

Abstract
Bone marrow (BM) mesenchymal stem cells (MSCs) are critical components of the BM microenvironment and play an essential role in supporting hematopoiesis. Dysfunction of MSCs is associated with the impaired BM microenvironment that promotes leukemia development. However, whether and how restoration of the impaired BM microenvironment can inhibit leukemia development remain unknown. Using an established leukemia model and the RNA-seq analysis, we discovered functional degeneration of MSCs during leukemia progression. Importantly, intra-BM instead of systemic transfusion of donor healthy MSCs restored the BM microenvironment, demonstrated by functional recovery of host MSCs, improvement of thrombopoiesis, and re-balance of myelopoiesis. Consequently, intra-BM MSC treatment reduced tumor burden and prolonged survival of the leukemia-bearing mice. Mechanistically, donor MSC treatment restored the function of host MSCs and reprogrammed host macrophages into arginase 1 positive phenotype with tissue repair features. Transfusion of MSC-reprogrammed macrophages largely recapitulated the therapeutic effects of MSCs. Taken together, our study reveals that donor MSCs reprogram host macrophages to restore the BM microenvironment and inhibit leukemia development.
Introduction
MSC therapy has been widely used in treating immune-related graft-vs-host disease (GVHD) and inflammation-related diseases [1, 2]. Over the decades, a lot of evidence demonstrates that MSCs regulate innate and adaptive immune responses largely by secreting distinct sets of cytokines, growth factors, and chemokines depending on different disease contexts [3–7]. Given the short lifespan of donor MSCs after transfusion [8], the underlying molecular and cellular mechanisms by which these cells produce therapeutic effects remain elusive. It is also completely unknown whether donor MSCs can restore the impaired BM microenvironment and consequently suppress disease progression in leukemia setting.
Macrophages are pivotal for maintenance of the tissue microenvironment, tissue repair, and even the tumor microenvironment [9–13]. BM resident macrophages maintain the homeostasis of HSCs and loss of these macrophages leads to mobilization of HSCs into peripheral blood (PB) [14]. The functions of macrophages are plastic and can be reshaped by distinct sets of soluble factors. When performing tissue repair, macrophages highly express arginase 1 (Arg1) [15], an enzyme that converts L-arginine to urea and L-ornithine. After co-culture with MSCs, macrophages can be polarized from pro-inflammation (M1) to anti-inflammation (M2) type, up-regulating IL-10 and CD206 and down-regulating IL-6 and IL-1β [16]. Upon stimulated by LPS or TNF-α, MSCs can cross-talk with lung macrophages and reprogram these macrophages to secrete IL-10 to alleviate sepsis [17]. Despite these knowledge, whether healthy MSCs can reprogram macrophages from leukemia-bearing host to repair the damaged BM microenvironment is not known.
Using the established mouse model mimicking chronic MPN/MDS diseases [18–20], we discovered that the deteriorating BM microenvironment was associated with disease progression. Intra-BM instead of systemic transfusion of healthy MSCs restored the local BM microenvironment, improved thrombopoiesis, reduced tumor burden, and prolonged survival of leukemia-bearing mice. Mechanistically, we found that MSCs suppress leukemia development through reprogramming resident macrophages. Our study demonstrates that intra-BM transfusion of MSCs can restore the local BM microenvironment to systemically prevent leukemia progression.
Materials and Methods
Mice.
All mouse strains were maintained on C57BL/6 genetic background. Mice expressing the conditional oncogenic NrasG12D mutation (a gift from Dr. Jing Zhang lab at University of Wisconsin-Madison, Wisconsin, USA) were crossed to Vav-Cre mice to generate LSL Nras/+; Vav-Cre compound mice (NV mice). Genotyping of the adult mice was performed as described previously [18]. Vav-Cre strain (CD45.2), wild-type CD45.2, and CD45.1 strain (C57BL/6) strain were purchased from Jackson lab. GFP strain (CD45.2) was gifted by Guangdong Laboratory Animals Monitoring Institute. All mice were maintained within the SPF grade animal facility of Guangzhou Institutes of Biomedicine and Health, Chinese Academy of Sciences (GIBH, CAS, China). All animal experiments were approved by the Institutional Animal Care and Use Committee of Guangzhou Institutes of Biomedicine and Health (IACUC-GIBH).
NrasG12D leukemia model.
White blood cells (CD45.2+, 0.3 million) after depletion of stromal cells from NrasG12D compound mice (LSL Nras/+; Vav-Cre) or control mice (CD45.2 strain) were sorted and transplanted into sublethally (6.5 Gy, RS2000, Rad Source Inc) irradiated CD45.1 recipient by retro-orbital intravenous injection. Mice were fed with trimethoprim-sulfamethoxazole-treated water for two weeks to prevent infection. Hematopoietic lineages in PB were assessed monthly by flow cytometry. During the development of NrasG12D-induced leukemia, the CD11b+ percentage in PB indicated the tumor burden (CD11b+%).
MSC treatment for leukemia-bearing mice.
For MSC transfusion, multiple approaches including retro-orbital, tail intravenous, and local intra-BM transfusion were applied independently. For tail vein transfusion, each leukemia-bearing mouse was injected with 2.5 × 10^7 MSCs/kg (Passage 2) in 100 μl DPBS by tail vein transfusion. For retro-orbital transfusion, each leukemia-bearing mouse was injected with 2.5 × 10^7 MSCs/kg (Passage 2) in 200 μl DPBS by retro-orbital transfusion. For local intra-BM transfusion, tibia of each leukemia-bearing mouse was injected with 2.5 × 10^7 MSCs/kg (Passage 2) in 20 μl DPBS by local intra-BM transfusion. MSCs were injected once every two weeks and continued in a time window of 16 weeks. Every tibia was treated once per month by switching the injection site every other dose. The control mice were injected with DPBS following the same treatment procedure as MSCs. Analysis of platelets and CD11b+ cells in PB was performed monthly.
Results
Deterioration of BM MSCs accompanies the development of Nras-mutant-induced leukemia
Mice carrying an endogenous mutant Nras allele develop myelodysplastic/myeloproliferative neoplasms (MDS/MPN)-like leukemia with a long latency [18–21]. Here we found the primary BM leukemic cells failed to accelerate the disease in the secondary recipient mice, implying a role of the BM microenvironment in disease etiology (Supplementary Fig. S1A–B). Leukemic MSCs in a mouse T-ALL model suppressed normal hematopoiesis [22]. We hypothesized that the BM microenvironment is impaired by Nras-mutant leukemic cells, which in return impedes normal hematopoiesis and accelerates leukemia progression. Indeed, we observed quantitative decreases and functional degeneration of MSCs (Ter119−CD45−CD31−Sca1+CD51+CD146+) during disease development and progression (Fig. 1a–c). To further characterize the residual MSCs in mice with leukemia, we performed RNA-Seq analysis of the residual MSCs from leukemia-bearing mice at an early disease phase (CD11b+% in PB: 35%−45%). Under leukemia condition, the residual MSCs secreted much less soluble factors, including Il6, Il11, Ccl2, Ccl7, Cxcl12, Cxcl13 and Cxcl14 (padj < 0.05, fold change > 2), compared with MSCs from normal wild-type mice (Supplementary Fig. S1C). Collectively, these results show that the MSCs dramatically deteriorate during the disease development and progression of Nras-mutation-caused leukemia.
Fig. 1. Impaired bone marrow MSCs in mice with NrasG12D mutation-induced leukemia.
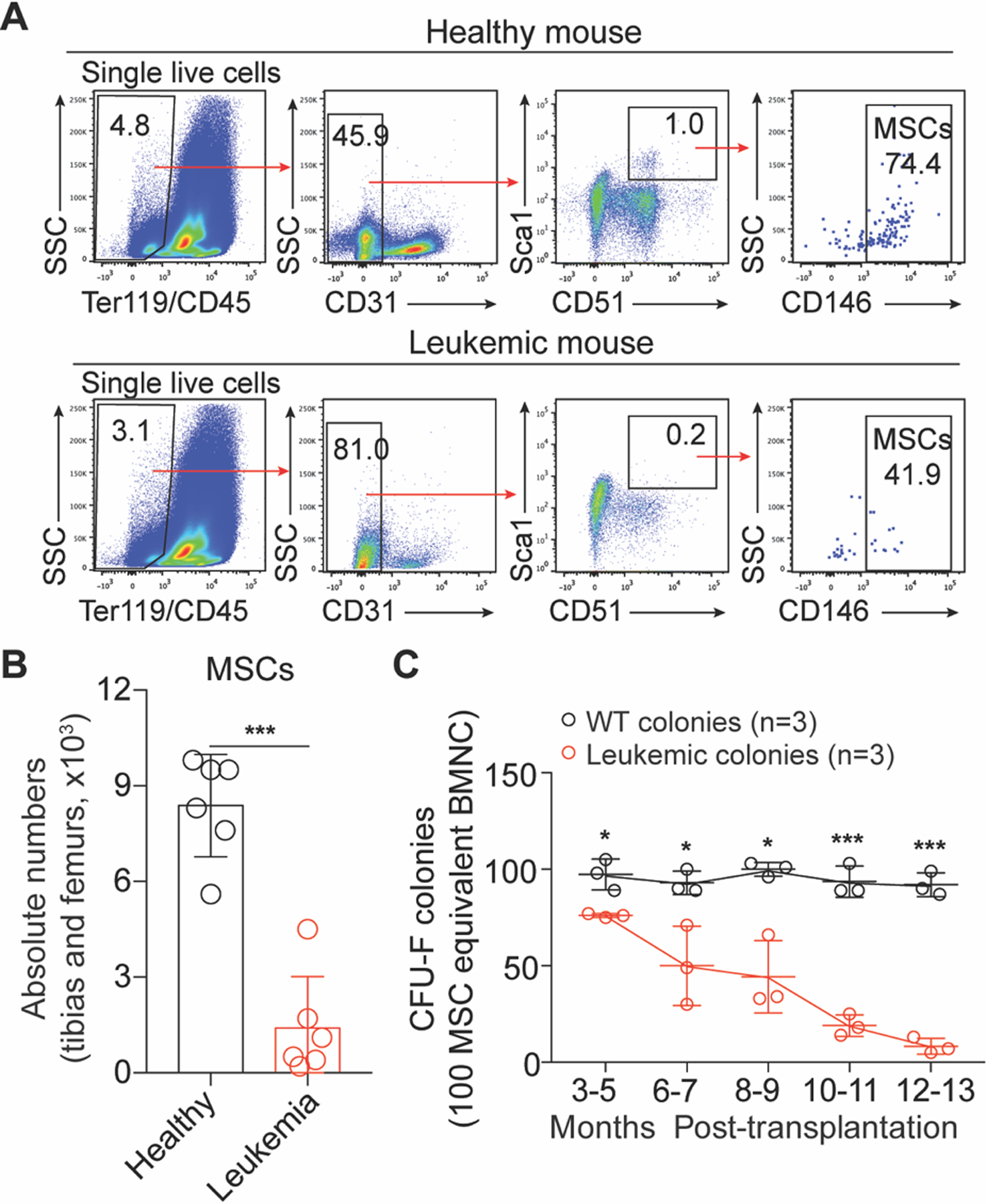
a Gating strategies for BM MSCs (Ter119−CD45−CD31−Sca1+CD51+ CD146+). b Statistical analysis of the absolute numbers of BM MSCs (mean ± SD, n = 6). c Kinetic analysis of functional MSCs in CFU-F assay (mean ± SD, n = 3). Asterisks indicate *p < 0.05, ***p < 0.001 (unpaired student’s t-test (two-tailed)).
Intra-BM transfusion of healthy MSCs improves thrombopoiesis, reduces tumor burden and improves survival of the leukemia-bearing mice
We hypothesized that restoration of the impaired BM microenvironment in leukemia-bearing mice might suppress/delay the disease progression. To test this hypothesis, we attempted healthy MSC treatment using GFP-tagged MSCs isolated from the tibias and femurs of healthy mice as previously reported [23]. The isolated primary MSCs were expanded shortly in vitro to passage two (P2) and cryopreserved. For MSC treatment, the cryopreserved P2 MSCs were recovered and cultured for five days, phenotypically identified (CD45−Ter119−CD31−CD51+CD105+LepR+PDGFRα+PDGFRβ+Sca1+) (Supplementary Fig. S2A), and suspended in DPBS (2.5 × 10^7/ml) for transfusion. Initially, we adopted a direct delivery procedure by injecting donor MSCs every two weeks either via tail vein (dose: 2.5 × 10^7 MSCs/kg in 100 μL DPBS) (Supplementary Fig. S2B) or retro-orbital (dose: 2.5 × 10^7 MSCs/kg in 200 μL DPBS) transfusion (Supplementary Fig. S2C) into the leukemia-bearing mice at a late disease phase (CD11b+ cells > 60% in PB). However, these delivery approaches failed to produce therapeutic effects. In vitro cultured MSCs lose their natural homing feature [24], the retro-orbital and intravenous transfusion of cultured MSCs failed to home to bone marrow (Supplementary Fig. S2D–E). Thus, we attempted intra-BM transfusion to overcome the homing defect caused by in vitro culture. A sequential doses of MSCs (2.5 × 10^7 MSCs/kg per dose in 20 μL DPBS) were injected into the tibia cavities of leukemia-bearing mice with two-week intervals for up to 16 weeks (Fig. 2a). Strikingly, the tumor burden continuously decreased during MSC treatment (Fig. 2b). Consequently, the survival of MSC-treated leukemia-bearing mice was significantly prolonged (Untreated: 51.5 days, MSC-treated: >115 days, p < 0.001) (Fig. 2c). Therefore, intra-BM transfusion of healthy donor MSCs improves the survival of leukemia-bearing mice.
Fig. 2. Intra-BM transfusion of donor MSCs prolongs survival of leukemia-bearing mice.

a Schematic diagram of MSC transfusion strategy. b Kinetic analysis of tumor burden (CD11b+) of MSC-treated leukemia-bearing mice. MSC-treated leukemia-bearing mice (blue line): n = 6. Untreated leukemia-bearing mice were used as disease control (n = 6; red line), PBS-treated leukemia-bearing mice were used as injected control (n = 3; green line), and MSC-treated WT mice were used as treatment control (n = 4; black line). c Kaplan-Meier survival of MSC-treated leukemia-bearing mice. Kaplan-Meier survival curves of untreated (red line, n = 6, Median survival = 51.5 days) and MSC-treated (blue line, n = 6, Median survival = 115 days) leukemia-bearing mice are shown (Log-rank (Mantel-Cox) test: p < 0.001). MSC treatment was terminated after 16 weeks. d Statistical analysis of white blood cells (WBC) and platelets (PLT) counts in PB (mean ± SD, n =6–8).
MSC-treatment systemically re-balances myelopoiesis and activates megakaryopoiesis
We next investigated the underlying mechanisms associated with the systemically decreased tumor burden. We found that the hematopoiesis in the MSC-treated leukemia-bearing mice was re-balanced, demonstrated by significant decreases of white blood cells (Untreated vs. MSC-treated: 23.04 vs 8.876, p = 0.009), and significant elevation of platelets (Untreated vs. MSC-treated: 2.64 vs. 6.01, p = 0.004) (Fig. 2d) in PB. On the contrary, the PBS-treated leukemia-bearing mice exhibited neither improved hematopoiesis (Supplementary Fig. S2F) nor prolonged survival. Collectively, these results indicate that intra-BM transfusion of healthy donor MSCs systemically improves hematopoiesis and prolongs the survival of leukemia-bearing mice.
To further investigate the systemic effects of the local MSC-treatment on hematopoiesis in leukemia-bearing mice, we analyzed the ratios of myeloid progenitor subpopulations in MSC-treated leukemia-bearing mice. Consistent with the elevated platelet levels and reduced myeloid cells in PB, the MSC-treated leukemia-bearing mice showed increased proportions of megakaryocyte-erythroid progenitors (MEP) (> 1.6 folds) (p < 0.001) and decreased ratios of granulocyte-macrophage progenitors (GMP) (> 1.5 folds) (p < 0.001) in both injected and non-injected sites than those sites in PBS-treated leukemia-bearing mice (Supplementary Fig. S3A and S3B). In addition, we observed increased (> 1.3 folds) ratios of mature megakaryocytes (≥ 8N) in both injected and non-injected sites in MSC-treated leukemia-bearing mice (Supplementary Fig. S3C and S3D) in comparison with PBS-treated leukemia-bearing control mice (p < 0.001). Thus, these data demonstrate that MSC-treatment systemically re-balances myelopoiesis and activates megakaryopoiesis in leukemia-bearing mice.
Recovered host MSCs are functional as healthy counterparts
To investigate whether the improved hematopoiesis is associated with restoration of the BM microenvironment, we analyzed the MSC-treated tibias eight weeks after MSC treatment. Interestingly, host MSCs (GFP negative) were partially recovered (Fig. 3a), but restricted to the locally treated tibias (Supplementary Fig. S4A–B). Functionally, the recovered host MSCs formed markedly more CFU-F colonies than the residual MSCs from untreated leukemia-bearing mice (> 3.8 folds) (p < 0.001) (Fig. 3b). To characterize the recovered MSCs at the transcriptome level, we sorted the recovered MSCs for RNA-Seq analysis. Unsupervised hierarchical clustering analysis showed that the recovered MSCs clustered closer to healthy MSCs (Fig. 3c). Further, the expression of cytokines and chemokines, including Il6, Ccl2, Ccl7, Ccl19, Cxcl12, Cxcl13, and Cxcl14, was restored in the recovered MSCs compared to that in MSCs from untreated leukemia-bearing control mice (padj < 0.05, fold change > 2) (Supplementary Fig. S4C). Therefore, donor MSC-treatment results in local functional restoration of host MSCs.
Fig. 3. Characterization of recovered host MSCs from MSC-treated leukemia-bearing mice.

a Statistical analysis of the absolute numbers of host MSCs (GFP−, host-derived MSCs) in tibias from untreated and MSC-treated leukemia-bearing mice (mean ± SD, n = 5). b Statistical analysis of CFU-F colonies (mean ± SD, n = 5). c Unsupervised hierarchical clustering of RNA-Seq data of healthy MSCs, leukemic MSCs, and recovered MSCs (GFP−, recipient-derived) (n = 3–6). Asterisks indicate ***p < 0.001 (unpaired student’s t-test (two-tailed)).
The donor MSCs reprogram macrophages to execute tissue-repair function
We further investigated the cellular mechanism underlying the restored BM microenvironment mediated by donor MSCs under leukemia condition. BM macrophages play a pivotal role in maintaining the BM niche [9]. To study whether donor MSCs reprogram BM macrophages, we performed co-culture assay of healthy MSCs with BM macrophages (L-Mac) sorted from the leukemia-bearing mice in vitro for twelve hours and re-sorted the macrophages (E-Mac) for RNA-Seq analysis. GSEA illustrated that angiogenesis-related genes, including Vegfa, Hif1a, Serpine1, Eng and Thbs1 [25], were enriched among the differentially expressed genes in E-Mac (Fig. 4a and Supplementary Fig. S5A). Genes associated with cell migration, including Sirpa [26], were also enriched in E-Mac (Fig. 4a and Supplementary Fig. S5B). Further, gene-ontology analysis demonstrated features of positive regulation of cell migration and angiogenesis in E-Mac (Supplementary Fig. S5C). RNA-Seq analysis showed that the expression of arginase 1 (Arg1), an indicator of tissue repair function [15], was dramatically up-regulated over thousand folds in E-Mac (Fig. 4b) after direct co-culture with MSCs in vitro. However, the Arg1 expression in macrophages after transwell co-culture was barely elevated (Fig. 4b), indicating that direct cell-cell interaction instead of MSC-secreted soluble factors is essential for the functional reprogramming. Consistent with the observation in vitro, the expression of Arg1 was also significantly increased in BM macrophages directly isolated from MSC-treated leukemia-bearing mice (Fig. 4c). Furthermore, intracellular flow cytometry staining confirmed that the ratios of Arg1high macrophage subpopulation significantly increased in E-Mac at the MSC-treated sites (p < 0.01) (Fig. 4d, e). Collectively, these results indicate that the donor MSCs reprogram BM macrophages from leukemia-bearing mice to execute tissue-repair function.
Fig. 4. Characterization of MSC-reprogrammed BM resident macrophages isolated from leukemia-bearing mice.
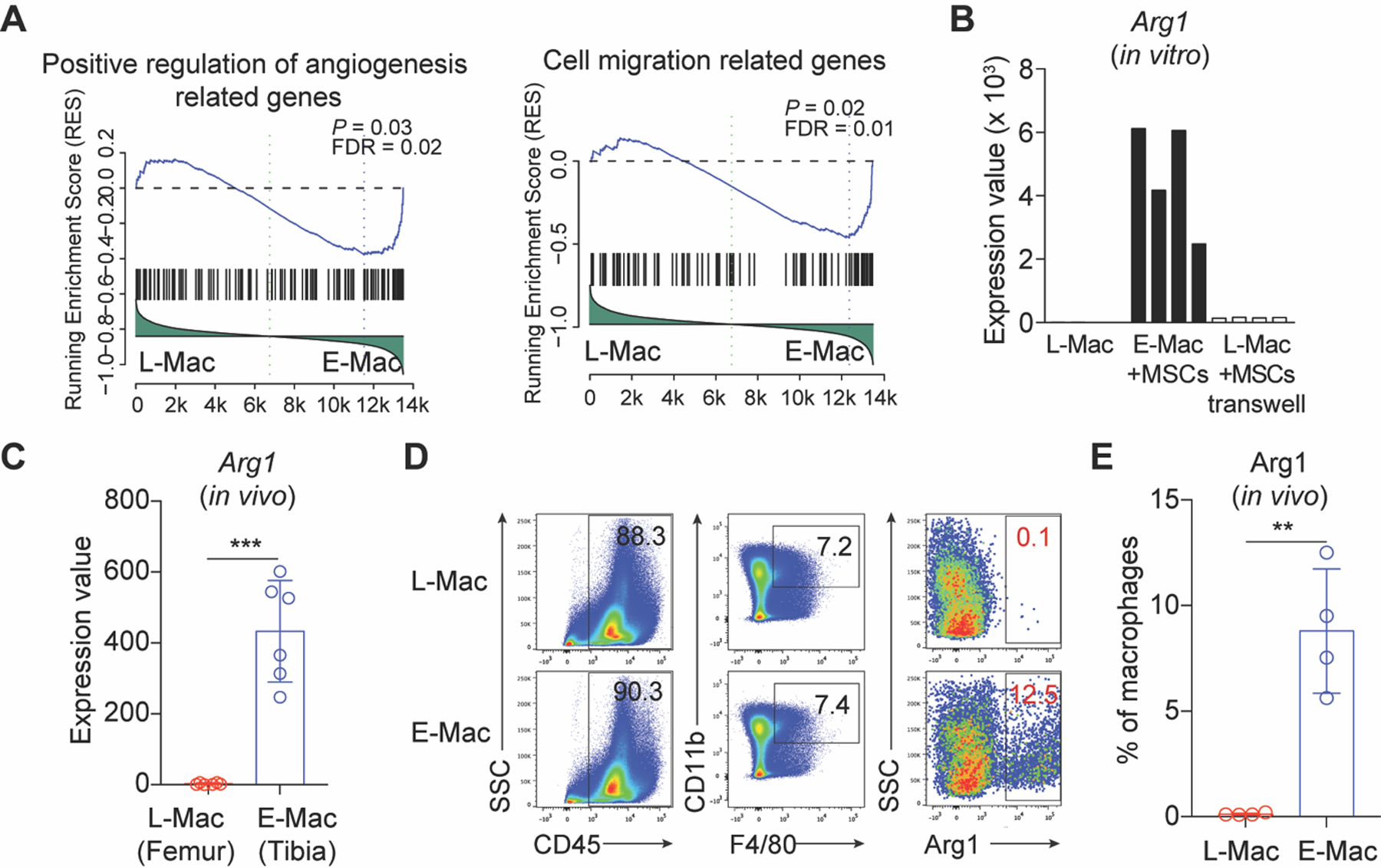
a Gene set enrichment analysis (GSEA) of the positive regulation of angiogenesis and cell migration-related genes in L-Mac and E-Mac. L-Mac indicates leukemic macrophages. E-Mac indicates MSC-reprogrammed leukemic macrophages, which were co-cultured with MSCs in vitro for 12 h. b RNA-Seq analysis of Arg1 in L-Mac and E-Mac in vitro. c RNA-Seq analysis of Arg1 in leukemic macrophages sorted from MSC-treated leukemic mice in vivo. d Representative intracellular staining plots of Arg1 proteins in L-Mac and E-Mac. e Statistical analysis of the ratios of Arg1high macrophage subpopulation in L-Mac and E-Mac (mean ± SD, n = 4). Asterisks indicate **p < 0.01, ***p < 0.001 (unpaired student’s t-test (two-tailed)).
The E-Mac treatment largely recapitulates the therapeutic effects of MSC treatment
Given the short lifespan of donor MSCs in vivo [8], we speculated that MSCs mediate the restoration of the BM microenvironment of leukemia-bearing mice by reprogramming macrophages. We isolated macrophages from leukemia-bearing mice and co-cultured them with healthy MSCs for 12h, and then transplanted these E-Mac back into leukemia-bearing mice by intra-BM transfusion (Fig. 5a). We indeed found that the thrombopoiesis was significantly improved (> 6 folds) after E-Mac treatment (p < 0.001) (Fig. 5b, c). Host MSCs were also significantly increased (> 3 folds) in E-Mac-treated leukemia-bearing mice (p < 0.001) (Fig. 5d, e). Collectively, these results demonstrate that MSC-reprogrammed macrophages largely recapitulate the therapeutic effects of MSCs.
Fig. 5. Intra-BM transfusion of MSC-reprogrammed macrophages largely rescues the therapeutic effects of MSC-treatment in leukemic mice.

a Schematic diagram of MSC-reprogrammed macrophages transfusion strategy. b Representative dot plots of platelet populations and quantitative gating. c Kinetic analysis of platelets in PB of leukemia-bearing mice treated with PBS or E-Mac (n = 6). d Flow cytometry analysis of MSCs in leukemia-bearing mice post PBS/E-Mac treatment. e Statistical analysis of the absolute numbers of host MSCs in tibias from PBS/E-Mac treated leukemic mice (mean ± SD, n = 6). Asterisk indicates ***p < 0.001 (unpaired student’s t-test (two-tailed)).
Discussion
Deteriorating BM microenvironment accompanies chronic leukemia progression. Here we unravel a de novo approach of reverting the impaired BM microenvironment by intra-BM injection of donor MSCs. Upon injection, the donor MSCs quickly reprogrammed local host BM macrophages to repair the niche, thus improving normal hematopoiesis and suppressing leukemia development. Our studies reveal de novo mechanisms underlying MSC-mediated local BM microenvironment restoration that systemically suppress leukemia development.
Given the short-term lifespan of the exogenous MSCs in vivo, it is surprising that local injection of donor MSCs results in long-term improvement of thrombopoiesis and reduction of tumor burden. Following injection, exogenous donor MSCs immediately reprogram host resident macrophages that further organize the overhaul of local BM microenvironment, including restoring the functions of host MSCs. There is a lot of evidence supporting the pivotal roles of macrophages in tissue repair [27, 28]. Donor MSCs can transiently release a key wave of tissue-repair factors, such as CCL7 [29] and CXCL12 [30], and reprogram host macrophages, subsequently resulting in the recovery of host MSCs. Recovered host MSCs further secreted much higher level of CCL7 and CXCL12 that can further facilitate BM niche repair. Donor MSCs could also directly modulate the other niche cells, in addition to macrophages, to participate in BM niche repair [3]. Consequently, the restored local BM microenvironment outputs abundant hematopoiesis-improving cytokines. Thus, despite the short life-span, donor MSCs provide long-term thrombopoiesis improvement and tumor burden reduction through the stepwise microenvironment restoration.
Of note, reprogramming host macrophages by MSCs is required for MSC-mediated microenvironment restoration and leukemia inhibition. The molecular features involved in angiogenesis and cell migration signaling demonstrated by MSC-reprogrammed macrophages suggest that the impaired bone marrow macrophages might be therapeutic targets for improving normal hematopoiesis and suppressing leukemia in MDS/MPN patients with NRAS mutations.
In addition, many studies have reported that MSCs-derived extracellular vesicles are being examined for their biological effects in MSC-based cellular therapy. For example, in models of kidney regeneration, biological effect of systemic delivery of MSCs could be replaced by systemic infusion of microparticles/exosomes isolated from MSCs [31, 32]. Thus, to further investigate the potential mechanisms, the therapeutic effects of systemic transfusion of MSCs-derived microparticles/exosomes will be tested in the future.
MSC treatment inhibits leukemia development in the Nras mutation-induced MPN/MDS-like disease model. We also attempted to broaden MSC treatment for acute leukemia in the MLL-AF9-initiated model (Supplementary Fig. S6A), in which impaired MSCs results in the reduction of osteogenesis and CXCL12 production [33]. Despite a mild elevation of platelet level, the intra-BM transfusion of donor MSCs failed to significantly improve normal hematopoiesis or suppress acute leukemia development (Supplementary Fig. S6B–H). Therefore, the intra-BM MSC treatment might be beneficial for MPN/MDS leukemias, such as JMML and CMML, but insufficient for suppressing acute leukemia.
In conclusion, we establish a novel approach of reverting the deteriorated bone marrow microenvironment by intra-BM transfusion of healthy MSCs under leukemia condition. This study also unveils de novo mechanisms underlying MSC-mediated local BM microenvironment restoration that systemically suppress leukemia development.
Supplementary Material
Acknowledgments
This work was supported by grants from the National Key R&D Program of China (2019YFA0110200), the Strategic Priority Research Program of Chinese Academy of Sciences (XDA16010601), the Chinese Ministry of Science and Technology (2015CB964401, 2016YFA0100601, 2016YFA0100600, 2017YFA0103401, and 2015CB964902), the Major Research and Development Project of Guangzhou Regenerative Medicine and Health Guangdong Laboratory (2018GZR110104006, 2018GZR0201008), the CAS Key Research Program of Frontier Sciences (QYZDB-SSW-SMC057), the Health and Medical Care Collaborative Innovation Program of Guangzhou Scientific and Technology (201803040017), CAMS Innovation Fund for Medical Sciences (2016-12M-1-002), the General Program from Guangzhou Scientific and Technological Project (201707010157), the Science and Technology Planning Project of Guangdong Province (2017B030314056, 2017B020230004), the grants from the National Natural Science Foundation of China (Grant No 81925002, 81970099, 31471117, 31271457, 81470281, 81421002, 81730006, 31600948, and 81861148029), the CAMS Initiative for Innovative Medicine (2016-I2M-1-017) and the grants from NIH, USA (AI079087, D.W. and HL130724 D.W.)
Footnotes
Conflict of Interests The authors declare no competing financial interests.
References
- 1.Ren G, Zhang L, Zhao X, Xu G, Zhang Y, Roberts AI, et al. Mesenchymal stem cell-mediated immunosuppression occurs via concerted action of chemokines and nitric oxide. Cell Stem Cell 2008. February 7; 2(2): 141–150. [DOI] [PubMed] [Google Scholar]
- 2.Prockop DJ. Inflammation, fibrosis, and modulation of the process by mesenchymal stem/stromal cells. Matrix Biol 2016. April; 51: 7–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shi Y, Wang Y, Li Q, Liu K, Hou J, Shao C, et al. Immunoregulatory mechanisms of mesenchymal stem and stromal cells in inflammatory diseases. Nat Rev Nephrol 2018. August; 14(8): 493–507. [DOI] [PubMed] [Google Scholar]
- 4.Le Blanc K, Mougiakakos D. Multipotent mesenchymal stromal cells and the innate immune system. Nat Rev Immunol 2012. April 25; 12(5): 383–396. [DOI] [PubMed] [Google Scholar]
- 5.Mittal M, Tiruppathi C, Nepal S, Zhao YY, Grzych D, Soni D, et al. TNFalpha-stimulated gene-6 (TSG6) activates macrophage phenotype transition to prevent inflammatory lung injury. Proc Natl Acad Sci U S A 2016. December 13; 113(50): E8151–E8158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang G, Cao K, Liu K, Xue Y, Roberts AI, Li F, et al. Kynurenic acid, an IDO metabolite, controls TSG-6-mediated immunosuppression of human mesenchymal stem cells. Cell Death Differ 2018. July; 25(7): 1209–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Du L, Lin L, Li Q, Liu K, Huang Y, Wang X, et al. IGF-2 Preprograms Maturing Macrophages to Acquire Oxidative Phosphorylation-Dependent Anti-inflammatory Properties. Cell Metab 2019. February 4. [DOI] [PubMed] [Google Scholar]
- 8.Eggenhofer E, Benseler V, Kroemer A, Popp FC, Geissler EK, Schlitt HJ, et al. Mesenchymal stem cells are short-lived and do not migrate beyond the lungs after intravenous infusion. Front Immunol 2012; 3: 297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ehninger A, Trumpp A. The bone marrow stem cell niche grows up: mesenchymal stem cells and macrophages move in. J Exp Med 2011. March 14; 208(3): 421–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen J, Yao Y, Gong C, Yu F, Su S, Chen J, et al. CCL18 from tumor-associated macrophages promotes breast cancer metastasis via PITPNM3. Cancer Cell 2011. April 12; 19(4): 541–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gubin MM, Esaulova E, Ward JP, Malkova ON, Runci D, Wong P, et al. High-Dimensional Analysis Delineates Myeloid and Lymphoid Compartment Remodeling during Successful Immune-Checkpoint Cancer Therapy. Cell 2018. November 1; 175(4): 1014–1030 e1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen CC, Wang L, Plikus MV, Jiang TX, Murray PJ, Ramos R, et al. Organ-level quorum sensing directs regeneration in hair stem cell populations. Cell 2015. April 9; 161(2): 277–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu C, Wu C, Yang Q, Gao J, Li L, Yang D, et al. Macrophages Mediate the Repair of Brain Vascular Rupture through Direct Physical Adhesion and Mechanical Traction. Immunity 2016. May 17; 44(5): 1162–1176. [DOI] [PubMed] [Google Scholar]
- 14.Winkler IG, Sims NA, Pettit AR, Barbier V, Nowlan B, Helwani F, et al. Bone marrow macrophages maintain hematopoietic stem cell (HSC) niches and their depletion mobilizes HSCs. Blood 2010. December 2; 116(23): 4815–4828. [DOI] [PubMed] [Google Scholar]
- 15.Bosurgi L, Cao YG, Cabeza-Cabrerizo M, Tucci A, Hughes LD, Kong Y, et al. Macrophage function in tissue repair and remodeling requires IL-4 or IL-13 with apoptotic cells. Science 2017. June 9; 356(6342): 1072–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cho DI, Kim MR, Jeong HY, Jeong HC, Jeong MH, Yoon SH, et al. Mesenchymal stem cells reciprocally regulate the M1/M2 balance in mouse bone marrow-derived macrophages. Exp Mol Med 2014. January 10; 46: e70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nemeth K, Leelahavanichkul A, Yuen PS, Mayer B, Parmelee A, Doi K, et al. Bone marrow stromal cells attenuate sepsis via prostaglandin E(2)-dependent reprogramming of host macrophages to increase their interleukin-10 production. Nat Med 2009. January; 15(1): 42–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang J, Liu Y, Li Z, Du J, Ryu MJ, Taylor PR, et al. Endogenous oncogenic Nras mutation promotes aberrant GM-CSF signaling in granulocytic/monocytic precursors in a murine model of chronic myelomonocytic leukemia. Blood 2010. December 23; 116(26): 5991–6002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang J, Liu Y, Li Z, Wang Z, Tan LX, Ryu MJ, et al. Endogenous oncogenic Nras mutation initiates hematopoietic malignancies in a dose- and cell type-dependent manner. Blood 2011. July 14; 118(2): 368–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang JY, Kong GY, Liu YG, Du J, Chang YI, Tey SR, et al. Nras(G12D/+) promotes leukemogenesis by aberrantly regulating hematopoietic stem cell functions. Blood 2013. June 27; 121(26): 5203–5207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li Q, Haigis KM, McDaniel A, Harding-Theobald E, Kogan SC, Akagi K, et al. Hematopoiesis and leukemogenesis in mice expressing oncogenic NrasG12D from the endogenous locus. Blood 2011. February 10; 117(6): 2022–2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lim M, Pang Y, Ma S, Hao S, Shi H, Zheng Y, et al. Altered mesenchymal niche cells impede generation of normal hematopoietic progenitor cells in leukemic bone marrow. Leukemia 2016. January; 30(1): 154–162. [DOI] [PubMed] [Google Scholar]
- 23.Zhu H, Guo ZK, Jiang XX, Li H, Wang XY, Yao HY, et al. A protocol for isolation and culture of mesenchymal stem cells from mouse compact bone. Nat Protoc 2010. March; 5(3): 550–560. [DOI] [PubMed] [Google Scholar]
- 24.Rombouts WJ, Ploemacher RE. Primary murine MSC show highly efficient homing to the bone marrow but lose homing ability following culture. Leukemia 2003. January; 17(1): 160–170. [DOI] [PubMed] [Google Scholar]
- 25.Medina RJ, O’Neill CL, O’Doherty TM, Knott H, Guduric-Fuchs J, Gardiner TA, et al. Myeloid angiogenic cells act as alternative M2 macrophages and modulate angiogenesis through interleukin-8. Mol Med 2011. Sep-Oct; 17(9–10): 1045–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ohnishi H, Kobayashi H, Okazawa H, Ohe Y, Tomizawa K, Sato R, et al. Ectodomain shedding of SHPS-1 and its role in regulation of cell migration. J Biol Chem 2004. July 2; 279(27): 27878–27887. [DOI] [PubMed] [Google Scholar]
- 27.Wynn TA, Vannella KM. Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity 2016. March 15; 44(3): 450–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu C, Wu CA, Yang QF, Gao J, Li L, Yang DQ, et al. Macrophages Mediate the Repair of Brain Vascular Rupture through Direct Physical Adhesion and Mechanical Traction. Immunity 2016. May 17; 44(5): 1162–1176. [DOI] [PubMed] [Google Scholar]
- 29.Schenk S, Mal N, Finan A, Zhang M, Kiedrowski M, Popovic Z, et al. Monocyte chemotactic protein-3 is a myocardial mesenchymal stem cell homing factor. Stem Cells 2007. January; 25(1): 245–251. [DOI] [PubMed] [Google Scholar]
- 30.Kato T, Khanh VC, Sato K, Takeuchi K, Carolina E, Yamashita T, et al. SDF-1 improves wound healing ability of glucocorticoid-treated adipose tissue-derived mesenchymal stem cells. Biochem Biophys Res Commun 2017. November 18; 493(2): 1010–1017. [DOI] [PubMed] [Google Scholar]
- 31.Bruno S, Grange C, Deregibus MC, Calogero RA, Saviozzi S, Collino F, et al. Mesenchymal stem cell-derived microvesicles protect against acute tubular injury. J Am Soc Nephrol 2009. May; 20(5): 1053–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhou Y, Xu H, Xu W, Wang B, Wu H, Tao Y, et al. Exosomes released by human umbilical cord mesenchymal stem cells protect against cisplatin-induced renal oxidative stress and apoptosis in vivo and in vitro. Stem Cell Res Ther 2013. April 25; 4(2): 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hanoun M, Zhang D, Mizoguchi T, Pinho S, Pierce H, Kunisaki Y, et al. Acute myelogenous leukemia-induced sympathetic neuropathy promotes malignancy in an altered hematopoietic stem cell niche. Cell Stem Cell 2014. September 4; 15(3): 365–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.