

Abstract
Apolipoprotein (apo) A-I, the major structural protein of high-density lipoprotein (HDL), is present in human and mouse cerebrospinal fluid (CSF) despite its lack of expression in brain cells. To identify the origin of apoA-I in CSF, we generated intestine-specific and liver-specific Apoa1 knockout mice (Apoa1ΔInt and Apoa1Δliv mice, respectively). Lipoprotein profiles of Apoa1ΔInt and Apoa1ΔLiv mice resembled those of control littermates, whereas knockout of Apoa1 in both intestine and liver (Apoa1ΔIntΔLiv) resulted in a 60-percent decrease in HDL-cholesterol levels, thus strongly mimicking the Apoa1−/− mice. Immunoassays revealed that mouse apoA-I was not present in the CSF of the Apoa1ΔIntΔLiv mice. Furthermore, apoA-I levels in CSF were highly correlated with plasma spherical HDL levels, which were regulated by ABCA1 and LCAT. Collectively, these results suggest that apoA-I protein in CSF originates in liver and small intestine and is taken up from the plasma.
Keywords: apoA-I, cerebrospinal fluid, liver, intestine, mice, ABCA1, LCAT, plasma, adrenal gland, ELISA
Introduction
Apolipoprotein (apo)A-I, the major structural protein of HDL, has various potential beneficial effects on the prevention of cardiovascular disease, including coronary heart diseases and atherosclerosis [1–6]. ApoA-I expression is restricted to hepatocytes and small intestinal epithelial cells; however, apoA-I is relatively abundant in cerebrospinal fluid (CSF) in both humans and various animal models [7,8]. ApoA-I is not likely expressed in neuronal or glial cells in the brain. Consequently, most studies on lipoproteins in the central nerve system (CNS) have focused on other apolipoproteins that are synthesized in the brain, e.g. apoE, apoJ, or apoD [9,10]. The most abundant apolipoprotein in the brain is apoE[11] and there is robust data showing a neuroprotective role for apoE-containing lipoproteins [9,11,12]. ApoE-HDL is also generated by interacting with the ABCA1 transporter and LCAT to become spherical HDL. Since apoE has a binding motif to LDL receptor, LRP1, and VLDL receptor[13], apoE-null mice have a significant increase in middle size lipoprotein in the plasma due to the loss receptor-mediated clearance[14,15]. In the neuronal cells and glial cells, apoE-HDL particles are taken up by LDL receptors and/or LRP1[16]. Recently, it has been shown that apoE is also a ligand for sortilin, and when disturbed, such as in those patients with the apoE4 isoform, it may lead to decrease delivery of the anti-inflammatory lipid, docosahexaenoic acid (DHA) to the brain in Alzheimer’s disease [17]. It has also been shown that excess cholesterol and phospholipid can efflux from glial cells to apoA-I and form discoidal preβ-HDL by an ABCA1-dependent process [18–23]. Accordingly, similar to its putative beneficial effects on cells in the peripheral circulation, cholesterol efflux mediated by apoA-I and ABCA1 in CSF along with its high-affinity receptor, scavenger receptor class BI (SR-BI), may also have neuroprotective benefits for several different brain disorders.
ApoA-I-bound lipoproteins are favorably interacting with SR-BI and effectively deliver HDLcholesterylester (CE) into target cells such as hepatocytes and steroidogenic tissue in the circulation [24,25]. Neurosteroids [26,27] are steroids synthesized in CNS. Alike the steroidogenic cells in the peripheral nervous system (such as the adrenal cortex and gonadal), cholesterol is the starting substrate for steroid synthesis by CYP11A1 [28–30] in neuronal and glial cells, however, HDL-C in the CNS are not currently well studied as a carrier for neuronsteroid substrates [31,32]. ApoA-I is also known to co-function with apoE in neuronal regeneration and remyelination [33,34]. Furthermore, apoA-I deficiency in an Alzheimer mouse model, APP/PS/DeltaE9 mice, was found to exacerbate the vascular injury and cognitive decline due to amyloid deposition [35]. It was also shown that the various features of CNS degeneration also occurs in a separate mouse model of Alzheimer’s disease, APP/PS1 mice, and degeneration were alleviated by overexpression of apoA-I in the liver [36]. More recent studies revealed that mice deficient apoAI (Apoa1−/−) with the APP/PS1 transgene show more amyloid deposits in the cerebral cortex [37]. Elevation of several inflammatory factors was also observed in this study, consistent with the known anti-inflammatory effects of HDL found in other disease models [37–40]. A clinical study evaluating the link between lipoproteins and multiple sclerosis found that plasma HDL-cholesterol (HDL-C) levels and apoA-I levels were inversely correlated with neurodegeneration [41]. Despite the potential importance of apoA-I in several neurodegenerative diseases, the exact process by which apoA-I enters the CSF has not been thoroughly investigated and results are still controversial [42–45]. A study using Abca1 brain-specific knockout mice found an increased leveled of CSF apoA-I, which may have occurred because of the upregulation of the translocation of plasma apoA-I to satisfy the demand of apoA-I-HDL in the brain compartment[46].
The purpose of this study is to better understand apoA-I brain metabolism by identifying the origin of apoA-I detected in CSF using tissue-specific apoA-I-deficient mice. We show that both the liver and intestine contribute to apoA-I levels in CSF through a process that involves LCAT and ABCA1 in the generation of spherical HDL in plasma.
Materials and Methods
Experimental animals.
Abca1 heterozygote knockout mice (DBA/1-Abca1tm1Jdm/J) were purchased from Jackson Laboratory (JAX) (Bar Harbor, Maine USA, #003897) and back-crossed to C57BL/6N for 8 generations. Cytomegalovirus enhancer/first exon and the first intron of chicken beta-actin/splice acceptor in rabbit beta-globin element (CAG)-cre transgenic mice were a generous gift from the Center for Experimental Animal Science, Nagoya City University Graduate School of Medical Sciences. Lcat-null mice were obtained from Dr. Edward Rubin (Lawrence Berkeley National Laboratory, USA) and back-crossed to C57BL/6N for 8 generations. The Apoa1-floxed (Apoa1fl/fl) mice were generated at NHLBI, NIH, Bethesda, MD., USA, and transferred to the Center for Experimental Animal Science, Nagoya City University Graduate School of Medical Sciences, Nagoya, Japan for experimental studies and for generating additional mouse lines. They were maintained with MFG standard feeding chow (Oriental Yeast Co., Tokyo, Japan) at 25°C room temperature with 12 h light–dark cycles. The experimental procedures were approved by the Animal Welfare Committee of Nagoya City University Graduate School of Medical Sciences according to the institutional guidelines (approval number H28M-01, MA20–016, MA20–019).
Generation of tissue-specific Apoa1-deficient mice.
Apoa1-floxed (Apoa1fl/fl) mice were generated using the CRISPR/Cas9 technology by sequentially inserting the two loxP sites in intron 1 and intron 3 [47]. Briefly, a single guide RNA (sgRNA) for intron 1 (CTAAAGGTGCGAGATATCCA) was generated using their custom in vitro transcription service (Thermo Fisher). This sgRNA (20 ng/μl) was co-microinjected with a donor single-strand oligonucleotide (100 ng/μl) and Cas9 mRNA (100 ng/μL) into the cytoplasm of zygotes collected from B6D2F1/J mice (JAX #100006). Microinjection buffer was: 10 mM Tris–HCl (pH 7.5), 0.1 mM EDTA, 100 mM NaCl. The injected in 1st round oligonucleotide (TGGCTCCATAGCGCCTCCCAGTTGATGCTCCACTGTCCAAATCAATACCGTGGAATAACTTCGTATAGCATACATTATACGAAGTTATGAATTCTATCTCGCACCTTTAGCCATTCTAGCCAATGCTTCCATGGGCTTGAATTGTGTGTGGAGCC) had the loxP sequence (shown in bold) and EcoRI site (shown in italic and underlined). The EcoRI site was inserted to facilitate genotyping of born mice. Injected embryos were cultured overnight in M16 medium (MilliporeSigma) in a 37°C incubator with 6% CO2. After 24 h, embryos that reached the 2-cell stage of development were implanted into the oviducts of pseudopregnant foster mothers (CD-1 mice from Charles River Laboratories). For genotyping, DNA was isolated from tail clips or blood with the Maxwell16 System (Promega) and analyzed by PCR amplification of the target site (forward primer: 3’-CCA GGC TGA GCT TAT CAG TC-5’; reverse primer: 3’-AGC ACC ACA GCT TTC ATC-5’). PCR conditions were as follows: 94°C, 5 min; 35 cycles: 94°C for 30 s, 58°C for 40 s, 72°C for 45 s; and 72°C for 5 min. DNA from mice harboring the inserted intron 1 oligonucleotide with loxP and EcoRI sites in the heterozygous condition, generated 340 and 380 bp bands, for wild-type and mutant alleles, respectively. By crossing the mice, loxP site in intron 1 was placed in homozygous condition. The insertion was confirmed by Sanger sequencing of the 380 bp band. Positive mice with a perfect loxP sequence in intron 1 were mated to produce zygotes, which were used for microinjection of the Intron 3 loxP. The intron 3 sgRNA (GGGTGCTGTCTGACCAGTAC) and donor oligonucleotide (CCACAGCCCCTCATTCAGCCTATGAGTGCCAAATCCCTTTTCCTTGGTAACCCCCAGTACATAACTTCGTATAGCATACATTATACGAAGTTATGGATCCTGGTCAGACAGCACCCAAAACAAAACAAAACAAAACAAAAAACAAAAAACGGGACTGGCCTTGTAACCAGC; loxP sequence is in bold and an inserted BamHI site is in italic and underlined) were generated and microinjected using the same procedures as described above. As before, a Bam H1 site was inserted to facilitate genotyping of post-natal mice. After the 2nd round of microinjections, offspring were genotyped for the presence of both intron 1 and intron 3 loxP sites. As before, the correct insertion of the oligonucleotide with the 2nd loxP site was demonstrated by PCR amplification of the target site (forward primer: 3’-GAT GGG ATT CAT CTG GCT GTT G-5’; reverse primer: 3’-CCC TAA TGT GCA GTT GCT ATG G-5’). PCR conditions were the same, as described above, for genotyping loxP site in Intron 1, except the temperature of annealing was 60°C. DNA from mice harboring the intron 3 inserted oligonucleotide with loxP and BamHI sites in the heterozygous condition, generated 280 and 320 bp bands, for wild-type and mutant alleles, respectively. The insertion was confirmed by sequencing of the 320 bp band. In order to demonstrate the presence of both loxP sites on the same chromosome, separate PCR analyses were performed. In this case, forward (3’-CGA AGT TAT GAA TTC TAT CTC G-5’) and reverse (3’-CAG GAT CCA TAA CTT CGT ATA ATG-5’) primers were located near loxP sites, and restriction sites for EcoRI and Bam HI were included in the forward and reverse primers, respectively. PCR conditions were similar as for genotyping the presence of the loxP site in intron 1, except elongation time at 72°C was 1 min 20 sec. The allele with both loxP sites produced a 751 bp band; with the wild-type allele, there was no band. Upon establishment of the floxed Apoa1 allele routine genotyping of the mice was performed by Transnetyx, Inc. (Cordova, TN, USA). Apoa1-floxed (Apoa1fl/fl) mice were crossed to villin (Vil)-cre transgenic, albumin (Alb)-cre transgenic, or cytomegalovirus enhancer/first exon and the first intron of chicken beta-actin/splice acceptor in the rabbit beta-globin element (CAG)-cre transgenic mice to generate Apoa1fl/fl;Vil-cre (designated Apoa1ΔInt), Apoa1fl/fl;Alb-cre (designated Apoa1ΔLiv), Apoa1fl/fl;Vil-cre;Alb-cre (designated Apoa1ΔIntΔLiv), and Apoa1fl/fl;CAG-cre (designated Apoa1−/−). Tail DNA was collected from weaned pups at 3 wks and used as a template DNA. The genotypes were detected using a real-time PCR system, StepOnePlus® and StepOne® Software v2.0 (Thermo Fisher Scientific Inc. USA), examined by the amplification plot during the PCR and the melt curve analysis of the product DNA (qPCR-MCA). PowerUP® SYBR® master mix: autoclaved Milli-Q® water: primers (35 μL: 66 μL: 0.5 μL x 2 per 8 wells) were applied to MicroAmp® Fast Optical 96-Well Reaction Plate and diluted 1 μL of tail DNA was mixed into the individual well. PCR were performed as follows: 95°C for 10 min (stage 1);95°C 30 s, 58°C for 45 s, 65°C for 1min, 15 cycles (stage 2). Then 95°C 30 s, 55°C for 45 s, 65°C for 1min, for 30 to 45 cycles (stage 3) for amplification. Melt curve analyses was performed to identify the amplicons from 60°C to 95°C by increasing 0.3°C per min. These PCR condition with the primer sets (Fig.1B) showed only a single peak by melt curve analysis. The peak temperature of Apoa1-wild allele, Apoa1-floxed, Apoa1-ablated, villin-cre, albumin-cre, CAG-cre, Abca1-wild (forward primer 5’-CCT TTC TCA TAG GGT TGG TCA-3’and reverse primer 5’-ACT GTT CCT CAG GAG CCA GA-3’), Abca1-null (5’-TTT CTC ATA GGG TTG GTC A-3’ and 5’-TGC AAT CCA TCT TGT TCA AT-3’), Lcat-wild (5’-TGA ACT CAG TAA CCA CAC ACG GCC TG-3’ and 5’-GTC CTC TGT CTT ACG GTA GCA CAT CC-3’), and Lcat-null (5’-AAC GAG ATC AGC AGC CTC TGT TCC AC-5’ and 5’-GTC CTC TGT CTT ACG GTA GCA CAT CC-3’) for qPCR-MCA were 86.6, 86.5, 85, 84.7, 84.5, 83, 84, 87.2, 86.6, and 86.5 °C, respectively. Selected amplicons were sequenced by a capillaryDNA-sequencer, Prism 3130xl (Applied Biosystems Inc., USA), to conform its sequence as designed.
Figure 1.
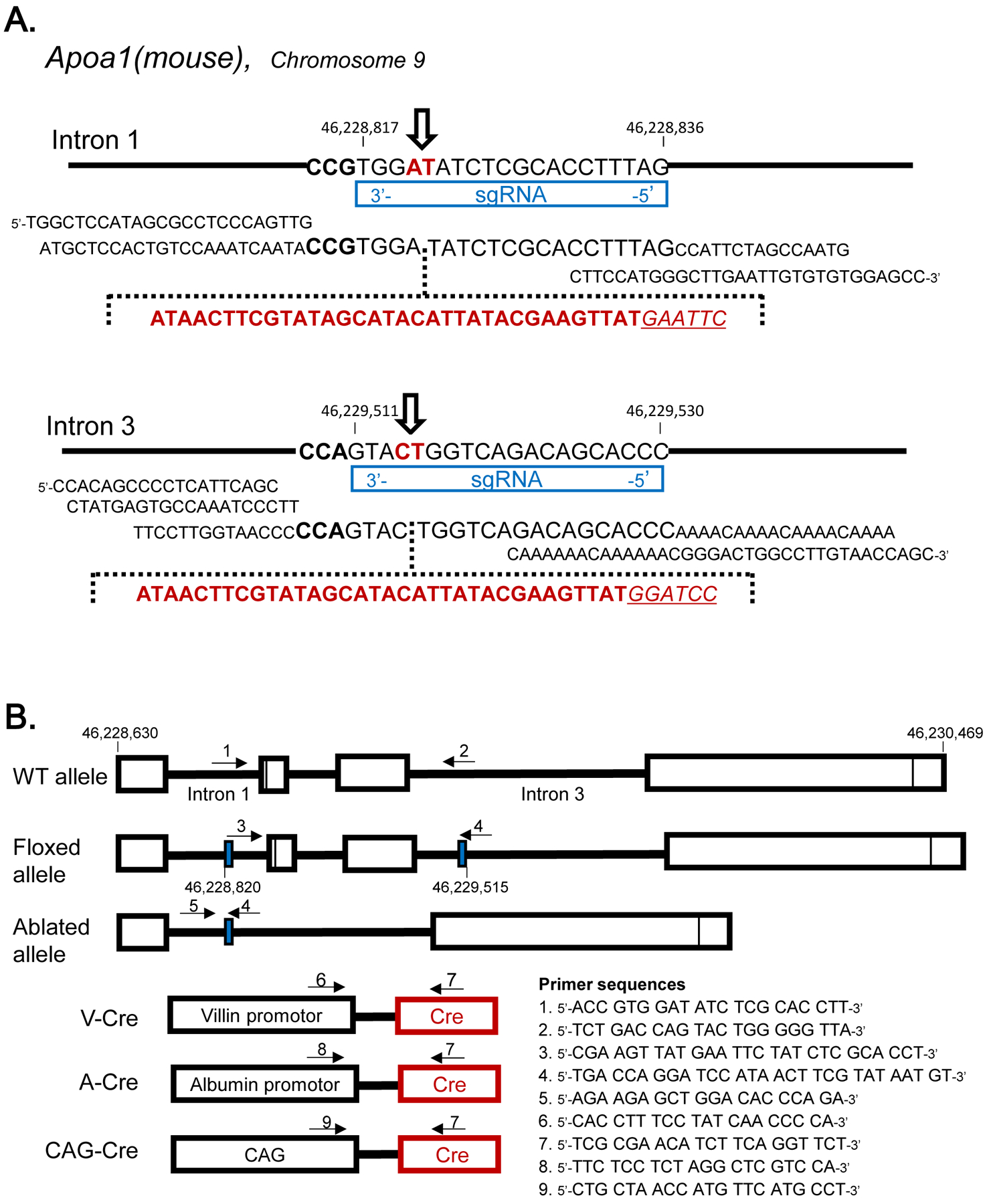
Genetic Design A: Insertion of loxP in mouse Apoa1 allele at Intron 1 and Intron 3 using CRISPR/Cas9 technique. The sequence indicated at genome alleles are complementally sequence for sgRNA and PAM sequences. Donor oligonucleotides including loxP sequence were described, respectively. The open arrow indicated the cleavage sites by cas9. B: The genotyping primers positions of Apoa1fl/fl mice and cre transgenic mice. Solid blue boxes indicated the loxP insertion sites. The primer sets for each allele’s genotyping were indicated in arrows.
Tissue and biofluid collection
To evaluate Apoa1 mRNA levels, a fragment of liver and jejunum tissue were collected from mice perfused with PBS (10 mM Phosphate buffer, pH 7.4, 0.15 M NaCl) containing 5 mM EDTA and immediately frozen in liquid nitrogen and stored at −80°C until analyzed. The frozen tissue samples were homogenized in ISOGEN (Nippon gene, Ltd., Japan), using a polytron (PT1200C/CL, Central Scientific Commerce, Inc., Japan). The total RNA fractions were isolated according to the standard protocol. The first strand DNA was generated using 5 μg of RNA (A260/A280: 2.0 ± 0.7, A260/A230: 1.4 ± 0.3; NanoDrop One® v1.4.2, Thermo Fisher Scientifict, Inc., USA) as a template with random hexamers (Superscript® First-Strand Synthesis System, Thermo Fisher Scientific, Inc., USA). The template cDNAs were examined in qPCR-MCA system using Apoa1 primers (forward primer on Exon 3, 5’-ACG TAT GGC AGC AAG ATG AAC-3’ and reverse primer on Exon 4, 5’-AGA GCT CCA CAT CCT CTT TCC-3’) and a housekeeping gene, Gapdh, primers (5’-GCC AGC CTC GTC CCG TAG ACA −3’ and 5’-ACC CGT TTG GCT CCA CCC TTC −3’) with the same qPCR-MCA condition described above. For morphological observation of adrenal glands, mice were killed by decapitation immediately after taking them from their breeding cage. The adrenal glands were then rapidly collected and placed into PBS + 5 mM EDTA to remove excess blood. The image of the whole adrenal gland was photographed with Canon EOS 3x-digital camera equipped with EF100 mm f/2.8L Macro IS USM micro-lens. For the collection of mouse CSF, mice were anesthetized and perfused thoroughly with 5 mM EDTA containing PBS. Mouse CSF within the cisterna magma (2–25 μL per mouse) was suctioned using a 29G-insulin syringe (SS-05M2913, TERUMO Co. Inc., Japan)[48–50].
Mouse plasma lipid analyses
Mice were fasted over-night and blood samples were collected via retroorbital venous plexus, using heparin coated hematocrit capillary tubes (Drunmond®, USA), under anesthesia. Plasma was obtained as the supernatant after centrifugation at 12 krpm for 5 min to precipitate blood cells and debris. Plasma samples were stored at 4°C until analysis. Plasma was applied to gel-permeation high performance liquid chromatography for lipoprotein separation and lipid content was determined by enzymatic colorimetric detection for total cholesterol and triglyceride levels (Skylight Biotech, Inc., Japan).
Cryo-treatment of mouse brain
ApoA-I conditional KO mice at 20-weeks of age or older when indicated were administrated 100 uL of 45%w/v D-glucose solution immediately before anesthesia. Frostbite injury of the right hemisphere of the brain cortex was achieved by 30 second application of cotton Q-tips dipped in liquid nitrogen gently pressed over the right hemisphere of the surgically exposed skull bone. After the liquid nitrogen treatment, the apparent surface frostbite wound was examined at 2 weeks and scored (0–10) by the size of the damaged area.
Lipoprotein Analysis by Transmission Electron Microscopy
High-Density Lipoproteins (HDL) were purified from pooled mouse or rat CSF by KBr density-gradient ultracentrifugation at 1.063 −1.21 g/mL and dialyzed against PBS. HDL particles were negatively stained with 2% uranyl acetate solution and imaged using a transmission electron microscope (JEM-1011J, JEOL, Japan) HDL particle diameters were calibrated by DigitalMicrograph® (Gatan, Inc., USA).
Mouse apoA-I and mouse apoE measurement by ELISA
Mouse apoA-I and apoE concentrations in plasma and CSF were quantified by the Mouse Apolipoprotein A1 ELISAPRO kits[51], and Mouse Apolipoprotein E ELISAPRO kits[52], (Mabtech AB, Sweden). Briefly, mouse plasma and mouse CSF were diluted 200,000X and 100X for mouse apoA-I assays, and 10,000X and 1,000X for apoE assays, respectively.
Statistical analysis
Differences between multiple groups were evaluated by one-way analysis of variance (ANOVA) using Prism® v8.4.3 (GraphPad Software LLC, USA). The adjusted P values were evaluated in Tukey’s multiple comparisons test and p <0.05 were considered significant.
Results:
Ablation of Apoa1 gene expression in the target organs.
Tissue-specific Apoa1 knockout mice were generated through a CRISPR/Cas9 approach to create Apoa1-floxed mice that were crossed with either villin-promotor-cre, albumin-promotor-cre, or CAGcre transgenic mice to generate Apoa1fl/fl, Apoa1ΔInt, Apoa1ΔLiv, Apoa1ΔIntΔLiv, Apoa1−/− lines (Figure 1A). Real-time PCR was used to confirm Apoa1-disruption in specific tissues, including the small intestine and liver. Expression of Apoa1 mRNA in small intestine tissue was calculated by Apoa1 Ct compared to Gapdh Ct were −8.8, NA (not applicable, no amplification), −9.3, and NA in Apoa1fl/fl, Apoa1ΔInt, Apoa1ΔLiv, Apoa1ΔIntΔLiv, respectively. The Apoa1 Ct to Gapdh Ct in the liver was 6.5, 4.6, NA, and NA respectively, indicating Apoa1 transcription was significantly decreased in the targeted tissues.
Characterizations of tissue-specific Apoa1-null mice.
Plasma lipoprotein profiles were examined by a gel-permeation HPLC followed by colorimetric assays (Figure 2). Apoa1fl/fl mice showed the typical lipoprotein profile observed in wild-type (WT) mice (Figure 2A). Unexpectedly, despite the lack of apoA-I synthesis in small intestines or livers, both Apoa1ΔInt and Apoa1ΔLiv mice retained similar lipoprotein levels to the control Apoa1fl/fl mice (Figures 2B, 2C). Moreover, tissue-specific Apoa1-null mice were able to maintain apoA-I protein levels in plasma (Figure 5A). Consistent with earlier work [53], this may be due to compensatory up-regulation of apoA-I expression from the other tissue. Total cholesterol levels in HDL fractions (corresponding to 8 to 16 nm in diameter) in this HPLC analyses were reduced to 37.4% and 39.5% in Apoa1ΔIntΔLiv (Figure 2D) and in Apoa1−/− (Figure 2E), respectively. The major HDL particle sizes, which were estimated from the peak retention times, were Apoa1fl/fl (11.5), Apoa1ΔInt (11.4), Apoa1ΔLiv (11.5), Apoa1ΔIntΔLiv (13.5), and Apoa1−/− (13.2 nm), respectively. These results suggest that HDL in Apoa1ΔIntΔLiv and Apoa1−/− mice may indicate that HDL in these fractions were generated using nonapoA-I apolipoproteins, e.g. apoA-II, apoE, apoA-IV and/or apoC’s[54]. ApoA-I protein levels in plasma were not detected in Apoa1ΔIntΔLiv or Apoa1−/− mice (Figure 5A). Figure 2F and 2G show the lipoprotein profiles of Abca1-null and Lcat-null mice. Despite the lack of HDL-C, a small amount of apoA-I is detected, which is likely present as lipid-poor apoA-I or as apoA-I containing discoidal HDL(preβ-HDL).
Figure 2.
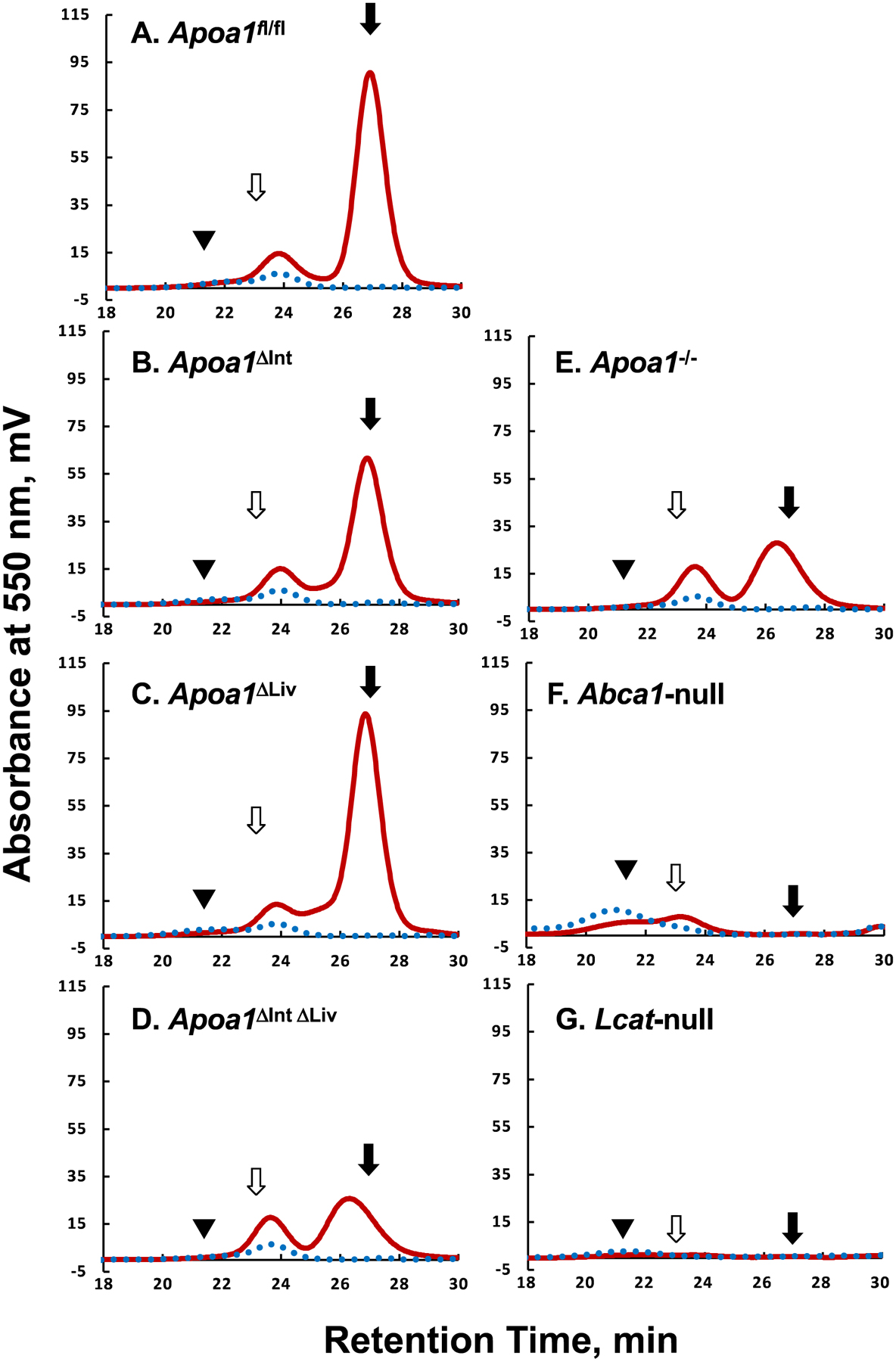
Typical Lipoprotein profiles of mice plasma. The apoA-I floxed (Apoa1fl/fl) mice without cre expression(A), with villin-cre expression, Apoa1ΔInt (B), with albumin-cre expression, Apoa1ΔLiv (C), with both villin-cre and albumin-cre expression, Apoa1ΔIntΔLiv (D), with CAG-cre expression, Apoa1/- (E), Abca1-null mice(F), and Lcat-null mice(G) plasma were analyzed by the gel-permeation HPLC equipped with an online enzymatic colorimetric lipid assay system (Skylight Biotech Inc, Japan). Solid burgundy line, total cholesterol. The dotted blue line, triacylglycerol. Solid triangles, open arrows, and solid arrows point to the typical retention time of human VLDL, human LDL, and human HDL, respectively.
Figure 5.
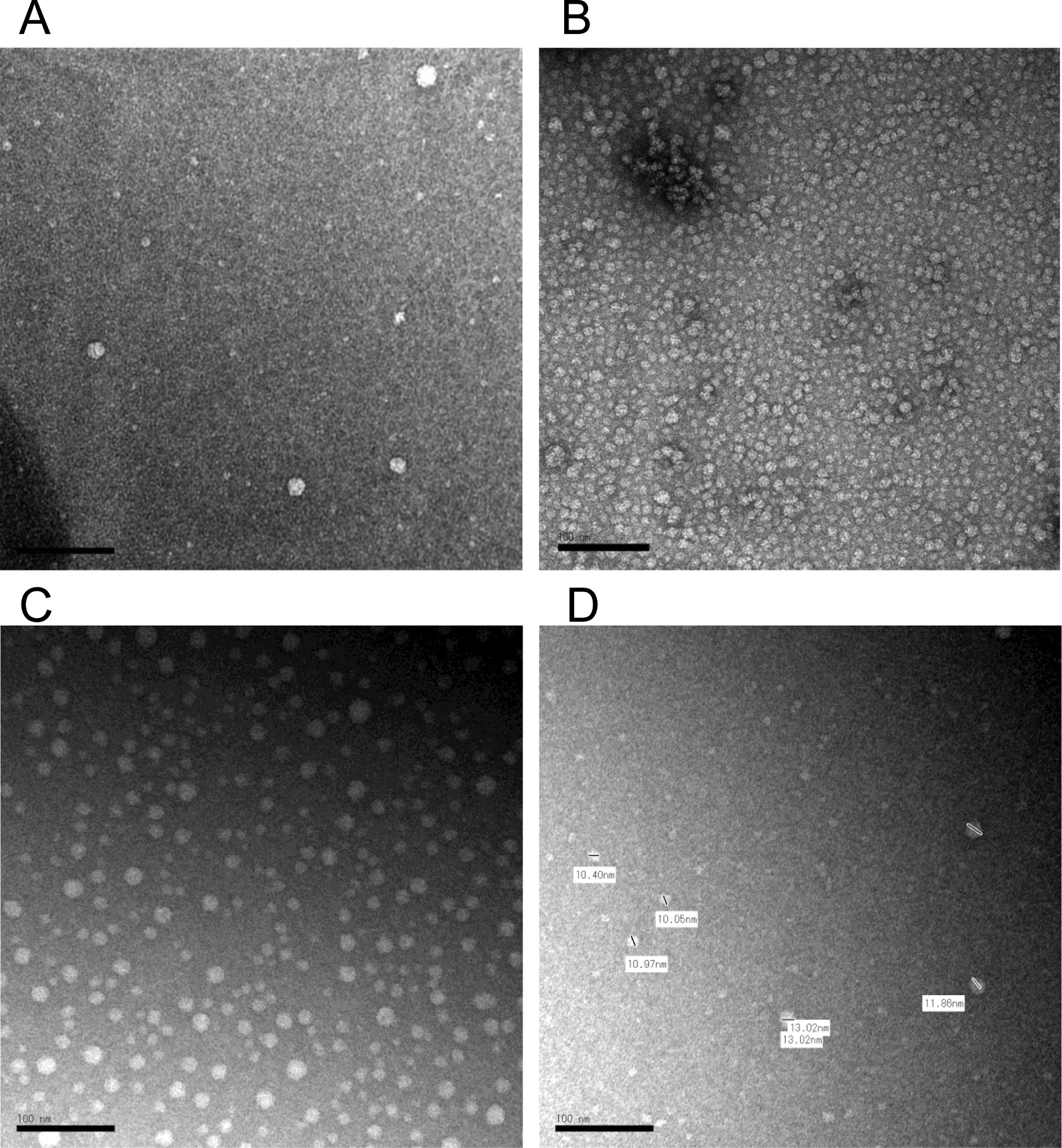
Negative staining transmission electron microscopy (TEM) image of HDL in cerebrospinal fluid (CSF) and in plasma. The contrast was obtained by a 2% uranyl acetate solution. The typical HDL particles in mouse CSF (A), in human plasma (B), and in rat CSF (C, D). The scale bar indicates 100 nm. Short bars in the image D was used for calibrations of each particle diameter lengths which indicated by side.
In mice, HDL is the major carrier and source of cholesterol in circulation and Abca1-null mice or Lcat-null mice are known to be depleted of cholesteryl esters in the adrenal cortex [55]. Loss of HDLcholesterylester (CE) uptake in adrenal glands was apparent in excised tissue from Abca1-null and Lcat-null mice, specifically, increased reddish-brown appearance compared to adrenal glands excised from C57BL/6N WT mice that appeared opaque-whitish (Figure 3). Among tissue-specific Apoa1 knockout mouse, the adrenal glands of Apoa1fl/fl, Apoa1ΔInt, and Apoa1ΔLiv more closely resembled the appearance of WT mice, i.e. opaque-whitish, compared to the reddish-brown appearance of adrenal glands from Apoa1ΔIntΔLiv, and Apoa1−/− mice (Figure 3) These results suggest the observed large apoAI-deficient HDL particles present in Apoa1ΔIntΔLiv and Apoa1−/− mice are inefficient as CE delivery vehicles for adrenal glands. This would be expected to lead to a defect in acute steroid synthesis due to lack of storage of substrates for enzyme like Cyp11a1 in the steroid synthesis pathway [55]. Neurosteroids have a genomic action that promotes neuron dendrite extension, synapse formation, neural circuit construction, etc. as well as a nongenomic effect that regulates signal transduction at synapses of constructed neural circuits[26,27]. ApoA-I-HDL in CSF may contribute as cholesterol provider to neuronal cells thus healing injury in the brain. To determine if the loss of apoA-I-HDL in general circulation affects brain repair to injury, cryoinjury tests were performed in the right hemisphere of the cerebral cortex of Apoa1fl/fl, Apoa1ΔInt, Apoa1ΔLiv, Apoa1ΔIntΔLiv, and Apoa1−/− mice (Figure 4). Frostbite damage, an indicator of cryoinjury, was scored after 2 weeks. Apparent injury scores were higher in Apoa1ΔIntΔLiv, and Apoa1−/− compared to Apoa1fl/fl control mice, but not to Apoa1ΔInt nor Apoa1ΔLiv mice (Figure 4).
Figure 3.
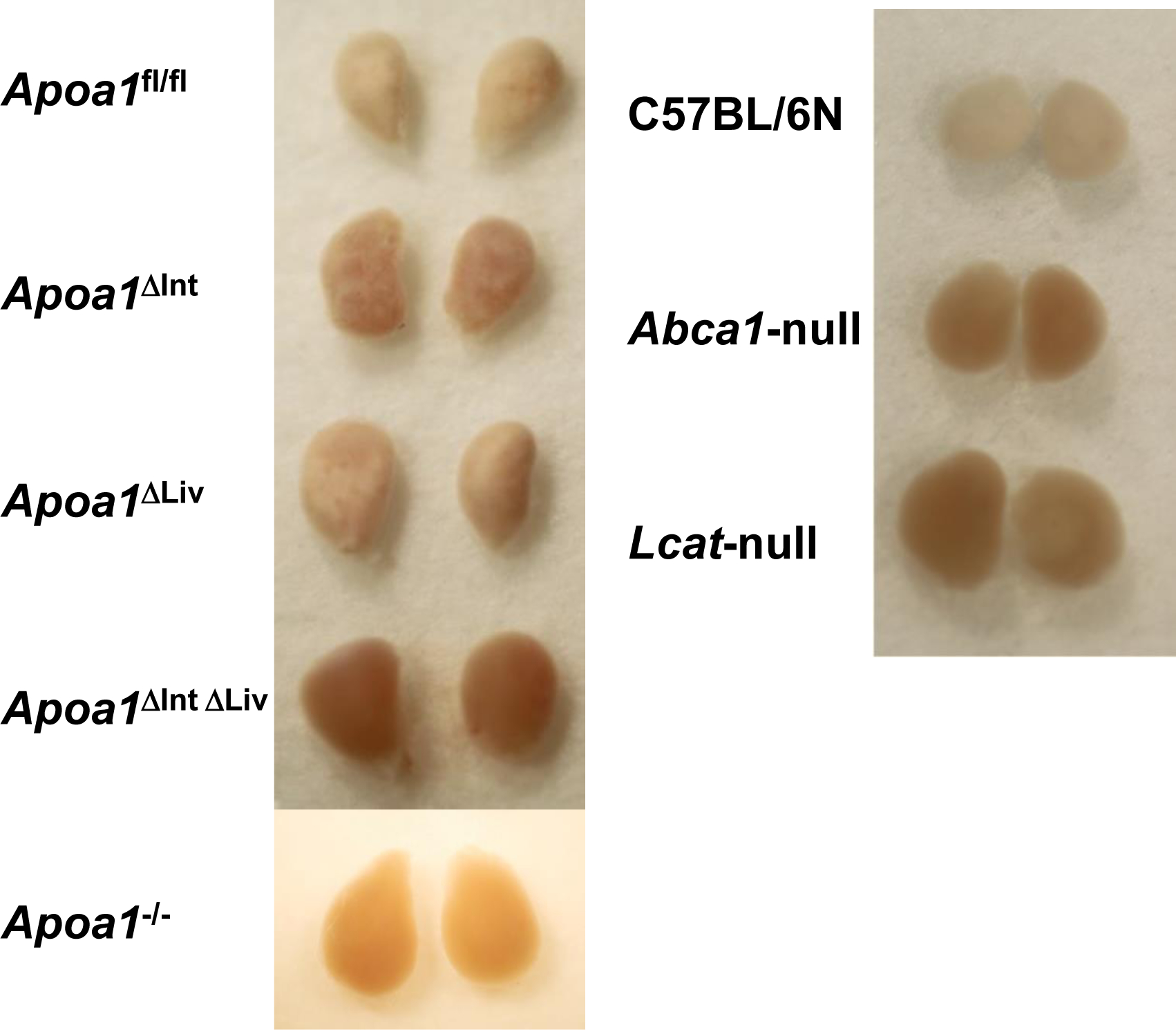
The adrenal gland of apoA-I conditional knockout mice. Adrenal glands store cholesterol in the cortex to supply cholesterol at the acute steroid generations. Apoa1fl/fl mice with Vil-cre Alb-cre expressed mice and with CAG-cre expressed mice showed reddish adrenal gland as hypoalphalipoproteinemia mice model, Abca1-null, and Lcat-null mice.
Figure 4.
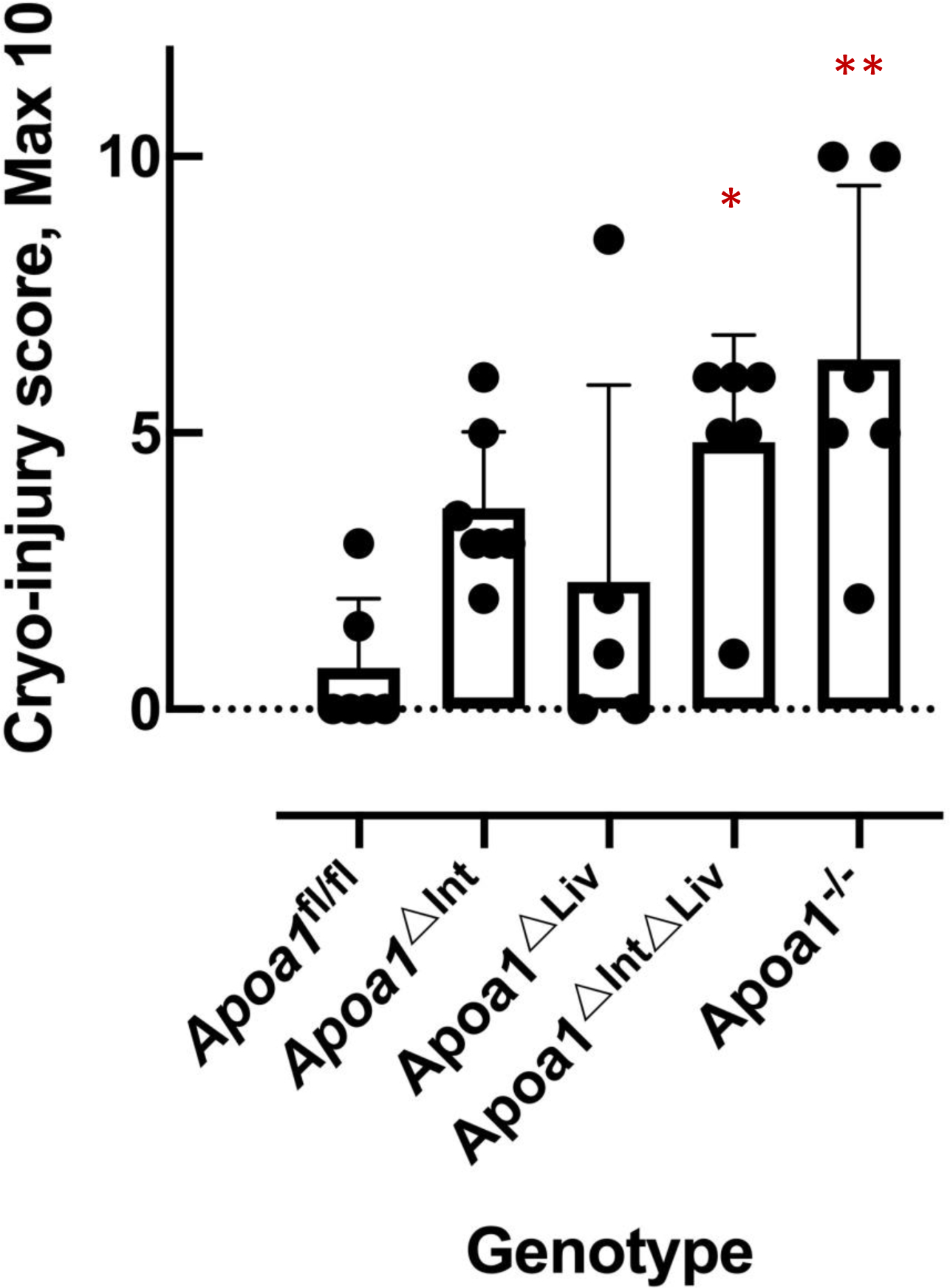
The cryo-injury score of the mouse brain. P values were obtained by a two-tailed t-test. Asterisks indicate the statistical difference compared to the Apoa1fl/fl mice (*: p<0.05, **: p<0.01).
CSF apoA-I originates from general circulation.
To define the morphology of CSF-HDL, HDL was isolated from rodent CSF by DGUC, and observed by transmission electron microscopy. TEM and negative staining of CSF HDL identified spherical mature HDL particles of various sizes (10.05 to 13.02 nm in diameter), which is greater in size than HDL from general circulation (Figure 5A–D) To quantify apoA-I (and apoE) proteins levels in CSF and plasma from tissue-specific Apoa1-null mice, ELISAs were performed (Mabtech AB, Sweden). ApoA-I levels in plasma from Apoa1ΔInt mice were significantly reduced by 63.3% compared to control plasma from Apoa1fl/fl mice (Figure 6A). In contrast, apoE levels were significantly increased by 1.7-fold in Apoa1ΔInt mice compared to control Apoa1fl/fl mice (Figure 6B). For Apoa1ΔLiv mice, plasma apoA-I levels were also affected by the loss of apoA-I expression in livers. And similarly, apoE levels were significantly increased in plasma by 3.7-fold compared to control Apoa1fl/fl mice (Figures 6A, B). Ablation of apoA-I in both the intestine and liver of Apoa1ΔIntΔLiv mice completely abolished apoA-I protein levels in plasma (P<0.0001, Figure 6A) and apoE protein levels were significantly increased >6-fold (P<0.001, Figure 6B). The global Apoa1−/− mice showed similar lipoprotein profiles to Apoa1ΔIntΔLiv mice (Figure 6A). Since Abca1-null and Lcat-null mice are on the C57BL/6N background, the statistical analyses were performed separately. As expected, plasma apoAI levels were significantly reduced in Abca1-null and Lcat-null mice (P<0.0001, P<0.01, respectively) compared to C57BL/6N mice (Figure 6A). Plasma apoE levels were significantly reduced in Abca1null mice but were increased in plasma from Lcat-null mice. Most importantly, apoA-I and apoE protein levels were quantified in mouse CSF. ApoA-I levels in CSF from in Apoa1ΔIntΔLiv and Apoa1−/− mice were undetected (P<0.01, P<0.01), even after an extended incubation period of primary antibody incubation to 24 h instead of 1 h. Neither tissue-specific disruption of Apoa1 in the small intestine (Apoa1ΔInt) or the liver (Apoa1ΔLiv) resulted in a significant decrease in CSF apoA-I protein levels compared to control Apoa1fl/fl mice (Figure 6C). Although apoA-I expression is normal in Abca1-null mice, little to no apoA-I protein was detected in mouse CSF from these mice, a significant reduction compared to C57BL/6N WT control mice (P<0.0001, Figure 6C). In Lcat-null mice, apoA-I protein levels were also significantly decreased in CSF as compared to C57BL/6N WT mice (P<0.0001, Figure 5C). In contrast, apoE levels in CSF were relatively unaffected by these various genetic mouse models, except for the Apoa1−/− global knockout strain, which showed a significant decrease as compared to C57BL/6N WT mice (P<0.0001, Figure 6D). Since the loss of plasma apoA-I coincides with the loss of CSF apoA-I, correlation analyses were conducted, and we found that apoA-I protein levels in CSF were positively correlated to apoA-I protein levels in plasma among Apoa1fl/fl, Apoa1ΔInt, Apoa1ΔLiv, Apoa1ΔIntΔLiv, and Apoa1−/− mice groups (R2 = 0.96, Figure 7). WT C57BL/6N mice were found to have a 2.22-fold increase in CSF/plasma apoA-I protein ratio compared to Apoa1fl/fl mice group (Figure 7). This result implicates other factors than plasma apoA-I level existing to facilitate CSF apoA-I level in mice. Conversely, CSF apoE protein levels and plasma apoE protein levels correlation was not found among Apoa1fl/fl, Apoa1Δint, Apoa1Δliv, Apoa1ΔintΔLiv, and Apoa1−/− mice (R2 = 0.007, Figure 7). Collectively, these results strongly suggest that apoA-I protein in CSF was derived from general circulation and originated from both liver and small intestine.
Figure 6.
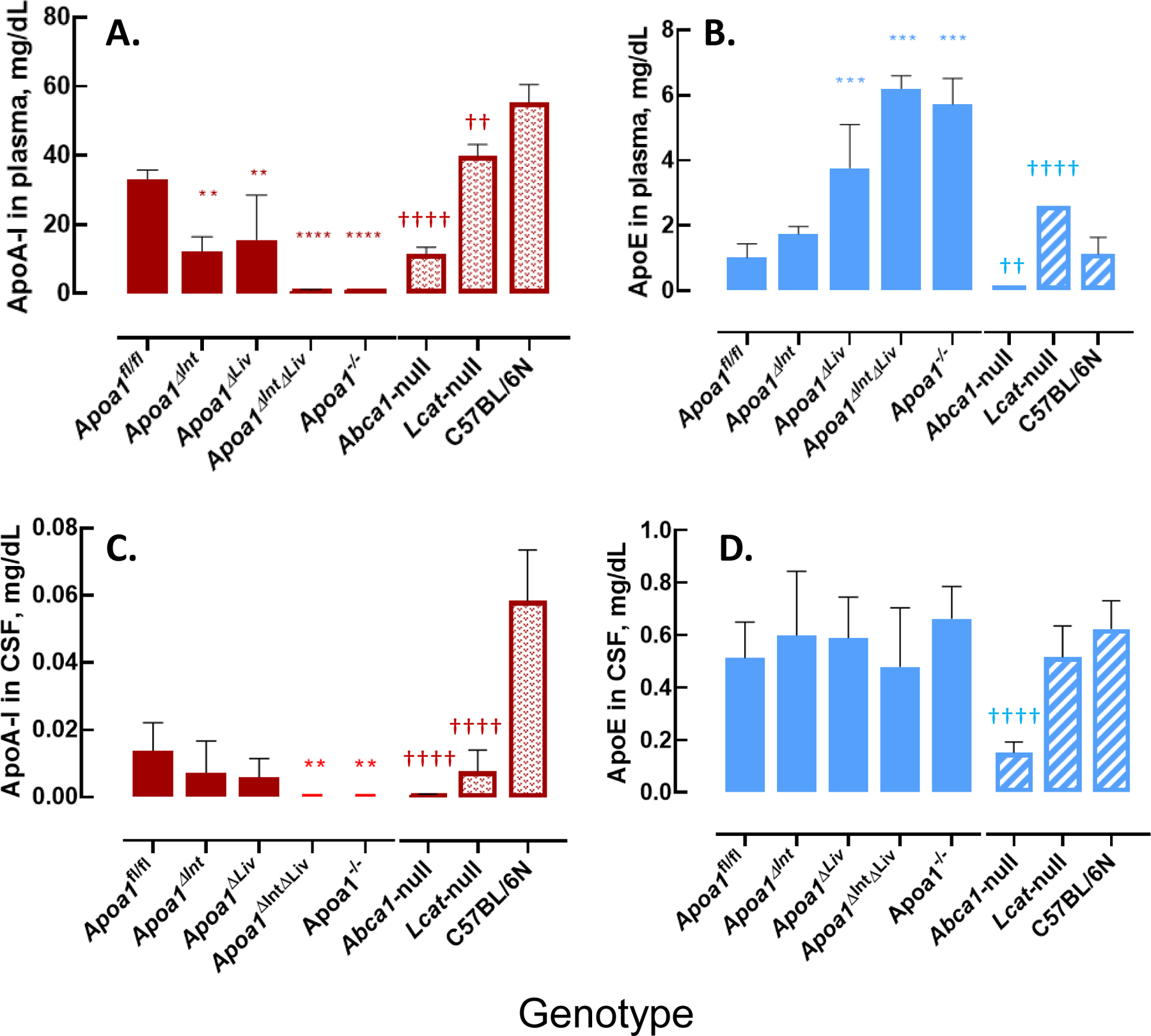
ApoA-I and apoE level in mouse plasma and mouse CSF. A: apoA-I level in mouse plasma. B: apoE level in mouse plasma. C: apoA-I level in mouse CSF. D: apoE level in mouse CSF. Asterisks indicate the statistical difference compared to the Apoa1fl/fl mice (**: p<0.01, ***: p<0.001, ****:p<0.0001). Daggers indicate the statistical difference compared to the C57BL/6 N mice (††: p<0.01, ††††: p<0.0001).
Figure 7.
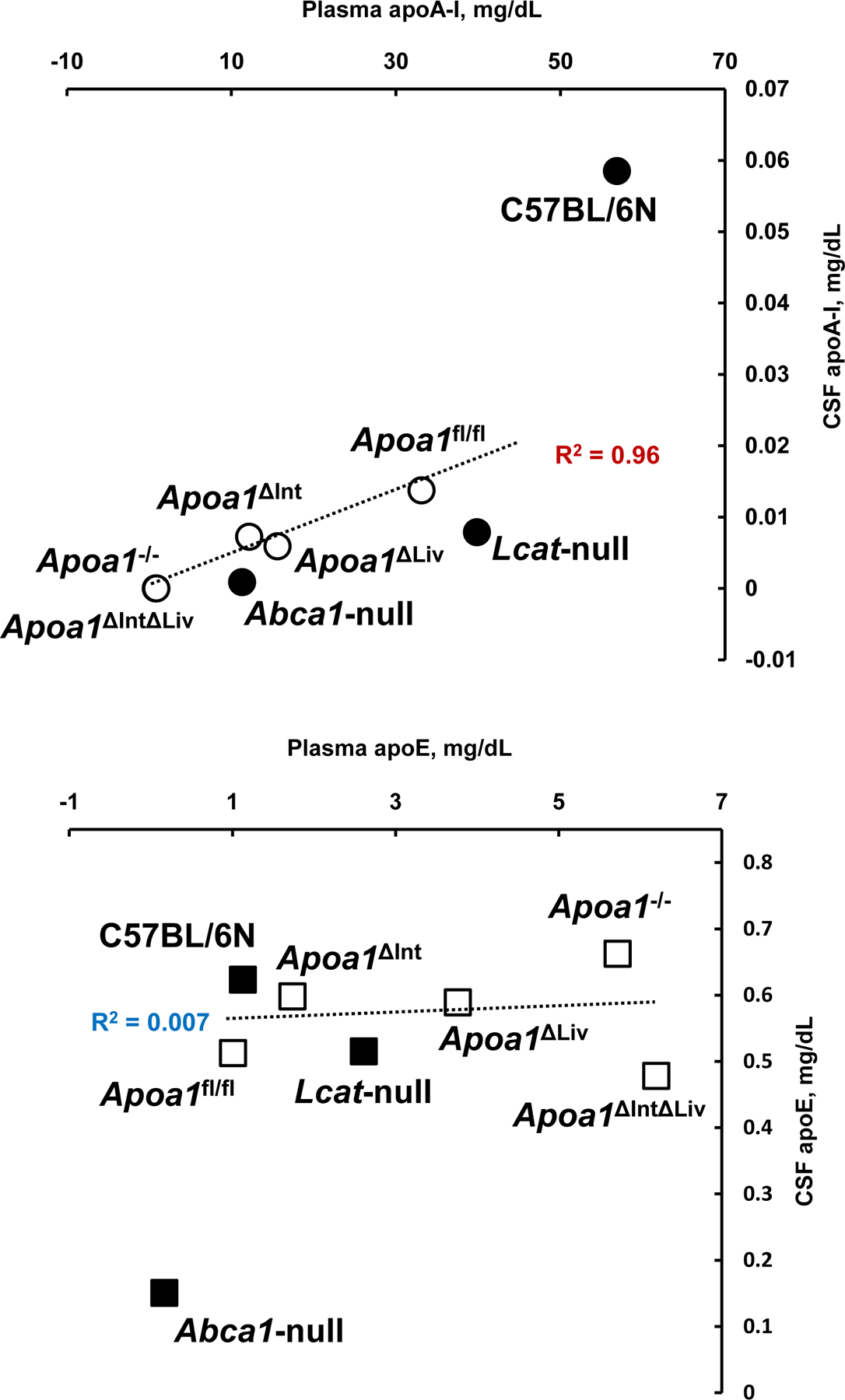
Scattered plots of plasma and CSF apolipoproteins. Mean values of apoA-I and apoE in plasma and in CSF (Figure 5A and 5C, Figure 5B and 5D) were replotted. Open symbol; apoA-I floxed mouse group. Solid symbol; mouse group of C57BL/6N background.
Discussion
In this study, using the CRISPR/Cas9 technology, we generated Apoa1fl/fl mice that were then crossed with cre transgenic mice driven by villin and albumin promoters to obtain tissue-specific deletion of ApoA-I in the small intestine (Apoa1Δint) and liver (Apoa1Δliv), respectively. While these mice were similar to their control littermates, combinatorial deletion of apoA-I in both the small intestine and liver (Apoa1ΔintΔLiv mice), markedly decreased not only plasma apoA-I protein levels but also CSF apoA-I protein levels. These results support that apoA-I in CSF likely originates from plasma, and probably from both the small intestine and liver. Based on the distribution of total protein in HPLC fractions, Apoa1ΔIntΔLiv mice likely have larger HDL particles than control mice, which agrees with a previous report on global Apoa1−/− mice [54]. Total HDL cholesterol contents of these mice were reduced by 62.6% and 60.5% compared to Apoa1fl/fl mice, which indicates that other apolipoproteins (e.g. apoA-II, apoA-IV, or apoE) likely interact and facilitate the formation of these large HDL particles (Figure 2D, 2E), potentially through ABCA1mediated efflux to generate preβ-HDL particles[56,57]. Plasma apoE levels were found to be elevated 6-fold in Apoa1ΔIntΔLiv and Apoa1−/− mice, as compared to Apoa1fl/fl mice, which may be due to reduced HDL levels as related to a faster catabolic rate of apoE-HDL than apoA-I-HDL mediated by receptors for apoE-containing lipoproteins, e.g. the chylomicron remnant receptor and LRP1[15]. Changes to HDL function in Apoa1ΔIntΔLiv and Apoa1−/− mice were also supported by reduced HDL-CE uptake in the adrenal cortex, as evidenced by the increased reddish-brown appearance of the adrenal glands in these mice (Figure 3). The HDL receptor, SR-BI is highly expressed in the adrenal cortex. Adrenal glands from Apoa1ΔIntΔLiv mice resemble those isolated from Abca1-null or Lcat-null mice, which lack the HDL-CE in their plasma [55,58,59]. These mice may have a defect in acute stress responses, e.g. low corticosterone synthesis or defective propagations, as observed in Abca1-null or Lcat-null mice[55] and presumed to be the case in Apoa1−/− mice[60]. These results are consistent with a model whereby defects in SR-BI-mediated uptake of lipoproteins in neuronal and glial cells due to lack of apoA-I containing lipoproteins[46,61] accounts for our findings.
ApoA-I has been proposed as a protective factor after neuronal damage [35–37,62]. The severity of cryo-injuries (brain frostbite) was relatively larger in Apoa1ΔIntΔLiv and Apoa1−/− mice, as compared to Apoa1fl/fl mice (Figure 4). Apoa1ΔInt mice brain injury scores were also slightly higher, thus suggesting decreased HDL function in the brain. This is supported by evidence that Apoa1ΔInt mice have reduced plasma HDL and reduced HDL function, as indicated by reddish-brown adrenal glands (Figure 3). In plasma, apoA-I protein levels, as detected by ELISAs were reduced in Apoa1ΔInt mice compared to control Apoa1fl/fl mice. In CSF, apoA-I protein levels were similar across Apoa1fl/fl, Apoa1ΔInt, and Apoa1ΔLiv mice. It was previously reported that apoA-I levels in CSF were significantly reduced in Abca1-null mice [63] and Lcat-null mice [64], and results from this study agree with these previous studies. Plasma apoA-I levels and CSF apoA-I levels were highly correlated among Apoa1fl/fl, Apoa1ΔInt, Apoa1ΔLiv, Apoa1ΔIntΔLiv, and Apoa1−/− mice. Conversely, the association between plasma and CSF apoA-I levels in Abca1-null mice and Lcat-null mice were not as strong, which suggests that Abca1-null and Lcat-null mice likely do not transfer as efficiently as WT mice plasma-derived apoA-I into CSF(Figure 7). However, low levels of apoA-I were detected in Abca1-null and Lcat-null mice CSF. These results indicate that the presence of spherical HDL is not crucial for the transfer apoA-I into the CSF[44].
Lipid-free apoA-I can also interact with the blood-brain barrier (BBB) model endothelial cell line, hCMEC/D3, which form the BBB but not the blood–cerebrospinal fluid barrier (BCSFB). It has been proposed that lipid-free apoA-I is transported in a clathrin-independent manner, and requires a cholesterol-rich plasma membrane[45]. Since SR-BI is known to transport HDL-cholesterol into cells, SR-BI localized at brain microvascular endothelial cells could also participate in the transcytosis of apoA-I into CSF[43]. ApoA-I is a prototypical “exchangeable apolipoprotein” and is in a dynamic equilibrium between being bound to HDL versus being free [65,66]. Potentially, apoA-I on spherical HDL is liberated by a triggering event like interacting with SR-BI[43], and/or PLTP[67] and then further interacts with some unknown factor(s) as a lipid-free form when it crosses the BBB[45]. One possible candidate in this process may be cubilin. It is ubiquitously expressed by cells that make up the BBB and is known to bind to apoA-I, as well as albumin and vitamin B12[68]. In heterozygote cubilin-KO mice, a relatively slow catabolic rate of HDL2 was reported[69] that may involve decreased translocation of apoA-I into the abluminal side of cells.
Once, the lipid-free apoA-I translocated into the brain interstitial fluid, it may interact with neuronal and glial ABCA1 before entry into the CSF compartment (Figure 8) via CSF-interstitial fluid exchange like the glymphatic flux pathway [70–73]. In support of this hypothesis, apoE in CSF was reduced by approximately 75% in Abca1-null mice compared to control WT (C57BL/6N), despite the fact that substantial apoE is synthesized in neuronal and glial cells in the brain. Abca1-null mice CSF had less apoA-I possibly due to a reduction of spherical HDL from the luminal blood side of BBB, and decreased lipidation within the brain itself leading to increased catabolism. In the presence of Abca1 expression, it is expected that increased levels of apoA-I will be provided at the blood side of BBB, and that any lipid-free apoA-I transferred into brain interstitial fluid will be converted to disc-shape, preβ-HDL[74]. This could occur in neuronal and glial cellular lipids by interacting ABCA1 [18,46,63] and followed by LCAT reaction [64] to generate spherical mature HDL. LCAT is known to be ubiquitously expressed in neuronal and glial cells (Figure 8).
Figure 8.
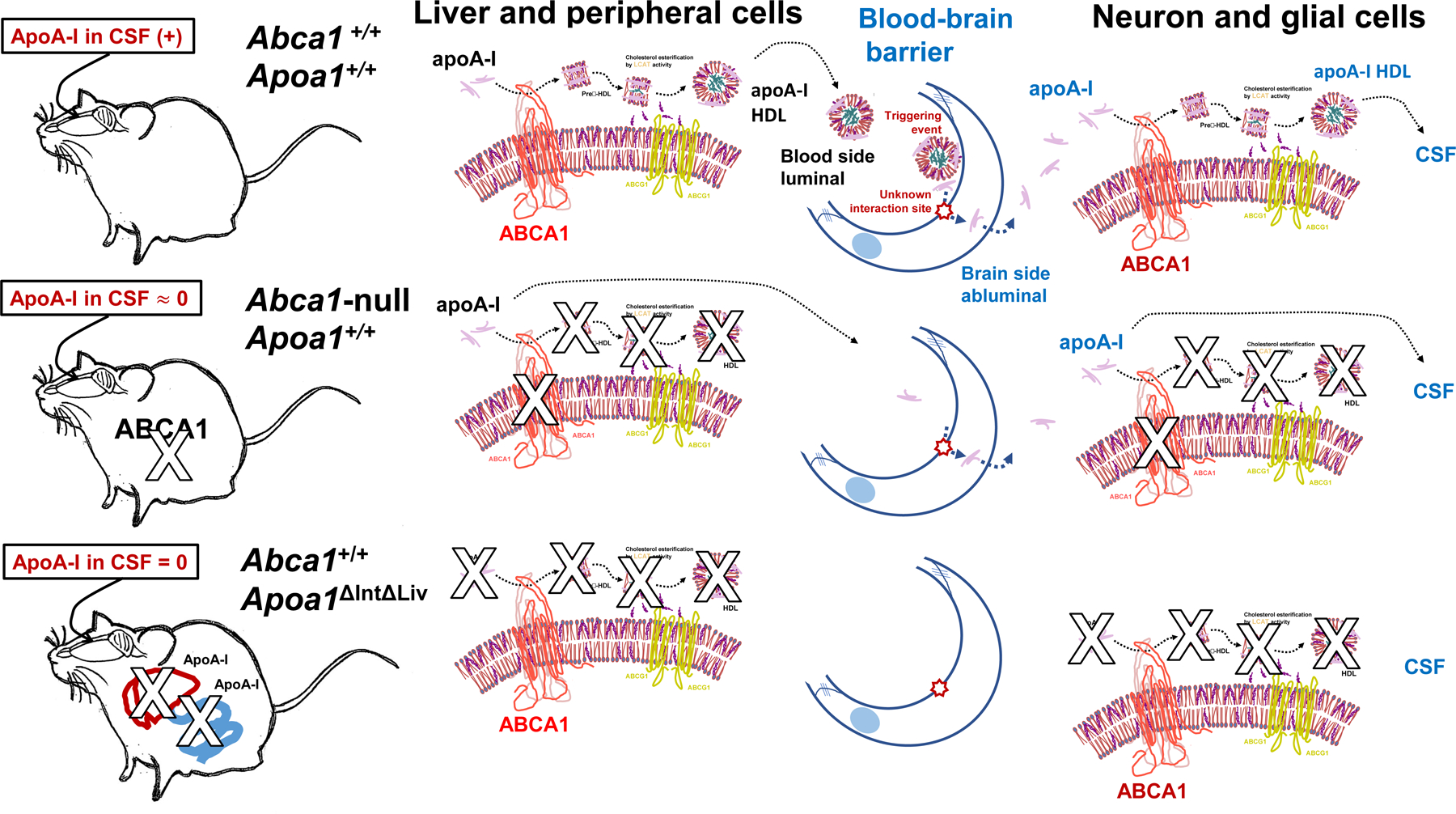
Illustrated summary sketch of apoA-I fates in biofluid and the mouse genotypes.
In conclusion, we demonstrated that Apoa1ΔIntΔLiv mice showed complete loss of apoA-I protein levels in CSF, as well as in plasma. Moreover, apoA-I levels in CSF from Abca1-null mice was found to be reduced by approximately 98% compared to control WT mice, despite that apoA-I is robustly expressed in these mice. Thus, ABCA1 is likely necessary for the maintenance of apoA-I content in both the plasma and CSF compartments. This is critically important as apoA-I/HDL in CSF likely serves to protect against neuronal insults and damage in the brain[34,37]. Based on these results, the main findings of the current study are that 1) CSF apoA-I likely originates from plasma, 2) both the liver and small intestine tissues have the ability to generate sufficient HDL despite the loss of apoA-I expression in the other tissue which may have occurred by compensatory changes in genes that modulate HDL-C levels, and 3) ABCA1 activity and mature HDL formation lead to increased apoA-I levels in plasma and other body fluids that ultimately lead to its transfer into the CSF. These findings support key roles for the liver and small intestine in maintaining proper brain function and protection or repairing from neural injury through the regulation of CSF apoA-I content.
Acknowledgements:
Authors are grateful to Mr. Takumi Kishida and Mr. Yasuyuki Maekawa, medical students at Nagoya City University, and Ms. Marina Tachibana, a PhD student at Tokushima University for their excellent technical assistance. Authors thank Mr. Hiroshi Takase, a Core Lab, Nagoya City University, for his outstanding technical support on TEM images. Authors sincerely thank Dr. Frank J. Gonzalez, Center for Cancer Research, NCI, NIH for his helpful advice and critical editing of this manuscript.
Funding
This project was supported by Grant-in-Aid from Ministry of Education, Culture, Science and Technology of Japan (MEXT) (23591338, 15K08615, 15K15349, 16K08236), Takahashi Industrial and Economic Research Foundation (08-00303038), Nagoya City University Grant-in-Aid for Encouragement of Scientists (2018-17, 1913007, 1943006, and 2050002). Research by ATR and BV was supported by intramural DIR research funds by NHLBI. And this work was also supported by awards from the National Institutes of Health, USA (KCV, HL128996, HL116263).
Abbreviations:
- ABCA1
ATP binding cassette subfamily A member 1
- apoA-I
apolipoprotein A-I
- apoE
apolipoprotein E
- APP
amyloid precursor protein
- CAG
modified chicken β-actin promoter with CMV-IE enhancer and β-globin gene
- Cas9
CRISPR-associated protein 9
- Cre
carbapenem-resistant Enterobacteriaceae
- CRISPR
clustered regularly interspaced short palindromic repeats
- CSF
cerebrospinal fluid
- DGUC
density gradient ultra-centrifuge
- EDTA
ethylenediaminetetraacetic acid
- ELISA
enzyme-linked immunosorbent assay
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- HDL
high-density lipoprotein
- LCAT
lecithin–cholesterol acyltransferase
- LRP1
low-density lipoprotein receptor-related protein 1
- loxP
locus of cross-over in P1
- PAM
protospacer adjacent motif
- PS1
presenilin-1
- SR-BI
scavenger receptor class B type 1
- TEM
transmission electron microscopy
Footnotes
Disclosure of conflicts of interest:
The authors declare no financial or other conflicts of interest.
References:
- [1].Gordon T, Castelli WP, Hjortland MC, Kannel WB and Dawber TR (1977). High density lipoprotein as a protective factor against coronary heart disease. The Framingham Study. Am J Med 62, 707–14. [DOI] [PubMed] [Google Scholar]
- [2].Michell DL and Vickers KC (2016). HDL and microRNA therapeutics in cardiovascular disease. Pharmacol Ther 168, 43–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Gordon SM et al. (2018). High density lipoprotein proteome is associated with cardiovascular risk factors and atherosclerosis burden as evaluated by coronary CT angiography. Atherosclerosis 278, 278–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Barrett TJ et al. (2019). Apolipoprotein AI) Promotes Atherosclerosis Regression in Diabetic Mice by Suppressing Myelopoiesis and Plaque Inflammation. Circulation 140, 1170–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Sacks FM et al. (2009). Selective delipidation of plasma HDL enhances reverse cholesterol transport in vivo. J Lipid Res 50, 894–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Wu BJ, Chen K, Shrestha S, Ong KL, Barter PJ and Rye KA (2013). High-density lipoproteins inhibit vascular endothelial inflammation by increasing 3β-hydroxysteroid-Δ24 reductase expression and inducing heme oxygenase-1. Circ Res 112, 278–88. [DOI] [PubMed] [Google Scholar]
- [7].Roheim PS, Carey M, Forte T and Vega GL (1979). Apolipoproteins in human cerebrospinal fluid. Proc Natl Acad Sci U S A 76, 4646–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Koch S, Donarski N, Goetze K, Kreckel M, Stuerenburg HJ, Buhmann C and Beisiegel U (2001). Characterization of four lipoprotein classes in human cerebrospinal fluid. J Lipid Res 42, 1143–51. [PubMed] [Google Scholar]
- [9].Borghini I, Barja F, Pometta D and James RW (1995). Characterization of subpopulations of lipoprotein particles isolated from human cerebrospinal fluid. Biochim Biophys Acta 1255, 192–200. [DOI] [PubMed] [Google Scholar]
- [10].Danik M, Champagne D, Petit-Turcotte C, Beffert U and Poirier J (1999). Brain lipoprotein metabolism and its relation to neurodegenerative disease. Crit Rev Neurobiol 13, 357–407. [DOI] [PubMed] [Google Scholar]
- [11].Elliott DA, Weickert CS and Garner B (2010). Apolipoproteins in the brain: implications for neurological and psychiatric disorders. Clin Lipidol 51, 555–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Flowers SA and Rebeck GW (2020). APOE in the normal brain. Neurobiol Dis 136, 104724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Ruiz J et al. (2005). The apoE isoform binding properties of the VLDL receptor reveal marked differences from LRP and the LDL receptor. J Lipid Res 46, 1721–31. [DOI] [PubMed] [Google Scholar]
- [14].Pászty C, Maeda N, Verstuyft J and Rubin EM (1994). Apolipoprotein AI transgene corrects apolipoprotein E deficiency-induced atherosclerosis in mice. J Clin Invest 94, 899–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Ishibashi S, Herz J, Maeda N, Goldstein JL and Brown MS (1994). The two-receptor model of lipoprotein clearance: tests of the hypothesis in “knockout” mice lacking the low density lipoprotein receptor, apolipoprotein E, or both proteins. Proc Natl Acad Sci U S A 91, 4431–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Safina D, Schlitt F, Romeo R, Pflanzner T, Pietrzik CU, Narayanaswami V, Edenhofer F and Faissner A (2016). Low-density lipoprotein receptor-related protein 1 is a novel modulator of radial glia stem cell proliferation, survival, and differentiation. Glia 64, 1363–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Asaro A, Carlo-Spiewok AS, Malik AR, Rothe M, Schipke CG, Peters O, Heeren J and Willnow TE (2020). Apolipoprotein E4 disrupts the neuroprotective action of sortilin in neuronal lipid metabolism and endocannabinoid signaling. Alzheimers Dement [DOI] [PubMed] [Google Scholar]
- [18].Hirsch-Reinshagen V et al. (2004). Deficiency of ABCA1 impairs apolipoprotein E metabolism in brain. J Biol Chem 279, 41197–207. [DOI] [PubMed] [Google Scholar]
- [19].Ito J, Zhang LY, Asai M and Yokoyama S (1999). Differential generation of high-density lipoprotein by endogenous and exogenous apolipoproteins in cultured fetal rat astrocytes. J Neurochem 72, 2362–9. [DOI] [PubMed] [Google Scholar]
- [20].Koldamova RP, Lefterov IM, Ikonomovic MD, Skoko J, Lefterov PI, Isanski BA, DeKosky ST and Lazo JS (2003). 22R-hydroxycholesterol and 9-cis-retinoic acid induce ATP-binding cassette transporter A1 expression and cholesterol efflux in brain cells and decrease amyloid beta secretion. J Biol Chem 278, 13244–56. [DOI] [PubMed] [Google Scholar]
- [21].Yokoyama S (2006). Assembly of high-density lipoprotein. Arterioscler Thromb Vasc Biol 26, 20–7. [DOI] [PubMed] [Google Scholar]
- [22].Yokoyama S (2006). ABCA1 and biogenesis of HDL. J Atheroscler Thromb 13, 1–15. [DOI] [PubMed] [Google Scholar]
- [23].Yokoyama S (2000). Release of cellular cholesterol: molecular mechanism for cholesterol homeostasis in cells and in the body. Biochim Biophys Acta 1529, 231–44. [DOI] [PubMed] [Google Scholar]
- [24].Cao G, Garcia CK, Wyne KL, Schultz RA, Parker KL and Hobbs HH (1997). Structure and localization of the human gene encoding SR-BI/CLA-1. Evidence for transcriptional control by steroidogenic factor 1. J Biol Chem 272, 33068–76. [DOI] [PubMed] [Google Scholar]
- [25].Trigatti B, Rigotti A and Krieger M (2000). The role of the high-density lipoprotein receptor SR-BI in cholesterol metabolism. Curr Opin Lipidol 11, 123–31. [DOI] [PubMed] [Google Scholar]
- [26].Reddy DS (2010). Neurosteroids: endogenous role in the human brain and therapeutic potentials. Prog Brain Res 186, 113–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Baulieu EE (1997). Neurosteroids: of the nervous system, by the nervous system, for the nervous system. Recent Prog Horm Res 52, 1–32. [PubMed] [Google Scholar]
- [28].Tsutsui K and Yamazaki T (1995). Avian neurosteroids. I. Pregnenolone biosynthesis in the quail brain. Brain Res 678, 1–9. [DOI] [PubMed] [Google Scholar]
- [29].Usui M, Yamazaki T, Kominami S and Tsutsui K (1995). Avian neurosteroids. II. Localization of a cytochrome P450scc-like substance in the quail brain. Brain Res 678, 10–20. [DOI] [PubMed] [Google Scholar]
- [30].Tsutsui K, Ukena K, Takase M, Kohchi C and Lea RW (1999). Neurosteroid biosynthesis in vertebrate brains. Comp Biochem Physiol C Pharmacol Toxicol Endocrinol 124, 121–9. [DOI] [PubMed] [Google Scholar]
- [31].Diotel N et al. (2018). Steroid Transport, Local Synthesis, and Signaling within the Brain: Roles in Neurogenesis, Neuroprotection, and Sexual Behaviors. Front Neurosci 12, 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Provost AC, Péquignot MO, Sainton KM, Gadin S, Sallé S, Marchant D, Hales DB and Abitbol M (2003). Expression of SR-BI receptor and StAR protein in rat ocular tissues. C R Biol 326, 841–51. [DOI] [PubMed] [Google Scholar]
- [33].Boyles JK et al. (1989). A role for apolipoprotein E, apolipoprotein A-I, and low density lipoprotein receptors in cholesterol transport during regeneration and remyelination of the rat sciatic nerve. J Clin Invest 83, 1015–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Bahrami A, Barreto GE, Lombardi G, Pirro M and Sahebkar A (2019). Emerging roles for high-density lipoproteins in neurodegenerative disorders. Biofactors 45, 725–739. [DOI] [PubMed] [Google Scholar]
- [35].Lefterov I, Fitz NF, Cronican AA, Fogg A, Lefterov P, Kodali R, Wetzel R and Koldamova R (2010). Apolipoprotein A-I deficiency increases cerebral amyloid angiopathy and cognitive deficits in APP/PS1DeltaE9 mice. J Biol Chem 285, 36945–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Lewis TL et al. (2010). Overexpression of human apolipoprotein A-I preserves cognitive function and attenuates neuroinflammation and cerebral amyloid angiopathy in a mouse model of Alzheimer disease. J Biol Chem 285, 36958–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Button EB et al. (2019). ApoA-I deficiency increases cortical amyloid deposition, cerebral amyloid angiopathy, cortical and hippocampal astrogliosis, and amyloid-associated astrocyte reactivity in APP/PS1 mice. Alzheimers Res Ther 11, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Thacker SG, Zarzour A, Chen Y, Alcicek MS, Freeman LA, Sviridov DO, Demosky SJ and Remaley AT (2016). High-density lipoprotein reduces inflammation from cholesterol crystals by inhibiting inflammasome activation. Immunology 149, 306–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Gordon SM, McKenzie B, Kemeh G, Sampson M, Perl S, Young NS, Fessler MB and Remaley AT (2015). Rosuvastatin Alters the Proteome of High Density Lipoproteins: Generation of alpha-1-antitrypsin Enriched Particles with Anti-inflammatory Properties. Mol Cell Proteomics 14, 3247–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Schwendeman A et al. (2015). The effect of phospholipid composition of reconstituted HDL on its cholesterol efflux and anti-inflammatory properties. J Lipid Res 56, 1727–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Murali N et al. (2020). Cholesterol and neurodegeneration: longitudinal changes in serum cholesterol biomarkers are associated with new lesions and gray matter atrophy in multiple sclerosis over 5 years of followup. Eur J Neurol 27, 188–e4. [DOI] [PubMed] [Google Scholar]
- [42].Stukas S et al. (2014). Intravenously injected human apolipoprotein A-I rapidly enters the central nervous system via the choroid plexus. J Am Heart Assoc 3, e001156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Fung KY, Wang C, Nyegaard S, Heit B, Fairn GD and Lee WL (2017). SR-BI Mediated Transcytosis of HDL in Brain Microvascular Endothelial Cells Is Independent of Caveolin, Clathrin, and PDZK1. Front Physiol 8, 841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Dal Magro R et al. (2019). The Extent of Human Apolipoprotein A-I Lipidation Strongly Affects the βAmyloid Efflux Across the Blood-Brain Barrier. Front Neurosci 13, 419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Zhou AL, Swaminathan SK, Curran GL, Poduslo JF, Lowe VJ, Li L and Kandimalla KK (2019). Apolipoprotein A-I Crosses the Blood-Brain Barrier through Clathrin-Independent and Cholesterol-Mediated Endocytosis. J Pharmacol Exp Ther 369, 481–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Karasinska JM et al. (2009). Specific loss of brain ABCA1 increases brain cholesterol uptake and influences neuronal structure and function. J Neurosci 29, 3579–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Liu Y, Du Y, Xie W, Zhang F, Forrest D and Liu C (2019). Generation of Conditional Knockout Mice by Sequential Insertion of Two loxP Sites In Cis Using CRISPR/Cas9 and Single-Stranded DNA Oligonucleotides. Methods Mol Biol 1874, 191–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Sano O et al. (2016). ABCG1 and ABCG4 Suppress γ-Secretase Activity and Amyloid β Production. PLoS One 11, e0155400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].DeMattos RB, Bales KR, Parsadanian M, O’Dell MA, Foss EM, Paul SM and Holtzman DM (2002). Plaque-associated disruption of CSF and plasma amyloid-beta (Abeta) equilibrium in a mouse model of Alzheimer’s disease. J Neurochem 81, 229–36. [DOI] [PubMed] [Google Scholar]
- [50].Liu L and Duff K (2008). A technique for serial collection of cerebrospinal fluid from the cisterna magna in mouse. J Vis Exp [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Aboumsallem JP, Mishra M, Amin R, Muthuramu I, Kempen H and De Geest B (2018). Successful treatment of established heart failure in mice with recombinant HDL (Milano). Br J Pharmacol 175, 4167–4182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Centa M et al. (2018). Acute Loss of Apolipoprotein E Triggers an Autoimmune Response That Accelerates Atherosclerosis. Arterioscler Thromb Vasc Biol 38, e145–e158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Rubin DC, Swietlicki EA, Wang JL, Dodson BD and Levin MS (1996). Enterocytic gene expression in intestinal adaptation: evidence for a specific cellular response. Am J Physiol 270, G143–52. [DOI] [PubMed] [Google Scholar]
- [54].Plump AS, Azrolan N, Odaka H, Wu L, Jiang X, Tall A, Eisenberg S and Breslow JL (1997). ApoA-I knockout mice: characterization of HDL metabolism in homozygotes and identification of a post-RNA mechanism of apoA-I up-regulation in heterozygotes. J Lipid Res 38, 1033–47. [PubMed] [Google Scholar]
- [55].Tsujita M, Akita N, Yokota T, Kobayashi F and Yokoyama S (2020). Selective Correction of Genotype Yield by Probucol in HDL-Deficient Mice Propagation. J Atheroscler Thromb 27, 25–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Hara H and Yokoyama S (1992). Role of apolipoproteins in cholesterol efflux from macrophages to lipid microemulsion: proposal of a putative model for the pre-beta high-density lipoprotein pathway. Biochemistry 31, 2040–6. [DOI] [PubMed] [Google Scholar]
- [57].Duka A et al. (2013). ApoA-IV promotes the biogenesis of apoA-IV-containing HDL particles with the participation of ABCA1 and LCAT. J Lipid Res 54, 107–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Tomimoto S et al. (2001). Effect of probucol in lecithin-cholesterol acyltransferase-deficient mice: inhibition of 2 independent cellular cholesterol-releasing pathways in vivo. Arterioscler Thromb Vasc Biol 21, 394–400. [DOI] [PubMed] [Google Scholar]
- [59].Ng DS, Francone OL, Forte TM, Zhang J, Haghpassand M and Rubin EM (1997). Disruption of the murine lecithin:cholesterol acyltransferase gene causes impairment of adrenal lipid delivery and up-regulation of scavenger receptor class B type I. J Biol Chem 272, 15777–81. [DOI] [PubMed] [Google Scholar]
- [60].Rebholz SL, Melchior JT, Davidson WS, Jones HN, Welge JA, Prentice AM, Moore SE and Woollett LA (2018). Studies in genetically modified mice implicate maternal HDL as a mediator of fetal growth. FASEB J 32, 717–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Chang EH, Rigotti A and Huerta PT (2009). Age-related influence of the HDL receptor SR-BI on synaptic plasticity and cognition. Neurobiol Aging 30, 407–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Sengupta MB and Mukhopadhyay D (2016). Possible role of apolipoprotein A1 in healing and cell death after neuronal injury. Front Biosci (Elite Ed) 8, 460–77. [DOI] [PubMed] [Google Scholar]
- [63].Wahrle SE, Jiang H, Parsadanian M, Legleiter J, Han X, Fryer JD, Kowalewski T and Holtzman DM (2004). ABCA1 is required for normal central nervous system ApoE levels and for lipidation of astrocytesecreted apoE. J Biol Chem 279, 40987–93. [DOI] [PubMed] [Google Scholar]
- [64].Hirsch-Reinshagen V et al. (2009). LCAT synthesized by primary astrocytes esterifies cholesterol on gliaderived lipoproteins. J Lipid Res 50, 885–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Okuhira K, Tsujita M, Yamauchi Y, Abe-Dohmae S, Kato K, Handa T and Yokoyama S (2004). Potential involvement of dissociated apoA-I in the ABCA1-dependent cellular lipid release by HDL. J Lipid Res 45, 645–52. [DOI] [PubMed] [Google Scholar]
- [66].Handa D, Kimura H, Oka T, Takechi Y, Okuhira K, Phillips MC and Saito H (2015). Kinetic and thermodynamic analyses of spontaneous exchange between high-density lipoprotein-bound and lipid-free apolipoprotein A-I. Biochemistry 54, 1123–31. [DOI] [PubMed] [Google Scholar]
- [67].Chirackal Manavalan AP et al. (2014). Phospholipid transfer protein is expressed in cerebrovascular endothelial cells and involved in high density lipoprotein biogenesis and remodeling at the blood-brain barrier. J Biol Chem 289, 4683–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Kozyraki R et al. (1999). The intrinsic factor-vitamin B12 receptor, cubilin, is a high-affinity apolipoprotein AI receptor facilitating endocytosis of high-density lipoprotein. Nat Med 5, 656–61. [DOI] [PubMed] [Google Scholar]
- [69].Aseem O, Smith BT, Cooley MA, Wilkerson BA, Argraves KM, Remaley AT and Argraves WS (2014). Cubilin maintains blood levels of HDL and albumin. J Am Soc Nephrol 25, 1028–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Strazielle N and Ghersi-Egea JF (2013). Physiology of blood-brain interfaces in relation to brain disposition of small compounds and macromolecules. Mol Pharm 10, 1473–91. [DOI] [PubMed] [Google Scholar]
- [71].Iliff JJ, Lee H, Yu M, Feng T, Logan J, Nedergaard M and Benveniste H (2013). Brain-wide pathway for waste clearance captured by contrast-enhanced MRI. J Clin Invest 123, 1299–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Nedergaard M (2013). Neuroscience. Garbage truck of the brain. Science 340, 1529–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Mestre H, Mori Y and Nedergaard M (2020). The Brain’s Glymphatic System: Current Controversies. Trends Neurosci 43, 458–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Tsujita M, Wolska A, Gutmann DAP and Remaley AT (2018). Reconstituted Discoidal High-Density Lipoproteins: Bioinspired Nanodiscs with Many Unexpected Applications. Curr Atheroscler Rep 20, 59. [DOI] [PubMed] [Google Scholar]