

Abstract
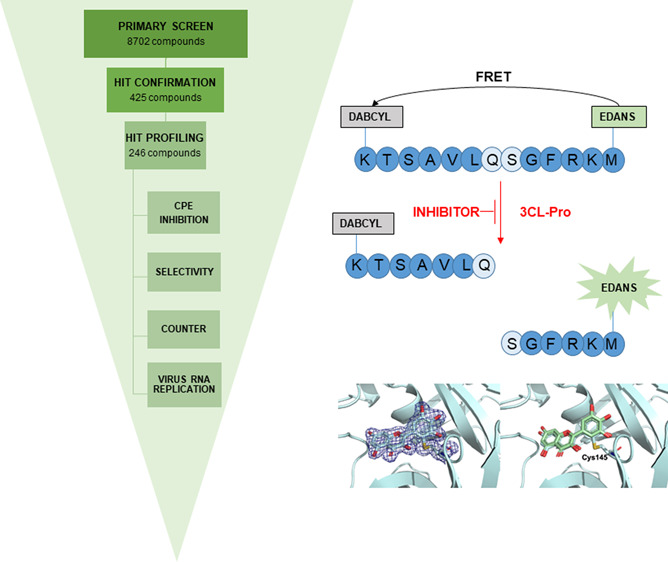
Compound repurposing is an important strategy for the identification of effective treatment options against SARS-CoV-2 infection and COVID-19 disease. In this regard, SARS-CoV-2 main protease (3CL-Pro), also termed M-Pro, is an attractive drug target as it plays a central role in viral replication by processing the viral polyproteins pp1a and pp1ab at multiple distinct cleavage sites. We here report the results of a repurposing program involving 8.7 K compounds containing marketed drugs, clinical and preclinical candidates, and small molecules regarded as safe in humans. We confirmed previously reported inhibitors of 3CL-Pro and have identified 62 additional compounds with IC50 values below 1 μM and profiled their selectivity toward chymotrypsin and 3CL-Pro from the Middle East respiratory syndrome virus. A subset of eight inhibitors showed anticytopathic effect in a Vero-E6 cell line, and the compounds thioguanosine and MG-132 were analyzed for their predicted binding characteristics to SARS-CoV-2 3CL-Pro. The X-ray crystal structure of the complex of myricetin and SARS-Cov-2 3CL-Pro was solved at a resolution of 1.77 Å, showing that myricetin is covalently bound to the catalytic Cys145 and therefore inhibiting its enzymatic activity.
Keywords: SARS-CoV-2, main protease, screening, FRET, repurposing
Coronaviruses are enveloped viruses with large (27–31 kb) single-stranded positive-sense RNA genomes. Genes encoding structural and accessory proteins are located at the 3′ end which terminates with an UTR and a poly-A tail. The 5′ end consists of a 5′ cap structure and a noncoding region untranslated region (UTR) followed by open reading frames (ORF) coding for 14–16 nonstructural proteins.1 Past outbreaks of severe acute respiratory syndrome coronavirus (SARS-CoV) and Middle East respiratory syndrome (MERS) have demonstrated the zoonotic potential of coronaviruses.2 The coronavirus SARS-CoV-2 was initially isolated and identified in December 2019 in Wuhan, China and within less than six months developed into a pandemic, leading to severe impacts on global health and economic systems. To date (November 24, 2020), WHO reports that 64 million people are infected and almost 1.5 million have died.3 SARS-CoV-2 belongs to the β-coronavirus genus of Coronaviridae family of the order Nidovirales.2 Nonstructural proteins of SARS-CoV-2 take up two-thirds of the viral genome and are prominent targets for drugs modulating the replication machinery of the virus.4 This group includes viral replication and transcription enzymes (RNA-dependent RNA polymerase [RdRp], helicase, and 2′O-MTase), the papain-like protease (PLpro), as well as the 3C-like protease (3CL-Pro or M-Pro), the focus of this study.
Role of 3CL-Pro and Qualification as an Antiviral Target
As a single-stranded positive-sense RNA virus, SARS-CoV-2 uses cellular translation machinery directly after infection of the cell to produce viral polyproteins pp1a and pp1ab. The generation of individual viral proteins involves proteolytic cleavage of 3CL-Pro and PLpro, whereby 3CL-Pro is cleaved autocatalytically from the polyprotein before itself processing the 13 nonstructural proteins which go on to initiate further viral replication.1 Therefore, it plays a central role in progression of infection and generation of new viral particles. Structurally, the SARS-CoV-2 3CL-Pro forms a homodimer, wherein each monomer has three domains.5 Domains I (residues 8–100) and II (residues 101–183) are located at the N-terminal and are chymotrypsin-like, with six antiparallel beta-sheets forming the substrate binding site located in the cleft between them. Domain III (residues 200–303) at the C-term consists of five helices and is responsible for regulation of dimer formation. Domains I and II are connected to domain III through a loop structure (184–199). The active site of SARS-CoV-2 3CL-Pro is formed by a catalytic dyad of Cys-145 and His-41, which is uncommon for Cys protease family enzymes. In common with SARS-CoV 3CL-Pro, a water molecule forms a hydrogen bond with His-41 and can act as a third component in the active site.5 The SARS-CoV-2 3CL-Pro cleaves at least 11 sites in the polyproteins with preference for Leu-Gln↓(Ser, Ala, Gly) (↓ marks the cleavage position) and a high specificity for the Gln in P1 position, a function only shared with enteroviruses. Sequence analysis shows that SARS-CoV-2 3CL-Pro shares 96.1 and 50.3% sequence identity with the 3CL-Pro of SARS-CoV and MERS, respectively.6 The similarity in full protein sequence across the three viruses raises the possibility of successful inhibition of SARS-CoV-2 3CL-Pro using compounds previously identified in studies on SARS-CoV and MERS and opportunities for identification of compounds with broad-spectrum activity.
Known Inhibitors of 3CL-Pro and Examples from SARS-CoV and MERS
Peptidomimetic inhibitors of several viral main proteases have previously been designed according to natural substrate sequences, in which the peptide bonds are chemically modified to be noncleavable7 using designed chemical warheads including carbonyls, hydroxy- and chloromethyl ketones, and diketo moieties. These classes of inhibitors often contain a Michael acceptor moiety and act as reactive substrates. A prominent example is rupintrivir (AG7088), an irreversible inhibitor of the human rhinovirus (HRV) 3C protease, although it lacks activity against SARS-CoV 3CL-Pro, most probably due to the planar fluorophenylalanine at P2.8 In general, this group of inhibitors exhibit limitations in some relevant medicinal chemistry and pharmacokinetic features such as half-life and stability in human plasma as well as high binding to plasma proteins which serves to reduce their bioavailability. A second group of 3CL-Pro inhibitors are the alpha-ketoamides peptidomimetics which reversibly inhibit SARS-CoV 3CL-Pro through nucleophilic attachment of the catalytic cysteine to the alpha-keto group. Representatives of this group have been shown to inhibit the MERS 3CL-Pro at submicromolar, and SARS-CoV 3CL-Pro at low micromolar, concentrations.5 A third group is represented by nonpeptidic reversible inhibitors, among them benzoquinoline compounds, designed using computational modeling. The compounds contain two benzoquinolinones connected to N-phenyl tetrazole moiety through a sulfur atom. The binding mode is proposed to be based on formation of hydrogen bonds and hydrophobic interaction between the S1 and S2 side of 3CL-Pro and benzoquinolinones and N-phenyltetrazole groups, respectively.9 Pyrazoles have also been shown to inhibit the SARS-CoV 3CL-Pro as well as 3CL-Pro of type 14 rhinovirus (RV14) at low micromolar level, raising the possibility for broad spectrum antiviral compounds to be identified.10
In vitro screening of compound libraries and molecular docking based on the crystal structure of the SARS-CoV and MERS 3CL-Pro have revealed inhibitors with potentially different modes of actions against the SARS-CoV-2 3CL-Pro. Among them are metal (zinc)-conjugates (e.g., zinc-pyrithione, PDB: 6YT8),8,11 natural products of the isoflavone family12 (e.g., 5,7,3′,4′-tetrahydroxy-2′-(3,3-dimethylallyl)-isoflavone), drug candidates (e.g., ebselen), and approved drugs (e.g., disulfiram and carmofur).6 To date, virtual screening of large libraries of compounds has been reported (extended list of references and data: https://ghddi-ailab.github.io/Targeting2019-nCoV/computational/), and several in vitro screens have been published mainly in preprint form against SARS-CoV-2 3CL-Pro.13 Most recently, a number of studies have shown that inhibitors of the protease of hepatitis-C, including boceprevir (IC50 = 1.6 μM) and telaprevir (IC50 = 55 μM), are active against the SARS-CoV-2 3CL-Pro and act by forming a covalent bond with the sulfur of the catalytic Cys145.14−16
Compound Repurposing
In the case of emerging diseases such as COVID-19, where limited therapeutic or prevention options in the form of vaccines or prophylaxis exist, compound repurposing can allow for fast identification of drug candidates, if the selected therapeutics are efficacious, well-tolerated and have suitable safety profiles. Considerable additional insights into anti-Coronaviridae therapeutic discovery accumulated through past outbreaks of SARS-CoV (2002/2003) and MERS (2012) which informed multiple drug repurposing-based clinical studies following the emergence of SARS-CoV-2. Clinical trials with antivirals (lopinavir, ritonavir)17 and antimalarials (chloroquine and hydroxychloroquine)18 have yet to show consistent safety and efficacy. However, other studies including the glucocorticoid, dexamethasone,19 and the viral RNA-polymerase inhibitor remdesivir20 have led to regulatory approvals for their use in COVID-19 patients. Nevertheless, more effective treatments are still required, in particular for patients at earlier stages of infection, where pharmacological interventions have not been approved so far. Classical antiviral drug development can take many years and require high financial investments. Therefore, repurposing of known drugs represents a cost and time saving alternative especially when combined with high-throughput screening against relevant disease models or target mechanisms as part of the candidate identification process.
Part of the response to the above need is the project (“EXaSCale smArt pLatform Against paThogEns for CoronaVirus, Exscalate4CoV or E4C”, http://www.exscalate4cov.eu), which is funded through EU’s H2020-SC1-PHE-CORONAVIRUS-2020 emergency call (Grant 101003551). It collects 18 European public institutions and a private company (DOMPE SpA) in the leading role and brings together high-end computational facilities, drug discovery, and BSL-3 high-throughput screening resources. The first aim of E4C is to identify repurposed candidates for a fast track entrance on clinical trials, and the second phase is to provide the foundation for a European platform for rapid response to pandemics with novel drug candidates. In this study, we report on identification of 3CL-Pro inhibitors using repurposed drugs and annotated preclinical compounds together with a comparative discussion on similar results obtained by cheminformatics, modeling,21 and structural biology approaches.
Results and Discussion
Assay Development
Enzymatic Parameters
Results covering key SARS-CoV-2 3CL-Pro enzymatic properties are shown in Figure 1. Substrate turnover was directly proportional to enzyme concentrations up to 60 nM and assay incubation times up to 15 min post substrate addition (Figures 1A and 1B). The relative enzymatic activity was not influenced by the presence of DTT (Figure 1C), with Vmax = 63 070 RFU/min in the presence of 1 mM DTT and 58 111 RFU/min without DTT. The corresponding Km values were 19 μM with 1 mM DTT and 16 μM without DTT. The assay was not sensitive to DMSO up to a concentration of 5% v/v (Figure S2).
Figure 1.

Determination of key enzymatic parameters for SARS-CoV-2 3CL-Pro: (A) fluorescence signal versus time for enzyme concentrations from 0 to 0.25 μM (substrate concentration = 15 μM); (B) replotting the t = 15 min data against enzyme concentration; (C) determination of Km and Vmax in the presence and absence of DTT ([SARS-CoV-2 3CL-Pro] = 60 nM). Reaction rate calculations used increase in signal between 0 and 2 min.
Compound Preincubation Conditions
Reference inhibitors were calpeptin, a close analogue of GC376, and zinc pyrithione, which binds the SARS-CoV-2 3CL-Pro catalytic cysteine.8,22 The effects of time and temperature conditions during compound preincubation with SARS-CoV-2 3CL-Pro were determined. Compounds were preincubated with the enzyme for 15, 30, and 60 min (at 25 °C and 20 μM concentration) before substrate addition. The inhibitory effect of zinc pyrithione increased with preincubation time, while calpeptin inhibition was unchanged (Figure S-3). Similarly, zinc pyrithione was 3-fold more potent after preincubation at 37 °C versus preincubation at 25 °C (Figure S-4). Therefore, to increase assay sensitivity, the preincubation step in the screening protocol was performed for 60 min at 37 °C.
Effect of DTT
The effect of DTT (0 and 1 mM) on the properties of previously proposed inhibitors of SARS-CoV-2 3CL-Pro was tested at 20 μM compound concentration (Figure S-5). Zinc pyrithione,8 lost its inhibitory effects in the presence of DTT. Chlorhexidine, predicted by docking studies to bind SARS-CoV-2 3CL-Pro,1 was not an effective inhibitor at 20 μM concentration, regardless of DTT. The protease inhibitor calpeptin18 retained its inhibitory effect independent of DTT. To account for any DTT dependent effects, primary screening was performed without DTT in the assay buffer.
Identification of SARS-CoV-2 3CL-Pro Inhibitors
The workflow for primary, hit confirmation and profiling stages is shown in Figure 2A. 8702 compounds from three collections were screened at 20 μM. Detected activities were normalized to the inhibitory activity of Zn-Pyrithione that was used as positive control and set to 100% (positive control) and to values measured for the DMSO control, set to 0% inhibition (negative control). In individual examples, compounds showed >100% inhibition (Figure 2C) which is indicative of the underlying variation on single concentration measurements in compound and control wells.
Figure 2.

Overview of primary and hit confirmation screening: (A) screening workflow; (B) primary screen, frequency distribution of inhibition for test compounds and controls; (C) inhibition at primary versus hit confirmation for 425 selected inhibitors, R2 = 0.65; (D) compound with annotated primary target classes for screened set (LHS) and 328 confirmed hit compounds (RHS).
Primary Screen Overview
Primary screen quality control was achieved by analyzing the performance of intraplate controls (Z′-factor) and the reproducibility of duplicate compounds located in the three source libraries (Figure S-6). The Z′-factor values in the primary screen all exceeded 0.68 with an average of 0.86 ± 0.05, indicating acceptable assay performance (Table S-1). The frequency distribution of SARS-CoV-2 3CL-Pro inhibition (%) of the 8702 primary screen compounds is shown in Figure 2B. Individual compound inhibition data and associated annotations are in Table S-2 and can be accessed at ChEMBLDB 28 using the AID code CHEMBL4495564.23 Comparing duplicate test compounds which originated from different sources in the primary screen gave an R2 = 0.77 (Figure S-6), indicating consistency in compound potency between different source libraries (Figure S-1). Compounds on each plate were classified as hits if: (test compound inhibition) > (mean + 6× standard deviations of corresponding plate DMSO control inhibition values). A total of 425 compounds with inhibition in primary screening were prioritized for hit confirmation (HC) in the original assay format (in triplicate). Compounds which showed optical interference (elevated fluorescence intensities at 60 min) were removed from the workflow.
Hit Confirmation
All Z′-values at HC exceeded 0.6, indicating good assay quality. Comparison of primary and HC inhibition values (Figure 2C) gave R2 = 0.65. The primary annotated targets of the 8702 screened compounds and the 328 confirmed hits (hit rate = 3.8%) were compared to identify possible enrichment effects (Figure 2D). However, the distribution of target classes associated with the confirmed hit population was similar to that of the screened set.
Hit Profiling Overview
A set of 246 compounds with mean SARS-CoV-2 3CL-Pro inhibition exceeding 50% at HC were then progressed to hit profiling (HP). First, the effects of DTT on compound inhibition levels were assessed at 20 μM test compound. Some 156 compounds showed a relative reduction in SARS-CoV-2 3CL-Pro inhibition of more than 30% in the presence of 1 mM DTT, and these were designated as “DTT sensitive”. The remaining 89 compounds gave less than 30% change in inhibition in the presence of 1 mM DTT, and these were designated as “DTT insensitive”. The sensitivity to DTT is likely due to its effects on compound reactivity toward the Cys-145 moiety in the SARS-CoV-2 3CL-Pro catalytic domain. Multiple enzymatic studies on 3CL-Pro5 have used DTT to reduce protein oxidation and maintain consistency with those protocols, DTT was used in this work, although it is recognized that other reducing agents such as TCEP are also effective.
All 246 compounds were profiled in dose response in the absence of DTT, and the 89 DTT insensitive compounds were also tested in the presence of DTT (Table S-3). Without DTT, some 119 of 246 compounds gave IC50 values below 5 μM for SARS-CoV-2 3CL-Pro inhibition. In the presence of 1 mM DTT, 40 compounds gave an IC50 below 5 μM for SARS-CoV-2 3CL-Pro inhibition (Table S-3).
A subset of mainly DTT insensitive compounds was then analyzed for selectivity toward chymotrypsin and the MERS 3CL-Pro (Table S-3). Assay buffers for chymotrypsin contained 1 mM DTT. In general, this group of compounds was not active against chymotrypsin, with only 7 compounds showing inhibition >30%. Seven compounds showed inhibitory effect on MERS 3CL-Pro with IC50 values <10 μM, among them PX-12 (IC50 = 7.6 μM) for which an IC50 of 21 μM has been reported.6 In the Vero-E6 phenotypic assay only 22 compounds from the 246 selected SARS-CoV-2 3CL-Pro inhibitors showed >10% inhibition of CPE at 10 μM. Further studies on two unique compounds (MG-132 and thioguanosine) gave an IC50 < 20 μM in CPE inhibition assay. These compounds have also been shown to be active against Caco2 cells in a recent study.24 Interestingly, MG-132 was more potent in the Vero-E6 CPE assay (IC50 = 0.4 μM), and viral replication assays (EC50 = 0.1 μM) (Figure S7), that against SARS-CoV-2 3CL-Pro (IC50 = 13.1 μM).
Compounds Previously Reported as Inhibitors of SARS-CoV-2 3CL-Pro
The screen identified 6 compounds (TDZD-8, carmofur, tideglusib, ebselen, disulfiram, and baicalein) with IC50 values <1 μM and which have been previously reported as inhibitors of SARS-CoV-2 3CL-Pro (Figure 3 and Table S-3). In general, these compounds were 10-fold more potent in our hands compared to literature reports, which may be due to the extended preincubation of compounds with SARS-CoV-2 3CL-Pro for 60 min at 37 °C. The compounds TDZD-8 and carmofur were insensitive to DTT, while the other four compounds in this group lost activity in the presence of 1 mM DTT (Figure 3, Table S-3). In the selectivity screening, carmofur was also weakly active against chymotrypsin (14% inhibition).
Figure 3.

Hit profiling for known SARS-CoV-2 3CL-Pro inhibitors identified in the primary screen, n.t. = not tested. (Only DTT insensitive compounds were tested in the presence of DTT.)
In our hands, these compounds did not show activity in the CPE Vero-E6 CPE phenotypic assay and they were also inactive in a recently reported study which evaluated SARS-CoV-2 anti-CPE phenotypes in a Caco2 cell line.24 Interestingly, epigallocatechin gallate, a compound structurally related to baicalein, was also shown to be active against SARS-CoV-2 3CL-Pro but did not show DTT dependence (IC50 without DTT = 1.6 μM, with DTT 0.4 μM), (Table S-3). * = ref (5), ** = ref25
SARS-CoV-2 3CL-Pro Inhibitors with Moderate Anti-CPE Effect
The second group of confirmed hits is formed by compounds not previously reported as inhibitors of SARS-CoV-2 3CL-Pro but which showed moderate inhibition (between 10 and 19%) of CPE in Vero-E6 cells (Figure 4 and Table S-3). With the exception of MG-132, inhibition of SARS-CoV-2 3CL-Pro for this group was not observed in the presence of DTT (Table S-3). Of the 8 compounds, also only thioguanosine and MG-132 produced dose responses in the Vero-E6 CPE assay giving IC50 values of 3.9 and 0.4 μM, respectively. Corresponding cytotoxicity CC50 values in Vero-E6 imaging assay were >20 and 2.9 μM, respectively. MG-132 showed a similar reduction in vRNA with an IC50 value of 0.1 μM (Figure S7). Thioguanosine has previously been shown to have anti-SARS-CoV-2 CPE effects in a Caco-2 cell-based assay.24
Figure 4.

Dose dependent effects of SARS-CoV-2 3CL-Pro inhibitors which also demonstrated a moderate anticytopathic effect in Vero-E6 cells. n.t. = not tested. n.a. = not active.
Compounds Inhibiting SARS-CoV-2 3CL-Pro without Anti-CPE Effects
The third group of compounds act as inhibitors of SARS-CoV-2 3CL-Pro, both in the presence and absence of DTT (IC50 < 5 μM in both conditions). Here, we highlight compounds which are selective also against chymotrypsin (inhibition <10% at 10 μM) and exhibit promising medicinal chemistry properties (Figure 5 and Table S-3). The group contains mainly preclinical compounds which are not ready for immediate repurposing, with the exception being the anti-Parkinson’s disease compound benserazide.
Figure 5.

Dose dependent effects of 3CL-Pro inhibitors which demonstrated inhibitory effect independent from the presence of DTT in the assay media. n.t. = not tested.
Myricetin: MPro X-ray Crystallography Results
The X-ray crystal structure of the complex of myricetin and SARS-Cov-2 3CL-Pro was solved at a resolution of 1.77 Å (Figure 6, lhs and PDB 7B3E) (Table S-4). The 2Fo–Fc map unambiguously shows that myricetin is covalently bound to the catalytic Cys145, with a bond length of 1.7 Å. The orientation found is determined in large part by the covalent bond between Cys145 sulfur and the 2′ position of the flavonoid, leading to unprecedented binding for a flavonoid scaffold. Consistent with the measured complex structure, the noncovalent in silico docking calculations for myricetin docking21 led to a similar pose (Figure 6, rhs). Moreover, the 2Fo–Fc map of the myricetin:MPro X-ray structure showed that the binding pocket is only partially occupied by myricetin, and voids are filled by solvent (ethylene glycol and water) molecules, signaling an opportunity for future structure-based drug design efforts.
Figure 6.

In panels a and b, the 2D diagrams of myricetin and of myricetin covalently bound to the catalytic Cys145 are shown. Panel c shows the X-ray crystal structure of myricetin covalently bound to the catalytic Cys145 of 3CL-Pro with 2Fo–Fc electron density map contoured at 1 sigma (PDB: 7B3E). In panel d, the noncovalent docking pose21 of myricetin (green) is superimposed onto the X-ray crystal structure. https://www.rcsb.org/structure/7B3E. (e) Possible mechanism of myricetin oxidation. Reproduced from ref (21).
Discussion
The protease 3CL-Pro is responsible for accurately processing the SARS CoV-2 polyprotein at 11 distinct cleavage sites. With the unique exception of the specificity for Gln in P1, the high number of different cleavage sites recognized suggests a certain degree of promiscuity with regard to cleavage site selectivity on other subpockets. This is consistent both with the diversity of peptide-like compounds found as inhibitors so far and with recent comparison of crystal structures between the 3CL-Pro apo-form obtained at room temperature with the corresponding low-temperature apo- and inhibitor bound forms, which show large changes in catalytic cavity dimensions upon ligand binding.26 Molecular dynamics simulations also predict high flexibility in solution, especially of the loop regions adjacent to binding sites.21,27 The protein is suggested to work as a dimer with N- and C-terminal regions of each monomer actively involved in ligand binding. The 3CL-Pro catalytic cavity includes four exposed cysteines, of which Cys145 appears predisposed to reacting with covalent ligands, and His41 that forms hydrogen bonds with a water molecule, which is assumed to be the third part of the catalytic side.28 This differs from other Cys-proteases where a triad (Cys-His-Asp/Glu) is typically involved.29 In addition, there are also four exposed histidine residues which may be responsible for other 3CL-Pro biochemical characteristics observed in this study, including DTT sensitivity. These structural properties, combined with the absence of homologous human proteases, point toward anti-3CL-Pro compounds, alone or in combination with other therapies, as having the capacity to exert antiviral effects and thus be a potential treatment of COVID-19 infection.
Reference Compounds
In this study, zinc pyrithione, an antifungal and bacteriostatic compound, was used as the positive control (IC50 = 0.1 μM) and was independently identified as a screening hit. This compound was previously shown to inhibit SARS-CoV 3CL-Pro (IC50 = 0.8 μM)8,11 by binding to its catalytic cavity30 (PDB: 6YT8), suggesting a similar interaction mode for this compound with SARS-CoV-2 3CL-Pro. We reconfirmed the activity of published inhibitors, including TDZD-8, carmofur, tideglusib, ebselen, disulfiram, and baicalein, which gave IC50 values between 20 and 220 nM. Analogues of ebselen (IC50 = 30 nM) carrying the same N-phenyl-1,2-benzothiazol-3-ones scaffold also stood out in the confirmed hit population, and the preclinical compounds PD08290 (IC50 = 20 nM), MLS0315771 (IC50 = 20 nM), PBIT (IC50 = 20 nM), and ML345 (IC50 = 20 nM) showed higher potencies than ebselen itself (Table S-3). The 60 min preincubation time at 37 °C facilitated the identification of slowly binding putative cysteine-reactive inhibitors. This type of inhibitory mechanism is supported by the loss of inhibitory capacity for ebselen, disulfiram, tideglusib, and baicalein in the presence of reducing conditions (1 mM DTT). Moreover, we confirmed the Caspase 3 inhibitor, Z-DEVD-FMK, as active (IC50 = 10 nM) with 600-fold higher potency than observed previously.13 However, this compound did not influence CPE in phenotypic assays.
Compounds with Anticytopathic Effects
A general correlation between 3CL-Pro and CPE inhibition in the Vero-E6 model was not found (Table S-3). Of the anti-CPE compounds, only thioguanosine and MG-132 produced a measurable dose response in the CPE assay (IC50 values of 3.9 and 0.4 μM, respectively). Previously, thioguanosine has been reported to be an inhibitor of the PL-Pro’s of SARS-COV31 and PEDV32 (PDB: 5XU8) and to be anticytopathic against SARS-CoV-2.33 Structural evidence of catalytic Cys145 sulfur involvement in thioguanine inhibition of PL-Pro34 may be mechanistically similar to the effects it exerts in the SARS-CoV-2 3CL-Pro cavity.
The lack of anti-CPE effect for the great majority of identified SARS-CoV-2 3CL-Pro inhibitors may be due to modest cell membrane permeability, poor metabolic stability or efflux effect from Vero-E6 cells which express PgP. A second possible reason might reside on the chemical reactivity of SARS-CoV-2 3CL-Pro inhibitors within cells. For example, the antifungal phenylmercuric acetate (IC50 = 10 nM) or antiarthritis auranofin (IC50 = 210 nM) both contain chemical moieties prone to react with nucleophiles such as mercury, zinc or gold.11 Other compounds have a Michael acceptor, either already present into the structure (bonaphthone with IC50 = 40 nM) or generated in situ, such as in the case of the molecule HEAT25 (PDB: 6YNQ). The observation that several compounds with similar structures showed measurable IC50 values against SARS CoV-2 3CL-Pro in a biochemical assay readouts, however, without influencing the CPE, casts doubts on scaffold selectivity and constitutes a warning for medicinal chemistry lead design.
Compounds Inhibiting SARS-CoV-2 3CL-Pro
This screen identified PR-619 to be active against 3CL-Pro from SARS-CoV-2 (IC50 = 0.4 μM) and MERS (IC50 = 2.3 μM). This compound contains two isothiocyanate groups and is a known inhibitor of several deubiquitinating enzymes with IC50 values ranging 2–10 μM.35,36 PR-619 is inactive in cellular phenotypic assay either in Vero-E6 cells and Caco-2,24 suggesting its potential as a lead compound against SARS-CoV-2 may be limited. Among peptide-like molecules, calpeptin (IC50 = 4 μM) and MG-132 (IC50 = 7.4 μM) were identified, while the associated derivative boceprevir gave below 50% inhibition in HC. These molecules are predicted to act by binding Cys145 in the catalytic domain. Nevertheless, calpeptin and MG-132 should be regarded as reversible inhibitors, as both contain aldehyde warhead groups. The recently discovered CDK degrader CR8-(R)37 was moderately potent against SARS-CoV-2 3CL-Pro (IC50 = 0.8 μM), but with no anti-CPE effect. However, its activity on CDK machinery could influence the cell phenotype challenged by viral infection, and hence, there might be a safety concern for further development of CDK inhibitors. Among natural product-based compounds, isatin derivatives have been identified as potential inhibitors;38 however, this was not confirmed in this study, notwithstanding that the isatin-like “1,2-dicarbonyl” moiety is present in the antiviral bonaphthone (IC50 = 40 nM). Recent interest as an anti-influenza agent has been raised for this molecule, although no further development has been recorded.39 Interestingly, the hypotheses that flavonoids could act as a privileged scaffold for both SARS CoV and SARS CoV-2 3CL-Pro ligands40,12 was confirmed in our investigations. In addition to baicalein, a series of flavonoids with IC50 ranging 180 nM to 3.6 μM (Table S-3) were identified, and within this group, myricetin derivatives were well-represented.
The presence of polyhydroxy phenolic moiety seems to provide some interaction advantages, as suggested by the reported SARS CoV-2 3CL-Pro crystal structure with baicalein (PDB: 6M2N). The related dopamine decarboxylase inhibitor benserazide (IC50 = 0.1 μM), IGF-1 inhibitor NT-157 (IC50 = 0.5), nootropic agent exifone (IC50 = 4.38 μM) have in common an identical trihydroxy phenolic moiety. Moreover, a similar ortho dihydroxy phenol moiety is present in the dye hematoxylin (IC50 = 0.2 μM), dopamine receptor agonists apomorphine (IC50 = 0.5 μM), dihydrexidine (IC50 = 1.1 μM), and SKF-38393 (IC50 = 2.6 μM) and lipoxygenase inhibitor nordihydroguaiaretic acid (IC50 = 2.59 μM). Unfortunately, all of these compounds showed DTT sensitivity, likely due to the high oxidation propensity of polyhydroxy phenols.41 For instance, two polyhydroxy phenol-containing compounds, myricetin and benserazide, have been successfully predicted to have nanomolar affinities based on structural modeling.21 Polyhydroxy phenolic moieties are, however, considered promiscuous and are found as frequent hitters in large scale assessments of screening library performance.42 This may not only be due to their redox features but also to the presence of a high number of closely arranged promiscuous hydrogen-bond acceptor/donor sites, which raises the number of protein 3D-pharmacophores satisfiable by such molecular structures. Notwithstanding, these “promiscuity” features, curcumin, benserazide, apomorphine, flavopiridol, anthracycline-based antibiotics, and other products have reached approval for clinical use across several indications.
To visualize how hit compounds may bind to the protein, we exemplarily docked MG-132 and thioguanosine, the two molecules shown to inhibit viral CPE, into the main catalytic cavity of 3CL-Pro, as well as myricetin for which confirmatory structural information was obtained (Figure 7). While for MG-132 the docking pose is straightforward, as many peptide-like structures are oriented in a similar direction, for the smaller and more compact thioguanosine, the optimal pose was not as evident. The observation that corresponding compound guanosine did not exert the same inhibition potency in the screen prompted us to focus on the sulfur atom. Through extensive DFT calculations, we assessed that, in contrast to guanosine, the most probable tautomer for thioguanosine involves a thiol group rather than a thio-ketonic group (Figure S-8). When docked, this tautomer clearly shows its sulfur atom in proximity to the catalytic Cys145, allowing the ribose moiety to make hydrogen-bond interactions with Glu166 backbone atoms (Figure 7). When in such high proximity, thiol-redox reactions to generate S–S species are possible, not only for thioguanosine but for the other potent sulfur-containing compounds which were identified (Table S-3). Myricetin appears bound to 3CL-Pro in an opposite orientation to that previously seen with baicalein43 (PDB: 6M2N). The unprecedented sulfur addition to the 2′ position of myricetin suggest the presence of a quinoid species apt to be attacked by a strong nucleophile with consequent oxidative loss of an electron or a hydride. This event could be supported by the lack of reducing agents in the crystallization medium like DTT.
Figure 7.

Thioguanosine (magenta), MG-132 (white), and myricetin21 (green) docked into monomer 3CL-Pro (see also ref (21)). Catalytic Cys145 are highlighted behind the water-accessible surfaces in ball and stick, while ligands are in stick representation. Protein cavity water-accessible surface is colored by hydrophilicity. The most probable thioguanosine tautomer with the thiol form has been considered according to QM calculations (see Figure S-8). Reproduced from ref (21).
Only 20% of the 66 compounds identified with IC50 < 1 μM had molecular weights (MWs) above 400 Da. The average MW of the screened set was 364.9 ± 143.4, with 3114 having MW > 400 (35.8%). The hypothesized 3CL-Pro cavity flexibility could therefore reward smaller compounds compared to larger ones, either because the protease induced fit requires more time to successfully complete, or because catalytic Cys145 needs more time to address adjacent electrophiles. In this direction, structural differences in cavity binding between nanomolar and micromolar affinity ligands have been illuminating.21 We postulated therefore that SARS CoV-2 3CL-Pro analyzed in our biochemical assay setup may reward smaller ligands.
The submicromolar inhibition level achieved by myricetin (IC50 = 0.2 μM) against SARS-CoV-2 3CL-Pro, however, was not reflected in anti-CPE or inhibition of viral replication in Vero-E6 cells (Table S-3) even though the similar compound baicalein did show anticytopathic effect in Vero-E6 cells and relative RNA reduction in the cell after 24 h incubation. The longer cellular readout we used (72 h) might allow metabolic and/or efflux processes to play a role in diminishing the internal molecular concentration of myricetin. However, the information collected with this protein complex can be valuable to drive future structure-based design attempt for flavonoid scaffolds.
Promising Candidates for New Lead Generation
This study identified several novel potent inhibitors of SARS CoV-2 3CL-Pro, including calpeptin, CR8-(R), bonaphthone, myricetin, MLS0315771, ML311, and thioguanosine, which show potential for further investigation. Among these compounds, there are known scaffolds including peptide derivatives, flavonoids, and kinase and protease inhibitors. The diverse structural characteristics of identified actives can provide new opportunities to medicinal chemists as part of further compound property optimization. A promising candidate for further studies is the peptide-like proteasome inhibitor MG-132 (IC50 = 7.4 μM). This compound has lower potency on 3CL-Pro compared to the anticytopathic (IC50 = 0.4 μM) and antiviral RNA replication (IC50 = 0.1 μM) effects in VERO-E6 cells. The greater potency seen in the cellular readouts would indicate that MG-132’s antiviral properties may involve polypharmacology and are likely not solely mediated by its inhibition of 3CL-Pro function. The compound is known to be active against coronaviruses,44,45 and further chemoproteomic and confirmatory in vivo studies would be valuable.
Repurposed Candidates
Focusing on already commercialized actives and more promising candidates for clinical developments, the aromatic-l-amino-acid decarboxylase inhibitor benserazide, antiviral riodoxol, dopamine agonist and phosphodiesterase inhibitor apomorphine, and proton pump inhibitor rabeprazole may be interesting to study in more detail. All of these molecules can be administered orally with measured acute toxicity in rodents higher than 5 mg/kg dose. Benserazide and apomorphine, for instance, can ensure exposure in the brain, and riodoxol has already been used as an antiseptic and disinfectant and in ointments in the treatment of chronic skin diseases and eczema of a subacute character,46 while rabeprazole has been used in the clinic as antibacterial against Helicobacter pylori. In-vitro assays determining their limited effects on viral CPE and replication may not fully reflect the infection pathology. We would suggest that additional investigation in other screening formats using, for example, human derived lung tissue samples may establish if the efficacy observed against the SARS-CoV-2 3CL-Pro can be reflected in antiviral phenotypes for these compounds.
Materials and Methods
Library Composition
The screened compounds were sourced from three collections. First, was the Dompe “Safe-In-Man” (SIM) proprietary collection containing 607 drug candidates, which have undergone at least successful Phase I studies. Second, was the EU-OPENSCREEN collection of 2463 compounds annotated in line with the Drugs & Probes database (https://www.probes-drugs.org/compoundsets), which samples drugs and drug candidates in several development phases together, along with preclinical probes with high affinity for their primary targets. Finally was the Fraunhofer repurposing collection, assembled based on the design feature the Broad Repurposing collection47 (Drug Repurposing Hub https://www.broadinstitute.org/drug-repurposing-hub). The Fraunhofer Repurposing Library contains 5632 compounds including 3400 compounds that have reached clinical use across 600 indications as well as 1582 preclinical compounds with varying degrees of validation. Overall, 8702 compounds were available for screening. The compound collections overlapped in terms of identity, as shown in Figure S1. This overlap was useful in determining the consistency in compound response for material sourced from different collections (Figure S1). In general, all test compounds were quality controlled by LC/MS for purity and identity (purity >90%) and were stored at −20 °C in 100% DMSO prior to use.
Source of Proteins
The 3CL-Pro of SARS-CoV-2 was synthesized using the ORF1ab polyprotein residues 3264–3569, (GenBank code: MN908947.3). Gene synthesis, protein production, and purification were as reported by Zhang et al.,5 where eluted fractions containing the target protein were pooled and subjected to buffer exchange in 20 mM Tris-HCl, 150 mM NaCl, 1 mM EDTA, and 1 mM DTT, pH 7.8. The 3CL-Pro MERS gene (NCBI reference Sequence: NC_019843.3) was purchased from GenScript. The 3CL-Pro MERS protein was also expressed as described in Zhang et al.5 Briefly, the 3CL-Pro MERS expression pellets were clarified by ultracentrifugation and purified using a Ni-Sepharose column and by HiTrap Q HP column. Eluted fractions containing the target protein were pooled and subjected to buffer exchange in 20 mM Tris-HCl, 150 mM NaCl, 1 mM EDTA, and 1 mM DTT, pH 7.8. Protein purity was confirmed by SDS-PAGE analysis.
Primary Assay Development
The detection of enzymatic activity of the SARS-CoV-2 3CL-Pro was performed under conditions similar to those reported by Zhang et al.5 Enzymatic activity was measured by a Förster resonance energy transfer (FRET), using the dual-labeled substrate, DABCYL-KTSAVLQ↓SGFRKM-EDANS (Bachem #4045664) containing a protease specific cleavage site after the Gln. In the intact peptide, EDANS fluorescence is quenched by the DABCYL group. Following enzymatic cleavage, generation of the fluorescent product was monitored (Ex/Em = 340/460 nm) (EnVision, PerkinElmer). The assay buffer contained 20 mM Tris (pH 7.3), 100 mM NaCl, and 1 mM EDTA. The assay was established in an automated screening format (384 well black microplates, Corning, #3820) and optimized with respect to assay volume (10 μL), enzyme concentration (60 nM), substrate concentration (15 μM), incubation time (60 min with compounds, 15 min with substrate) and temperature (37 °C for incubation with compounds, 25 °C for incubation with substrate), DMSO tolerance (up to 5 v/v%), response to inhibition with known compounds as zinc pyrithione,8 and the effects of reducing agents (DTT).
Primary Screen, Hit Confirmation, and Profiling
In the primary screen, test compounds (stock at 10 mM in 100% DMSO), positive (zinc pyrithione (medchemexpress, #HY-B0572) 10 mM in 100% DMSO), and negative (100% DMSO) controls were transferred to 384-well assay microplates by acoustic dispensing (Echo, Labcyte). Plate locations were: test compounds at 20 μM final (columns 1 to 22); positive control zinc pyrithione at 10 μM final (column 23); and negative control 0.2% v/v (column 24). Five microliters of SARS-CoV-2 3CL-Pro stock (120 nM) in assay buffer were added to compound plates and incubated for 60 min at 37 °C. After addition of 5 μL of substrate (30 μM in assay buffer), the final concentrations were: 15 μM substrate; 60 nM SARS-CoV-2 3CL-Pro; 20 μM compound; and 0.2% DMSO in a total volume of 10 μL/well. The fluorescence signal was then measured at 15 min, and inhibition (%) was calculated relative to controls (Envision, PerkinElmer). Results were normalized to the 100% (positive control) and 0% (negative control) inhibition. To flag possible optical interference effects, primary assay plates were also read 60 min after substrate addition, when the reaction was complete.
For hit confirmation (HC), the putative hits from the primary screen were picked and retested in the same primary assay format in triplicate at 20 μM compound. Confirmed compounds were then profiled in triplicate in 11 point concentration responses, starting from 20 μM top concentration with 1:2 dilution steps. While the primary screening assay did not contain DTT, the HC and profiling steps were performed ± DTT (@ 1 mM) to identify any DTT-dependent effects.
Selectivity Assays (MERS 3CL-Pro and Chymotrypsin)
A MERS 3CL-Pro inhibition assay was performed to observe potential broad inhibition effects of compounds against related coronaviruses. The substrate was identical to that used in the SARS-CoV-2 3CL-Pro primary assay. The assay buffer contained 20 mM Tris (pH 7.3), 100 mM NaCl, and 1 mM EDTA. Test compounds were preincubated with 2.1 μM of enzyme (2 μL) and 0.5% DMSO for 60 min at 37 °C prior to addition of substrate to obtain a final concentration of 15 μM substrate and 20 μM test compound in a 20 μL total volume. The signal was monitored after 30 min of incubation. A dose response curve of the compound GC376 was used as a positive control,48 and DMSO (0.5% final) was used as a negative control. Although, showing low similarity to human proteases and being a cysteine protease, SARS-CoV-2 3CL-Pro is structurally related to the serine protease of the chymotrypsin family with a cysteine replacing the serine in the active site.8,49 Therefore, a chymotrypsin activity assay kit (#K352-100, Biovision) was used according to manufacturer’s instructions to profile the relative selectivity of confirmed Hits. Here, the assay buffer contained 1 mM DTT, which was essential to observe chymotrypsin enzymatic activity. Cleaved substrate was detected at Ex/Em = 380/460 nm. The positive control (100% inhibition) was nafamostat (final concentration 5 μM), and the negative control was DMSO at 0.5%.
Anticytopathic Effect and Virus Yield Reduction Assays
The CPE assay used the African green monkey kidney cell line (Vero-E6) which had previously been engineered to constitutively express GFP.50 Cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM; Gibco) supplemented with 10% fetal calf serum (FCS; Biowest), 0.075% sodium bicarbonate (7.5% solution, Gibco), and 1× Pen–strep (Gibco) and kept under 5% CO2 at 37 °C. Assay medium contained 2% FCS. SARS-CoV-2 strain BetaCov/Belgium/GHB-03021/2020 recovered from a nasopharyngeal swab taken from an asymptomatic patient returning from Wuhan, China in the beginning of February 2020 was sequenced on a MinION platform (Oxford Nanopore). After serial passaging on Huh7 and Vero-E6 cells, infectious content of the virus stock was determined by titration on Vero-E6 cells using the Spearman–Kärber method. To measure inhibition of the SARS-CoV-2 cytopathic effect, 384-well plates (Greiner #781092) were spotted with test compounds using an acoustic dispenser (Echo, Labcyte) to yield 10 μM final concentration at 0.1% DMSO in primary screening. The day before infection (Day −1), plates were equilibrated to room temperature, and 30 μL of Vero-E6 EGFP cells were added to give 8000 cells/well. On the day of infection (Day 0), plates were transported to the CAPS-IT robotic system for the addition of virus dilution (MOI = 0.001) using a liquid handler (EVO 100, Tecan) to a final well volume of 60 μL, and left for incubation at 37 °C, 5% CO2. At 4 days postinfection, GFP signal was captured using wide field fluorescence imaging by exciting at 485–20 nm and emitting with the BGRFRN filter set. A 5× objective captured 80% of an entire well on a 384 plate at once. The optimal exposure time was determined based on fluorescence intensity and was set on 0.023 s. A 2 × 2 binning was used, and autofocus plane count was reduced to increase image acquisition speed. An image analysis protocol was developed in-house by using the SpotDetector bioapplication (Cellomics, Thermofisher). After background reduction on the raw image files, a fixed fluorescent intensity threshold was determined for the identification of GFP cells. Afterward, the total area (“total amount of surface covered by fluorescent cells”) was calculated for the detected cells and compared to the positive (remdesivir 20 μM) and negative (DMSO) control. This parameter directly correlates to cell confluence. Compound profiling used the screening primary protocol as above, with compounds arrayed in 8 point concentration responses in triplicate (max 20 μM, 1:3 dilution steps). Assessment of the underlying cytotoxicity of the compounds was performed as described above in dose response but without virus infection and using sodium-selenite (20 μM final) as the cytotoxicity control.
For selected compounds, efficacy was verified in a virus yield reduction assay. Briefly, VeroE6 cells were seeded in a 96-well cell culture plate in a density of 40 000 cells per well. After 24 h, compounds were added to the medium, starting at a highest concentration of 25 μM and then serially diluted 1:3 (8 steps). Each compound was carried out in duplicate. After 2 h, cells were inoculated with virus (MOI = 0.1) and left for incubation. Two hours later, the virus-containing medium was washed away with PBS and replaced with fresh medium and compound. At 2 days post infection, cells were visually inspected for cytopathic effects, and supernatant was collected in the lysis buffer. RNA extraction was carried out using the NucleoSpin kit (Macherey-Nagel) according to the manufacturer’s instructions. Subsequent RT-qPCR was performed on a LightCycler96 platform (Roche) based on SARS-CoV-2 N gene RNA amplification using forward (5′-GACCCCAAAATCAGCGAAAT) and reverse (5′-TCTGGTTACTGCCAGTTGATCTG) primers and probe (5′- FAM-ACCCCGCATTACGTTTGGTGGACC-BHQ1) designed by CDC (United States Centers for Disease Control and Prevention). Standard of infectious virus with a known titer was used to quantify the amount of viral RNA (vRNA) as TCID50 (50% tissue culture infectious dose) per mL. Effect of the compound was expressed as log reduction of vRNA.
Protein Expression and Purification for Crystallization Studies
The 3CLpro of SARS-CoV-2 plasmid was kindly provided by the research group of Prof. Rolf Hilgenfeld from the Institute of Biochemistry, Center for Structural and Cell Biology in Medicine, University of Lübeck (Germany) (ORF1ab polyprotein residues 3264–3569, GenBank code: MN908947.3). Protein production and purification was as reported by Zhang et al., where eluted fractions containing the target protein were pooled and subjected to buffer exchange in 20 mM Tris-HCl, 150 mM NaCl, 1 mM EDTA, and 1 mM DTT, pH 7.8.5 Protein was flash frozen in LN2 at a concentration of 10–20 mg/mL and stored in aliquots at −80 °C.
Crystallization, Data Collection, Data Reduction, Structure Determination, Refinement, and Final Model Analysis
Prior to crystallization, each protein aliquot was thawed in ice and buffer exchanged against 20 mM Tris-HCl, 150 mM NaCl, and 1 mM EDTA, pH 7.8 to remove DTT excess. Crystals of myricetin:SARS-CoV-2 3CL-Pro were obtained with the vapor diffusion technique, in sitting drops, using seeding. The protein at 5 mg/mL was incubated with 30-fold molar excess of myricetin and mixed in equal volume with 0.1 M dl-glutamic acid monohydrate, 0.1 M dl-alanine, 0.1 M gycine, 0.1 M dl-lysine monohydrochloride, 0.1 M dl-serine, 0.1 M HEPES/MOPS pH 7.5, 20% v/v ethylene glycol, and 10% w/v PEG 8000 as a precipitant solution. Diffraction data were collected at 100 K at the XDR2 beamline of the Elettra Sincrotrone Trieste51 using a 0.9717 Å wavelength. The collected data set was processed with XDS52 and Aimless53 from the CCP4 suite.54 The structure was solved by molecular replacement with Phaser55 using as a search model 7ALH (PDB ID). The initial model was refined alternating cycles of manual model building in COOT56,57 and automatic refinement using Phenix58 (version 1.18.2_3874).
Data Analysis
Data analysis of assay development results was performed using GraphPad Prism 8. In test compound screening assays, data was analyzed using commercial software (Activitybase, IDBS). Test compound results were normalized relative to respective controls and control well outliers eliminated according to the three-sigma method. Dose response curves were fitted to 4-parameter logistic functions in the ActivityBase software (ActivityBase Version 8.0.5.4) Assay quality was assessed using the Z′-factor calculation with Z′ > 0.5 as threshold for acceptance.59 TIBCO Spotfire Analyst 7.11 was used for visualization/selection. PDB website ligand interaction scripts (or CCG-MOE version 2019) were used for structural visualizations. By applying a molecular mechanics method integrated with Wave function Spartan ’18 parallel suite [Version 18.4.1], the equilibrium geometry among the thioguanosine tautomers has been obtained.
The ligands were screened against the conformations using OpenEye FRED60 and Schrödinger Suite Glide Version 85012.61 Another geometrical docking method was used for the identification of the best binding mode.62,63
Acknowledgments
Funding for this study was received from Exscalate4Cov under the European Union’s Horizon 2020 research and innovation programme under Grant 101003551. Preparation of data for release was funded by H2020 Grant 824087 (EOSC-LIFE), and we thank Emma Manners, Anna Gaulton, and Andrew Leach at ChEMBL for excellent support in organizing data sets. The EU-OPENSCREEN bioactive compound collection was provided by the EU-OPENSCREEN ERIC (Berlin, Germany). The 3CL-Pro of SARS-CoV-2 was kindly provided by Dr. Alke Meents from DESY (Hamburg, Germany). We thank Yulia Gerhardt and Peter Maas of SPECS and Joshua Bitker (ex-Broad) for input into the selection and quality control of the Fraunhofer compound library. This work was supported by the Fraunhofer InternaI Programs under Grant No. Anti-Corona 840260 (DRECOR).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsptsci.0c00216.
Figures for compound library composition (Figure S-1), DMSO tolerance of the primary assay (Figure S-2), assay optimization regarding incubation time, temperature, and response to known inhibitors (Figures S-3–S-5), confirmation of activity for compounds sourced from different libraries (Figure S-6), antiviral activity of MG-132 in Vero-E6 cells (Figure S-7), modeling of thioguanosine analogues (Figure S-8), tables for quality control of the primary screen reported as Z prime values (Table S-1), primary screening results as a link to raw data (Table S-2), and crystallographic data collection and refinement statistics (Table S-4) (PDF)
Summary of the primary screen data (SuppT3-PS), hit confirmation (SuppT3-HC) and profiling data (SuppT3-HP) (Table S-3) (XLSX)
Accession Codes
Coordinates and structure factors were deposited to the Protein Data Bank with accession code 7B3E.
Author Present Address
▲ Y.Y.: State Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai, 201203, China; School of Life Science and Technology, ShanghaiTech University, Shanghai, 201210, China; Eskitis-SIMM Joint Laboratory for Drug Discovery, Australia
The authors declare no competing financial interest.
Supplementary Material
References
- Wu C.; Liu Y.; Yang Y.; Zhang P.; Zhong W.; Wang Y.; Wang Q.; Xu Y.; Li M.; Li X.; Zheng M.; Chen L.; Li H. (2020) Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin. B 10, 766–788. 10.1016/j.apsb.2020.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]; Hilgenfeld R. (2014) From SARS to MERS: crystallographic studies on coronaviral proteases enable antiviral drug design. FEBS J. 281, 4085–4096. 10.1111/febs.12936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zumla A.; Chan J. F. W.; Azhar E. I.; Hui D. S. C.; Yuen K.-Y. (2016) Coronaviruses — drug discovery and therapeutic options. Nat. Rev. Drug Discovery 15 (5), 327–347. 10.1038/nrd.2015.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO COVID19 website https://covid19.who.int/ (latest seen on Nov 18th, 2020).
- Cannalire R., Cerchia C., Beccari A. R., Di Leva F. S., and Summa V. (2020) Targeting SARS-CoV-2 Proteases and Polymerase for COVID-19 Treatment: State of the Art and Future Opportunities. J. Med. Chem. 10.1021/acs.jmedchem.0c01140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L.; Lin D.; Sun X.; Curth U.; Drosten C.; Sauerhering L.; Becker S.; Rox K.; Hilgenfeld R. (2020) Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 368 (6489), 409–412. 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- https://www.uniprot.org/blast/(using 3CL-Pro sequences: 7CBT - SARS-CoV-2, 5WKK-MERS, 3M3V-SARS-CoV) (accessed 2021-01-27).
- Sirois S.; Zhang R.; Gao W.; Gao H.; Li Y.; Zheng H.; Wei D.-Q. (2007) Discovery of Potent Anti-SARS-CoV MPro Inhibitors. Curr. Comput.-Aided Drug Des. 3, 191. 10.2174/157340907781695440. [DOI] [Google Scholar]
- Ramajayam R.; Tan K.-P.; Liang P.-H. (2011) Recent development of 3C and 3CL protease inhibitors for anti-coronavirus and anti-picornavirus drug discovery. Biochem. Soc. Trans. 39 (5), 1371–1375. 10.1042/BST0391371. [DOI] [PubMed] [Google Scholar]
- Ahn T.-Y.; Kuo C.-J.; Liu H.-G.; Ha D.-C.; Liang P.-H.; Jung Y.-S. (2010) Synthesis and Evaluation of Benzoquinolinone Derivatives as SARS-CoV 3CL Protease Inhibitors. Bull. Korean Chem. Soc. 31 (1), 87–91. 10.5012/bkcs.2010.31.01.087. [DOI] [Google Scholar]
- Kuo C.-J.; Liu H.-G.; Lo Y.-K.; Seong C.-M.; Lee K.-I.; Jung Y.-S.; Liang P.-H. (2009) Individual and common inhibitors of coronavirus and picornavirus main proteases. FEBS Lett. 583, 549. 10.1016/j.febslet.2008.12.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C.-C.; Kuo C.-J.; Hsu M.-F.; Liang P.-H.; Fang J.-M.; Shie J.-J.; Wang A. H.-J. (2007) Structural basis of mercury- and zinc-conjugated complexes as SARS-CoV 3C-like protease inhibitors. FEBS Lett. 581 (28), 5454–5458. 10.1016/j.febslet.2007.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo S.; Kim S.; Kim D. Y.; Kim M.-S.; Shin D. H. (2020) Flavonoids with inhibitory activity against SARS-CoV-2 3CLpro. J. Enzyme Inhib. Med. Chem. 35 (1), 1539–1544. 10.1080/14756366.2020.1801672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W.; Xu M.; Chen C. Z.; Guo H.; Shen M.; Hu X.; Shinn P.; Klumpp-Thomas C.; Michael S. G.; Zheng W. (2020) Identification of SARS-CoV-2 3CL Protease Inhibitors by a Quantitative High-Throughput Screening. ACS Pharmacology & Translational Science 3 (5), 1008–1016. 10.1021/acsptsci.0c00108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu L.; Ye F.; Feng Y.; Yu F.; Wang Q.; Wu Y.; Zhao C.; Sun H.; Huang B.; Niu P.; Song H.; Shi Y.; Li X.; Tan W.; Qi J.; Gao G. F. (2020) Both Boceprevir and GC376 efficaciously inhibit SARS-CoV-2 by targeting its main protease. Nat. Commun. 11 (1), 4417. 10.1038/s41467-020-18233-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kneller D. W.; Galanie S.; Phillips G.; O’Neill H. M.; Coates L.; Kovalevsky A. (2020) Malleability of the SARS-CoV-2 3CL Mpro Active-Site Cavity Facilitates Binding of Clinical Antivirals. Structure 28 (12), 1313–1320. 10.1016/j.str.2020.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oerlemans R., Ruiz-Moreno A. J., Cong Y., Kumar N. D., Velasco-Velazquez M. A., Neochoritis C. G., Smith J., Reggiori F., Groves M. R., and Dömling A. (2021) Repurposing the HCV NS3–4A protease drug boceprevir as COVID-19 therapeutics. RSC Med. Chem. 10.1039/D0MD00367K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young B. E.; Xiang Ong S. W.; Kalimuddin S. (2020) Epidemiologic Features and Clinical Course of Patients Infected With SARS-CoV-2 in Singapore. JAMA 323 (15), 1488–1494. 10.1001/jama.2020.3204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oscanoa T. J.; Romero-Ortuno R.; Carvajal A.; Savarino A. (2020) A pharmacological perspective of chloroquine in SARS-CoV-2 infection: An old drug for the fight against a new coronavirus?. Int. J. Antimicrob. Agents 56 (3), 106078. 10.1016/j.ijantimicag.2020.106078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RECOVERY Collaborative Group , Horby P., Lim W. S., and Emberson J. R. (2020) Dexamethasone in Hospitalized Patients with Covid-19 - Preliminary Report. N Engl J. Med. 10.1056/NEJMoa2021436. [DOI] [PMC free article] [PubMed]
- Beigel J. H.; Tomashek K. M.; Dodd L. E.; Mehta A. K.; Zingman B. S.; Kalil A. C.; Hohmann E.; Chu H. Y.; Luetkemeyer A.; Kline S.; Lopez de Castilla D.; Finberg R. W.; Dierberg K.; Tapson V.; Hsieh L.; Patterson T. F.; Paredes R.; Sweeney D. A.; Short W. R.; Touloumi G.; Lye D. C.; Ohmagari N.; Oh M.-d.; Ruiz-Palacios G. M.; Benfield T.; Fatkenheuer G.; Kortepeter M. G.; Atmar R. L.; Creech C. B.; Lundgren J.; Babiker A. G.; Pett S.; Neaton J. D.; Burgess T. H.; Bonnett T.; Green M.; Makowski M.; Osinusi A.; Nayak S.; Lane H. C. (2020) Remdesivir for the Treatment of Covid-19 — Final Report. N. Engl. J. Med. 383, 1813–1826. 10.1056/NEJMoa2007764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gossen J., Albani S., Hanke A., Joseph B. P., Bergh C., Kuzikov M., Costanzi E., Manelfi C., Storici P., Gribbon P., Beccari A. R., Talarico C., Spyrakis F., Lindahl E., Zaliani A., Carloni P., Wade R. C., Musiani F., Kokh D. B., and Rossetti G. (2021) Blueprint for High Affinity SARS-CoV-2 Mpro Inhibitors from Activity-Based Compound Library Screening Guided by Analysis of Protein Dynamics. ACS Pharmacol. Transl. Sci., 2021. 10.1021/acsptsci.0c00215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C.; Sacco M. D.; Hurst B.; Townsend J. A.; Hu Y.; Szeto T.; Zhang X.; Tarbet B.; Marty M. T.; Chen Y.; Wang J. (2020) Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell Res. 30, 678–692. 10.1038/s41422-020-0356-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Data can be retrieved in ChEMBLDB version 28 https://www.ebi.ac.uk/chembl/document_report_card/CHEMBL4495564/.
- Ellinger B., Bojkova D., Zaliani A., Cinatl J., Claussen C., Westhaus S., Reinshagen J., Kuzikov M., Wolf M., Geisslinger G., Gribbon P., and Ciesek S.. Identification of inhibitors of SARS-CoV-2 in-vitro cellular toxicity in human (Caco-2) cells using a large scale drug repurposing collection. ResearchSquare 2021, in print. 10.1038/s41597-021-00848-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su H., Yao S., Zhao W., Li M., Liu J., Shang W. J., Xie H., Ke C., Gao M., Yu K., Liu H., Shen J., Tang W., Zhang L., Zuo J., Jiang H., Bai F., Wu Y., Ye Y., and Xu Y. (2020) Discovery of baicalin and baicalein as novel, natural product inhibitors of SARS-CoV-2 3CL protease in vitro. bioRxiv 2020. 10.1101/2020.04.13.038687. [DOI] [Google Scholar]
- Kneller D. W.; Phillips G.; O’Neill H. M.; Jedrzejczak R.; Stols L.; Langan P.; Joachimiak A.; Coates L.; Kovalevsky A. (2020) Structural plasticity of SARS-CoV-2 3CL Mpro active site cavity revealed by room temperature X-ray crystallography. Nat. Commun. 11, 3202. 10.1038/s41467-020-16954-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grottesi A.; Bešker N.; Emerson A.; Manelfi C.; Beccari A. R.; Frigerio F.; Lindahl E.; Cerchia C.; Talarico C. (2020) Computational Studies of SARS-CoV-2 3CLpro: Insights from MD Simulations. Int. J. Mol. Sci. 21 (15), 5346. 10.3390/ijms21155346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kneller D. W.; Phillips G.; Weiss K. L.; Pant S.; Zhang Q.; O’Neill H. M.; Coates L.; Kovalevsky A. (2020) Unusual zwitterionic catalytic site of SARS-CoV-2 main protease revealed by neutron crystallography. J. Biol. Chem. 295 (50), 17365–17373. 10.1074/jbc.AC120.016154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbalenya A. E.; Snijder E. J. (1996) Viral cysteine proteinases. Perspect. Drug Discovery Des. 6, 64–86. 10.1007/BF02174046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guenther S., Reinke P., Oberthuer D., Yefanov O., Ginn H., Meier S., Lane T., Lorenzen K., Gelisio L., Brehm W., Dunkel I., Domaracky M., Saouane S., Lieske J., Ehrt C., Koua F., Tolstikova A., White T., Groessler M., Fleckenstein H., Trost F., Galchenkova M., Gevorkov Y., Li C., Awel S., Peck A., Xavier P. L., Barthelmess M., Schlünzen F., Werner N., Andaleeb H., Ullah N., Falke S., Franca B. A., Schwinzer M., Brognaro H., Seychell B., Gieseler H., Melo D., Zaitsev-Doyle J. J., Norton-Baker B., Knoska J., Pena Murillo G. E., Mashhour A. R., Guicking F., Hennicke V., Fischer P., Rogers C., Monteiro D. C. F., Hakanpää J., Meyer J., Noei H., Gribbon P., Ellinger B., Kuzikov M., Wolf M., Zhang L., Sun X., Pletzer-Zelgert J., Wollenhaupt J., Feiler C., Weiss M., Schulz E.-C., Mehrabi P., Schmidt C., Schubert R., Han H., Krichel B., Fernández-García Y., Escudero-Pérez B., Günther S., Turk D., Uetrecht C., Beck T., Tidow H., Chari A., Zaliani A., Rarey M., Cox R., Hilgenfeld R., Chapman H. N., Pearson A. R., Betzel C., and Meents A. (2020) Catalytic cleavage of HEAT and subsequent covalent binding of the tetralone moiety by the SARS-CoV-2 main protease. bioRxiv 10.1101/2020.05.02.043554. [DOI] [Google Scholar]
- Báez-Santos Y. M.; St. John S. E.; Mesecar A. D. (2015) Antiviral Res. 115, 21–38. 10.1016/j.antiviral.2014.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu H.-F.; Chen C.-C.; Moses D. C.; Chen Y.-H.; Lin C.-H.; Tsai Y.-C.; Chou C.-Y. (2018) Antiviral Res. 158, 199–205. 10.1016/j.antiviral.2018.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang S.; Cheng S. C.; Tang H. C.; Sun C. Y.; Chou C. Y. (2018) 6-Thioguanine is a noncompetitive and slow binding inhibitor of human deubiquitinating protease USP2. Sci. Rep. 8, 3102. 10.1038/s41598-018-21476-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaim C. D., Perng Y.-C., Zhao X., Canadeo L. A., Harastani H. H., Darling T. L., Boon A. C. M., Lenschow D. J., and Huibregtse J. M. (2020) 6-Thioguanine blocks SARS-CoV-2 replication by inhibition of PLpro protease activities bioRxiv 10.1101/2020.07.01.183020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altun M.; Kramer H. B.; Willems L. I.; McDermott J. L.; Leach C. A.; Goldenberg S. j.; Kumar K. G. S.; Konietzny R.; Fischer R.; Kogan E.; Mackeen M. M.; McGouran J.; Khoronenkova S. V.; Parsons J. L.; Dianov G. L.; Nicholson B.; Kessler B. M. (2011) Activity-Based Chemical Proteomics Accelerates Inhibitor Development for Deubiquitylating Enzymes. Chem. Biol. 18 (11), 1401–1412. 10.1016/j.chembiol.2011.08.018. [DOI] [PubMed] [Google Scholar]
- Tian X.; Isamiddinova N. S.; Peroutka R. J.; Goldenberg S. J.; Mattern M. R.; Nicholson B.; Leach C. (2011) Characterization of selective ubiquitin and ubiquitin-like protease inhibitors using a fluorescence-based multiplex assay format Assay. Assay Drug Dev. Technol. 9 (2), 165–173. 10.1089/adt.2010.0317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Słabicki M.; Kozicka Z.; Petzold G.; Li Y.-D.; Manojkumar M.; Bunker R. D.; Donovan K. A.; Sievers Q. L.; Koeppel J.; Suchyta D.; Sperling A. S.; Fink E. C.; Gasser J. A.; Wang L. R.; Corsello S. M.; Sellar R. S.; Jan M.; Gillingham D.; Scholl C.; Fröhling S.; Golub T. R.; Fischer E. S.; Thomä N. H.; Ebert B. L. (2020) The CDK inhibitor CR8 acts as a molecular glue degrader that depletes cyclin K. Nature 585, 293–297. 10.1038/s41586-020-2374-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P.; Liu H.; Sun Q.; Liang H.; Li C.; Deng X.; Liu Y.; Lai L. (2020) Potent inhibitors of SARS-CoV-2 3C-like protease derived from N-substituted isatin compounds. Eur. J. Med. Chem. 206, 112702. 10.1016/j.ejmech.2020.112702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zastavnyĭ I. V. (1979) Comparative study of the anti-influenzal effectiveness of bonaphthon, oxoline, florenal and rimantadine. Mikrobiol Zh 41 (1), 69–74. [PubMed] [Google Scholar]
- Chauhan A., and Kalra S. (2020) Identification of potent COVID-19 main protease (MPRO) inhibitors from flavonoids. PREPRINT (Version 1) available at Research Square. 10.21203/rs.3.rs-34497/v1. [DOI] [Google Scholar]
- Pandey K. B.; Rizvi S. I. (2009) Plant polyphenols as dietary antioxidants in human health and disease. Oxid. Med. Cell. Longevity 2 (5), 270–278. 10.4161/oxim.2.5.9498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baell J. B. (2016) Feeling Nature’s PAINS: Natural Products, Natural Product Drugs, and Pan Assay Interference Compounds (PAINS). J. Nat. Prod. 79 (3), 616–628. 10.1021/acs.jnatprod.5b00947. [DOI] [PubMed] [Google Scholar]
- Su H. X.; Yao S.; Zhao W. F.; Li M. J.; Liu J.; Shang W. J.; Xie H.; Ke C. Q.; Hu H. C.; Gao M. N.; Yu K. Q.; Liu H.; Shen J. S.; Tang W.; Zhang L. K.; Xiao G. F.; Ni L.; Wang D. W.; Zuo J. P.; Jiang H. L.; Bai F.; Wu Y.; Ye Y.; Xu Y. C. (2020) Anti-SARS-CoV-2 activities in vitro of Shuanghuanglian preparations and bioactive ingredients. Acta Pharmacol. Sin. 41 (9), 1167–1177. 10.1038/s41401-020-0483-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider M.; Ackermann K.; Stuart M.; Wex C.; Protzer U.; Schatzl H. M.; Gilch S. (2012) Severe acute respiratory syndrome coronavirus replication is severely impaired by MG132 due to proteasome-independent inhibition of M-calpain. J. Virol. 86 (18), 10112–10122. 10.1128/JVI.01001-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longhitano L.; Tibullo D.; Giallongo C.; Lazzarino G.; Tartaglia N.; Galimberti S.; Volti G. L.; Palumbo G. A.; Liso A. (2020) Proteasome Inhibitors as a Possible Therapy for SARS-CoV-2. Int. J. Mol. Sci. 21 (10), 3622. 10.3390/ijms21103622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grinev A. N.; Pershin G. N.; Stebaeva L. F.; Nikolaeva I. S.; Shvedov V. I.; Kharizomenova I. A.; Botkina E. M. (1984) A new antiviral chemotherapeutant, riodoxol. Pharm. Chem. J. 18, 208–211. 10.1007/BF00759009. [DOI] [Google Scholar]
- Corsello S.; Bittker J.; Liu Z.; Gould J.; McCarren P.; Hirschman J. E.; Johnston S. E.; Vrcic A.; Wong B.; Khan M.; Asiedu J.; Narayan R.; Mader C. C.; Subramanian A.; Golub T. R. (2017) The Drug Repurposing Hub: a next-generation drug library and information resource. Nat. Med. 23, 405–408. 10.1038/nm.4306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y.; Liu H.; Galasiti Kankanamalage A. C.; Weerasekara S.; Hua D. H.; Groutas W. C.; Chang K. O.; Pedersen N. C. (2016) Reversal of the Progression of Fatal Coronavirus Infection in Cats by a Broad-Spectrum Coronavirus Protease Inhibitor. PLoS Pathog. 12 (3), e1005531 10.1371/journal.ppat.1005531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi R. S.; Jagdale S. S.; Bansode S. B.; Shiva Shankar S.; Tellis M. B.; Pandya V. K.; Chugh A.; Giri A. P.; Kulkarni M. J. (2020) Discovery of potential multi-target-directed ligands by targeting host-specific SARS-CoV-2 structurally conserved main protease. J. Biomol. Struct. Dyn. 1. 10.1080/07391102.2020.1760137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivens T.; Van den Eynde C.; Van Acker K.; Nijs E.; Dams G.; Bettens E.; Ohagen A.; Pauwels R.; Hertogs K. (2005) Development of a homogeneous screening assay for automated detection of antiviral agents active against severe acute respiratory syndrome-associated coronavirus. J. Virol. Methods 129 (1), 56–63. 10.1016/j.jviromet.2005.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lausi A.; Polentarutti M.; Onesti S.; Plaisier J. R.; Busetto E.; Bais G.; Barba L.; Cassetta A.; Campi G.; Lamba D.; Pifferi A.; Mande S. C.; Sarma D. D.; Sharma S. M.; Paolucci G. (2015) Status of the crystallography beamlines at Elettra. Eur. Phys. J. Plus 130 (43), 1–8. 10.1140/epjp/i2015-15043-3. [DOI] [Google Scholar]
- Kabsch W. (2010) XDS. Acta Crystallogr., Sect. D: Biol. Crystallogr. D66, 125–132. 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans P. R.; Murshudov G. N. (2013) How Good Are My Data and What Is the Resolution. Acta Crystallogr., Sect. D: Biol. Crystallogr. D69, 1204–1214. 10.1107/S0907444913000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn M. D.; Ballard C. C.; Cowtan K. D.; Dodson E. J.; Emsley P.; Evans P. R.; Keegan R. M.; Krissinel E. B.; Leslie A. G.; McCoy A.; McNicholas S. J.; Murshudov G. N.; Pannu N. S.; Potterton E. A.; Powell H. R.; Read R. J.; Vagin A.; Wilson K. S. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr., Sect. D: Biol. Crystallogr. D67, 235–242. 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy A. J.; Grosse-Kunstleve R. W.; Adams P.; Winn M. D.; Storoni L. C.; Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674. 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. (2004) Coot: model-building tools for molecular graphics.. Acta Crystallogr., Sect. D: Biol. Crystallogr. D60, 2126–2132. 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Emsley P.; Lohkamp B.; Scott W. G.; Cowtan K. (2010) Features and Development of Coot.. Acta Crystallogr., Sect. D: Biol. Crystallogr. D66, 486–501. 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebschner D.; Afonine P. V.; Baker M. L.; Bunkóczi G.; Chen V. B.; Croll T. I.; Hintze B.; Hung L.-W.; Jain S.; McCoy J.; Moriarty N. W.; Oeffner R. D.; Poon B. K.; Prisant M. G.; Read R. J.; Richardson J. S.; Richardson D. C.; Sammito M. D.; Sobolev O. V.; Stockwell D. H.; Terwilliger T. C.; Urzhumtsev A. G.; Videau L. L.; Williams C. J.; Adams P. (2019) Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. D75, 861–877. 10.1107/S2059798319011471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iversen P. W.; Eastwood B. J.; Sittampalam G. S.; Cox K. L. (2006) A Comparison of Assay Performance Measures in Screening Assays: Signal Window, Z′ Factor, and Assay Variability Ratio. J. Biomol. Screening 11 (3), 247–252. 10.1177/1087057105285610. [DOI] [PubMed] [Google Scholar]
- McGann M. (2011) FRED Pose Prediction and Virtual Screening Accuracy. J. Chem. Inf. Model. 51 (3), 578–596. 10.1021/ci100436p. [DOI] [PubMed] [Google Scholar]
- Friesner R. A.; Banks J. L.; Murphy R. B.; Halgren T. A.; Klicic J. J.; Mainz D. T.; Repasky M. P.; Knoll E. H.; Shelley M.; Perry J. K.; Shaw D. E.; Francis P.; Shenkin P. S. (2004) Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 47 (7), 1739–1749. 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- Beccari A. R.; Cavazzoni C.; Beato C.; Costantino G. (2013) LiGen: a high performance workflow for chemistry driven de novo design. J. Chem. Inf. Model. 53 (6), 1518–1527. 10.1021/ci400078g. [DOI] [PubMed] [Google Scholar]
- Beato C.; Beccari A. R.; Cavazzoni C.; Lorenzi S.; Costantino G. (2013) Use of experimental design to optimize docking performance: the case of LiGenDock, the docking module of LiGen, a new de novo design program. J. Chem. Inf. Model. 53 (6), 1503–1517. 10.1021/ci400079k. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.