

Abstract
Predicting compound–protein affinity is beneficial for accelerating drug discovery. Doing so without the often-unavailable structure data is gaining interest. However, recent progress in structure-free affinity prediction, made by machine learning, focuses on accuracy but leaves much to be desired for interpretability. Defining intermolecular contacts underlying affinities as a vehicle for interpretability; our large-scale interpretability assessment finds previously used attention mechanisms inadequate. We thus formulate a hierarchical multiobjective learning problem, where predicted contacts form the basis for predicted affinities. We solve the problem by embedding protein sequences (by hierarchical recurrent neural networks) and compound graphs (by graph neural networks) with joint attentions between protein residues and compound atoms. We further introduce three methodological advances to enhance interpretability: (1) structure-aware regularization of attentions using protein sequence-predicted solvent exposure and residue–residue contact maps; (2) supervision of attentions using known intermolecular contacts in training data; and (3) an intrinsically explainable architecture where atomic-level contacts or “relations” lead to molecular-level affinity prediction. The first two and all three advances result in DeepAffinity+ and DeepRelations, respectively. Our methods show generalizability in affinity prediction for molecules that are new and dissimilar to training examples. Moreover, they show superior interpretability compared to state-of-the-art interpretable methods: with similar or better affinity prediction, they boost the AUPRC of contact prediction by around 33-, 35-, 10-, and 9-fold for the default test, new-compound, new-protein, and both-new sets, respectively. We further demonstrate their potential utilities in contact-assisted docking, structure-free binding site prediction, and structure−activity relationship studies without docking. Our study represents the first model development and systematic model assessment dedicated to interpretable machine learning for structure-free compound–protein affinity prediction.
Graphical Abstract
INTRODUCTION
Current drug–target interactions are predominantly represented by the interactions between small-molecule compounds as drugs and proteins as targets.1 The enormous chemical space to screen compounds is estimated to contain 1060 drug-like compounds.2 These compounds act in biological systems of millions or more protein species or “proteoforms” (considering genetic mutations, alternative splicing, and post-translation modifications of proteins).3,4 Facing such a combinatorial explosion of compound–protein pairs, drug discovery calls for efficient characterization of compound efficacy and toxicity, and computational prediction of compound–protein interactions (CPI) addresses the need.
Classical physics-driven methods model atomic-level energetics using cocrystallized or docked 3D structures of compound–protein pairs,5,6 such as molecular mechanical and quantum mechanical force fields, potentials of mean force, and empirical and statistical scoring. Over the years of development, these methods are increasingly accurate7–9 for applications including quantitative structure–activity relationship (QSAR). Moreover, their affinity predictions are intrinsically interpretable toward revealing mechanistic principles, with the consideration of atomic contacts, dynamics, and energetics, as well as solvent effects. Recently, thanks to increasingly abundant molecular data and advanced computing power, data-driven machine learning (especially deep learning) methods are also developed using the input structures of compound–protein complexes10–12 or proteins alone (see a related task of classifying binding13,14), albeit with less focus on interpretability. However, these structure-based methods, physics- or data-driven, are limited by the availability of structure data. Indeed, 3D structures are often not available for compound–protein pairs or even proteins alone and their prediction through docking is still a computationally demanding and challenging task.
To overcome the data limitation of structure-based affinity-prediction methods and broaden the applicability to more chemical–proteomic pairs without structures, our focus of the study is structure-free prediction of compound–protein affinities. Recent developments only use identities of compounds (SMILES15,16 or graphs16,17) and proteins (amino acid sequences15,17 or shorter, predicted structural property sequences16) as inputs. Compared to these recent work, our goals are two folds: improved generalizability to “new” molecules unseen in training data as well as improved interpretability to a level that data supports (not yet the level of mechanical principles that can be revealed by physics-driven structure-based methods). In particular, interpretability remains a major gap between the capability of current structure-free machine-learning models and the demand for rational drug discovery. The central question about interpretability is whether and how methods (including machine learning models) could explain why they make certain predictions (affinity level for any compound–protein pair in our context). This important topic is rarely addressed in structure-free machine learning models. DeepAffinity16 has embedded joint attentions over compound–protein component pairs and uses such joint attentions to assess origins of affinities (binding sites) or specificities. Additionally, attention mechanisms have been used for predictions of CPI,18 chemical stability,19 and protein secondary structures.20 Assessment of interpretability for all these studies was either lacking or limited to a few case studies. We note a recent work proposing post-hoc attribution-based test to determine whether a model learns binding mechanisms.21
We raise reasonable concerns on how much attention mechanisms can reproduce native contacts in compound–protein interactions. Attention mechanisms were originally developed to boost the performance of seq2seq models for neural machine translations.22 They have gained popularity for interpreting deep learning models in visual question answering,23 natural language processing,24 and healthcare.25 However, they were also found to work differently from human attentions in visual question answering.26
Representing the first effort dedicated to the interpretability of structure-free compound–protein affinity predictors (in particular, deep-learning models), our study is focused on how to define, assess, and enhance interpretability for these methods as follows.
How to Define Interpretability for Affinity Prediction.
Interpretable machine learning is increasingly becoming a necessity27 for fields beyond drug discovery. Unlike interpretability in a generic case,27 what interpretability actually means and how it should be evaluated is much less ambiguous for compound–protein affinity prediction. To make explanations conform with scientific knowledge, human understanding, and drug-discovery needs; we define interpretability of affinity prediction as to the ability to explain predicted affinity through underlying atomic interactions (or contacts). Specifically, atomic contacts of various types are known to constitute the physical basis of intermolecular interactions,28 modeled in force fields to estimate interaction energies,6 needed to explain mechanisms of actions for drugs,29,30 and relied upon to guide structure–activity research in drug discovery.31,32 Therefore, we use the ability to replicate such corresponding contacts while predicting affinities as a vehicle for interpretability. The current definition of interpretability (residue-atom pairs in contact) is primitive compared to mechanistic principles in structure-based classical methods. However, it is expected to serve as a vehicle to help fill the mechanistic void in structure-free affinity predictors (especially deep-learning models). We emphasize that simultaneous prediction of affinity and contacts does not necessarily make the affinity predictors intrinsically interpretable unless predicted contacts form the basis for predicted affinities.
How to Assess Interpretability for Affinity Prediction.
Once interpretability of affinity predictors is defined first through atomic contacts, it can be readily assessed against ground truth known in compound–protein structures, which overcomes the barrier for interpretable machine learning without ground truth.33 In our study, we have curated a data set of compound–protein pairs, all of which are labeled with pKd/pKi values and contact details, and we have split them into training, test, new-compound, new-protein, and both-new sets. We measure the accuracy of contact prediction over various sets using area under the precision–recall curve (AUPRC) which is suitable for binary classification (contacts/noncontacts) with highly imbalanced classes (far fewer contacts than noncontacts). We have performed large-scale assessments of attention mechanisms in various molecular data representations (protein amino-acid sequences and structure–property annotated sequences16 as well as compound SMILES and graphs) and corresponding neural network architectures (convolutional and recurrent neural networks [CNN and RNN] as well as graph convolutional and isomorphism networks [GCN and GIN]). We have found that current attention mechanisms inadequate for interpretable affinity prediction, as their AUPRCs, were merely slightly more than chance (0.004).
How to Enhance Interpretability for Affinity Prediction.
We have made three main contributions to enhance interpretability for structure-free deep-learning models.
The first contribution is to incorporate physical constraints into data representations, model architectures, and model training. (1) To respect the sequence nature of protein inputs and to overcome the computational bottlenecks of RNNs, inspired by protein folding principles, we represent protein sequences as hierarchical k-mers and model them with hierarchical attention networks (HANs). (2) To respect the structural contexts of proteins, we predict from protein sequence solvent exposure over residues and contact maps over residue pairs, and we introduce novel structure-aware regularizations for structured sparsity of model attentions.
The second contribution is to supervise attentions with native intermolecular contacts available to training data and to accordingly teach models how to pay attention to pairs of compound atoms and protein residues while making affinity predictions. We have formulated a hierarchical multiobjective optimization problem where contact predictions form the basis for affinity prediction. We utilize contact data available to training compound–protein pairs and design hierarchical training strategies accordingly.
The last contribution is to design intrinsic explainability into the architecture of a deep “relational” network. Inspired by physics, we explicitly model and learn various types of atomic interactions (or “relations”) through deep neural networks with joint attentions embedded. This was motivated by relational neural networks first introduced to learn to reason in computer vision34,35 and subsequent interaction networks to learn the relations and interactions of complex objects and their dynamics.36,37 Moreover, we combine such deep relational modules in a hierarchy to progressively focus attention from putative protein surfaces, binding-site k-mers and residues, to putative residue-atom binding pairs.
The rest of the paper is organized as follows. The aforementioned contributions in defining, measuring, and enhancing interpretable affinity prediction will be detailed in the Methods section. In the Results section, we first show over established affinity-benchmark data sets that the original DeepAffinity16 and its variants (with various molecular representations and neural networks) have comparable or better accuracy in affinity prediction, compared to current noninterpretable structure-free methods. We then describe a data set newly curated for both affinity and contact prediction. The data set is designed to be diverse and challenging with the generalizability test in mind. Using this data set, we incrementally introduce the three contributions to DeepAffinity and compare the resulting DeepAffinity+ (using the first two contributions) and DeepRelations (using all three contributions) to a competing interpretable method. Both methods produce remarkably improved interpretability (now defined as accuracy of contacts predicted by joint attentions) while maintaining accurate and generalizable affinity prediction. Importantly, compared to the competing method and their reduced version without supervising attentions, they show that sufficiently better interpretability (much more accurate contact predictions) can help improve accuracy in affinity prediction. Lastly, we use various focused studies to show the spatial patterns of top-10 predicted contacts, the benefit of these predictions to contact-assisted protein–ligand docking, and the additional utilities of aggregating attentions and decomposing predicted affinities for binding site prediction and QSAR.
METHODS
Toward genome-wide prediction of compound–protein interactions (CPI), we assume that proteins are only available in 1D amino-acid sequences, whereas compounds are available in 1D SMILES or 2D chemical graphs. We start the section with the curation of a data set of compound–protein pairs with known pKd/pKi values, which is also of known intermolecular contacts. We will introduce the state-of-the-art and our newly-adopted neural networks to predict from such molecular data. These neural networks will be first adopted in our previous framework of DeepAffinity16 (supervised learning with joint attention) so that the interpretability of attention mechanisms can be systematically assessed in CPI prediction. We will then describe our physics-inspired, intrinsically explainable architecture of deep relational networks where aforementioned neural networks are used as basis models. With carefully designed regularization terms, we will explain multistage deep relational networks that increasingly focus attention on putative binding-site k-mers, binding-site residues, and residue-atom interactions, for the prediction and interpretation of compound–protein affinity. We will also explain how the resulting model can be trained strategically.
Benchmark Set with Compound–Protein Affinities and Contacts.
We have previously curated affinity-labeled compound–protein pairs16 based on BindingDB.38 In this study, we used those pKi/pKd-labeled data with amino-acid sequence length no more than 1000 and curated a subset with known complex-protein cocrystal structures. We further merge the data with the refined set of PDBbind (v. 2019),39 leading to 4446 pairs between 3672 compounds and 1287 proteins. More details about procedures are provided in the Supporting Information, Section 1.1. Resulting data characteristics, including compound property distributions and protein class statistics, are described in the Results section.
The compound data are in the format of canonical SMILES as provided in PubChem,40 and the protein data are in the format of FASTA sequences (UniProt canonical). Compound SMILES were also converted to graphs with RDKit.41 Ionization states of compounds defined in PubChem were validated using the software OpenBabel, and the compounds were further sanitized and standardized using “chem.Sanitize-Mol( )” in the software RDKit. More details are provided in the Supporting Information, Section 1.2. Atomic-level intermolecular contacts (or “relations”) were derived from compound–protein cocrystal structures in PDB,42 as ground truth for the interpretablity of affinity prediction. Specifically, we cross-referenced aforementioned compound–protein pairs in PDBsum43 and used its LigPlot service to collect high-resolution atomic contacts or relations. These direct, first-shell contacts are given in the form of contact types (hydrogen bond or hydrophobic contact), atomic pairs, and atomic distances.
The data set was randomly split into fourfolds where fold 1 did not overlap with fold 2 in compounds, did not do so with fold 3 in proteins and with fold 4 in either compounds or proteins. Folds 2, 3, and 4 are referred to as new-compound, new-protein, and both-new sets for generalizability tests, and they contain 521, 795, and 205 pairs, respectively. Fold 1 was randomly split into training (2334) and test (591) sets. More procedural details about data splitting are summarized in the Supporting Information, Algorithm 1. The split of the whole data set is illustrated in Figure 1 below. The similarity profiles between training molecules and those in the test and generalization sets are analyzed in the Results section later.
Figure 1.
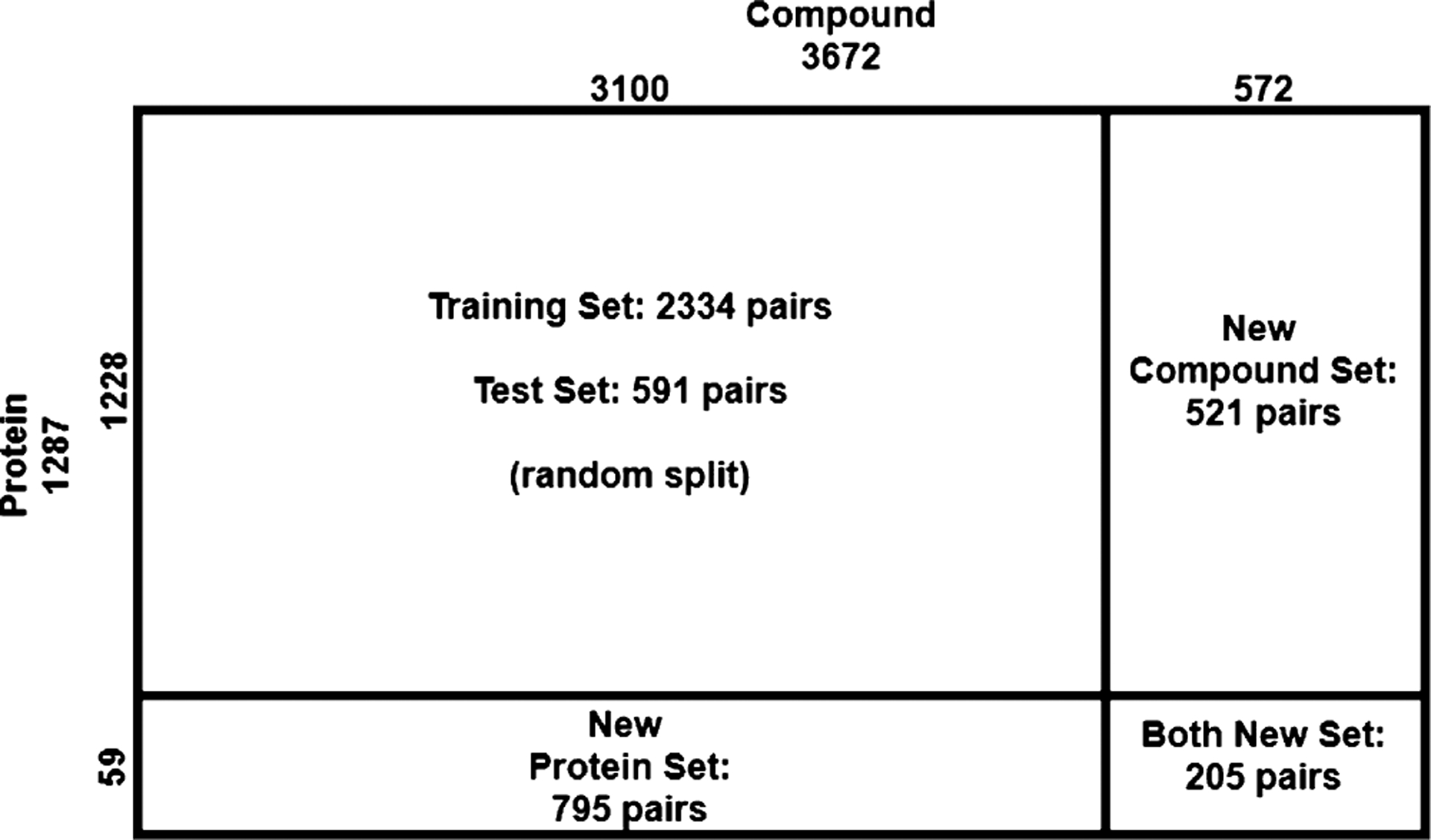
Complete data set consists of training, test, compound-unique, protein-unique, and double unique sets with compound–protein counts provided.
Although monomer structures of proteins are often unavailable, their structural features can be predicted from protein sequences alone with reasonable accuracy. We have predicted the secondary structure and solvent accessibility of each residue using the latest SCRATCH44,45 and contact maps for residue pairs using RaptorX-contact46 (see details in the Supporting Information, Section 1.3). These data provide additional structural information to regularize our machine learning models. If protein structures are available, actual rather than predicted, such data can be used instead.
Data Representation and Corresponding Basis Neural Networks.
Baseline: CNN and RNN for 1D Protein and Compound Sequences.
When molecular data are given in 1D sequences, these inputs are often processed by convolutional neural networks (CNN)15,47 and by recurrent neural networks (RNN) that are more suitable for sequence data with long–term interactions.16
Challenges remain in RNN for compound strings or protein sequences. For compounds in SMILES strings, the descriptive power of such strings can be limited. In this study, we overcome the challenge by representing compounds in chemical formulae (2D graphs) and using two types of graph neural networks (GNN). For proteins in amino-acid sequences, the often-large lengths demand deep RNNs that are hard to be trained effectively (gradient vanishing or exploding and nonparallel training).48 We previously overcame the second challenge by predicting structure properties from amino-acid sequences and representing proteins as a much shorter structure property sequences where each four-letter tuple corresponds to a secondary structure.16 This treatment however limits the resolution of interpretability to be at the level of protein secondary structures (multiple neighboring residues) rather than individual residues. In this study, we overcome the second challenge while achieving residue-level interpretability by using biologically motivated hierarchical RNN (HRNN).
Notation Summary.
Scalars, vectors, and matrices are denoted in normal lowercase, bold-faced lowercase, and uppercase characters, respectively. Subscripts i, t, and j are for the ith protein residue, tth protein k-mer, and jth compound atom, respectively. Subscript it represents the ith residue in the tth k-mer (where i can be regarded as a global residue index). Therefore, the jth atom of compound described in dg features is denoted xj and its learned representation (embedded through GNN) is denoted zj. The ith residue of protein with dp features is denoted by yi, and its learned representation (embedded through HRNN) is denoted by hit, where t is the index of the k-mer containing residue i. These residue representations hit within the k-mer are then aggregated to obtain the k-mer representation ht, and all k-mer representations are concatenated to reach the protein representation.
Superscripts r, (l), and [s] indicate the rth relation about molecular features, the lth layer of graph neural networks, and the sth stage of DeepRelations, respectively.
Proposed: GCN and GIN for 2D Compound Graphs.
Compared to 1D SMILES strings, chemical formulae (2D graphs) of compounds have more descriptive power and are increasingly used as inputs to predictive models.16–19,49 In this study, compounds are represented as 2D graphs in which vertices are atoms and edges are covalent bonds between atoms. Suppose that n is the maximum number of atoms in our compound set (compounds with smaller number of atoms are padded to reach size n), let us consider a graph , where is the set of n vertices (each with dg features), that of vertex features , that of edges, and is unweighted symmetric adjacency matrix. Let and be the degree matrix (the diagonals of ).
We used the graph convolutional network (GCN)50 and graph isomorphism network (GIN)51 which are the state-of-the-art for graph embedding and inference. GCN consists of multiple layers, and at layer l, the model can be written as follows
| (1) |
where is the output, are the trainable parameters, and is the number of features, all at layer l. Initial conditions (when l = 0) are and .
GIN is the most powerful graph neural network in theory: its discriminative or representational power is equal to that of the Weisfeiler–Lehman graph isomorphism test.52 Similar to GCN, GIN consists of multiple layers, and at layer l, the model can be written as a multilayer perceptron (MLP)
| (2) |
where , ϵ(l) can be either a trainable parameter or a fixed hyperparameter. Each GIN layer has several nonlinear layers compared to GCN layer with just a ReLU per layer, which might improve predictions but suffer in interpretability.
The final representation for a compound is if GCN or GIN has L layers. In this study, vertex features are as in ref 19, with few additional features detailed later in physics-inspired relational modules. A summary of these features is provided in the Supporting Information, Table S2.
Proposed: HRNN for 1D Protein Sequences.
We aim to keep the use of RNN that respects the sequence nature of protein data and mitigate the difficulty of training RNN for long sequences. To that end, inspired by the hierarchy of protein structures, we model protein sequences using hierarchical attention networks (HANs). Specifically, during protein folding, sequence segments may fold separately into secondary structures and the secondary structures can then collectively pack into a tertiary structure needed for protein functions. We exploit such hierarchical nature by representing a protein sequence of length easily in thousands as tens or hundreds of k-mers (consecutive sequence segments) of length k (hyperparameter in this study). Accordingly, we process the hierarchical data with hierarchical attention networks (HANs)53 which have been proposed for natural language processing. We also refer to it as hierarchical RNN (HRNN). Although the inter-k-mer attentions might overcome potential issues brought by k-mer definition as they do in natural language processing,53 it would be interesting to examine the potential benefit of using other domain-relevant definition of k-mers, such as (predicted or actual) secondary structure elements.
Given , a protein sequence described with dp features for each residue , we partition it into T consecutive, nonoverlapping k-mers. We use two types of RNNs in hierarchy for modeling within and across k-mers. We first use an embedding layer to represent the ith residue in the tth k-mer as a vector eit. We use a shared RNN for all k-mers for the latent representation of the residue: hit = RNN(eit) (t = 1, …, T). We then summarize each k-mer as kt with an intra-k-mer attention mechanism
| (3) |
With another RNN for kt, we reach the representation of the tth k-mer: ht = RNN(kt) (t = 1, …, T).
The final representation for a protein sequence is the collection of ht.
Joint Attention over Protein–Compound Atomic Pairs for Interpretability.
Once the learned representation of protein sequences (H = [h1, …, ht, …, hT], where t is the index of protein k-mer) and that of compound sequences or graphs , where j is the index of compound atom) are defined, they are processed with a joint k-mer–atom attention mechanism to interpret any downstream prediction
| (4) |
With , the joint attention between the tth k-mer and the jth atom, we can combine it with the intra-k-mer attention over each residue i in the tth k-mer and reach , the joint attention between the ith protein residue and the jth compound atom
| (5) |
This joint attention mechanism is an extension of our previous work,16 where a protein sequence was represented as a single, “flat” RNN rather than multiple, hierarchical RNNs.
Given learned representations hi for protein residue i (the k-mer index is ignored for simplicity) and zj for compound atom j as well as the joint attention over the pair, we further jointly embed the pair and aggregate over all pairs to reach f— the joint embedding of protein , compound , and their residue-atom “interactions” captured by
| (6) |
where Θ3, Θ4, and b2 are learnable parameters. The joint embedding f is fed to a CNN and two multilayer perceptrons (MLP) to make affinity prediction as before.16 In other words, for contact prediction directly forms the basis of f for affinity prediction.
In comparison, Gao et al.’s method18 also uses joint attention for contact prediction. However, the joint attention matrix is marginalized for either the compound or the protein; and the separately processed compound or protein representations were used for affinity prediction. More specifically
| (7) |
The separate final representations for the compound (od) and the protein (op) were fed to downstream layers for affinity prediction, with much of information lost on the joint attention (the basis of contact prediction).
DeepRelations.
Overall Architecture.
We have developed an end-to-end “by-design” interpretable architecture named DeepRelations for joint prediction and interpretation of compound–protein affinity. The overall architecture is shown in Figure 2.
Figure 2.
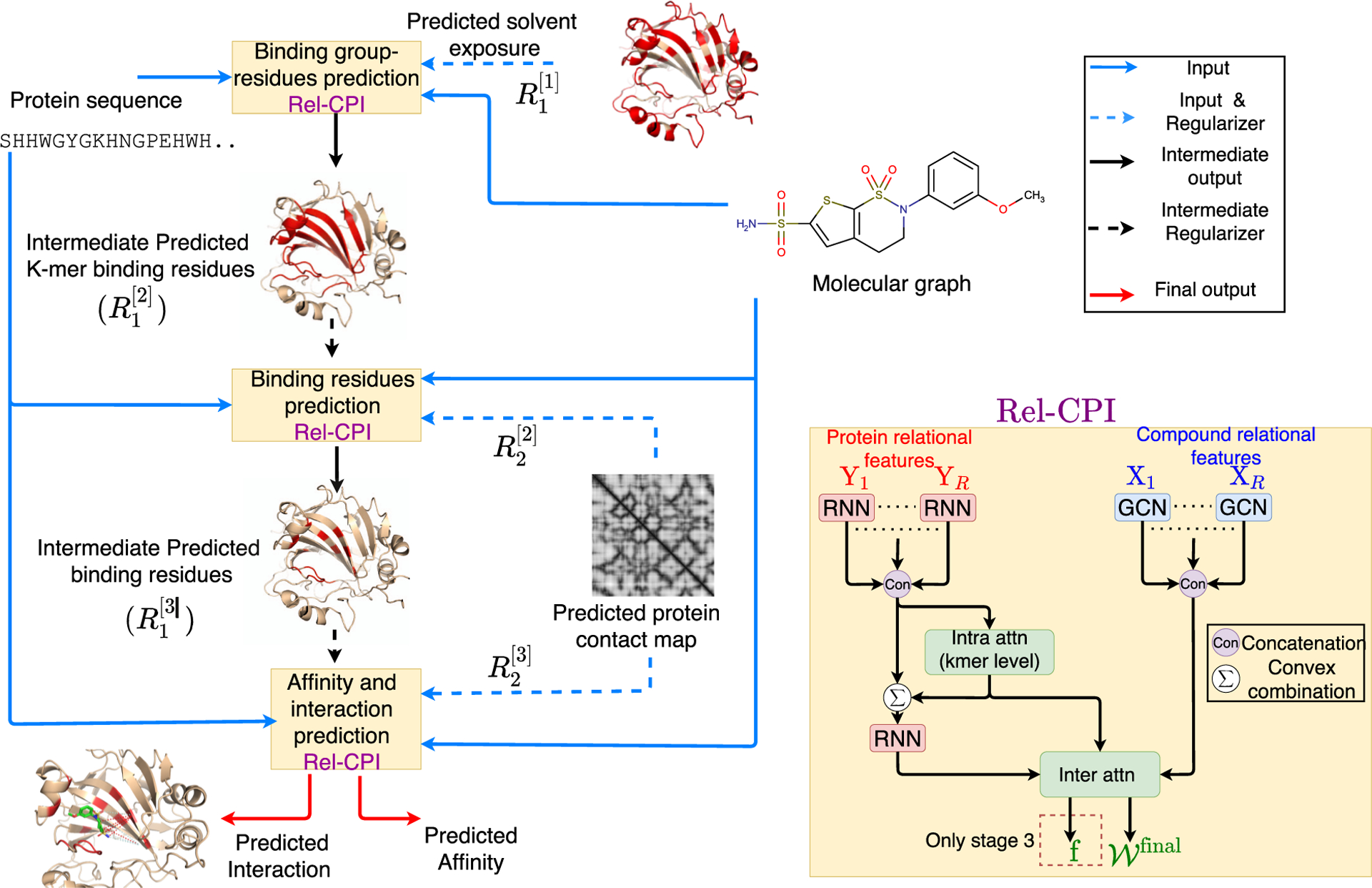
Schematic illustration of DeepRelations, an intrinsically explainable neural network architecture for predicting compound–protein interactions. Three linked relational modules (Rel-CPI in the small yellow boxes) correspond to three stages of attention focusing. Each module embeds relational features with joint attentions over pairs of protein residues and compound atoms (details on the right). In comparison, DeepAffinity+ has a single module with all relational features lumped together. Both methods are structure-free, and protein structures are just for illustration.
There are three relational modules (Rel-CPI) corresponding to three stages. Their attentions are trained to progressively focus on putative binding k-mers, residues, and pairs; and earlier-stage attentions guide those in the next stage through regularization. In each Rel-CPI module, there are K = 10 types of atomic “relational” features for proteins or compounds [nine relation (sub)types are described next, and the last is the union of all nine types of features]. All types of relational features are individually fed to aforementioned neural network pairs (for instance, HRNN for protein sequences and GCN for compound graphs, or HRNN-GCN in short), concatenated, and jointly embedded for proteins and compounds with attentions over residue–compound pairs. The embedding output (based on joint attentions for contact prediction) of the last module is fed to CNN and MLP layers for affinity prediction. All three modules are trained end-to-end as a single model. In contrast, DeepAffinity+ only has one module without multistage focusing, and its module only uses the last type of relational features (the union of the first nine types).
Physics-Inspired Relational Modules.
The relational modules are inspired by physics. Specifically, atomic “relations” or interactions constitute the physical bases and explanations of compound–protein interaction affinities and are often explicitly modelled in force fields. We have considered the following six types of relations with attentions paid on and additional input data defined for.
Electrostatic interactions: the ion feature of a protein residue is its net charge as in the force field CHARMM36 and that of a compound atom is its formal charge. The dipole feature of a protein residue is 1 for polar residues (S, T, C, Y, N, Q, and H54) or 0 for others and that of a compound atom is its Gasteiger partial charge. The electrostatics thus include all four combinations (subtypes) of residue-atom relations: ion–ion, ion–dipole, dipole–ion, and dipole–dipole.
Hydrogen bond: noncovalent interaction (A···H−D) between an electronegative atom as a hydrogen “acceptor” (“A”) and a hydrogen atom that is covalently bonded to an electronegative atom called a hydrogen “donor” (“D”). Therefore, if a protein residue or compound atom could provide a hydrogen acceptor/donor, its hydrogen-bond feature is −1/+1; otherwise, the feature value is 0. A protein residue is allowed to be both hydrogen-bond donor and acceptor. Specifically, for protein residues, amino acids of hydrogen-bond acceptors are N, D, Q, E, H, S, T, and Y and those of hydrogen-bond donors are Y, W, T, S, K, H, Q, N, and R.55 For compound atoms, the hydrogen-bond acceptor or donor is defined as in the base features factory file (atom types “SingleAtomAcceptor” and “SingleAtom-Donor” in the file “BaseFeatures.fdef”) of the software RDKit v. 2018.03.4.
Halogen bond: a halogen bond (A···X−D) is very similar to hydrogen bond except that a halogen “X” (rather than hydrogen) atom (often found in drug compounds) is involved in such interactions. As standard amino acids do not contain halogen atoms, a protein residue can only be a halogen bond acceptor (“A” in A···X−D) and assigned a nonzero halogen-bond feature of −1, only if it is amino acid S, T, Y, D, E, H, C, M, F, W,56 N, or Q. On the compound side, only a halogen atom is assigned a nonzero feature value. Specifically, halogen-bond features of iodine, bromine, chlorine, and fluorine atoms are assigned at +4, +3, +2, and +1, respectively, for decreasing halogen-bonding strengths.56
Hydrophobic interactions: the interactions between hydrophobic protein residues and compound atoms contribute significantly to the binding energy between them. This feature is only nonzero and set at 1 for hydrophobic residues of proteins or nonpolar atoms of compounds (atoms whose absolute values of partial atomic charges are less than 0.2 units57,58).
Aromatic interactions: aromatic rings in histidine, tryptophan, phenylalanine, and tyrosine participate in “stacking” interactions with aromatic moieties of a compound (π−π stacking). Therefore, if a protein residue has an aromatic ring, its aromatic feature is set at 1 and otherwise at 0. Similarly, if a compound atom is part of an aromatic ring, the feature is set at 1 and otherwise at 0.
VdW interactions: van der Waals are weaker interactions compared to others. However, the large amount of these interactions contribute significantly to the overall binding energy between a protein and a compound. We consider the amino-acid type and the atom element as their features and use an embedding layer to derive their continuous representations.
For each (sub)type of atomic relations, corresponding protein and compound features are fed into basis neural network models such as HRNN for protein sequences and GNN for compound graphs. The embeddings over all types are concatenated for protein residues or compound atoms and then jointly embedded with joint attentions over residue-atom pairs.
Physical Constraints as Attention Regularization.
The joint attention matrices in each Rel-CPI module, for individual relations or overall, are regularized with the following two types of physical constraints. We note that, aiming at the general case where protein structures may not be available, we use sequence-predicted rather than actual structure properties (solvent exposure and residue contacts) when introducing these physical constraints.
Focusing regularization in the first regularization, a constraint input is given as a matrix to penalize the attention matrix if it is focused on undesired regions of proteins. In addition, an L1 sparsity regularization is on the attention matrix to promote interpretability as a small portion of protein residues interact with compounds. Therefore, this “focusing” penalty can be formalized as follows
| (8) |
where the term, a parameter, can be considered as soft thresholding, and the matrix norms are element-wise. The L1 regularization term in R1(·) is only included in the first module (stage 1), where R1(·) is the only regularization term. It is then moved to another term in the second, and the last modules, where multiple regularization terms, are used together.
The first regularization is used for all three Rel-CPI modules or stages with increasingly focusing . Let be the constraint matrix and be the learned attention matrix in the sth stage. In the first stage, , being binary, is one only for any residue i predicted to be solvent-exposed (relative solvent-accessible area predicted above 0.25 by SCRATCH44,45) in order to focus on potential surfaces. In the second stage, to focus on putative binding residues hierarchically learned for k-mers and residues at module/stage 1. In the third and last stage, focuses on putative contacts between protein residues and compound atoms based on the learned binding residues at module/stage 2.
Structure-aware sparsity regularization over protein contact maps. We further develop a structure aware sparsity constraints based on known or RaptorX-predicted contact maps of the unbound protein. As sequentially distant residues might be close in 3D and form binding sites for compounds, we define overlapping groups of residues where each group consists of a residue and its spatially close neighboring residues. Just in the second stage, we introduce Group Lasso for spatial groups and the Fused Sparse Group Lasso (FSGL) for sequential groups on the overall, joint attention matrix
| (9) |
The group Lasso penalty will encourage a structured group-level sparsity so that few clusters of spatially close residues share similar attentions within individual clusters. The fused sparsity will encourage local smoothness of the attention matrix so that sequentially close residues share similar attentions with compound atoms. The L1 term again maintains the sparsity of the attention matrix . This regularization is only introduced in the second and third stages for and , after the first-stage attention matrix is supposedly focused on protein surfaces. The attention matrix in the last stage, , is used for predicting residue-atom contacts.
Supervised Attentions.
It has been shown in visual question answering that attention mechanisms in deep learning can differ from human attentions.26 As will be revealed in our results, they do not necessarily focus on actual atomic contacts (relations) in compound-protein interactions either. We have thus curated a relational subset of our compound–protein pairs with affinities, for which known ground-truth atomic contacts or relations are available. We summarize actual contacts of a pair in a matrix of length m × n, which is a binary pairwise interaction matrix padded with 0 to reach the maximum number of protein residues or compound atoms and then normalized by the total number of nonzero entries. We have accordingly introduced an additional third regularization term to supervise attention matrix in the second and third stages
| (10) |
In the case of DeepAffinity+ with a single module, all three regularization terms are included as in the last module of DeepRelations.
Training Strategy for Hierarchical Multiobjectives.
Accuracy and interpretability are the two objectives we pursue at the same time. In our case, the two objectives are hierarchical: compound–protein affinity originates from atomic-level interactions (or “relations”) and better interpretation in the latter potentially contributes to better prediction of the former.
Challenges remain in solving the hierarchical multiobjective optimization problem. Optimizing for both objectives simultaneously (for instance, through weighted sum of them) does not respect that the two objectives do no perfectly align with each other and are of different sensitivities to model parameters. Therefore, we consider the problem as multilabel machine learning, and we design hierarchical training strategies to solve the corresponding hierarchical multiobjective optimization problem, which is detailed next.
Take DeepAffinity+ as an example. We first “pre-trained” it to minimize mean-squared error (MSE) of pKi/pKd regression alone, with physical constraints turned on; in other words, attentions were regularized [through R1(·) and R2(·)] but not supervised in this stage. We tuned combinations of all hyperparameters except λbind in the discrete set of {10−4, 10−3, and 10−2}, with 200 epochs at a learning rate of 0.001. Over the validation set, we recorded the lowest RMSE for affinity prediction and chose the hyperparameter combination with the highest AUPRC for contact prediction such that the corresponding affinity RMSE (root-mean-square error) does not deteriorate from the lowest by more than 10%.
With the optimal values of all hyperparameters but λbind fixed, we then loaded the corresponding optimized model in the first stage and “fine-tuned” the model to minimize MSE additionally regularized by supervised attentions [through R1(·), R2(·), and R3(·)]. We used the same learning rate (0.001) and training epochs (200) in fine-tuning; and we tuned λbind in the set of {100, …, 105} following the same strategy as in pretraining.
The tuned hyperparameters for all DeepAffinity+ variants are summarized as follows. For HRNN-GCN_cstr [modeling protein sequences with HRNN and compound graphs with GCN, regularized by physical constraints in R2(·)], we chose λgroup = 10−4, λfused = 10−3, and λL1-overall = 10−2; and for its supervised version HRNN-GCN_cstr_sup, the additional λbind = 104. For HRNN-GIN_cstr [modeling protein sequences with HRNN and compound graph with GIN, regularized by physical constraints in R2(·)], we chose λgroup = 10−4, λfused = 10−3, and λL1-overall = 10−4; and for its supervised version HRNN-GIN_cstr_sup, the additional λbind = 103. R1(·) was for attentions on individual relations in DeepRelations and not applicable for DeepAffinity+ variants, although a surface-focusing regularization on overall attentions could be introduced.
We did similarly for hyperparameter tuning for Deep-Relations while constraining (and supervising) attentions. The whole DeepRelations model, including the three Rel-CPI modules, is trained end-to-end.59 To save computational resources, we used the same hyperparameters in R2(·) (λL1-overall, λfused, and λgroup) as those optimally tuned in HRNN-GCN_cstr_sup. We then tuned the rest of the hyperparameters (λL1, λrelation, andλbind) following the aforementioned process of pretraining and fine-tuning. In the end, we chose λrelation = 10−4, λL1 = 10−5, λgroup = 10−4, λfused = 10−3, λL1-overall = 10−2, and λbind = 103 for DeepRelations. λbind is usually larger because it is multiplied to the attention-supervision term that can be orders of magnitude smaller than other terms.
RESULTS
We first assess the accuracy of compound–protein affinity predictions made by state-of-the-art noninterpretable methods and our interpretable DeepAffinity framework16 (with new variants), using three established benchmark sets. After establishing that DeepAffinity achieves the state-of-the-art accuracy in affinity prediction, we then describe a newly curated data set with both affinities and contacts of compound–protein interactions and assess the interpretability of various DeepAffinity versions and a competing interpretable method adapted to affinity prediction. We find that current attention-based interpretable models are not adequate for interpreting affinity (i.e., predicting contacts). Thus, we proceed to regularize and supervise attentions in DeepAffinity to make DeepAffinity+ models. We additionally use a novel, physics-inspired, and intrinsically interpretable deep relational architecture to make DeepRelations models.
Over the curated data set, we compare our methods with a competing, structure-free interpretable method in accuracy, generalizability, and interpretability. Using a series of case studies, we also analyze the accuracy levels and spatial patterns of their top-predicted contacts, which are shown to benefit protein–ligand docking. We end the section by introducing analytics to aggregate joint attentions and decompose predicted affinity and by demonstrating their potential utilities toward binding site prediction for proteins and SAR for compounds (scoring and lead optimization).
DeepAffinity with Interpretable Attentions Achieves the State-of-the-Art Accuracy in Compound–Protein Affinity Prediction.
As the starting point of interpretability assessment and improvement, our previous interpretable DeepAffinity framework16 is first compared to current methods based on prediction accuracy for established benchmark sets.
For affinity benchmark data sets, we adopt three established ones of increasing difficulty, the Davis,60 the kinase inhibitor BioActivity (KIBA)61 and the refined set of PDBbind (v. 2019).39 We filtered and partitioned the first two data sets consistently with earlier studies.15,61–63 The Davis data set62 contains all 30,056 Kd-labeled pairs between 68 kinase inhibitors (including FDA-approved drugs) and 442 kinases, randomly split into 25,046 for training and 5010 for testing (the widely used “S1” setting62). The filtered KIBA data set61,62 contains 118,254 pairs between 2111 kinase inhibitors and 229 kinases, including 98,545 for training and 19,709 for testing (S1 split again). Other split settings were not pursued because published performances in such settings are not always available and comparable. The KIBA scores combine ki, kd, and IC50 sources for consistency and are further processed.15,62 As to the refined PDBbind data set (v. 2019), we filtered and processed it (see details in the Supporting Information Section S1.1) to reach 3505 pairs with ki or kd labeled between 1149 proteins and 2870 compounds. Compared to Davis and KIBA, the PDBbind data set contains more diverse protein classes: 2157 interactions with enzymes including 72 with kinases, 62 with nuclear receptors, 33 with G protein-coupled receptors (GPCRs), and 106 with ion channels. The portion of labeled compound–protein pairs is much lower than that of Davis and KIBA. We randomly split the PDBbind data set into 2921 pairs for training and 584 for testing.
For our framework of DeepAffinity,16 we adopt various data representations and corresponding state-of-the-art neural network architectures as detailed in the Methods section. To model proteins, we have adopted RNN using protein SPS16 as input data as well as CNN and newly developed HRNN using protein amino-acid sequences. To model compounds, we have adopted RNN using SMILES as input data as well as GCN and GIN using compound graphs with node features and edge adjacency.19 In the end, we have tested five DeepAffinity variants (including four new) for protein–compound pairs, including RNN–RNN,16 RNN–GCN, CNN–GCN, HRNN–GCN, and HRNN–GIN. Names before and after hyphens indicate models to embed proteins and compounds, respectively; and embeddings of a pair of protein and compound are passed through joint attentions in eq 6 before being fed to a convolutional neural network (CNN) and multilayer perceptrons (MLP).16 For instance, the first one, RNN–RNN indicates that protein SPS sequences are modeled by RNN and compound SMILES or graphs are modeled by RNN. This is essentially our previous method16 except that no unsupervised pretraining or ensemble averaging is used here. We have tuned hyperparameters for DeepAffinity variants including learning rate ({10−3, 10−4}), batch size ({64, 128} (16 for CNN-GCN because of the limit of GPU memory) and dropout rate ({0.1, 0.2}) using random 10% of training data as validation sets. When HRNN was used to model protein sequences, we have also tuned k-mer lengths and group sizes in pairs [{(40,30), (48,25), (30,40), (25,48), (15,80), (80,15)} for Davis and {(40,25), (50,20), (25,40), (20,50)} for KIBA and PDBbind] using the validation sets.
For comparison, we use published current methods that are not structure-based, including DeepDTA,15 KronRLS,64 and WideDTA,65 all of which are noninterpretable. Their results for the Davis and KIBA sets were self-reported in individual studies and summarized in a comparison study.63 Their results for the PDBbind set are derived by retraining released source codes with published hyperparameter grids and individual training sets (except wideDTA whose codes are not available). In addition, we compare to structure-free methods that are interpretable. Except DeepAffinity, the only other interpretable method published so far (Gao et al.) was for predicting binary compound–protein interaction.18 As its codes are not publicly available, we have implemented the method, revised its model’s last layer (sigmoid), and retrained the model for affinity prediction using each training set. To ensure fair comparison, all deep-learning models including our DeepAffinity variants here are trained for 100 epochs or until convergence (the validation loss does not improve within 15 epochs), as competing methods previously did.63
We compare aforementioned competing methods and DeepAffinity variants in accuracy using two assessment metrics: RMSE (root-mean-squared error; see Table 1) and CI (concordance index; see Table 2). Although RMSE evaluates the proximity between predictions are to corresponding native values, CI,66 often used for virtual screening, measures the probability of correctly ordering nonequal pairs. We summarize the results in Tables 1 and 2.
Table 1.
Comparing Current Methods (Noninterpretable Except Gao et al.) and Interpretable DeepAffinity Variants in Prediction Accuracy (Measured by RMSE, the Lower the Better) for the Davis, KIBA, and PDBbind Benchmark Setsa
| deepaffinity | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| RMSE | deepDTA | kronRLS | wideDTA | Gao et al.c | RNN-RNN16 | RNN-GCN | CNN-GCN | HRNN-GCN | HRNN-GIN |
| Davis | 0.5109b | 0.6080b | 0.5119b | 0.7864 | 0.5032 | 0.5095 | 0.8106 | 0.5019 | 0.6604 |
| KIBA | 0.4405b | 0.6200b | 0.4230b | 0.7368 | 0.4335 | 0.5367 | 0.8244 | 0.4480 | 0.6669 |
| PDBbind | 2.0631 | 1.8005 | 1.8071 | 1.4524 | 1.4277 | 1.5580 | 1.4743 | 1.4858 | |
The best performance in each data set is bold-faced.
Self-reported and published results as summarized in Thafar et al.63
Originally a binary classifier, it was implemented and revised by us for affinity prediction.
Table 2.
Comparing Current Methods (Noninterpretable Except Gao et al.) and Interpretable DeepAffinity Variants in Prediction Accuracy (Measured by the Concordance Index or CI, the Larger the Better) for the Davis, KIBA, and PDBbind Benchmark Setsa
| deepaffinity | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| RMSE | deepDTA | kronRLS | wideDTA | Gao et al.c | RNN-RNN16 | RNN-GCN | CNN-GCN | HRNN-GCN | HRNN-GIN |
| Davis | 0.8780b | 0.8830b | 0.8860b | 0.7824 | 0.9000 | 0.8808 | 0.7373 | 0.8814 | 0.8224 |
| KIBA | 0.8630b | 0.7820b | 0.8750b | 0.7335 | 0.8423 | 0.7968 | 0.5761 | 0.8420 | 0.6893 |
| PDBbind | 0.7125 | 0.7197 | 0.7610 | 0.8042 | 0.7543 | 0.7119 | 0.7544 | 0.7398 | |
The best performance in each data set is bold-faced.
Self-reported and published results as summarized in Thafar et al.63
Originally a binary classifier, it was implemented and revised by us for affinity prediction.
From both tables, we conclude that the original DeepAffinity method16 (RNN–RNN; RNN for protein SPS; and RNN for compound SMILES) and its variants compared favorably to the state-of-the-art. Specifically, the DeepAffinity variants achieved the best performances in RMSE and CI for both the Davis data set and the most diverse and sparse data set of PDBbind. It closely followed the best performances (WideDTA) for the KIBA data set. In particular, the newly introduced HRNN models for protein sequences (higher-resolution than SPS) and graph models GCN and GIN for compound graphs achieved the best or close-to-the-best performances, which enables interpreting affinity prediction at the level of protein residues and compound atoms without sacrificing the accuracy. Considering that other methods are not interpretable and the only exception Gao et al. did not perform as well, the performances of interpretable DeepAffinity variants are particularly impressive.
Our New Data Set for Both Affinity and Contact Prediction is Diverse and Challenging.
To support systematic assessment and development of explainable affinity prediction, we have constructed a data set of 4446 compound–protein pairs (between 1287 proteins and 3672 compounds) with both affinity values (pKi or pKd) and atomic contacts (available in cocrystal structures). More details are included in the Methods section and the Supporting Information, Section 1.1.
The data set contains diverse proteins and compounds. Among the 4446 pairs, there are 2913 interactions with enzymes including 114 with kinases, 105 with nuclear receptors, 89 with GPCRs, and 111 with ion channels. The enzymes are across all seven enzyme commission classes (see details including EC class breakdowns in the Supporting Information, Section 1.1). The 3672 compounds cover wide ranges of physicochemical properties (log P, molecular weight, and affinity values) as seen in Figure 3.
Figure 3.
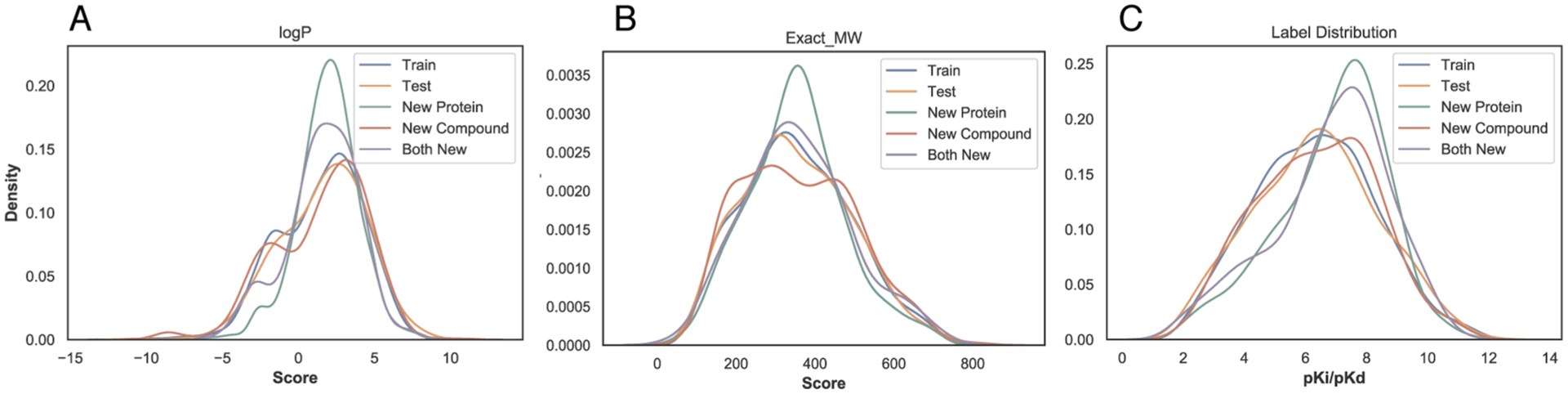
Distributions of compound properties across various subsets: (A) log P; (B) exact molecule weight; and (C) pKi/pKd labels.
The data set is split into training including validation (2334), test (591), new-protein (795), new-compound (521), and both-new sets (205), as illustrated in Figure 1. Compared to the test set, the three generalization sets not only contain new proteins or/and compounds but also mainly consist of very dissimilar proteins or/and compounds compared to the training set, which suggest their challenges for machine learning. For instance, the new-protein set only contains proteins not present in the training set. 454 (57.1%) pairs in the set involve new proteins whose global sequence identities to the closest training proteins are below 30%, and 452 (56.8%) pairs involve new proteins whose local binding k-mer identities are below 30% (note that only around 10% residues of an average binding k-mer are binding residues). Similarly, 414 (79.5%) new-compound pairs involve new compounds whose Tanimoto scores (details in the Supporting Information, Section 1.4) to the closest training compounds are below 0.5. The both-new set only contains pairs of new proteins and new compounds with similarly low resemblance to the training set. 98 (47.8%) pairs involve new proteins with sequence identity below 30% and new compounds with Tanimoto scores below 0.5. Therefore, the both-new set is expected to be the most challenging set among the four for the generalizability of machine learning models. Pair breakdowns are visualized in part of Figure 6 (counts). In addition, Jensen–Shannon distances between compound properties of training and those of the other sets are given in Table S1, similarly revealing the most challenging both-new set.
Figure 6.
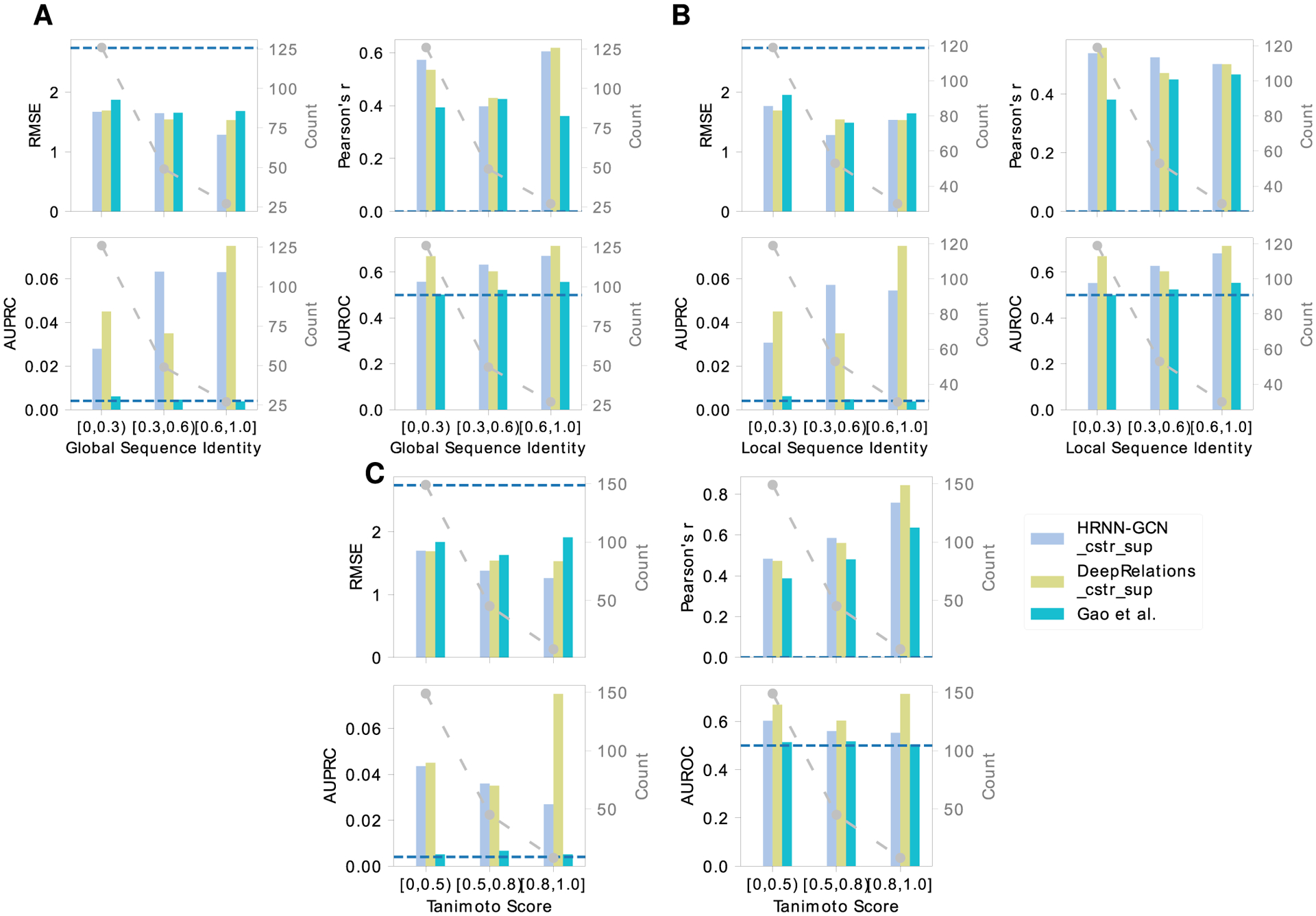
Comparing DeepAffinity+, DeepRelations, and Gao’s method in the generalizability of affinity prediction (RMSE and Pearson’s r) and contact prediction (AUPRC and AUROC) to molecules unlike training data. A horizontal dashed line indicates the performance of a random predictor.
Attentions alone are Inadequate for Interpreting Compound–Protein Affinity Prediction.
Now that we have established the accuracy of attention-embedded DeepAffinity and constructed a suitable data set, our first task for interpretability is to systematically assess the adequacy of attention mechanisms for interpreting model-predicted compound–protein affinities. To that end, using our newly curated benchmark set for both affinity and contact prediction, we have tested six DeepAffinity variants for protein–compound pairs (including RNN–RNN, RNN–GCN, CNN–GCN, HRNN–RNN, HRNN–GCN, and HRNN–GIN) as well as the only other interpretable method (Gao et al.) that is also attention-based and adapted by us from a classifier to a regressor. All models are retrained using the new training set with details in the Methods section. The first two DeepAffinity (RNN–RNN and RNN–GCN) models’ attentions on proteins are at the secondary structure levels. Their joint attentions were thus converted to residue-atom matrices, using equal weights across all residues within a secondary structure, in the postanalysis of interpretability. The rest have joint attentions at the level of pairs of protein residues and compound atoms.
The accuracy of affinity prediction, measured by RMSE and Pearson’s r in pKi/pKd, is summarized for the DeepAffinity variants in the top panel of Figure 4 and Table S3. Overall, all variants have shown affinity RMSE (Pearson’s r) around 1.5 (0.65), 1.6 (0.50), 1.4 (0.70), and 1.7 (0.50) for the default test, new-protein, new-compound, and both-new sets, respectively. In particular, the HRNN–GCN version achieved an RMSE (Pearson’s r) of 1.47 (0.70), 1.46 (0.56), 1.34 (0.73), and 1.49 (0.61) for the four sets, respectively, showing a robust accuracy profile. In contrast, the competing method (Gao et al.) has worse RMSE values between 1.72 and 1.87 and worse Pearson’s r between 0.42 and 0.58.
Figure 4.
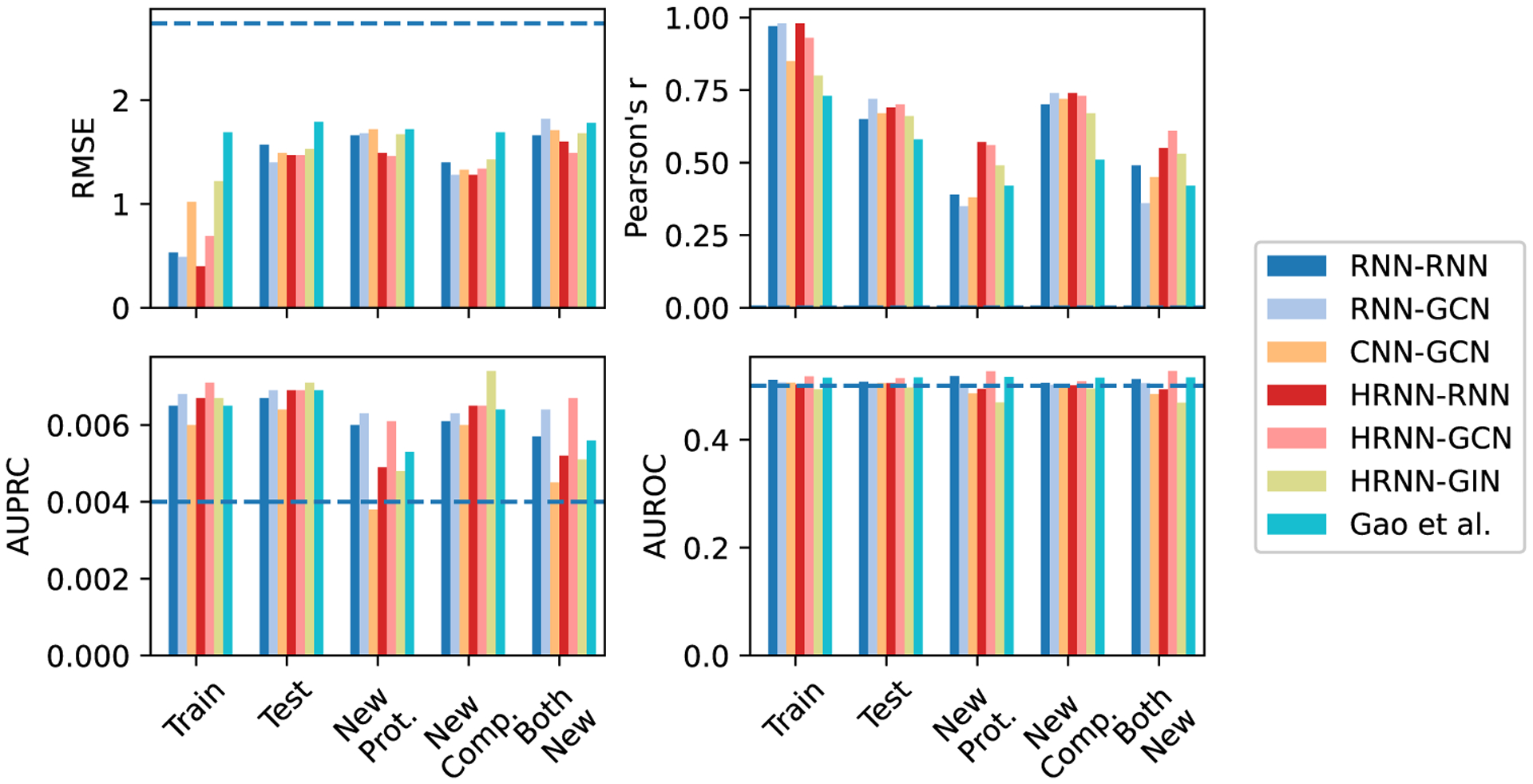
Comparing accuracy and interpretability among various versions of DeepAffinity with unsupervised joint attention mechanisms as well as another interpretable method (Gao et al.). Separated by hyphens in legends are neural network models for proteins and compounds respectively. A horizontal dashed line indicates the performance of a random predictor.
The interpretability of affinity prediction is assessed against ground truth of intermolecular residue–atom contacts, as shown in the bottom panel of Figure 4 and Table S3. Specifically, we use joint attention scores to classify all possible residue-atom pairs into contacts or noncontacts. As contacts only represent a tiny portion 0.0040 ± 0.0029 in our data set of all possible pairs, we use the area under the precision–recall curve (AUPRC) as the major metric and the area under the receiver operating characteristic curve (AUROC) as a reference, to assess such binary classification. Here, AUPRC/AUROC is averaged over all pairs involved in the corresponding set. Interestingly, compared to chance (AUPRC = 0.004 and AUROC = 0.5), all attention-based models including DeepAffinity variants and Gao et al. only had slightly better AUPRC (around 0.006 albeit a 50% improvement) except CNN–GCN for the new-protein set. The best DeepAffinity variant, HRNN–GCN, did improve against Gao et al.
From the results above, we conclude that attention mechanisms alone are inadequate for the interpretability of compound–protein affinity predictors, regardless of the choice of commonly used, generic neural network architectures.
Regularizing Attentions with Physical Constraints Modestly Improves Interpretability.
Our next task is to enhance the interpretability of compound–protein affinity prediction beyond the level achieved by attention mechanisms alone. The first idea is to incorporate domain-specific physical constraints into model training. The rationale is that, by bringing in the (predicted) structural contexts of proteins and protein–compound interactions, attention can be guided in their sparsity patterns accordingly for better interpretability.
We start with the two best-performing DeepAffinity variants so far (HRNN–GCN and HRNN–GIN), where protein amino-acid sequences are modeled by hierarchical RNN and compound graphs by various GNNs (including GCN and GIN). We introduce structure-aware sparsity regularization R2(·) to the two models to make “DeepAffinity+” variants. The resulting HRNN-GCN_cstr and HRNN-GIN_cstr models with physical constraints are assessed in Figure 5 and Table S4. Compared to the nonregularized counterparts in Figure 4 and Table S3, both models achieved similar accuracy levels across various test sets for affinity prediction. As to their interpretability, HRNN–GCN_cstr had similar AUPRC as before regularization (0.006) and HRNN–GIN_cstr slightly improved AUPRC to around 0.008, although both were still close to the baseline (0.004). These results suggest that incorporating physical constraints to structurally regularize the sparsity of attentions is useful for improving interpretability but may not be enough.
Figure 5.
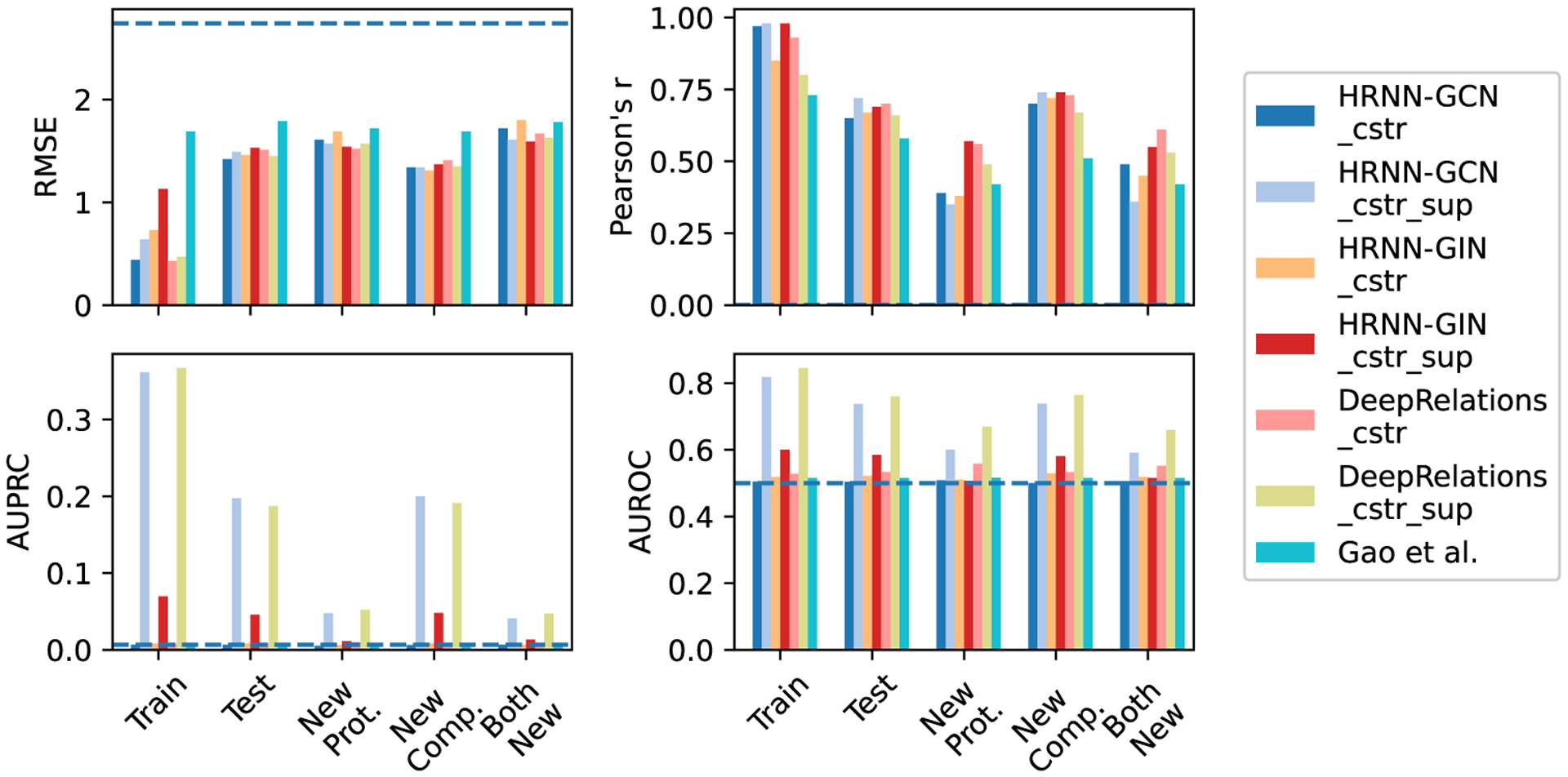
Comparing accuracy and interpretability among various versions of DeepAffinity+ (DeepAffinity with regularized and supervised attentions) and DeepRelations. “cstr” in legends indicates physical constraints imposed on attentions through regularization term R2(·), whereas “sup” indicates supervised attentions through regularization term R3(·). A horizontal dashed line indicates the performance of a random predictor.
Supervising Attentions Significantly Improves Interpretability.
As regularizing attentions with physical constraints was not enough to enhance interpretability, our next idea is to additionally supervise attentions with ground-truth contact data available to training examples. Again, we introduce “DeepAffinity+” models starting with HRNN–GCN and HRNN–GIN, by both regularizing and supervising attentions (using R2(·) and R3(·)).
The performances of resulting HRNN–GCN_cstr_sup and HRNN–GIN_cstr_sup models are shown in Figure 5. Importantly, HRNN–GCN_cstr_sup (light blue) significantly improved interpretability of affinity prediction without the sacrifice of accuracy. The average AUPRC improved to 0.197, 0.048, 0.200, and 0.041 for the default test, new-protein, new-compound, and both-new sets, representing a 30.4-, 9.2-, 31.2-, and 6.3-fold increase, respectively, compared to the version with just regularization but not supervision of attentions (HRNN–GCN_cstr). The performances also represented a 32.9-, 9.9-, 35.1-, and 8.6-fold increase, respectively, compared to Gao et al. Interestingly, supervising attentions in HRNN–GIN did not lead to significant improvement in interpretability.
Building Explainability into DeepRelations Architecture Further Improves Interpretability.
Toward better interpretability, besides regularizing and supervising attentions, we have further developed an explainable, deep relational neural network named DeepRelations. Here, atomic “relations” constituting physical bases and explanations of compound–protein affinities are explicitly modeled in the architecture with multistage gradual “zoom-in” to focus attention. In other words, the model architecture itself is intrinsically explainable by design.
The performances of the resulting DeepRelations (with both regularized and supervised attentions) are shown in Figure 5 (yellow-green “DeepRelations_cstr_sup”). With equally competitive accuracy in affinity prediction as all previous models, DeepRelations achieved further improvements in interpretability. The AUPRC values were similar to the best DeepAffinity+ model (HRNN-GCN_cstr_sup): 0.187, 0.052, 0.191, and 0.047 for the default test, new-protein, new-compound, and both-new sets, respectively. The AUROC values improved to 0.76, 0.67, 0.76, and 0.66 for the four sets, representing an increase of 0.03, 0.07, 0.03, and 0.07 compared to those of the best DeepAffinity+, respectively.
To disentangle various components of DeepRelations and understand their relative contributions to DeepRelations’ improved interpretability, we removed components from DeepRelations for the ablation study. Besides regularized and supervised attentions, we believe that the main contributions in the architecture itself are (1) the multistage “zoom-in” mechanisms that progressively focus attentions from surface, binding k-mers, binding residues to binding residue-atom pairs; and (2) the explicit modeling of atomic relations that can explain the structure feature-affinity mappings consistently with physics principles.
We thus made three DeepRelations variants: DeepRelations without multistage focusing, without explicit atomic relations, or without both. We compare them with DeepRelations in Figure S1. Consistent with our conjecture, we found that, the explicit modeling of atomic relations was the main contributor as its removal led to worse affinity and contact predictions in new-protein and both-new sets. The multistage focusing also contributes as its removal led to worse affinity prediction for both new-compound and both-new sets.
Validation of Affinity Prediction.
To validate the affinity accuracy of our two final models HRNN–GCN_cstr_sup (DeepAffinity+ hereinafter) and DeepRelations_cstr_sup (DeepRelations hereinafter), we have performed several randomization tests. First, using random sampling of the training set would lead to affinity RMSEs above 2.7 and Pearson’s r around 0; whereas using the sample mean would lead to affinity RMSEs between 1.85 and 2.02 and an undefined Pearson’s r. Both random affinity predictors performed considerably worse than DeepAffinity+ and DeepRelations (RMSE between 1.3 and 1.6 and Pearson’s r between 0.5 and 0.7). Second, Y-randomization tests67 of DeepAffinity+ and DeepRelations (20 trials each) led to much worse affinity prediction (RMSE between 2.20 and 2.45 and Pearson’s r around 0). Compound-randomization tests of our two models had similar results (RMSE between 1.95 and 2.22 and Pearson’s r around 0 for new proteins). More details can be found in Tables S5–7. Therefore, we conclude that our models’ affinity accuracy is significantly better than chance correlations.
To further improve the accuracy of affinity prediction, we have constructed ensembles of DeepAffinity+, DeepRelations, and both, by using combinations of hyperparameters (such as the dropout ratio, λbind, and the width of fully-connected layers). More details can be found in the Supporting Information, Section 2.5. Notably, the DeepAffinity+ ensemble decreased affinity RMSE from 1.49 to 1.29, increased Pearson’s r from 0.68 to 0.77, and increased predictive R2 from 0.45 to 0.59 for the test set. It similarly improved the accuracy of affinity prediction, albeit to a lesser extent, for other sets involving new molecules. More results are reported in Table S8.
Better Interpretability Helps Better Accuracy and Generalizability of Affinity Prediction.
To examine whether the more interpretable affinity predictors are also more accurate in affinity prediction, we compare our two final models HRNN–GCN_cstr_sup (DeepAffinity+ hereinafter) and DeepRelations_cstr_sup (DeepRelations hereinafter) to the competing interpretable affinity predictor Gao et al. Re-examining earlier results (Figure 5 and Table S4) shows that DeepAffinity+ and DeepRelations with much better interpretability (AUPRC increase between 8.6 and 59-fold) than Gao et al. are also more accurate in affinity prediction (RMSE drop between 0.15 and 0.42 and Pearson’s r increase around 0.25) over all sets considered. Even when we compare DeepAffiity+ and DeepRelations to their attention-unsupervised counterparts (HRNN–GCN_cstr and DeepRelations_cstr), we find that better interpretability (contact prediction) leads to better accuracy (lower RMSE and higher Pearson’s r for affinity prediction) in 6 of 8 cases where the only exceptions occurred when AUPRC values were low.
Here, we further compare DeepAffinity+ and DeepRelations to Gao et al. in affinity and contact prediction over multiple difficulty ranges (measured by protein global sequence identity, protein local binding k-mer sequence identity, or compound Tanimoto scores) of the new-compound, new-protein, and both-new sets. The results are reported in Figure 6 as well as Figures S2–7 and Table S9. We find that the same conclusion (better interpretability leads to better accuracy) also applies, where model generalizability is needed the most: pairs involving very dissimilar proteins (global or local sequence identity below 30%) or/and compounds (Tanimoto scores below 0.5) compared to training molecules. Importantly, in those cases demanding generalizability the most, DeepAffinity+ and DeepRelations have much better accuracy (affinity-prediction RMSE decrease between 0.14 and 0.40 and Pearson’s r increase between 0.10 and 0.18) as well as significantly improved interpretability (contact-prediction AUPRC increase between 5.9 and 33.3-fold) compared to Gao et al.
DeepAffinity+ and DeepRelations also showed competitive generalizability in both affinity and contact prediction. From the most similar proteins (sequence identity above 60%) to the least (sequence identity below 30%), affinity-prediction RMSE values of DeepAffinity+ (DeepRelations) only increased 0.13 (0.08) for the new-compound set and increased 0.00 (0.16) for the most challenging both-new set. From the most similar compounds (Tanimoto scores above 0.8) to the least (Tanimoto scores below 0.5), affinity-prediction RMSE values of DeepAffinity+ (DeepRelations) only increased 0.14 (0.08) for the new-compound set and increased 0.43 (0.48) for the most challenging both-new set. Similar conclusions can be made about their generalizability in contact prediction.
Case Studies.
Now that we have established and explained how DeepAffinity+ and DeepRelations significantly improve the interpretability of compound–protein affinity prediction, we went on to delve into their affinity and contact predictions in comparison to Gao et al. using a series of cases studies of increasing difficulty. Summary performances of the five cases are reported in Table 3. DeepAffinity+ and DeepRelations had better affinity and contact prediction in all cases compared to the competing method whose top-10 predicted contacts failed to produce any native contacts. In order to understand model behaviors, our analysis next would focus on the patterns of top-10 contacts predicted by DeepAffinity+ and DeepRelations compared to Gao et al.
Table 3.
Performance Summary of Three Interpretable Methods for Five Case Studies
| DeepAffinity+ | DeepRelations | Gao et al. | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| protein | ligand | affinity error | contact AUROC | contact AUPRC | top-10 contact precision | affinity error | contact AUROC | contact AUPRC | top-10 contact precision | affinity error | contact AUROC | contact AUPRC | top-10 contact precision |
| Two Compounds Bind to the Same Pocket of a New, Nonhomologous Protein (Different Affinity-Prediction Quality) | |||||||||||||
| CA2 | AL1 | 1.89 | 0.658 | 0.284 | 0.5 | 2.70 | 0.828 | 0.075 | 0.6 | 3.28 | 0.500 | 0.006 | 0.0 |
| IT2 | 2.92 | 0.601 | 0.034 | 0.3 | 3.03 | 0.780 | 0.309 | 0.5 | 3.09 | 0.630 | 0.009 | 0.0 | |
| Two New Compounds Bind to Distinct Pockets of a Protein | |||||||||||||
| PYGM | CPB | 0.10 | 0.552 | 0.006 | 0.1 | 0.39 | 0.513 | 0.005 | 0.0 | 0.61 | 0.522 | 0.001 | 0.0 |
| T68 | 0.68 | 0.944 | 0.675 | 1.0 | 0.66 | 0.908 | 0.610 | 1.0 | 1.80 | 0.635 | 0.006 | 0.0 | |
| A New Compound Very Dissimilar to Training Compounds Binds to a New Protein Nonhomologous to Training Proteins | |||||||||||||
| LCK | LHL | 2.12 | 0.500 | 0.053 | 0.4 | 1.30 | 0.702 | 0.053 | 0.4 | 2.89 | 0.540 | 0.005 | 0.0 |
Two Compounds Bind to the Same Pocket of a New Protein Nonhomologous to Training Examples.
Our first case study involves a protein from the new-protein set, human carbonic anhydrase II (CA2, UniProt ID: P00918), that has no close homologue in the training set. Specifically, the closest training protein would be human carbonic anhydrase IV (CA4, UniProt ID: P22748) with a sequence identity below the 30% threshold (29%). We choose two compounds (HET IDs: AL1 and IT2) that bind to the same pocket of CA2 with distinct sizes (AL1 is larger by 14 heavy atoms) and affinity-prediction quality (see Table 3).
We compare in Figure 7 the top-10 contacts between protein residues and compound atoms that are predicted by three methods. Top-predicted contacts by Gao et al. were scattered across protein residues that are far from the binding site, failing to match any native contact. In contrast, those top-10 contacts predicted by DeepAffinity+ and DeepRelations were more focused in or near the binding site, containing 3–6 native contacts that are direct, first-shell contacts. Between our two models, DeepRelations showed better contact prediction in these two cases: its top-10 predictions were more focused in the binding site and contained 60 and 50% native contacts for compounds AL1 and IT2, respectively. The more focused contact prediction of our methods could be attributed to structure-aware regularization using protein residue–residue contact maps. DeepRelations had better focus than DeepAffinity+, possibly because of the multistage focusing strategy.
Figure 7.
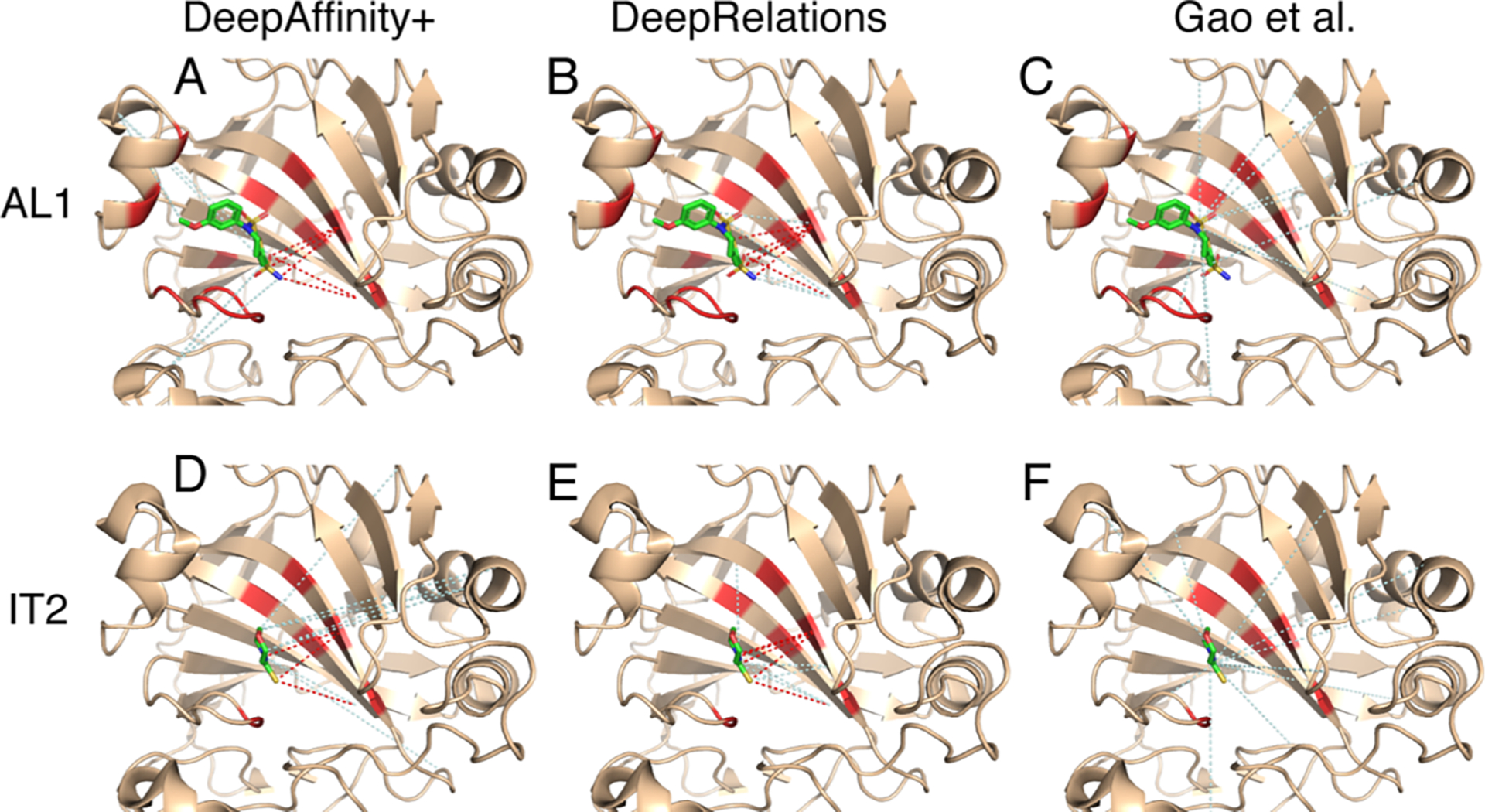
Structural visualization of top-10 intermolecular contacts predicted by DeepAffinity+ (left), DeepRelations (middle), and Gao et al. (right) for two test cases. Here, two compounds [AL1: top panels (A–C) and IT2: bottom panels (D–F); stick representations] bind to the same pocket of the human carbonic anhydrase II that is new and nonhomologous to training data (wheat cartoons where binding residues are highlighted in red). Shown in dashed lines are top-10 predicted contacts (interactions between protein residues and compound atoms). The dashed lines in red and pale cyan highlight correct and incorrect predictions, respectively, according to native, direct contacts retrieved by LigPlot.
Even the incorrect predictions of DeepRelations can correspond to residue-atom pairs that are close (but above the 4 Å-cutoff used in the first-shell contact definition). For instance, in the case of compound AL1, the four incorrect predictions all corresponded to correct binding residues that were paired to wrong compound atoms. In the case of compound IT2, the five incorrect predictions included two that paired correct binding-site residues to wrong atoms and three that included (the very next) sequence neighbors of correct binding-site residues.
These two cases also provided examples to interpret the values of AURPC and top-10 contact precision. A seemingly “low” AUPRC value of 0.075 can lead to 5 of 10 top predictions being correct. The reason is that native contacts represent a rare minority (0.004) among all possible residue-atom pairs, and an AUPRC value of 0.075 actually represents over 18-fold increase compared to the baseline AUPRC by chance. Meanwhile, a top-10 contact precision of 0.4 predicted by our structure-free methods is close to the average level (0.44) achieved by a popular structure-based protein–ligand docking program, AutoDock Vina,68 under default settings.69
Two New Compounds Bind to Distinct Pockets of a Protein.
Our next case study involves two compound–protein pairs from the new-compound set, where two compounds (HET ID: CPB and T68) not present in the training set bind to two distinct pockets of the rabbit glycogen phosphorylase (PYGM, UniProt ID: P00489). The protein is present in the training set with 38 ligands (all but one are occupying the same pocket as T68). In addition, the compound CPB does not resemble its closest training example interacting with the same protein (HET ID: 62N) when 62N rather occupies the same pocket as T68. Therefore, contact prediction for the CPB case would be much more challenging. Indeed, our results supported the conjecture (Table 3). In their top-10 contact predictions, our both models achieved 100% native contacts for T68 but just 10% (DeepAffinity+) or even 0% (DeepRelations) for CPB. They had good estimation of binding affinity for both cases.
A closer look into their contact predictions reveal more insights. As seen in Figure 8, consistent with our earlier observations, Gao et al.’s contact predictions are dispersed across the whole protein, whereas ours are focused. In the case of T68, our predictions are focused in the correct binding site (and even the correct binding residues). However, in the case of CPB, our predictions are actually still focused in the same site as they did for T68, only being wrong this time. Interestingly many falsely-predicted contacts for CPB were not only in the other binding site (circled area) but also with the T68 binding residues. This model behavior is understandable when almost all training examples, including a very similar compound, are indicating a different site. It also reveals a situation that would challenge more generalizability and demand more explainability from machine learning methods. Intriguingly, DeepAffinity+ still managed to make one correct contact prediction (pointed at by a red arrow).
Figure 8.
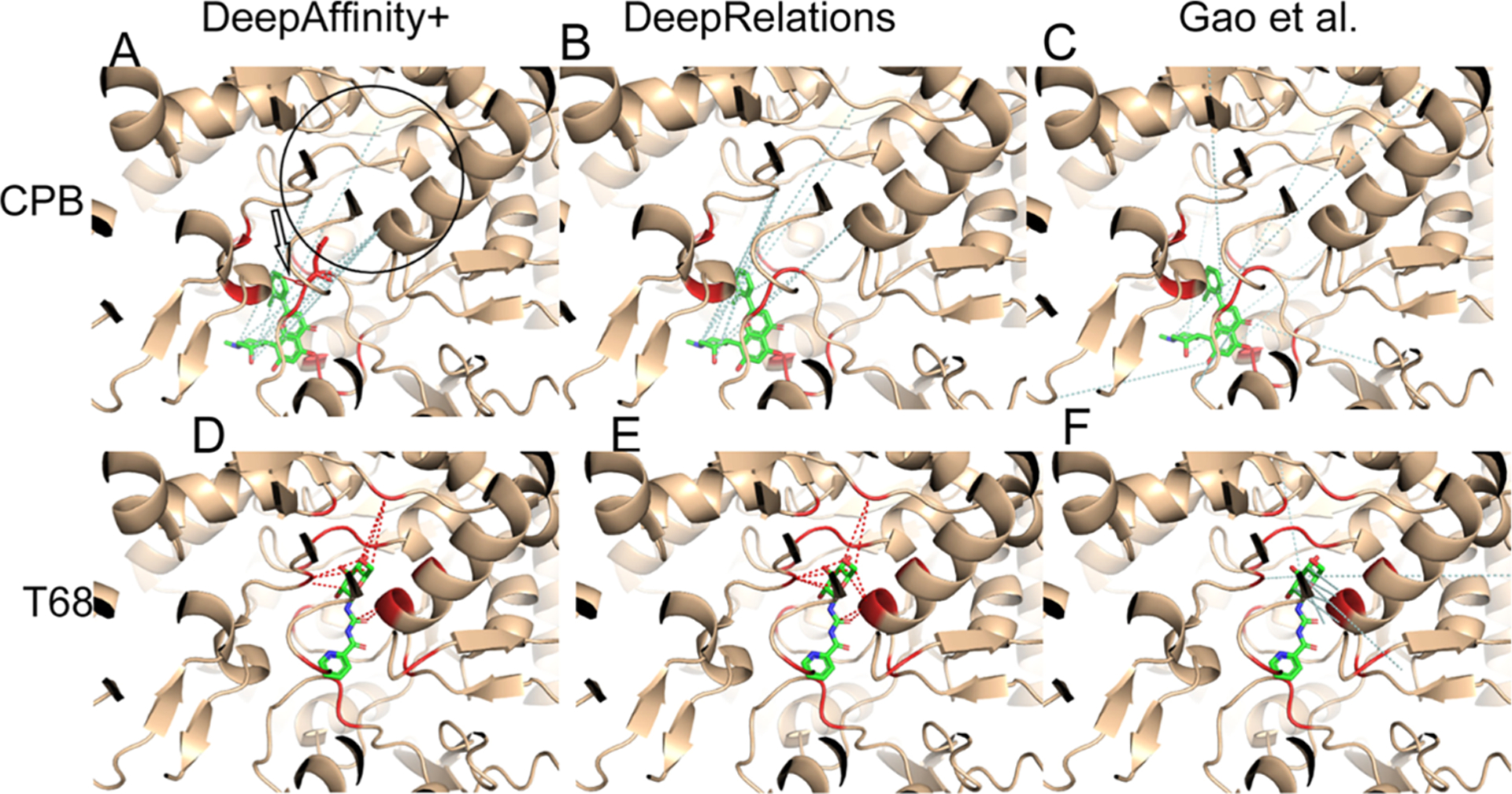
Structural visualization of top-10 intermolecular contacts predicted by DeepAffinity+ (left), DeepRelations (middle) and Gao et al. (right) for another two test cases. Here, two compounds that are new to training data [CPB: top panels (A–C) and T68: bottom panels (D–F); stick representations] bind to distinct pockets of the human glycogen phosphorylase (wheat cartoons where binding residues are highlighted in red). Shown in dashed lines are top-10 predicted contacts (interactions between protein residues and compound atoms), including correct (red) and incorrect (pale cyan) ones according to LigPlot’s definition of native, direct contacts. The black hollow arrow in panel A points to the only correct prediction by DeepAffinity+ and the black circle there indicates the binding site for T68. Interestingly, many incorrect predictions by DeepAffinity+ and DeepRelations for CPB were with binding residues to T68.
Pair of New Protein and New Compound Very Dissimilar to Training Examples.
Our last case study is even more challenging in that both the protein (the human tyrosine-protein kinase Lck, LCK in short, UniProt ID: P06239) and the compound (HET ID: LHL) are new and they do not even resemble training examples. Specifically, the most similar training protein would be the human tyrosine-protein kinase BTK, BTK in short, UniProt ID: Q06239) with sequence identity at 28%. The most similar training compound would be K60 (HET ID) with the Tanimoto score at 0.12. Indeed, our results (Table 3) showed that contact AUPRC is just around 0.053. Given the explanation to interpret AUPRC and top-10 contact precision in the first case study, one would notice that the AUPRC value is 14-fold of the baseline (0.004) and 40% of our top-10 contact predictions were true positives (a level close to average protein–ligand docking performances).
As seen in Figure 9, again, our contact predictions are more focused in or near the binding site compared to the competing methods, which can be attributed to our structure-aware attention regularization (and supervision). A closer look into the false positives reveal more into our methods. Take DeepAffinity+ as an example. Among the six false-positive contact predictions, four were pairing correct binding residues with wrong compound atoms, one was paired to a protein residue that is a close sequence neighbor (two residues away) of a correct binding residue, and one was paired to a protein residue that is not present in the cocrystal structure but predicted to be spatially close to a correct binding residue. In other words, the origins of false positives in contact prediction include (but are not limited to) pairing with other (nearby) compound atoms and pairing with sequential or predicted spatial neighbors of protein binding-residues. When the criterion of native contacts is relaxed from direct, first-shell contacts within 4 Å to more contacts within longer distance cutoffs, the precision level would further increase, which is detailed next.
Figure 9.
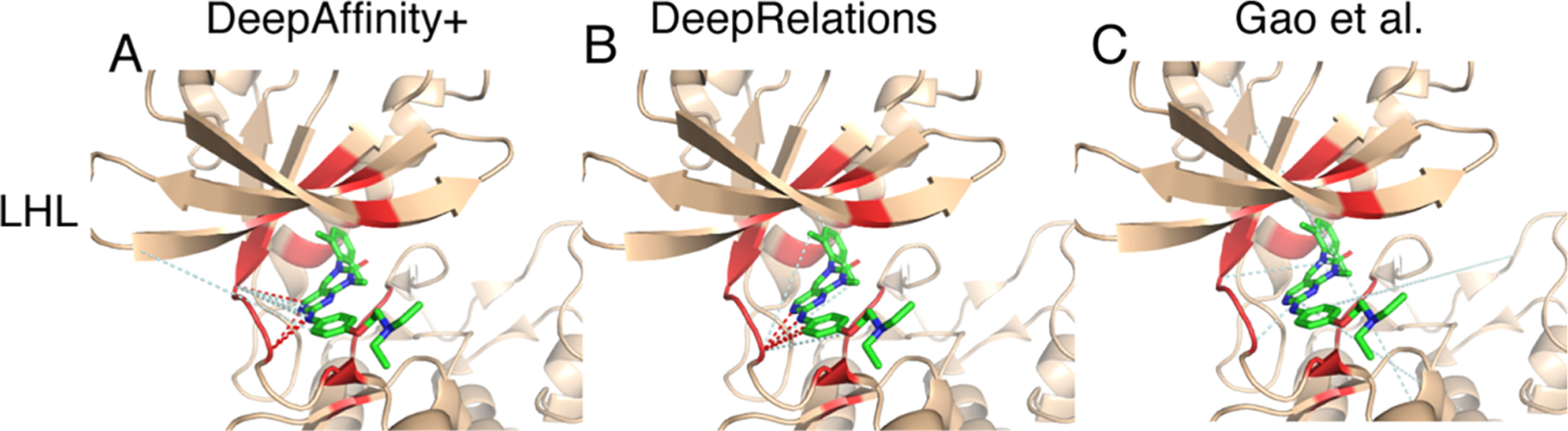
Structural visualization of top-10 intermolecular contacts predicted by (A) DeepAffinity+, (B) DeepRelations, and (C) Gao et al. for a difficult test case. Here, both the compound (LHL, in sticks) and the protein (tyrosine-protein kinase Lck, in wheat cartoons with binding residues highlighted in red) are new and very dissimilar to training data. The red and pale cyan dashed lines represent correct and incorrect top-10 predicted contacts, respectively. DeepAffinity+ and DeepRelations still managed to achieve the precision of 40% in their top-10 contact predictions.
Global Patterns of Top-10 Predicted Contacts.
We extended the analysis of the patterns of predicted contacts to all test cases. Considering that the native contacts are defined strictly as direct, first-shell contacts within 4 Å, we assess 4–10 Å distance distributions of residue-atom pairs predicted by DeepAffinity+ (HRNN–GCN_cstr_sup) and DeepRelations in comparison with Gao et al. As seen in the global analysis in Figure 10 and Table S10, DeepAffinity+ and DeepRelations significantly outperform the competing method in all distance ranges over all test sets. Specifically, among their top-10 contact predictions, around 40% for the default test and new-compound sets were first-shell contacts within 4 Å and the ratios increased to about 70% when considering contacts within 10 Å. For the more challenging cases of new-protein and both-new sets, the ratios of predicted contacts within 4 Å and 10 Å were around 20 and 50%, respectively. These results significantly outperformed the competing method whose ratios were merely 4–6% over all sets. Between our two models, DeepRelations behaved similarly as DeepAffinity+ and had more top-10 predictions falling in the long range of 8–10 Å.
Figure 10.

Distributions of top-10 contacts, predicted by DeepAffinity+, DeepRelations, and Gao’s method, in various distance ranges.
Predicted Contacts Assist and Improve Protein–Ligand Docking.
From the case studies and the global analysis above, we have concluded that top-10 contact predictions by our methods are enriched with native contacts within 4 Å (20–40%) as well as dominated by longer-range “contacts” within 10 Å (50–70%). We therefore test how much the top-10 contact predictions, including false positives, could make a positive impact in the drug discovery process. Picking a typical task—protein–ligand docking and a popular tool—AutoDock Vina,68 we assess how our contact predictions could assist the task by reducing the search space.
Specifically, we chose the five case studies (except the case where DeepRelations made no correct contact prediction) and performed unbound protein–ligand docking (all protein structures are unbound except PYGM whose structure is cocrystallized with its cognate phosphate AMP). Each pair (rigid protein and flexible ligand) is docked twice: one with the default procedure to define a search “box” covering the entire protein and the other using a restricted box that barely covers all residues in the top-10 DeepRelations contact predictions (including false positives) and then has 20 Å-padding. All the other docking parameters in AutoDock Vina are default, including a total of nine protein–ligand complex models ordered and reported at the end. Docking performances were evaluated by ligand rmsd of the top few models using the software DOCKRMSD.70
Results in Table 4 show that AutoDock Vina assisted by DeepRelations top-10 contact predictions had much improved docking performance compared to otherwise. When the top-10 contact precision was 40, 50, 50, and 100%, respectively, the best ligand rmsd (among all nine complex models) reduced from 2.77, 4.01, 16.62, and 18.75 Å down to 2.45, 1.59, 4.73, and 1.88 Å, respectively. The quality of the top-1 models also drastically improved in three of four cases. Although the way to incorporate predicted contacts into protein–ligand docking remains to be optimized, these results have proved that the precision and spatial pattern of our structure-free contact prediction is at a level useful to assist and improve structure-based protein–ligand docking for pose prediction.
Table 4.
Ligand Docking Performances for Case Studiesa
| rmsd (Å)—vina | rmsd (Å)—contact-assisted vina | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| protein (UniProt, PDB) | ligand | complex PDB | top-10 contact precision (%) | top 1 | top 3 | top 5 | best | top 1 | top 3 | top 5 | best |
| LCK (P06239, 3LCK) | LHL | 3KMM | 40 | 3.02 | 3.02 | 2.77 | 2.77 | 4.65 | 2.98 | 2.45 | 2.45 |
| CA2 (P00918, 2CBA) | AL1 | 1BNN | 60 | 18.55 | 16.62 | 16.62 | 16.62 | 4.78 | 4.78 | 4.73 | 4.73 |
| CA2 (P00918, 2CBA) | IT2 | 3P5A | 50 | 15.98 | 15.98 | 4.01 | 4.01 | 3.65 | 3.65 | 3.65 | 1.59 |
| PYGM (P00489, 8GPB) | T68 | 3ZCU | 100 | 36.40 | 18.75 | 18.75 | 18.75 | 9.08 | 2.23 | 2.23 | 1.88 |
The default Autodock Vina is compared with that assisted by DeepRelations top-10 contact predictions.
Affinity Prediction for Target Prioritization.
Using the two aforementioned CA2 (human carbonic anhydrase II) compounds (AL1 and IT2) in the first case study, we also explore the utility of our models for target prioritization for given compounds. As no affinity data were observed in our data set for AL1 or IT2 with proteins other than CA2, we approximate the set of “off-targets” with all the 1286 non-CA2 proteins in our data set. For either compound AL1 or IT2, we assessed the distribution of its off-target affinities predicted by DeepAffinity+ and compared the distribution (see Figure S8) to its predicted on-target (CA2) affinity. As shown in Figure S8, 83.1% (100%) and 88.2% (99.7%) of predicted off-target affinities are weaker than the predicted (actual) affinity to the target CA2, for compounds AL1 and IT2, respectively. Removing CA2 homologues (4 in total) from the non-CA2 proteins led to nearly the same results (data not shown). We note that this case is particularly challenging because no homologues of the target CA2 are in the training set, and the errors of target affinity prediction are higher than average. More systematic and dedicated studies are needed for this topic in future.
More Utilities from Explainable Affinity Prediction.
In the last part of the results, we explore additional utilities of our methods toward facilitating drug discovery: binding-site prediction for proteins and structure–activity relationship (SAR) for compounds. Our methods do not demand protein structures or protein–ligand docking to make these predictions. Rather, they simply aggregate predicted attentions (or predicted weights of residue-atom contacts) or/and decompose predicted affinities. Although not directly designed or optimized for these tasks, our explainable models have shown promising potentials in the tasks toward rational drug discovery.
Binding Site Prediction.
The first extended utility we aim at is structure-free and ligand-specific binding-site prediction for proteins. To this end, we feed an arbitrary pair of protein and compound to the trained DeepAffinity+ and DeepRelations models and predict the weights of residue-atom pairs ( where i and j are the indices of a protein residue and a compound atom, respectively). We then calculate the max-marginal attention () for each residue i as a weight for ranking. The performances of the residue weights toward ligand-specific binding site prediction are summarized in Figure 11 and Table S11. Here, binding-site residues of a protein are strictly defined as those making direct, first-shell contacts with a paired compound. Without the help of protein structures, predicted residue-contact maps, or protein–ligand docking, our methods on average achieved AUPRC (AUROC) of around 0.43 (0.77) for the default test and new-compound sets as well as AUPRC (AUROC) of around 0.18 (0.69) for the more challenging new-protein and both-new sets. In contrast, the competing method had AUPRC and AUROC close to the random performances of 0.004 and 0.50, respectively.
Figure 11.
Comparing three interpretable methods (DeepAffinity+, DeepRelations, and Gao et al.) in binding-site prediction.
Between DeepAffinity+ and DeepRelations, we noticed that the latter had better performance in predicting binding sites for new proteins. Specifically, the AUPRC (AUROC) increased from 0.17 (0.65) to 0.21 (0.73) for the new-protein set and did from 0.16 (0.65) to 0.20 (0.72) for the both-new set.
Structure Activity Relationship (SAR).
The second extended utility we aim at is SAR for compounds. To test the utility, we choose two subchallenges (SC3 and SC4) from Grand Challenge 3 of D3R:71 Janus kinase 2 (JAK2) and Angiopoietin-1 receptor (TIE2) that were excluded in our training set (thus new proteins). The most similar proteins to JAK2 and TIE2 in our training set are calcium/calmodulin-dependent protein kinase kinase 2 (CAMKK2, sequence identity 48%) and cyclin-dependent kinase 2 (CDK2, sequence identity 39%), respectively. The two data sets include 17 and 18 congeneric compounds, respectively, with Kd values measured. They were meant to “detect large changes in affinity because of small changes in chemical structure” (https://drugdesigndata.org/about/grand-challenge-3). In other words, the data sets focus on the sensitivity of methods targeting SAR. Chemical graphs, actual pKd, and DeepRelations-predicted pKd of the JAK2 and TIE2 compounds are in Figures S9 and S10, respectively.
Here, we compare our DeepAffinity+ and DeepRelations not only to structure-free Gao et al. but also to 18 structure-based methods from the community that participated in the subchallenges. The assessment metrics for affinity ranking are Kendall’s τ and Spearman’s ρ as in D3R. A summary of the performances is in Table 5. In the case of JAK2, the 18 structure-based methods had τ ranging from 0.71 to −0.56 and ρ ranging from 0.86 to −0.70, including eight methods with negative τ and ρ (see details in Table S13). As to the structure-free affinity predictors, Gao et al. had τ = −0.42 and ρ = −0.54, whereas our DeepAffinity+ had slightly better τ = −0.36 and ρ = −0.47, both outperforming just one structure-based method. However, our DeepRelations achieved τ = 0.15 and ρ = 0.21, outperforming 12 (two-thirds) of the structure-based methods. In the case of TIE2, the 18 structure-based methods had τ ranging from 0.57 to −0.57 and ρ ranging from 0.76 to −0.69, including eight methods with negative τ and ρ (see details in Table S14). Interestingly, the best structure-based method for JAK2 was only placed 12th among 18 with slightly negative τ and ρ for TIE2. In contrast, all the structure-free affinity predictors performed well for TIE2: Gao et al., DeepAffinity+, and DeepRelations had τ (ρ) reaching 0.60 (0.74), 0.65 (0.79), and 0.61 (0.72), respectively, and they all outperformed the best structure-based method. The scatter plots of actual versus our predicted pKd are in Figure S11. We note that all 18 structure-based methods used crystal structures of proteins and often-expensive ligand docking, whereas structure-free methods did not. Our methods only cost a fraction of a second when making quality predictions for tens to hundreds of compound–protein pairs, thus a useful complement to structure/docking-based methods toward virtual screening.
Table 5.
Summary of Scoring Performances Among Three Structure-free Methods (including Our DeepAffinity+ and DeepRelations and Eighteen Structure-based Methods
| JAK2 (subchallenge 3) | TIE2 (subchallenge 4) | ||||||
|---|---|---|---|---|---|---|---|
| rankinga | method(s) | T | ρ | ranking | method(s) | τ | ρ |
| 1–5 | 5 structure-based methods in D3R | 0.16–0.71 | 0.25–0.86 | 1 | structure-free DeepAffinity+ (ours) | 0.65 | 0.79 |
| 6 | structure-free DeepRelations (ours) | 0.15 | 0.21 | 2 | structure-free DeepRelations (ours) | 0.61 | 0.72 |
| 6 | 1 structure-based method in D3R | 0.13 | 0.32 | 2 | structure-free Gao et al. | 0.60 | 0.74 |
| 8–18 | 11 structure-based methods in D3R | −0.31–0.05 | −0.50–0.05 | 2 | 2 structure-based methods in D3R | 0.57 | 0.74–0.76 |
| 19 | structure-free DeepAffinity+ | −0.36 | −0.47 | 6–21 | 16 structure-based methods in D3R | −0.57–0.50 | −0.69–0.67 |
| 20 | structure-free Gao et al. | −0.42 | −0.54 | ||||
| 21 | 1 structure-based method in D3R | −0.56 | −0.70 | ||||
The 18 structure-based methods participated in the D3R subchallenges and were assessed officially. The three structure-free methods were assessed post hoc.
Beyond affinity scoring, we further examine DeepRelations in extracting SAR knowledge toward drug discovery. A central question in lead optimization is where and how to modify a lead compound to improve its property (affinity here). As a stepping stone, we construct predictors from our DeepRelations in order to anticipate the affinity changes when a functional-group substituent is introduced to a lead. Specifically, we regard our predicted as estimated binding energy for a compound–protein pair and our predicted joint-attention as the fraction of contribution between protein residue i and compound atom j. Borrowing the idea of energy decomposition, we calculate the binding-energy contribution of a functional group R as the product of the predicted binding-energy and the sum-marginals of joint attention: . In this way, the difference of this R-group contribution, , can be a predictor of affinity change when introducing a substituent R-group to a compound.
To test our predictor for lead optimization, we use the JAK2 data set involving 17 compounds that share a common scaffold and have distinct combinations of two functional groups (3 choices for R1 and 10 for R2; see Figure 12A and S9). We construct 121 pairs of compounds between a weaker binder (origin) and a stronger binder (end). 7, 36, and 78 of the structural changes from the origin to the end compound involve R1, R2, and both-R substitutions, respectively. We compare three methods in predicting these 121 affinity changes with assessment metrics including Pearson’s r (main assessment), Spearman’s ρ, and Kendall’s τ (Figure 12B–D). A straightforward predictor using DeepRelations’ ΔpKd without decomposition had r = 0.218, whereas the decomposed affinity-change predictor improved r to 0.361. If one has access to the protein in complex with a previously discovered compound and can have an accurate estimate of the binding residues, the summation of protein residue i in can be just over binding residues rather than all residues. In that case, the new can slightly improve r further to 0.363. Our decomposed affinity-change predictor similarly improved ρ and τ. Compared to the 18 structure-based competing methods that participated in the D3R JAK2 subchallenge, our structure-free predictor with decomposition outperformed 15 (five-sixths) of them in r, ρ, and τ, as detailed in Tables S15 and S16.
Figure 12.
Actual (x-axis) vs DeepRelations-predicted (y-axis) affinity changes when introducing functional-group substitutions [R1, R2, or both in (A)] to lead compounds for JAK2. The three predictors are as follows: (B) predicted affinity change ; (C) group-decomposed affinity change using all protein residues and the substituent group R alone; and (D) group-decomposed affinity change using estimated protein binding residues and the substituent group R alone.
When we split the analysis into three series involving R1, R2, and both-R separately, we observed that improved r from 0.267 to 0.753, −0.081 to 0.137, and 0.244 to 0.377, respectively (Figure S13). Using the binding-residue information could slightly improve the correlation further. Interestingly, when both R-groups are substituted (78 cases), had a better Pearson’s correlation (0.405) with the actual affinity changes than (−0.121) and even (0.375) did (Figure S14), potentially suggesting that the R1 group could be explored first for affinity optimization. Once a functional group R is chosen, affinity changes upon any proposed substitution can be predicted using our group-decomposed .
CONCLUSIONS
Toward accurate and interpretable machine learning for structure-free prediction of compound–protein interactions, we have curated compound–protein interaction data set annotated with both affinities and intermolecular atom-contacts, assessed the adequacy of current attention-based deep learning models for both accuracy and interpretability, and developed novel machine-learning models (in particular, DeepAffinity+ and DeepRelations) to remarkably enhance interpretability without sacrificing accuracy. We have also shown that our methods’ accuracy for affinity prediction is comparable or better than competing (noninterpretable) methods using established benchmark data sets. This is the first study with dedicated model development and systematic model assessment for interpretability in affinity prediction.
Our study has found that commonly-used attention mechanisms alone, although better than chance in most cases, are not satisfying in interpretability. The most attended protein–ligand contacts in affinity prediction do not reveal native contacts underlying affinities at a useful level. The conclusion maintains regardless of the representation of molecules (sequences/strings or graphs) or the architecture of neural networks. We have tackled the challenge with three innovative, methodological advances. First, we introduce structure-aware constraints to regularize attentions (or guide their sparsity patterns), using sequence-predicted structural contexts such as protein surfaces and protein residue–residue contact maps. Second, we exploit available native contacts to supervise novel joint attentions, that is, to teach neural network how to weigh residue-atom pairs when making affinity predictions. Lastly, we build intrinsically explainable model architecture where various atomic relations, reflecting physics laws, are explicitly modeled and aggregated for affinity prediction. Joint attentions are embedded over residue-atom pairs for their relations. A multistage hierarchy, trained end-to-end, progressively focuses attentions on protein surfaces, binding k-mers and residues, and residue-atom contact pairs. The first two advances are introduced in both DeepAffinity+ and DeepRelations; and the last is additionally introduced in DeepRelations. Their best versions involve hierarchical recurrent neural networks (HRNN) to embed protein sequences and graph convolutional networks (GCN) to embed compound graphs.
Empirical results demonstrate the superiority of DeepAffinity+ and DeepRelations in interpretable and accurate prediction of compound–protein interactions. Their affinity prediction shows generalizability to compounds or/and proteins that are new or even dissimilar to training data. Compared to a competing interpretable method, they boosted the AUPRC for contact prediction (a measure of interpretability) by around 33-, 10-, 35-, and 9-fold for the default test, new-protein, new-compound, and both-new sets, respectively. Importantly, improved model interpretability has shown to contribute to improve model accuracy and generalizability.
Case studies suggest that DeepAffinity+ and DeepRelations predict not only more correct but also more well-patterned contacts that are focused in or near binding sites, thanks to the structure-aware regularization and supervision of joint attentions. A global analysis indicates that around 40% (20%) of our top-10 predicted contacts are native contacts that are direct and first-shell for the test and the new-compound set (the new-protein and both-new set). Many “incorrect” predictions because of the strict definition of native contacts were within reasonable ranges—in fact, around 70% (50%) of the top-10 predicted contacts correspond to residue-atom pairs within 10 Å when the set does not (does) involve a new protein. With the precision level and the focused pattern, our top-10 contact predictions (including false positives) have demonstrated their value in assisting and improving protein–ligand docking, while the protocol to incorporate the predictions into docking remains to be optimized.
By aggregating joint attention and decomposing predicted affinities, we also demonstrate additional utilities of our explainable affinity and contact predictor, toward drug-discovery tasks such as binding site prediction, SAR (scoring), and SAR (lead optimization). Although not directly designed nor optimized for these tasks, our methods and analyses have shown great potentials in these tasks toward facilitating drug discovery.
An additional benefit of our structure-free methods is their broad applicability toward the vast chemical and proteomic spaces. They do not rely on 3D structures of compound–protein complexes or even proteins alone when such structures are often unavailable. The only inputs needed are protein sequences and compound graphs. Meanwhile, they adopt the latest technology to predict structural contexts from protein sequences (such as surfaces, secondary structures, and residue–residue contact maps). They introduce structure-aware regularization to incorporate the predicted structural contexts into affinity and contact predictions. When structure data are available, DeepRelations can readily integrate such data by using actual rather than predicted structural contexts. We tested the use of actual versus predicted protein residue–residue contact maps and did not observe significant performance differences in our cases (Table S12).
Our study demonstrates that it is much more effective to directly teach explainability to machine learning models (such as our structure-aware regularization and supervision of joint attentions) and build explainability into model architectures (such as our explicit modeling of atomic relations in DeepRelations) than to demand explainability from general-purpose models (such as seeking contact-interpretation from unsupervised, generic attention mechanisms). In other words, designing intrinsically interpretable machine learning models incorporated with domain knowledge, although more difficult, can be much more desired than pursuing interpretability in a post hoc manner.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by the National Institutes of Health (R35GM124952) and the National Science Foundation (CCF-1943008). Part of the computing time was provided by the Texas A&M High Performance Research Computing.
Footnotes
The authors declare no competing financial interest.
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jcim.0c00866.
Data curation, compound preprocessing, protein residue–residue contact map prediction, compound similarity calculation, compound property distributions, and atom features for compounds, numeric details, ablation study for DeepRelations, SAR performance details of individual methods, compounds in JAK2 and TIE2 subchallenges, and lead-optimization performance details (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jcim.0c00866
Contributor Information
Mostafa Karimi, Department of Electrical and Computer Engineering and TEES-AgriLife Center for Bioinformatics and Genomic Systems Engineering, Texas A&M University, College Station, Texas 77843, United States.
Di Wu, Department of Electrical and Computer Engineering, Texas A&M University, College Station, Texas 77843, United States.
Zhangyang Wang, Department of Computer Science and Engineering, Texas A&M University, College Station, Texas 77843, United States; Department of Electrical and Computer Engineering, The University of Texas at Austin, Austin, Texas 78712, United States.
Yang Shen, Department of Electrical and Computer Engineering and TEES-AgriLife Center for Bioinformatics and Genomic Systems Engineering, Texas A&M University, College Station, Texas 77843, United States;.
REFERENCES
- (1).Santos R; Ursu O; Gaulton A; Bento AP; Donadi RS; Bologa CG; Karlsson A; Al-Lazikani B; Hersey A; Oprea TI; Overington JP A comprehensive map of molecular drug targets. Nat. Rev. Drug Discovery 2017, 16, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Bohacek RS; McMartin C; Guida WC The art and practice of structure-based drug design: A molecular modeling perspective. Med. Res. Rev 1996, 16, 3–50. [DOI] [PubMed] [Google Scholar]
- (3).Ponomarenko EA; Poverennaya EV; Ilgisonis EV; Pyatnitskiy MA; Kopylov AT; Zgoda VG; Lisitsa AV; Archakov AI The Size of the Human Proteome: The Width and Depth. Int. J. Anal. Chem 2016, 2016, 7436849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Aebersold R; Agar JN; Amster IJ; Baker MS; Bertozzi CR; Boja ES; Costello CE; Cravatt BF; Fenselau C; Garcia BA; Ge Y; Gunawardena J; Hendrickson RC; Hergenrother PJ; Huber CG; Ivanov AR; Jensen ON; Jewett MC; Kelleher NL; Kiessling LL; Krogan NJ; Larsen MR; Loo JA; Ogorzalek Loo RR; Lundberg E; MacCoss MJ; Mallick P; Mootha VK; Mrksich M; Muir TW; Patrie SM; Pesavento JJ; Pitteri SJ; Rodriguez H; Saghatelian A; Sandoval W; Schlüter H; Sechi S; Slavoff SA; Smith LM; Snyder MP; Thomas PM; Uhlén M; Van Eyk JE; Vidal M; Walt DR; White FM; Williams ER; Wohlschlager T; Wysocki VH; Yates NA; Young NL; Zhang B How many human proteoforms are there? Nat. Chem. Biol 2018, 14, 206–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Muegge I; Rarey M Small molecule docking and scoring. Rev. Comput. Chem 2001, 17, 1–60. [Google Scholar]
- (6).Gilson MK; Zhou H-X Calculation of protein-ligand binding affinities. Annu. Rev. Biophys. Biomol. Struct 2007, 36, 21–42. [DOI] [PubMed] [Google Scholar]
- (7).Kim R; Skolnick J Assessment of programs for ligand binding affinity prediction. J. Comput. Chem 2008, 29, 1316–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Gathiaka S; Liu S; Chiu M; Yang H; Stuckey JA; Kang YN; Delproposto J; Kubish G; Dunbar JB; Carlson HA; et al. D3R grand challenge 2015: Evaluation of protein-ligand pose and affinity predictions. J. Comput.-Aided Mol. Des 2016, 30, 651–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Wagner JR; Churas CP; Liu S; Swift RV; Chiu M; Shao C; Feher VA; Burley SK; Gilson MK; Amaro RE Continuous Evaluation of Ligand Protein Predictions: A Weekly Community Challenge for Drug Docking. Structure 2019, 27, 1326–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Wallach I; Dzamba M; Heifets A AtomNet: a deep convolutional neural network for bioactivity prediction in structure-based drug discovery. 2015, arXiv:1510.02855. arXiv preprint.
- (11).Gomes J; Ramsundar B; Feinberg EN; Pande VS Atomic convolutional networks for predicting protein-ligand binding affinity. 2017, arXiv:1703.10603. arXiv preprint.
- (12).Jiménez J;Škalič M; Martínez-Rosell G; De Fabritiis G KDEEP: Protein-Ligand Absolute Binding Affinity Prediction via 3D-Convolutional Neural Networks. J. Chem. Inf. Model 2018, 58, 287–296. [DOI] [PubMed] [Google Scholar]
- (13).Torng W; Altman RB Graph Convolutional Neural Networks for Predicting Drug-Target Interactions. J. Chem. Inf. Model 2019, 59, 4131–4149. [DOI] [PubMed] [Google Scholar]
- (14).Lim J; Ryu S; Park K; Choe YJ; Ham J; Kim WY Predicting Drug-Target Interaction Using a Novel Graph Neural Network with 3D Structure-Embedded Graph Representation. J. Chem. Inf. Model 2019, 59, 3981–3988. [DOI] [PubMed] [Google Scholar]
- (15).Öztürk H;Özgür A; Ozkirimli E DeepDTA: deep drug-target binding affinity prediction. Bioinformatics 2018, 34, i821–i829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Karimi M; Wu D; Wang Z; Shen Y DeepAffinity: interpretable deep learning of compound-protein affinity through unified recurrent and convolutional neural networks. Bioinformatics 2019, 35, 3329–3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Feng Q; Dueva EV; Cherkasov A; Ester M PADME: A Deep Learning-based Framework for Drug-Target Interaction Prediction. CoRR 2018, arXiv:1807.09741. [Google Scholar]
- (18).Gao KY; Fokoue A; Luo H; Iyengar A; Dey S; Zhang P Interpretable Drug Target Prediction Using Deep Neural Representation. International Joint Conference on Artificial Intelligence, 2018; pp 3371–3377. [Google Scholar]
- (19).Li X; Yan X; Gu Q; Zhou H; Wu D; Xu J DeepChemStable: Chemical Stability Prediction with an Attention-Based Graph Convolution Network. J. Chem. Inf. Model 2019, 59, 1044–1049. [DOI] [PubMed] [Google Scholar]
- (20).Uddin MR; Mahbub S; Rahman MS; Bayzid MS SAINT: Self-Attention Augmented Inception-Inside-Inception Network Improves Protein Secondary Structure Prediction. Bioinformatics 2020, 36, 4599–4608. [DOI] [PubMed] [Google Scholar]
- (21).McCloskey K; Taly A; Monti F; Brenner MP; Colwell LJ Using attribution to decode binding mechanism in neural network models for chemistry. Proc. Natl. Acad. Sci 2019, 116, 11624–11629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Bahdanau D; Cho K; Bengio Y Neural machine translation by jointly learning to align and translate. 2014, arXiv:1409.0473. arXiv preprint.
- (23).Lu J; Yang J; Batra D; Parikh D Hierarchical question-image co-attention for visual question answering. Advances in Neural Information Processing Systems, 2016; pp 289–297. [Google Scholar]
- (24).Xu K; Ba J; Kiros R; Cho K; Courville A; Salakhudinov R; Zemel R; Bengio Y Show, attend and tell: Neural image caption generation with visual attention. International Conference on Machine Learning, 2015; pp 2048–2057. [Google Scholar]
- (25).Choi E; Bahadori MT; Sun J; Kulas J; Schuetz A; Stewart W Retain: An interpretable predictive model for healthcare using reverse time attention mechanism. Advances in Neural Information Processing Systems, 2016; pp 3504–3512. [Google Scholar]
- (26).Das A; Agrawal H; Zitnick L; Parikh D; Batra D Human attention in visual question answering: Do humans and deep networks look at the same regions? Comput. Vis. Image Understand 2017, 163, 90–100. [Google Scholar]
- (27).Doshi-Velez F; Kim B Towards A Rigorous Science of Interpretable Machine Learning. 2017, arXiv:1702.08608.
- (28).Dill K; Bromberg S Molecular Driving Forces: Statistical Thermodynamics in Biology, Chemistry, Physics, and Nanoscience; Garland Science, 2012. [Google Scholar]
- (29).Brzozowski AM; Pike ACW; Dauter Z; Hubbard RE; Bonn T; Engström O;Öhman L; Greene GL; Gustafsson J-Å; Carlquist M Molecular basis of agonism and antagonism in the oestrogen receptor. Nature 1997, 389, 753. [DOI] [PubMed] [Google Scholar]
- (30).Congreve M; Murray CW; Blundell TL Keynote review: Structural biology and drug discovery. Drug discovery today 2005, 10, 895–907. [DOI] [PubMed] [Google Scholar]
- (31).Wlodawer A; Vondrasek J Inhibitors of HIV-1 protease: a major success of structure-assisted drug design. Annu. Rev. Biophys. Biomol. Struct 1998, 27, 249–284. [DOI] [PubMed] [Google Scholar]
- (32).Brik A; Wong C-H HIV-1 protease: mechanism and drug discovery. Org. Biomol. Chem 2003, 1, 5–14. [DOI] [PubMed] [Google Scholar]
- (33).Yang F; Du M; Hu X Evaluating explanation without ground truth in interpretable machine learning. 2019, arXiv:1907.06831. arXiv preprint.
- (34).Santoro A; Raposo D; Barrett DG; Malinowski M; Pascanu R; Battaglia P; Lillicrap T A simple neural network module for relational reasoning. Advances in Neural Information Processing Systems, 2017; pp 4967–4976. [Google Scholar]
- (35).Lu C; Krishna R; Bernstein M; Fei-Fei L Visual relationship detection with language priors. European Conference on Computer Vision, 2016; pp 852–869. [Google Scholar]
- (36).Battaglia P; Pascanu R; Lai M; Rezende DJ Interaction networks for learning about objects, relations and physics. Advances in Neural Information Processing Systems, 2016; pp 4502–4510. [Google Scholar]
- (37).Hoshen Y Vain: Attentional multi-agent predictive modeling. Advances in Neural Information Processing Systems, 2017; pp 2701–2711. [Google Scholar]
- (38).Liu T; Lin Y; Wen X; Jorissen RN; Gilson MK BindingDB: a web-accessible database of experimentally determined protein-ligand binding affinities. Nucleic Acids Res. 2007, 35, D198–D201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Liu Z; Li Y; Han L; Li J; Liu J; Zhao Z; Nie W; Liu Y; Wang R PDB-wide collection of binding data: current status of the PDBbind database. Bioinformatics 2015, 31, 405–412. [DOI] [PubMed] [Google Scholar]
- (40).Kim S; Chen J; Cheng T; Gindulyte A; He J; He S; Li Q; Shoemaker BA; Thiessen PA; Yu B; Zaslavsky L; Zhang J; Bolton EE PubChem 2019 update: improved access to chemical data. Nucleic Acids Res. 2018, 47, D1102–D1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).RDKit: Open-Source Cheminformatics. http://www.rdkit.org (Online; accessed April 2019).
- (42).Berman HM; Westbrook J; Feng Z; Gilliland G; Bhat TN; Weissig H; Shindyalov IN; Bourne PE The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Laskowski RA; Jabłońska J; Pravda L; Vařeková RS; Thornton JM PDBsum: Structural summaries of PDB entries. Protein Sci. 2018, 27, 129–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Cheng J; Randall AZ; Sweredoski MJ; Baldi P SCRATCH: a protein structure and structural feature prediction server. Nucleic Acids Res. 2005, 33, W72–W76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Magnan CN; Baldi P SSpro/ACCpro 5: almost perfect prediction of protein secondary structure and relative solvent accessibility using profiles, machine learning and structural similarity. Bioinformatics 2014, 30, 2592–2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Wang S; Sun S; Li Z; Zhang R; Xu J Accurate de novo prediction of protein contact map by ultra-deep learning model. PLoS Comput. Biol 2017, 13, No. e1005324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Lee I; Keum J; Nam H DeepConv-DTI: Prediction of drug-target interactions via deep learning with convolution on protein sequences. PLoS Comput. Biol 2019, 15, No. e1007129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Trinh TH; Dai AM; Luong M-T; Le QV Learning longer-term dependencies in rnns with auxiliary losses. 2018, arXiv:1803.00144. arXiv preprint.
- (49).Cortés-Ciriano I; Bender A KekuleScope: prediction of cancer cell line sensitivity and compound potency using convolutional neural networks trained on compound images. J. Cheminf 2019, 11, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Kipf TN; Welling M Semi-supervised classification with graph convolutional networks. 2016, arXiv:1609.02907. arXiv preprint.
- (51).Xu K; Hu W; Leskovec J; Jegelka S How powerful are graph neural networks? 2018, arXiv:1810.00826. arXiv preprint.
- (52).Weisfeiler B; Lehman AA A reduction of a graph to a canonical form and an algebra arising during this reduction. Nauchno-Technicheskaya Informatsia 1968, 2, 12–16. [Google Scholar]
- (53).Yang Z; Yang D; Dyer C; He X; Smola A; Hovy E Hierarchical attention networks for document classification. Proceedings of the 2016 Conference of the North American Chapter of the Association for Computational Linguistics: Human Language Technologies, 2016; pp 1480–1489. [Google Scholar]
- (54).Branden CI; Tooze J Introduction to Protein Structure; Garland Science, 2012. [Google Scholar]
- (55).Pommié C; Levadoux S; Sabatier R; Lefranc G; Lefranc M-P IMGT standardized criteria for statistical analysis of immunoglobulin V-REGION amino acid properties. J. Mol. Recognit 2004, 17, 17–32. [DOI] [PubMed] [Google Scholar]
- (56).Sirimulla S; Bailey JB; Vegesna R; Narayan M Halogen Interactions in Protein-Ligand Complexes: Implications of Halogen Bonding for Rational Drug Design. J. Chem. Inf. Model 2013, 53, 2781–2791. [DOI] [PubMed] [Google Scholar]
- (57).Roy K; Kar S; Das RN Understanding the Basics of QSAR for Applications in Pharmaceutical Sciences and Risk Assessment; Academic Press, 2015. [Google Scholar]
- (58).Todeschini R; Consonni V Handbook of Molecular Descriptors; John Wiley & Sons, 2008; Vol. 11. [Google Scholar]
- (59).Wang Z; Chang S; Yang Y; Liu D; Huang TS Studying very low resolution recognition using deep networks. IEEE CVPR; IEEE, 2016; pp 4792–4800. [Google Scholar]
- (60).Davis MI; Hunt JP; Herrgard S; Ciceri P; Wodicka LM; Pallares G; Hocker M; Treiber DK; Zarrinkar PP Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol 2011, 29, 1046. [DOI] [PubMed] [Google Scholar]
- (61).Tang J; Szwajda A; Shakyawar S; Xu T; Hintsanen P; Wennerberg K; Aittokallio T Making sense of large-scale kinase inhibitor bioactivity data sets: a comparative and integrative analysis. J. Chem. Inf. Model 2014, 54, 735–743. [DOI] [PubMed] [Google Scholar]
- (62).He T; Heidemeyer M; Ban F; Cherkasov A; Ester M SimBoost: a read-across approach for predicting drug–target binding affinities using gradient boosting machines. J. Cheminf 2017, 9, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Thafar M; Raies AB; Albaradei S; Essack M; Bajic VB Comparison Study of Computational Prediction Tools for Drug-Target Binding Affinities. Front. Chem 2019, 7, 782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Pahikkala T; Okser S; Airola A; Salakoski T; Aittokallio T Wrapper-based selection of genetic features in genome-wide association studies through fast matrix operations. Algorithm Mol. Biol 2012, 7, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Öztürk H; Ozkirimli E;Özgür A WideDTA: prediction of drug-target binding affinity. 2019, arXiv:1902.04166. arXiv preprint. [DOI] [PMC free article] [PubMed]
- (66).Gönen M; Heller G Concordance probability and discriminatory power in proportional hazards regression. Biometrika 2005, 92, 965–970. [Google Scholar]
- (67).Veerasamy R; Rajak H; Jain A; Sivadasan S; Varghese CP; Agrawal RK Validation of QSAR models-strategies and importance. Int. J. Drug Des. Discovery 2011, 3, 511–519. [Google Scholar]
- (68).Trott O; Olson AJ AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem 2010, 31, 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Feinstein WP; Brylinski M Calculating an optimal box size for ligand docking and virtual screening against experimental and predicted binding pockets. J. Cheminf 2015, 7, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Bell EW; Zhang Y DockRMSD: an open-source tool for atom mapping and RMSD calculation of symmetric molecules through graph isomorphism. J. Cheminf 2019, 11, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Gaieb Z; Parks CD; Chiu M; Yang H; Shao C; Walters WP; Lambert MH; Nevins N; Bembenek SD; Ameriks MK; Mirzadegan T; Burley SK; Amaro RE; Gilson MK D3R Grand Challenge 3: blind prediction of protein-ligand poses and affinity rankings. J. Comput.-Aided Mol. Des 2019, 33, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.