

Abstract
Enantiopure secondary alcohols are fundamental high‐value synthetic building blocks. One of the most attractive ways to get access to this compound class is the catalytic hydroboration. We describe a new concept for this reaction type that allowed for exceptional catalytic turnover numbers (up to 15 400), which were increased by around 1.5–3 orders of magnitude compared to the most active catalysts previously reported. In our concept an aprotic ammonium halide moiety cooperates with an oxophilic Lewis acid within the same catalyst molecule. Control experiments reveal that both catalytic centers are essential for the observed activity. Kinetic, spectroscopic and computational studies show that the hydride transfer is rate limiting and proceeds via a concerted mechanism, in which hydride at Boron is continuously displaced by iodide, reminiscent to an SN2 reaction. The catalyst, which is accessible in high yields in few steps, was found to be stable during catalysis, readily recyclable and could be reused 10 times still efficiently working.
Keywords: ammonium salts, asymmetric catalysis, chiral alcohols, cooperative catalysis, hydroboration
A new concept for asymmetric hydroboration of ketones is shown, in which an ammonium halide initiates a hydride transfer to a Lewis acid activated ketone. Advantages of the bifunctional catalyst include remarkable turnover numbers (up to 15 400, 50 ppm catalyst) and high enantioselectivity. The catalyst was recycled 10 times still providing high efficiency. Mechanistic and DFT studies confirm the cooperative mechanism.

Introduction
Catalytic enantioselective reductions of ketones are among the most important asymmetric reactions owing to the significance of enantiopure secondary alcohols as building blocks for the synthesis of bioactive compounds. [1] An attractive class of catalytic asymmetric reductions is the hydroboration which has been described using various catalyst types.[ 2 , 3 ] A number of them furnished products with high enantioselectivities. Probably the most popular method of this type is the Corey‐Bakshi‐Shibata (CBS) reduction of ketones which uses readily available oxazaborolidine catalysts and H3B*L (L=THF, DMS) as stoichiometric reducing agent and is applicable to a broad substrate range. [4] However, despite the great progress achieved with a number of previously reported catalyst concepts, a current limitation still is that highly active catalysts allowing for turnover numbers (TONs) >500—while still acting highly enantioselectively—remained elusive.
Some years ago we introduced the concept of asymmetric bifunctional Lewis acid/aprotic onium salt catalysis and since then this concept has demonstrated its potential in various reaction classes such as [2+2] cycloadditions, [5] SN reactions [6] and 1,2‐additions. [7] The synthetic transformations investigated were either previously not viable or not very efficient in terms of catalytic activity. Herein we report that this concept allows for extraordinary catalytic activity combined with high enantioselectivity in the hydroboration of ketones, allowing for TONs up to 15 400, being equivalent to an increase of activity of about 1.5 to >3 orders of magnitude compared to the most efficient asymmetric hydroboration catalysts. Our development was driven by the idea that a Lewis acidic oxophilic metal center could activate a ketone substrate, whereas a borane reagent might be activated by an appended ammonium halide moiety via a boron/halide interaction (Scheme 1). The activated borane would thus be quasi‐intramolecularly directed towards the ketone. This simultaneous activation of both reactants was considered as promising tool to attain high catalytic activity,[ 8 , 9 , 10 ] while the reactants were expected to be precisely spatially preorganizable within the chiral environment of the bifunctional active site thus enabling high levels of enantioselectivity.
Scheme 1.

Visualization of the concept of bifunctional Lewis Acid (L.A.)—ammonium salt catalyzed enantioselective hydroboration of ketones.
Results and Discussion
Development and Optimization Studies
As model reaction the hydroboration of acetophenone 1 a by pinacolborane (HBPin) was studied at 25 °C using Al catalyst C1 (Table 1). [11] Initial experiments conducted in CH2Cl2 proceeded disappointingly, because only traces of racemic product were formed like in entry 1. [12] A subsequent solvent screening (Supporting Information) not only revealed an accelerating effect by THF, but also allowed for high enantioselectivity (entry 2). The use of pure THF was more efficient than a mixture of THF and CH2Cl2 (entry 3). The use of other common hydroboration agents led to inferior results (Supporting Information).
Table 1.
Development of the title reaction.

|
# |
C1 [mol %] |
solvent |
[1 a] [mol/L] |
HBPin [equiv] |
iPrOH [equiv] |
yield[a] [%] |
ee [b] [%] |
TON |
|---|---|---|---|---|---|---|---|---|
|
1 |
5 |
CH2Cl2 |
0.14 |
1.0 |
– |
2 |
2 |
0.4 |
|
2 |
5 |
THF |
0.14 |
1.0 |
– |
24 |
94 |
4.8 |
|
3 |
5 |
CH2Cl2/THF (2:1) |
0.14 |
1.0 |
– |
16 |
93 |
3.2 |
|
4 |
5 |
THF |
0.14 |
1.0 |
1.0 |
52 |
92 |
10.4 |
|
5 |
5 |
THF |
0.14 |
2.0 |
1.0 |
72 |
92 |
14.4 |
|
6 |
0.5 |
THF |
1.0 |
2.0 |
1.0 |
99 |
95 |
198 |
|
7 |
0.05 |
THF |
1.0 |
2.0 |
1.0 |
99 |
97 |
1980 |
|
8 |
0.05 |
THF |
1.0 |
1.0 |
1.0 |
84 |
93 |
1680 |
|
9 |
0.01 |
THF |
1.0 |
2.0 |
1.0 |
93 |
92 |
9300 |
|
10 |
0.005 |
THF |
1.6 |
2.0 |
1.0 |
77 |
91 |
15 400 |
|
11 |
0.005 |
THF |
neat |
2.0 |
1.0 |
72 |
91 |
14 400 |
[a] Yield determined by 1H‐NMR of the crude product using an internal standard. [b] Enantiomeric excess determined by GC.
Continuous reaction monitoring by 1H‐NMR under the conditions of entry 2 revealed that the free alcohol 2 a is generated as catalytic intermediate, which is subsequently borylated. This implicated that a proton source is required for catalytic turnover. In the initial studies residual water was probably the proton source. Various (sub)stoichiometric proton sources were evaluated identifying iPrOH as the most efficient one of those examined (see Supporting Information). By this the yield was strongly improved (entry 4). Additional improvements were achieved by an excess of HBPin (entry 5) and an increased concentration. This allowed to significantly reduce the catalyst loading to technically interesting values (entries 6–11). With 0.05 mol % of C1 an ee of 97 % in combination with a nearly quantitative product yield was attained (entry 7). [13] However, under these optimized conditions, attractive results were also attained with just one equivalent of HBPin (entry 8). Also, with as little as 0.01 and 0.005 mol % C1—that is, 100 ppm and 50 ppm catalyst, respectively—high ee values (92 % and 91 %) and good yields (93 % and 77 %) were noticed (entries 9 & 10), corresponding to TONs of 9300 and 15 400, respectively. These values are substantially higher than for all previously reported highly enantioselective catalysts in this reaction type. [2] The reaction was also performed under neat conditions and similar results were attained (entry 11).
Reaction Scope
The reaction conditions of Table 1/entry 7 were then applied to different prochiral ketones (Table 2). Ortho‐, meta‐ and para‐chloro substituted aryl rings within alkyl aryl ketones were all well tolerated and provided 2 b–2 d. Next to other σ‐acceptors like p‐fluoro (→ 2 e) and π‐acceptors like p‐nitro (→ 2 f), p‐cyano (→ 2 g), a p‐methylester (→ 2 h) and a p‐dimethylamide group (→ 2 i), also σ‐donors like p‐methyl (→ 2 j) as well as π‐donors like p‐methoxy (→ 2 k) and even unprotected p‐amino (→ 2 l) were well accepted on the aromatic moieties. In case of 2 k a basic reaction work‐up was required to avoid racemization of the product. Chemoselectivity problems were not found with functional groups that are also susceptible to reductions. In addition, extended π‐systems such as 2‐naphthyl (→ 2 m) were successfully used. Next to methyl ketones, other alkyl ketones (→ 2 n–2 r) as well as heteroaryl ketones (→ 2 s & 2 t) were accommodated and allowed for good to high enantioselectivity.
Table 2.
Investigation of different ketone substrates 1.[a,b]

|
|

[a] Yields of isolated products. [b] The enantiomeric excess was determined by HPLC or GC. [c] 0.1 mol % of C1 were used. [d] 0.2 mol % of C1 were used. [e] 0.5 mol % of C1 were used. [f] 1.0 mol % of C1 were used. [g] Basic work‐up using sat. NaHCO3. [h] Neutral work‐up using water.
Noteworthy is also that dialkyl ketones can be efficiently used, if the difference of the steric demand of both residues is sufficiently large. 2 u was thus formed in almost quantitative yield with 98 % ee. Like expected with decreasing size difference the enantioselectivity decreased (→ 2 v & 2 w). A similar effect was observed for enones like shown for 2 x, 2 y and 2 z. The latter has been reported as intermediate toward a number of carotenoid‐derived odorants and bioactive terpenes including α‐damascone. [14]
Upscaling and Catalyst Recycling
The model reaction was also investigated on a gram scale (Table 3). With 0.05 mol % of catalyst and around 1 g of substrate, the product was formed with high yield (97 %) and enantioselectivity (ee=96 %, entry 1). To enforce a quantitative yield, a reaction on 10 g scale was run with prolonged reaction time and provided excellent results (entry 2). A gram scale experiment was also performed employing only 0.02 mol % of catalyst (entry 3). Also in this case, a quantitative yield (TON 4950) and high enantioselectivity were attained (ee=96 %). These results demonstrate the practical utility of this method.
Table 3.
Scale‐up results.

|
# |
scale 1 a [g] ([mmol]) |
C1 [mol %] |
t [d] |
yield[a] [%] |
amount 2 a [g] |
ee [b] [%] |
TON |
|---|---|---|---|---|---|---|---|
|
1 |
1.00 (8.35) |
0.05 |
1 |
97 |
0.99 |
96 |
1860 |
|
2 |
1.00 (8.35) |
0.02 |
4.5 |
99 |
1.01 |
96 |
4950 |
|
3 |
10.03 (83.50) |
0.05 |
4.5 |
99 |
10.14 |
95 |
1980 |
[a] Yield of isolated product. [b] Enantiomeric excess determined by GC.
In addition, the possibility to recycle the catalyst was examined (Scheme 2). In total 11 runs were conducted. Taking advantage of the ammonium salt moiety within the catalyst, n‐pentane was added to the reaction mixture after 22 h in order to precipitate C1, which was then washed, dried under high vacuum, and reused. For the first 9 runs, nearly quantitative yields were achieved. The ee slowly dropped from 95 to 91 %. Starting from the tenth run, the yield started to decrease (81 % for run 10, 75 % for run 11). Nevertheless, the enantiomeric excess stayed above 90 %. Albeit these results have not been optimized regarding the catalyst reisolation, they show that C1 is remarkably stable during the reaction and also during recovery thus further increasing the practicality of the title reaction.
Scheme 2.

Catalyst recycling studies. Yield determined by 1H‐NMR of the crude product using an internal standard. Enantiomeric excess determined by GC.
Mechanistic Studies
a) Control Experiments
To learn more about the role and impact of the catalytically relevant groups in C1, experiments with several control catalyst systems were conducted. Regarding the Lewis acidic center, catalysts with different anionic ligands were investigated (Table 4). Next to C1 bearing a chloride ligand also the corresponding fluoride (C2) and methyl (C3) containing complexes were employed. It was found that C1 was significantly more efficient regarding productivity and enantioselectivity. In particular C3 was found to be a poor catalyst. In combination with the solvent effect, an explanation of the exceptionally high activity of C1 might be that the catalytically active species makes use of a cationic Al center by displacing the metal bound chloride with THF (see also DFT calculations below). Cationic Al salen and salphen catalysts lacking an ammonium functional group and bearing two coordinating THF molecules at the Al center were previously reported and structurally characterized by Coates et al. [15] Because the Al−F bond in C2 is much stronger than the Al−Cl bond, [7] the generation of the cationic species is less favored. In addition, neither Me in C3, nor the isopropoxide formed by protonation of C3 with isopropanol are expected to readily form a cationic Al center.
Table 4.
Comparison of anionic ligands at the Al center.

|
# |
C |
yield[a] [%] |
ee [b] [%] |
|---|---|---|---|
|
1 |
C1 |
99 |
97 |
|
2 |
C2 |
42 |
80 |
|
3 |
C3 |
14 |
27 |
[a] Yield determined by 1H‐NMR of the crude product using an internal standard. [b] The enantiomeric excess was determined by GC.
Upon treatment of C1 with an excess of HBPin, in situ recorded IR, 1H‐NMR and UV/Vis spectra did not result in significant changes. A similar result was also found for catalyst treatment with iPrOH in UV/Vis experiments (Supporting Information). The formation of significant amounts of Al‐H or Al‐OiPr species thus seems unlikely.
To learn more about the importance of the ammonium halide moiety, different sets of experiments were performed. In the initial set, various tetrabutylammonium salts were examined as catalysts to study the effect of the anion. Using the conditions of Table 1, entry 7, but in the absence of a catalyst, racemic product was formed in 13 % yield after 22 h at 25 °C (Table 5, entry 1). Employing different halide salts, catalytic activity increased with increasing size and thus higher polarizability of the anion (entries 3–5). In contrast to this trend, the highest activity was found with fluoride. However, we found that treatment of HBPin with TBAF results in partial formation of BF3 and TBA[B(Pin)2]. This outcome was confirmed by X‐ray crystal structure analysis (see Supporting Information).[ 16 , 17 ] BF3 might thus act as a Lewis acid cocatalyst which increases the catalytic activity.
Table 5.
Control experiments with TBAX salts.

|
# |
TBAX, X= |
TBAX [mol %] |
yield[a] [%] |
|---|---|---|---|
|
1 |
–[b] |
– |
13 |
|
2 |
F |
1 |
99 |
|
3 |
Cl |
1 |
37 |
|
4 |
Br |
1 |
55 |
|
5 |
OTf |
1 |
38 |
|
6 |
I |
1 |
81 |
|
7 |
I |
0.05 |
37 |
[a] Yield determined by 1H‐NMR of the crude product using an internal standard. [b] No catalyst was used.
A lower yield was found for less nucleophilic anions like triflate. Decreasing the TBAI loading from 1.0 to 0.05 mol % resulted in decreased activity. These results demonstrate that ammonium halides have a catalytic activity in the reduction. Nevertheless, with TBAI the catalytic activity is significantly lower than for catalyst C1 also bearing an iodide counterion (compare Table 5, entry 6 to Table 1, entry 6–9). This confirms the importance of the Lewis acidic Al center.
The interaction between halide ions and the boron center ‐maybe resulting in a more pronounced hydride character of HBPin‐ was studied by 1H‐NMR (for details see the Supporting Information). The methyl signal of the pinacolate framework was used as probe, because the hydride signal itself was quite broad. For the same reason, also 11B‐NMR was not employed. By adding the corresponding TBA halide salt (1 equiv) to HBPin in DCM‐d2 at 25 °C, the signals were slightly upfield‐shifted, arguably as a result of a higher electron density at the B center. The softer the halide ion, the smaller should be the interaction with the relatively hard B center. According to these expectations the highest shift was found for chloride. However, the observed effect is small (around 0.05 ppm), probably due to the remote position of the investigated Me group.
Using 0.5 equiv of TBACl, there was no second signal for free HBPin, but the observed shift was smaller pointing to a dynamic behaviour for the chloride coordination. Also at −20 °C there was a single signal, but the observed shift was larger, pointing to more halide adduct in the equilibrium.
In addition, we investigated the variation of the ammonium counterion in the bifunctional complexes under the optimized conditions (Table 6). C1 (entry 1) offered the best productivity for this catalyst series. Using C1‐Br and C1‐Cl bearing bromide and chloride counterions, respectively, the product yields were reduced by around 30 %. Using a “non”‐nucleophilic ion the productivity was further decreased but remained noticeable. This seems contra‐intuitive, but might be explained by the generation of a cationic Al center thus releasing a nucleophilic chloride, while the non‐nucleophilic anion is not expected to strongly interact with the cationic Al center.
Table 6.
Study of the impact of different counterions in catalysts C1.

|
# |
Catalyst |
yield[a] [%] |
ee [b] [%]% |
|---|---|---|---|
|
1 |
C1 |
99 |
97 |
|
2 |
C1‐Br |
69 |
95 |
|
3 |
C1‐Cl |
67 |
93 |
|
4[c] |
C1‐PF6 |
57 |
92 |
[a] Yield determined by 1H‐NMR of the crude product using an internal standard. [b] The enantiomeric excess was determined by GC. [c] The corresponding triethylammonium salt was used as the catalyst.
Since the catalytic activity employing the different halide ions is not in agreement with the proposed binding tendencies of chloride, bromide and iodide to HBPin, it seems that the formation of an anionic halide/HBPin adduct intermediate cannot account for the catalytic activity differences. DFT calculations disclosed below suggest that interaction of the halide anion with the B center and the transfer of the hydride to the carbonyl group is a concerted process, reminiscent to an SN2 reaction thus continuously displacing hydride by halide. Formation of an anionic intermediate is thus not necessary. Like in an SN2 reaction, polarizability is a decisive factor (see also Figure 5).
Figure 5.

IBOs that relate to the p electron pairs of the iodide in A. Two electron pairs have two equally sized lobes (one of them is shown on the left) and one is polarized towards the borane (right). For a color Figure see the Supporting Information.
Additional control experiments were performed with the widely used salen catalyst C4 [18] (Table 7). As shown in entry 1, in the absence of any catalyst and iPrOH, only traces of product were formed. In the presence of iPrOH (1 equiv), the yield raised to 13 % (entry 2). The same conditions, but using 0.05 mol % of catalyst C4, 16 % of 2 a were formed with an ee of 72 % (entry 3). With 0.005 mol %, 13 % of product were obtained (entry 4) with an ee of 49 %, probably because the background reaction gained importance (compare to entry 2). These experiments show that catalyst C4 lacking an internal ammonium moiety is significantly less active and acts less enantioselectively than the bifunctional catalyst C1. C4 is also less active than TBAI (0.05 mol %, entry 5). With the binary system of C4 and TBAI (entry 6), results became slightly better as compared to the use of C4 only (entry 3). Nevertheless, the productivity was lower than with the TBAI alone (entry 5). These results show that the bifunctional catalyst is substantially more efficient than monofunctional Lewis acid or ammonium catalysts and the corresponding binary catalyst system, probably as result of an intramolecular double activation pathway.
Table 7.
Control experiments with catalyst C4.

|
# |
C4 [mol %] |
TBAI [mol %] |
iPrOH [equiv] |
yield[a] [%] |
ee [b] [%] |
|---|---|---|---|---|---|
|
1 |
0 |
0 |
0 |
2 |
– |
|
2 |
0 |
0 |
1 |
13 |
– |
|
3 |
0.050 |
0 |
1 |
16 |
72 |
|
4 |
0.005 |
0 |
1 |
13 |
49 |
|
5 |
0 |
0.05 |
1 |
37 |
– |
|
6 |
0.050 |
0.05 |
1 |
25 |
74 |
[a] Yield determined by 1H‐NMR of the crude product using an internal standard. [b] The enantiomeric excess was determined by GC.
b) Kinetic Investigations
Reaction monitoring and kinetic investigations were performed via 1H‐NMR spectroscopy in THF‐d8 at 25 °C using 0.1 mol % of C1 (for details of the kinetic studies see the Supporting Information).
Catalyst robustness and a possible product inhibition were investigated by Blackmond's Reaction Progress Kinetic Analysis (RPKA) “same excess” protocol. [19] Three experiments were performed using different initial concentrations (Table 8, Figure 1). [20]
Table 8.
Initial reaction conditions for the RPKA “same excess” experiments.

|
Exp. |
[1 a] [mol L−1] |
[HBPin] [mol L−1] |
[iPrOH] [mol L−1] |
[2 a] [mol L−1] |
|---|---|---|---|---|
|
1 |
0.84 |
1.68 |
0.84 |
0 |
|
2 |
0.42 |
1.26 |
0.42 |
0 |
|
3 |
0.42 |
1.26 |
0.42 |
0.42 |
Figure 1.

Reaction profiles of 1 a using Blackmond's “same excess” protocol under the conditions of Table 8. [19]
The decay of the concentration of 1 a over the course of the reaction is shown. Experiment 1 (Table 8) serves as reference reaction. In experiment 2 the starting concentration of 1 a corresponded to that of the reference reaction experiment when the latter had reached 50 % conversion. [19] A time shift shows that in experiment 2 the reaction proceeded slower than in experiment 1. In a further experiment 3 the starting point of experiment 2 was used except that 50 mol % of product 2 a was added, because in reference experiment 1 also 50 % product were present after 50 % conversion. The good overlay of the reaction profiles of experiment 1 and 3 and the acceleration of experiment 3 compared to experiment 2 demonstrate two things:
There is apparently no significant catalyst decomposition taking place, because otherwise the reaction rate of experiment 2 and 3 should be higher than that of experiment 1.
As the reaction rate of experiment 3 is higher than that of experiment 2, the product 2 a has an accelerating effect. It is likely that 2 a also acts as a proton source to release the product (see below).
The empirical rate law was determined by the Variable Time Normalization Analysis (VTNA) described by Burés. [21] Again, the model reaction of 1 a was examined at 25 °C in THF‐d8 using catalyst C1. Six reactions with different initial concentrations of 1 a, C1, HBPin, iPrOH and 2 a were used monitoring the concentration of all reagents (for details see the Supporting Information).
The best fit for the normalization of the time scale axis was achieved for the following empirical rate:
| (1) |
The reaction rate thus follows a first order kinetic dependence for catalyst C1 [22] and the substrate 1 a, whereas for HBPin, iPrOH and 2 a orders between 0 and 1 were found. [23] The first order kinetic in catalyst indicates that a single catalyst molecule is probably involved in the turnover‐limiting step. To probe this interpretation we took account of a non‐linear effect. [24] As expected, a linear correlation between catalyst ee and product ee values were found thus confirming this claim (see Supporting Information).
As both alcohols accelerate the reaction, they are likely to cause the protonation step during the catalytic reaction. By the concentration profiles of 1 a and the corresponding boric‐acid esters 2 a‐BPin and i PrO‐BPin this assumption is reinforced (see Supporting Information). There is also an uncatalyzed side reaction taking place, in which HBPin reacts with iPrOH to form the corresponding boric acid ester, which constantly reduces the concentration of both iPrOH and HBPin. This event thus slows down the overall reaction progress.
Based on the described experimental results, we propose the simplified catalytic cycle described in Scheme 3.Acetophenone 1 a is expected to coordinate to the Lewis acidic aluminum center, which is suggested to be cationic in THF in the catalytically most active form A in agreement with studies of Coates and the above described solvent effect. [15] A cationic Al complex lacking the coordinated ketone substrate was indeed detected by ESI mass spectrometry using catalyst reisolated after a catalytic run. Moreover, our DFT studies presented below show that the activation barrier is significantly lower with a cationic compared to a neutral Al center. The iodide counterion of A (alternatively a released chloride ion) could activate and direct HBPin quasi‐intramolecularly towards the keto moiety to generate Al‐alcoholate B and IBPin (alternatively ClBPin). Protonation of B by iPrOH could release alcohol 2 a, whereas the generated isopropanolate could be trapped by IBPin (alternatively ClBPin). Upon accumulation of 2 a, it can also serve as proton source like iPrOH to release more of 2 a, while itself being transformed to the final boric ester product 2 a‐BPin. Coordination of another acetophenone molecule would close the catalytic cycle.
Scheme 3.

Proposed simplified catalytic cycle.
c) Computational Investigations
To gain more insight into the reaction mechanism, the reduction of 1 a with HBPin was investigated by density functional theory (DFT) at the M06‐2X/def2‐TZVP level of theory on M06‐2X/def2‐SVP geometries with solvent effects accounted for by the conductor‐like screening model (COSMO, see Computational Details). The catalytic mechanism with C1S (S for simplified) was studied, where the two tBu groups of C1 in para‐position to the oxygen were removed to simplify the model.
The quantum chemical investigations predict a mechanism proposed in Figure 3. It turns out that step I, in which the hydride of the borane is transferred to the ketone and the borane binds to the free iodide, is rate determining. With the Al‐Cl catalyst C1S, this step has a barrier of 102 kJ mol−1, which is too high to explain the observed kinetics. Thus, C1 is not expected to be the active catalytic species. In agreement with the experimental finding that THF is required for the reaction to proceed, we found that a replacement of the chloride by THF is necessary to form an active catalyst C1+. Figure 2 shows a simplified geometry of this complex (C1SS +), where the tBu groups and the onium moiety are removed (SS for twice simplified). The exchange releases the chloride, which could subsequently also take over the role of the nucleophilic iodide in the catalytic cycle. This explains that some activity was observed with C4, lacking the onium moiety and the iodide, as well as catalyst systems with “non”‐nucleophilic ions like C1‐PF6.
Figure 3.

Details of the catalytic steps as obtained from DFT with relative free‐energy profile of the reaction steps A → B (step I) and B+iPrOH → C (step II). Dashed lines represent bimolecular steps.
Figure 2.

The binding IBO for [C1SS +]Cl− (left) and C1SS (right). For a color Figure see the Supporting Information.
The reaction barrier with the THF‐activated catalyst C1S + is significantly lowered to 62 kJ mol−1. It should be noted that according to DFT, C1S is lower in energy by 20 kJ mol−1 than C1S +. However, the high excess of the solvent shifts the equilibrium towards the active form.
To explain the observed reactivity, structurally simplified complexes of the active (C1SS +) and the original (C1SS) catalyst (see Figure 2) were investigated with the help of DFT and Intrinsic Bond Orbitals (IBOs) (see Computational Details). IBOs are helpful to interpret quantum chemical calculations, as they often provide chemically meaningful orbitals.
For C1SS +, the bond length Al‐O (THF) was 1.87 Å. The localized IBO, which is responsible for this bond, is located at aluminum by only 12 % of its charge. In C1SS the corresponding Al‐Cl bond length is 2.17 Å. In this case 19 % of the bond charge is located at aluminum. As a consequence, the aluminum center carries a higher partial charge (IBO charge) of +1.19 in C1SS + than in C1SS (+1.08). Therefore, as expected the Lewis acidity of the aluminum is increased by the exchange of chloride with THF.
The resulting mechanism with C1S + and its energetics are illustrated in Figure 3. The structures of the respective transition states can be found in Figure 4. We provide the energies for the unimolecular steps only, since the free‐energy of association is rather ill‐defined at our level of treatment of the solvation (COSMO) and in any case depends on the concentration of the reaction partners.
Figure 4.
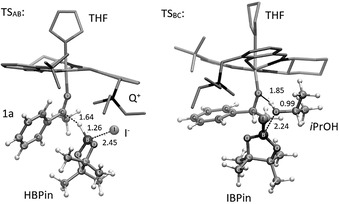
Geometries of the transition states obtained by DFT. Lengths of the bonds that are formed or broken during the transition are given in Å. For a color Figure see the Supporting Information.
As already mentioned, step I from A to B represents the rate determining step of the catalytic mechanism with a barrier of 62 kJ mol−1. It describes the carbonyl reduction via a hydride transfer from the borane to the electrophilic center of the ketone, which is activated by the Lewis‐acidic aluminum. In a concerted reaction the borane simultaneously binds to the free iodide.
An electronic effect of the iodide on the borane was also studied. The B‐I distance in A (before the bond formation) is 3.56 Å and decreases to 2.45 Å in TSAB and further to 2.14 Å once the B−I bond is formed in B. An analysis of the IBOs reveals that there already is a slight interaction between the iodide and the borane in A. Figure 5 shows that the electron density of one IBO, that relates to the free electron pair at the iodide, is polarized towards the borane. The role of the iodide, through the help of the onium moiety, is not only to be in the proximity of the HBPin to act as a binding partner as soon as the hydride is transferred, but might also push electron density towards the borane activating the hydride.
As the enantioselectivity is here defined in the rate determining step, the formation of the enantiomer with minor yield is also studied. It yields a barrier of 74 kJ mol−1. The enantiomeric excess can be calculated based on these results to be 98 % which is in very good agreement with the measured ee value of 97 %.
In step II an iPrOH molecule is added (B+iPrOH) and the alcoholate that is coordinated to the aluminum center is protonated. As a consequence, the Al−O bond length increases from 1.80 Å to 2.00 Å.
The step possesses an early transition state with a small barrier of 8 kJ mol−1. To confirm the experimentally found kinetics, where the product alcohol takes over the role of a protonating agent during the course of the reaction, step II was also investigated with the product alcohol 2 a instead of iPrOH. Apart from slightly more steric hindrance the reaction is qualitatively the same. The barrier increases to 16 kJ mol−1 but is still very small. When iPrOH is consumed in the course of the reaction and the concentration of the product alcohol increases, it is likely that the latter will be the more and more dominating proton source.
To estimate the reliability of the DFT results, single point calculations have been repeated with the functionals TPSS and PBE0 (see Supporting Information). They result in overall smaller barriers but the conclusions drawn from DFT are unaltered.
Conclusion
In summary, we have reported a concept for the catalytic asymmetric hydroboration of ketones, which allows for extraordinarily high turnover numbers (up to 15 400) that are around 1.5–3 orders of magnitude higher than with the most efficient catalysts previously reported. The chiral secondary alcohols—high‐value‐added products—were typically formed with high yields and high enantioselectivity. In our concept an aprotic ammonium halide moiety and an oxophilic Lewis acid work in concert and cooperate with each other within the same catalyst molecule. This was confirmed by a number of control experiments showing that both catalytic centers are indispensable for the observed activity. Moreover, kinetic, spectroscopic and computational studies revealed that the hydride transfer is most likely rate limiting. According to our calculations, it proceeds via a concerted mechanism, in which hydride is continuously displaced by iodide at the B center, reminiscent to an SN2 reaction. Simultaneously, the hydride attacks the ketone, which is activated by a cationic AlIII center. Further practical value is added by the fact that the catalyst, which is readily accessible in high yields in few steps, is stable during catalysis and readily recyclable by taking advantage of the ammonium salt moiety. This allowed to reuse the catalyst 10 times, while still efficiently working.
Computational Details
Molecular geometries were optimized with the DL‐FIND [25] optimization library in Chemshell. [26] The density functional theory (DFT) calculations were performed with the Turbomole V 7.0.1 and V 7.4.1 program package [27] using (DFT) with the M06‐2X functional [28] and the def2‐SVP basis set. [29] Frequencies were calculated at the same level of theory. Numerical integration was carried out on a m4 grid and SCF energies were converged for an energy difference of less than 10−8 atomic units. All transition structures were verified to possess only a single mode with imaginary frequency. IRC (internal reaction coordinate) calculations starting from the transition structures were performed and verified the reactants and products. The free energy G was calculated at 298.15 K within the RRHO (rigid rotor harmonic oscillator) approximation. Vibrational frequencies less than 100 cm−1 were raised to this threshold. At fixed geometries, the energy was calculated using the def2‐TZVP basis set [29] and the solvent effects were accounted for with the conductor‐like screening model (COSMO) [30] using a dielectric constant of ϵ=7.39 for THF at 298.15 K. For the atomic radii in the cavity the default values for COSMO were used. Intrinsic bond orbitals (IBOs) [31] were calculated based on orbitals of M06‐2X/def2‐TZVP level of theory with exponent 2 in the localization method. The visualization of the IBOs was realized using the IboView program by Knizia (http://www.iboview.org/).
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was financially supported by the Deutsche Forschungsgemeinschaft (DFG, PE 818/8‐1, project 404194277). We thank Dr. Wolfgang Frey for an X‐ray crystal structure analysis (see ref. [17]). J.H. acknowledges financial support in the form of a PhD scholarship from the Studienstiftung des Deutschen Volkes (German National Academic Foundation). We thank the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) for supporting this work by funding EXC 2075‐390740016 under Germany's Excellence Strategy. The authors acknowledge support by the state of Baden‐Württemberg through bwHPC and the German Research Foundation (DFG) through grant no INST 40/467‐1 FUGG (JUSTUS cluster). Open access funding enabled and organized by Projekt DEAL.
M. Titze, J. Heitkämper, T. Junge, J. Kästner, R. Peters, Angew. Chem. Int. Ed. 2021, 60, 5544.
Contributor Information
Prof. Dr. Johannes Kästner, Email: kaestner@theochem.uni-stuttgart.de.
Prof. Dr. René Peters, Email: rene.peters@oc.uni-stuttgart.de.
References
- 1.
- 1a. Noyori R., Ohkuma T., Angew. Chem. Int. Ed. 2001, 40, 40–73; [PubMed] [Google Scholar]; Angew. Chem. 2001, 113, 40–75; [Google Scholar]
- 1b. Noyori R., Koizumi M., Ishii D., Ohkuma T., Pure Appl. Chem. 2001, 73, 227–232; [Google Scholar]
- 1c. Ohkuma T., Kitamura M., Noyori R., New Frontiers in Asymmetric Catalysis, Wiley, Hoboken, 2006, pp. 1–32; [Google Scholar]
- 1d. Zaidlewicz M., Pakulski M. M. in Science of Synthesis Stereoselective Synthesis, Vol. 2 (Eds.: De Vries J. G., Molander G. A., Evans P. A.), Thieme, Stuttgart, 2011, pp. 59–131; [Google Scholar]
- 1e.N. Arai, T. Ohkuma in Science of Synthesis Stereoselective Synthesis, Vol. 2 (Eds.: J. G. De Vries, G. A. Molander, P. A. Evans), Thieme, Stuttgart, 2011, pp. 9–57;
- 1f. Gröger H., Borchert S., Krausser M., Hummel W. in Encyclopedia of Industrial Biotechnology, Vol. 3 (Ed.: Flickinger M. C.), Wiley, Hoboken, 2010, pp. 2094–2110; [Google Scholar]
- 1g. Shende V. S., Singh P., Bhanage B. M., Catal. Sci. Technol. 2018, 8, 955–969; [Google Scholar]
- 1h. Yang Z. H., Zeng R., Yang G., Wang Y., Li L. Z., Lv Z. S., Yao M., Lai B., J. Ind. Microbiol. Biotechnol. 2008, 35, 1047–1051; [DOI] [PubMed] [Google Scholar]
- 1i. Chen B. S., Ribeiro de Souza F. Z., RSC Adv. 2019, 9, 2102–2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reviews:
- 2a. Cho B. T., Chem. Soc. Rev. 2009, 38, 443–452; [DOI] [PubMed] [Google Scholar]
- 2b. Shegavi M. L., Bose S. K., Catal. Sci. Technol. 2019, 9, 3307–3336; see also: [Google Scholar]
- 2c. Brown H. C., Ramachandran P. V., Acc. Chem. Res. 1992, 25, 16–24; [Google Scholar]
- 2d. Ramachandran P. V., Brown H. C., ACS Symposium Series, Vol. 641, ACS Publications, Washington, 1996, pp. 84–97. [Google Scholar]
- 3.Reviews on boron chemistry:
- 3a. Kaur P., Khatik G., Nayak S., Curr. Org. Synth. 2016, 14, 665–682; [Google Scholar]
- 3b. Hall D. G. in Boronic Acids: Preparation and Applications in Organic Synthesis and Medicine, Wiley-VCH, Weinheim, 2006, pp. 1–99; [Google Scholar]
- 3c. Defrancesco H., Dudley J., Coca A., ACS Symp. Ser. 2016, 1236, 1–25. [Google Scholar]
- 4.
- 4a. Corey E. J., Helal C. J., Angew. Chem. Int. Ed. 1998, 37, 1986–2012; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 1998, 110, 2092–2118; for highly enantioselective hydroborations using HBPin, see: [Google Scholar]
- 4b. Guo J., Chen J., Lu Z., Chem. Commun. 2015, 51, 5725–5727; [DOI] [PubMed] [Google Scholar]
- 4c. Vasilenko V., Blasius C. K., Wadepohl H., Gade L. H., Angew. Chem. Int. Ed. 2017, 56, 8393–8397; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 8513–8517; [Google Scholar]
- 4d. Blasius C. K., Vasilenko V., Gade L. H., Angew. Chem. Int. Ed. 2018, 57, 10231–10235; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 10388–10392; [Google Scholar]
- 4e. Vasilenko V., Blasius C. K., Gade L. H., J. Am. Chem. Soc. 2018, 140, 9244–9254; [DOI] [PubMed] [Google Scholar]
- 4f. Magre M., Maity B., Falconnet A., Cavallo L., Rueping M., Angew. Chem. Int. Ed. 2019, 58, 7025–7029; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 7099–7103; [Google Scholar]
- 4g. Lebedev Y. N., Polishchuk I., Maity B., Cavallo L., Rueping M., J. Am. Chem. Soc. 2019, 141, 19415–19423; [DOI] [PubMed] [Google Scholar]
- 4h. Vasilenko V., Blasius C. K., Wadepohl H., Gade L. H., Chem. Commun. 2020, 56, 1203–1206; [DOI] [PubMed] [Google Scholar]
- 4i. Liu W., Guo J., Xing S., Lu Z., Org. Lett. 2020, 22, 2532–2536; [DOI] [PubMed] [Google Scholar]
- 4j.for a recent review, see: Wenbo L., Zhan L., Chin. J. Org. Chem. 2020, 40, 3596–3604. [Google Scholar]
- 5.
- 5a. Kull T., Peters R., Angew. Chem. Int. Ed. 2008, 47, 5461–5464; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 5541–5544; [Google Scholar]
- 5b. Kull T., Cabrera J., Peters R., Chem. Eur. J. 2010, 16, 9132–9139; [DOI] [PubMed] [Google Scholar]
- 5c. Meier P., Broghammer F., Latendorf K., Rauhut G., Peters R., Molecules 2012, 17, 7121–7150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Broghammer F., Brodbeck D., Junge T., Peters R., Chem. Commun. 2017, 53, 1156–1159. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Brodbeck D., Broghammer F., Meisner J., Klepp J., Garnier D., Frey W., Kästner J., Peters R., Angew. Chem. Int. Ed. 2017, 56, 4056–4060; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 4115–4119; [Google Scholar]
- 7b. Brodbeck D., Álvarez-Barcia S., Meisner J., Broghammer F., Klepp J., Garnier D., Frey W., Kästner J., Peters R., Chem. Eur. J. 2019, 25, 1515–1524; [DOI] [PubMed] [Google Scholar]
- 7c. Junge T., Titze M., Frey W., Peters R., ChemCatChem, DOI: 10.1002/cctc.202001921R1. [Google Scholar]
- 8. Cooperative Catalysis—Designing Efficient Catalysts for Synthesis (Ed.: Peters R.), Wiley-VCH, Weinheim, 2015. [Google Scholar]
- 9.For the cleavage of B-H Bonds by metal-ligand cooperation, see: Higashi T., Kusumoto S., Nozaki K., Chem. Rev. 2019, 119, 10393–10402. [DOI] [PubMed] [Google Scholar]
- 10.Selected related polyfunctional catalysts developed by our group:
- 10a. Miskov-Pajic V., Willig F., Wanner D. M., Frey W., Peters R., Angew. Chem. Int. Ed. 2020, 59, 19873–19877; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10b. Willig F., Lang J., Hans A. C., Ringenberg M. R., Pfeffer D., Frey W., Peters R., J. Am. Chem. Soc. 2019, 141, 12029–12043; [DOI] [PubMed] [Google Scholar]
- 10c. Schmid J., Junge T., Lang J., Frey W., Peters R., Angew. Chem. Int. Ed. 2019, 58, 5447–5451; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 5501–5505; [Google Scholar]
- 10d. Mechler M., Peters R., Angew. Chem. Int. Ed. 2015, 54, 10303–10307; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 10442–10446; [Google Scholar]
- 10e. Schmid J., Frey W., Peters R., Organometallics 2017, 36, 4313–4324; [Google Scholar]
- 10f. Mechler M., Frey W., Peters R., Organometallics 2014, 33, 5492–5508; [Google Scholar]
- 10g. Mechler M., Latendorf K., Frey W., Peters R., Organometallics 2013, 32, 112–130. [Google Scholar]
- 11.Non-enantioselective Al-catalyzed hydroboration of ketones:
- 11a. Woodside A. J., Smith M. A., Herb T. M., Manor B. C., Carroll P. J., Rablen P. R., Graves C. R., Organometallics 2019, 38, 1017–1020; [Google Scholar]
- 11b. Jakhar V. K., Barman M. K., Nembenna S., Org. Lett. 2016, 18, 4710–4713; [DOI] [PubMed] [Google Scholar]
- 11c. Franz D., Sirtl L., Pöthig A., Inoue S., Z. Anorg. Allg. Chem. 2016, 642, 1245–1250; [Google Scholar]
- 11d. Pollard V. A., Orr S. A., McLellan R., Kennedy A. R., Hevia E., Mulvey R. E., Chem. Commun. 2018, 54, 1233–1236; [DOI] [PubMed] [Google Scholar]
- 11e. Lemmerz L. E., McLellan R., Judge N. R., Kennedy A. R., Orr S. A., Uzelac M., Hevia E., Robertson S. D., Okuda J., Mulvey R. E., Chem. Eur. J. 2018, 24, 9940–9948; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11f. Yang Z., Zhong M., Ma X., De S., Anusha C., Parameswaran P., Roesky H. W., Angew. Chem. Int. Ed. 2015, 54, 10225–10229; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 10363–10367; Al-catalyzed hydroboration of alkynes and/or alkenes: [Google Scholar]
- 11g. Bismuto A., Thomas S. P., Cowley M. J., Angew. Chem. Int. Ed. 2016, 55, 15356–15359; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 15582–15585; [Google Scholar]
- 11h. Li F., Bai X., Cai Y., Li H., Zhang S. Q., Liu F. H., Hong X., Xu Y., Shi S. L., Org. Process Res. Dev. 2019, 23, 1703–1708. [Google Scholar]
- 12.Review on Al catalysts in reduction chemistry: Nikonov G. I., ACS Catal. 2017, 7, 7257–7266. [Google Scholar]
- 13.Under these conditions we also investigated the use of the corresponding C2-symmetric ligand bearing two ammonium moieties. However, both yield (11 %) and ee (18 %) were poor.
- 14. Mori K., Synlett 1995, 1097–1109. [Google Scholar]
- 15.
- 15a. Mahadevan V., Getzler Y. D. Y. L., Coates G. W., Angew. Chem. Int. Ed. 2002, 41, 2781–2784; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2002, 114, 2905–2908; [Google Scholar]
- 15b. Mulzer M., Ellis W. C., Lobkovsky E. B., Coates G. W., Chem. Sci. 2014, 5, 1928–1933; [Google Scholar]
- 15c. Getzler Y. D. Y. L., Mahadevan V., Lobkovsky E. B., Coates G. W., J. Am. Chem. Soc. 2002, 124, 1174–1175. [DOI] [PubMed] [Google Scholar]
- 16.For a KF catalyzed hydroboration, see: Kuciński K., Hreczycho G., Eur. J. Org. Chem. 2020, 552–555. [Google Scholar]
- 17. Deposition Number 2030886 contains the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service www.ccdc.cam.ac.uk/structures.
- 18.
- 18a. Sigman M. S., Jacobsen E. N., J. Am. Chem. Soc. 1998, 120, 5315; [Google Scholar]
- 18b. Myers J. K., Jacobsen E. N., J. Am. Chem. Soc. 1999, 121, 8959. [Google Scholar]
- 19.
- 19a. Baxter R. D., Sale D., Engle K. M., Yu J.-Q., Blackmond D. G., J. Am. Chem. Soc. 2012, 134, 4600–4606; [DOI] [PubMed] [Google Scholar]
- 19b. Blackmond D. G., J. Am. Chem. Soc. 2015, 137, 10852–10866. [DOI] [PubMed] [Google Scholar]
- 20.[iPrOH] determined by 1H-NMR for experiment 1 in Table 8 after 50 % conversion is in good agreement with the calculated value used in Table 8.
- 21.
- 21a. Burés J., Angew. Chem. Int. Ed. 2016, 55, 16084–16087; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 16318–16321; [Google Scholar]
- 21b. Nielsen C. D.-T., Burés J., Chem. Sci. 2019, 10, 348–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Also for catalyst C4 lacking an onium moiety a first order kinetic dependence was found.
- 23.For the BPin ester of 2 a, no accelerating effect was found.
- 24. Satyanarayana T., Abraham S., Kagan H. B., Angew. Chem. Int. Ed. 2009, 48, 456–494; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 464–503. [Google Scholar]
- 25. Kästner J., Carr J. M., Keal T. W., Thiel W., Wander A., Sherwood P., J. Phys. Chem. A 2009, 113, 11856–11965. [DOI] [PubMed] [Google Scholar]
- 26. Metz S., Kästner J., Sokol A. A., Keal T. W., Sherwood P., Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2014, 4, 101–110. “ChemShell, a Computational Chemistry Shell” can be found under http://www.chemshell.org. [Google Scholar]
- 27.“TURBOMOLE V7 2015, a development of the University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007, TURBOMOLE GmbH, since 2007; can be found under http://www.turbomole.com.
- 28. Zhao Y., Truhlar D., Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- 29.
- 29a. Weigend F., Ahlrichs R., Phys. Chem. Chem. Phys. 2005, 7, 3297–3305; [DOI] [PubMed] [Google Scholar]
- 29b. Weigend F., Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [DOI] [PubMed] [Google Scholar]
- 30. Klamt A., Schüürmann G., J. Chem. Soc. Perkin Trans. 2 1993, 799–805. [Google Scholar]
- 31. Knizia G., J. Chem. Theory Comput. 2013, 9, 4834–4843. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary