

Abstract
The kidney has extraordinary metabolic demands to sustain the active transport of solutes that is critical to renal filtration and clearance. Mitochondrial health is vital to meet those demands and maintain renal fitness. Decades of studies have linked poor mitochondrial health to kidney disease. Key regulators of mitochondrial health - 5-AMP-activated protein kinase (AMPK), sirtuins, and peroxisome proliferator-activated receptor γ coactivator-1alpha (PGC1α) - have all been shown to play significant roles in renal resilience against disease. This review will summarize the latest research into the activities of those regulators and evaluate the roles and therapeutic potential of targeting those regulators in acute kidney injury, glomerular kidney disease, and renal fibrosis.
Keywords: mitochondria, acute kidney injury, diabetic nephropathy, fibrosis, proteinuria
Introduction
In order to maintain a normal serum sodium level, the human kidney must filter and reabsorb nearly 20,000 milliequivalents of sodium daily, which is over a pound of sodium actively reabsorbed against a hundred-fold gradient. Unsurprisingly, the specialized cells that undertake that task require a tremendous amount of energy. Because of this energy dependency, a large body of literature has asked whether the kidney is particularly susceptible to metabolic insults and if those susceptibilities may offer therapeutic opportunities. Peroxisome proliferator-activated receptor γ (PPARγ) coactivator-1alpha (PGC1α), as a master regulator of mitochondrial biogenesis, and the enzymes that regulate PGC1α offer tremendous potential in that regard.
PGC1α is a member of the PGC1 family of transcriptional coactivators, along with PGC1β and PGC1α-related coactivator (PRC), that play a regulatory role in metabolic homeostasis by “boosting” the activity of nuclear receptors. PGC1α specifically acts as a master regulator of mitochondrial biogenesis. PGC1α was first described in 1998 when a group interested in obesity and regulation of brown fat uncoupling proteins identified this protein as a potent transcriptional coactivator for PPARγ.1 They showed that PGC1α is highly expressed after cold exposure with a simultaneous increase in the expression of uncoupling proteins, which disrupt the mitochondrial proton gradient to generate heat. Soon after, a much broader role of PGC1α was described. In addition to “wasting” metabolic heat energy via uncoupling, PGC1α was shown to control a multifaceted program to maintain total energy balance by increasing expression of oxidative phosphorylation genes and increasing mitochondrial biogenesis through upregulated mitochondrial DNA proliferation and increased expression of key mitochondrial transcription factors, nuclear respiratory factor (NRF1/2) and mitochondrial transcription factor A (TFAM).2 All actions combined, PGC1α emerged as a potent inducer of respiratory capacity despite energy loss from uncoupling.
PGC1α Regulation and Effectors
In addition to cold exposure, many other stimuli have been shown to induce PGC1α expression including exercise via AMPK,3 calorie restriction and oxidative stress via SIRT1,4 and hypoxia via AMPK (Figure 1).5 The downstream effects of PGC1α involve nearly every facet of mitochondrial function. Most notably, it stimulates mitochondrial biogenesis via increased translation of TFAM and NRF1/2.6, 7 But PGC1α also contributes to defense against oxidative stress by activating SIRT 3,4, 8 improves the health of the mitochondrial pool by activating estrogen related receptor-α (ERRα) and transcription factor EB (TFEB) to optimize mitochondrial dynamics,9 stimulates the biosynthesis of nicotinamide adenine dinucleotide (NAD+),10, 11 and stimulates β oxidation of fatty acids through PPARs and retinoid X receptors (RXRs).12, 13 PGC1α can be downregulated by many factors traditionally implicated in pro-inflammatory and pro-fibrotic cascades such as tumor necrosis factor-α (TNFα), transforming growth factor-β (TGFβ), and toll-like receptor 4 (TLR4).14–16 PGC1α is upregulated by a variety of metabolically significant transcription factors including hepatocyte nuclear factor 1 homeobox-β (HNF1β), TFEB, and pyruvate kinase M2 (PKM2).17–19 Once expressed, the PGC1α protein can undergo post-translational modifications that alter its activity. The most well understood, and perhaps most significant, PGC1α post-translational modifications are phosphorylation by 5-AMP-activated protein kinase (AMPK) and deacetylation by Sirtuin1 (SIRT1). A detailed discussion of PGC1α regulation is outside the scope of this review as this article will focus on PGC1α effects in the kidney. We will briefly discuss AMPK and sirtuins, mainly as they work through PGC1α, but more thorough discussions of PGC1α regulation have been published previously.20
Figure 1:
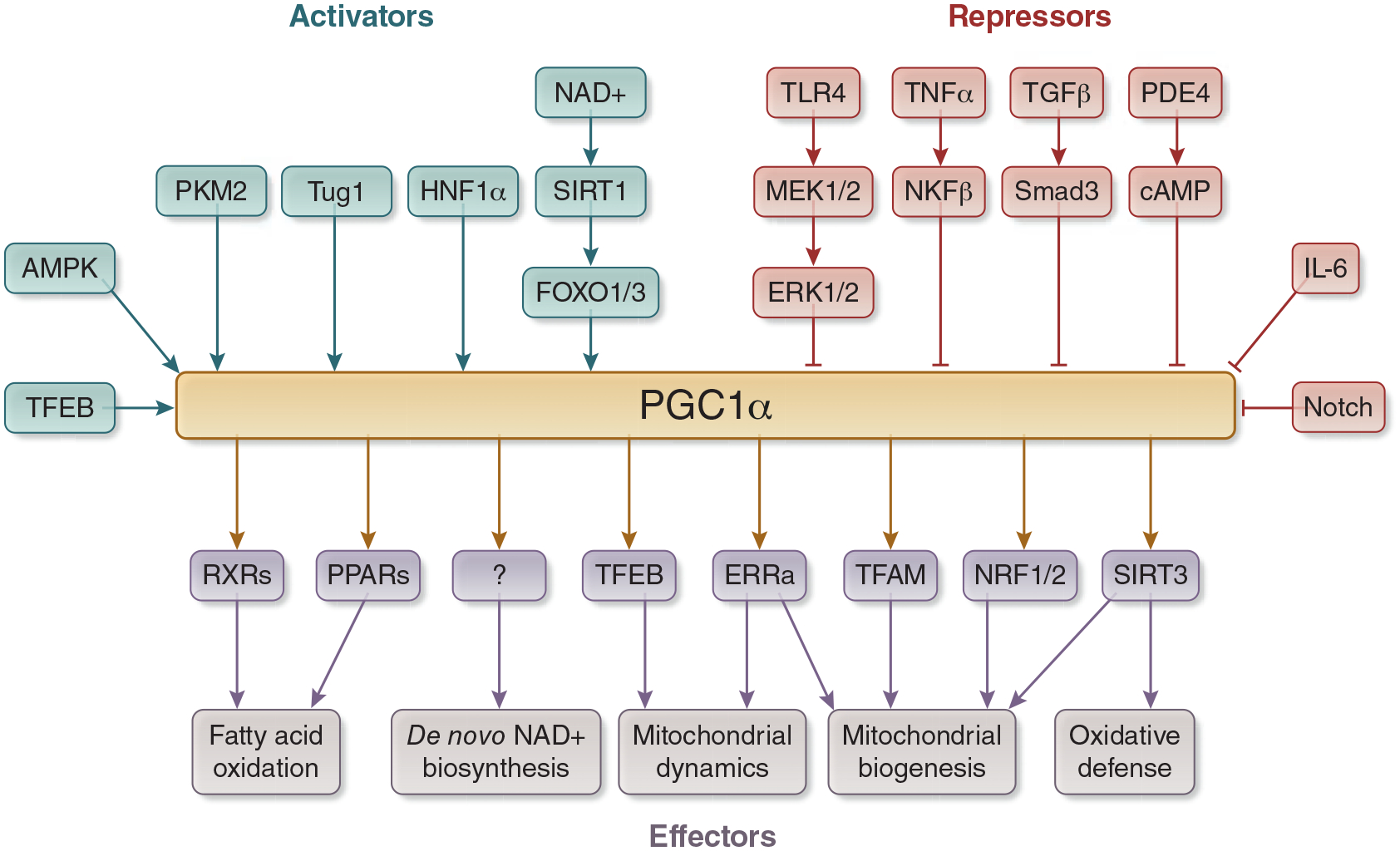
Regulators and Effectors of PGC1α. PGC1α is activated by many transcription factors, including SIRT1, AMPK, TFEB, PKM2, Tug1, and HNF1α. Transcription factors typically involved in inflammatory and pro-fibrotic pathways repress PGC1α, including TLR4, TNFα, TGFβ, PDE4, IL-6. PGC1α activates fatty acid oxidation through PPARs and RXRs. It enhances de novo biosynthesis of NAD+ through mechanisms that have yet to be delineated, and it controls mitochondrial health through activation of ERRα, TFAM, NRF1/2, and SIRT3. Abbreviations: PGC1α - Peroxisome proliferator activated receptor gamma coactivator 1 alpha, SIRT - sirtuin, AMPK - 5’ AMP-activated protein kinase, TFEB - transcription factor EB, PKM2 - pyruvate kinase M2, Tug1 - Taurine Up-Regulated 1, HNF1α - hepatocyte nuclear factor 1 alpha, TLR4 - toll-like receptor 4, TNFα - tumor necrosis factor alpha, TGFβ - transforming growth factor beta, PDE4 - phosphodiesterase 4, IL-6 - interleukin 6, PPARs - Peroxisome proliferator-activated receptors, RXRs - retinoid X receptors, ERRα - estrogen-related receptor alpha, TFAM - transcription factor A, mitochondrial, NRF - nuclear respiratory factor.
AMPK
AMPK acts as a monitor and protector of cellular energy needs through two critical functions: inhibition of energy consumption and stimulation of energy production. Anabolic pathways that consume energy to construct and store larger molecules are broadly inhibited by AMPK. For example, HMG-CoA reductase catalyzes the rate-limiting step in cholesterol synthesis and is inhibited upon phosphorylation by AMPK.21 Likewise, AMPK phosphorylates and inhibits acetyl-CoA carboxylases ACC1 and ACC2, which initiate the first steps in lipid synthesis.22 AMPK inhibits glycogen storage by inactivating glycogen synthases GYS1 and GYS223 and gluconeogenesis by inactivating CREB Regulated Transcription Coactivator 2 (CRTC2), a potent activator of many gluconeogenic genes.24 AMPK also works to conserve energy by inhibiting cell growth and protein translation, processes that consume cellular ATP stores. The mammalian target of rapamycin (mTOR) is a central kinase that stimulates cellular growth. AMPK inhibits mTOR by deactivating Raptor, a protein that complexes with mTOR to activate protein translation and cellular growth.25 AMPK additionally phosphorylates and activates Eukaryotic Elongation Factor 2 Kinase (eEF2K), which inhibits protein elongation.26
To make energy available, AMPK promotes the catabolism of macromolecules, increases glucose utilization, and mobilizes lipid stores. By deactivating Raptor, AMPK removes a natural brake on autophagy, the process whereby cellular components are broken down and recycled. With active Raptor, mTOR complexes inhibit autophagy by phosphorylating Unc-51 Like Autophagy Activating Kinase 1 (ULK1).25 AMPK also directly activates ULK1 to stimulate autophagy.27 To increase cellular glucose entry and utilization, AMPK stimulates membrane localization of glucose transporters GLUT1 and GLUT428 and stimulates glucose flux through glycolysis by activating the rate-limiting enzyme in glycolysis, phosphofructokinase-1.29 AMPK also activates lipases to release fatty acids from triglyceride stores30 and induces fatty acid import into mitochondria by inactivating malonyl-CoA production, thereby alleviating inhibition of Carnitine palmitoyltransferase I (CPT1), the rate-limiting enzyme for mitochondrial fatty acid oxidation (Figure 2).31
Figure 2:
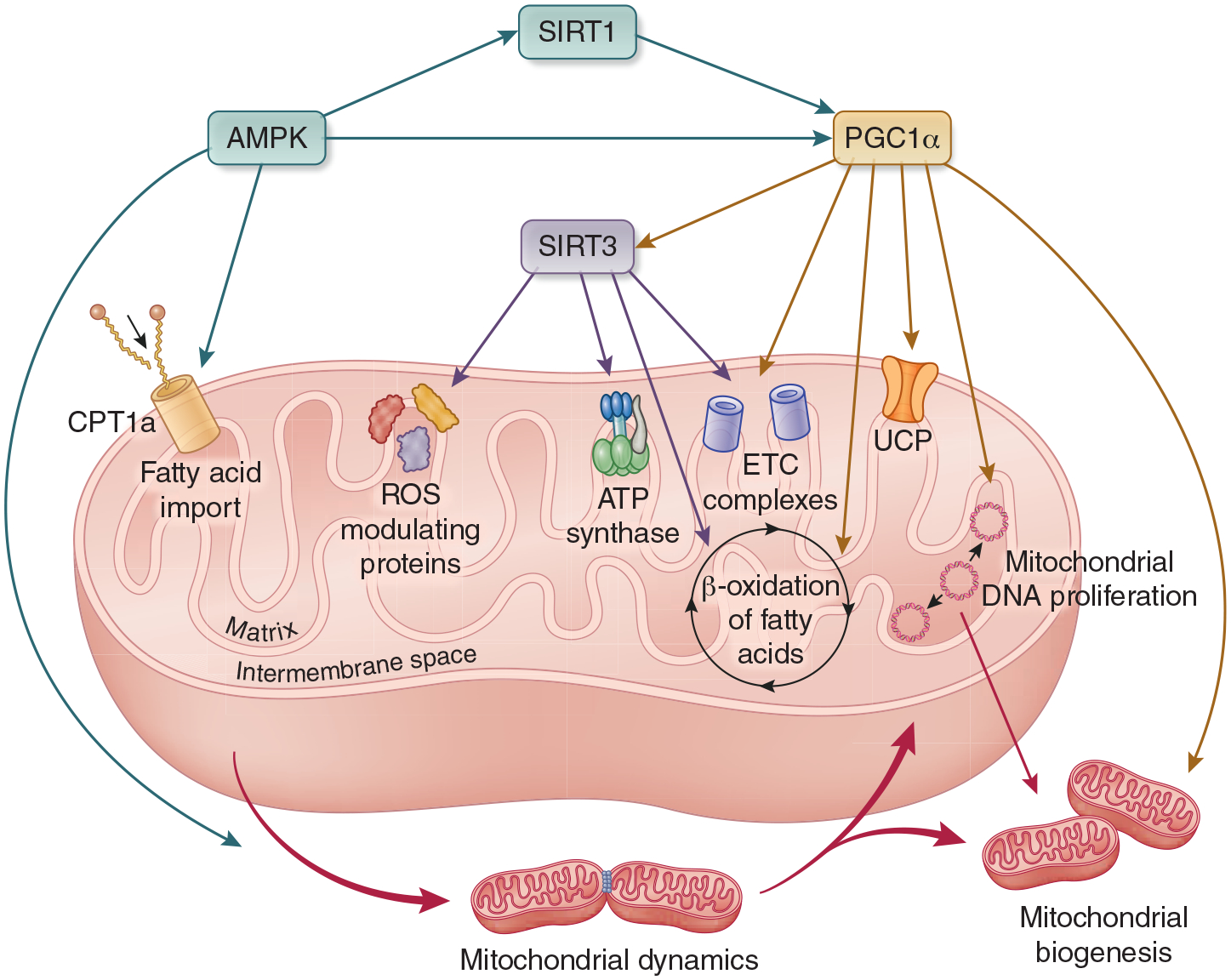
Direct effects of AMPK, Sirtuins, and PGC1α on mitochondrial function. AMPK activates SIRT1 and PGC1α. It also alleviates inhibition of CPT1, which stimulates fatty acid import into the mitochondria, and it activates the primary receptor for DRP1, which initiates mitochondrial fission and impacts mitochondrial dynamics. PGC1α stimulates mitochondrial biogenesis and mitochondrial DNA proliferation through NRF1/2 and TFAM. It increases expression of all complexes of the ETC and increases fatty acid flux through β-oxidation. In the liver and skeletal muscle, PGC1α stimulates expression of UCP. It also activates SIRT3. SIRT3 deacetylases and activates ETC complexes, including ATP Synthase. It activates many mitochondrial enzymes responsible for ROS scavenging, and it deacetylates LCAD, which initiates β oxidation to enhance energy substrate availability in the mitochondria. Abbreviations: AMPK - 5’ AMP-activated protein kinase, PGC1α - Peroxisome proliferator activated receptor gamma coactivator 1 alpha, SIRT - sirtuin, CPT1 - carnitine palmitoyltransferase I, DRP1 - dynamin-1-like protein, NRF - nuclear respiratory factor, TFAM - transcription factor A, mitochondrial, ETC - electron transport chain, UCP - uncoupling protein, ATP - adenosine triphosphate, ROS - reactive oxygen species, LCAD - long-chain acyl-CoA dehydrogenase.
Finally, AMPK plays a broad role in maintaining mitochondrial homeostasis and optimizing oxidative phosphorylation, the cell’s most efficient energy producing process. Most notably, AMPK stimulates mitochondrial biogenesis through activation of PGC1α. AMPK phosphorylates at least two unique PGC1α sites32 and also indirectly activates PGC1α via activation of SIRT1 and TFEB.18, 33 As mentioned above, AMPK releases CPT1 inhibition. This stimulates transport of fatty acids into the mitochondria where they undergo sequential β-oxidation to become critical fuel substrate for the Krebs cycle and the electron transport chain (ETC).31 AMPK also modulates mitochondrial dynamics, the coordinated process where mitochondria undergo cycles of fission and fusion to maintain appropriate size, shape, and overall health. In the presence of overwhelming mitochondrial stress, the normal dynamic process cannot keep pace, and mitochondria undergo fragmentation.34 AMPK activates the primary receptor for dynamin-like protein (DRP1), a fundamental component of mitochondrial fission.35 Similarly, the AMPK-activated ULK1 autophagy pathway induces mitochondrial autophagy—termed mitophagy—to remove damaged mitochondria.36 In summary, AMPK acts as a true guardian of energy expenditure by reducing anabolism, enhancing fuel catabolism to generate energy, and multidimensionally optimizing mitochondrial fitness.
In kidney disease, AMPK has been shown to protect against fibrosis, diabetic kidney disease (DKD), and acute kidney injury (AKI). AMPK expression mitigated progressive renal fibrosis from high-fat diet, aging, folic acid nephropathy, and ureteral obstruction in mice.37–40 Activation of AMPK also attenuated diabetic nephropathy in mice and reduced hyperglycemia-associated expression of fibrosis genes in podocytes.41, 42 AMPK activation reduced acute tubular injury in rats and mice exposed to cisplatin.43–45 Preconditioning proximal tubule cells and mice with AMPK activators reduced the severity of ischemic renal injury.46 Through its broad actions, AMPK activity is generally associated with improved outcomes after diverse experimental renal insults. While this review focuses on PGC1α activation, the other AMPK-driven effects also warrant further study as potential pathways to modulate kidney disease.
Sirtuins
Sirtuins are a class of NAD+ consuming deacetylases that exhibit a broad range of cellular functions. Two of the most studied sirtuins, and the two most pertinent to this review, are SIRT1 and SIRT3. SIRT1 resides in the nucleus where it regulates the activity of key transcription factors. By inhibiting NFκB via deacetylation, SIRT1 diminishes TNFα signaling to exert an anti-inflammatory effect.47 SIRT1 also reduces cellular senescence, cell death, and susceptibility to oxidative stress by deacetylating p5348 and forehead box type O (FoxO) transcription factors.49 SIRT1 stimulates erythropoietin expression in response to hypoxia by regulating hypoxia-inducible factor-2α.50 It also stimulates mitochondrial biogenesis by deacetylating and activating PGC1α.51 (Figure 2)
SIRT3 resides in the mitochondrial matrix where it deacetylates integral complexes of the ETC, including adenosine triphosphate (ATP) synthase, to boost ATP production.52, 53 SIRT3 also reduces oxidative stress by directly binding and altering the function of many mitochondrial enzymes involved in production or scavenging of reactive oxygen species (ROS) including α-Ketoglutarate dehydrogenase (αKGDH),54 electron flavoprotein dehydrogenase (ETFDH),55 and superoxide dismutase 2 (SOD2).8 Lastly, SIRT3 increases mitochondrial fuel supply by deacetylation of long-chain acyl coenzyme A dehydrogenase (LCAD), which enhances fatty acid β-oxidation.56 In perhaps a useful oversimplification, SIRT1 broadly counteracts stress via anti-inflammatory actions that also increase PGC1α expression, while SIRT3 works specifically to boost mitochondrial function and cellular respiration.
Extensive studies have elucidated a protective role for sirtuins in renal disease.57 Expression of both SIRT1 and SIRT3 was decreased in aging kidneys.58–60 Loss of SIRT1 accelerated glomerulosclerosis and albuminuria in aging mice,61 while upregulation was associated with longevity.58–62 SIRT1 attenuated renal fibrosis in mice with ureteral obstruction63 and 5/6 nephrectomy64, 65 while SIRT3 mitigated hypertension-associated fibrosis.66 In AKI, sirtuins are downregulated by unknown mechanisms. SIRT3 knockout led to more severe injury in a mouse sepsis model.67 Experimental activation of SIRT1 protected against ischemia-reperfusion injury (IRI);68 SIRT3 overexpression protected against sepsis-associated AKI;69 and both overexpression of SIRT1 or pharmacologic activation of SIRT3 protected against cisplatin-induced injury.70, 71 In human glomeruli, diabetes reduced SIRT1, which worsened albuminuria and disease progression in mice.72
Outside of SIRT1 and SIRT3, other sirtuins play a role in renal disease, though perhaps not in direct concert with PGC1α. SIRT6 was upregulated in a mouse fibrosis model, and inhibition or genetic knockout led to more severe fibrosis73 and more severe age-related kidney disease.74 In cellular models, SIRT6 interacted with β-catenin to deacetylate histones and prevent fibrogenic gene expression.73 SIRT7 and SIRT5 may not be renoprotective like other sirtuins. For example, knockout of SIRT7, another histone deacetylase, led to reduced expression of TNFα and protected against cisplatin-induced AKI.75 Knockout of SIRT5, a lysine deacylase, mitigated renal injury after IRI or cisplatin treatment by impairing the balance between mitochondrial and peroxisomal β-oxidation.76 In cellular models, SIRT5 appeared more protective as overexpression alleviated cisplatin injury.77 As with AMPK, the broad role of sirtuins in the search for renal therapies is an active area of study.
PGC1α and Kidney Disease
In the kidney, PGC1α is highly expressed in the renal cortex and the corticomedullary junction, regions where cellular respiration is highest.78 PGC1α has been widely studied as part of a broader effort to understand the role of mitochondrial health in acquired kidney diseases. Abnormal-appearing mitochondria have been associated with human kidney injury since early electron microscopy studies from the 1970s.79 More recent studies have shown that cells of the renal tubule enduring AKI develop mitochondrial dysfunction with less efficient oxygen consumption,80 accumulation of fatty acids,81 impaired biosynthesis of NAD+,82 decreased mitochondrial biogenesis, and a shift of mitochondrial dynamics away from the fused state. Each of these changes diminishes the healthy pool of mitochondria necessary for normal renal health and function.83 A growing body of literature has demonstrated that PGC1α may play a significant role in mitigating not only AKI, but also glomerular kidney disease and renal fibrosis.
PGC1α and Acute Kidney Injury
Within the kidney, the renal tubular cells are most abundant in mitochondria, the most oxidative, and the most affected by AKI. Yet, AKI-associated mitochondrial dysfunction is not merely a corollary of decreased renal blood flow and oxygen delivery; indeed tissue oxygen levels do not appear to change even in a setting of reduced oxygen delivery.78 The mitochondrial pool actually becomes “sick” in response to ischemic, inflammatory, or toxic stress. Identifying the mechanisms whereby mitochondria are affected in AKI may lead to strategies that optimize the health of the renal mitochondrial pool to prevent and combat AKI.
Nearly every facet of mitochondrial function is impaired in AKI regardless of the etiology (Figure 3). Mitochondrial swelling is an early feature of human renal ischemia, even when AKI does not ensue;84 it is a common finding across experimental models of AKI and is considered structural evidence of mitochondrial depolarization and dysfunction.85, 86 Toxic, inflammatory, and ischemic forms of AKI result in cortical accumulation of triglycerides, which can become peroxidated and further exacerbate renal injury.87, 88 Mitochondrial dynamics are also altered by AKI, resulting in decreased fusion, increased fragmentation, increased release of cytochrome C and ROS, and increased apoptosis.45, 71, 89 Interventions that shift mitochondrial dynamics away from fragmentation, either broadly by SIRT1 activation or specifically by suppression of the fission mediator DRP1, have been shown to reduce the severity of AKI.71, 90 Cellular respiration is also notably impaired in AKI with reduced oxygen consumption, reduced expression of ETC components including ATP synthase, depolarization of the mitochondrial membrane, decreased mitochondrial NAD+ levels, and decreased ATP production.10, 78, 91, 92
Figure 3:
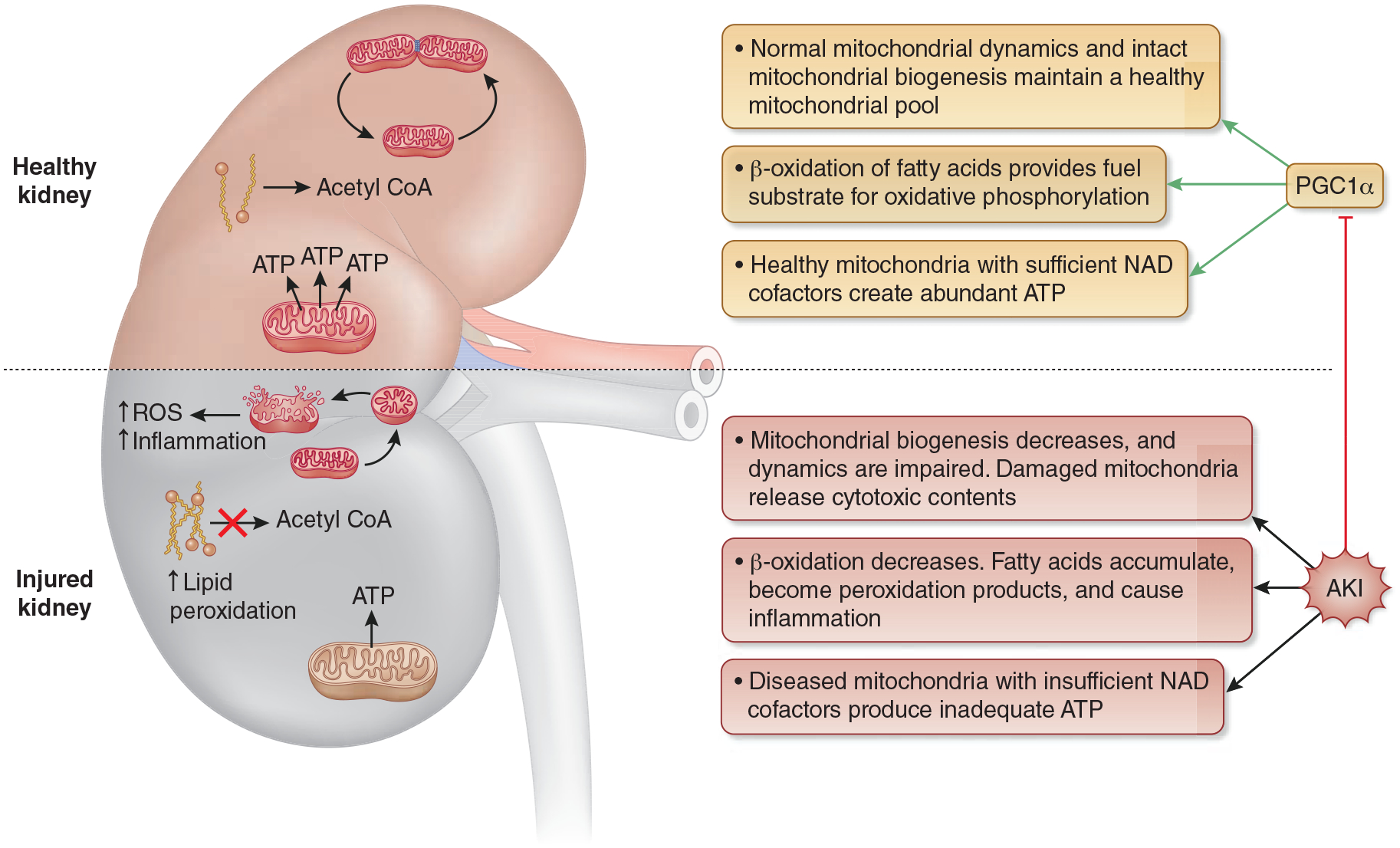
The Role of PGC1α in AKI. With AKI-induced suppression of PGC1α, mitochondrial biogenesis decreases, and mitochondrial dynamics are altered. The result is an insufficient quantity and lower quality pool of mitochondria. When mitochondrial injury is too significant to be safely disposed through normal mitochondrial dynamics, injured mitochondria undergo fragmentation and release damaging contents into the cells, which leads to increased ROS production and inflammation. The decreased availability of healthy mitochondria combined with reduced expression of ETC complexes, leads to decreased ATP production and cellular energy deprivation. Finally, PGC1α suppression leads to reduced fatty acid flux through β-oxidation with a resultant fatty acid build up that can also be damaging to cellular health. Abbreviations: PGC1α - Peroxisome proliferator activated receptor gamma coactivator 1 alpha, ROS - reactive oxygen species, ETC - electron transport chain, ATP - adenosine triphosphate, NAD - nicotinamide adenine dinucleotide.
Evidence from IRI and sepsis models showed that mitochondrial health impairment led energy production to shift from oxidative phosphorylation to glycolysis in the tubular epithelial cells that worsened injury, though the triggering mechanisms of that shift have not been elucidated.93, 94 The cells that shifted back to oxidative respiration ultimately recovered, while those that could not restore oxidative phosphorylation atrophied and contributed to fibrosis development.94 Inhibiting the glycolytic shift with 2-deoxyglucose (2DG) in cells led to improved mitochondrial function,93 while inhibiting the last step of glycolysis via renal tubular specific knockout of PKM2 led to resistance against IRI.95 Other interventions that protect the mitochondrial membrane potential, preserve ATP synthase and ATP production, or supplement NAD+ levels have all been shown to mitigate renal injury.10, 96, 97
PGC1α, along with its downstream target genes, are widely reported to be downregulated in AKI.10, 78, 98–100 In a septic mouse model, both the expression of PGC1α itself and the expression of downstream ETC components and β-oxidation enzymes were suppressed in direct proportion to renal injury severity. Moreover, transcript levels of PGC1α and its targets rebounded with injury resolution.78 PGC1α knockout mice were more susceptible to AKI from sepsis, folic acid, and cisplatin,11, 78, 99 while mice with renal tubule specific PGC1α overexpression were more resilient against IRI and cisplatin induced AKI.10, 11 Modulating upstream regulators of PGC1α has shown similar effects: AICAR and resveratrol—respectively, small molecule activators of AMPK and sirtuins—reduced severity of cisplatin AKI and renal ischemic injury44, 101–103 Interestingly, PGC1α overexpression can become detrimental in AKI when its downstream mediators are inhibited, potentially through oxidative free radicals and other toxic effects of an augmented population of injured mitochondria. For example, inhibiting lysosomes with chloroquine and thereby inhibiting PGC1α’s positive effect on mitophagy induction not only eliminated the protective effect of PGC1α, but made PGC1α overexpression toxic with worsened cisplatin-induced nephrotoxicity and increased oxidative stress.11
There is still much to learn regarding the mechanisms that link AKI and PGC1α suppression. It is well understood that sirtuins can be downregulated in AKI;67 and also that biosynthesis of NAD+, a necessary cofactor for sirtuin function, is decreased in AKI.10, 82 There is also evidence that inflammatory pathways linked to TLR4, IL-6, NKκβ, and ERK1/2—all of which inhibit PGC1α—are upregulated in AKI; blocking these inflammatory pathways either genetically or with pharmacologic targeting can protect PGC1α levels and alleviate kidney injury in animal models.16, 98, 104 Further elucidating the complex interplay of PGC1α regulators and effectors will be paramount for developing targeted AKI therapies.
PGC1α and Glomerular Disease
While in situ hybridization imaging does not show strong localization of PGC1α to the glomerulus,78 multiple studies have delineated an important role for this transcriptional coactivator in maintaining glomerular health. That role has been studied best through the lens of DKD.105 (Figure 4) Metabolomic sampling of human kidneys showed a panel of differentially abundant metabolites that implicated suppression of mitochondrial activity in diabetic kidneys. Likewise, renal cortex from patients with DKD showed reduced expression of PGC1α.106 This is at least in part due to downregulation of key PGC1α regulators including sirtuins72, 107 and FoxO1.108 In fact, there is evidence that the podocyte’s ability to regulate mitochondrial biogenesis is impaired in diabetes. RNA sequencing of glomeruli revealed that a long noncoding RNA (lncRNA) called taurine-upregulated gene 1 (Tug1) is decreased in diabetic glomeruli. Tug1 was then shown to bind upstream of the PPARGC1A locus to enhance PGC1α expression. Furthermore, Tug1 interacted with PGC1α to further increase PGC1α expression. Podocyte specific overexpression of Tug1 in diabetic mice led to increased PGC1α expression, improved hyperglycemia-associated histology, and decreased albuminuria.109 An additional study showed that diabetic aberrations in the glycolytic pathway affected PGC1α in clinically significant ways. Proteomic analysis of dissected glomeruli from diabetic humans with and without kidney disease showed that diabetic patients without kidney disease had increased activity of metabolic pathways. Specifically, PKM2, the final enzyme of the glycolytic pathway, was upregulated. Hyperglycemia was shown to decrease PKM2 while podocyte specific PKM2 knock out mice developed worse nephropathy. Intriguingly, a small-molecule PKM2 activator led to increased PGC1α expression and reversal of hyperglycemia-induced metabolic abnormalities and mitochondrial dysfunction.19
Figure 4:

The effects of hyperglycemia on glomerular health. Hyperglycemia inhibits PGC1α by suppressing PGC1α activators PKM2, AMPK, SIRT1, and Tug1 while upregulating PGC1α inhibitors, TLR4 and NFκβ. With decreased PGC1α expression, the glomerulus experiences decreased expression of key podocyte genes, which leads to albuminuria. There is also lipid accumulation and reduction in antioxidant enzymes stimulated by PGC1α with resultant accumulation of oxidative stress, which leads to podocyte apoptosis and mesangial expansion. Abbreviations: PGC1α - Peroxisome proliferator activated receptor gamma coactivator 1 alpha, PKM2 - pyruvate kinase M2, AMPK - 5’ AMP-activated protein kinase, SIRT - sirtuin, Tug1 - Taurine Up-Regulated 1, TLR4 - toll-like receptor 4, NFκβ - nuclear factor kappa-light-chain-enhancer of activated B cells.
Downregulation of PGC1α in hyperglycemia led to impaired mitochondrial dynamics with increased mitochondrial fragmentation and impaired cellular respiration.110 Increasing PGC1α through overexpression or pharmacologic stimulation of SIRT1 reduced albuminuria in diabetic mice and protected podocytes from glucose-mediated reduction of respiratory complex activity, alterations in mitochondrial membrane potential, and impaired autophagy.72, 107, 111, 112 Overexpression of PGC1α targets using the PPARγ agonist, rosiglitazone, protected diabetic mouse kidneys and cultured podocytes against oxidative damage and glomerulosclerosis. Analogously, stimulation with metformin or AICAR, (both of which activate AMPK) or resveratrol reduced renal expression of pro-fibrotic TGFβ1 and alpha smooth muscle actin (αSMA).91, 92, 111, 112 There is also evidence from diabetic rats and cultured podocytes that PGC1α expression promoted expression of key podocyte genes, nephrin and podocalyxin, that maintain filtration barrier integrity.107, 113 In a type 2 diabetes mouse model, hyperglycemia also led to accumulation of lipids in diabetic kidneys114 with resultant lipotoxicity and oxidative stress which has long been associated with DKD progression.115 Diabetic mice receiving fenofibrate, a PPARα agonist, exhibited increased AMPK, PGC1α, and PGC1α target gene expression; these changes were associated with reduced albuminuria, renal fatty acid accumulation, mesangial expansion, and inflammatory infiltrate.114
Outside of DKD, the potentially protective role of PGC1α in nephrotic syndrome is an exciting new opportunity that is only beginning to be studied. In rats, pioglitazone treatment in an FSGS model protected against glomerulosclerosis.116 And, as reported in hyperglycemia studies, activating the PGC1α pathway decreased proteinuria with increased expression of nephrin and synaptopodin in an acute nephrotic syndrome mouse model.117 The most promising data regarding PGC1α target genes ameliorating nephrosis come from trials of PPARγ agonists in patients with nephrotic syndrome. In a phase I trial of rosiglitazone, five out of eleven patients with primary focal segmental glomerulosclerosis (FSGS) had delayed deterioration of renal function during a 16 month follow up period compared to pre-treatment trends.118 More recently, there are emerging case series of children with steroid resistant nephrotic syndrome whose disease has responded to pioglitazone therapy.119 With the DKD and nephrotic syndrome data in mind, a mouse model of podocyte specific PGC1α overexpression was generated. Surprisingly, PGC1α overexpression led to albuminuria, azotemia, and histology consistent with collapsing FSGS that was gene-dose responsive. PGC1α overexpression also switched the podocyte’s energy preference from glucose to fatty acid oxidation.120 It is not clear why indirect PGC1α stimulation through activation of upstream regulators or agonism of the PGC1α downstream target, PPARγ, is protective of podocyte health while direct transgenic overexpression is detrimental. There is still much to be learned, but there are hopeful signals that at least certain targets in the PGC1α pathway hold promise for glomerular targeted therapies.
PGC1α and Renal Fibrosis
PGC1α and its activators have also been implicated in the progression of fibrotic kidney disease. Transcriptomic analysis of human and mouse kidney tissue with and without fibrosis revealed that fibrotic tissue had lower expression of PGC1α and β-oxidation enzymes and higher lipid content than controls.15 Further investigation showed that the pro-fibrotic cytokine, TGFβ1, suppressed PGC1α.15 Another study examining renal biopsy samples from diabetic patients with and without DKD showed that TLR4 and NFκβ were both highly expressed in diseased kidneys and were associated with PGC1α suppression. Inhibiting TLR4 or NFκβ restored PGC1α and improved the fibrotic phenotype in db/db diabetic mice.121 Yet another study identified increased phosphodiesterase 4 (PDE4) expression in fibrotic renal tissue from a mouse ureteral obstruction model as a driver of PGC1α suppression via cAMP signaling and showed that inhibition of PDE4 with siRNA or selective PDE4 inhibitor, rolipram, attenuated fibrosis.122 Finally, sustained signaling of developmental pathways such as Notch, has been shown to promote fibrosis by suppressing PGC1α and other genes involved in fatty acid oxidation. Overexpression of Notch signaling led to severe fibrosis in mouse models whereas overexpression of PGC1α mitigated Notch-induced fibrosis.123
Independent of external suppression, PGC1α deficiency in knockout mice led to spontaneous tubulointerstitial inflammation with increased expression of inflammatory cytokines and receptors such as IL-6 and TNFα, which are independently linked to fibrosis development.99 Likewise, genetic overexpression of PGC1α, pharmacologic activation of AMPK, and pharmacologic agonism of PGC1α targets, PPARα or PPARγ, protected against fibrosis progression.15, 91, 124, 125
There is little known about the mechanisms utilized by PGC1α to attenuate fibrosis progression. In cultured cells, PGC1α overexpression repressed TGFβ1/Smad signaling, a major pathway associated with fibrosis progression.126 Notably, TGFβ1 itself has been reported to suppress PGC1α, likely creating a reinforcing feedback loop for normal health that switches in response to profibrotic stimuli.127 Although debated, the epithelial-mesenchymal transition (EMT) may play a role in the pathogenesis of renal fibrosis. EMT has been associated with broad-ranging mitochondrial dysfunction including increased cellular oxidative stress, loss of mitochondrial membrane potential, decreased mitochondrial DNA, and decreased expression of ETC complexes—all functions that are regulated by PGC1α.128 Inhibiting mitochondrial replication induced EMT while PGC1α overexpression prevented EMT in cells treated with aldosterone.128
There is optimism that PGC1α related pathways may be clinically useful targets to prevent renal fibrosis. However, fibrotic diseases are less likely to be resolved with acute therapies, and there is currently a severe paucity of data studying the long-term sequelae of PGC1α-associated alterations in mitochondrial metabolism. That will need to be addressed before chronic therapies targeting fibrosis are developed for clinical use.
PGC1α and NAD+ Metabolism
PGC1α regulates NAD+ biosynthesis. As a redox cofactor, NAD+ is involved in most major energy metabolism pathways including glycolysis, the citric acid cycle, β-oxidation of fatty acids, and the ETC. NAD+ also plays a critical role as substrate for enzymes that execute post-translational modifications to proteins. NAD+ can be biosynthesized through three pathways. Most NAD+ is created through the salvage pathway, which recycles NAD+ via niacinamide (NAM), but NAD+ is also synthesized from niacin through the Preiss-Handler pathway or from tryptophan through the de novo biosynthesis pathway. Metabolomic analysis examined which metabolites were differentially abundant in ischemic kidneys and in PGC1α knockout kidneys. Both ischemic kidneys and PGC1α knockout kidneys were deficient in NAM and NAD+. Conversely, mice with tubular specific PGC1α overexpression had increased NAM. Furthermore, supplementing NAM in PGC1α knockout mice was sufficient to restore normal NAD+ levels, prevent ischemic AKI, and normalize AKI-associated renal fat accumulation.10 RNA sequencing comparing PGC1α overexpressing kidneys to injured post-ischemic kidneys and PGC1α knockout kidneys revealed that PGC1α expression was associated with increased transcript levels of nearly all enzymes in the de novo NAD+ biosynthetic pathway while those enzymes were suppressed in injured kidneys and in PGC1α knockout kidneys.10 Urine metabolomic sampling of human AKI samples subsequently showed evidence of de novo NAD+ biosynthetic impairment with specific suppression of quinolinate phosphoribosyltransferase (QPRT), a bottleneck enzyme of the pathway.82 Mouse QPRT knockouts were NAD+ deficient and suffered worse renal injury after IRI. Supplementing NAD+ levels with NAM through the salvage pathway, and thus bypassing the suppressed pathway, mitigated AKI in both QPRT deficient mice and in a small pilot randomized placebo-controlled trial of humans undergoing cardiac surgery.82
In addition to biosynthetic impairment, NAD+ is likely consumed at higher rates during AKI, contributing to NAD+ depletion. Poly ADP-ribose polymerases (PARPs) respond to stress as a DNA repair mechanism and cleave NAD+. PARPs were upregulated in a rabbit septic AKI model and were associated with decreased ATP and NAD+. Inhibiting PARPs improved NAD+ and ATP levels and mitigated AKI.129 Likewise, sirtuins consume NAD+, a relationship that may explain many of the similar findings between NAD+ augmentation and sirtuin activation promoting longevity in experimental systems. Sirtuins activate PGC1α, which in turn stimulates NAD+ biosynthesis, thus creating a delicate balance of consumption and production that tightly regulates NAD+ levels (Figure 5).
Figure 5:
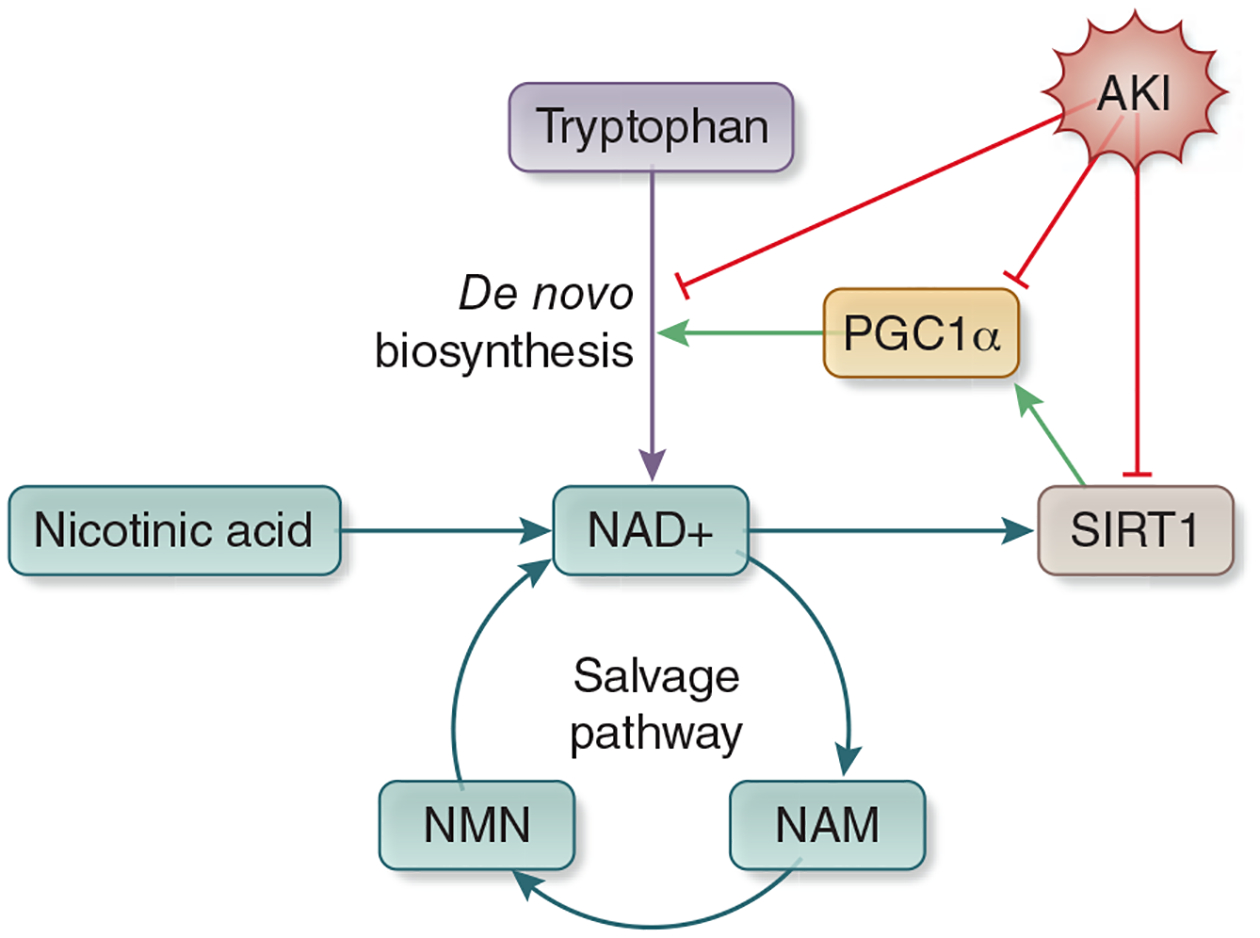
PGC1α regulation of NAD Biosynthesis. NAD+ is metabolized through three pathways. The salvage pathway recycles NAD+ after it is converted to NAM. The Priess-Handler Pathway creates NAD+ from nicotinic acid, and the de novo biosynthesis pathway metabolizes NAD+ from tryptophan. PGC1α expression upregulates the enzymes that comprise the de novo biosynthesis pathway. SIRT1 cleaves NAD to NAM to order to perform its actions, which include stimulation of PGC1α, creating a positive feedback loop. AKI reduces expression of PGC1α, SIRT1, and enzymes of the de novo NAD+ biosynthesis pathway with a resultant reduction in total NAD+. Abbreviations: : PGC1α - Peroxisome proliferator activated receptor gamma coactivator 1 alpha, NAD - nicotinamide adenine dinucleotide, NAM - niacinamide, SIRT - sirtuin, AKI - acute kidney injury, NMN - nicotinamide mononucleotide.
While it is clear that NAD+ levels are important for cellular health and resilience against renal injury and that PGC1α plays a critical role in that regulation, there is still much to be learned about NAD+ metabolism and regulation. The evolutionary redundancy in the NAD+ biosynthetic pathways hint toward a critical need for this cofactor to be robustly produced, but also to exhibit precisely tunable production. It is interesting that the de novo NAD+ biosynthetic pathway only accounts for roughly 10% of total NAD+ production,130 yet in AKI, that pathway is specifically downregulated to a degree that becomes clinically relevant. Likewise, mutations in this otherwise minor pathway have been linked to renal developmental anomalies,131–133 while mutations in nicotinamide mononucleotide adenylyltransferase 1 (NMNAT1) of the salvage pathway are not associated with renal anomalies despite that pathway’s more significant contribution to NAD+ stores and other clinical findings in those patients consistent with NAD+ deficiency.134 Untangling the individual impacts of the different NAD+ synthesis pathways, despite their redundancy, and elucidating the etiology of the de novo pathway’s critical influence on the kidney, will be essential to understanding the full reach of PGC1α.
Pharmacologic Targeting of the AMPK/ Sirtuin/PGC1α Pathway
As discussed above, there are many pharmacologic agents that impact upstream or downstream effectors of PGC1α. AICAR and resveratrol are two of the more widely studied agents that stimulate PGC1α activators. AICAR, an AMPK agonist increases PGC1α expression and has been shown to reduce severity of cisplatin mediated AKI,44 IRI,103 fibrotic myofibroblast activation,124 and hyperglycemia-associated autophagy dysfunction.92 Resveratrol is a notable natural product that activates sirtuins. In models of kidney disease, resveratrol increased AMPK and PGC1α expression and mitgated diabetic nephropathy.107, 111, 112 Similarly, resveratrol decreased profibrotic signalling47 and renal scarring in a mouse folic acid model.64 In an AKI model, resveratrol reduced severity of toxic injury or IRI101, 102, 135 and restored mitochondrial respiratory capacity after hemorrhagic shock.136
NAD+ supplementation has also been demonstrated as a method to achieve PGC1α-like renoprotection by augmenting downstream effects. NAD+ levels decrease in renal injury due to imparied biosynthesis and increased NAD+ consumption. Treatment with NAM not only restored NAD+ levels in mice after ischemic injury, but also alleviated the renal fat accumulation and renal insufficiency associated with ischemic and cisplatin injury.10 In a small placebo-controlled randomized clinical trial, oral NAM enhanced NAD+ biosynthesis and reduced AKI after cardiac surgery.82 Another NAD+ precursor, nicotinamide mononucleotide (NMN), protected mouse kidneys from age-associated AKI susceptibility by restoring SIRT1 activity,60 while NAM protected mice from ureteral obstruction-related fibrosis and reduced the expression of fibrotic proteins in TGFβ-stimulated cells.137
Many other agents have been shown to impact PGC1α activity through less clear mechanisms. Salidroside, the active component of the Rhodiola rosea plant, and glycyrrhic acid, from liquorice root both led to increased SIRT1 and PGC1α and protected mice from diabetic nephropathy.138, 139 Another study showed that melatonin could also stimulate AMPK activity and PGC1α expression to protect mice from diabetic kidney injury.140 Agonists of the 5HT1F serotonin receptor were shown to increase mitochondrial proteins, increase PGC1α, and accelerate renal recovery after IRI.141 Green tea extracts also increased PGC1α and protected mice against cyclosporine-induced renal injury.142
However, despite the many available and promising options for modulating these energy pathways to protect against acute and chronic renal disease, no treatment has advanced to clinical practice. Many of the above approaches lack the necessary specificity for modualting PGC1α. Moreover, only limited data are available about the long-term effects of activating PGC1α. The kidney offers a prime example of this challenge. Renal tubular cells respond well to PGC1α overexpression with acute protection from injury, while podocytes within the same organ apparently experience a detrimental hyperproliferation in response to PGC1α overexpression. Yet, those same podocytes that do not respond well to PGC1α overexpression do seem to respond well to upstream PCG1α activators or agonism of PPARγ. The challenges with targeting PGC1α in the kidney also extend to considerations of other organs. For example, cardiac PGC1α overexpression reduces pathological remodeling of aging hearts in some studies,143 while it led to dilated cardiomyopathy144 and reduced tolerance to cardiac ischemia145 in others. The long-term cardiac effects of a systemically administered PGC1α activator targeting renal disease may thus impede clinical development. These examples emphasize the importance of continuing to identify mechanisms underlying the AMPK-Sirtuin-PGC1α axis.
Conclusions
Through bountiful studies, a clear link has been established between mitochondrial health and renal resilience against AKI, glomerular disease, and fibrosis. Key regulators mitochondrial pool health, namely AMPK, sirtuins, and PGC1α, are emerging as promising theraputic targets. The hurdle that must now be overcome is translating these insights safely from the bench to the bedside. To do that, the intricate web connecting all of these regulators must continue to be unwound. Perhaps rather than targeting master regulators such as AMPK and PGC1α, safer therapies may emerge from increased understanding of downstream effectors. There is also a great need to understand the organ and cell specific roles of these effectors as individual tissues respond differently to metabolic-based therapies based on their innate requirements for fuel and energy production. Overall, the goal of prescribing metabolic rehabilitation as a form of renal therapy revolving around PGC1α and its regulators holds enormous promise.
Energy Pathways and Emerging Renal Drugs: SGLT2 inhibitors and HIF stabilizers
While not yet definitely linked to the AMPK-Sirtuin-PGC1α axis, two emerging drug classes are likely to impact renal metabolism in beneficial ways. Sodium glucose co-transporter 2 (SGLT2) inhibitors are now widely used to treat type 2 diabetes by inhibiting renal glucose reabsorption. SGLT2 inhibitors have been shown to reduce AKI, progression to end-stage renal disease, and death.146 In addition to improved glycemic control, kidney protection may be afforded by metabolic alterations in renal tubular cells. A recent study showed that diabetes led to hyperactivation of mTOR complex 1, which caused ATP production to shift from lipolysis to ketolysis in proximal tubules. SGLT2 inhibitors increased ketone body levels, which ameliorated diabetes-associated reduction in renal ATP and nephropathy in mice.147 Another study used imaging mass spectrometry to show that Krebs cycle metabolites abnormally accumulated in the renal cortex from diabetic mice, reflecting increased utilization of this pathway to metabolize excess glucose. SGLT2 inhibition converted the metabolite profile back to wild type ratios in which the largest fractions of metabolites were from glycolysis.148 Finally, data from renal tubule cells showed that both insulin and glucose reabsorption suppressed gluconeogenesis by reducing SIRT1-mediated PGC1α activation, and SGLT2 inhibition abrogated this effect.149
Less widely used currently but emerging as a powerful drug class to treat CKD-related anemia, hypoxia-inducible factor (HIF) prolyl hydroxylase inhibitors, called HIF stabilizers, also exert metabolic effects in the kidney. In renal tubule cell culture and kidneys of streptozotocin-treated rats, HIF stabilizers reduced Krebs cycle flux and increased basal glycolysis.150 Functionally, HIF stabilizers have been shown to reduce albuminuria in diabetic rodent models and to reduce accumulation of lipid peroxidation products.150, 151
Targeting energy pathways of renal tubule cells may prove beneficial for treating an array of kidney diseases. Future studies for SGLT2 inhibitors and HIF stabilizers may identify novel connections to the AMPK-SIRT-PGCα axis. Regardless, the emergence of these two drug classes suggests promise for applying insights from energy metabolism to renal pharmacology.
Financial Support:
AJC is supported by NIH grant K12-HD000850. SMP’s laboratory is supported by grants from the National Institutes of Health: R35-HL139424; R01-DK095072; R01-AG027002; and R01-HL125275
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Statement: SMP is listed as an inventor on patent filings from Beth Israel Deaconess Medical Center. SMP holds equity in Raksana Therapeutics. SMP has received consulting fees from Astellas, Cytokinetics, Mission Therapeutics, and Aerpio where he serves on its Scientific Advisory Board.
References
- 1.Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. March 20 1998;92(6):829–39. doi: 10.1016/s0092-8674(00)81410-5 [DOI] [PubMed] [Google Scholar]
- 2.Wu Z, Puigserver P, Andersson U, et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. July 9 1999;98(1):115–24. doi: 10.1016/s0092-8674(00)80611-x [DOI] [PubMed] [Google Scholar]
- 3.Terada S, Goto M, Kato M, Kawanaka K, Shimokawa T, Tabata I. Effects of low-intensity prolonged exercise on PGC-1 mRNA expression in rat epitrochlearis muscle. Biochemical and Biophysical Research Communications. 2002/08/16/ 2002;296(2):350–354. doi: 10.1016/S0006-291X(02)00881-1 [DOI] [PubMed] [Google Scholar]
- 4.Anderson RM, Barger JL, Edwards MG, et al. Dynamic regulation of PGC-1alpha localization and turnover implicates mitochondrial adaptation in calorie restriction and the stress response. Aging Cell. 2008;7(1):101–111. doi: 10.1111/j.1474-9726.2007.00357.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhu L, Wang Q, Zhang L, et al. Hypoxia induces PGC-1α expression and mitochondrial biogenesis in the myocardium of TOF patients. Cell research. June 2010;20(6):676–87. doi: 10.1038/cr.2010.46 [DOI] [PubMed] [Google Scholar]
- 6.Larsson NG, Wang J, Wilhelmsson H, et al. Mitochondrial transcription factor A is necessary for mtDNA maintenance and embryogenesis in mice. Nature genetics. March 1998;18(3):231–6. doi: 10.1038/ng0398-231 [DOI] [PubMed] [Google Scholar]
- 7.Scarpulla RC. Nuclear control of respiratory chain expression by nuclear respiratory factors and PGC-1-related coactivator. Annals of the New York Academy of Sciences. December 2008;1147:321–34. doi: 10.1196/annals.1427.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tao R, Coleman MC, Pennington JD, et al. Sirt3-mediated deacetylation of evolutionarily conserved lysine 122 regulates MnSOD activity in response to stress. Molecular cell. December 22 2010;40(6):893–904. doi: 10.1016/j.molcel.2010.12.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Soriano FX, Liesa M, Bach D, Chan DC, Palacín M, Zorzano A. Evidence for a Mitochondrial Regulatory Pathway Defined by Peroxisome Proliferator-Activated Receptor-γ Coactivator-1α, Estrogen-Related Receptor-α, and Mitofusin 2. Diabetes. 2006;55(6):1783. doi: 10.2337/db05-0509 [DOI] [PubMed] [Google Scholar]
- 10.Tran MT, Zsengeller ZK, Berg AH, et al. PGC1α drives NAD biosynthesis linking oxidative metabolism to renal protection. Nature. 2016;531(7595):528–532. doi: 10.1038/nature17184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lynch MR, Tran MT, Ralto KM, et al. TFEB-driven lysosomal biogenesis is pivotal for PGC1α-dependent renal stress resistance. JCI Insight. April/18/2019;4(8)doi: 10.1172/jci.insight.126749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vega RB, Huss JM, Kelly DP. The coactivator PGC-1 cooperates with peroxisome proliferator-activated receptor alpha in transcriptional control of nuclear genes encoding mitochondrial fatty acid oxidation enzymes. Molecular and cellular biology. March 2000;20(5):1868–76. doi: 10.1128/mcb.20.5.1868-1876.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huss JM, Levy FH, Kelly DP. Hypoxia inhibits the peroxisome proliferator-activated receptor alpha/retinoid X receptor gene regulatory pathway in cardiac myocytes: a mechanism for O2-dependent modulation of mitochondrial fatty acid oxidation. The Journal of biological chemistry. July 20 2001;276(29):27605–12. doi: 10.1074/jbc.M100277200 [DOI] [PubMed] [Google Scholar]
- 14.Palomer X, Alvarez-Guardia D, Rodríguez-Calvo R, et al. TNF-alpha reduces PGC-1alpha expression through NF-kappaB and p38 MAPK leading to increased glucose oxidation in a human cardiac cell model. Cardiovascular research. March 1 2009;81(4):703–12. doi: 10.1093/cvr/cvn327 [DOI] [PubMed] [Google Scholar]
- 15.Kang HM, Ahn SH, Choi P, et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat Med. 2015/01/01 2015;21(1):37–46. doi: 10.1038/nm.3762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smith JA, Stallons LJ, Collier JB, Chavin KD, Schnellmann RG. Suppression of mitochondrial biogenesis through toll-like receptor 4-dependent mitogen-activated protein kinase kinase/extracellular signal-regulated kinase signaling in endotoxin-induced acute kidney injury. The Journal of pharmacology and experimental therapeutics. February 2015;352(2):346–57. doi: 10.1124/jpet.114.221085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Casemayou A, Fournel A, Bagattin A, et al. Hepatocyte Nuclear Factor-1β Controls Mitochondrial Respiration in Renal Tubular Cells. J Am Soc Nephrol. November 2017;28(11):3205–3217. doi: 10.1681/asn.2016050508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang J, Wang X, Zhu Y, et al. Exercise activates lysosomal function in the brain through AMPK-SIRT1-TFEB pathway. CNS Neuroscience & Therapeutics. 2019;25(6):796–807. doi: 10.1111/cns.13114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Qi W, Keenan HA, Li Q, et al. Pyruvate kinase M2 activation may protect against the progression of diabetic glomerular pathology and mitochondrial dysfunction. Nat Med. June 2017;23(6):753–762. doi: 10.1038/nm.4328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miller KN, Clark JP, Anderson RM. Mitochondrial regulator PGC-1a-Modulating the modulator. Current opinion in endocrine and metabolic research. March 2019;5:37–44. doi: 10.1016/j.coemr.2019.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carling D, Zammit VA, Hardie DG. A common bicyclic protein kinase cascade inactivates the regulatory enzymes of fatty acid and cholesterol biosynthesis. FEBS letters. November 2 1987;223(2):217–22. doi: 10.1016/0014-5793(87)80292-2 [DOI] [PubMed] [Google Scholar]
- 22.Munday MR, Campbell DG, Carling D, Hardie DG. Identification by amino acid sequencing of three major regulatory phosphorylation sites on rat acetyl-CoA carboxylase. European journal of biochemistry. August 1 1988;175(2):331–8. doi: 10.1111/j.14321033.1988.tb14201.x [DOI] [PubMed] [Google Scholar]
- 23.Bultot L, Guigas B, Von Wilamowitz-Moellendorff A, et al. AMP-activated protein kinase phosphorylates and inactivates liver glycogen synthase. The Biochemical journal. April 1 2012;443(1):193–203. doi: 10.1042/bj20112026 [DOI] [PubMed] [Google Scholar]
- 24.Koo SH, Flechner L, Qi L, et al. The CREB coactivator TORC2 is a key regulator of fasting glucose metabolism. Nature. October 20 2005;437(7062):1109–11. doi: 10.1038/nature03967 [DOI] [PubMed] [Google Scholar]
- 25.Gwinn DM, Shackelford DB, Egan DF, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Molecular cell. April 25 2008;30(2):214–26. doi: 10.1016/j.molcel.2008.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leprivier G, Remke M, Rotblat B, et al. The eEF2 kinase confers resistance to nutrient deprivation by blocking translation elongation. Cell. May 23 2013;153(5):1064–79. doi: 10.1016/j.cell.2013.04.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nature cell biology. February 2011;13(2):132–41. doi: 10.1038/ncb2152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chavez JA, Roach WG, Keller SR, Lane WS, Lienhard GE. Inhibition of GLUT4 translocation by Tbc1d1, a Rab GTPase-activating protein abundant in skeletal muscle, is partially relieved by AMP-activated protein kinase activation. The Journal of biological chemistry. April 4 2008;283(14):9187–95. doi: 10.1074/jbc.M708934200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Doménech E, Maestre C, Esteban-Martínez L, et al. AMPK and PFKFB3 mediate glycolysis and survival in response to mitophagy during mitotic arrest. Nature cell biology. October 2015;17(10):1304–16. doi: 10.1038/ncb3231 [DOI] [PubMed] [Google Scholar]
- 30.Ahmadian M, Abbott MJ, Tang T, et al. Desnutrin/ATGL is regulated by AMPK and is required for a brown adipose phenotype. Cell metabolism. June 8 2011;13(6):739–48. doi: 10.1016/j.cmet.2011.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cho YS, Lee JI, Shin D, et al. Molecular mechanism for the regulation of human ACC2 through phosphorylation by AMPK. Biochemical and Biophysical Research Communications. 2010/01/01/ 2010;391(1):187–192. doi: 10.1016/j.bbrc.2009.11.029 [DOI] [PubMed] [Google Scholar]
- 32.Jäger S, Handschin C, St-Pierre J, Spiegelman BM. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proceedings of the National Academy of Sciences of the United States of America. July 17 2007;104(29):12017–22. doi: 10.1073/pnas.0705070104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Settembre C, De Cegli R, Mansueto G, et al. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nature cell biology. June 2013;15(6):647–58. doi: 10.1038/ncb2718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cho B, Choi SY, Cho HM, Kim HJ, Sun W. Physiological and pathological significance of dynamin-related protein 1 (drp1)-dependent mitochondrial fission in the nervous system. Experimental neurobiology. September 2013;22(3):149–57. doi: 10.5607/en.2013.22.3.149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Toyama EQ, Herzig S, Courchet J, et al. Metabolism. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science (New York, NY). January 15 2016;351(6270):275–281. doi: 10.1126/science.aab4138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Egan DF, Shackelford DB, Mihaylova MM, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science (New York, NY). January 28 2011;331(6016):456–61. doi: 10.1126/science.1196371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Declèves A-E, Mathew AV, Cunard R, Sharma K. AMPK mediates the initiation of kidney disease induced by a high-fat diet. J Am Soc Nephrol. 2011;22(10):1846–1855. doi: 10.1681/ASN.2011010026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dong D, Cai GY, Ning YC, et al. Alleviation of senescence and epithelial-mesenchymal transition in aging kidney by short-term caloric restriction and caloric restriction mimetics via modulation of AMPK/mTOR signaling. Oncotarget. March 7 2017;8(10):16109–16121. doi: 10.18632/oncotarget.14884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cavaglieri RC, Day RT, Feliers D, Abboud HE. Metformin prevents renal interstitial fibrosis in mice with unilateral ureteral obstruction. Molecular and Cellular Endocrinology. 2015/09/05/ 2015;412:116–122. doi: 10.1016/j.mce.2015.06.006 [DOI] [PubMed] [Google Scholar]
- 40.Lee M, Katerelos M, Gleich K, et al. Phosphorylation of Acetyl-CoA Carboxylase by AMPK Reduces Renal Fibrosis and Is Essential for the Anti-Fibrotic Effect of Metformin. J Am Soc Nephrol. September 2018;29(9):2326–2336. doi: 10.1681/asn.2018010050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jin Y, Liu S, Ma Q, Xiao D, Chen L. Berberine enhances the AMPK activation and autophagy and mitigates high glucose-induced apoptosis of mouse podocytes. European journal of pharmacology. January 5 2017;794:106–114. doi: 10.1016/j.ejphar.2016.11.037 [DOI] [PubMed] [Google Scholar]
- 42.Lim JH, Kim HW, Kim MY, et al. Cinacalcet-mediated activation of the CaMKKβ-LKB1-AMPK pathway attenuates diabetic nephropathy in db/db mice by modulation of apoptosis and autophagy. Cell death & disease. February 15 2018;9(3):270. doi: 10.1038/s41419-018-0324-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li J, Gui Y, Ren J, et al. Metformin Protects Against Cisplatin-Induced Tubular Cell Apoptosis and Acute Kidney Injury via AMPKα-regulated Autophagy Induction. Scientific reports. April 7 2016;6:23975. doi: 10.1038/srep23975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tsogbadrakh B, Ryu H, Ju KD, et al. AICAR, an AMPK activator, protects against cisplatin-induced acute kidney injury through the JAK/STAT/SOCS pathway. Biochem Biophys Res Commun. February 12 2019;509(3):680–686. doi: 10.1016/j.bbrc.2018.12.159 [DOI] [PubMed] [Google Scholar]
- 45.Bao H, Zhang Q, Liu X, et al. Lithium targeting of AMPK protects against cisplatin-induced acute kidney injury by enhancing autophagy in renal proximal tubular epithelial cells. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. December 2019;33(12):14370–14381. doi: 10.1096/fj.201901712R [DOI] [PubMed] [Google Scholar]
- 46.Lieberthal W, Tang M, Lusco M, Abate M, Levine JS. Preconditioning mice with activators of AMPK ameliorates ischemic acute kidney injury in vivo. American journal of physiology Renal physiology. October 1 2016;311(4):F731–f739. doi: 10.1152/ajprenal.00541.2015 [DOI] [PubMed] [Google Scholar]
- 47.Zhu X, Liu Q, Wang M, et al. Activation of Sirt1 by resveratrol inhibits TNF-α induced inflammation in fibroblasts. PloS one. 2011;6(11):e27081. doi: 10.1371/journal.pone.0027081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Langley E, Pearson M, Faretta M, et al. Human SIR2 deacetylates p53 and antagonizes PML/p53-induced cellular senescence. The EMBO journal. May 15 2002;21(10):2383–96. doi: 10.1093/emboj/21.10.2383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brunet A, Sweeney LB, Sturgill JF, et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science (New York, NY). March 26 2004;303(5666):2011–5. doi: 10.1126/science.1094637 [DOI] [PubMed] [Google Scholar]
- 50.Dioum EM, Chen R, Alexander MS, et al. Regulation of Hypoxia-Inducible Factor 2α Signaling by the Stress-Responsive Deacetylase Sirtuin 1. Science (New York, NY). 2009;324(5932):1289. doi: 10.1126/science.1169956 [DOI] [PubMed] [Google Scholar]
- 51.Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1α and SIRT1. Nature. 2005/03/01 2005;434(7029):113–118. doi: 10.1038/nature03354 [DOI] [PubMed] [Google Scholar]
- 52.Ahn B-H, Kim H-S, Song S, et al. A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(38):14447–14452. doi: 10.1073/pnas.0803790105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rahman M, Nirala NK, Singh A, et al. Drosophila Sirt2/mammalian SIRT3 deacetylates ATP synthase β and regulates complex V activity. The Journal of cell biology. July 21 2014;206(2):289–305. doi: 10.1083/jcb.201404118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tretter L, Adam-Vizi V. Generation of Reactive Oxygen Species in the Reaction Catalyzed by α-Ketoglutarate Dehydrogenase. The Journal of Neuroscience. 2004;24(36):7771. doi: 10.1523/JNEUROSCI.1842-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rodrigues JV, Gomes CM. Mechanism of superoxide and hydrogen peroxide generation by human electron-transfer flavoprotein and pathological variants. Free Radical Biology and Medicine. 2012/07/01/ 2012;53(1):12–19. doi: 10.1016/j.freeradbiomed.2012.04.016 [DOI] [PubMed] [Google Scholar]
- 56.Hirschey MD, Shimazu T, Goetzman E, et al. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature. March 4 2010;464(7285):121–5. doi: 10.1038/nature08778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Morigi M, Perico L, Benigni A. Sirtuins in Renal Health and Disease. J Am Soc Nephrol. 2018;29(7):1799–1809. doi: 10.1681/ASN.2017111218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Braidy N, Guillemin GJ, Mansour H, Chan-Ling T, Poljak A, Grant R. Age related changes in NAD+ metabolism oxidative stress and Sirt1 activity in wistar rats. PloS one. 2011;6(4):e19194–e19194. doi: 10.1371/journal.pone.0019194 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 59.Kwon Y, Kim J, Lee CY, Kim H. Expression of SIRT1 and SIRT3 varies according to age in mice. Anatomy & cell biology. March 2015;48(1):54–61. doi: 10.5115/acb.2015.48.1.54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Guan Y, Wang SR, Huang XZ, et al. Nicotinamide Mononucleotide, an NAD(+) Precursor, Rescues Age-Associated Susceptibility to AKI in a Sirtuin 1-Dependent Manner. J Am Soc Nephrol. August 2017;28(8):2337–2352. doi: 10.1681/asn.2016040385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chuang PY, Cai W, Li X, et al. Reduction in podocyte SIRT1 accelerates kidney injury in aging mice. American journal of physiology Renal physiology. September 1 2017;313(3):F621–f628. doi: 10.1152/ajprenal.00255.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Benigni A, Corna D, Zoja C, et al. Disruption of the Ang II type 1 receptor promotes longevity in mice. The Journal of clinical investigation. March 2009;119(3):524–30. doi: 10.1172/jci36703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.He W, Wang Y, Zhang MZ, et al. Sirt1 activation protects the mouse renal medulla from oxidative injury. The Journal of clinical investigation. April 2010;120(4):1056–68. doi: 10.1172/jci41563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li J, Qu X, Ricardo SD, Bertram JF, Nikolic-Paterson DJ. Resveratrol inhibits renal fibrosis in the obstructed kidney: potential role in deacetylation of Smad3. The American journal of pathology. September 2010;177(3):1065–71. doi: 10.2353/ajpath.2010.090923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang Y, Connelly KA, Thai K, et al. Sirtuin 1 Activation Reduces Transforming Growth Factor-β1-Induced Fibrogenesis and Affords Organ Protection in a Model of Progressive, Experimental Kidney and Associated Cardiac Disease. The American journal of pathology. January 2017;187(1):80–90. doi: 10.1016/j.ajpath.2016.09.016 [DOI] [PubMed] [Google Scholar]
- 66.Li N, Zhang J, Yan X, et al. SIRT3-KLF15 signaling ameliorates kidney injury induced by hypertension. Oncotarget. June 13 2017;8(24):39592–39604. doi: 10.18632/oncotarget.17165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhao W-Y, Zhang L, Sui M-X, Zhu Y-H, Zeng L. Protective effects of sirtuin 3 in a murine model of sepsis-induced acute kidney injury. Scientific reports. 2016/09/13 2016;6(1):33201. doi: 10.1038/srep33201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fan H, Yang HC, You L, Wang YY, He WJ, Hao CM. The histone deacetylase, SIRT1, contributes to the resistance of young mice to ischemia/reperfusion-induced acute kidney injury. Kidney international. March 2013;83(3):404–13. doi: 10.1038/ki.2012.394 [DOI] [PubMed] [Google Scholar]
- 69.Zhao W, Zhang L, Chen R, et al. SIRT3 Protects Against Acute Kidney Injury via AMPK/mTOR-Regulated Autophagy. Front Physiol. 2018;9:1526–1526. doi: 10.3389/fphys.2018.01526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hasegawa K, Wakino S, Yoshioka K, et al. Kidney-specific overexpression of Sirt1 protects against acute kidney injury by retaining peroxisome function. The Journal of biological chemistry. April 23 2010;285(17):13045–56. doi: 10.1074/jbc.M109.067728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Morigi M, Perico L, Rota C, et al. Sirtuin 3-dependent mitochondrial dynamic improvements protect against acute kidney injury. The Journal of clinical investigation. February 2015;125(2):715–26. doi: 10.1172/jci77632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hong Q, Zhang L, Das B, et al. Increased podocyte Sirtuin-1 function attenuates diabetic kidney injury. Kidney international. June 2018;93(6):1330–1343. doi: 10.1016/j.kint.2017.12.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cai J, Liu Z, Huang X, et al. The deacetylase sirtuin 6 protects against kidney fibrosis by epigenetically blocking β-catenin target gene expression. Kidney international. January 2020;97(1):106–118. doi: 10.1016/j.kint.2019.08.028 [DOI] [PubMed] [Google Scholar]
- 74.Huang W, Liu H, Zhu S, et al. Sirt6 deficiency results in progression of glomerular injury in the kidney. Aging. March 28 2017;9(3):1069–1083. doi: 10.18632/aging.101214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Miyasato Y, Yoshizawa T, Sato Y, et al. Sirtuin 7 Deficiency Ameliorates Cisplatin-induced Acute Kidney Injury Through Regulation of the Inflammatory Response. Scientific reports. April 12 2018;8(1):5927. doi: 10.1038/s41598-018-24257-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chiba T, Peasley KD, Cargill KR, et al. Sirtuin 5 Regulates Proximal Tubule Fatty Acid Oxidation to Protect against AKI. J Am Soc Nephrol. December 2019;30(12):2384–2398. doi: 10.1681/asn.2019020163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Li W, Yang Y, Li Y, Zhao Y, Jiang H. Sirt5 Attenuates Cisplatin-Induced Acute Kidney Injury through Regulation of Nrf2/HO-1 and Bcl-2. BioMed research international. 2019;2019:4745132. doi: 10.1155/2019/4745132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tran M, Tam D, Bardia A, et al. PGC-1α promotes recovery after acute kidney injury during systemic inflammation in mice. The Journal of clinical investigation. October 2011;121(10):4003–14. doi: 10.1172/jci58662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Trump BF, Valigorsky JM, Jones RT, Mergner WJ, Garcia JH, Cowley RA. The application of electron microscopy and cellular biochemistry to the autopsy. Observations on cellular changes in human shock. Human pathology. July 1975;6(4):499–516. doi: 10.1016/s0046-8177(75)80068-2 [DOI] [PubMed] [Google Scholar]
- 80.Swärd K, Valsson F, Sellgren J, Ricksten SE. Differential effects of human atrial natriuretic peptide and furosemide on glomerular filtration rate and renal oxygen consumption in humans. Intensive care medicine. January 2005;31(1):79–85. doi: 10.1007/s00134-004-2490-3 [DOI] [PubMed] [Google Scholar]
- 81.Simon N, Hertig A. Alteration of Fatty Acid Oxidation in Tubular Epithelial Cells: From Acute Kidney Injury to Renal Fibrogenesis. Frontiers in medicine. 2015;2:52. doi: 10.3389/fmed.2015.00052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Poyan Mehr A, Tran MT, Ralto KM, et al. De novo NAD(+) biosynthetic impairment in acute kidney injury in humans. Nat Med. 2018;24(9):1351–1359. doi: 10.1038/s41591-018-0138z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhan M, Brooks C, Liu F, Sun L, Dong Z. Mitochondrial dynamics: regulatory mechanisms and emerging role in renal pathophysiology. Kidney international. 2013/04/01/ 2013;83(4):568–581. doi: 10.1038/ki.2012.441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Parekh DJ, Weinberg JM, Ercole B, et al. Tolerance of the Human Kidney to Isolated Controlled Ischemia. Journal of the American Society of Nephrology. 2013;24(3):506. doi: 10.1681/ASN.2012080786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mukhopadhyay P, Horváth B, Zsengellér Z, et al. Mitochondrial-targeted antioxidants represent a promising approach for prevention of cisplatin-induced nephropathy. Free Radic Biol Med. 2012;52(2):497–506. doi: 10.1016/j.freeradbiomed.2011.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hall AM, Rhodes GJ, Sandoval RM, Corridon PR, Molitoris BA. In vivo multiphoton imaging of mitochondrial structure and function during acute kidney injury. Kidney international. 2013/01/01/ 2013;83(1):72–83. doi: 10.1038/ki.2012.328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zager RA, Johnson AC, Hanson SY. Renal tubular triglyercide accumulation following endotoxic, toxic, and ischemic injury. Kidney international. January 2005;67(1):111–21. doi: 10.1111/j.1523-1755.2005.00061.x [DOI] [PubMed] [Google Scholar]
- 88.Ratliff BB, Abdulmahdi W, Pawar R, Wolin MS. Oxidant Mechanisms in Renal Injury and Disease. Antioxid Redox Signal. 2016;25(3):119–146. doi: 10.1089/ars.2016.6665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Qin N, Cai T, Ke Q, et al. UCP2-dependent improvement of mitochondrial dynamics protects against acute kidney injury. The Journal of Pathology. 2019/03/01 2019;247(3):392–405. doi: 10.1002/path.5198 [DOI] [PubMed] [Google Scholar]
- 90.Tang WX, Wu WH, Qiu HY, Bo H, Huang SM. Amelioration of rhabdomyolysis-induced renal mitochondrial injury and apoptosis through suppression of Drp-1 translocation. Journal of nephrology. Nov-Dec 2013;26(6):1073–82. doi: 10.5301/jn.5000268 [DOI] [PubMed] [Google Scholar]
- 91.Zhang L, Liu J, Zhou F, Wang W, Chen N. PGC-1α ameliorates kidney fibrosis in mice with diabetic kidney disease through an antioxidative mechanism. Molecular medicine reports. March 2018;17(3):4490–4498. doi: 10.3892/mmr.2018.8433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lee SY, Kang JM, Kim DJ, et al. PGC1α Activators Mitigate Diabetic Tubulopathy by Improving Mitochondrial Dynamics and Quality Control. Journal of diabetes research. 2017;2017:6483572. doi: 10.1155/2017/6483572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ji R, Chen W, Wang Y, et al. The Warburg Effect Promotes Mitochondrial Injury Regulated by Uncoupling Protein-2 in Septic Acute Kidney Injury. Shock (Augusta, Ga). June 2 2020;doi: 10.1097/shk.0000000000001576 [DOI] [PubMed] [Google Scholar]
- 94.Lan R, Geng H, Singha PK, et al. Mitochondrial Pathology and Glycolytic Shift during Proximal Tubule Atrophy after Ischemic AKI. J Am Soc Nephrol. November 2016;27(11):3356–3367. doi: 10.1681/asn.2015020177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zhou H-L, Zhang R, Anand P, et al. Metabolic reprogramming by the S-nitroso-CoA reductase system protects against kidney injury. Nature. 2019/01/01 2019;565(7737):96–100. doi: 10.1038/s41586-018-0749-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Szeto HH, Liu S, Soong Y, et al. Mitochondria-targeted peptide accelerates ATP recovery and reduces ischemic kidney injury. J Am Soc Nephrol. 2011;22(6):1041–1052. doi: 10.1681/ASN.2010080808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Suzuki T, Yamaguchi H, Kikusato M, et al. Mitochonic Acid 5 Binds Mitochondria and Ameliorates Renal Tubular and Cardiac Myocyte Damage. Journal of the American Society of Nephrology. 2016;27(7):1925. doi: 10.1681/ASN.2015060623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ruiz-Andres O, Suarez-Alvarez B, Sánchez-Ramos C, et al. The inflammatory cytokine TWEAK decreases PGC-1α expression and mitochondrial function in acute kidney injury. Kidney international. February 2016;89(2):399–410. doi: 10.1038/ki.2015.332 [DOI] [PubMed] [Google Scholar]
- 99.Fontecha-Barriuso M, Martín-Sánchez D, Martinez-Moreno JM, et al. PGC-1α deficiency causes spontaneous kidney inflammation and increases the severity of nephrotoxic AKI. J Pathol. September 2019;249(1):65–78. doi: 10.1002/path.5282 [DOI] [PubMed] [Google Scholar]
- 100.Portilla D, Dai G, McClure T, et al. Alterations of PPARα and its coactivator PGC-1 in cisplatin-induced acute renal failure. Kidney international. 2002;62(4):1208–1218. doi: 10.1111/j.1523-1755.2002.kid553.x [DOI] [PubMed] [Google Scholar]
- 101.Chander V, Chopra K. Protective effect of nitric oxide pathway in resveratrol renal ischemia-reperfusion injury in rats. Archives of medical research. January 2006;37(1):19–26. doi: 10.1016/j.arcmed.2005.05.018 [DOI] [PubMed] [Google Scholar]
- 102.Kim DH, Jung YJ, Lee JE, et al. SIRT1 activation by resveratrol ameliorates cisplatin-induced renal injury through deacetylation of p53. American journal of physiology Renal physiology. August 2011;301(2):F427–35. doi: 10.1152/ajprenal.00258.2010 [DOI] [PubMed] [Google Scholar]
- 103.Lempiäinen J, Finckenberg P, Levijoki J, Mervaala E. AMPK activator AICAR ameliorates ischaemia reperfusion injury in the rat kidney. Br J Pharmacol. 2012;166(6):1905–1915. doi: 10.1111/j.1476-5381.2012.01895.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zhao Y, Nie M, Xu P, et al. Nitrosporeusine A attenuates sepsis-associated acute kidney injury through the downregulation of IL-6/sIL-6R axis activation-mediated PGC-1α suppression. Biochem Biophys Res Commun. July 30 2019;515(3):474–480. doi: 10.1016/j.bbrc.2019.05.151 [DOI] [PubMed] [Google Scholar]
- 105.Galvan DL, Green NH, Danesh FR. The hallmarks of mitochondrial dysfunction in chronic kidney disease. Kidney international. November 2017;92(5):1051–1057. doi: 10.1016/j.kint.2017.05.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sharma K, Karl B, Mathew AV, et al. Metabolomics reveals signature of mitochondrial dysfunction in diabetic kidney disease. J Am Soc Nephrol. 2013;24(11):1901–1912. doi: 10.1681/ASN.2013020126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zhang T, Chi Y, Ren Y, Du C, Shi Y, Li Y. Resveratrol Reduces Oxidative Stress and Apoptosis in Podocytes via Sir2-Related Enzymes, Sirtuins1 (SIRT1)/Peroxisome Proliferator-Activated Receptor γ Co-Activator 1α (PGC-1α) Axis. Medical science monitor : international medical journal of experimental and clinical research. February 15 2019;25:1220–1231. doi: 10.12659/msm.911714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wu L, Wang Q, Guo F, et al. Activation of FoxO1/ PGC-1α prevents mitochondrial dysfunction and ameliorates mesangial cell injury in diabetic rats. Mol Cell Endocrinol. September 15 2015;413:1–12. doi: 10.1016/j.mce.2015.06.007 [DOI] [PubMed] [Google Scholar]
- 109.Long J, Badal SS, Ye Z, et al. Long noncoding RNA Tug1 regulates mitochondrial bioenergetics in diabetic nephropathy. The Journal of clinical investigation. 2016;126(11):4205–4218. doi: 10.1172/JCI87927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Guo K, Lu J, Huang Y, et al. Protective role of PGC-1α in diabetic nephropathy is associated with the inhibition of ROS through mitochondrial dynamic remodeling. PloS one. 2015;10(4):e0125176–e0125176. doi: 10.1371/journal.pone.0125176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kitada M, Kume S, Imaizumi N, Koya D. Resveratrol improves oxidative stress and protects against diabetic nephropathy through normalization of Mn-SOD dysfunction in AMPK/SIRT1-independent pathway. Diabetes. February 2011;60(2):634–43. doi: 10.2337/db10-0386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zhao YH, Fan YJ. Resveratrol improves lipid metabolism in diabetic nephropathy rats. Frontiers in bioscience (Landmark edition). June 1 2020;25:1913–1924. [DOI] [PubMed] [Google Scholar]
- 113.Bao L, Cai X, Dai X, et al. Grape seed proanthocyanidin extracts ameliorate podocyte injury by activating peroxisome proliferator-activated receptor-γ coactivator 1α in low-dose streptozotocin-and high-carbohydrate/high-fat diet-induced diabetic rats. Food & function. August 2014;5(8):1872–80. doi: 10.1039/c4fo00340c [DOI] [PubMed] [Google Scholar]
- 114.Hong YA, Lim JH, Kim MY, et al. Fenofibrate improves renal lipotoxicity through activation of AMPK-PGC-1α in db/db mice. PloS one. 2014;9(5):e96147–e96147. doi: 10.1371/journal.pone.0096147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Murea M, Freedman BI, Parks JS, Antinozzi PA, Elbein SC, Ma L. Lipotoxicity in Diabetic Nephropathy: The Potential Role of Fatty Acid Oxidation. Clinical Journal of the American Society of Nephrology. 2010;5(12):2373. doi: 10.2215/CJN.08160910 [DOI] [PubMed] [Google Scholar]
- 116.Yang HC, Ma LJ, Ma J, Fogo AB. Peroxisome proliferator-activated receptor-gamma agonist is protective in podocyte injury-associated sclerosis. Kidney international. May 2006;69(10):1756–64. doi: 10.1038/sj.ki.5000336 [DOI] [PubMed] [Google Scholar]
- 117.Zuo Y, Yang H-C, Potthoff SA, et al. Protective effects of PPARγ agonist in acute nephrotic syndrome. Nephrol Dial Transplant. 2012;27(1):174–181. doi: 10.1093/ndt/gfr240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Peyser A, MacHardy N, Tarapore F, et al. Follow-up of phase I trial of adalimumab and rosiglitazone in FSGS: III. Report of the FONT study group. BMC Nephrology. 2010/01/29 2010;11(1):2. doi: 10.1186/1471-2369-11-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Agrawal S, Chanley MA, Westbrook D, et al. Pioglitazone Enhances the Beneficial Effects of Glucocorticoids in Experimental Nephrotic Syndrome. Scientific reports. 2016/05/04 2016;6(1):24392. doi: 10.1038/srep24392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Li S-Y, Park J, Qiu C, et al. Increasing the level of peroxisome proliferator-activated receptor γ coactivator-1α in podocytes results in collapsing glomerulopathy. JCI Insight. 2017;2(14):e92930. doi: 10.1172/jci.insight.92930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Yuan S, Liu X, Zhu X, et al. The Role of TLR4 on PGC-1α-Mediated Oxidative Stress in Tubular Cell in Diabetic Kidney Disease. Oxidative medicine and cellular longevity. 2018;2018:6296802. doi: 10.1155/2018/6296802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ding H, Bai F, Cao H, et al. PDE/cAMP/Epac/C/EBP-β Signaling Cascade Regulates Mitochondria Biogenesis of Tubular Epithelial Cells in Renal Fibrosis. Antioxid Redox Signal. September 1 2018;29(7):637–652. doi: 10.1089/ars.2017.7041 [DOI] [PubMed] [Google Scholar]
- 123.Han SH, Wu MY, Nam BY, et al. PGC-1α Protects from Notch-Induced Kidney Fibrosis Development. J Am Soc Nephrol. November 2017;28(11):3312–3322. doi: 10.1681/asn.2017020130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Chen K-H, Hsu H-H, Lee C-C, et al. The AMPK agonist AICAR inhibits TGF-β1 induced activation of kidney myofibroblasts. PloS one. 2014;9(9):e106554–e106554. doi: 10.1371/journal.pone.0106554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Makled MN, El-Kashef DH. Saroglitazar attenuates renal fibrosis induced by unilateral ureteral obstruction via inhibiting TGF-β/Smad signaling pathway. Life sciences. July 15 2020;253:117729. doi: 10.1016/j.lfs.2020.117729 [DOI] [PubMed] [Google Scholar]
- 126.Choi H-I, Park JS, Kim D-H, et al. PGC-1α Suppresses the Activation of TGF-β/Smad Signaling via Targeting TGFβRI Downregulation by let-7b/c Upregulation. Int J Mol Sci. 2019;20(20):5084. doi: 10.3390/ijms20205084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Sohn EJ, Kim J, Hwang Y, Im S, Moon Y, Kang DM. TGF-β suppresses the expression of genes related to mitochondrial function in lung A549 cells. Cellular and molecular biology (Noisy-le-Grand, France). October 8 2012;Suppl.58:Ol1763–7. [PubMed] [Google Scholar]
- 128.Yuan Y, Chen Y, Zhang P, et al. Mitochondrial dysfunction accounts for aldosterone-induced epithelial-to-mesenchymal transition of renal proximal tubular epithelial cells. Free Radic Biol Med. July 1 2012;53(1):30–43. doi: 10.1016/j.freeradbiomed.2012.03.015 [DOI] [PubMed] [Google Scholar]
- 129.Wang YM, Han RL, Song SG, Yuan XP, Ren XS. Inhibition of PARP overactivation protects acute kidney injury of septic shock. European review for medical and pharmacological sciences. September 2018;22(18):6049–6056. doi: 10.26355/eurrev_201809_15942 [DOI] [PubMed] [Google Scholar]
- 130.Fukuwatari T, Shibata K. Nutritional aspect of tryptophan metabolism. International journal of tryptophan research : IJTR. 2013;6(Suppl 1):3–8. doi: 10.4137/ijtr.S11588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Shi H, Enriquez A, Rapadas M, et al. NAD Deficiency, Congenital Malformations, and Niacin Supplementation. New England Journal of Medicine. 2017/08/10 2017;377(6):544–552. doi: 10.1056/NEJMoa1616361 [DOI] [PubMed] [Google Scholar]
- 132.Cuny H, Rapadas M, Gereis J, et al. NAD deficiency due to environmental factors or gene-environment interactions causes congenital malformations and miscarriage in mice. Proceedings of the National Academy of Sciences. 2020;117(7):3738. doi: 10.1073/pnas.1916588117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Szot JO, Campagnolo C, Cao Y, et al. Bi-allelic Mutations in NADSYN1 Cause Multiple Organ Defects and Expand the Genotypic Spectrum of Congenital NAD Deficiency Disorders. Am J Hum Genet. 2020;106(1):129–136. doi: 10.1016/j.ajhg.2019.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Bedoni N, Quinodoz M, Pinelli M, et al. An Alu-mediated duplication in NMNAT1, involved in NAD biosynthesis, causes a novel syndrome, SHILCA, affecting multiple tissues and organs. Human molecular genetics. June 12 2020;doi: 10.1093/hmg/ddaa112 [DOI] [PubMed] [Google Scholar]
- 135.Hong YA, Bae SY, Ahn SY, et al. Resveratrol Ameliorates Contrast Induced Nephropathy Through the Activation of SIRT1-PGC-1α-Foxo1 Signaling in Mice. Kidney & blood pressure research. 2017;42(4):641–653. doi: 10.1159/000481804 [DOI] [PubMed] [Google Scholar]
- 136.Wang H, Guan Y, Karamercan MA, et al. Resveratrol Rescues Kidney Mitochondrial Function Following Hemorrhagic Shock. Shock (Augusta, Ga). August 2015;44(2):173–80. doi: 10.1097/shk.0000000000000390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Zheng M, Cai J, Liu Z, et al. Nicotinamide reduces renal interstitial fibrosis by suppressing tubular injury and inflammation. J Cell Mol Med. 2019;23(6):3995–4004. doi: 10.1111/jcmm.14285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Xue H, Li P, Luo Y, et al. Salidroside stimulates the Sirt1/PGC-1α axis and ameliorates diabetic nephropathy in mice. Phytomedicine : international journal of phytotherapy and phytopharmacology. February 15 2019;54:240–247. doi: 10.1016/j.phymed.2018.10.031 [DOI] [PubMed] [Google Scholar]
- 139.Hou S, Zhang T, Li Y, Guo F, Jin X. Glycyrrhizic Acid Prevents Diabetic Nephropathy by Activating AMPK/SIRT1/PGC-1α Signaling in db/db Mice. Journal of diabetes research. 2017;2017:2865912. doi: 10.1155/2017/2865912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Li J, Li N, Yan S, et al. Melatonin attenuates renal fibrosis in diabetic mice by activating the AMPK/PGC1α signaling pathway and rescuing mitochondrial function. Molecular medicine reports. February 2019;19(2):1318–1330. doi: 10.3892/mmr.2018.9708 [DOI] [PubMed] [Google Scholar]
- 141.Garrett SM, Whitaker RM, Beeson CC, Schnellmann RG. Agonism of the 5hydroxytryptamine 1F receptor promotes mitochondrial biogenesis and recovery from acute kidney injury. The Journal of pharmacology and experimental therapeutics. August 2014;350(2):257–64. doi: 10.1124/jpet.114.214700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Rehman H, Krishnasamy Y, Haque K, et al. Green tea polyphenols stimulate mitochondrial biogenesis and improve renal function after chronic cyclosporin a treatment in rats. PloS one 2014;8(6):e65029. doi: 10.1371/journal.pone.0065029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Whitehead N, Gill JF, Brink M, Handschin C. Moderate Modulation of Cardiac PGC-1α Expression Partially Affects Age-Associated Transcriptional Remodeling of the Heart. Front Physiol. 2018;9:242–242. doi: 10.3389/fphys.2018.00242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Lehman JJ, Barger PM, Kovacs A, Saffitz JE, Medeiros DM, Kelly DP. Peroxisome proliferator-activated receptor gamma coactivator-1 promotes cardiac mitochondrial biogenesis. The Journal of clinical investigation. October 2000;106(7):847–56. doi: 10.1172/jci10268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Lynn EG, Stevens MV, Wong RP, et al. Transient upregulation of PGC-1alpha diminishes cardiac ischemia tolerance via upregulation of ANT1. J Mol Cell Cardiol. 2010;49(4):693–698. doi: 10.1016/j.yjmcc.2010.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Neuen BL, Young T, Heerspink HJL, et al. SGLT2 inhibitors for the prevention of kidney failure in patients with type 2 diabetes: a systematic review and meta-analysis. The lancet Diabetes & endocrinology. November 2019;7(11):845–854. doi: 10.1016/s2213-8587(19)30256-6 [DOI] [PubMed] [Google Scholar]
- 147.Tomita I, Kume S, Sugahara S, et al. SGLT2 Inhibition Mediates Protection from Diabetic Kidney Disease by Promoting Ketone Body-Induced mTORC1 Inhibition. Cell metabolism. July 19 2020;doi: 10.1016/j.cmet.2020.06.020 [DOI] [PubMed] [Google Scholar]
- 148.Tanaka S, Sugiura Y, Saito H, et al. Sodium-glucose cotransporter 2 inhibition normalizes glucose metabolism and suppresses oxidative stress in the kidneys of diabetic mice. Kidney international. November 2018;94(5):912–925. doi: 10.1016/j.kint.2018.04.025 [DOI] [PubMed] [Google Scholar]
- 149.Sasaki M, Sasako T, Kubota N, et al. Dual Regulation of Gluconeogenesis by Insulin and Glucose in the Proximal Tubules of the Kidney. Diabetes. September 2017;66(9):2339–2350. doi: 10.2337/db16-1602 [DOI] [PubMed] [Google Scholar]
- 150.Hasegawa S, Tanaka T, Saito T, et al. The oral hypoxia-inducible factor prolyl hydroxylase inhibitor enarodustat counteracts alterations in renal energy metabolism in the early stages of diabetic kidney disease. Kidney international. May 2020;97(5):934–950. doi: 10.1016/j.kint.2019.12.007 [DOI] [PubMed] [Google Scholar]
- 151.Sugahara M, Tanaka S, Tanaka T, et al. Prolyl Hydroxylase Domain Inhibitor Protects against Metabolic Disorders and Associated Kidney Disease in Obese Type 2 Diabetic Mice. Journal of the American Society of Nephrology. 2020;31(3):560–577. doi: 10.1681/asn.2019060582 [DOI] [PMC free article] [PubMed] [Google Scholar]