 7; 8; 10; 14; 20; 22; 26; 35; 36; 40 | Whole brain with focus on limbic system and BS |
| RNA foci | 2 | 32; 41 |
CC; HPC; CB; thalamus; BS: SN WM: subcortical, callosal |
| Other accumulations | 6 | 14; 25; 28; 30; 35; 36 | CC: temporal; thalamus; CB; BG; BS: pons (locus coeruleus), medulla, SN |
| 2. Cellular alterations | |||
| Neuronal alterations | 21 | 1; 2; 4; 8; 9; 10; 12; 13; 15; 16; 17; 18; 22; 23; 24; 25; 27; 29; 30; 36; 40 |
Limbic system (CC: entorhinal; HPC) CC; BG; NBM; BS: SN, raphe nuclei, hypoglossal nucleus; medullary arcuate nucleus, MRF; CB WM |
| Glial alterations | 15 | 2; 4; 6; 8; 9; 16; 17; 22; 25; 30; 36; 40 |
Limbic system (CC: entorhinal cortex; HPC; olfactory bulb; amygdala) CC; thalamus; BG; NBM; CB; BS: raphe nuclei, medullary pyramids, SN, LC; hypothalamus WM |
| 3. White matter pathology | 14 | 4; 6; 9; 10; 11; 13; 22; 25; 27; 31; 32; 34; 36; 40 | WM: deep; subcortical; callosal |
The most/best studied brain regions are indicated in bold font.
BG, basal ganglia; BS, brain stem; CB, cerebellum; CC, cerebral cortex; HPC, hippocampus; LC, locus coeruleus; MRF, medullary reticular formation; NBM, nucleus basalis of Meynert; SN, substantia nigra; WM, white matter.
PROTEIN AND NUCLEOTIDE DEPOSITS
Intracytoplasmic eosinophilic thalamic inclusion bodies
A total of 14 studies examined post‐mortem neuropathological samples of 66 patients for the presence of thalamic inclusion bodies (TI) (Table 2). These thalamic inclusion bodies are round to oval structures located in the cytoplasm of neurons that stain homogeneously eosinophilic with HE staining. Anti‐ubiquitin antibody positivity and tau negativity have been demonstrated with immunohistochemical methods. 31 No more than one to two TI were present per neuron. They were not found in post‐mortem brains of patients with congenital DM1 that died at a young age. 17 , 23 They were found in a variable number (18–100%) of adult DM1 patients and in a variable percentage of thalamic cells (3.5–35%), depending on the study. TI were not specific to DM1 as has been found in unaffected controls and controls with other degenerative brain diseases, although the percentage of affected neurons in DM1 exceeded that in the control populations. 19 The latter finding was not present in a previous study on TI, in which the number of inclusions was higher in healthy controls than in one DM1 patient. 17
Marinesco bodies in the substantia nigra
A total of six studies evaluated the presence of Marinesco bodies (MB) in the substantia nigra neurons in post‐mortem brains of 31 patients. 19 , 26 , 34 , 42 , 45 , 48 Marinesco bodies are eosinophilic intranuclear bodies in the pigmented neurons of the substantia nigra (Fig. 3). MBs are not specific to DM1, as they have been observed in other brain diseases, such as Alzheimer's and amyotrophic lateral sclerosis, and in physiological aging. 19 , 42 MB may be accompanied by eosinophilic intracytoplasmic inclusion bodies that highly resemble thalamic inclusion bodies at the structural and ultrastructural (i.e. electron microscopy) level. 19 As with these intracytoplasmic antibodies, immunohistochemistry has demonstrated anti‐ubiquitin antibody positivity of MB. Whereas one study found a higher load of MB in DM1 patients compared to controls with systemic or neurodegenerative disease, 19 equal loads in patients versus controls were found in substantia nigra neurons in another study. 42
Fig 3.
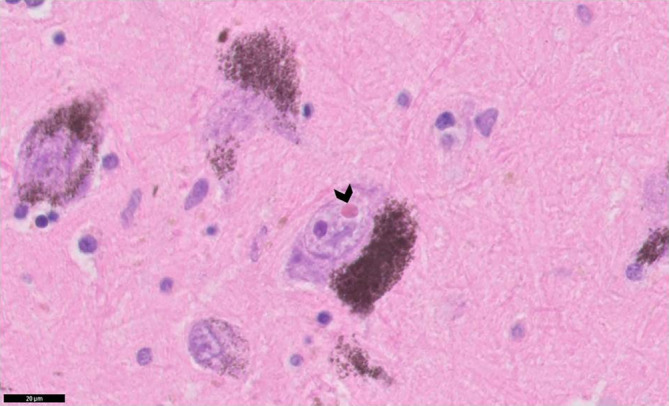
Hitological features of an MB in the substantia nigra. Black arrowhead depicts a Marinesco body (an eosinophilic structure) adjacent to a similar‐sized nucleolus (a basophilic structure). This image is kindly provided by Dr. Kusters.
Neurofibrillary tangles
A total of 19 studies reported on the presence or absence of tau pathology in 104 DM1 patients (Table 2). Most studies detected neurofibrillary tangles (NFT) in all or most of the patients that were evaluated. 22 , 43 , 45 , 47 , 49 Reportedly, the brain areas that demonstrated the highest burden of neurofibrillary tangles (NFTs) were the entorhinal cortex, (subdivisions of) the hippocampus, the parahippocampal cortex, the temporal cortex, and the amygdala. Other, often less affected areas included the olfactory bulb, 20 brain stem, 45 , 47 basal ganglia, 20 , 47 cerebellum, 45 and other (non‐temporal lobe) parts of the neocortex 20 , 34 , 43 , 45 , 49 The load of NFTs was higher in DM1 patients than in controls in three studies. 22 , 45 , 47 In contrast to the aforementioned studies, Moriuchi et al. 24 found NFT in only two out of 17 DM1 patients, and Ono et al. 19 found no or few NFTs in the hippocampus in DM1 patients. Studies reported no or incidental “plaques” accompanying the tau‐related neuropathological changes. 20 , 22 , 32 , 34 , 45 , 47 Studies demonstrated that the NFT in DM1 are preferentially composed of tau proteins devoid of the amino acids encoded by exons 2 and 3 of the microtubule associated protein tau (MAPT) gene. 32 , 45 , 49
Nuclear accumulations
Two studies demonstrated the presence of abnormal, mutant RNA accumulations in the nucleus of brain cells using fluorescence in situ hybridization techniques. 44 , 53 These accumulations have been coined “RNA foci.” Jiang et al. 44 observed these foci diffusely throughout the brain of adult DM1 patients, in neurons of the cortex, hippocampus, thalamus, and brainstem, including the substantia nigra. Of note, the cerebellum displayed small foci in the Purkinje cells but not in neurons of the molecular or granule cell layer. Foci were sometimes multiple and were also occasionally observed in oligodendrocytes of white matter in subcortical and callosal areas. 44 More recently, Michel et al. 53 demonstrated RNA foci GVD in the temporal cortex that coincided in the human brain in the earliest (i.e. fetal and embryonal) stages of development.
Other accumulations
Four studies (total n = 17) noted the presence of granulovacuolar degeneration (GVD), involving electron‐dense granules within double membrane‐bound cytoplasmic vacuoles, in the temporal lobe/hippocampal area of DM1 patients. 19 , 26 , 50 , 51 GVD were also found in other neurodegenerative diseases. 50 Ono et al. 19 (n = 4 patients) found few GVD in the hippocampal area, and two out of five patients from the study by Oyanagi et al. 26 had “several” GVD in the temporal cortex that coincided with NFTs and Hirano bodies. In hippocampal pyramidal neurons, one group demonstrated a strong relationship between GVD and tau pathology. 50 , 51 Skein‐like ubiquitin‐positive inclusions in the striatum were observed in aging controls and in patients with various neurodegenerative disease, including five out six patients with myotonic dystrophy type 1. 40
CELLULAR ALTERATIONS
Neuronal loss
In four post‐mortem cases of children with congenital onset DM1, no neuronal loss was found in the brain on autopsy. 21 In adults, severe neuronal loss was demonstrated in a proportion of patients as follows: (1) dorsal raphe nucleus and superior central nucleus of the midbrain; (2) pontine raphe and pontine reticular formation; and (3) dorsal medullary central nucleus, ventral medullary central nucleus, medullary arcuate nucleus, and subtrigeminal medullary nucleus in the medulla. 28 , 29 , 30 , 41 The authors noted this neuronal loss in the brainstem in association with hypersomnia and hypoventilation. 28 , 30 In contrast, Mizukami et al., 37 Itoh et al., 48 and Lidang Jensen et al. 27 reported no loss of neurons in the (pontine and medullary) brain stem. In the substantia nigra, mild neuronal loss was observed in two case reports 34 , 37 ; two case series demonstrated neuronal loss in zero out of seven and two out of 11 patients in this region. 42 , 48 Similarly, neuronal loss was observed in the cerebellum in the case in Mizukami et al. 37 but absent in two case series on 11 and nine patient evaluating cerebellar granule and Purkinje cells, respectively. 48 , 52 In the cerebral cortex, Maurage et al. 45 (n = 3), Abe et al. 25 (n = 1), Ono et al. 41 (n = 8), Watanabe et al. 34 (n = 1), and Lidang Jensen et al. 27 (n = 2) observed no neuronal loss. Other studies observed cortical neuronal loss in a variable proportion of patients: Mizukami et al. 37 (one out of one), Itoh et al. 48 (six out of 11), and Jinnai et al. 52 (seven out of nine). Cortical neuronal atrophy was observed by Yoshimura et al. 20 and Watanabe et al. 34 The basal ganglia did not demonstrate neuronal loss. 25 , 34 , 48 We did not find studies that evaluated neuronal loss in the thalamus. In the hippocampus, mild neuronal loss was found by Watanabe et al. 34 (n = 1) but not by Ono et al. 41 (n = 8) and Itoh et al. 48 (n = 11). In the adjacent entorhinal cortex, however, neuronal loss was observed by Jinnai et al. 52 (9 out of 9) and Itoh et al. 48 (6 out of 11). Neuronal loss was also found in the nucleus basalis of Meynert, 34 , 37 and in the locus coeruleus. 37 , 48 With two exceptions, 30 , 41 most studies did not report how neuronal loss was specifically evaluated. Neuronal loss was quantified qualitatively or semi‐quantitatively. The study by Ono et al., 41 in which design‐based stereological techniques were used, is a notable exception.
Neuronal heterotopia
Heterotopic neurons were found in the white matter in three studies 13 , 21 , 34 that evaluated the brains from nine patients. Four brains were from patients with congenital DM1 that died shortly after birth; the other five were from DM1 patients that deceased at ages 36 to 51. Heterotopic neurons were found both in subcortical and deep white matter.
Reactions of glial cells
Eight studies evaluated the presence of gliosis in the brain of 37 DM1 patients. In the two largest studies (n = 11 and n = 9), gliosis was observed diffusely throughout the deep white matter. 48 , 52 Itoh et al. 48 also found astrogliosis in two out of 11 brains in the locus coeruleus and substantia nigra. In contrast, Kumada et al. 42 observed no gliosis in the substantia nigra in any of seven brains. Ono et al. found gliosis in six out of eight patients in the raphe nuclei and superior central nucleus of the brainstem that accompanied neural loss. 28 , 29 Yoshimura et al. 20 and Mizukami et al. 37 observed cortical gliosis. In the case described by Yoshimura et al., 20 cortical gliosis was accompanied by diffuse gliosis throughout the brain, including the cerebellum, the brain stem, and the limbic system. In another case report, no cortical gliosis was found. 34 Studies did not typically specify the type of gliosis (e.g. reactive astrogliosis, microgliosis/activated microglia cells, or oligodendrocyte response).
WHITE MATTER PATHOLOGY
Various neuropathological findings in white matter were reported in 14 studies (Tables 1 and 2). There was loss of myelin from deep and subcortical white matter, and widened perivascular spaces (Figs 4, 5), giving the white matter an “état criblé” appearance. The largest study (n = 11) reported loss of axons accompanying the myelin loss, 48 whereas in two case reports myelin loss coincided with “relative axonal sparing.” 25 , 37 Itoh et al. 48 also found capillary hyalinization and fibrillary gliosis.
Fig 4.
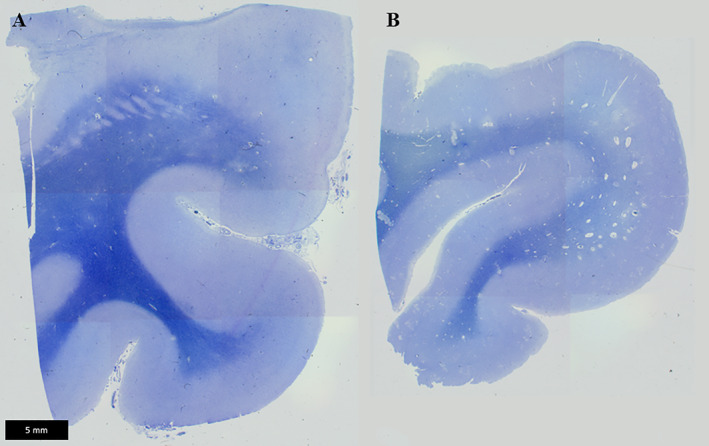
Perivascular dilatation in the cerebrum. In the temporal lobe, enlarged perivascular spaces are observed on sections, stained Klüver–Barrera, from a DM1 patient (B) but not a control individual (A).
Fig 5.
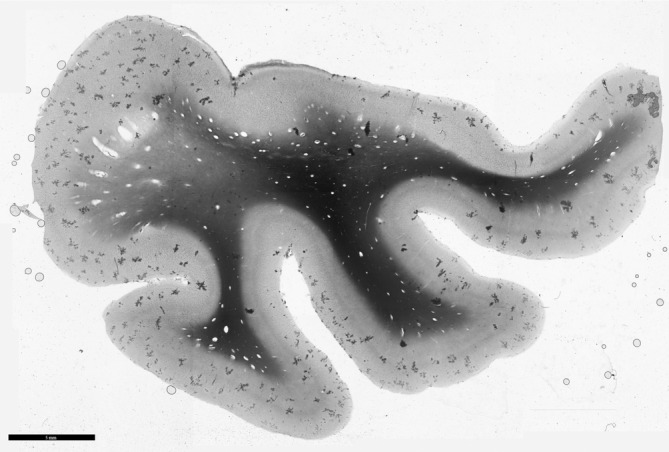
Perivascular dilatation in the cerebrum. Enlarged perivascular spaces are identified by polarized light imaging in the same patient as in Figure 4.
DISCUSSION
The various and anatomically diffuse microscopic brain alterations in myotonic dystrophy type 1 are a reflection of the heterogeneous clinical features and imaging characteristics of brain involvement of the disease (Fig. 6). On the basis of our findings, we classified microscopic brain pathology in DM1 as: (1) protein and nucleotide deposits; (2) cellular anomalies; and (3) white matter pathology (which will be discussed in more detail in the next sections).
Fig 6.
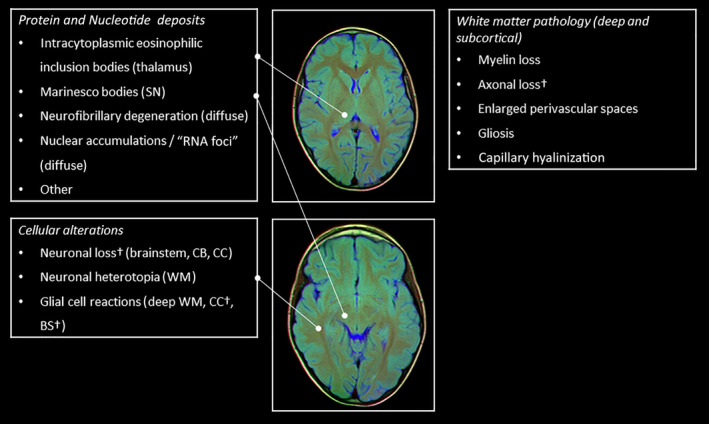
Summary of brain imaging findings. †Inconsistent findings across studies. BS, brain stem; CB, cerebellum; CC, cerebral cortex; HPC, hippocampus; SN, substantia nigra; WM, white matter. Brain MRI slice images are provided by Nevit Dilmen, used under Attribution‐ShareAlike 3.0 Unported (CC BY‐SA 3.0) license [https://creativecommons.org/licenses/by‐sa/3.0/deed.en], downloaded from commons.wikimedia.org.
Protein and nucleotide deposits
The presence of various deposits has been noted in histopathologic studies in DM1. The first descriptions include the intracytoplasmic eosinophilic bodies in the thalamus and the MBs in the substantia nigra. Their clinical relevance is uncertain, as MB could be found in persons as young as 21 years of age and are not associated with a specific disease. 55 However, their prevalence in the substantia nigra increases with age and they could be considered as a marker for neurodegeneration. 56 Immunohistochemically, MB are generally positive for proteins of the ubiquitin–proteasome system. 57 Their increased occurrence in the substantia nigra in DM1, if validated in controlled, larger studies, might imply a relationship between protein degradation systems, abnormal proteins resulting from aberrant splicing in DM1, and neurodegeneration. Interestingly, echography studies have demonstrated hyperechogenicity of the substantia of the substantia nigra in a proportion of DM1 patients, in comparison to controls. 58 Furthermore, the presence of accumulations and neuronal loss in the brainstem (see below) is interesting in light of abnormal levels of dopamine and serotonin metabolites in the cortex and the brain stem, respectively, in animals studies. 59 These findings invite the study of in vivo (i.e. CSF) and ex vivo neurotransmitter metabolism in DM1‐affected individuals.
More recently, the demonstration of RNA “foci,” toxic accumulations of mutant RNA, in the brain provide a direct link between the molecular pathophysiology of DM1 and visible histopathological alterations. 44 , 53 The CTG repeat expansion in the 3′‐untranslated region of the DMPK is transcribed into long CUG‐containing mRNAs that remain in the nucleus and may sequestrate RNA‐binding proteins and, consequently, lead to transcriptional dysregulation. 60 The formation of these alterations is likely located upstream in the cascade of pathophysiological brain changes in DM1, and they have been shown to start very early in the disease process. Interestingly, the development of brain “foci” is well captured in animal models and, consequently, is an aspect that is open to future therapeutic study in DM1.
Tau protein is encoded by a single gene (MAPT) on chromosome 17q21 and represented by different isoforms generated by alternative splicing from the MAPT gene. An expanding spectrum of diseases now classified as “tauopathies” is characterized by NFTs that consist of aggregates of hyperphosphorylated and abnormally phosphorylated isoforms of tau proteins. 61 , 62 In several of the tauopathies, aberrant splicing of the MAPT RNA has been demonstrated, and aberrant alternative tau splicing could promote tau pathology. 63 Tau missplicing results in isoform‐specific impairment in normal physiological function and enhanced recruitment of excessive tau isoforms into the pathological process. 63 In DM1, recent work has suggested an interplay between toxic RNA, disordered splicing, and the dysregulation of microtubule‐associated protein tau. 64 , 65 In DM1, the “foci,” consisting of accumulations of toxic RNA, are known sequester RNA‐binding proteins, such as splicing factors, consequently leading to dysregulated alternative splicing in neurons. 64 , 65 Accordingly, alterations of tau alternative splicing have been reported in DM1 with missplicing of exons 2 and 6 in the brain. Thus, DM1 is characterized by the preferential accumulation of the 0N3R isoform. 32 Moreover, several studies provide evidence of the effects of missplicing on tau aggregation, protein–protein interactions, microtubule stabilization, and axonal transport, which may be relevant to myotonic dystrophy type 1. 64 , 65
In DM1, neurofibrillary degeneration (NFD) is preferentially seen in parts of the limbic system including (parts of) the hippocampus and adjacent cortex, but other brain regions may also show signs of tau pathology. Clinical features of DM1, such as diminished olfaction, apathy, and learning and memory disorders may reflect involvement of the limbic system; 5 , 66 , 67 however, the exact relation with NFD in this area is unknown. It should be noted that tau pathology can be present in brains of individuals that have shown healthy aging from a clinical perspective, and that tau missplicing is not restricted to tauopathies but can also be present in non‐tau proteinopathies. 62 , 63 It remains to be seen to what extent tau pathology in DM1 is causal to the heterogeneous brain phenotypes of the disease. Besides this uncertainty about the clinicopathological correlations, the spatiotemporal progression of NFD in DM1 is undiscovered at present. PET‐Tau imaging offers a means to study such progression in vivo and could be applied to DM1 in future studies. 68 In addition, the relationship of tau pathology with other histopathological features of DM1 should be further studied. Understanding the pathophysiology of DM may increase our knowledge of the complexity of the pathological interplay among RNAopathy, spliceopathy, and proteinopathy implicated in tauopathies and other neurodegenerative diseases. 65
Cellular alterations
Neuronal loss in the brainstem was inconsistently present across studies. Neuronal loss in the brainstem may relate to clinical features of excessive daytime sleepiness and ventilatory dysfunction. 28 , 29 Brainstem neuronal loss was not captured in a DM mouse model with respiratory dysfunction. 69 In the cerebellum, neuronal loss has been the subject of limited study but does not seem to be a generalized finding. In common with other trinucleotide repeat expansion, somatic instability (i.e. increase in size) of the CTG repeat was lower in cerebellar tissue than in other brain regions. 70 Of note, most structural imaging studies (voxel‐based volumetry and diffusion tensor imaging) are less suitable for the study of the brain stem and the cerebellum in comparison to the hemispheral grey matter. However, alterations in brainstem raphe echogenicity in DM1, noted by Krogias and colleagues, may be a possible correlate to brainstem histopathological changes. 58 In the cerebral cortex, histopathological results were mixed, with some studies demonstrating no neuronal loss and others only in a proportion of patients. Cortical neuronal loss is in line with magnetic resonance spectroscopy studies that found decreased concentrations of N‐acetylaspartate, mostly in cortical grey matter. Possible explanations for the divergent findings between studies may include heterogeneous patient populations, (unrecognized) co‐morbidities, and technical reasons. Importantly, with few exceptions, neuronal loss was “quantified” in a qualitative (i.e. present/absent) or semi‐quantitative manner in all studies. Although gross changes may be detected with these methods, the number of a specific cell type cannot be simply deduced from the number of cross‐sectional profiles found in thin tissue sections, as this parameter also depends on cell volume, tissue orientation, and tissue atrophy. 71 , 72 Consequently, pending more advanced studies, these results on neuronal loss should be interpreted with caution. In contrast to studies on neuronal loss, we found no studies that evaluated synaptic density in the brain of DM1 patients. Such studies would be of interest, as imaging studies have found grey matter volume loss throughout the cortex in human DM1 patients. 73 , 74 One correlate of grey matter volume loss based on voxel‐based‐morphometry could be neuronal loss, but given the findings in this study, this is unlikely the only explanation. Reduced synaptic density may provide a complementary explanation that could be worth exploring. 75 In cell and animal models, there is evidence that the CTG repeat expansion of DM1 may affect synaptogenesis and synaptic function. 59 , 76 Finally, as a developmental alteration, heterotopic neurons may be found in the white matter of DM1 patients. Their clinical relevance in relation to central nervous system features of congenital and infantile‐onset DM1 is currently unknown.
White matter changes
Degenerative characteristics of white matter include loss of myelin in combination with a varying amount of axonal loss, dilation of perivascular spaces, gliosis, and capillary hyalinization in deep and subcortical white matter. These findings are in line with white matter alterations captured with in vivo neuroimaging: dilated perivascular spaces, T2/FLAIR hyperintensities, and alterations in microstructurem, as demonstrated with DTI imaging. 74 , 77 , 78 Although the white matter histological alterations in DM1 share characteristics with cerebral small‐vessel disease, other characteristics of the latter, such as microbleeds and lacunes, have not been observed in DM1. Consequently, chronic brain ischemia seems unlikely to be the major mechanism through which white matter lesions originate. Renard et al. (2018) proposed increased burden due to microvascular changes and lack of drainage of interstitial fluid and degraded protein products as a mechanism for anterior temporal white matter lesions in DM1. 79 This hypothesis was based on the co‐localization of dilated perivascular spaces and white matter hyperintensities in the anterior temporal pole, which DM1 shares with the monogenetic small vessel disease cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL). 79 , 80 Of interest, dilated perivascular spaces were recently demonstrated to be associated with the burden of neurofibrillary tangles in Alzheimer's disease. 81 At the basis of the association of dilated perivascular spaces with various tauopathies, including DM1 and Alzheimer's disease, could be a common pathophysiological mechanism (e.g. astrocytic dysfunction) worth further exploring.
CONCLUSION
Summarizing our results, clinical and neuroimaging heterogeneity of DM1 reflect the various histopathological changes in DM1. Arbitrarily, histopathological findings can be classified as: (1) protein and nucleotide deposits; (2) cellular alterations including neuronal loss and gliosis; and (3) white matter pathology. We observed both anomalies that could be considered developmental in nature (e.g. heterotopic neurons) and neurodegeneration. Indeed, many of the neuropathological findings in DM1 share similarities with other neurodegenerative diseases and can be seen in “physiological” aging (albeit to a lesser degree). The latter findings are compatible with the concept of DM1 as a progeroid/accelerated aging disease. 82 , 83 However, in common with the highly variable and unique admixture of clinical brain features in DM1, the specific combination of histopathological findings seems to be unique. For some aspects such as neurofibrillary degeneration, great progress has been made in linking the pathophysiological concepts of RNAopathy and spliceopathy to histopathological changes. 64 These interrelations remain elusive for other pathological findings at this time.
In a seminal editorial on “myotonic dystrophy as a brain disorder”, Ashizawa (1998, p. 291)noted that “availability of brain autopsy materials in DM is limited and the MR imaging data are difficult to correlate with the histological findings.” 84 Unfortunately, some 20 years later, we must conclude that this has not changed. Most published studies are case reports and small case series. These stand in sharp contrast to a large and increasingly sophisticated body of literature on neuroimaging in DM1. 8 , 9 However, studies that correlate in vivo neuroimaging with post‐mortem histopathological findings are virtually non‐existent. Clearly, there is great need for larger follow‐up studies that evaluate the associations between the many neuroimaging findings and microscopic alterations in relation to clinical DM1 features. Furthermore, more detailed morphometric studies of animal model brains would inform us of which aspects of the human DM1 histopathology are captured by the (mouse) models. To facilitate such studies, international collaboration with an established set of clinical and imaging variables and a logistical pipeline that allows identical handling (e.g. design‐based stereology for morphometric aspects) of brain autopsy samples is essential and should be high on the DM1 scientific research agenda. 11 , 12
DISCLOSURE
The authors declare no conflict of interest.
In the original study protocol, we only included studies with patients with genetically proven DM1. This criterion was modified to the criterion presented in Appendix III. Because a lot of our studies were published before or shortly after the discovery of the DMPK gene in 1992, we felt that this item was too restrictive as we had to exclude approximately 50% of otherwise eligible studies based on this criterion.
In the original study protocol, we included studies that obtained results exclusively by macroscopic and/or genetic examinations. This criterion was modified to the criterion presented in Appendix III. We felt that the inclusion of studies on each study level (i.e. macroscopic, microscopic, and genetic) would be too extensive. We decided to focus on microscopic alterations and excluded those that did not present at least one microscopic outcome.
| PubMed (836 hits) | EMBASE (1263 hits) |
|---|---|
|
(“Myotonic Dystrophy”[Mesh] OR Curschmann*[tiab] OR Dystrophia Myotoni*[tiab] OR Dystrophic Myoton*[tiab] OR Myotonia Atrophica*[tiab] OR Myotonia Dystrophi*[tiab] OR Myotonic Dystroph*[tiab] OR Steinert*[tiab] OR Steinert's[tiab]) AND (“Brain”[Mesh] OR “Myelin Sheath”[Mesh] OR Amygdal*[tiab] OR Brain[tiab] OR Brains[tiab] OR Brainstem*[tiab] OR Caeruleus[tiab] OR Callos*[tiab] OR Cerebr*[tiab] OR Ceruleus[tiab] OR Coeruleus[tiab] OR Corpus Callos*[tiab] OR Cortex*[tiab] OR Cortical[tiab] OR Cortices[tiab] OR Corticospinal*[tiab] OR Diencephal*[tiab] OR Encephal*[tiab] OR Epithalam*[tiab] OR Fascicul*[tiab] OR Glia[tiab] OR Glial[tiab] OR Gray Matter*[tiab] OR Grey Matter*[tiab] OR Hippocamp*[tiab] OR Hypothalam* [tiab] OR Limbic*[tiab] OR Lymbic*[tiab] OR Mesencephal*[tiab] OR Mesolimbic*[tiab] OR Metencephal*[tiab] OR Midbrain*[tiab] OR Myelencephal*[tiab] OR Olfactory Pathway*[tiab] OR Parahippocampal*[tiab] OR Perforant Nerve Tract*[tiab] OR Perforant Path*[tiab] OR Telencephal*[tiab] OR Ventricl*[tiab] OR Ventricul*[tiab] OR White Matter*[tiab]) |
(myotonic dystrophy/ OR (Curschmann* OR Dystrophia Myotoni* OR Myotonia Atrophica* OR Myotonia Dystrophica* OR Myotonic Atroph* OR Myotonic Dystroph* OR Steinert* OR Steinert's).ti,ab,kw.) AND (exp brain/ OR exp. brain tissue/ OR exp. brain ventricle/ OR myelin/ OR myelin sheath/ OR perforant nerve tract/ OR (Amygdal* OR Brain OR Brains OR Brainstem* OR Caeruleus OR Callos* OR Cerebr* OR Ceruleus OR Coeruleus OR Corpus callos* OR Cortex* OR Cortical OR Cortices OR Corticospinal* OR Diencephal* OR Encephal* OR Epithalam* OR Fascicul* OR Glia OR Glial OR Gray Matter* OR Grey Matter* OR Hippocamp* OR Hypothalam* OR Limbic* OR Lymbic* OR Mesencephal* OR Mesolimbic* OR Metencephal* OR Midbrain* OR Myelencephal* OR Olfactory Pathway* OR Parahippocampal* OR Perforant Nerve Tract* OR Perforant Path* OR Telencephal* OR Ventricl* OR Ventricul* OR White Matter*).ti,ab,kw.) |
| Number | First author | Year | Journal | Notes |
|---|---|---|---|---|
| Language restrictions | ||||
| 1 | Tomi H | 1986 | Clinical Neurology | Article in Japanese |
| 2 | Mitake S | 1989 | Rinsho Shinkeigaku | Article in Japanese |
| 3 | Yoneyama S | 1992 | Rinsho Shinkeigaku | Article in Japanese |
| 4 | Haranaka M | 2000 | No To Hattatsu | Article in Japanese |
| 5 | Iwata T | 2009 | Rinsho Shinkeigaku | Article in Japanese |
| Conference abstracts | ||||
| 1 | Dhaenens C | 2009 | Medizinische Genetik | |
| 2 | Gourdon G | 2009 | Medizinische Genetik | |
| 3 | Kang Y | 2009 | Medizinische Genetik | |
| 4 | Kuru S | 2010 | Neuropathology | |
| 5 | Saito Y | 2010 | Neuropathology | |
| 6 | Caillet‐Boudin M | 2011 | Alzheimer's and Dementia | |
| 7 | Ikeda K | 2011 | Neuropathology | |
| 8 | Jinnai K | 2011 | Neuropathology | |
| 9 | Kuru S | 2011 | Neuropathology | |
| 10 | Matsuda M | 2012 | Neuropathology | |
| 11 | Imanishi A | 2013 | Sleep | |
| 12 | Komene T | 2013 | Neuropathology | |
| 13 | Oya Y | 2013 | Neuropathology | |
| 14 | Servais L | 2015 | Neuromuscular Disorders | |
| 15 | Iwasaki Y | 2016 | Clinical Neurology | |
| 16 | Sano T | 2016 | Clinical Neurology | |
| 17 | Shioya A | 2016 | Clinical Neurology | |
| Cell/tissue culture studies | ||||
| 1 | Hernandez‐Hernandez O | 2006 | Journal of Neuroscience Research | |
| 2 | Ghanem D | 2009 | FEBS Letters | |
| 3 | Marteyn A | 2011 | Cell Stem Cell | |
| 4 | Velazquez‐Bernardinho P | 2012 | Molecular Biology Reports | |
| 5 | Reddy K | 2014 | Nucleic Acids Research | |
| Animal studies | ||||
| 1 | Balasubramanyam A | 1998 | Journal of Comparative Neurology | |
| 2 | Seznec H | 2001 | Human Molecular Genetics | |
| 3 | Sato S | 2002 | Human Molecular Genetics | |
| 4 | Westerlaken J | 2003 | Brain Research | |
| 5 | Garcia‐Lopez A | 2008 | PLoS One | |
| 6 | Oude Ophuis R | 2009 | Muscle Nerve | |
| 7 | Charizanis K | 2012 | Neuron | |
| 8 | Wang E | 2012 | Cell | |
| 9 | Hernandez‐Hernandez O | 2013 | Rare Diseases | |
| 10 | Poulos M | 2013 | Human Molecular Genetics | |
| 11 | Choi J | 2016 | Scientific Reports | |
| 12 | Wang P | 2017 | Human Molecular Genetics | |
| Genetic studies | ||||
| 1 | Ishii S | 1996 | Human Genetics | |
| 2 | Gennarelli M | 1999 | Neuromuscular Disorders | |
| 3 | Korade‐Mirnics Z | 1999 | Human Molecular Genetics | |
| 4 | Sergeant N | 2001 | Human Molecular Genetics | |
| 5 | Leroy O | 2006 | Biochimica et Biophysica Acta | |
| 6 | Leroy O | 2006 | Journal Neuroscience Research | |
| 7 | Dhaenens C | 2008 | Experimental Neurology | |
| 8 | Axford M | 2011 | Journal of Medical Genetics | |
| 9 | Lopez Castel A | 2011 | Human Molecular Genetics | |
| 10 | Suenaga K | 2012 | PLoS One | |
| 11 | Axford M | 2013 | PLoS Genetics | |
| 12 | Hernandez‐Hernandez O | 2013 | Brain | |
| 13 | Goodwin M | 2015 | Cell Reports | |
| CSF studies | ||||
| 1 | Hirase T | 1984 | Brain and Development | |
| 2 | Martinez‐Rodriguez J | 2003 | Sleep | |
| 3 | Ciafaloni E | 2008 | Neurology | |
| 4 | Winblad S | 2008 | European Journal of Neurology | |
| Double reporting | ||||
| 1 | Ono S | 1987 | Journal of the Neurological Sciences | |
| 2 | Yoshimura N | 1990 | Clinical Neuropathology | |
| Significant neurological comorbidities | ||||
| 1 | Kobayashi S | 2005 | Journal of the Neurological Sciences | |
| No patient tissue used | ||||
| 1 | Gennarelli M | 1995 | Biochemical and Biophysical Research Communications | |
| 2 | Whiting E | 1995 | Human Molecular Genetics | |
| 3 | Wang J | 2007 | Journal of Neurochemistry | |
| 4 | Cleary J | 2010 | Nature Structural & Molecular Biology | |
REFERENCES
- 1. Harper PS. Myotonic Dystrophy, 3rd edn. London: W.B. Saunders, 2001. [Google Scholar]
- 2. Landfeldt E, Nikolenko N, Jimenez‐Moreno C et al. Disease burden of myotonic dystrophy type 1. J Neurol 2019; 266: 998–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wahbi K, Porcher R, Laforet P et al. Development and validation of a new scoring system to predict survival in patients with Myotonic dystrophy type 1. JAMA Neurol 2018; 75: 573–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gourdon G, Myotonic dystrophies MG. State of the art of new therapeutic developments for the CNS. Front Cell Neurosci 2017; 11: 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Okkersen K, Buskes M, Groenewoud J et al. The cognitive profile of myotonic dystrophy type 1: A systematic review and meta‐analysis. Cortex 2017; 95: 143–155. [DOI] [PubMed] [Google Scholar]
- 6. van der Velden BG, Okkersen K, Kessels RP et al. Affective symptoms and apathy in myotonic dystrophy type 1 a systematic review and meta‐analysis. J Affect Disord 2019; 250: 260–269. [DOI] [PubMed] [Google Scholar]
- 7. Laberge L, Begin P, Montplaisir J, Mathieu J. Sleep complaints in patients with myotonic dystrophy. J Sleep Res 2004; 13: 95–100. [DOI] [PubMed] [Google Scholar]
- 8. Minnerop M, Gliem C, Kornblum C. Current Progress in CNS imaging of Myotonic dystrophy. Front Neurol 2018; 9: 646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Okkersen K, Monckton DG, Le N, Tuladhar AM, Raaphorst J, van Engelen BGM. Brain imaging in myotonic dystrophy type 1: A systematic review. Neurology 2017; 89: 960–969. [DOI] [PubMed] [Google Scholar]
- 10. Serra L, Silvestri G, Petrucci A et al. Abnormal functional brain connectivity and personality traits in myotonic dystrophy type 1. JAMA Neurol 2014; 71: 603–611. [DOI] [PubMed] [Google Scholar]
- 11. Axford MM, Pearson CE. Illuminating CNS and cognitive issues in myotonic dystrophy: Workshop report. Neuromuscul Disord 2013; 23: 370–374. [DOI] [PubMed] [Google Scholar]
- 12. Bosco G, Diamanti S, Meola G, on behalf of the DM‐CNS Group . Workshop report: Consensus on biomarkers of cerebral involvement in myotonic dystrophy, 2–3 December 2014, Milan, Italy. Neuromuscul Disord 2015; 25: 813–823. [DOI] [PubMed] [Google Scholar]
- 13. Rosman NP, Rebeiz JJ. The cerebral defect and myopathy in myotonic dystrophy. A comparative clinicopathological study. Neurology 1967; 17: 1106–1012. [DOI] [PubMed] [Google Scholar]
- 14. Culebras A, Feldman RG, Merk FB. Cytoplasmic inclusion bodies within neurons of the thalamus in myotonic dystrophy: A light and electron microscope study. J Neurol Sci 1973; 19: 319–329. [DOI] [PubMed] [Google Scholar]
- 15. Wisniewski HM, Berry K, Spiro AJ. Ultrastructure of thalamic neuronal inclusions in myotonic dystrophy. J Neurol Sci 1975; 24: 321–329. [DOI] [PubMed] [Google Scholar]
- 16. Sarnat HB, O'Connor T, Byrne PA. Clinical effects of Myotonic dystrophy on pregnancy and the neonate. JAMA Neurol 1976; 33: 459–465. [DOI] [PubMed] [Google Scholar]
- 17. Pena CE. Intracytoplasmic neuronal inclusions in the human thalamus. Light‐microscopic, histochemical, and ultrastructural observations. Acta Neuropathol 1980; 52: 157–159. [DOI] [PubMed] [Google Scholar]
- 18. Young RS, Gang DL, Zalneraitis EL, Krishnamoorthy KS. Dysmaturation in infants of mothers with myotonic dystrophy. Arch Neurol 1981; 38: 716–719. [DOI] [PubMed] [Google Scholar]
- 19. Ono S, Inoue K, Mannen T, Mitake S et al. Intracytoplasmic inclusion bodies of the thalamus and the substantia nigra, and Marinesco bodies in myotonic dystrophy: A quantitative morphological study. Acta Neuropathol 1989; 77: 350–356. [DOI] [PubMed] [Google Scholar]
- 20. Yoshimura N, Otake M, Igarashi K, Matsunaga M, Takebe K, Kudo H. Topography of Alzheimer's neurofibrillary change distribution in myotonic dystrophy. Clin Neuropathol 1990; 9: 234–239. [PubMed] [Google Scholar]
- 21. Garcia‐Alix A, Cabanas F, Morales C et al. Cerebral abnormalities in congenital myotonic dystrophy. Pediatr Neurol 1991; 7: 28–32. [DOI] [PubMed] [Google Scholar]
- 22. Kiuchi A, Otsuka N, Namba Y, Nakano I, Tomonaga M. Presenile appearance of abundant Alzheimer's neurofibrillary tangles without senile plaques in the brain in myotonic dystrophy. Acta Neuropathol 1991; 82: 1–5. [DOI] [PubMed] [Google Scholar]
- 23. Hageman AT, Gabreels FJ, Liem KD, Renkawek K, Boon JM. Congenital myotonic dystrophy; a report on thirteen cases and a review of the literature. J Neurol Sci 1993; 115: 95–101. [DOI] [PubMed] [Google Scholar]
- 24. Moriuchi T, Kagawa N, Mukoyama M, Hizawa K. Autopsy analyses of the muscular dystrophies. Tokushima J Exp Med 1993; 40: 83–93. [PubMed] [Google Scholar]
- 25. Abe K, Fujimura H, Toyooka K et al. Involvement of the central nervous system in myotonic dystrophy. J Neurol Sci 1994; 127: 179–185. [DOI] [PubMed] [Google Scholar]
- 26. Oyanagi K, Ogawa H, Nakajima T. Rod‐like intracytoplasmic inclusions in large neurons of the caudate nucleus: Frequent appearance in myotonic dystrophy. Clin Neuropathol 1994; 13: 134–138. [PubMed] [Google Scholar]
- 27. Lidang Jensen M, Rix M, Schroder HD, Teglbjaerg PS, Ebbesen F. Fetal akinesia‐hypokinesia deformation sequence (FADS) in 2 siblings with congenital myotonic dystrophy. Clin Neuropathol 1995; 14: 105–108. [PubMed] [Google Scholar]
- 28. Ono S, Kanda F, Takahashi K et al. Neuronal cell loss in the dorsal raphe nucleus and the superior central nucleus in myotonic dystrophy: A clinicopathological correlation. Acta Neuropathol 1995; 89: 122–125. [DOI] [PubMed] [Google Scholar]
- 29. Ono S, Kurisaki H, Sakuma A, Nagao K. Myotonic dystrophy with alveolar hypoventilation and hypersomnia: A clinicopathological study. J Neurol Sci 1995; 128: 225–231. [DOI] [PubMed] [Google Scholar]
- 30. Ono S, Kanda F, Takahashi K et al. Neuronal loss in the medullary reticular formation in myotonic dystrophy: A clinicopathological study. Neurology 1996; 46: 228–231. [DOI] [PubMed] [Google Scholar]
- 31. Ono S, Takahashi K, Kanda F et al. Immunohistochemical study of intracytoplasmic inclusion bodies of the thalamus in myotonic dystrophy. J Neurol Sci 1996; 140: 96–100. [DOI] [PubMed] [Google Scholar]
- 32. Vermersch P, Sergeant N, Ruchoux MM et al. Specific tau variants in the brains of patients with myotonic dystrophy. Neurology 1996; 47: 711–717. [DOI] [PubMed] [Google Scholar]
- 33. Ono S, Takahashi K, Fukuoka Y et al. Intracytoplasmic inclusion bodies of the substantia nigra in myotonic dystrophy. Immunohistochemical observations. J Neurol Sci 1997; 148: 193–198. [DOI] [PubMed] [Google Scholar]
- 34. Watanabe C, Katayama S, Noda K, Kaneko M, Inai K, Nakamura S. Heterotopic neurons in congenital myotonic dystrophy with mental retardation. Neuropathology 1997; 17: 243–247. [Google Scholar]
- 35. Ono S, Takahashi K, Jinnai K et al. Loss of serotonin‐containing neurons in the raphe of patients with myotonic dystrophy: A quantitative immunohistochemical study and relation to hypersomnia. Neurology 1998; 50: 535–538. [DOI] [PubMed] [Google Scholar]
- 36. Ono S, Takahashi K, Jinnai K et al. Loss of catecholaminergic neurons in the medullary reticular formation in myotonic dystrophy. Neurology 1998; 51: 1121–1124. [DOI] [PubMed] [Google Scholar]
- 37. Mizukami K, Sasaki M, Baba A, Suzuki T, Shiraishi H. An autopsy case of myotonic dystrophy with mental disorders and various neuropathologic features. Psychiatry Clin Neurosci 1999; 53: 51–55. [DOI] [PubMed] [Google Scholar]
- 38. Spillantini MG, Tolnay M, Love S, Goedert M. Microtubule‐associated protein tau, heparan sulphate and alpha‐synuclein in several neurodegenerative diseases with dementia. Acta Neuropathol 1999; 97: 585–594. [DOI] [PubMed] [Google Scholar]
- 39. Endo A, Motonaga K, Arahata K, Harada K, Yamada T, Takashima S. Developmental expression of myotonic dystrophy protein kinase in brain and its relevance to clinical phenotype. Acta Neuropathol 2000; 100: 513–520. [DOI] [PubMed] [Google Scholar]
- 40. Kawashima T, Furuta A, Doh‐ura K, Kikuchi H, Iwaki T. Ubiquitin‐immunoreactive skein‐like inclusions in the neostriatum are not restricted to amyotrophic lateral sclerosis, but are rather aging‐related structures. Acta Neuropathol 2000; 100: 43–49. [DOI] [PubMed] [Google Scholar]
- 41. Ono S, Takahashi K, Kanda F et al. Decrease of neurons in the medullary arcuate nucleus in myotonic dystrophy. Acta Neuropathol 2001; 102: 89–93. [DOI] [PubMed] [Google Scholar]
- 42. Kumada S, Uchihara T, Hayashi M et al. Promyelocytic leukemia protein is redistributed during the formation of intranuclear inclusions independent of polyglutamine expansion: An immunohistochemical study on Marinesco bodies. J Neuropathol Exp Neurol 2002; 61: 984–991. [DOI] [PubMed] [Google Scholar]
- 43. Maurage CA, Sergeant N, Ruchoux MM, Hauw JJ, Delacourte A. Phosphorylated serine 199 of microtubule‐associated protein tau is a neuronal epitope abundantly expressed in youth and an early marker of tau pathology. Acta Neuropathol 2003; 105: 89–97. [DOI] [PubMed] [Google Scholar]
- 44. Jiang H, Mankodi A, Swanson MS, Moxley RT, Thornton CA. Myotonic dystrophy type 1 is associated with nuclear foci of mutant RNA, sequestration of muscleblind proteins and deregulated alternative splicing in neurons. Hum Mol Genet 2004; 13: 3079–3088. [DOI] [PubMed] [Google Scholar]
- 45. Maurage CA, Udd B, Ruchoux MM et al. Similar brain tau pathology in DM2/PROMM and DM1/Steinert disease. Neurology 2005; 65: 1636–1638. [DOI] [PubMed] [Google Scholar]
- 46. Yamazaki M, Hasegawa M, Mori O et al. Tau‐positive fine granules in the cerebral white matter: A novel finding among the tauopathies exclusive to parkinsonism‐dementia complex of Guam. J Neuropathol Exp Neurol 2005; 64: 839–846. [DOI] [PubMed] [Google Scholar]
- 47. Oyamada R, Hayashi M, Katoh Y et al. Neurofibrillary tangles and deposition of oxidative products in the brain in cases of myotonic dystrophy. Neuropathology 2006; 26: 107–114. [DOI] [PubMed] [Google Scholar]
- 48. Itoh K, Mitani M, Kawamoto K et al. Neuropathology does not correlate with regional differences in the extent of expansion of CTG repeats in the brain with Myotonic dystrophy type 1. Acta Histochem Cytochem 2010; 43: 149–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Dhaenens CM, Tran H, Frandemiche ML et al. Mis‐splicing of tau exon 10 in myotonic dystrophy type 1 is reproduced by overexpression of CELF2 but not by MBNL1 silencing. Biochim Biophys Acta 2011; 1812: 732–742. [DOI] [PubMed] [Google Scholar]
- 50. Yamazaki Y, Matsubara T, Takahashi T et al. Granulovacuolar degenerations appear in relation to hippocampal phosphorylated tau accumulation in various neurodegenerative disorders. PLoS One 2011; 6: e26996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Nakamori M, Takahashi T, Yamazaki Y, Kurashige T, Yamawaki T, Matsumoto M. Cyclin‐dependent kinase 5 immunoreactivity for granulovacuolar degeneration. Neuroreport 2012; 23: 867–872. [DOI] [PubMed] [Google Scholar]
- 52. Jinnai K, Mitani M, Futamura N, Kawamoto K, Funakawa I, Itoh K. Somatic instability of CTG repeats in the cerebellum of myotonic dystrophy type 1. Muscle Nerve 2013; 48: 105–108. [DOI] [PubMed] [Google Scholar]
- 53. Michel L, Huguet‐Lachon A, Gourdon G. Sense and antisense DMPK RNA foci accumulate in DM1 tissues during development. PLoS One 2015; 10: e0137620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. De Antonio M, Dogan C, Hamroun D et al. Unravelling the myotonic dystrophy type 1 clinical spectrum: A systematic registry‐based study with implications for disease classification. Rev Neurol 2016; 172: 572–580. [DOI] [PubMed] [Google Scholar]
- 55. Yuen P, Baxter DW. The morphology of Marinesco bodies (paranucleolar corpuscles) in the melanin‐pigmented nuclei of the brain‐stem. J Neurol Neurosurg Psychiatry 1963; 26: 178–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Beach TG, Walker DG, Sue LI, Newell A, Adler CC, Joyce JN. Substantia nigra Marinesco bodies are associated with decreased striatal expression of dopaminergic markers. J Neuropathol Exp Neurol 2004; 63: 329–337. [DOI] [PubMed] [Google Scholar]
- 57. Odagiri S, Tanji K, Mori F et al. Immunohistochemical analysis of Marinesco bodies, using antibodies against proteins implicated in the ubiquitin‐proteasome system, autophagy and aggresome formation. Neuropathology 2012; 32: 261–266. [DOI] [PubMed] [Google Scholar]
- 58. Krogias C, Bellenberg B, Prehn C et al. Evaluation of CNS involvement in myotonic dystrophy type 1 and type 2 by transcranial sonography. J Neurol 2014; 262: 365–374. [DOI] [PubMed] [Google Scholar]
- 59. Hernandez‐Hernandez O, Guiraud‐Dogan C, Sicot G et al. Myotonic dystrophy CTG expansion affects synaptic vesicle proteins, neurotransmission and mouse behaviour. Brain 2013; 136: 957–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Pettersson OJ, Aagaard L, Jensen TG, Damgaard CK. Molecular mechanisms in DM1 ‐ a focus on foci. Nucleic Acids Res 2015; 43: 2433–2441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kovacs GG. Neuropathology of Neurodegenerative Diseases: A Practical Guide. Cambridge and New York: Cambridge University Press, 2015. [Google Scholar]
- 62. Kovacs GG. Tauopathies. Handb Clin Neurol 2017; 145: 355–368. [DOI] [PubMed] [Google Scholar]
- 63. Park SA, Ahn SI, Gallo JM. Tau mis‐splicing in the pathogenesis of neurodegenerative disorders. BMB Rep 2016; 49: 405–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Caillet‐Boudin ML, Fernandez‐Gomez FJ, Tran H, Dhaenens CM, Buee L, Sergeant N. Brain pathology in myotonic dystrophy: When tauopathy meets spliceopathy and RNAopathy. Front Mol Neurosci 2014; 6: 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Fernandez‐Gomez F, Tran H, Dhaenens CM et al. Myotonic dystrophy: An RNA toxic gain of function Tauopathy? Adv Exp Med Biol 2019; 1184: 207–216. [DOI] [PubMed] [Google Scholar]
- 66. Gallais B, Montreuil M, Gargiulo M, Eymard B, Gagnon C, Laberge L. Prevalence and correlates of apathy in myotonic dystrophy type 1. BMC Neurol 2015; 15: 148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Masaoka Y, Pantelis C, Phillips A et al. Markers of brain illness may be hidden in your olfactory ability: A Japanese perspective. Neurosci Lett 2013; 549: 182–185. [DOI] [PubMed] [Google Scholar]
- 68. Villemagne VL, Fodero‐Tavoletti MT, Masters CL, Rowe CC. Tau imaging: Early progress and future directions. Lancet Neurol 2015; 14: 114–124. [DOI] [PubMed] [Google Scholar]
- 69. Panaite PA, Kuntzer T, Gourdon G, Lobrinus JA, Barakat‐Walter I. Functional and histopathological identification of the respiratory failure in a DMSXL transgenic mouse model of myotonic dystrophy. Dis Model Mech 2013; 6: 622–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ishii S, Nishio T, Sunohara N et al. Small increase in triplet repeat length of cerebellum from patients with myotonic dystrophy. Hum Genet 1996; 98: 138–140. [DOI] [PubMed] [Google Scholar]
- 71. Kipp M, Kiessling MC, Hochstrasser T, Roggenkamp C, Schmitz C. Design‐based stereology for evaluation of histological parameters. J Mol Neurosci 2017; 61: 325–342. [DOI] [PubMed] [Google Scholar]
- 72. Schmitz C, Hof PR. Design‐based stereology in neuroscience. Neuroscience 2005; 130: 813–831. [DOI] [PubMed] [Google Scholar]
- 73. Antonini G, Mainero C, Romano A et al. Cerebral atrophy in myotonic dystrophy: A voxel based morphometric study. J Neurol Neurosurg Psychiatry 2004; 75: 1611–1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Minnerop M, Weber B, Schoene‐Bake JC et al. The brain in myotonic dystrophy 1 and 2: Evidence for a predominant white matter disease. Brain 2011; 134: 3530–3546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Henstridge CM, Sideris DI, Carroll E et al. Synapse loss in the prefrontal cortex is associated with cognitive decline in amyotrophic lateral sclerosis. Acta Neuropathol 2018; 135: 213–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Marteyn A, Maury Y, Gauthier MM et al. Mutant human embryonic stem cells reveal neurite and synapse formation defects in type 1 myotonic dystrophy. Cell Stem Cell 2011; 8: 434–444. [DOI] [PubMed] [Google Scholar]
- 77. Zanigni S, Evangelisti S, Giannoccaro MP et al. Relationship of white and gray matter abnormalities to clinical and genetic features in myotonic dystrophy type 1. Neuroimage Clin 2016; 11: 678–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Fukuda H, Horiguchi J, Ono C, Ohshita T, Takaba J, Ito K. Diffusion tensor imaging of cerebral white matter in patients with myotonic dystrophy. Acta radiologica 2005; 46: 104–109. [DOI] [PubMed] [Google Scholar]
- 79. Renard D, Menjot de Champfleur N. MRI hydrographic 3D sequences: Myotonic dystrophy type 1 meets CADASIL. Acta Neurol Belg 2018; 118: 307–308. [DOI] [PubMed] [Google Scholar]
- 80. Kim H, Lim YM, Oh YJ, Lee EJ, Kim KK. Comparison of brain magnetic resonance imaging between myotonic dystrophy type 1 and cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. PLoS One 2018; 13: e0208620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Boespflug EL, Simon MJ, Leonard E et al. Targeted assessment of enlargement of the perivascular space in Alzheimer's disease and vascular dementia subtypes implicates Astroglial involvement specific to Alzheimer's disease. J Alzheimers Dis 2018; 66: 1587–1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Meinke P, Hintze S, Limmer S, Schoser B. Myotonic dystrophy‐a Progeroid disease? Front Neurol 2018; 9: 601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Mateos‐Aierdi AJ, Goicoechea M, Aiastui A et al. Muscle wasting in myotonic dystrophies: A model of premature aging. Front Aging Neurosci 2015; 7: 125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Ashizawa T. Myotonic dystrophy as a brain disorder. Arch Neurol 1998; 55: 291–293. [DOI] [PubMed] [Google Scholar]