

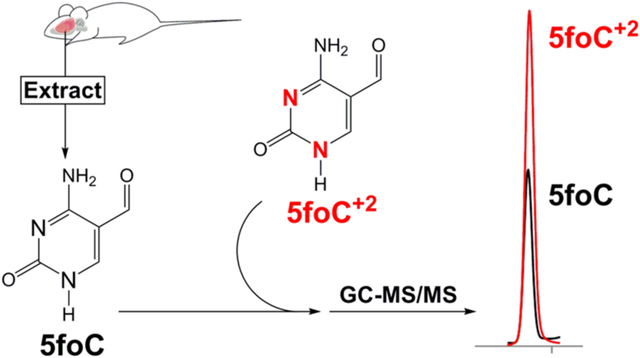
Abstract
The DNA of all organisms is metabolically active due to persistent endogenous DNA damage, repair, and enzyme-mediated base modification pathways important for epigenetic reprogramming and antibody diversity. The free bases released from DNA either spontaneously or by base excision repair pathways constitute DNA metabolites in living tissues. In this study, we have synthesized and characterized the stable-isotope standards for a series of pyrimidines derived from the normal DNA bases by oxidation and deamination. We have used these standards to measure free bases in small molecule extracts from rat brain. Free bases are observed in extracts, consistent with both endogenous DNA damage and 5-methylcytosine demethylation pathways. The most abundant free base observed is uracil, and the potential sources of uracil are discussed. The free bases measured in tissue extracts constitute the end product of DNA metabolism and could be used to reveal metabolic disturbances in human disease.
Graphical Abstract
INTRODUCTION
The DNA of higher organisms is composed predominantly of the canonical bases adenine, thymine, guanine, and cytosine. In 1948, 5-methylcytosine (5mC) was identified in calf thymus DNA.1 It is formed by enzymatic methylation of a cytosine residue in DNA following DNA replication.2,3 Subsequent studies established that 5mC codes as cytosine during DNA replication,4 but the presence of 5mC profoundly alters DNA–protein interactions.5,6 The location of 5mC residues in DNA establishes a cytosine methylation pattern that is one of the key elements of the epigenetic programming of gene transcription in eukaryotes.7,8
Enzymatic demethylation of 5mC in DNA has been proposed by multiple groups.9 However, evidence in favor of such a pathway was recently supported with the identification of 5-hydroxymethylcytosine (5hmC) in the DNA of rodent cerebellar Purkinje neurons.10 Members of the TET family of α-ketoglutarate-dependent dioxygenases have been shown to convert 5mC to 5hmC in DNA.11 Further details of this pathway are less clear, but evidence has been reported12–14 indicating further oxidation of 5hmC to 5-formylcytosine (5foC) and 5-carboxycytosine (5caC), as well as deamination to the corresponding uracil analogues. The uracil analogues include 5-hydroxymethyluracil (5hmU), 5-formyluracil (5foU), and 5-carboxyuracil (5caU), as shown in Figure 1.
Figure 1.
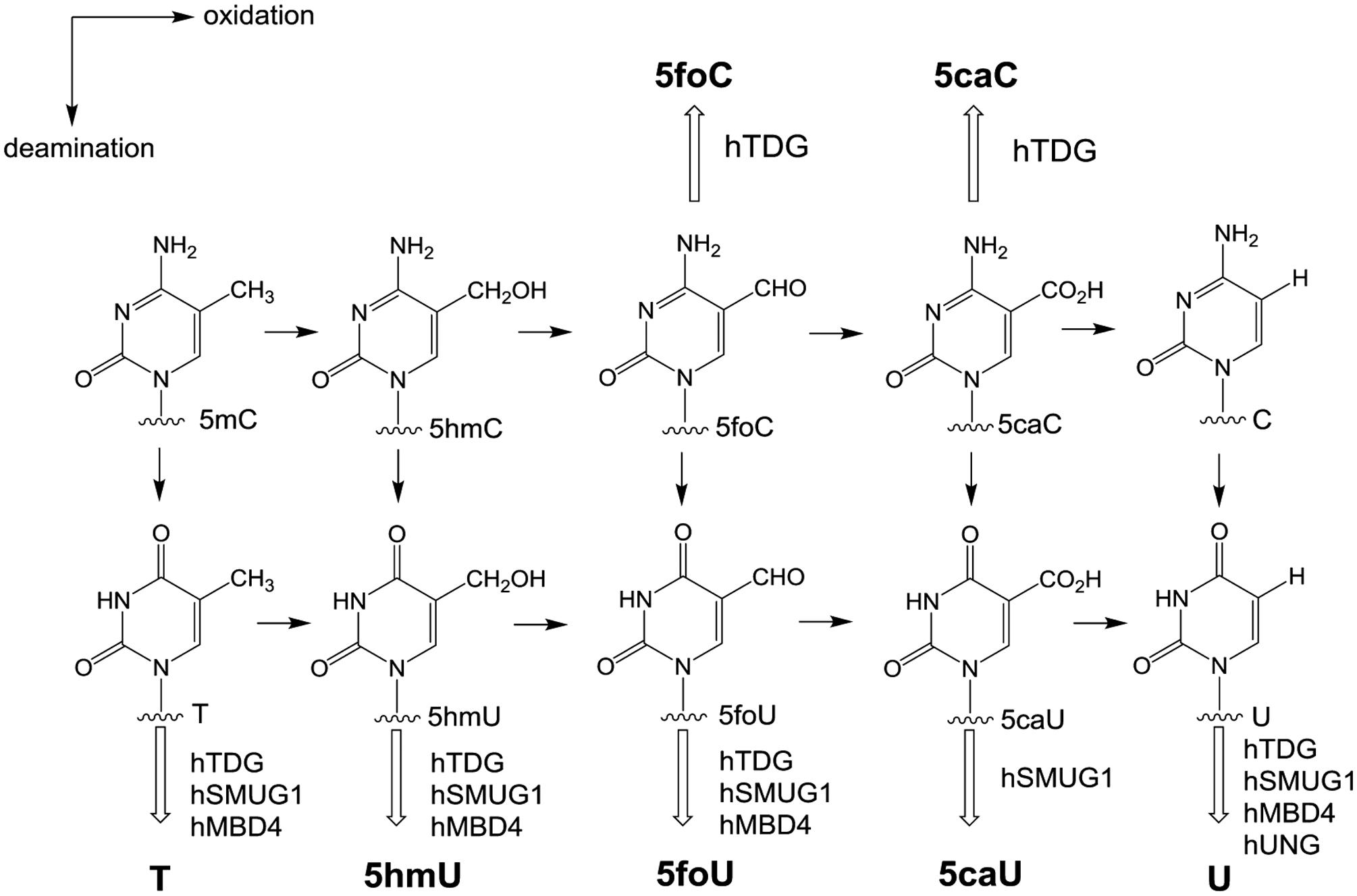
Potential reactions in the 5-methylcytosine oxidation/deamination pathways. Free base products released by known glycosylases are shown in bold. Glycosylases known to remove these bases are shown adjacent to the hollow arrows.
DNA bases can also be modified by chemical reactions including both oxidation and deamination.15 One of the more frequent endogenous DNA damage events is the hydrolytic deamination of cytosine, which results in the formation of uracil.16 Recent studies have also demonstrated that cytosine residues in DNA can be enzymatically deaminated by activation-induced deaminases (AID) and apolipoprotein B mRNA editing enzyme catalytic polypeptide 1 (APOBEC-1).17–19 The enzymatic conversion of cytosine to uracil facilitates somatic hypermutation in immunoglobulin variable genes in mammals, which results in increased antibody diversity. While uracil is a normal component of RNA, it is recognized as a damaged base when it is in DNA and is cleaved by members of the uracil-DNA glycosylase family.20–24 Products of enzymatic modification of 5mC, with the exception of 5hmC,25 such as 5foC, 5caC, and the deamination products 5hmU, 5foU, and 5caU, can also be removed from DNA by glycosylases of the base excision repair (BER) pathway (Figure 1).
While DNA has been considered historically to be a repository of genetic information that can be passed unmodified to progeny cells, recent evidence suggests that DNA can undergo multiple enzymatic modifications that can provide epigenetic regulation of gene transcription, diversity in immunological cells, and, potentially, other functions. The emerging pattern is that DNA can be metabolically active with the generation of modified bases that are subject to removal by glycosylases of the BER pathway. Multiple methods have been reported for measuring DNA modifications as the corresponding 2′-deoxynucleosides;26,27 however, substantially fewer studies have been devoted to examining modified free bases. As the free bases can be considered to be the end products of multiple DNA metabolic pathways, examination of free bases in human cell extracts and tissues could help to reveal further details of these pathways in normal tissues as well as defective pathways associated with human disease. In this article, we describe the synthesis and characterization of the stable-isotope analogues of the potential enzymatic products of the DNA pyrimidines. Furthermore, we present initial studies in which these analogues have been used to examine DNA metabolites in the rat brain.
MATERIALS AND METHODS
Materials.
Triethyl orthoformate, ethyl cyanoacetate, diethyl malonate, sodium metal, lithium aluminum hydride solution, triethylamine, potassium persulfate, silver nitrate, and 15N2-urea were obtained from Sigma-Aldrich (St. Louis, MO). Potassium perruthenate, paraformaldehyde, N-tert-butyldimethylsilyl-N-methyltrifluoroacetamide + 1% tert-butyldimetheylchlorosilane (MTBSTFA + 1% TBDMCS), reagent grade solvents, and anhydrous solvents were purchased from Fisher Scientific (Pittsburgh, PA). 15N2-Orotic acid, 2H4-thymine, DMSO-d6, and methanol-d4 were purchased from Cambridge Isotope Laboratory (Cambridge, MA). 2H4-5-methylcytosine and 2H2-cytosine were obtained from CDN Isotopes (Quebec, Canada). 15N2-uracil was purchased from Euriso-top (Saint-Aubin, France).
Analytical Methods.
NMR spectra were acquired with a Bruker UltraShield 300 MHz spectrometer (Billerica, MA) using DMSO-d6 or methanol-d4 as the solvent.
UV spectra were obtained with a Varian Cary 300 Bio UV/vis spectrophotometer (Santa Clara, CA).
HPLC analysis was performed with a ThermoFinnigan Surveyor HPLC system with a photodiode array detector (Waltham, MA) using a reverse-phase HPLC column (SUPELCOSIL-LC-18-S, 15 cm × 4.6 mm, 5 μm). The column was equilibrated with 10 mM ammonium acetate buffer (pH 6.8; flow rate, 1 mL/min). Mobile phases consisted of (A) 10 mM ammonium acetate at pH 6.8 and (B) methanol. The method was run isocratic for the first 10 min with mobile phase (A), and then a 20 min linear gradient was applied running from 0 to 50% B followed by column re-equilibration with 0% B at a flow rate of 1 mL/min. Analysis was conducted using a photodiode array detector (wavelength, 200–800 nm).
GC-MS data for analytical characterization of the free bases was obtained with an Agilent Technologies 5975C inert XL EI Triple-Axis detector coupled to an Agilent Technologies 7890A GC. For each free base standard, 50 μL of a 2.5 mM solution was placed into a 12 × 32 mm vial containing a 250 μL fused silica insert (Thermo Scientific; Rockwood, TN), and the solvent was evaporated under reduced pressure. The dried samples were reconstituted with 20 μL of acetonitrile and 20 μL of MTBSTFA + 1% TBDMCS, sealed, and heated at 140 °C for 40 min to make their tert-butyl-dimethylsilyl (TBDMS) derivatives. Each silylated sample was injected (1 μL) onto a GC-MS equipped with a Hewlett-Packard silica capillary column (30 m × 0.25 mm) coated with cross-linked 5% phenyl/95% methylpolysiloxane (film thickness, 0.25 μm). The following temperatures were used: 250 °C for the injector and 260 °C for the detector interface. The initial oven temperature was 100 °C for 2 min and was ramped to 180 °C at 10 °C/min and then to 260 °C at 30 °C/min and held for 7 min.
High-resolution GC-MS spectra, of the TBDMS derivatives, were obtained with a Waters Micromass GCT (Milford, MA) coupled to an Agilent 6890 GC. Ultra-high-resolution microelectrospray mass spectra of the underivitized standards were obtained with a Bruker Solarix 12 T FT-ICR MS. Samples were constantly infused and ionized by positive ion microelectrospray.28
Synthetic Procedures.
Cytosine Analogues.
15N2-Ethyl Ureidomethylenecyanoacetate (1).
To a 25 mL dry round-bottomed flask were added the following: 15N2-urea (1 g, 16.66 mmol, 98% enriched), triethyl orthoformate (4.5 mL, 26.66 mmol), and ethyl cyanoacetate (1.7 mL, 16.66 mmol). The neat reaction mixture was refluxed for 10 h. The solid was filtered and washed with acetone to obtain 15N2-ethyl ureidomethylenecyanoacetate (1.7 g, 55% yield). 1H NMR (DMSO-d6, 300 MHz) δ (ppm): 10.54–10.58 (d, 1H), 8.07–8.11 (d, 1H), 7.61 (br s, 1H), 7.38 (br s, 1H), 4.20–4.27 (q, 2H), 1.24–1.29 (t, 3H).
15N2-5-Carboxyethylcytosine (2).
To the solution of sodium ethoxide, prepared by mixing 15 mL absolute ethanol and sodium (230 mg, 10 mmol), was added 15N2-ethyl ureidomethylenecyanoacetate (1.7 g, 9 mmol), and the reaction mixture was refluxed for 4 h. The alcohol was removed by filtration, and the collected solid was dissolved in 100 mL of water. This solution was filtered and acidified with glacial acetic acid to pH 5. The precipitate was collected on a Buchner funnel and washed with alcohol and then with ether to obtain 15N2-5-carboxyethylcytosine (0.8 g, 50% yield). 1H NMR (DMSO-d6) δ (ppm): 11.38 (s, 1H), 8.20 (s, 1H), 7.83 (br s, 1H), 7.63 (br s, 1H), 4.18–4.25 (q, 2H), 1.24–1.29 (t, 3H).
15N2-5-Hydroxymethylcytosine (3) and 15N2-5-Carboxycytosine (4).
To a 10 mL solution of LiAlH4 (1 M in THF) was added 15N2-5-carboxyethylcytosine (370 mg, 2 mmol) at room temperature. The reaction mixture was warmed to 40 °C and stirred for 2 h. After cooling the mixture to room temperature, water (5 mL) was added dropwise to quench excess LiAlH4. The solid was removed by filtration and washed with water (50 mL). The water extracts were combined and concentrated to a volume of 10 mL. GC-MS analysis of this crude mixture identified the presence of 15N2-5-hydroxymethylcytosine and 15N2-5-carboxycytosine. The crude product was purified by HPLC using a Hypersil prep HPLC column, which was eluted with up to 50% methanol in 10 mM ammonium acetate (pH 5.5) to obtain 15N2-5-hydroxymethylcytosine (84 mg, measured by UV with ε269 = 5700 M−1 cm−1) and 15N2-5-carboxycytosine (21 mg, measured by UV with ε275 = 5800 M−1 cm−1). 1H NMR for 15N2-5-hydroxymethylcytosine (DMSO-d6) δ (ppm): 10.40 (s, 1H), 7.26 (s, 1H), 6.47 (br s, 2H), 4.92–4.95 (br t, 1H), 4.14–4.16 (d, 2H). HRMS (ESI+): m/z calcd for C5H7N15N2O2 [M + H]+, 144.05508; found, 144.05518. 1H NMR for 15N2-5-carboxycytosine (CD3OD) δ (ppm): 12.34 (s, 1H), 8.38 (s, 1H). HRMS (ESI+): m/z calcd for C5H5N15N2O3 [M + H]+, 158.03438; found, 158.03448.
15N2-5-Formylcytosine (5).
A solution of 15N2-5-hydroxymethylcytosine (50 mg, 0.35 mmol) was prepared in 5 mL of 50 mM sodium hydroxide and cooled to 0 °C. To this solution was added potassium perruthenate (71 mg, 0.35 mmol), and the reaction was allowed to stir at 0 °C for 2 h and then at room temperature for 10 h. The fine black particles were filtered through a 3 mL syringe packed with 1 mL of silica and 0.5 mL of sand and then through a 0.45 μm syringe filter [From bottom to top: filter, 1 mL of silica, and 0.5 mL of sand]. The collected filtrate was concentrated and HPLC purified using a Hypersil prep HPLC column, which was eluted with up to 50% methanol in 10 mM ammonium acetate (pH 5.5) to obtain 15N2-5-formylcytosine (15.3 mg, measured by UV with ε276 = 6600 M−1 cm−1). 1H NMR (DMSO-d6) δ (ppm): 9.41 (s, 1H), 8.38 (s, 1H), 7.84–7.86 (br m, 3H). HRMS (ESI+): m/z calcd for C5H5N15N2O2 [M + H]+, 142.03948; found, 142.03952.
Uracil Analogues.
15N2-Ethyl Ureidomethylenemalonate (6).
To 25 mL dry round-bottomed flask were added the following: 15N2-urea (1 g, 16 mmol), triethyl orthoformate (4.5 mL, 26.66 mmol), and diethyl malonate (3.5 mL, 21 mmol). The neat reaction mixture was refluxed for 4 h. The solid was filtered and washed with acetone to obtain 15N2-ethyl ureidomethylenemalonate (1 g, 27% yield). 1H NMR (DMSO-d6) δ (ppm): 11.41 (s, 1H), 8.14 (s, 1H), 7.55 (br s, 1H), 7.30 (br s, 1H), 4.17–4.24 (q, 4H), 1.23–1.27 (t, 6H).
15N2-5-Carboxyethyluracil (7).
To a solution of sodium ethoxide, prepared by mixing 15 mL absolute ethanol and sodium (100 mg, 4.3 mmol), was added 15N2-ethyl ureidomethylenemalonate (0.5 g, 2.15 mmol), and the reaction mixture was refluxed for 3 h. The alcohol was removed by filtration, and the collected solid was dissolved in 10 mL of water. This solution was acidified with glacial acetic acid to pH 5 to precipitate the desired product. The precipitate was collected on a Buchner funnel and washed with alcohol and then with ether to obtain 15N2-5-carboxyethyluracil (0.353 g, 93% yield). 1H NMR (DMSO-d6) δ (ppm): 11.80 (s, 1H), 11.29 (s, 1H), 8.09 (s, 1H), 4.11–4.18 (q, 2H), 1.19–1.24 (t, 3H).
15N2-5-Carboxyuracil (8).
A solution of 15N2-5-carboxyethyluracil (100 mg, 0.53 mmol) in 10 mL of 1 M NaOH was heated at 60 °C for 12 h. At the end of reaction, the mixture was allowed to cool at room temperature, and pH of the solution was adjusted to 4.5 using dilute HCl. The solution was concentrated to the volume of 5 mL and allowed to stand at 4 °C overnight to precipitate 5-carboxyuracil. The precipitates were collected via filtration and washed with acetone (10 mL) to obtain 15N2-5-carboxyuracil (75 mg, 90% yield). 1H NMR (DMSO-d6) δ (ppm): 12.73 (s, 1H), 11.99 (s, 2H), 8.26 (s, 1H). HRMS (ESI+): m/z calcd for C5H415N2O4 [M + H]+, 159.01838; found, 159.01849.
15N2-5-Hydroxymethyluracil (9).
Triethylamine (0.5 mL, 3.3 mmol) was added to a suspension of 15N2-uracil (250 mg, 2.2 mmol) and paraformaldehyde (200 mg, 6.6 mmol) in 10 mL of water. The solution turned clear when heated at 65 °C and was allowed to stir for 12 h at 65 °C. The water was reduced to a volume of 5 mL, and 5 mL of 95% ethanol was added to the mixture. The mixture was allowed to stand at 4 °C overnight to precipitate 15N2-5-hydroxymethyluracil as a white solid (250 mg, 80% yield). 1H NMR (DMSO-d6) δ (ppm): 10.79–11.18 (br m, 2H), 7.22–7.24 (d, 1H), 4.83 (br s, 1H), 4.10–4.11 (m, 1H). HRMS (ESI+): m/z calcd for C5H615N2O3 [M + H]+, 145.03908; found, 145.03920.
15N2-5-Formyluracil (10).
A solution of 15N2-5-hydroxymethyluracil (100 mg, 0.69 mmol) in 5 mL of water was heated at approximately 90 °C. The solution was then cooled to 45 °C, and K2S2O8 (187 mg, 0.69 mmol) and AgNO3 (3.5 mg, 0.02 mmol) were added. The product began to slowly precipitate. The reaction was stirred for 20 min at 40 °C and was then cooled to room temperature over 15 min while stirring was continued. The suspension was filtered, and the collected solid was rinsed with 2 mL of cold water to yield 15N2-5-formyluracil (70 mg, 71% yield). 1H NMR (DMSO-d6) δ (ppm): 11.89 (s, 1H), 11.50 (s, 1H), 9.74 (s, 1H), 8.14 (s, 1H). HRMS (ESI+): m/z calcd for C5H415N2O3 [M + H]+, 143.02348; found, 143.02356.
Animals.
Animal studies were conducted in a facility approved by the American Association for the Accreditation of Laboratory Animal Care (AAALAC), and all experiments were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee (protocol no. 1312056) of the University of Texas Medical Branch (UTMB). All animals were housed at the UTMB animal care facility and maintained according to U.S. Department of Agriculture standards (12 h light/dark cycle, food and water ad libitum). Male Sprague–Dawley (Charles Rivers, Wilmington, MA) rats (400–500 g) were anesthetized and humanely euthanized, and then their brains were collected and immediately frozen.
Metabolite Extraction from Rat Brain Tissue.
Whole brain tissue (1.75 g) from a male Sprague–Dawley rat was combined with 4 mL of deionized water and homogenized using a tissue homogenizer (POLYTRON PT 3100) for 45 s (15 s × 3) on an ice bath.29 The homogenized tissue was pelleted by centrifugation for 30 min at 14 000 rpm at 4 °C. The clear supernatant (3.5 mL) was aspirated, and 1 mL of 80:20 methanol/water at dry ice temperature (−75 °C) was added to the pellets and mixed. After 15 min at −75 °C, the sample was centrifuged at 14 000 rpm for 10 min at 4 °C, and the soluble extract (supernatant) was removed and mixed with the previously aspirated fraction. The pellets were then resuspended in 1 mL of 80:20 methanol/water and placed on dry ice for 15 min. The solution was centrifuged at 14 000 rpm for 10 min at 4 °C to yield a second clear supernatant, which was aspirated and combined with the previous extracts. The pellets were resuspended in 1 mL of 80:20 methanol/water, and the resulting suspension was sonicated in an ice bath for 45 s (15 s × 3) using a Branson Sonifier 450. The suspension was then centrifuged at 14 000 rpm for 10 min at 4 °C to yield a third clear supernatant, which was aspirated and combined with previous fractions to give a total extract volume of 6 mL. The extract was then filtered through a 3000 Da cutoff centrifugal spin filter (Millipore UFC900324) at 14 000g for 30 min. The clear filtrate (5.5 mL) was collected and mixed with isotope-labeled internal standards for HPLC purification and GC-MS analysis.
HPLC Isolation and GC-MS/MS Quantification of Metabolites.
A portion of the rat brain tissue extract (2 mL) was mixed with an internal standard mixture (50 μL, 1.13 × 10−5 M, 11 labeled standards) and purified by HPLC over a SUPELCOSIL-LC-18-S (15 cm × 4.6 mm, 5 μm) column, which was isocratically eluted with 10 mM ammonium acetate, pH 6.8, and 50% methanol for 10 min at a flow rate of 1 mL/min. In 2 mL microfuge tubes, HPLC fractions (0.6–1.5 mL each) were collected based on the HPLC retention times of the free base standards. To ensure separation of 5caU and 6caU, fractions were collected between 2.8–3.4 and 3.5–4.1 min, respectively. Each collected fraction was transferred to an autosampler vial and dried, and pyrimidines were converted to their TBDMS derivatives as described earlier. GC-MS/MS was used for the quantification of pyrimidine free base metabolites in the extracted rat brain tissue. For quantification, 1 μL injections were made on an Agilent Technologies 7000C GC-MS Triple-Quad detector coupled to an Agilent Technologies 7890B GC. The following temperatures were used: 250 °C for the injector and 260 °C for the detector interface. The initial oven temperature was 100 °C for 2 min and was ramped to 260 °C at 30 °C/min and held for 10 min. Data were collected in multiple reaction monitoring mode (MRM) using transitions determined using commercially available and synthetic standards.
RESULTS AND DISCUSSION
Synthesis and Characterization of Stable-Isotope-Enriched Pyrimidine Analogues.
Cytosine Analogues.
Previously, Whitehead30 demonstrated that urea, triethylorthoformate, and ethyl cyanoacetate could be condensed to form ethyl ureidomethylene cyanoacetate (Figure 2) followed by ring closure to form 5-carboxyethylcytosine. We utilized the same strategy, but substituted 15N2-enriched urea, to yield 15N2-enriched 5-carboxyethylcytosine. The product obtained was pure, as indicated by 1H NMR, and no other pyrimidines were observed by GC-MS. Using this procedure, approximately 1 g of 15N2-enriched urea can generate 0.8 g of enriched 5-carboxyethylcytosine.
Figure 2.
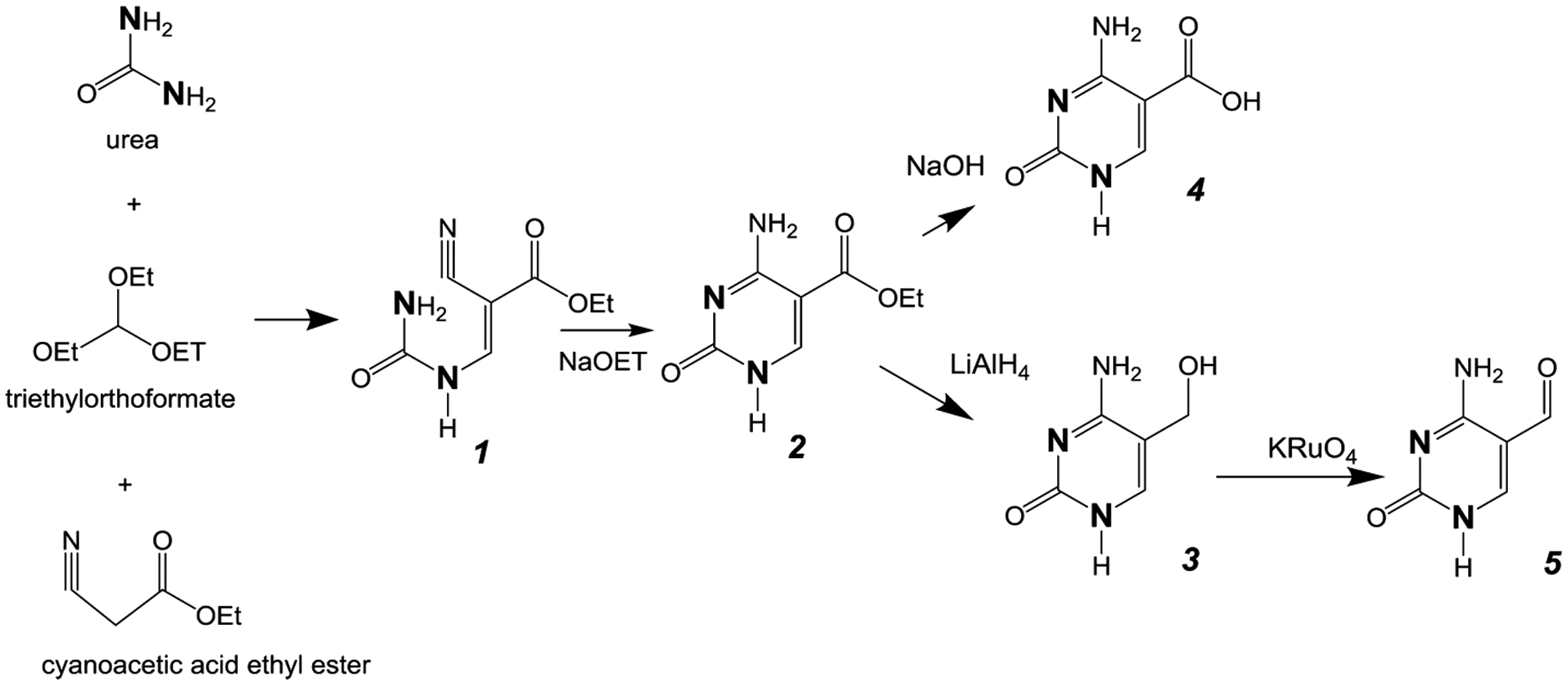
Synthetic pathway for cytosine analogs. The bold N denotes heavy isotope labeling (15N).
Acid hydrolysis of the ethyl ester of 5-carboxyethylcytosine yielding 5-carboxycytosine did not go to completion. However, alkaline hydrolysis in 1 N NaOH provided 15N2-5-carboxycytosine as the only pyrimidine product.
The reduction of the ethyl ester of 5-carboxyethylcytosine using LiAlH4 to form 5-hydroxymethylcytosine has been previously reported.31 In our hands, both 15N2-5-hydroxymethylcytosine and the hydrolysis product 15N2-5-carboxycytosine were generated in approximately equal amounts. We therefore used this method to generate the 15N2-analogues of the 5-hydroxymethyl and 5-carboxy analogues of cytosine, which were subsequently purified by HPLC. Product identity and purity were verified by GC-MS analysis.
The last product in the cytosine series, 5-formylcytosine, was generated by oxidation of enriched 5-hydroxymethylcytosine with potassium perruthenate.32 The product was purified by HPLC and characterized by GC-MS.
Uracil Analogues.
In the uracil series, the analogous condensation of urea, triethyl orothoformate, and diethylmalonate has been shown to yield ethylureidomethylene malonate, with ring closure generating 5-carboxyethyluracil,33 as shown in Figure 3. Using this procedure, approximately 1 g of 15N2-enriched urea can be converted to 350 mg of 15N2-enriched 5-carboxyethyluracil.
Figure 3.
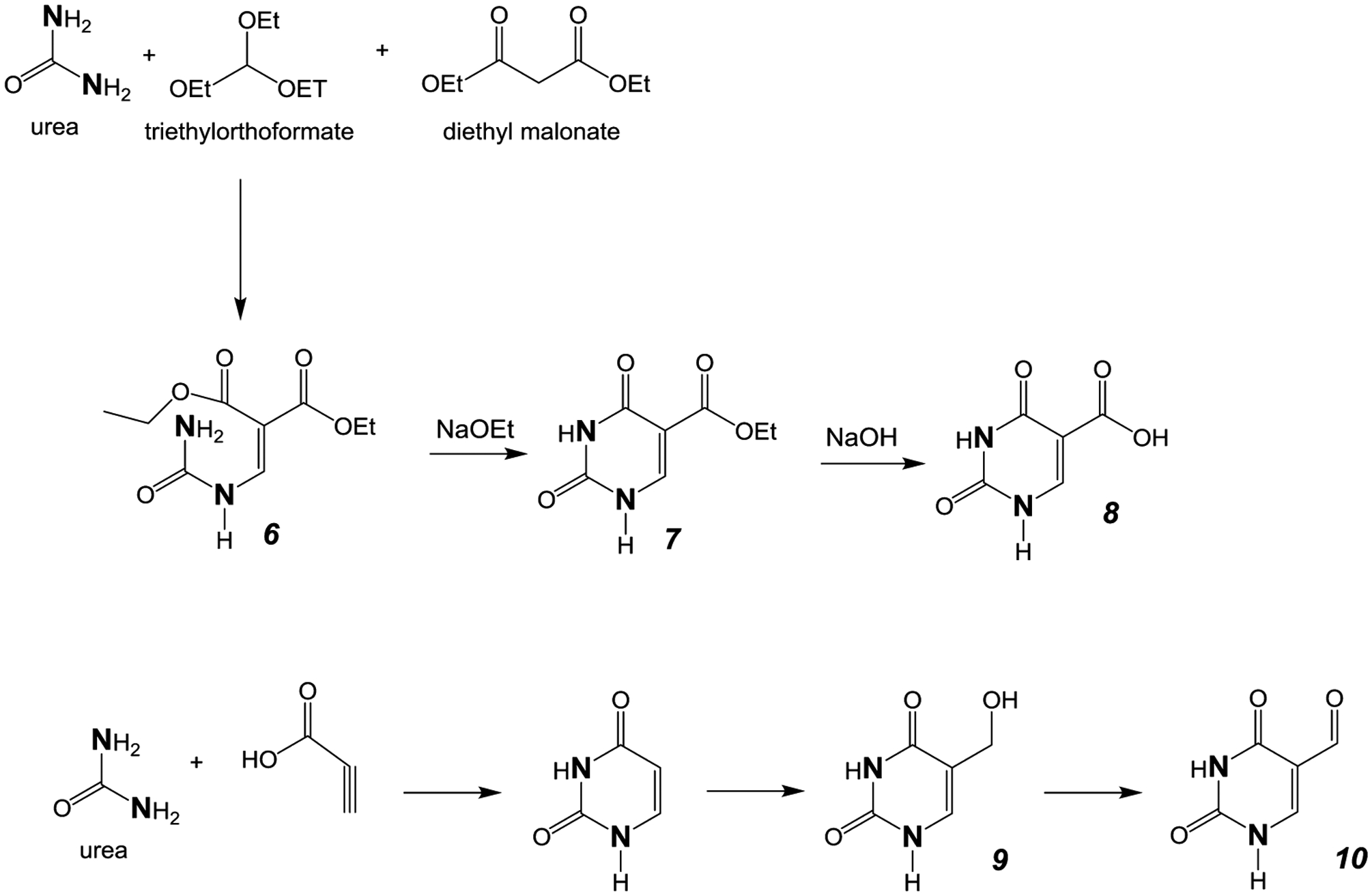
Synthetic pathway for uracil analogs. The bold N denotes heavy isotope labeling (15N).
The acid hydrolysis of 5-carboxyethyluracil yields 5-carboxyluracil; however, substantial amounts of uracil were also observed. Alternatively, alkaline hydrolysis of enriched 5-carboxyethyluracil generates enriched 5-carboxyluracil as the only pyrimidine product. The reduction of 5-carboxyethyluracil to 5-hydroxymethyluracil with LiAlH4, unlike the reduction of 5-carboxyethylcytosine described above, was unsuccessful. Similarly, reduction of 5-carboxyethyluracil with sodium borohydride did not proceed with significant yield.34 An alternative approach was needed, therefore, to generate 15N2-enriched 5-hydroxymethyluracil and 5-formyluracil.
Previously, it has been established that the condensation of 15N2-enriched urea with propiolic acid in polyphosphoric acid yields 15N2-enriched uracil.35,36 The conversion of uracil to 5-hydroxymethyluracil has been reported to proceed in high yield with aqueous formaldehyde and triethylamine.37 We confirmed the analogous formation of 15N2-5-hydroxymethyluracil conversion using enriched uracil.
The oxidation of 5-hydroxymethyluracil with silver nitrate and potassium persulfate in water yields 5-formyluracil,37 completing the synthesis of the uracil series. The 15N2-enriched uracil analogues were purified by HPLC and characterized by GC-MS, NMR, and UV spectroscopies as described below.
Characterization of the Pyrimidine Analogues.
Synthetic analogues could be separated from one another in sufficient quantity by HPLC, as shown in Figure 4. HPLC-purified analogues were characterized by GC-MS, as shown in Figure 5. The corresponding UV spectral characteristics and pKa values are shown in Table 1. GC-MS mass and retention times are provided in Table 2.
Figure 4.
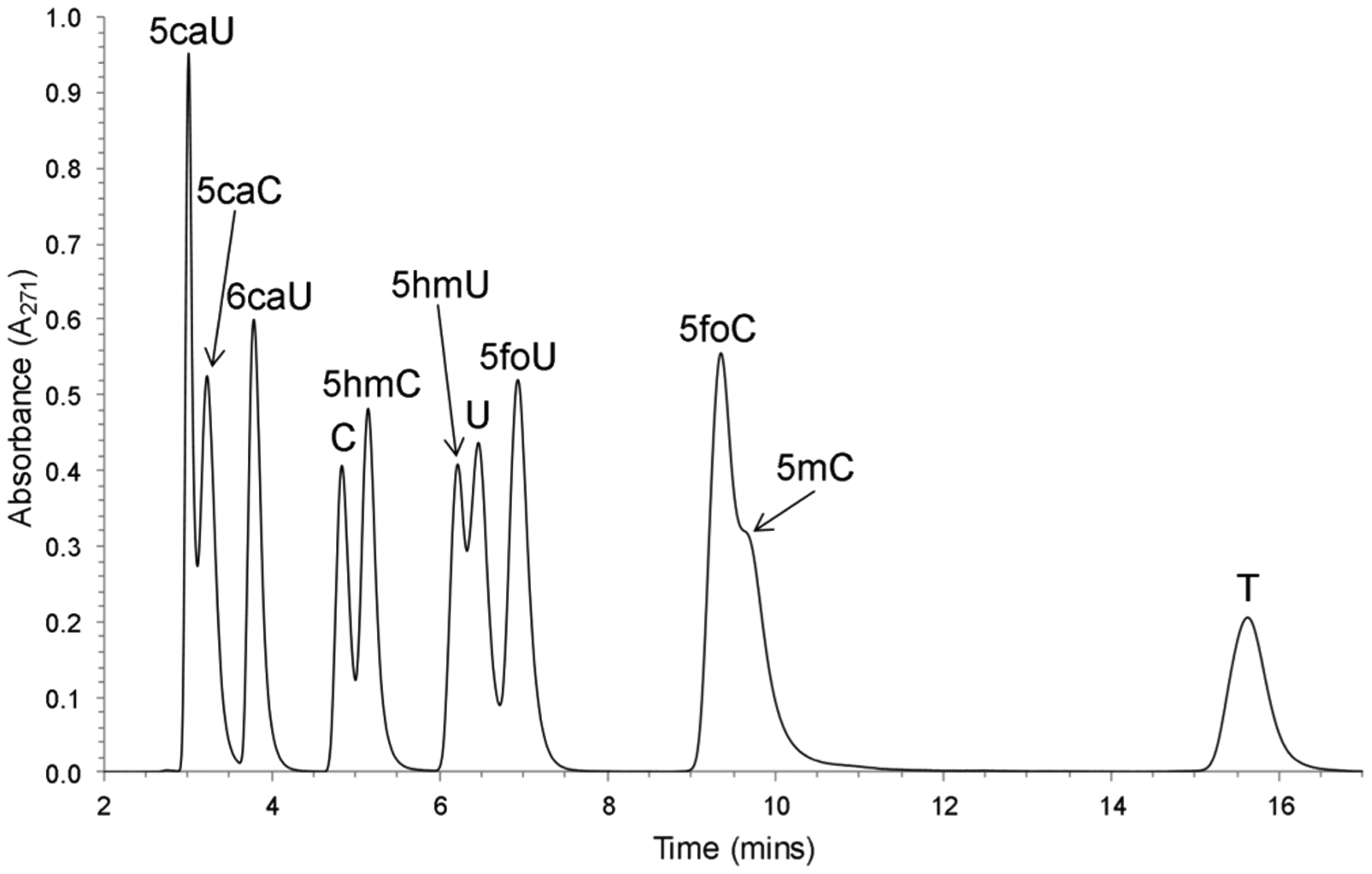
HPLC chromatogram of oxidized pyrimidine free bases obtained with UV absorbance. A mixture of oxidized pyrimidine free bases was separated by HPLC using a SUPELCOSIL-LC-18-S column eluted with 50% methanol in 10 mM ammonium acetate at pH 6.8 (flow rate, 1 mL/min).
Figure 5.
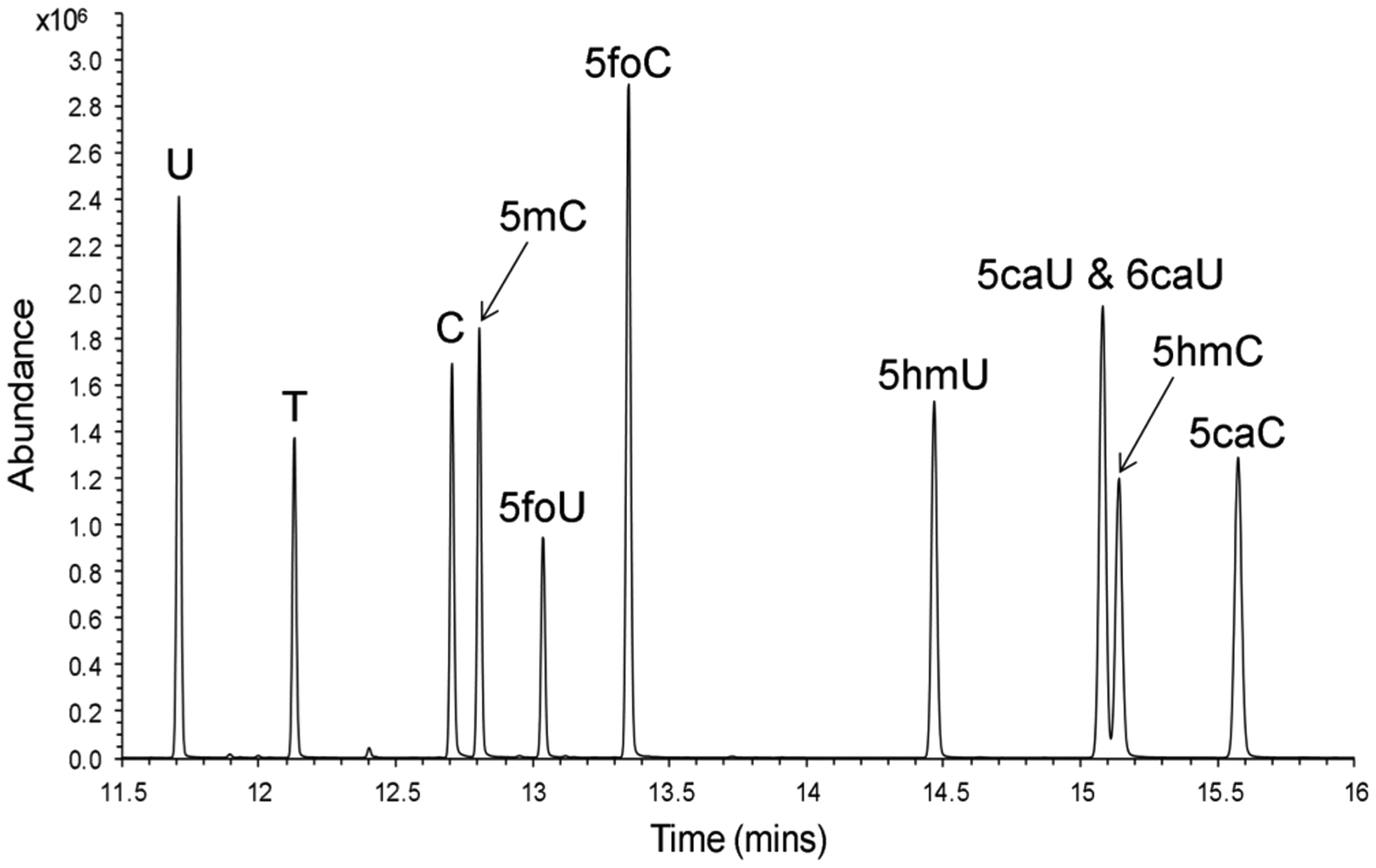
GC-MS chromatogram of free bases mixture. A mixture of oxidized pyrimidine free bases was derivatized to the TBDMS derivatives and injected onto an Agilent Technologies 7890A GC system that was coupled to an Agilent 5975C MSD with Triple-Axis detector. Data was acquired in the selected ion mode and is presented as the total ion chromatogram.
Table 1.
UV λmax, ε, pKa Values, and H6 Proton Chemical Shift for Pyrimidine Free Bases Examined in This Studya
| compound | λmax at pH 7 (nm) | ε at pH 7 (M−1 cm−1) | pKa | H6 proton chemical shift (ppm) |
|---|---|---|---|---|
| thymine | 265 | 7900b | 9.9, >13.0 | 7.25 |
| 5-hydroxymethyluracil | 261 | 8000b | 9.4, ~14.0 | 7.23 |
| 5-formyluracil | 293 | 12 000 | 6.8c, >9.4 | 8.14 |
| 5-carboxyuracil | 268 | 8300 | 4.1d, 9.0c | 8.26 |
| uracil | 2b0 | 8200b | 9.5, >13.0 | 7.38 |
| 5-methylcytosine | 273 | 6200b | 4.6, 12.4 | 7.69 |
| 5-hydroxymethylcytosine | 269 | 5700b | 4.3, ~13.0 | 7.26 |
| 5-formylcytosine | 276 | 6600 | 2.6e, 9.1 | 8.38 |
| 5-carboxycytosine | 275 | 5800 | 2.5f, 4.7f, >9.1 | 8.38 |
| cytosine | 267 | 6100b | 4.5, 12.2 | 7.30 |
| 6-carboxyuracil | 279 | 7700b | 2.4, 9.5 | 6.00 |
Values reported in this table were measured in this study for the free base analogues except where otherwise noted.
Values for λmax, ε, and pKa are from ref 40. Values for ε correspond to λmax.
The pKa values for 5-formyluracil and 5-carboxyuracil are from ref 41.
The pKa value reported correspond to the 2′-deoxynucleoside analogue from ref 42.
The reported pKa value corresponds to the 2′-deoxynucleoside analogue from ref 43.
The pKa value for 5-carboxycytosine is from ref 44.
Table 2.
HR-MS (GC-ToF-MS) Data, GC, and HPLC Retention Times of Stable Isotope Enriched Pyrimidine Free Bases Examined in This Study
| compound | chemical formula TBDMS derivatives | theoretical M-[tbutyloxy] | experimentala M-[tbutyloxy] | GCa tR (min) | HPLCb tR (min) |
|---|---|---|---|---|---|
| 2H4-thymine | C17H302H4N2O2Si2 | 301.17056 | 301.1697 | 12.13 | 15.63 |
| 15N2-5-hydroxymethyluracil | C23H4815N2O3Si3 | 429.22091 | 429.2229 | 14.47 | 6.22 |
| 15N2-5-formyluracil | C17H3215N2O3Si2 | 313.11879 | 313.1196 | 13.04 | 6.93 |
| 15N2-5-carboxyuracil | C23H4615N2O4Si3 | 443.20018 | 443.2021 | 15.08 | 3.01 |
| 15N2-uracil | C16H3215N2O2Si2 | 285.12387 | 285.1239 | 11.71 | 6.47 |
| 2H4-5-methylcytosine | C17H312H4N3OSi2 | 300.18654 | 300.1856 | 12.81 | 9.69 |
| 15N2-5-hydroxymethylcytosine | C23H49N15N2O2Si3 | 428.23690 | 428.2385 | 15.14 | 5.15 |
| 15N2-5-formylcytosine | C17H33N15N2O2Si2 | 312.13477 | 312.1355 | 13.35 | 9.36 |
| 15N2-5-carboxycytosine | C23H47N15N2O3Si3 | 442.21616 | 442.2178 | 15.58 | 3.24 |
| 2H2-cytosine | C16H312H2N3OSi2 | 284.15834 | 284.1571 | 12.71 | 4.84 |
| 15N2-6-carboxyuracil | C23H4615N2O4Si3 | 443.20018 | 443.2007 | 15.08 | 3.79 |
Mass and retention times for TBDMS derivatives were measured on a high-resolution single quadrupole GC-ToF-MS.
HPLC retention time for underivatized free bases (tR).
All of the DNA pyrimidine metabolites can be separated by both HPLC and GC, facilitating identification and quantification. In principle, a mixture of analytes and standards could be quantified without separation by examining appropriate mass transitions. Within the series of metabolites examined here, the nominal mass of the unenriched 5-hydroxymethyl analogues is the same as that of the 15N2-enriched 5-formyl analogue (isobars) in both the uracil and cytosine series. However, at higher mass resolution, the analogues can be distinguished without separation, as shown in Figure 6 and Table 3, based on their exact masses.
Figure 6.
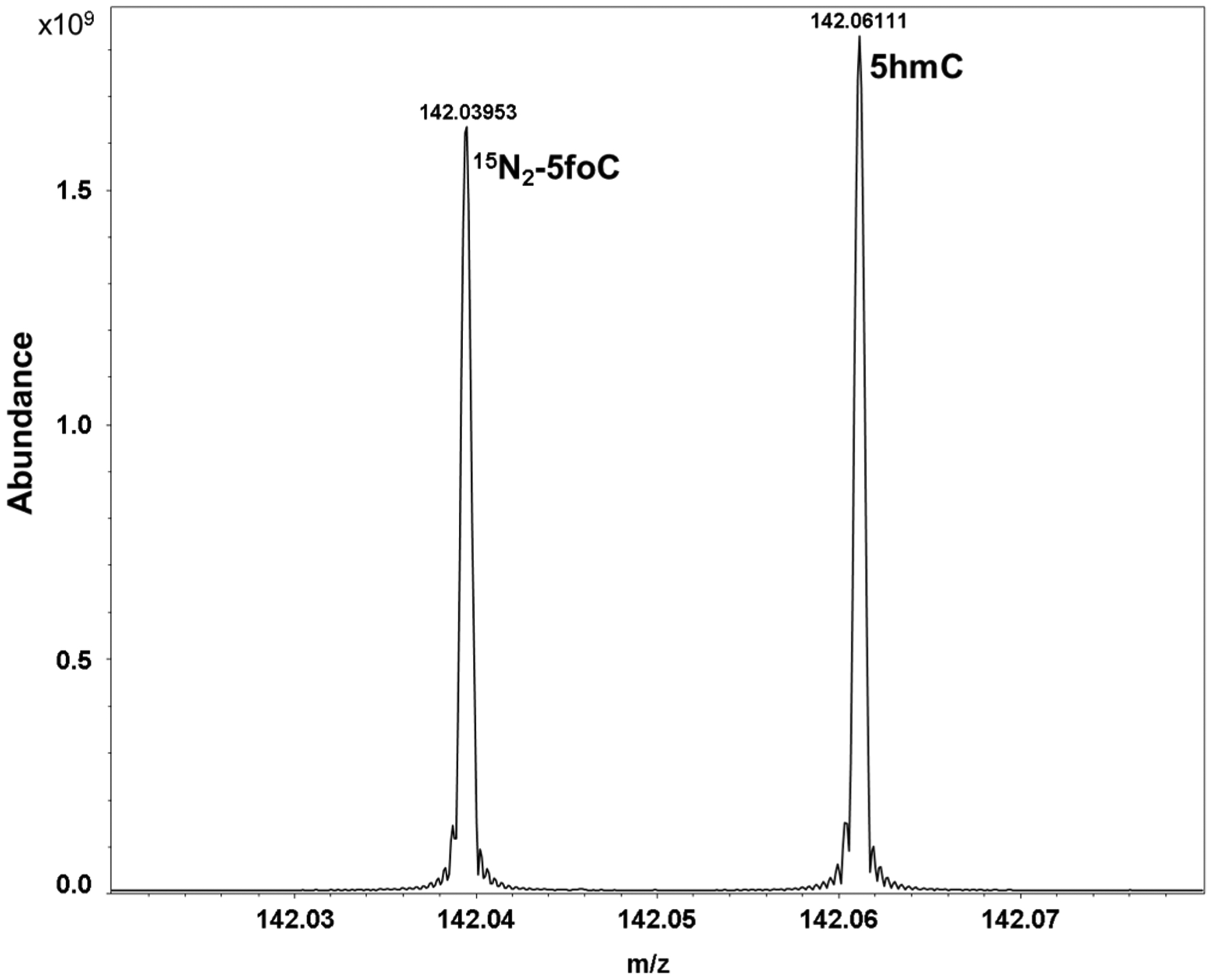
Ultra-high-resolution 12 T FT-ICR MS positive ion microelectrospray ionization spectrum of a mixture of 15N2-5foC and 5hmC showing a zoomed spectrum over a 60 mDa window. FT-ICR MS easily baseline resolves the biologically active 5hmC that differs in mass by only ~22 mDa from the isotope-labeled standard 15N2-5foC. Mass accuracy was ~350 ppb for 15N2-5foC and ~210 ppb for 5hmC. Spectra were acquired on a 12 T Bruker Solarix Fourier transform ion cyclotron resonance mass spectrometer (FT-ICR MS).
Table 3.
Ultra-High-Resolution FT-ICR MS Mass Measurements for Pyrimidine Free Bases Examined in This Study
| compound | formula | [M + H]+ (theoretical) | [M + H]+ (experimental) |
|---|---|---|---|
| thymine | C5H6N2O2 | 127.05018 | 127.05018 |
| 5-hydroxymethyluracil | C5H6N2O3 | 143.04508 | 143.04516 |
| 5-formyluracil | C5H4N2O3 | 141.02948 | 141.02950 |
| 5-carboxyuracil | C5H4N2O4 | 157.02438 | 157.02442 |
| uracil | C4H4N2O2 | 113.03458 | 113.03450 |
| 5-methylcytosine | C5H7N3O | 126.06618 | 126.06613 |
| 5-hydroxymethylcytosine | C5H7N3O2 | 142.06108 | 142.06108 |
| 5-formylcytosine | C5H5N3O2 | 140.04548 | 140.04543 |
| 5-carboxycytosine | C5H5N3O3 | 156.04038 | 156.04037 |
| cytosine | C4H5N3O | 112.05048 | 112.05044 |
| 15N2-5-hydroxymethyluracil | C5H615N2O3 | 145.03908 | 145.03920 |
| 15N2-5-formyluracil | C5H415N2O3 | 143.02348 | 143.02356 |
| 15N2-5-carboxyuracil | C5H415N2O4 | 159.01838 | 159.01849 |
| 15N2-5-hydroxymethylcytosine | C5H7N15N2O2 | 144.05508 | 144.05518 |
| 15N2-5-formylcytosine | C5H5N15N2O2 | 142.03948 | 142.03952 |
| 15N2-5-carboxycytosine | C5H5N15N2O3 | 158.03438 | 158.03448 |
Development of a Method To Measure Pyrimidine Frees Bases from Tissue Extracts.
Our initial attempts to measure pyrimidine standards by GC-MS revealed that 10 of the 11 pyrimidines could be resolved by GC alone (Figure 5). Only the isomeric 5- and 6-carboxyuracil analogues co-eluted. When we first attempted to measure the pyrimidines in tissue extracts by GC-MS, we observed substantial background noise. Peaks expected for stable-isotope analogues that were added into the extracts also displayed coeluting contaminants. Additionally, overall metabolite recovery from the tissue extracts was much lower than expected.
Our initial GC-MS results suggested that additional sample cleanup was necessary, so we turned to HPLC separation as an initial cleanup step. Our work with the synthesis of the stable-isotope analogues revealed that we could obtain substantial resolution of many of the pyrimidines by HPLC (Figure 4). Most importantly, we were able to separate the 5- and 6-carboxyuracil isomers that cannot be resolved by GC-MS.
Although all of the pyrimidines expected from DNA metabolism can be distinguished by exact mass, the isomer of 5-carboxyuracil, 6-carboxyuracil (orotic acid), is a normal metabolite in the de novo synthesis of pyrimidines38,39 and therefore its presence in biological extracts is expected. The 5- and 6-carboxyuracil analogues are inseparable by GC; however, they are easily separated by HPLC at pH 6.8 (Figure 4). The pKa values for this pyrimidine series40–44 are listed in Table 1. We note that the pKa values for the carboxyl groups of 5- and 6-carboxyuracil differ by 1 pH unit.
The next step in our method development was to combine the HPLC prepurification with GC-MS analysis, as shown in Figure 7. Freshly obtained animal tissue is homogenized, and cell debris is removed by centrifugation. The aqueous extract is then combined with water and methanol, chilled, and cleared by centrifugation, as described in Materials and Methods. The clear filtrate is then mixed with stable-isotope-enriched standards, and a portion of this mixture is injected onto the HPLC. Fractions are obtained containing one or more of each of the pyrimidine analytes, and these fractions are dried under reduced pressure, derivatized, and analyzed by GC-MS.
Figure 7.
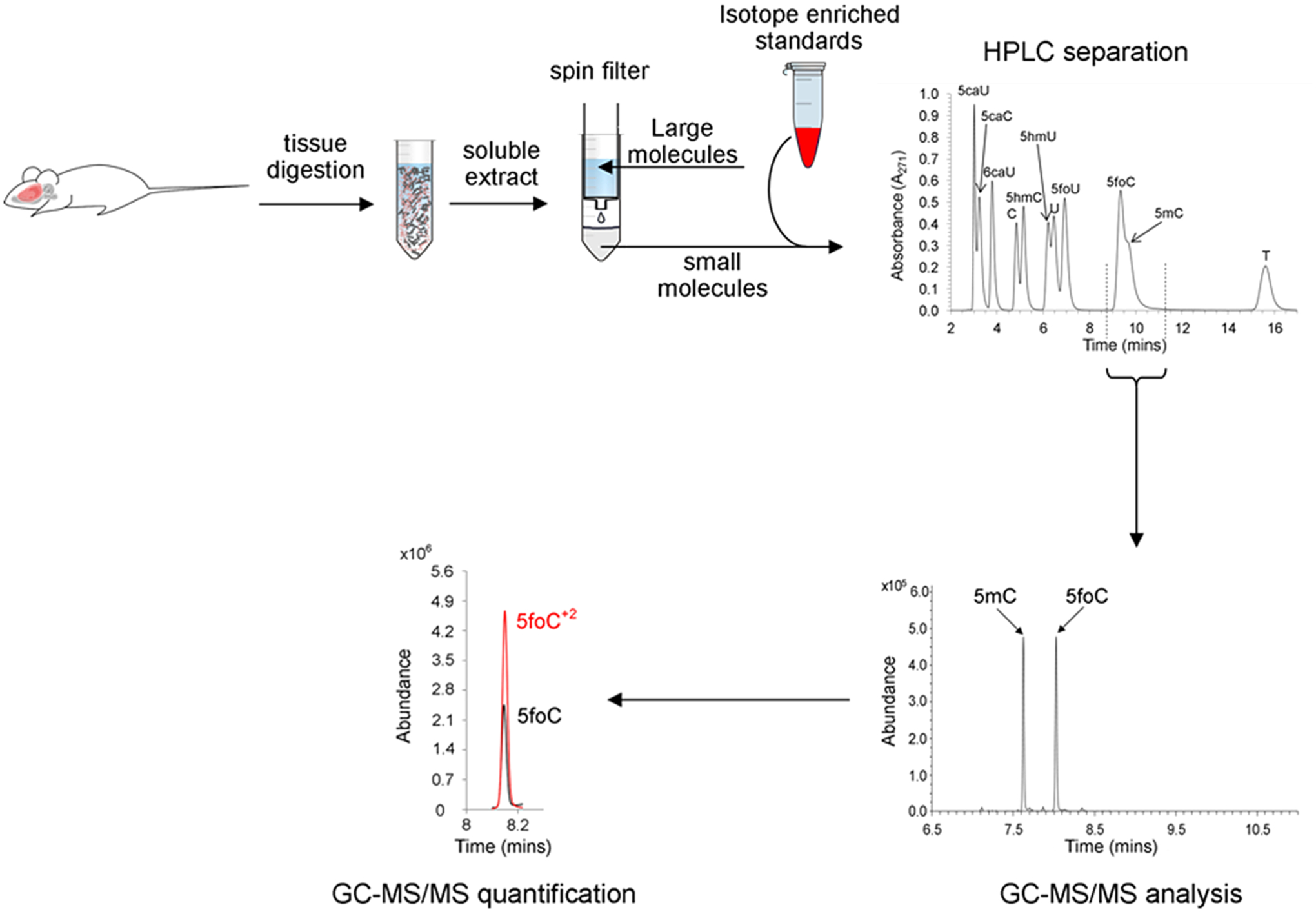
Workflow diagram for the isolation and analysis of pyrimidine free bases from a rat brain.
Our initial workup used GC separation and a single-quad mass spectrometer. Although selected ion monitoring on the single quad can be used for analyte identification and quantification, we observed substantial background peaks coeluting with the pyrimidines of interest. We therefore used a triple-quad mass spectrometer, which is able to substantially reduce background noise by trapping ions of interest and measuring selected fragments. Using this method, the results shown in Figure 8 are obtained. The level of each pyrimidine is then determined by measuring the size of the analyte peak relative to the size of the isotope standard peak. The limit of detection for this method is approximately 1 × 10−15 moles of pyrimidine injected. The volume of the derivatized sample is 40 μL, of which 1 μL is injected in splitless mode. Therefore, approximately 10−13 moles per sample can be detected with this method.
Figure 8.
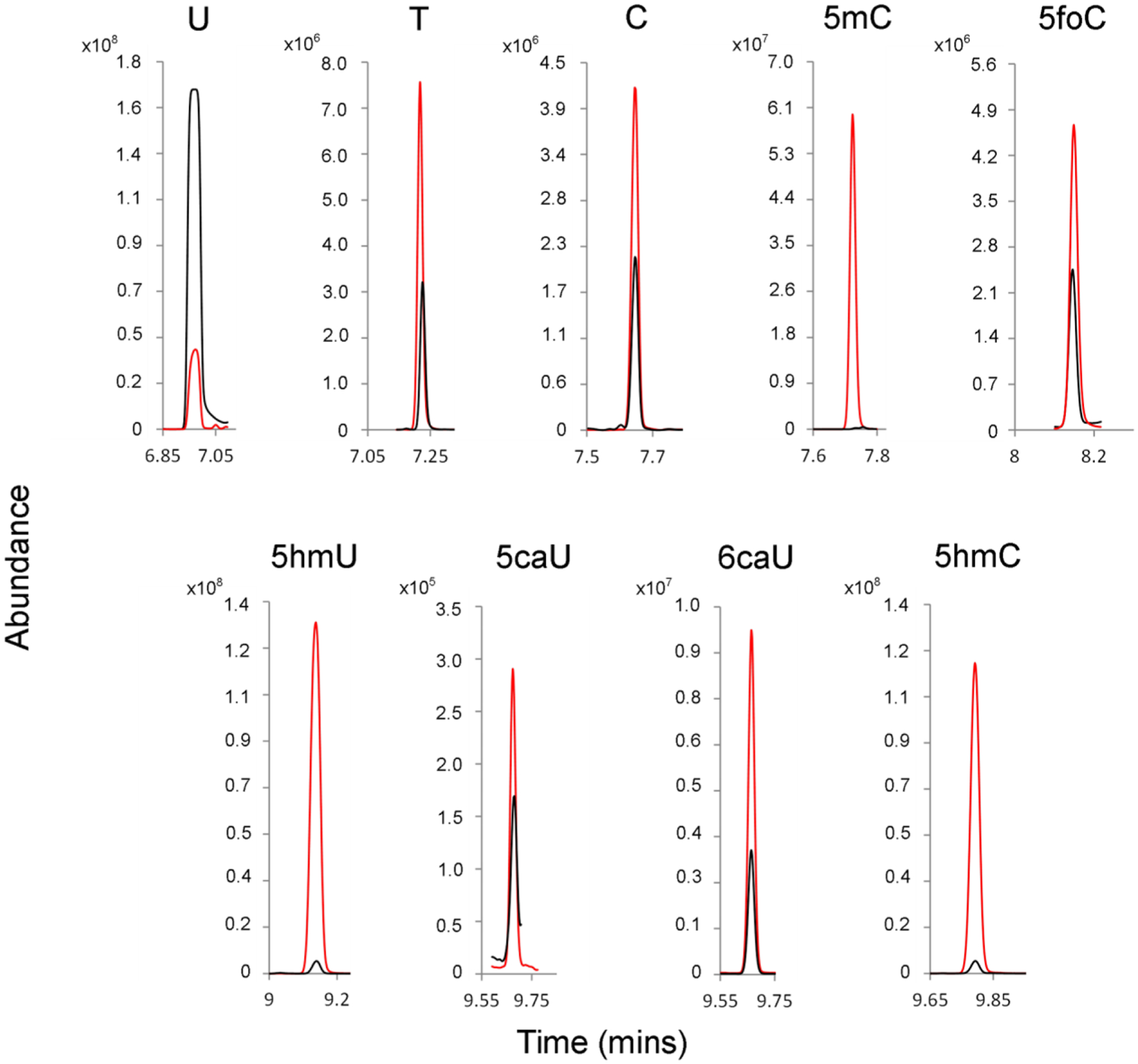
GC-MS/MS analysis of HPLC-purified pyrimidines obtained from a rat brain. Red, isotope-enriched internal standards; Black, unenriched metabolites from rat brain extract.
Measurement of Metabolites in Normal Rat Brain Extract.
The DNA of all living cells is persistently damaged by oxidation and hydrolysis.15 In addition to these chemically mediated pathways, DNA bases undergo enzyme-mediated modifications, including methylation, demethylation, and deamination of cytosine residues.10–14 Many of the chemically damaged or enzymatically modified bases are removed by proteins of the base excision repair (BER) pathway, and the damaged DNA is restored by repair synthesis.20–25 Collectively, these modifications constitute postreplicative DNA metabolism, and the end products of these pathways are the free base metabolites that are released into the cytoplasm and subsequently reutilized, excreted, or degraded.
Although substantial work from many laboratories has described the measurement of damaged bases in DNA, relatively little work has focused on measuring the free bases that are spontaneously or enzymatically released. In this work, we have prepared a series of isotopically enriched standards for a series of pyrimidine analogues known to be part of both endogenous DNA damage and enzyme-mediated DNA modification pathways. The isotope standards have been characterized by NMR and high-resolution mass spectrometry. We demonstrate here the feasibility of measuring the free bases in tissue extracts using stable-isotope-enriched standards. Following tissue homogenization and methanol–water extraction, isotope standards are added and small molecules are isolated using a spin filter. Free bases are separated by HPLC, and selected fractions are dried, derivatized, and examined by GC-MS/MS.
Nine of the 11 pyrimidines studied here are measurable in the rat brain extract by this method (Figure 8 and Table 4). Neither the potential metabolite 5foU nor its isotope standard are observed by GC-MS/MS, suggesting that 5foU undergoes chemical modification in the procedure described here. Potentially, the activated aldehyde of 5foU could condense with amines or sulfhydryl groups present in the biological extract. In contrast, the isotope standard of 5caC is observed. However, the level of unenriched 5caC in the tissue extract is below the limit of detection (~1 × 10−13 mol/g tissue).
Table 4.
Quantities (mol/g of Tissue) of Normal and Oxidized Pyrimidines in a Rat Brain Extract Measured Using Isotope-Labeled Standardsa
| compound | moles of free bases/g of tissue |
|---|---|
| uracil | 1.20 ± 0.11 × 10−08 |
| thymine | 9.01 ± 0.46 × 10−10 |
| cytosine | 1.18 ± 0.15 × 10−09 |
| 5-methylcytosine | 2.90 ± 0.13 × 10−11 |
| 5-formylcytosine | 1.21 ± 0.15 × 10−09 |
| 5-hydroxymethyluracil | 6.58 ± 0.37 × 10−11 |
| 5-carboxyuracil | 8.76 ± 0.05 × 10−10 |
| 6-carboxyuracil | 7.11 ± 0.08 × 10−10 |
| 5-hydroxymethylcytosine | 7.77 ± 0.10 × 10−11 |
| 5-carboxycytosine | <1.14 × 10−13 |
The data represent the average and standard deviation (SD) values from three different injections.
The amount of free thymine measured by this method is 9 × 10−10 mol/g, which corresponds to approximately 0.1% of the thymine found in DNA extracted from a tissue of similar size. In rat DNA, the ratio of thymine to cytosine is approximately 1.34,45 and the rate of spontaneous depyrimidination of thymine to cytosine is 1.28.46 Therefore, we would have expected more thymine than cytosine in the extracts resulting from depyrimidination. Surprisingly, the amount of thymine found was only about 80% that of cytosine.
Other potential mechanisms could also modulate thymine levels. Perhaps the deamination of 5mC to thymine, followed by glycosylase removal, could also contribute to the thymine levels;47 however, the ratio of 5mC to thymine in rat DNA is small, only about 1:30. Therefore, this pathway would not be expected to contribute substantially to the free thymine levels. DNA from dead and dying cells can be degraded to generate 5′-monophosphates that can be either reutilized for DNA synthesis or dephosphorylated to 2′-deoxynucleosides that can by degraded to free pyrimidines by phosphorylases. Uracil and thymine free bases can be generated and utilized by thymidine phosphorylase.48 In addition, uracil and thymine free bases analogues can be metabolized by dihydropyrimidine dehydrogenase and excreted.49 For example, the chemo-therapeutic agent 5-fluorouracil can be incorporated into the nucleotide pool by thymidine phosphorylase,50 which suggests a pathway for reducing thymine levels in the brain extracts. The observed levels of thymine in the extracts, therefore, most likely reflect thymine generated by spontaneous hydrolysis minus that which is salvaged or degraded.
Both cytosine and 5mC are observed in the tissue extracts. This result was unexpected, as most glycosylases do not target cytosine derivatives, although 5mC glycosylases have been reported in some species.51–53 In rat brain, approximately 4% of the cytosine residues are methylated.54 The ratio of 5mC to cytosine observed in our rat brain extracts was approximately 2.5%. Both cytosine and 5mC can be lost from the DNA by spontaneous hydrolysis of the glycosidic linkage, and the rates of depyrimidination for both are similar.46 Cytosine and 5-substituted cytosine bases are not enzymatically deaminated in animal cells, although the corresponding nucleoside and nucleotide analogues are rapidly deaminated in animal cells.55 For example, 5-fluorocytosine serves as a prodrug of 5-fluorouracil and is activated in mammalian tissues only by deamination in the presence of microbial cytosine deaminase.56 Unlike the uracil analogues discussed below, cytosine is not reutilized by salvage pathways, and it is not degraded by dihydropyrimidine dehydrogenase. Therefore, the cytosine and 5mC free bases observed in these extracts most likely reflect endogenous depyrimidination.
Among the oxidized bases, both 5hmU and 5hmC can arise by chemical oxidation of the corresponding thymine and 5mC methyl groups.57 An enzymatic pathway has also been identified for the conversion of 5mC to 5hmC in DNA.11–13 In mouse brain, the ratio of 5mC to 5hmC is approximately 10.58 The amount of thymine in rat DNA exceeds the amount of 5mC by a factor of 30. Therefore, the amount of 5hmU in the extracts would be expected to exceed that of 5hmC if only endogenous chemical pathways were considered. Furthermore, 5hmU is removed from DNA by the BER pathway, but 5hmC is not a substrate for TDG glycosylase, and no activities have been observed that remove 5hmC from DNA.25 The observation of similar levels of 5hmU and 5hmC in the extracts is, therefore, surprising and might suggest an as yet unidentified pathway for the removal of 5hmC from DNA.
Both 5hmU and 5hmC can be further oxidized to the corresponding 5-formyl analogues 5foU and 5foC.45 The enzymatic oxidation of 5hmC can also generate 5foC in DNA.59 Unlike 5hmC, 5foC paired with guanine is a preferred substrate for the TDG glycosylase,22 and 5foU is repaired by multiple members of the uracil-DNA glycosylase family.60 Neither 5foC nor 5foU would be expected to remain in DNA for long due to the BER pathway. In this study, neither 5foU nor the isotopomer standard was observed. It is known that 5foU can react with both amines and thiol groups, likely explaining its disappearance in this study.61,62 In contrast, substantial amounts of 5foC are observed. The observation of 5foC is consistent with enzymatic 5mC demethylation, where 5mC is first oxidized to 5hmC and then to 5foC and released from DNA by the BER pathway.
Further oxidation of 5foU and 5foC can generate the 5-carboxy analogues 5caU and 5caC.22 The human single-strand-selective mispaired uracil DNA-glycosylase (SMUG1) can remove 5caU from DNA,42 and human thymine-DNA glycosylase (hTDG) can remove 5caC.22 Although 5caU is observed in the extracts, 5caC is not observed. Unlike 5foU discussed above, the internal standard 5caC added to the extracts is measurable. Since the level of 5caC free base in the rat brain is below the level of detection in this study, it is unlikely to be a significant DNA metabolite.
The measurement of 5caU created an analytical challenge, as 6-carboxyuracil (6caU, orotic acid) is a normal metabolite in the de novo synthesis of pyrimidines.38,39 The isomeric 5- and 6-carboxyuracil analogues are inseparable by GC-MS; however, they can be separated by HPLC and subsequently measured in independent GC-MS/MS runs. Similar levels of both 5caU and 6caU are observed in this study.
The observed level of 5caU exceeds that of 5hmU by a factor of 10. As the analogues 5hmU, 5foU, and 5caU are generated by sequential oxidation, it is unlikely that the observed 5caU arose by endogenous oxidation. It has been proposed, however, that enzymatic oxidation of 5mC followed by enzymatic deamination to the corresponding uracil analogue is an additional pathway for 5mC demethylation.24 As 5foU is not observable by this method, the sequence of transformations leading from 5mC to 5caU cannot be established. However, the simultaneous observation of 5foC and 5caU suggests that multiple pathways exist for the modification of 5mC residues in DNA.
The most abundant pyrimidine observed in the tissue extract is uracil. As with thymine, uracil can be both a product of and substrate for thymidine phosphorylase,48 and both are degraded by dihydropyrimidine dehydrogenase.49 In addition, uracil and its analogues are both products and substrates of uridine phosphorylase.63 For example, exogenous 5-fluorouracil is incorporated into both DNA and RNA. Therefore, the phosphorylases would tend to decrease uracil levels, as opposed to increasing the level of uracil over that of thymine in the extracts.
There are two mechanisms by which uracil can be incorporated into DNA. In the first pathway, dUTP is incorporated by DNA polymerases during DNA replication. In the second pathway, cytosine residues in DNA are hydrolytically deaminated to uracil.64 In both pathways, the uracil in DNA is removed by members of the uracil-DNA glycosylase family, generating free uracil in the extract. The dUTP misincorporation pathway would predominate in replicating cells, whereas the deamination pathway would predominate in postmitotic cells. Both replicating and postmitotic cells would be present in the tissue extracts observed here. If the deamination pathway predominated, then we would expect uracil levels to be similar to cytosine levels in the extracts since the rates of DNA depyrimidination and cytosine deamination are similar. However, the level of uracil is more than an order of magnitude greater than that of cytosine. This suggests that uracil transiting through the DNA due to dUTP misincorporation might account for more of the released uracil. Because dUTP levels can be modulated by manipulation of one carbon metabolism, future studies using the methods presented here could resolve this question.65,66
CONCLUSIONS
While DNA is the repository of genetic information that is passed onto progeny cells, it is metabolically active in all cells due to persistent endogenous damage and repair. It is also subject to enzymatic modifications in cells that undergo differentiation, and both damage and enzymatic modification can occur simultaneously. When enzymatically removed from DNA, the modified free bases are released into the cytoplasm and are either excreted, further metabolized, or reutilized. Free bases can be recovered from cell and tissue extracts and analyzed. In this article, we describe the synthesis and characterization of a complete series of 15N2-enriched analogues of the possible base metabolites described to date. These analogues can be used to study DNA synthesis, modification, and base turnover in both normal tissues and cells with metabolic defects. Such studies should shed additional light on these increasingly important DNA metabolic pathways.
ACKNOWLEDGMENTS
We thank Maggie Parsley and Ian Bolding for their assistance with the animal experiments.
Funding
This work was supported in part by the National Institutes of Health, National Cancer Institute (CA184097), National Institute of Environmental Health Sciences (T32ES007254; J.L.H.), and as part of an interdisciplinary research team funded by The Moody Project for Translational Traumatic Brain Injury.
ABBREVIATIONS
- ε
molar absorptivity
- λmax
lambda max
- AAALAC
American Association for the Accreditation of Laboratory Animal Care
- AID
activation-induced deaminase
- APOBEC-1
apolipoprotein B mRNA editing enzyme catalytic polypeptide 1
- BER
base excision repair
- dUTP
2′-deoxyuridine 5′-triphosphate
- ESI+
electrospray ionization in positive ion mode
- FT-ICR MS
Fourier transform ion cyclotron resonance mass spectrometer
- GC-MS/MS
gas chromatography/tandem mass spectrometry
- GC-ToF-MS
gas chromatography time-of-flight mass spectrometry
- HRMS
high-resolution mass spectrometry
- hTDG
human thymine DNA glycosylase
- [M + H]+
protonated molecule
- MRM
multiple reaction monitoring mode
- MTBSTFA
N-tert-butyldimethylsilyl-N-methyltrifluoroacetamide
- TBDMCS
tert-butyldimetheylchlorosilane
- TBDMS
tert-butyl-dimethlylsilyl
- TDG
thymine DNA glycosylase
- TET
ten-eleven translocation
Footnotes
The authors declare no competing financial interest.
REFERENCES
- (1).Hotchkiss RD (1948) The quantitative separation of purines, pyrimidines, and nucleosides by paper chromatography. J. Biol. Chem 175, 315–332. [PubMed] [Google Scholar]
- (2).Ehrlich M, and Wang RY (1981) 5-Methylcytosine in eukaryotic DNA. Science 212, 1350–1357. [DOI] [PubMed] [Google Scholar]
- (3).Herring JL, Rogstad DK, and Sowers LC (2009) Enzymatic methylation of DNA in cultured human cells studied by stable isotope incorporation and mass spectrometry. Chem. Res. Toxicol 22, 1060–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Shen JC, Creighton S, Jones PA, and Goodman MF (1992) A comparison of the fidelity of copying 5-methylcytosine and cytosine at a defined DNA template site. Nucleic Acids Res 20, 5119–5125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Nan X, Tate P, Li E, and Bird A (1996) DNA methylation specifies chromosomal localization of MeCP2. Mol. Cell. Biol 16, 414–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Valinluck V, Tsai H-H, Rogstad DK, Burdzy A, Bird A, and Sowers LC (2004) Oxidative damage to methyl-CpG sequences inhibits the binding of the methyl-CpG binding domain (MBD) of methyl-CpG binding protein 2 (MeCP2). Nucleic Acids Res 32, 4100–4108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Razin A, and Riggs AD (1980) DNA methylation and gene function. Science 210, 604–610. [DOI] [PubMed] [Google Scholar]
- (8).Bird AP, and Wolffe AP (1999) Methylation-Induced Repression— Belts, Braces, and Chromatin. Cell 99, 451–454. [DOI] [PubMed] [Google Scholar]
- (9).Ooi SKT, and Bestor TH (2008) The colorful history of active DNA demethylation. Cell 133, 1145–1148. [DOI] [PubMed] [Google Scholar]
- (10).Kriaucionis S, and Heintz N (2009) The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science 324, 929–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, and Rao A (2009) Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324, 930–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Ito S, D’Alessio AC, Taranova OV, Hong K, Sowers LC, and Zhang Y (2010) Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature 466, 1129–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Wu SC, and Zhang Y (2010) Active DNA demethylation: many roads lead to Rome. Nat. Rev. Mol. Cell Biol 11, 607–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Globisch D, Münzel M, Müller M, Michalakis S, Wagner M, Koch S, Brückl T, Biel M, and Carell T (2010) Tissue distribution of 5-hydroxymethylcytosine and search for active demethylation intermediates. PLoS One 5, e15367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Mullaart E, Lohman PH, Berends F, and Vijg J (1990) DNA damage metabolism and aging. Mutat. Res., DNAging: Genet. Instab. Aging 237, 189–210. [DOI] [PubMed] [Google Scholar]
- (16).Lindahl T, and Nyberg B (1974) Heat-induced deamination of cytosine residues in deoxyribonucleic acid. Biochemistry 13, 3405–3410. [DOI] [PubMed] [Google Scholar]
- (17).Delker RK, Fugmann SD, and Papavasiliou FN (2009) A coming-of-age story: activation-induced cytidine deaminase turns 10. Nat. Immunol 10, 1147–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Peled JU, Kuang FL, Iglesias-Ussel MD, Roa S, Kalis SL, Goodman MF, and Scharff MD (2008) The biochemistry of somatic hypermutation. Annu. Rev. Immunol 26, 481–511. [DOI] [PubMed] [Google Scholar]
- (19).Fritz EL, and Papavasiliou FN (2010) Cytidine deaminases: AIDing DNA demethylation? Genes Dev 24, 2107–2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Lindahl T, Ljungquist S, Siegert W, Nyberg B, and Sperens B (1977) DNA N-glycosidases: properties of uracil-DNA glycosidase from Escherichia coli. J. Biol. Chem 252, 3286–3294. [PubMed] [Google Scholar]
- (21).Liu P, Burdzy A, and Sowers LC (2002) Substrate recognition by a family of uracil-DNA glycosylases: UNG, MUG, and TDG. Chem. Res. Toxicol 15, 1001–1009. [DOI] [PubMed] [Google Scholar]
- (22).Maiti A, and Drohat AC (2011) Thymine DNA glycosylase can rapidly excise 5-formylcytosine and 5-carboxylcytosine: potential implications for active demethylation of CpG sites. J. Biol. Chem 286, 35334–35338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Hashimoto H, Hong S, Bhagwat AS, Zhang X, and Cheng X (2012) Excision of 5-hydroxymethyluracil and 5-carboxylcytosine by the thymine DNA glycosylase domain: its structural basis and implications for active DNA demethylation. Nucleic Acids Res 40, 10203–10214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Guo JU, Su Y, Zhong C, Ming G, and Song H (2011) Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell 145, 423–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Rusmintratip V, and Sowers LC (2000) An unexpectedly high excision capacity for mispaired 5-hydroxymethyluracil in human cell extracts. Proc. Natl. Acad. Sci. U. S. A 97, 14183–14187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Liu S, Wang J, Su Y, Guerrero C, Zeng Y, Mitra D, Brooks PJ, Fisher DE, Song H, and Wang Y (2013) Quantitative assessment of Tet-induced oxidation products of 5-methylcytosine in cellular and tissue DNA. Nucleic Acids Res 41, 6421–6429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Bachman M, Uribe-Lewis S, Yang X, Williams M, Murrell A, and Balasubramanian S (2014) 5-Hydroxymethylcytosine is a predominantly stable DNA modification. Nat. Chem 6, 1049–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Emmett MR, and Caprioli RM (1994) Micro-electrospray mass spectrometry: Ultra-high-sensitivity analysis of peptides and proteins. J. Am. Soc. Mass Spectrom 5, 605–613. [DOI] [PubMed] [Google Scholar]
- (29).Hawkins BE, Cowart JC, Parsley MA, Capra BA, Eidson KA, Hellmich HL, Dewitt DS, and Prough DS (2013) Effects of trauma, hemorrhage and resuscitation in aged rats. Brain Res 1496, 28–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Whitehead CW (1953) The Reactions of Orthoesters with Ureas. A New Synthesis of Pyrimidines. J. Am. Chem. Soc 75, 671–675. [Google Scholar]
- (31).Miller CS (1955) Synthesis of 5-Hydroxymethylcytosine. J. Am. Chem. Soc 77, 752–753. [Google Scholar]
- (32).Booth MJ, Branco MR, Ficz G, Oxley D, Krueger F, Reik W, and Balasubramanian S (2012) Quantitative sequencing of 5-methylcytosine and 5-hydroxymethylcytosine at single-base resolution. Science 336, 934–937. [DOI] [PubMed] [Google Scholar]
- (33).Miyashita O, Matsumura K, Shimadzu H, and Hashimoto N (1981) Studies on fluorinated pyrimidines. I. A new method of synthesizing 5-fluorouracil and its derivatives. Chem. Pharm. Bull 29, 3181–3190. [DOI] [PubMed] [Google Scholar]
- (34).Zhu H-J, and Pittman CU (2003) Reductions of Carboxylic Acids and Esters with NaBH 4 in Diglyme at 162°C. Synth. Commun 33, 1733–1750. [Google Scholar]
- (35).Harada K, and Suzuki S (1976) The new synthesis of uracil and 1,3-dimethyluracil. Tetrahedron Lett 17, 2321–2322. [Google Scholar]
- (36).LaFrancois CJ, Fujimoto J, and Sowers LC (1998) Synthesis and characterization of isotopically enriched pyrimidine deoxynucleoside oxidation damage products. Chem. Res. Toxicol 11, 75–83. [DOI] [PubMed] [Google Scholar]
- (37).Hudson RH, and Wojciechowski F (2005) The detrimental effect of orotic acid substitution in the peptide nucleic acid strand on the stability of PNA2:NA triple helices. Can. J. Chem 83, 1731–1740. [Google Scholar]
- (38).Brusilow SW, and Hauser E (1989) Simple method of measurement of orotic acid and orotidine in urine. J. Chromatogr., Biomed. Appl 493, 388–391. [DOI] [PubMed] [Google Scholar]
- (39).D’Apolito O, Garofalo D, Paglia G, Zuppaldi A, and Corso G (2010) Orotic acid quantification in dried blood spots and biological fluids by hydrophilic interaction liquid chromatography tandem mass spectrometry. J. Sep. Sci 33, 966–973. [DOI] [PubMed] [Google Scholar]
- (40).(1968) CRC Handbook of Biochemistry: Selected Data for Molecular Biology (Sober H, Ed.) The Chemical Rubber Company, Cleveland, OH. [Google Scholar]
- (41).Privat EJ, and Sowers LC (1996) A proposed mechanism for the mutagenicity of 5-formyluracil. Mutat. Res., Fundam. Mol. Mech. Mutagen 354, 151–156. [DOI] [PubMed] [Google Scholar]
- (42).Darwanto A, Theruvathu JA, Sowers JL, Rogstad DK, Pascal T, Goddard W, and Sowers LC (2009) Mechanisms of base selection by human single-stranded selective monofunctional uracil-DNA glycosylase. J. Biol. Chem 284, 15835–15846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).LaFrancois CJ, Jang YH, Cagin T, Goddard WA, and Sowers LC (2000) Conformation and proton configuration of pyrimidine deoxynucleoside oxidation damage products in water. Chem. Res. Toxicol 13, 462–470. [DOI] [PubMed] [Google Scholar]
- (44).Maiti A, Michelson AZ, Armwood CJ, Lee JK, and Drohat AC (2013) Divergent mechanisms for enzymatic excision of 5-formylcytosine and 5-carboxylcytosine from DNA. J. Am. Chem. Soc 135, 15813–15822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Salganik RI, Drevich VF, and Vasyunina EA (1967) Isolation of ultraviolet-denatured regions of DNA and their base composition. J. Mol. Biol 30, 219–222. [DOI] [PubMed] [Google Scholar]
- (46).Lindahl T, and Karlström O (1973) Heat-induced depyrimidination of deoxyribonucleic acid in neutral solution. Biochemistry 12, 5151–5154. [DOI] [PubMed] [Google Scholar]
- (47).Schmutte C, Yang AS, Beart RW, and Jones PA (1995) Base excision repair of U:G mismatches at a mutational hotspot in the p53 gene is more efficient than base excision repair of T:G mismatches in extracts of human colon tumors. Cancer Res 55, 3742–3746. [PubMed] [Google Scholar]
- (48).Desgranges C, Razaka G, Rabaud M, Bricaud H, Balzarini J, and De Clercq E (1983) Phosphorolysis of (E)-5-(2-bromovinyl)-2′-deoxyuridine (BVDU) and other 5-substituted-2′-deoxyuridines by purified human thymidine phosphorylase and intact blood platelets. Biochem. Pharmacol 32, 3583–3590. [DOI] [PubMed] [Google Scholar]
- (49).Berger R, Stoker-de Vries SA, Wadman SK, Duran M, Beemer FA, de Bree PK, Weits-Binnerts JJ, Penders TJ, and van der Woude JK (1984) Dihydropyrimidine dehydrogenase deficiency leading to thymine-uraciluria. An inborn error of pyrimidine metabolism. Clin. Chim. Acta 141, 227–234. [DOI] [PubMed] [Google Scholar]
- (50).Longley DB, Harkin DP, and Johnston PG (2003) 5-fluorouracil: mechanisms of action and clinical strategies. Nat. Rev. Cancer 3, 330–338. [DOI] [PubMed] [Google Scholar]
- (51).Vairapandi M, and Duker NJ (1993) Enzymic removal of 5-methylcytosine from DNA by a human DNA-glycosylase. Nucleic Acids Res 21, 5323–5327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Zhu B, Zheng Y, Hess D, Angliker H, Schwarz S, Siegmann M, Thiry S, and Jost JP (2000) 5-methylcytosine-DNA glycosylase activity is present in a cloned G/T mismatch DNA glycosylase associated with the chicken embryo DNA demethylation complex. Proc. Natl. Acad. Sci. U. S. A 97, 5135–5139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Parrilla-Doblas JT, Ponferrada-Marín MI, Roldán-Arjona T, and Ariza RR (2013) Early steps of active DNA demethylation initiated by ROS1 glycosylase require three putative helix-invading residues. Nucleic Acids Res 41, 8654–8664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Gama-Sosa MA, Midgett RM, Slagel VA, Githens S, Kuo KC, Gehrke CW, and Ehrlich M (1983) Tissue-specific differences in DNA methylation in various mammals. Biochim. Biophys. Acta, Gene Struct. Expression 740, 212–219. [DOI] [PubMed] [Google Scholar]
- (55).Vilpo JA, and Vilpo LM (1991) Biochemical mechanisms by which reutilization of DNA 5-methylcytosine is prevented in human cells. Mutat. Res., DNAging: Genet. Instab. Aging 256, 29–35. [DOI] [PubMed] [Google Scholar]
- (56).Mullen CA, Kilstrup M, and Blaese RM (1992) Transfer of the bacterial gene for cytosine deaminase to mammalian cells confers lethal sensitivity to 5-fluorocytosine: a negative selection system. Proc. Natl. Acad. Sci. U. S. A 89, 33–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Burdzy A, Noyes KT, Valinluck V, and Sowers LC (2002) Synthesis of stable-isotope enriched 5-methylpyrimidines and their use as probes of base reactivity in DNA. Nucleic Acids Res 30, 4068–4074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Münzel M, Globisch D, Brückl T, Wagner M, Welzmiller V, Michalakis S, Müller M, Biel M, and Carell T (2010) Quantification of the sixth DNA base hydroxymethylcytosine in the brain. Angew. Chem., Int. Ed 49, 5375–5377. [DOI] [PubMed] [Google Scholar]
- (59).Shen L, and Zhang Y (2012) Enzymatic analysis of Tet proteins: key enzymes in the metabolism of DNA methylation. Methods Enzymol 512, 93–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Liu P, Burdzy A, and Sowers LC (2003) Repair of the mutagenic DNA oxidation product, 5-formyluracil. DNA Repair 2, 199–210. [DOI] [PubMed] [Google Scholar]
- (61).Armstrong VW, Sternbach H, and Eckstein F (1976) Affinity labeling of escherichia coli DNA-dependent RNA polymerase with 5-formyl-1-(α-D-ribofuranosyl)uracil 5′-triphosphate. Biochemistry 15, 2086–2091. [DOI] [PubMed] [Google Scholar]
- (62).Terato H, Morita H, Ohyama Y, and Ide H (1998) Novel modification of 5-formyluracil by cysteine derivatives in aqueous solution. Nucleosides, Nucleotides Nucleic Acids 17, 131–141. [DOI] [PubMed] [Google Scholar]
- (63).Pizzorno G, Cao D, Leffert JJ, Russell RL, Zhang D, and Handschumacher RE (2002) Homeostatic control of uridine and the role of uridine phosphorylase: a biological and clinical update. Biochim. Biophys. Acta, Mol. Basis Dis 1587, 133–144. [DOI] [PubMed] [Google Scholar]
- (64).Galashevskaya A, Sarno A, Vågbø CB, Aas P. a., Hagen L, Slupphaug G, and Krokan HE (2013) A robust, sensitive assay for genomic uracil determination by LC/MS/MS reveals lower levels than previously reported. DNA Repair 12, 699–706. [DOI] [PubMed] [Google Scholar]
- (65).Kruman II, Kumaravel TS, Lohani A, Pedersen W, Cutler RG, Kruman Y, Haughey N, Lee J, Evans M, and Mattson MP (2002) Folic acid deficiency and homocysteine impair DNA repair in hippocampal neurons and sensitize them to amyloid toxicity in experimental models of Alzheimer’s disease. J. Neurosci 22, 1752–1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Endres M, Biniszkiewicz D, Sobol RW, Harms C, Ahmadi M, Lipski A, Katchanov J, Mergenthaler P, Dirnagl U, Wilson SH, Meisel A, and Jaenisch R (2004) Increased postischemic brain injury in mice deficient in uracil-DNA glycosylase. J. Clin. Invest 113, 1711–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]