

Visual Abstract

Key Words: angiogenesis, angiopoietin, arteriovenous malformation, Glenn, von Willebrand factor
Abbreviations and Acronyms: ADAMTS-13, a disintegrin and metalloproteinase thrombospondin (motif) #13; AVM, arteriovenous malformation; EBM, endothelial basal media; EGM, endothelial growth media; HUVEC, human umbilical vein endothelial cell; IVC, inferior vena cava; LVAD, left ventricular assist device; PA, pulmonary artery; SVC, superior vena cava; vWF, von Willebrand factor
Highlights
-
•
Children with a bidirectional superior cavopulmonary connection (Glenn circulation) develop dysregulated angiogenesis and pulmonary angiodysplasia in the form of arteriovenous malformations (AVMs). No targeted therapy exists.
-
•
The von Willebrand factor (vWF)–angiopoietin axis plays a major role in normal angiogenesis, angiodysplasia, and AVM formation in multiple diseases.
-
•
vWF and angiopoietin-2 (which destabilizes vessel formation) were abnormal in children with a Glenn circulation versus control children. Within Glenn patients, angiopoietin-1 (which stabilizes vessel formation) and angiogenesis were different in the systemic versus pulmonary circulation. Plasma angiopoietin-1 was lower in the pulmonary circulation of Glenn patients with pulmonary AVMs than Glenn patients without AVMs.
-
•
In parallel, differences in multiple angiogenic and inflammatory signaling peptides were observed between Glenn patients and controls, which indicated derangements in multiple angiogenic pathways in Glenn patients.
-
•
These findings support the novel hypothesis that abnormal vWF metabolism and angiopoietin signaling dysregulate angiogenesis and contribute to pulmonary AVM formation in children with a Glenn circulation.
-
•
The vWF-angiopoietin axis may be a target to correct angiogenic imbalance and reduce pulmonary angiodysplasia in Glenn patients.
Summary
Children with a bidirectional superior cavopulmonary (Glenn) circulation develop angiodysplasia and pulmonary arteriovenous malformations (AVMs). The von Willebrand factor (vWF)–angiopoietin axis plays a major role in AVM formation in multiple diseases. We observed derangements in global angiogenic signaling, vWF metabolism, angiopoietins, and in vitro angiogenesis in children with a Glenn circulation versus controls and within Glenn pulmonary versus systemic circulations. These findings support the novel hypothesis that abnormalities in the vWF-angiopoietin axis may dysregulate angiogenesis and contribute to Glenn pulmonary AVMs. The vWF-angiopoietin axis may be a target to correct angiogenic imbalance in Glenn patients, for whom no targeted therapy exists.
Infants born with a single functional ventricle, a common and uniformly fatal form of congenital heart disease, require a series of staged operations that culminate in a total cavopulmonary connection (Fontan circulation) to survive. In the Fontan circulation, both superior vena cava (SVC) and inferior vena cava (IVC) blood flow is completely diverted to the pulmonary artery (PA) and lungs. Before the Fontan operation, infants undergo a bidirectional cavopulmonary connection (the Glenn operation) in which only SVC blood is diverted to the lungs and IVC blood continues to return to the heart, where it is pumped directly to the systemic circulation by the single ventricle. As such, the Glenn circulation is unique in that all pulmonary blood flow comes from the SVC and is therefore nonpulsatile and does not contain venous effluent from the liver.
For reasons that are unclear, when the SVC is the only source of pulmonary blood flow, pulmonary angiogenesis becomes dysregulated (1). Patients develop abnormal pulmonary microvessel density, which in its most severe form presents as macroscopic pulmonary arteriovenous malformations (AVMs) (2,3). As a result, all patients with a Glenn circulation develop some degree of intrapulmonary right-to-left shunting that may cause progressive cyanosis, hypoxia, exercise intolerance, and clinical deterioration (4,5). Currently no targeted medical therapy exists to prevent or treat pulmonary AVMs in infants with a Glenn circulation, in part because the mechanism of pulmonary angiodysplasia in Glenn patients is not understood.
von Willebrand factor (vWF), a large, multimeric glycoprotein composed of long chains of hundreds of vWF monomers, is the largest protein that circulates in the blood and participates in primary hemostasis (6). Emerging evidence demonstrates that vWF is an early regulator of angiogenesis that modulates angiopoietin-2, a potent destabilizer of blood vessels (7,8). Normal vWF multimers are necessary for normal angiogenesis. In contrast, abnormal metabolism of vWF may alter angiopoietin signaling and contribute to the development of angiodysplasia and AVMs in multiple human diseases (7,9,10). For example, patients with a nonpulsatile left ventricular assist device (LVAD) develop an acquired vWF deficiency and mucosal AVMs (11, 12, 13). High shear stress from the LVAD allows a disintegrin and metalloproteinase thrombospondin (motif) #13 (ADAMTS-13), the vWF protease produced by the liver, to degrade large vWF multimers into small vWF degradation fragments (14,15). In parallel, lack of pulsatile blood flow reduces endothelial secretion of new vWF multimers to replenish depleted vWF stores (16). As a result, LVAD patients acquire a vWF deficiency and develop angiodysplasia (11,17,18). Clinically, 18% to 40% of LVAD patients develop mucosal AVMs (12,13,17).
In contrast, mechanisms of angiodysplasia and AVM formation in Glenn patients are poorly understood. When the SVC is the only source of pulmonary blood flow, 2 potential mechanisms may directly alter vWF metabolism and thereby alter pulmonary angiogenesis (3). First, nonpulsatile pulmonary blood flow may reduce endothelial secretion of vWF in the pulmonary vasculature. Second, in a Glenn circulation, there is no direct blood flow from the liver, which produces ADAMTS-13 that influences degradation of vWF multimers into vWF degradation fragments. As a result, it is possible that abnormal levels of vWF multimers and fragments may change angiogenic signaling in the pulmonary vasculature and contribute to pulmonary AVM formation in patients with a Glenn circulation.
With this in mind, we investigated relationships between plasma levels of 60 angiogenic and inflammatory peptides, vWF metabolism, the vWF-angiopoietin signaling axis, and in vitro angiogenesis with plasma from children with a Glenn circulation with and without angiographic evidence of macroscopic pulmonary AVMs. We tested 4 hypotheses in patients with a Glenn circulation: 1) global angiogenic and inflammatory signaling is abnormal versus controls; 2) the vWF-angiopoietin axis is abnormal versus controls and different in systemic (IVC) plasma (hepatic venous effluent present) versus pulmonary (PA) plasma (hepatic venous effluent absent); 3) in vitro angiogenesis is dysregulated versus controls and different within Glenn systemic plasma versus pulmonary plasma; and 4) abnormalities correlate with the clinical presence of pulmonary AVMs.
Methods
This study was conducted in accordance with the American Heart Association journals’ implementation of the Transparency and Openness Promotion Guidelines. The data, analytic methods, and study materials are available to other researchers for purposes of reproducing the results or replicating the procedure.
Clinical study
Study approval was obtained from the Children’s Hospital of Philadelphia Institutional Review Board (#13-009936) and the Hospital of the University of Pennsylvania Institutional Review Board (#818944). Experiments were performed with blood samples collected from children with a Glenn circulation. Patients with a secondary source of pulmonary blood flow such as antegrade pulmonary blood flow or a Kawashima circulation were not included. Informed consent was obtained for each patient.
As a control, blood samples were collected from age-matched, normal children. These samples provided a physiologic reference point for normal plasma values and normal angiogenic responses in healthy children and set the basis for comparisons to Glenn patients.
Blood samples were collected between November 2016 and March 2019. Patient demographics were collected from patient medical records.
Cardiac catheterization and blood sample collection
At the time of pre-Fontan cardiac catheterization, fresh blood was obtained from children with a bidirectional Glenn circulation (n = 22). While anesthetized, paired whole blood samples were collected from the IVC and the PA below the superior cavopulmonary anastomosis. As a control, whole venous blood samples were obtained from age-matched (2- to 4-year old) children (n = 20). Blood was anticoagulated in sodium heparin and K2 EDTA blood tubes.
Angiographic determination of the presence of AVMs
The presence of macroscopic pulmonary AVMs was determined angiographically during cardiac catheterization. Standard criteria that included characteristic angiographic appearance, rapid pulmonary transit time of contrast dye, and pulmonary venous desaturation were used.
Plasma assays
The hypothesis that the vWF-angiopoietin axis is dysregulated in Glenn patients versus controls and different within Glenn PA plasma versus Glenn IVC plasma was tested with multiple assays. ADAMTS-13, vWF multimers and degradation fragments, and angiopoietin-1 and -2 levels were quantified with Glenn and control plasma as described below.
Quantification of plasma ADAMTS-13
Plasma ADAMTS-13, the vWF-specific protease, was measured with a quantitative solid-phase sandwich-based enzyme-linked immunoassay (ELISA) (R&D Systems, Minneapolis, Minnesota). Briefly, plasma samples were added to wells coated with a monoclonal antihuman ADAMTS-13 antibody. Wells were incubated with horseradish peroxidase (HRP)–conjugated polyclonal antihuman ADAMTS-13. Tetramethylbenzidine was added to elicit a colorimetric reaction that was quantified with spectrophotometry by a microplate reader (μQuant, Bio-Tek Instruments, Highland Park, Vermont). Data were analyzed with Gen5, version 2.05 (Bio-Tek). Plasma ADAMTS-13 values were interpolated from a standard curve.
Quantification of plasma vWF antigen
Plasma vWF antigen was measured with a SimpleStep ELISA (Abcam, Cambridge, Massachusetts). Briefly, plasma samples were added to wells coated with an antihuman vWF antibody. Wells were incubated with HRP-conjugated antihuman vWF antibody. Tetramethylbenzidine was added to elicit a colorimetric reaction that was quantified with spectrophotometry by a microplate reader (μQuant). Data were analyzed with Gen5, version 2.05 (Bio-Tek). Plasma ADAMTS-13 values were interpolated from a standard curve.
Gel electrophoresis and immunoblotting to quantify vWF multimers and fragments
High-molecular-weight plasma vWF multimers and vWF degradation fragments were resolved by standard electrophoresis and immunoblotting techniques (14,19, 20, 21). To resolve multimers, plasma was diluted in NuPAGE LDS Sample Buffer (Invitrogen, Carlsbad, California), heated, and loaded into vertical 1% agarose–sodium dodecyl sulfate (SDS) gels. Electrophoresis was performed in Tris-Acetate SDS running buffer (Invitrogen) in an XCell SureLock Mini-Cell Electrophoresis System (Invitrogen).
Proteins were transferred to polyvinylidene fluoride using the iBlot dry transfer device (Invitrogen). Membranes were blocked and incubated with rabbit antihuman vWF primary antibody (1:500, Dako, Carpinteria, California) overnight. Membranes were incubated with goat antirabbit immunoglobulin G HRP-conjugated secondary antibody (1:3,000, Cell Signaling, Danvers, Massachusetts), developed with Luminata Forte Western Blot HRP Substrate (Millipore, Billerica, Massachusetts), and imaged with an ImageQuant LAS 4000 (GE Healthcare, Piscataway, New Jersey).
To resolve degradation fragments, plasma was diluted in NuPAGE LDS Sample Buffer, heated, and loaded into NuPAGE 3% to 8% Tris-Acetate Polyacrylamide gels (Invitrogen). Electrophoresis was performed in Tris-Acetate SDS running buffer (Invitrogen). Protein was transferred, blocked, probed for vWF, and imaged as described above. As a loading control, each membrane was probed for human plasma albumin with a goat antihuman albumin HRP-conjugated antibody (1:10,000, Abcam).
Quantification of plasma vWF
Paired plasma samples (IVC and PA) from each Glenn patient plus a composite control were blotted in adjacent lanes. The difference in high-molecular-weight vWF multimers was quantified as the percent difference in total length of multimers in Glenn IVC or PA plasma versus controls (22). The density of vWF multimers and fragments was quantified as the mean difference in density of all multimers or all degradation fragments from Glenn IVC or Glenn PA plasma versus controls. ImageQuantTL (GE Healthcare) and ImageJ (National Institutes of Health) were used to generate and analyze densitometric plots, respectively.
Quantification of plasma angiogenic and inflammatory peptides
Sixty angiogenic and inflammatory peptides were quantified with a multiplexed sandwich ELISA-based quantitative protein microarray (Quantibody Human Angiogenesis Array 1000, RayBiotech, Norcross, Georgia). Capture antibodies for 60 human peptides were adhered to wells on a glass plate. Plasma samples were diluted 1:1 in a physiologic buffer and incubated with capture antibodies for 2 h at room temperature. Sample wells were washed, and 60 biotinylated detection antibodies were added. After washing, a streptavidin-labeled Cy3 fluorescent probe was added. Peptide quantification was performed by the manufacturer with a laser scanner (GenePix 4100A, Molecular Devices, San Jose, California), GenePix Pro7 (Molecular Devices), and Q-Analyzer Software (Raybiotech). Peptide levels were interpolated from standard curves.
Cell culture experiments
The hypothesis that angiogenesis in Glenn patients is dysregulated versus control children and different within Glenn IVC plasma (hepatic venous effluent present) versus PA plasma (no hepatic venous effluent present) was tested with in vitro endothelial cell culture experiments. These experiments were designed to interrogate endothelial cell proliferation and tubule and hub formation with in vitro endothelial cell culture. As such, these experiments reflected activities of angiogenesis, which when dysregulated, may contribute to angiodysplasia and AVM formation.
Human umbilical vein endothelial cells (HUVECs) (Lonza, Walkersville, Maryland) were cultured in buffered endothelial growth media (EGM-2, Lonza) at 37°C with 5% CO2. Third- or fourth-passage cells were used. Before experiments, HUVECs were trypsinized and washed with endothelial basal media (EBM, Lonza).
A fluorescent-labeled 5-ethynyl-2 deoxyuridine (EdU) uptake experiment was used to quantify endothelial cell proliferation, an established angiogenesis assay that evaluates the ability of endothelial cells to actively uptake and incorporate EdU, a nucleoside analogue, into newly synthesized DNA. Cells were incubated at a density of ∼9,000 cells/cm2 in 50/50 EBM/plasma from Glenn IVC, Glenn PA, or controls, exposed to 10 μM EdU for 2.5 h, and then fixed in 4% paraformaldehyde for 15 min. Nuclei were counterstained with 4′,6-diamidino-2-phenylindole. Endothelial cell proliferation was quantified by dividing counted EdU-positive nuclei by total nuclei per unit area.
A Matrigel assay was used to quantify the number of endothelial cell hubs and tubules, established metrics of angiogenesis that show the ability of endothelial cells to sprout and form vascular networks. HUVECs were cultured on Matrigel (Corning Lifesciences, Corning, New York) at a density of ∼30,000 cells/cm2 and incubated with 50/50 EBM/plasma from Glenn IVC, Glenn PA, or controls. Cells were imaged at baseline and after 12 h of growth. Cells were viewed with light microscopy and analyzed with ImageJ.
Statistics
GraphPad, version 6.00 (Prism, GraphPad Software, Inc. La Jolla, California) was used to perform statistical analyses and plot data. Normality of continuous data distributions was assessed with Kolmogorov-Smirnov tests. Parametric data were compared with one-way analysis of variance with Tukey post hoc test as well as paired or unpaired Student’s t tests as appropriate. Nonparametric data were compared with Wilcoxon signed rank tests for paired comparisons. Descriptive data in the text and tables are presented as mean ± standard deviation. Data in figures are presented as mean ± standard error. A value of p < 0.05 was considered statistically significant for all comparisons except those in the microarray assay. To account for multiple comparisons in the 60-peptide microarray data set, a Benjamini-Hochberg procedure was performed, and p < 0.01 was considered statistically significant.
Results
Patient demographics
Blood samples were collected from 22 children (32 ± 7 months of age) with a Glenn circulation and 20 healthy control children (24 to 48 months of age). The average body surface area of Glenn patients was 0.57 ± 0.6 m2, weight was 13 ± 2 kg, and 13 of 22 patients (59%) were female. Anatomic forms of single-ventricle heart disease in this study included: hypoplastic left heart syndrome (n = 11), pulmonary atresia with intact ventricular septum (n = 5), tricuspid atresia (n = 2), Ebstein’s anomaly (n = 2), double inlet right ventricle (n = 1), and double outlet right ventricle (n = 1).
Plasma ADAMTS-13 and vWF
Plasma ADAMTS-13 was significantly higher in both the IVC and PA of children with a Glenn circulation versus controls (control 589 ± 140 ng/ml, IVC 969 ± 281 ng/ml, PA 971 ± 226 ng/ml; p < 0.001) (Figure 1A). vWF antigen was significantly reduced in the PA and tended to decrease in the IVC (control 0.79 ± 0.27 IU/ml, IVC 0.69 ± 0.21 IU/ml; p = 0.172; PA 0.65 ± 0.17 IU/ml; p = 0.047 (Figure 1B).
Figure 1.

von Willebrand Factor Metabolism Is Abnormal in Patients With a Glenn Circulation
(A) In an enzyme-linked immunosorbent assay (ELISA), a disintegrin and metalloproteinase thrombospondin (motif) #13 (ADAMTS-13) was significantly higher in children with a Glenn circulation than controls. (B) In an ELISA, plasma von Willebrand factor (vWF) antigen was modestly lower in children with Glenn circulation than controls. (C) Representative gel electrophoresis for vWF showed a different pattern of high-molecular-weight (HMW) vWF multimers and vWF fragments in children with a Glenn circulation than controls. The top blot (green box) showed modestly decreased HMW vWF multimers in Glenn patients versus controls. The bottom blot (blue box) showed increases in multiple vWF fragment bands, especially bands at approximately 460, 365, and 310 kDa. (D,E) HMW vWF length and density were significantly lower in children with a Glenn circulation than controls. (F) The density of vWF degradation fragments trended toward an increase in children with a Glenn circulation compared to controls. ANOVA = analysis of variance; IVC = inferior vena cava; PA = pulmonary artery.
In parallel, the profile of vWF multimers and vWF degradation fragments was abnormal in Glenn patients (Figures 1C to 1F). In both the IVC and PA, there were fewer of the highest-molecular-weight vWF multimers (control 0 ± 0%, IVC −4 ± 4%, PA -3 ± 5%; p = 0.006) and a lower density of total high-molecular-weight vWF multimers (control 0 ± 0%, IVC −21 ± 29%, PA −20 ± 33%; p = 0.016) versus controls. A trend toward elevated vWF degradation fragments in the IVC and PA (control 0 ± 0%, IVC 13 ± 33%; p = 0.087; PA 13 ± 32%; p = 0.069) was also observed. Within Glenn patients, significant differences in ADAMTS-13 and vWF multimers and fragments were not observed in IVC versus PA plasma.
Plasma angiogenic and inflammatory peptides
Eighteen of 60 plasma angiogenic and inflammatory peptides were significantly lower in Glenn plasma versus control plasma (p < 0.01) (Supplemental Table 1). Of these, plasma angiopoietin-1, which stabilizes blood vessels, was significantly lower (control 39,529 ± 34,795 ng/ml, IVC 14,398 ± 9,565 ng/ml, PA 10,486 ± 7,298 ng/ml; p < 0.001) and angiopoietin-2, which destabilizes blood vessels, was significantly higher (control 898 ± 485 ng/ml, IVC 2,927 ± 1,917 ng/ml, PA 2,747 ± 2,149 ng/ml; p < 0.001) in both IVC and PA plasma from children with Glenn circulation versus controls (Figure 2B). Importantly, within Glenn patients, angiopoietin-1 was significantly lower in PA versus IVC plasma (p < 0.001) (Figure 2A).
Figure 2.

Plasma Angiopoietin Levels Are Abnormal in Patients With a Glenn Circulation
(A) In an ELISA-based quantitative protein microarray, angiopoietin-1 was significantly lower in children with a Glenn circulation than controls. Within Glenn patients, angiopoietin-1 was significantly lower in PA plasma versus IVC plasma. (B) Angiopoietin-2 was significantly higher in children with a Glenn circulation than controls. Abbreviations as in Figure 1.
Significant differences between Glenn IVC and PA plasma were also noted for CXCL16 (IVC 981 ± 372 ng/ml, PA 819 ± 295 ng/ml; p < 0.001) and leukemia inhibitory factor (LIF) (IVC 148 ± 64 ng/ml, PA 191 ± 80 ng/ml; p < 0.001) (Supplemental Table 1).
Plasma vascular endothelial growth factor (VEGF) was significantly lower in both IVC and PA plasma from Glenn patients versus controls (control, 3,348 ± 2,518 ng/ml, IVC 725 ± 703 ng/ml, PA 604 ± 589 ng/ml; p < 0.001) but was not significantly different between IVC versus PA plasma (Table 1).
Table 1.
Plasma Angiogenic Peptides
| Peptide (pg/ml) | Control (n = 19) |
IVC (n = 22) |
PA (n = 22) |
p Value |
|---|---|---|---|---|
| VEGF | 3,348 ± 578 | 724 ± 153∗ | 604 ± 126∗ | <0.001 |
| VEGF R2 | 9,724 ± 2,591 | 9,319 ± 1,164 | 9,062 ± 1,097 | 0.96 |
| VEGF R3 | 3,243 ± 934 | 2,049 ± 360 | 2,187 ± 293 | 0.29 |
| VEGF-D | 716 ± 208 | 643 ± 113 | 742 ± 150 | 0.90 |
| TIE-1 | 6,012 ± 8,554 | 2,479 ± 607 | 3,642 ± 1,186 | 0.18 |
| TIE-2 | 1,545 ± 405 | 1,164 ± 195 | 1,265 ± 245 | 0.64 |
Values are mean ± SD.
IVC = inferior vena cava; PA= pulmonary artery; TIE = tyrosine kinase with immunoglobulin-like and epidermal growth factor–like domains; VEGF = vascular endothelial growth factor.
p < 0.001, vs. control.
Plasma angiogenesis
Dysregulated angiogenesis was observed with both IVC and PA plasma from children with a Glenn circulation versus controls. Endothelial cell proliferation (control 1 ± 1%, IVC 32 ± 3%, PA 30 ± 4%; p < 0.001) (Figure 3) was significantly greater with plasma from both IVC and PA plasma from Glenn patients versus controls. Within Glenn patients, endothelial cell proliferation exhibited a trend toward higher proliferation with IVC than PA plasma (p = 0.061).
Figure 3.

Endothelial Cell Proliferation Is Abnormal in Patients With a Glenn Circulation
(A) In a 5-ethynyl-2 deoxyuridine (EdU) uptake experiment, endothelial cell proliferation was significantly greater with Glenn IVC and PA plasma than with control plasma. A trend toward a difference in proliferation was observed with IVC versus PA plasma. (B to D) Characteristic micrographics of endothelial cell proliferation with control plasma and Glenn IVC and PA plasma are shown. Abbreviations as in Figure 1.
Similarly, endothelial cells grown with plasma from both IVC and PA plasma from Glenn patients exhibited significantly lower endothelial cell hub count (control 49 ± 8 hubs/hpf, IVC 31 ± 9 hubs/hpf, PA 37 ± 5 hubs/hpf; p < 0.001), tubule count (control 95 ± 17 tubules/hpf, IVC 66 ± 15 tubules/hpf, PA 76 ± 10 tubules/hpf; p < 0.001), and total tubule length (control 16.4 ± 2.3 mm/hpf, IVC 14 ± 2 mm/hpf, PA 16.5 ± 2 mm/hpf; p = 0.009) versus controls (Figure 4). Within Glenn patients, endothelial cells cultured with Glenn PA plasma exhibited a significantly higher endothelial hub count (p = 0.032), tubule count (p = 0.049), and total tubule length (p = 0.014) than endothelial cells grown with IVC plasma (Figure 4).
Figure 4.

Endothelial Cell Tubule Formation is Abnormal in Patients With a Glenn Circulation
(A,B) In a Matrigel assay, endothelial cell hub and tubule counts were significantly lower with Glenn IVC and PA plasma than with control plasma. Glenn PA plasma significantly increased endothelial cell hub and tubule counts versus Glenn IVC plasma. (C to E) Characteristic micrographics of endothelial cells grown with control plasma and Glenn IVC and PA plasma are shown. Large endothelial hubs composed of many adherent cells and lower tubule counts were noted with plasma from Glenn IVC and PA plasma but not control plasma. Abbreviations as in Figure 1.
Patients with pulmonary AVMs
Four of 22 Glenn patients exhibited macroscopic pulmonary AVMs with characteristic angiographic appearance, rapid pulmonary transit time of contrast dye, and pulmonary venous desaturation. IVC angiopoietin-1 was significantly lower in patients with AVMs than in patients without AVMs (AVM+ 7,708 ± 2,120 ng/ml, AVM- 15,973 ± 9,985 ng/ml; p = 0.037) (Figure 5). Similarly, PA angiopoietin-1 trended toward being lower in patients with AVMs than in patients without AVMs (AVM+ 6,347 ± 2,444 ng/ml, AVM- 11,406 ± 7,733 ng/ml; p = 0.107). No significant differences in plasma ADAMTS-13, vWF multimers, vWF fragments, angiopoietin-2, or metrics of angiogenesis were observed between Glenn patients with and without pulmonary AVMs.
Figure 5.
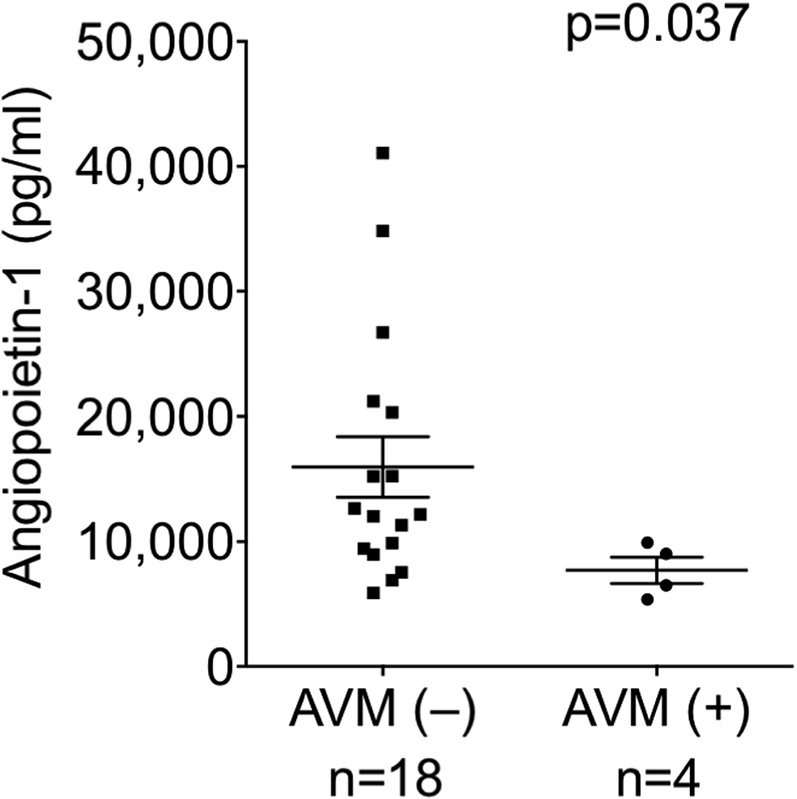
Angiopoietin-1 Is Low in Glenn Patients With Macroscopic Pulmonary AVMs
In Glenn patients with confirmed pulmonary AVMs, angiopoietin-1 levels were lower in the IVC than in patients without AVMs. Abbreviations as in Figure 1.
Discussion
In children with a Glenn circulation, we observed the following: 1) Multiple proangiogenic, antiangiogenic, and inflammatory peptide levels were different from controls. This suggested globally deranged angiogenic signaling in Glenn patients versus healthy children. 2) The vWF-angiopoietin axis was abnormal versus control children. Specifically, ADAMTS-13, vWF degradation fragments, and angiopoietin-2 (which destabilizes blood vessel formation) were higher, and vWF multimers were lower in both IVC plasma (hepatic venous effluent present) and PA plasma (hepatic venous effluent absent) versus controls but not different between the PA and IVC. In parallel, angiopoietin-1 (which stabilizes blood vessel formation) was significantly lower in Glenn PA plasma than IVC plasma. 3) Multiple metrics of in vitro angiogenesis were dysregulated with both IVC and PA plasma from Glenn patients versus controls and different between the IVC and PA. Endothelial cell proliferation was greater with both IVC and PA plasma versus controls. Differences in endothelial cell proliferation, tubule and hub counts, and total tubule length were observed with Glenn plasma from the PA versus the IVC. 4) Angiopoietin-1 levels were significantly lower in children that developed macroscopic pulmonary AVMs than children without AVMs.
These are the first data to support the novel hypothesis that altered angiogenic and inflammatory signaling plus abnormalities in vWF metabolism and angiopoietin signaling may contribute to dysregulated angiogenesis in Glenn patients. The major findings that angiopoietin-1 levels and angiogenesis are different in the Glenn pulmonary circulation than the systemic circulation and that angiopoietin-1 was lower in patients with macroscopic AVMs provide evidence for the role of angiopoietin-1 in pulmonary vascular signaling. Increased endothelial proliferation with impaired endothelial tubule formation may be an important contributor to dysregulated angiogenesis, which is the underlying basis for angiodysplasia and may result in AVM formation.
Together, these hypothesis-generating findings suggest a preliminary mechanism of AVM formation in Glenn patients in which a combination of global angiogenic and inflammatory imbalance, globally low vWF and high angiopoietin-2, plus low angiopoietin-1 in the pulmonary circulation may dysregulate angiogenesis and thereby promote angiodysplasia that results in clinical AVMs. Further investigation is needed to clarify these complex relationships and to determine whether the vWF-angiopoietin axis may be a target to correct angiogenic imbalance in Glenn patients with pulmonary AVMs, for whom no targeted therapies exist.
Clinical relationships between vWF and AVMs
Abnormal vWF metabolism is present in multiple patient populations that develop AVMs (12,23, 24, 25, 26, 27, 28). Patients with congenital and acquired von Willebrand disease frequently develop gastrointestinal AVMs (12,24, 25, 26, 27, 28, 29). For example, 12% of patients with type 2B von Willebrand disease develop AVMs (26). Similarly, patients with Heyde’s syndrome present with gastrointestinal bleeding from AVMs at a rate of 100-fold greater than the general population (28,30,31). In these patients, high shear stress through a stenotic aortic valve degrades vWF and produces an acquired vWF deficiency that is linked to the development of gastrointestinal AVMs (24). In a similar pathophysiology, high shear stress within nonpulsatile LVADs increases enzymatic degradation of large vWF multimers into vWF degradation fragments (14). As a result, approximately 18% to 40% of LVAD patients develop mucosal AVMs (12,13,17). Together, these observations from 3 diverse pathologies suggest a causal relationship between pathologic vWF metabolism, dysregulated angiogenesis, and AVMs.
vWF metabolism, angiogenesis, and AVMs
vWF metabolism is closely regulated by the rheologic conditions of the blood (6,32). Intravascular shear stress leads to continuous degradation of high-molecular-weight vWF multimers into small vWF degradation fragments by the vWF-specific protease, ADAMTS-13 (32, 33, 34, 35). To counterbalance vWF degradation, vWF is continuously replenished by endothelial cells in response to endothelial cell stretch from vascular pulsatility (16). As a result, the ratio of vWF multimers to vWF degradation fragments circulating in the blood is dependent on: 1) (ab)normal pulsatility; and 2) the (im)balance between degradation of vWF by ADAMTS-13 and replenishment of vWF by endothelial cells. Normally, the ratio of vWF multimers to vWF fragments is high. However, pathologic conditions that alter pulsatility and/or plasma ADAMTS-13 levels and activity may decrease the number of large vWF multimers and increase the number of vWF fragments. This affects not only hemostasis, but also angiogenic signaling, angiogenesis, and vascular architecture.
Indeed, vWF is a major regulator of angiogenesis and vascular remodeling (7,8). vWF plays regulatory roles in at least 20 molecular pathways involved in angiogenesis, vascular inflammation, endothelial cell life cycle, smooth muscle proliferation, and vascular remodeling (7,9,10). Importantly, vWF regulates angiogenesis in vivo by modulating angiopoietin-2 — normal vWF multimers are necessary for normal angiopoietin signaling and angiogenesis (7,9,36). Likewise, an abnormal profile of vWF multimers and vWF fragments may upset angiopoietin signaling and cause dysregulated angiogenesis.
AVMs are a poorly understood manifestation of dysregulated angiogenesis in which abnormal blood vessels form. Loss of balance between neovascular proliferation and stabilization during angiogenesis leads to dilated, dysfunctional, and friable vascular structures. Multiple signaling peptides have been implicated in the genesis of angiodysplasia and AVMs. For example, high levels of VEGF are strongly associated with angiodysplasia (37). We observed that 18 of 60 plasma angiogenic and inflammatory peptides, including VEGF, were significantly different in Glenn patients than in healthy, control children. However, VEGF was significantly lower in both systemic and pulmonary plasma of Glenn patients versus controls. Decreased VEGF suggests that the mechanism of pulmonary angiodysplasia in patients with a Glenn circulation may be independent of the VEGF pathway.
It is also well known that angiopoietins play an important role in angiodysplasia and AVM formation (8,38). Angiopoietin-1, which is produced in the liver, stabilizes blood vessels. In contrast, angiopoietin-2, which is produced primarily by endothelial cells, destabilizes blood vessels. Therefore, a simultaneous decrease in angiopoietin-1 and increase in angiopoietin-2, as we observed in our Glenn patients, produces 2 potent signals for dysregulated angiogenesis. These findings are similar to observations from patients with congenital von Willebrand disease who also exhibit abnormal levels of angiopoietin-1 and angiopoietin-2 and frequently develop gastrointestinal AVMs (26,39). Similarly, patients with a nonpulsatile LVAD develop an acquired vWF deficiency and exhibit increased angiopoietin-2 levels that are associated with mucosal AVMs (11, 12, 13,18). Together, these observations demonstrate important mechanistic and clinical relationships between vWF, angiopoietins, and AVMs. Our data suggest that these relationships may also be important in patients with a Glenn circulation.
The vWF-angiopoietin axis and angiogenesis in glenn patients
A Glenn circulation provides excellent hemodynamic stability in infants with single ventricle physiology (40). However, the durability of the Glenn circulation is limited by clinical manifestations of dysregulated angiogenesis. Glenn patients develop pulmonary AVMs that cause intrapulmonary right-to-left shunting (4) and progressive cyanosis (3, 4, 5) as well as collateral vessels from the systemic to the pulmonary circulation that increase hydrostatic pressure in the lungs and overload the single ventricle (3, 4, 5,41,42). Both AVMs and systemic-to-pulmonary collaterals cause clinical deterioration that affect outcomes after the Fontan operation (5,41,43).
Mechanisms of dysregulated angiogenesis in patients with a Glenn circulation are poorly understood. We observed that 18 of 60 plasma angiogenic and inflammatory peptides were significantly different from control children. The profile of changes was not entirely proangiogenic or antiangiogenic but rather reflected a complex combination of unbalanced pro- and antiangiogenic signals. Dysregulated angiogenic and inflammatory signaling in Glenn patients plus the complex interplay of nonpulsatile blood flow and lack of hepatic-derived factors in the Glenn pulmonary circulation may specifically alter vWF-angiopoietin signaling and lead to a unique pathophysiology in the pulmonary vascular bed that alters pulmonary angiogenesis and contributes to AVM formation as occurs in multiple other pathologies (7, 8, 9, 10).
With this in mind, in IVC and PA plasma from children with a Glenn circulation, we observed increased ADAMTS-13, decreased vWF multimers, and a trend toward increased vWF fragments. ADAMTS-13 is produced primarily by stellate cells in the liver (15). It is unclear why ADAMTS-13 was elevated. There is evidence that a Glenn circulation alters hepatic synthesis of clotting factors (44,45). We therefore speculate that altered hepatic synthetic function may have resulted in increased production of ADAMTS-13. The half-life of ADAMTS-13 is 2 to 3 days, which likely explains why differences were not observed between IVC and PA plasma. Clinically, there is significant evidence that vWF levels inversely correlate with ADAMTS-13 levels (46, 47, 48). An increase in circulating ADAMTS-13 likely increased basal degradation of vWF multimers into vWF fragments and accounted for an abnormal profile of vWF multimers and fragments that differed from controls. In parallel, absent pulsatility in the pulmonary circulation is expected to reduce endothelial secretion of vWF and decrease replenishment of vWF multimers (16). Taken together, these findings suggest parallel mechanisms that may alter the profile of circulating vWF multimers and fragments in patients with a Glenn circulation: 1) increased basal degradation of vWF by elevated ADAMTS-13 levels; and 2) decreased endothelial release of vWF into the pulmonary circulation due to absent pulmonary pulsatility.
vWF levels are closely linked to angiopoietin-2 levels (7,9,36). Therefore, it is not surprising that reduced vWF multimers were accompanied by elevated angiopoietin-2 levels in both IVC and PA plasma versus controls. These findings are consistent with data from an experimental Glenn model in rodents in which angiopoietin-2 gene expression and pulmonary microvessel density increased significantly (2,49).
Angiopoietin-1, an important stabilizer of blood vessel formation produced in the liver, was significantly reduced in both IVC and PA plasma versus controls as well as in PA versus IVC plasma within Glenn patients. We speculate that exclusion of hepatic venous effluent from the pulmonary circulation reduces available angiopoietin-1 in pulmonary arterial plasma. This finding is consistent with data from a porcine unidirectional Glenn model in which abnormal levels of the angiopoietin-1/Tie-2 (angiopoietin-1 receptor) complex were observed in the lung connected to the SVC but not the contralateral, control lung (50). There is also direct evidence that exclusion of hepatic effluent from the liver alters the profile of multiple other pro- and antiangiogenic factors in the lungs as well as indirect evidence for a “hepatic factor” responsible for pulmonary AVMs in Glenn patients — pulmonary AVMs typically regress after the Fontan operation in which the IVC is connected to the PA thereby reintroducing hepatic venous effluent into the pulmonary circulation (3,51). Our findings further highlight a major role for the liver in pulmonary vascular signaling.
Furthermore, angiopoietin-1 from the liver is important for normal lung development in vivo (52). Therefore, reduced angiopoietin-1 in the pulmonary circulation may destabilize angiogenesis in pediatric lungs, increase pulmonary microvessel density, and contribute to the formation of pulmonary AVMs (1). The observation that patients with macroscopic AVMs exhibited significantly lower levels of angiopoietin-1 supports this hypothesis.
In addition to angiopoietin-1, significant differences in CXCL16 and LIF were observed in Glenn IVC versus PA plasma. CXCL16, a chemokine involved in chemotaxis of immune cells, is not known to be involved in angiodysplasia. In contrast, LIF, a cytokine that inhibits cell differentiation, may be involved in brain AVM formation (53). It is conceptually appealing that LIF may play an ancillary role in angiodysplasia by inhibiting vascular cell differentiation. Further investigation is needed to test this hypothesis.
Study limitations
This study does not establish causality between abnormalities in vWF-angiopoietin signaling and pulmonary AVMs in patients with Glenn circulation. However, these data do suggest pathways that may be important in the genesis of angiodysplasia in patients with a Glenn circulation. Additional experimental and clinical studies are needed to further clarify these complex relationships.
AVMs are a poorly understood manifestation of dysregulated angiogenesis in which abnormal blood vessels form. Models of in vitro angiodysplasia do not exist. Although this study focused on angiodysplasia and clinical AVMs, our cell culture experiments measured in vitro angiogenesis. There are important distinctions between normal (physiologic) angiogenesis, pathologic angiogenesis in response to tissue injury, and dysregulated angiogenesis that results in angiodysplasia or clinical AVMs. This is a limitation of existing models for the study of angiogenesis versus angiodysplasia.
A potential limitation was the small number of patients studied. There is a risk that observed effects could be driven by chance findings from a few samples (type I statistical error). Encouragingly, patients showed consistent and robust biological changes across multiple overlapping assays, which suggest that this was not the case. However, with a small sample size, we may have failed to detect important effects (type II statistical error). Borderline statistically significant p values for the formation of vWF fragments (PA p = 0.06; IVC p = 0.08) and the relationship between angiopoietin-1 levels and pulmonary AVMs in PA plasma (n = 4; p = 0.13) suggested that this may have been the case.
Patients underwent angiography to identify pulmonary AVMs. Angiography is not the most sensitive method to diagnose pulmonary AVMs, especially small and subclinical AVMs. The observation that 4 of 22 patients showed angiographic evidence of macroscopic AVMs likely underestimates the true incidence of pulmonary angiodysplasia, which has been reported in 60% of Glenn patients with bubble echocardiography and may be a universal phenomenon in patients with a Glenn circulation (4,54).
Control blood was collected from healthy children without known congenital heart disease, hypoxia, or cardiovascular pathology. The purpose of the control group was to establish normal levels of angiogenic and inflammatory peptides, vWF metabolism, angiopoietins, and in vitro angiogenesis to understand how this physiology differs in Glenn patients. As such, control samples from normal children showed representative normal plasma values and normal angiogenic responses, which set the basis for comparison versus Glenn plasma. Values were measured from peripheral venous blood and not from the IVC or PA of control children.
AVMs typically regress after the Fontan operation (3). Samples were not collected from Fontan patients. It will be important to determine whether surgical construction of a Fontan circulation, which reintroduces hepatic venous effluent into the pulmonary circulation, reverses the abnormal findings observed in Glenn patients in this study.
Conclusions
In children with a Glenn circulation, global angiogenic and inflammatory signaling, vWF metabolism, angiopoietin levels, and angiogenesis are abnormal versus normal, control children. Within Glenn patients, angiogenesis is different in plasma from the PAs versus the IVC. Angiopoietin-1 (which stabilizes blood vessel formation) is significantly lower in the Glenn pulmonary circulation than in the Glenn systemic circulation. Angiopoietin-1 levels are significantly lower in children who develop pulmonary AVMs versus children who do not. These findings suggest a potential mechanism of AVM formation in which a combination of global angiogenic and inflammatory imbalance, reduced vWF and elevated angiopoietin-2, (which destabilizes blood vessel formation) plus low angiopoietin-1 levels in the pulmonary circulation dysregulate angiogenesis and thereby promote pulmonary angiodysplasia and AVM formation. Decreased VEGF suggests that the mechanism of pulmonary angiodysplasia in patients with Glenn circulation may be independent of VEGF.
These findings support the novel hypothesis that abnormalities in vWF metabolism and angiopoietin signaling superimposed on unbalanced pro- and antiangiogenic and inflammatory signaling contribute to dysregulated angiogenesis in patients with a Glenn circulation. Angiopoietin-1 may play a specific role in the genesis of pulmonary AVMs. Additional studies may lead to insights into whether targeting the vWF-angiopoietin axis may correct angiogenic imbalance in Glenn patients, for whom no targeted therapies exist. The findings of this study may also have important implications for other patient populations with abnormal vWF metabolism and angiogenesis such as those with congenital von Willebrand disease, Heyde’s syndrome, and nonpulsatile LVAD support.
Perspectives.
COMPETENCY IN MEDICAL KNOWLEDGE: Improved management of infants with complex congenital heart disease has produced an emerging population of patients with adult congenital heart disease. To improve long-term outcomes, mechanistic understanding and targeted management of pathologies that develop early in these patients are needed. In a clinical study, we observed dysregulated angiogenic and inflammatory signaling, vWF metabolism, angiopoietin levels, and in vitro angiogenesis in infants with a bidirectional Glenn circulation. These findings support the novel hypothesis that abnormal vWF metabolism and angiopoietin signaling superimposed on a background of deranged angiogenic and inflammatory signaling dysregulate angiogenesis in infants with a Glenn circulation. Resultant angiodysplasia may contribute to pulmonary AVM formation, which can cause clinical deterioration and affect outcomes.
TRANSLATIONAL OUTLOOK: In infants with a Glenn circulation, the development of pulmonary AVMs is common. Targeted therapies designed to preserve vWF multimers, normalize levels of plasma angiopoietins, and correct angiogenic imbalance may normalize angiogenesis and potentially reduce the incidence of pulmonary AVMs.
Funding Support and Author Disclosures
This project was performed with support from the Big Hearts to Little Hearts Foundation and the Congenital Heart Disease Coalition. The authors have reported that they have no relationships relevant to the contents of this paper to disclose.
Acknowledgment
The authors thank Abigail Waldron for assistance with patient recruitment throughout the project.
Footnotes
The authors attest they are in compliance with human studies committees and animal welfare regulations of the authors’ institutions and Food and Drug Administration guidelines, including patient consent where appropriate. For more information, visit the Author Center.
Appendix
For a supplemental table, please see the online version of this paper.
Appendix
References
- 1.Starnes S.L., Duncan B.W., Kneebone J.M. Pulmonary microvessel density is a marker of angiogenesis in children after cavopulmonary anastomosis. J Thorac Cardiovasc Surg. 2000;120:902–907. doi: 10.1067/mtc.2000.110248. [DOI] [PubMed] [Google Scholar]
- 2.Starnes S.L., Duncan B.W., Fraga C.H. Rat model of pulmonary arteriovenous malformations after right superior cavopulmonary anastomosis. Am J Physiol Heart Circ Physiol. 2002;283:H2151–H2156. doi: 10.1152/ajpheart.00368.2002. [DOI] [PubMed] [Google Scholar]
- 3.Srivastava D., Preminger T., Lock J.E. Hepatic venous blood and the development of pulmonary arteriovenous malformations in congenital heart disease. Circulation. 1995;92:1217–1222. doi: 10.1161/01.cir.92.5.1217. [DOI] [PubMed] [Google Scholar]
- 4.Vettukattil J.J., Slavik Z., Lamb R.K. Intrapulmonary arteriovenous shunting may be a universal phenomenon in patients with the superior cavopulmonary anastomosis: a radionuclide study. Heart. 2000;83:425–428. doi: 10.1136/heart.83.4.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kopf G.S., Laks H., Stansel H.C., Hellenbrand W.E., Kleinman C.S., Talner N.S. Thirty-year follow-up of superior vena cava-pulmonary artery (Glenn) shunts. J Thorac Cardiovasc Surg. 1990;100:662–670. discussion 670–1. [PubMed] [Google Scholar]
- 6.Sadler J.E. Biochemistry and genetics of von Willebrand factor. Annu Rev Biochem. 1998;67:395–424. doi: 10.1146/annurev.biochem.67.1.395. [DOI] [PubMed] [Google Scholar]
- 7.Starke R.D., Ferraro F., Paschalaki K.E. Endothelial von Willebrand factor regulates angiogenesis. Blood. 2011;117:1071–1080. doi: 10.1182/blood-2010-01-264507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Randi A.M., Smith K.E., Castaman G. von Willebrand factor regulation of blood vessel formation. Blood. 2018;132:132–140. doi: 10.1182/blood-2018-01-769018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Randi A.M., Laffan M.A., Starke R.D. Von Willebrand factor, angiodysplasia and angiogenesis. Mediterr J Hematol Infect Dis. 2013;5 doi: 10.4084/MJHID.2013.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lenting P.J., Casari C., Christophe O.D., Denis C.V. von Willebrand factor: the old, the new and the unknown. J Thromb Haemost. 2012;10:2428–2437. doi: 10.1111/jth.12008. [DOI] [PubMed] [Google Scholar]
- 11.Proudfoot A.G., Davidson S.J., Strueber M. von Willebrand factor disruption and continuous-flow circulatory devices. J Heart Lung Transplant. 2017;36:1155–1163. doi: 10.1016/j.healun.2017.06.004. [DOI] [PubMed] [Google Scholar]
- 12.Kang J., Hennessy-Strahs S., Kwiatkowski P. Continuous-flow LVAD support causes a distinct form of intestinal angiodysplasia. Circ Res. 2017;121:963–969. doi: 10.1161/CIRCRESAHA.117.310848. [DOI] [PubMed] [Google Scholar]
- 13.Patel S.R., Madan S., Saeed O. Association of nasal mucosal vascular alterations, gastrointestinal arteriovenous malformations, and bleeding in patients with continuous flow left ventricular assist devices. J Am Coll Cardiol HF. 2016;4:971–973. doi: 10.1016/j.jchf.2016.08.005. [DOI] [PubMed] [Google Scholar]
- 14.Bartoli C.R., Restle D.J., Zhang D.M., Acker M.A., Atluri P. Pathologic von Willebrand factor degradation with a left ventricular assist device occurs via two distinct mechanisms: mechanical demolition and enzymatic cleavage. J Thorac Cardiovasc Surg. 2015;149:281–289. doi: 10.1016/j.jtcvs.2014.09.031. [DOI] [PubMed] [Google Scholar]
- 15.Uemura M., Tatsumi K., Matsumoto M. Localization of ADAMTS13 to the stellate cells of human liver. Blood. 2005;106:922–924. doi: 10.1182/blood-2005-01-0152. [DOI] [PubMed] [Google Scholar]
- 16.Vincent F., Rauch A., Loobuyck V. Arterial pulsatility and circulating von Willebrand factor in patients on mechanical circulatory support. J Am Coll Cardiol. 2018;71:2106–2118. doi: 10.1016/j.jacc.2018.02.075. [DOI] [PubMed] [Google Scholar]
- 17.Bartoli C.R., Zhang D.M., Hennessy-Strahs S. Clinical and in vitro evidence that left ventricular assist device-induced von Willebrand factor degradation alters angiogenesis. Circ Heart Fail. 2018;11 doi: 10.1161/CIRCHEARTFAILURE.117.004638. [DOI] [PubMed] [Google Scholar]
- 18.Tabit C.E., Chen P., Kim G.H. Elevated angiopoietin-2 level in patients with continuous-flow left ventricular assist devices leads to altered angiogenesis and is associated with higher nonsurgical bleeding. Circulation. 2016;134:141–152. doi: 10.1161/CIRCULATIONAHA.115.019692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Restle D.J., Zhang D.M., Hung G. Preclinical models for translational investigations of left ventricular assist device-associated von Willebrand factor degradation. Artif Organs. 2015;39:569–575. doi: 10.1111/aor.12428. [DOI] [PubMed] [Google Scholar]
- 20.Bartoli C.R., Kang J., Restle D.J. Inhibition of ADAMTS-13 by doxycycline reduces von Willebrand factor degradation during supraphysiological shear stress: therapeutic implications for left ventricular assist device-associated bleeding. J Am Coll Cardiol HF. 2015;3:860–869. doi: 10.1016/j.jchf.2015.06.016. [DOI] [PubMed] [Google Scholar]
- 21.Dassanayaka S., Slaughter M.S., Bartoli C.R. Mechanistic pathway(s) of acquired von Willebrand syndrome with a continuous-flow ventricular assist device: in vitro findings. ASAIO J. 2013;59:123–129. doi: 10.1097/MAT.0b013e318283815c. [DOI] [PubMed] [Google Scholar]
- 22.Hennessey-Strahs S., Bermudez C.A., Acker M.A., Bartoli C.R. Toward a standard practice to quantify von Willebrand factor degradation during LVAD support. Ann Thorac Surg. 2020 doi: 10.1016/j.athoracsur.2020.09.039. Nov 20 [Epub head of print] [DOI] [PubMed] [Google Scholar]
- 23.Heilmann C., Trummer G., Beyersdorf F. Acquired von Willebrand syndrome in patients on long-term support with HeartMate II. Eur J Cardiothorac Surg. 2017;51:587–590. doi: 10.1093/ejcts/ezw348. [DOI] [PubMed] [Google Scholar]
- 24.Vincentelli A., Susen S., Le Tourneau T. Acquired von Willebrand syndrome in aortic stenosis. N Engl J Med. 2003;349:343–349. doi: 10.1056/NEJMoa022831. [DOI] [PubMed] [Google Scholar]
- 25.Castaman G., Federici A.B., Tosetto A. Different bleeding risk in type 2A and 2M von Willebrand disease: a 2-year prospective study in 107 patients. J Thromb Haemost. 2012;10:632–638. doi: 10.1111/j.1538-7836.2012.04661.x. [DOI] [PubMed] [Google Scholar]
- 26.Fressinaud E., Meyer D. International survey of patients with von Willebrand disease and angiodysplasia. Thromb Haemost. 1993;70:546. [PubMed] [Google Scholar]
- 27.Jehangir A., Pathak R., Ukaigwe A., Donato A.A. Association of aortic valve disease with intestinal angioectasia: data from the Nationwide Inpatient Sample. Eur J Gastroenterol Hepatol. 2018;30:438–441. doi: 10.1097/MEG.0000000000001068. [DOI] [PubMed] [Google Scholar]
- 28.Godino C., Lauretta L., Pavon A.G. Heyde's syndrome incidence and outcome in patients undergoing transcatheter aortic valve implantation. J Am Coll Cardiol. 2013;61:687–689. doi: 10.1016/j.jacc.2012.10.041. [DOI] [PubMed] [Google Scholar]
- 29.Duray P.H., Marcal J.M., Jr., LiVolsi V.A., Fisher R., Scholhamer C., Brand M.H. Gastrointestinal angiodysplasia: a possible component of von Willebrand's disease. Hum Pathol. 1984;15:539–544. doi: 10.1016/s0046-8177(84)80007-6. [DOI] [PubMed] [Google Scholar]
- 30.Loscalzo J. From clinical observation to mechanism—Heyde's syndrome. N Engl J Med. 2012;367:1954–1956. doi: 10.1056/NEJMcibr1205363. [DOI] [PubMed] [Google Scholar]
- 31.Warkentin T.E., Moore J.C., Anand S.S., Lonn E.M., Morgan D.G. Gastrointestinal bleeding, angiodysplasia, cardiovascular disease, and acquired von Willebrand syndrome. Transfus Med Rev. 2003;17:272–286. doi: 10.1016/s0887-7963(03)00037-3. [DOI] [PubMed] [Google Scholar]
- 32.Zhang X., Halvorsen K., Zhang C.Z., Wong W.P., Springer T.A. Mechanoenzymatic cleavage of the ultralarge vascular protein von Willebrand factor. Science. 2009;324:1330–1334. doi: 10.1126/science.1170905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dent J.A., Galbusera M., Ruggeri Z.M. Heterogeneity of plasma von Willebrand factor multimers resulting from proteolysis of the constituent subunit. Research Support, U.S. Gov't, P.H.S. J Clin Invest. 1991;88:774–782. doi: 10.1172/JCI115376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Turner N.A., Nolasco L., Ruggeri Z.M., Moake J.L. Endothelial cell ADAMTS-13 and VWF: production, release, and VWF string cleavage. Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov't. Blood. 2009;114:5102–5111. doi: 10.1182/blood-2009-07-231597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tsai H.M., Sussman, Nagel R.L. Shear stress enhances the proteolysis of von Willebrand factor in normal plasma. Blood. 1994;83:2171–2179. [PubMed] [Google Scholar]
- 36.Riba R., Oberprieler N.G., Roberts W., Naseem K.M. Von Willebrand factor activates endothelial nitric oxide synthase in blood platelets by a glycoprotein Ib-dependent mechanism. J Thromb Haemost. 2006;4:2636–2644. doi: 10.1111/j.1538-7836.2006.02195.x. [DOI] [PubMed] [Google Scholar]
- 37.Junquera F., Saperas E., de Torres I., Vidal M.T., Malagelada J.R. Increased expression of angiogenic factors in human colonic angiodysplasia. Am J Gastroenterol. 1999;94:1070–1076. doi: 10.1111/j.1572-0241.1999.01017.x. [DOI] [PubMed] [Google Scholar]
- 38.Holleran G., Hussey M., Smith S., McNamara D. Assessment of serum angiogenic factors as a diagnostic aid for small bowel angiodysplasia in patients with obscure gastrointestinal bleeding and anaemia. World J Gastrointest Pathophysiol. 2017;8:127–132. doi: 10.4291/wjgp.v8.i3.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Groeneveld D.J., Sanders Y.V., Adelmeijer J. Circulating angiogenic mediators in patients with moderate and severe von Willebrand disease: a multicentre cross-sectional study. Thromb Haemost. 2018;118:152–160. doi: 10.1160/TH17-06-0397. [DOI] [PubMed] [Google Scholar]
- 40.Day R.W., Etheridge S.P., Veasy L.G. Single ventricle palliation: greater risk of complications with the Fontan procedure than with the bidirectional Glenn procedure alone. Int J Cardiol. 2006;106:201–210. doi: 10.1016/j.ijcard.2005.01.039. [DOI] [PubMed] [Google Scholar]
- 41.Whitehead K.K., Harris M.A., Glatz A.C. Status of systemic to pulmonary arterial collateral flow after the fontan procedure. Am J Cardiol. 2015;115:1739–1745. doi: 10.1016/j.amjcard.2015.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Glatz A.C., Harrison N., Small A.J. Factors associated with systemic to pulmonary arterial collateral flow in single ventricle patients with superior cavopulmonary connections. Heart. 2015;101:1813–1818. doi: 10.1136/heartjnl-2015-307703. [DOI] [PubMed] [Google Scholar]
- 43.Glatz A.C., Rome J.J., Small A.J. Systemic-to-pulmonary collateral flow, as measured by cardiac magnetic resonance imaging, is associated with acute post-Fontan clinical outcomes. Circ Cardiovasc Imaging. 2012;5:218–225. doi: 10.1161/CIRCIMAGING.111.966986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Odegard K.C., McGowan F.X., DiNardo J.A. Coagulation abnormalities in patients with single-ventricle physiology precede the Fontan procedure. J Thorac Cardiovasc Surg. 2002;123:459–465. doi: 10.1067/mtc.2002.120010. [DOI] [PubMed] [Google Scholar]
- 45.Odegard K.C., McGowan F.X., Zurakowski D. Coagulation factor abnormalities in patients with single-ventricle physiology immediately prior to the Fontan procedure. Ann Thorac Surg. 2002;73:1770–1777. doi: 10.1016/s0003-4975(02)03580-4. [DOI] [PubMed] [Google Scholar]
- 46.Maino A., Siegerink B., Lotta L.A. Plasma ADAMTS-13 levels and the risk of myocardial infarction: an individual patient data meta-analysis. J Thromb Haemost. 2015;13:1396–1404. doi: 10.1111/jth.13032. [DOI] [PubMed] [Google Scholar]
- 47.Andersson H.M., Siegerink B., Luken B.M. High VWF, low ADAMTS13, and oral contraceptives increase the risk of ischemic stroke and myocardial infarction in young women. Blood. 2012;119:1555–1560. doi: 10.1182/blood-2011-09-380618. [DOI] [PubMed] [Google Scholar]
- 48.Scully M., Knöbl P., Kentouche K. Recombinant ADAMTS-13: first-in-human pharmacokinetics and safety in congenital thrombotic thrombocytopenic purpura. Blood. 2017;130:2055–2063. doi: 10.1182/blood-2017-06-788026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tipps R.S., Mumtaz M., Leahy P., Duncan B.W. Gene array analysis of a rat model of pulmonary arteriovenous malformations after superior cavopulmonary anastomosis. J Thorac Cardiovasc Surg. 2008;136:283–289. doi: 10.1016/j.jtcvs.2008.02.011. [DOI] [PubMed] [Google Scholar]
- 50.Kavarana M.N., Mukherjee R., Eckhouse S.R. Pulmonary artery endothelial cell phenotypic alterations in a large animal model of pulmonary arteriovenous malformations after the Glenn shunt. Ann Thorac Surg. 2013;96:1442–1449. doi: 10.1016/j.athoracsur.2013.05.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Starnes S.L., Duncan B.W., Kneebone J.M. Angiogenic proteins in the lungs of children after cavopulmonary anastomosis. J Thorac Cardiovasc Surg. 2001;122:518–523. doi: 10.1067/mtc.2001.115423. [DOI] [PubMed] [Google Scholar]
- 52.Grzenda A., Shannon J., Fisher J., Arkovitz M.S. Timing and expression of the angiopoietin-1-Tie-2 pathway in murine lung development and congenital diaphragmatic hernia. Dis Model Mech. 2013;6:106–114. doi: 10.1242/dmm.008821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Takagi Y., Aoki T., Takahashi J.C. Differential gene expression in relation to the clinical characteristics of human brain arteriovenous malformations. Neurol Med Chir (Tokyo) 2014;54:163–175. doi: 10.2176/nmc.oa2012-0422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bernstein H.S., Brook M.M., Silverman N.H., Bristow J. Development of pulmonary arteriovenous fistulae in children after cavopulmonary shunt. Circulation. 1995;92(suppl 9):II309–II314. doi: 10.1161/01.cir.92.9.309. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.