

Abstract
The functionalization of terpenes using cytochrome P450 enzymes is a versatile route to the production of useful derivatives that can be further converted to value-added products. Many terpenes are hydrophobic and volatile making their availability as a substrate for P450 enzymes significantly limited during microbial production. In this study, we developed a strategy to improve the accessibility of terpene molecules for the P450 reaction by linking terpene synthase and P450 together. As a model system, fusion proteins of 1,8-cineole synthase (CS) and P450cin were investigated and it showed an improved hydroxylation of the monoterpenoid 1,8-cineole up to 5.4-fold. Structural analysis of the CS-P450cin fusion proteins by SEC-SAXS indicated a dimer formation with preferred orientations of the active sites of the two domains. We also applied the enzyme fusion strategy to the oxidation of a sesquiterpene epi-isozizaene and the fusion enzymes significantly improved albaflavenol production in engineered E. coli. From the analysis of positive and negative examples of the fusion strategy, we proposed key factors in structure-based prediction and evaluation of fusion enzymes. Developing fusion enzymes for terpene synthase and P450 presents an efficient strategy toward oxidation of hydrophobic terpene compounds. This strategy could be widely applicable to improve the biosynthetic titer of the functionalized products from hydrophobic terpene intermediates.
Keywords: Oxidized terpenoid, fusion protein, terpene synthase, P450, SAXS, I-TASSER
Graphical abstract
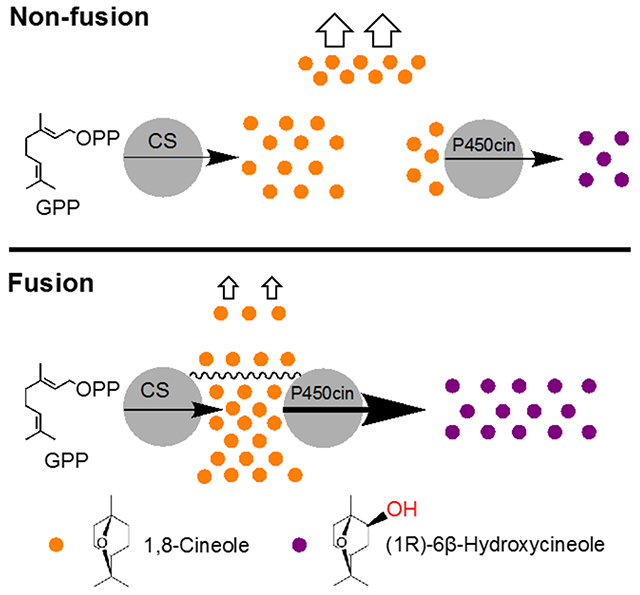
1. Introduction
Terpenes are a large class of natural products, primarily produced by plants and constitute the main components of essential oils. A typical monoterpene (C10), such as limonene, is a cyclic hydrocarbon molecule (C10H16) and can be used as a precursor of fuel additives, fragrances, insecticides, and pharmaceuticals (Aharoni et al., 2005). Production of terpenes in the microbial system is considered a more sustainable and stable alternative to the isolation from plants or via chemical synthesis. Functionalization of the terpene carbon backbone by enzymes such as cytochrome P450s could further expand the range of bio-based compounds which frequently can be converted to additional products of commercial interest (Bernhardt, 2006; Chang et al., 2007; Pateraki et al., 2015; Renault et al., 2014; Urlacher and Girhard, 2019). For example, limonene can be oxidized by P450 (CYP153) to perillyl alcohol, a precursor of promising anti-cancer agents (van Beilen et al., 2005). While P450s play an important role in the decoration and modification of terpenes essential for the new bioactivities, the hydrophobicity and volatility of terpene molecules might limit the availability of the substrate around the enzyme and result in low enzymatic conversion during microbial production, especially when a solvent overlay is used to reduce the loss of these volatile compounds via extraction of hydrophobic terpenes to the overlay (Alonso-Gutierrez et al., 2013). This makes the subsequent enzymatic reaction (which uses terpenes as substrates) less efficient and eventually lowers the titer of the final product.
To overcome the low availability of hydrophobic substrates for downstream enzymes such as P450s, one popular strategy is to create a spatial constraint that improves the proximity between the enzyme and the substrate (Conrado et al., 2008). Engineering of fusion proteins (Kourtz et al., 2005; Meynial Salles et al., 2007), protein scaffolds (Dueber et al., 2009), and compartmentalization of metabolic pathways (Avalos et al., 2013) have been explored to achieve the proximity effect. Among these approaches, engineering synthetic fusion proteins have been extensively used to modify enzymes toward efficient metabolic catalysis due to their simplicity and effectiveness (Yu et al., 2015). Using a short peptide linker sequence, two or more enzymes are combined and generate a single polypeptide that exhibits more than one activity or increases the reaction rate for consecutive enzymes. In the microbial production of isoprenoids, a higher pinene production level was reported by linking terpene synthase with geranyl pyrophosphate (GPP) synthase to overcome product inhibition from GPP (Sarria et al., 2014). Similarly, an engineered fusion of isopentenyl diphosphate (IPP) isomerase and isoprene synthase showed a 3.3-fold increase of isoprene titer (Gao et al., 2016). For P450 enzymes, fusions of P450 with a heterologous cytochrome P450 reductase have also proven successful in various instances. For example, a P450 TxtE was linked to the reductase domain of P450BM3 for improved activity and regio-promiscuity in aromatic nitration (Zuo et al., 2017).
Although engineering a fusion of P450 with a cytochrome P450 reductase is widely studied, there are fewer reports for engineering a fusion between P450 and a terpene synthase. Given that the considerable loss of the terpene substrate from the cell is a critical limitation for the subsequent P450 reaction during the microbial production (Alonso-Gutierrez et al., 2013), engineering a fusion protein by linking terpene synthase and P450 to form a chimeric protein could improve the proximity of P450 and the terpene substrate, which in turn would improve the substrate availability for P450. In this study, we selected the hydroxylation of monoterpene 1,8-cineole as a model system to demonstrate this approach in the microbial system via engineering the recombinant enzyme fusion between terpene synthase-P450 enzyme fusion (Figure 1). Guided by structural modeling, we engineered a series of fusion proteins between 1,8-cineole synthase and P450cin (CYP176A1) to investigate the hydroxylation of 1,8-cineole in both in vitro and in vivo conditions in comparison to non-fused enzymes. Structural analysis of fusion proteins revealed that different linker length changes the flexibility of the fusion enzymes. We also applied this enzyme fusion strategy for the oxidation of several other terpenes, and the data from both experiments and the modeling analysis, of positive and negative results suggested key factors (e.g. linker length, enzyme orientation, etc.) for designing a fusion protein. Our results demonstrated that engineering fusion enzyme is a feasible strategy for the efficient production of functionalized products from hydrophobic terpene intermediates.
Figure 1.
Engineering enzyme fusions by linking terpene synthase (CS, EizS) and P450 enzyme (CYP) for the production of oxidized terpenoids. GPP, geranyl pyrophosphate; FPP, farnesyl pyrophosphate; CS, 1,8-cineole synthase; EizS, epi-isozizaene synthase; GPPS, geranyl pyrophosphate synthase.
2. Material and methods
2.1. Strains and plasmid construction
All strains and plasmids used in this study are listed in Table 1. Strains and plasmids along with their associated information have been deposited in the public domain of the JBEI Registry (https://public-registry.jbei.org; entries JPUB_016968 to JPUB_017025) and are available from the authors upon request. E. coli DH1 strain was used for terpene and oxidized terpene production, and E. coli DH5α was used for genetic cloning. Genes of CinA (P450cin, CYP176A1; GenBank ID: AF456128) and CinC (Cindoxin, Cdx, GenBank ID: AF456128) from Citrobacter braakii, and CYP170A1 (sco5223; GenBank ID: NC_003888) from Streptomyces coelicolor A3(2) were codon-optimized and synthesized by Integrated DNA Technologies, Inc. (San Diego, CA, USA). Fpr (GenBank ID: CP032667) was cloned from E. coli genomic DNA. CS from Streptomyces clavuligerus was cloned from plasmid JBEI-15065 (Table 1). All peptide linkers were introduced onto the plasmid by PCR using primers listed in Supplementary Table S1.
Table 1.
Strains and plasmids used in this study
| Strains | Description | Reference |
|---|---|---|
| CS | E. coli BL21 (DE3) with pSKB3-CS | This study |
| CinA | E. coli BL21 (DE3) with pSKB3-CinA | This study |
| CinC | E. coli BL21 (DE3) with pSKB3-CinC | This study |
| G1 | E. coli BL21 (DE3) with pSKB3-CS-(GSG)1-CinA | This study |
| G2 | E. coli BL21 (DE3) with pSKB3-CS-(GSG)2-CinA | This study |
| G3 | E. coli BL21 (DE3) with pSKB3-CS-(GSG)3-CinA | This study |
| G4 | E. coli BL21 (DE3) with pSKB3-CS-(GSG)4-CinA | This study |
| G5 | E. coli BL21 (DE3) with pSKB3-CS-(GSG)5-CinA | This study |
| Fpr | E. coli BL21 (DE3) with pSKB3-Fpr | This study |
| 2pCin_Non-fusion | E. coli DH1 with JBEI-3122 + pTrc99a-trGPPS-CS-RBS-CinA-CinC | This study |
| CS-(GSG)1-P450cin | E. coli DH1 with JBEI-3122 + pTrc99a-trGPPS-CS-(GSG)1-CinA-CinC | This study |
| CS-(GSG)2-P450cin | E. coli DH1 with JBEI-3122 + pTrc99a-trGPPS-CS-(GSG)2-CinA-CinC | This study |
| CS-(GSG)3-P450cin | E. coli DH1 with JBEI-3122 + pTrc99a-trGPPS-CS-(GSG)3-CinA-CinC | This study |
| CS-(GSG)4-P450cin | E. coli DH1 with JBEI-3122 + pTrc99a-trGPPS-CS-(GSG)4-CinA-CinC | This study |
| CS-(GSG)5-P450cin | E. coli DH1 with JBEI-3122 + pTrc99a-trGPPS-CS-(GSG)5-CinA-CinC | This study |
| P450cin-(GSG)3- CS | E. coli DH1 with JBEI-3122 + pTrc99a-trGPPS-CinA-(GSG)3-CS-CinC | This study |
| 2pEiz | E. coli DH1 with JBEI-2704 + JBEI-15862 | This study |
| 2pEiz_Non-fusion | E. coli DH1 with JBEI-2704 + pTrc99a-EizS-RBS-CYP170A1-CinC | This study |
| EizS-(GSG)1-CYP170A1 | E. coli DH1 with JBEI-2704 + pTrc99a-EizS-(GSG)1-CYP170A1-CinC | This study |
| EizS-(GSG)2-CYP170A1 | E. coli DH1 with JBEI-2704 + pTrc99a-EizS-(GSG)2-CYP170A1-CinC | This study |
| EizS-(GSG)3-CYP170A1 | E. coli DH1 with JBEI-2704 + pTrc99a-EizS-(GSG)3-CYP170A1-CinC | This study |
| EizS-(GSG)4-CYP170A1 | E. coli DH1 with JBEI-2704 + pTrc99a-EizS-(GSG)4-CYP170A1-CinC | This study |
| EizS-(GSG)5-CYP170A1 | E. coli DH1 with JBEI-2704 + pTrc99a-EizS-(GSG)5-CYP170A1-CinC | This study |
| Plasmids | Description | Reference |
| pSKB3 | Modified pET-28a | (Kang et al., 2016) |
| JBEI-3122 | pBbA5c-MTSA-T1-MBI | (Alonso-Gutierrez et al., 2013) |
| JBEI-15065 | pTrc99a-GPPS-CSStr | (Mendez-Perez et al., 2017) |
| JBEI-2704 | pBbA5c-MevT-T1-MBIS | (Redding-Johanson et al., 2011) |
| JBEI-15862 | pTrc99a-coEizS | (Liu et al., 2018) |
| JPUB_016968 | pSKB3-CS | This study |
| JPUB_016970 | pSKB3-CinA | This study |
| JPUB_016972 | pSKB3-CinC | This study |
| JPUB_016974 | pSKB3-CS-G1-CinA | This study |
| JPUB_016976 | pSKB3-CS-G2-CinA | This study |
| JPUB_016978 | pSKB3-CS-G3-CinA | This study |
| JPUB_016980 | pSKB3-CS-G4-CinA | This study |
| JPUB_016982 | pSKB3-CS-G5-CinA | This study |
| JPUB_016984 | pSKB3-Fpr | This study |
| JPUB_016986 | pTrc99a-trGPPS-CS-RBS-CinA-CinC | This study |
| JPUB_016988 | pTrc99a-trGPPS-CS-(GSG)1-CinA-CinC | This study |
| JPUB_016990 | pTrc99a-trGPPS-CS-(GSG)2-CinA-CinC | This study |
| JPUB_016992 | pTrc99a-trGPPS-CS-(GSG)3-CinA-CinC | This study |
| JPUB_016994 | pTrc99a-trGPPS-CS-(GSG)4-CinA-CinC | This study |
| JPUB_016996 | pTrc99a-trGPPS-CS-(GSG)5-CinA-CinC | This study |
| JPUB_016998 | pTrc99a-trGPPS-CinA-(GSG)3-CS-CinC | This study |
| JPUB_017000 | pTrc99a-EizS-RBS-CYP170A1-CinC | This study |
| JPUB_017002 | pTrc99a-EizS-(GSG)1-CYP170A1-CinC | This study |
| JPUB_017003 | pTrc99a-EizS-(GSG)2-CYP170A1-CinC | This study |
| JPUB_017005 | pTrc99a-EizS-(GSG)3-CYP170A1-CinC | This study |
| JPUB_017007 | pTrc99a-EizS-(GSG)4-CYP170A1-CinC | This study |
| JPUB_017009 | pTrc99a-EizS-(GSG)5-CYP170A1-CinC | This study |
2.2. Protein expression and purification
A plasmid pSKB3 encoding interested proteins with N-terminal His-tag was transformed into E. coli BL21 (DE3). BL21 (DE3) strains bearing pSKB3 plasmids were cultured in Lysogeny Broth (LB) medium containing 50 μg/mL kanamycin at 37°C until the optical density of the culture at 600 nm (OD600) reached to 0.5 – 0.8. The culture was then supplemented with 0.4 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) for induction and transferred to 18°C for culturing overnight. Cells were collected by centrifugation and resuspended in 25 mM Tris-HCl (pH 8.0) buffer containing 300 mM NaCl and 10 mM imidazole (pH 8.0). Cells were lysed by sonication and proteins were purified using Ni-NTA Agarose (QIAGEN, Hilden, Germany). All purified proteins were desalted in 25 mM Tris-HCl (pH 8.0) buffer containing 100 mM NaCl, and 10% glycerol, and stored at −80°C. A Bradford assay (Sigma-Aldrich, St. Louis, MO, USA) was used to quantify the protein concentration of purified proteins, with bovine serum albumin (BSA) as a standard. The quantification of P450cin was determined by UV absorption at 415 nm (ε = 150 cm−1mM−1) (Slessor et al., 2012), and for redox proteins: Cdx at 456 nm (ε = 10825 cm−1M−1) (Hawkes et al., 2010), Fpr at 456 nm (ε = 7100 cm−1M−1) (Jenkins and Waterman, 1994). Both quantification results were shown in the Supplementary Table S4.
2.3. In vitro production of hydroxycineole
Using purified proteins, Bradford-quantified non-fusion (5 μM CS + 5 μM CinA) or fusion proteins (5 μM) were used for the in vitro production of hydroxycineole with 40 μM CinC and 10 μM Fpr in 50 mM Tris-HCl buffer (pH 7.4) containing 5 mM MgCl2 (Hawkes et al., 2010; Shaw et al., 2015). NADPH (2 mM) and 1 mM geranyl pyrophosphate (GPP, Sigma-Aldrich 19533) were added to start the reaction. The reaction was conducted in a total volume of 600 μL reaction in a 1.7-mL microcentrifuge tube at 25°C for 5 hours.
2.4. In vivo production of hydroxycineole
E. coli DH1 bearing two plasmids (JBEI-3122 + JPUB_016986 to JPUB_010998, Table 1) was used for hydroxycineole production. Starter cultures of all production strains were prepared by growing single colonies in LB medium containing 30 μg/mL chloramphenicol and 100 μg/mL carbenicillin at 37°C with 200-rpm shaking for overnight. The starter cultures were diluted in 5 mL EZ-Rich defined medium (Teknova, CA, USA) containing 10 g/L glucose (1%, w/v), 30 μg/mL chloramphenicol, 100 μg/mL carbenicillin, and 0.5 mM IPTG in 50-mL test tubes. 0.5 mL nonane (10%, v/v) was added when required as a solvent overlay. The E. coli cultures were incubated in rotary shakers (200 rpm) at 30°C for 48 hours.
2.5. In vivo production of oxidized epi-isozizaene
E. coli DH1 bearing two plasmids (JBEI-2704 + JPUB_017000 to JPUB_017009, Table 1) was used for oxidized epi-isozizaene production. Starter cultures of all production strains were prepared by growing single colonies in LB medium containing 30 μg/mL chloramphenicol and 100 μg/mL carbenicillin at 37°C with 200-rpm shaking for overnight. The starter cultures were diluted in 5 mL EZ-Rich defined medium (Teknova, CA, USA) containing 10 g/L glucose (1%, w/v), 30 μg/mL chloramphenicol, 100 μg/mL carbenicillin, and 65 mg/L δ-aminolevulinic acid, 0.5 mM IPTG in 50-mL culture tubes. 0.5 mL nonane (10%, v/v) was added when required as a solvent overlay. The E. coli cultures were incubated in rotary shakers (200 rpm) at 30°C for 72 hours.
2.6. Protein structure prediction
Structures of fusion proteins were predicted by Iterative Threading ASSEmbly Refinement (I-TASSER) server (Roy et al., 2012; Yang and Zhang, 2015; Zhang, 2009). The predicted models with the highest C-score were used for further analysis. For the non-fusion enzyme CS (PDB: 5NX6) (Karuppiah et al., 2017), P450cin (PDB: 1T2B) (Meharenna et al., 2004), EizS (PDB: 3KB9) (Aaron et al., 2010), CYP170A1 (PDB: 3DBG) (Zhao et al., 2009), LS (PDB: 2ONG) (Hyatt et al., 2007), and CYP153A6 (similar sequence to CYP153A7, PDB: 3RWL) (Pham et al., 2012), their structural models or homology model were retrieved from PDB and aligned with the predicted structures of correlated fusion proteins. Active sites of subunits in the fusion proteins were annotated according to literature. PyMOL was used for visualization and image generation.
2.7. Small Angle X-ray Scattering (SAXS)
Size Exclusion Chromatography (SEC) coupled to Small Angle X-ray Scattering (SAXS) data were collected at the beamline 12.3.1 at the Advanced Light Source synchrotron (Lawrence Berkeley National Laboratory, Berkeley, CA, USA) (Classen et al., 2013). Proteins were separated on an SEC Shodex KW-803 column at 20°C in 25 mM Tris HCl, pH 7.5, 50 mM NaCl and eluted directly into the SAXS sample cell at a flow rate of 0.5 mL/min. Three second X-ray exposures were collected continuously over the 30 minutes SEC elution at 1.127Å wavelength on a Dectris PILATUS3 × 2M detector. Initial data were processed at the beamline (Dyer et al., 2014; Hura et al., 2009). The sample to detector distance was 2.105 m. The SAXS eluent was split 4 to 1 between the SAXS sample cell and UV, multi-angle light scattering (MALS), and refractometer, measured on an 18-angle DAWN HELEOS II light scattering detector and Optilab refractive index concentration detector (Wyatt Technology, CA, USA). The system was calibrated with bovine serum albumin (BSA). Background subtraction from SAXS frames after the protein peaks and evolving factor analysis (EFA) was done using the program RAW. Representing tetrameric, dimeric, and monomeric population, three components were derived from the peak eluting at ~20 minutes. SAXS frames after the main peak were used for buffer subtraction. The targeted elution peak was deconvoluted into three components, representing monomer, dimer, and tetramer. Further SAXS analysis was done using RAW (Volume of Correlation MW) (Rambo and Tainer, 2013), SCÅTTER (Porod component, (https://bl1231.als.lbl.gov/scatter/), ATSAS (Guinier, Real space, GASBOR) (Franke et al., 2017). In the Guinier plots, all SAXS curves were linear, consistent with no aggregation present. Based on the crystal structures of the subunits (PDB: 5NX6 and 1T2B), SAXS models were built using MODELER (Fiser et al., 2000), followed by BilBOMD/FOXS (Pelikan et al., 2009; Schneidman-Duhovny et al., 2016, 2013). SAXS data collection and data analysis details are provided in Table 2.
Table 2.
SAXS Data collection and metrics
| SEC-SAXS Data | G3 monomer | G3 dimer | G4 monomer | G4 dimer |
|---|---|---|---|---|
| Nomenclature | 222 | 221 | 232 | 231 |
| Porod Debye (Px) | 3.9 | 3.4 | 3.3 | 3.3 |
| Low q (Å−1) | 0.0137 | 0.0158 | 0.0209 | 0.0137 |
| High q (Å−1) | 0.366 | 0.366 | 0.366 | 0.366 |
| Reciprocal Rg (Å) | 33 | 44 | 36 | 46 |
| Reciprocal I(0) (detector units) | 111 | 191 | 374 | 2,940 |
| Real space Rg (Å) | 33 | 44 | 35 | 46 |
| Real space I(0) (detector units) | 111 | 191 | 374 | 2,940 |
| Dmax (Å) | 96 | 150 | 106 | 158 |
| Vc Molecular Mass (kDa) | 85 | 147 | 85 | 153 |
| Theoretical Mass (kDa) [dimer] | 86.6 | [173] | 86.7 | [173] |
2.8. Gas chromatography-mass spectrometry (GC-MS) analysis
For 1,8-cineole, hydroxycineole, and perillyl alcohol, samples were extracted by an equal volume of ethyl acetate containing β-pinene (5 mg/L) as an internal standard. For epi-isozizaene and oxidized products, samples were extracted by an equal volume of ethyl acetate containing guaiazulene (5 mg/L) as an internal standard. The mixture of ethyl acetate and in vitro reaction solution or cell culture was vigorously shaken for 15 min and subsequently centrifuged at 21,130 g for 3 min to separate ethyl acetate from the aqueous phase. The ethyl acetate layer was collected and 1 μL was analyzed by Agilent GC-MS equipped with HP-5 column (Agilent, USA). The GC oven was programmed from 40°C (held for 3 min) to 295°C at 15°C/min. The solvent delay was set at 3.4 min. 1,8-cineole and perillyl alcohol were quantified using authentic standards. Hydroxycineole concentration was estimated using total ion chromatogram (TIC) areas with respect to the TIC areas of alternative standard 1,8-cineole, and β-pinene (a monoterpene) as an internal standard to normalize the GC results. Similarly, the concentrations of epi-isozizaene, albaflavenol, and albaflavenone were estimated using their TIC areas with respect to the TIC areas of alternative standard (-)-trans-caryophyllene, and guaiazulene (a sesquiterpene) was used as an internal standard to normalize the GC results. MS full scan spectrum of hydroxycineole and oxidized epi-isozizaene were shown in Supplementary Figures S1 and S2. When nonane overlay was used during the production, the solvent delay was set at 6.8 min. Both the nonane overlay and the aqueous phase of the culture were sampled for the GC-MS measurement, respectively. The production titers were the sum of both measured values after applying the dilution factor.
3. Results
3.1. Designing fusion enzymes of 1,8-cineole synthase and P450cin
1,8-Cineole, or eucalyptol, is a monoterpene (C10) naturally found in essential oils from Eucalyptus globulus and other plants. (Klocke et al., 1987; Shaw et al., 2015). 1,8-Cineole is also a potential precursor for high energy-density molecules used as jet fuels (Bergman and Siewers, 2016; Yang et al., 2017), and therefore E. coli was engineered previously to overproduce 1,8-cineole using the mevalonate (MVA) pathway (Mendez-Perez et al., 2017). Hydroxylation of 1,8-cineole introduces a functional group to this compound and allows further derivatization to more valuable products, such as p-cymene (Leita et al., 2010). P450cin (CYP176A1) from C. braakii has shown a specific activity of 1,8-cineole hydroxylation to produce (1R)-6β-hydroxycineole (or hydroxycineole) (Hawkes et al., 2002), and its redox partners, NADPH-dependent flavodoxin reductase and a flavodoxin, have also been reported (Hawkes et al., 2010).
To investigate the hydroxylation of 1,8-cineole as a model system for engineering the enzyme fusion between terpene synthase and P450, fusion proteins were designed between 1,8-cineole synthase (CS) and P450cin using a flexible peptide linker Gly-Ser-Gly (GSG) (Guo et al., 2017). First, we have predicted the structure of CS-P450cin fusion proteins linked by (GSG)n linkers using the I-TASSER server (Roy et al., 2012; Yang and Zhang, 2015; Zhang, 2009). The modeling provided a possible orientation of the CS and P450cin fusion proteins (Figure 2), and the results showed that the two active sites of CS and P450cin were proximally oriented. Based on the preliminary structural analysis of CS-P450cin fusion proteins, we engineered five CS-P450cin fusion proteins with different linker lengths by adjusting the repeat number (n) of (GSG)n linker (n = 1–5), and the resultant CS-P450cin fusion proteins were named G1 to G5, respectively according to their number of GSG linker repeats.
Figure 2.
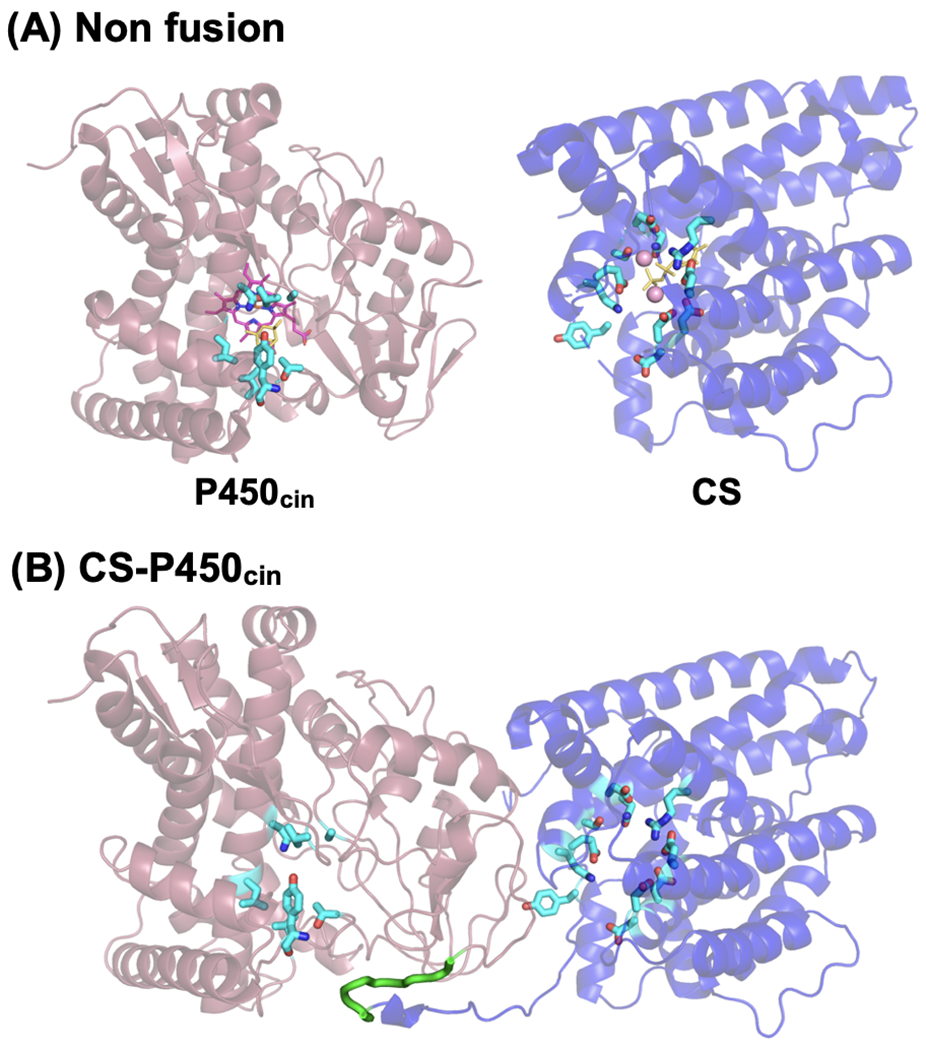
Structural prediction of CS-P450cin fusion protein. (A) Structures of non-fused enzymes of P450cin and CS monomer aligned with the predicted structure of CS-P450cin fusion. (B) Predicted structure of the CS-P450cin fusion protein with a (GSG)3 peptide linker (G3), C-score of I-TASSER = −2.74. P450cin, red cartoon; CS, blue cartoon; (GSG)3 linker, green loop; Heme, pink stick molecule; Residues of active sites, color stick molecules.
3.2. In vitro production of hydroxycineole with CS-P450cin fusions from GPP
To investigate the hydroxylation of 1,8-cineole by various CS-P450cin fusions with different linker lengths, equal moles of purified proteins were used for in vitro production of hydroxycineole from GPP substrate (Figure 3). In a 5-hour reaction, most CS-P450cin fusions except G1 showed a higher level of hydroxycineole production than non-fused individual CS and P450cin. The G1 fusion did not show a substantial difference relative to the non-fusion control (Figure 3C). The highest level of hydroxycineole production was observed from the G4 fusion and showed a 5.4-fold increase over non-fused CS and P450cin after 5 hours. The highest hydroxycineole production rate in G4 was reached after 2 hours at 0.051 μM/min, which is 6.4-fold faster than that of the non-fusion enzymes (0.008 μM/min). The overall hydroxylation ratio of the G4 fusion was 2.3% after 5 hours, which is 5.4-fold higher than that of the non-fusion enzymes (0.4%) (Figure 3D). All 5 fusion enzymes also showed up to a 2.7-fold increase of 1,8-cineole level over the non-fused enzymes during the first 3 hours (Figure 3C), suggesting that enzyme fusion also improves the activity of the cineole synthase and eventually more terpene substrate is available for the P450 reaction with the fusion proteins. Likely due to the volatility of 1,8-cineole, it was also observed that 1,8-cineole eventually decreased to a similar level from all different samples after a 3-hour reaction (Figure 3C).
Figure 3.
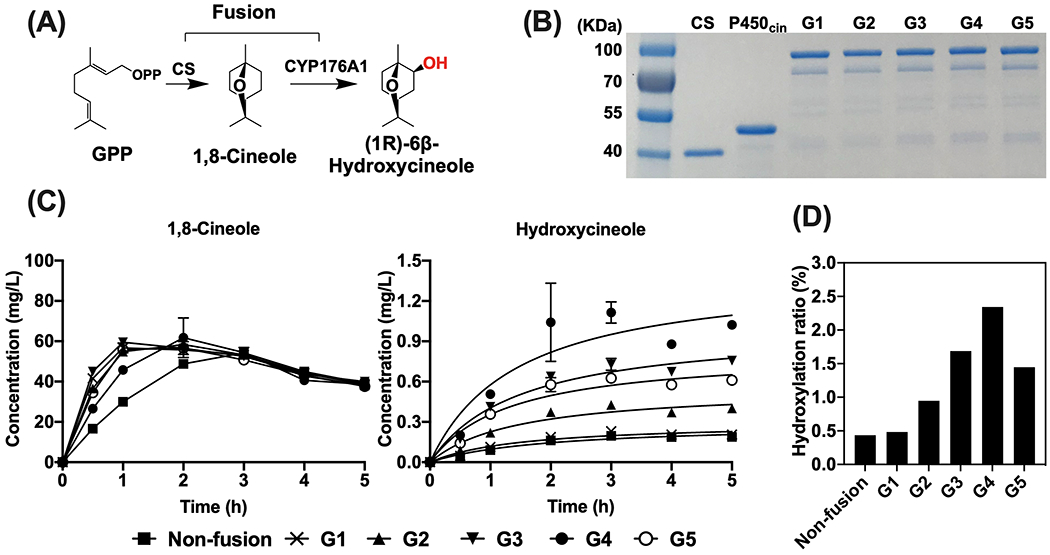
In vitro production of hydroxycineole. (A) In vitro two-step reaction from GPP. (B) SDS-PAGE gel of purified non-fusion and fusions for CS and P450cin. Fusions of CS and P450cin (G1 to G5) were engineered with 1 to 5 repeats of GSG peptide linker. Size of purified proteins: CS, 40.71 KDa; P450cin (CinA), 48.25 KDa; G1, 86.16 KDa; G2, 86.37 KDa; G3, 86.57 KDa; G4, 86.77 KDa; G5, 86.97 KDa. (C) In vitro time-course production of cineole and hydroxycineole from GPP substrate with purified proteins. Hydroxycineole concentrations are reported based on the equivalent concentration of 1,8-cineole. Error bars indicate one standard deviation (n = 3). (D) Hydroxylation ratio of in vitro reaction after 5 hours. The hydroxylation ratio is the molar ratio of hydroxycineole out of the total generated terpenes (1,8-cineole and hydroxycineole).
The in vitro production results with the engineered CS-P450cin fusions showed higher hydroxycineole production than non-fused CS and P450cin. The improved 1,8-cineole hydroxylation from fusion proteins could be attributed to the proximity of P450cin to its hydrophobic substrate 1,8-cineole, which shows the advantage of linking a P450 enzyme to a terpene synthase, particularly when a hydrophobic and volatile terpene is the intermediate and served as the substrate for a consequential P450 reaction during a multi-step terpene oxidation.
3.3. In vivo assessment of CS-P450cin fusions for hydroxycineole production from glucose
To in vivo assess hydroxycineole production using fusion enzymes, the CS-P450cin fusions were introduced into E. coli. Informed from the previous report (Mendez-Perez et al., 2017), E. coli strain containing 2 separate plasmids was used for hydroxycineole production by inserting the genes encoding the P450cin (CinA) and its redox partner Cdx (CinC) downstream of CS on the 2nd plasmid JBEI-15065 (Table 1). For the expression of the non-fused CS and P450cin, an RBS sequence (5’-TTTAAGAAGGAGATATAC-3’) was used for individual expression of CS and P450cin, respectively (Table 1 and Supplementary Table S1). For the CS-P450cin fusions, the same RBS was used for the entire fused gene sequence. As solvent overlay is usually used to prevent evaporation of the product during production, we used the overlay to evaluate the performance of fusion enzymes at the in vivo conditions. While dodecane was used as the overlay for 1,8-cineole production previously (Mendez-Perez et al., 2017), it has a similar molecular weight to hydroxycineole (MW=170), as well as a close retention time in chromatography. Therefore, we used nonane instead of dodecane as the overlay to obtain a better signal of hydroxycineole by GC-MS.
As shown in Figure 4, the use of solvent overlay generally facilitated 1,8-cineole production, but it did not help hydroxycineole levels. For both non-fusion and fusion proteins, hydroxycineole production without using overlay was 1.7 to 3.3-fold higher than that with an overlay. When nonane overlay was used, all fusions except G4 detected more hydroxycineole than the non-fusion control strain (Figure 4C), while generally less amount of 1,8-cineole (except for G2), suggesting an inefficient hydroxylation in the non-fusion control strain. When overlay was not used during the production, all 5 strains with fusion protein produced more hydroxycineole than the non-fusion control strain (Figure 4C). The highest hydroxycineole titer was observed from G3 (56 mg/L), which is 3.1-fold higher than that of the non-fusion control (18 mg/L). In contrast to the in vitro results, G4 did not show any significant advantage compared with the other fusions with different linker lengths. Similar to our previous work of a P450 conversion perillyl alcohol from limonene (Alonso-Gutierrez et al., 2013), the addition of solvent overlay facilitated the in situ extraction of 1,8-cineole and prevents the evaporation from the culture, but it also competed with the subsequent P450 enzymatic reaction for terpene substrates and eventually lower the P450 bioconversion efficiency. Overall, the use of fusion proteins improved the production of hydroxycineole with and without added solvent overlay, which suggested a higher efficiency during in vivo production of the oxidized terpene.
Figure 4.
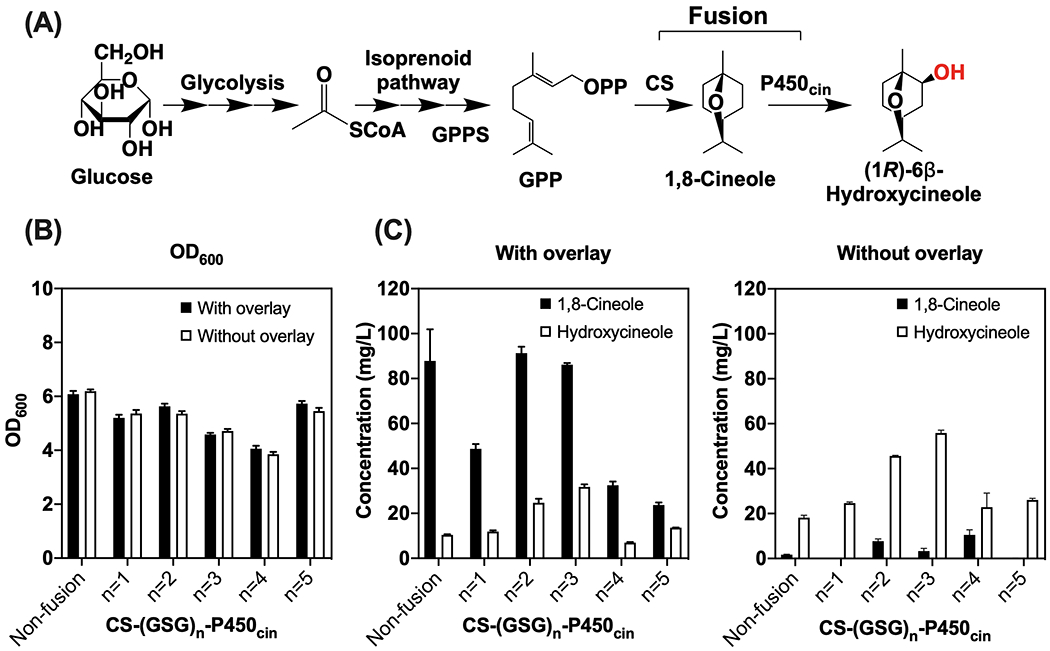
In vivo production of hydroxycineole by E. coli DH1 strains with engineered enzyme fusions. (A) Metabolic pathway of hydroxycineole production from glucose using enzyme fusions of CS and P450cin, CS-(GSG)n-P450cin (n = 1–5). (B) OD600 of production strains after 48 hours. (C) Production of 1,8-cineole and hydroxycineole with or without the solvent overlay. Hydroxycineole concentrations are reported based on the equivalent concentration of 1,8-cineole. Error bars indicate one standard deviation of triplicates.
In addition to the linker length, the enzyme orientation is another key factor for the activity of a fusion enzyme (Sarria et al., 2014). Given that G3 showed high production of hydroxycineole both in vitro and in vivo conditions, we used the (GSG)3 linker to construct a fusion enzyme P450cin-(GSG)3-CS by switching the order of CS and P450cin and to test hydroxycineole production. Compared to the CS-(GSG)3-P450cin fusion (G3), this fusion enzyme with reversed domain order produced 51% and 76% less 1,8-cineole and hydroxycineole, respectively when the overlay was used (Table 3). These trends were also significant when the overlay was not used. In this case, it produced 80% and 63% less 1,8-cineole and hydroxycineole, respectively, than in the G3 fusion protein (Table 3), indicating the inefficiency when CS and P450cin were linked in the reversed orientation. This P450cin-(GSG)3-CS fusion enzyme did not even result in a significant improvement of hydroxycineole but less 1,8-cineole production compared with the non-fusion control strain (Table 3). Structural modeling results suggested that the two active sites of P450cin and CS in this fusion protein are no longer proximally oriented (Supplementary Figure S3). As shown in Figure S3A, the active site of CS faces outside of the fusion complex surface, whereas the active site of P450cin is located in the middle of the fusion complex. Thus, the simulated fusion structure shows an unfavorable orientation for the two subunits and may explain its less efficiency as a fusion protein.
Table 3.
Characterization of the fusion protein with the reversed orientation of enzymes (P450cin-CS) during the in vivo hydroxycineole production
| 1,8-Cineole (mg/L) | Hydroxycineole (mg/L) * | |||
|---|---|---|---|---|
| With overlay | Without overlay | With overlay | Without overlay | |
| Non-fusion | 88 ± 14 | 2 ± 0 | 10 ± 0 | 18 ± 1 |
| CS-P450cin | 86 ± 1 | 3 ± 1 | 32 ± 1 | 56 ± 1 |
| P450cin-CS | 42 ± 10 | 1 ± 0 | 8 ± 0 | 21 ± 1 |
Data are represented as mean ± standard deviation of triplicates.
Hydroxycineole concentrations are reported based on the equivalent concentration of 1,8-cineole.
3.4. Structural characterization of CS-P450cin enzyme fusions
To experimentally determine the orientation of CS and P450cin, we used Size Exclusion Chromatography coupled to Small Angle X-ray Scattering (SEC-SAXS) to characterize the G3 and G4 fusion proteins in solution as these two fusion enzymes showed the best activities at the in vitro experiments. SAXS measures electron pair distances of proteins in solution and can reveal protein stoichiometry, flexibility, and overall orientation, in cases such as ours where the individual subunits have been crystallized (Putnam et al., 2007). For both G3 and G4 fusion proteins in the size exclusion chromatography profile, the proteins eluted in multiple peaks, with the earliest peak representing large aggregates and the second major peak representing a mixture of monomer, dimer, and tetramer, based on SAXS molecular mass calculation and in agreement with in-line Multi-Angle Light Scattering (MALS) measurements (Figure 5A).
Figure 5.
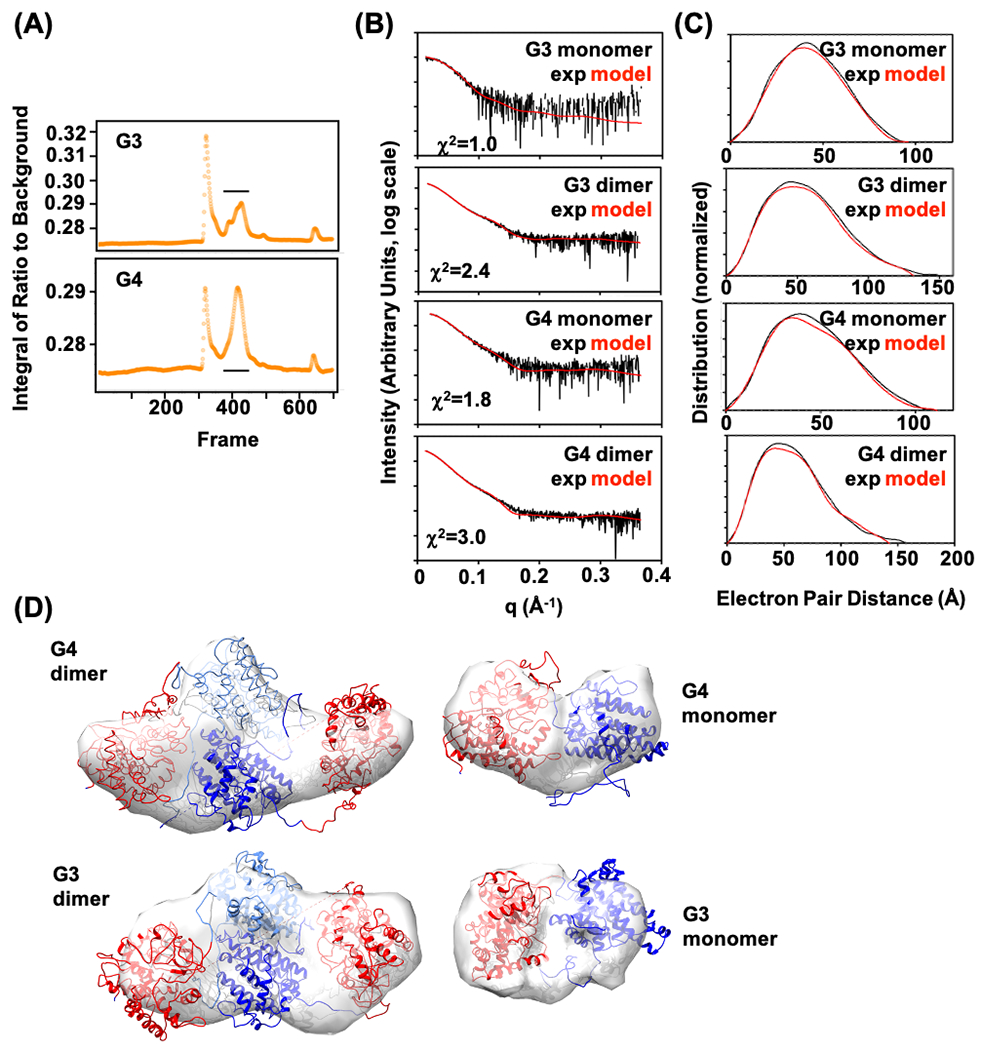
SAXS analysis of enzyme fusions. (A) SEC-SAXS chromatogram of G3 and G4 fusion proteins. The horizontal line indicates peak(s) that were further analyzed. (B) Reciprocal space SAXS curves after SVD, representing scattering from monomer and dimer for G3 and G4 enzyme fusions, overlaid with curves predicted from models that best fit the experimental data (red curves). χ2 fit of models is shown. (C) Corresponding real space SAXS curves with experimental and model curves overlaid. (D) Ab initio shape predictions overlaid with the best-fitting model. Models were based on 1,8-cineole synthase (PDB ID: 5NX6, blue) and P450cin (PDB ID:1T2B, red).
For the G3 fusion protein, the aggregate peak was higher in the UV signal than the monomer/dimer peak, but the second peak was larger than the aggregate peak in the G4 fusion protein (Figure 5A). The different proportions of peaks suggested that the G3 fusion protein may be more constrained than the G4 fusion protein. The SAXS frames from the second peaks were deconvoluted into three SAXS components, representing monomer, dimer, and tetramer. For both G3 and G4 fusion proteins, the dimer population was greater than monomer or tetramer, based on MALS and relative intensity of the scattering profile, even taking into account the greater scattering of the dimer compared to the monomer. A measure of the protein density and relative flexibility, the Porod Debye number for the G4 monomer and dimer were both 3.3 (Table 2), consistent with the protein behaving like beads on a string (Brosey et al., 2013; Rambo and Tainer, 2011). The number for the G3 monomer and dimer were 3.9 and 3.4 (Table 2), respectively, indicating greater rigidity in the G3 fusion protein.
Using two different modeling programs followed by molecular dynamics, we generated models of the G3 and G4 monomer and dimer to match against the experimental SAXS data (Figure 5B and 5C). The analysis compares single models and ensembles of models, the latter of which are usually found to best fit experimental SAXS data when there is significant flexibility. Surprisingly, we found that the experimental SAXS data curves fit well against single models, despite the flexibility indicated by the Porod Debye number in three of the SAXS curves. This SAXS modeling result suggests the two domains of CS and P450cin are shifting relative to each other but not completely swinging free (Figure 5D). For the monomer of fusion proteins, the SAXS structural analysis matched the predicted structure by I-TASSER for the fusion protein sequence (Figure 2), and the formalized dimers of fusion protein could also intensify the proximity effect.
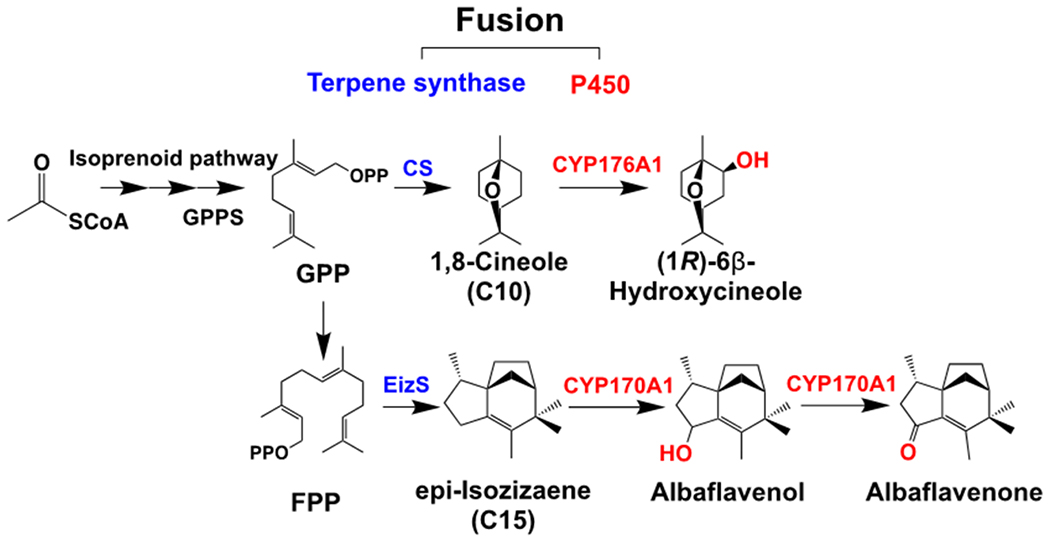
3.5. Production of oxidized epi-isozizaene using enzyme fusions.
We applied the enzyme fusion strategy to the biosynthetic pathway for oxidized epi-isozizaene products (Figure 1, Figure 6) such as albaflavenol, and the subsequently oxidized product albaflavenone, a sesquiterpene antibiotic found in S. coelicolor A3(2) (Zhao et al., 2008). A bacterial P450 (CYP170A1) has been identified to catalyze the oxidation of epi-isozizaene in S. coelicolor A3(2). Recently, epi-isozizaene biosynthesis was successfully engineered in E. coli to produce a novel jet-fuel precursor using the MVA pathway (Liu et al., 2018). To build the fusion protein for epi-isozizaene synthase (EizS) and CYP170A1, we predicted the possible structure of an EizS-CYP170A1 fusion protein, and it showed the proximal orientation for the two active sites of EizS and CYP170A1 in the fusion (Supplementary Figure S4). Following the fusion enzyme engineering strategy used in hydroxycineole biosynthesis, we constructed fusions for EizS and CYP170A1 with 1 to 5 repeats of Gly-Ser-Gly (GSG) peptide linker. An RBS sequence (5’-TTTCACACAGGAAACAGACC-3’) was used for the expression of EizS and CYP170A1 individually in the non-fusion control strain similar to the 1,8-cineole oxidation case. For the EizS-CYP170A1 fusions, the same RBS was used for the entire fused gene sequence (Table 1, Supplementary Table S1).
Figure 6.
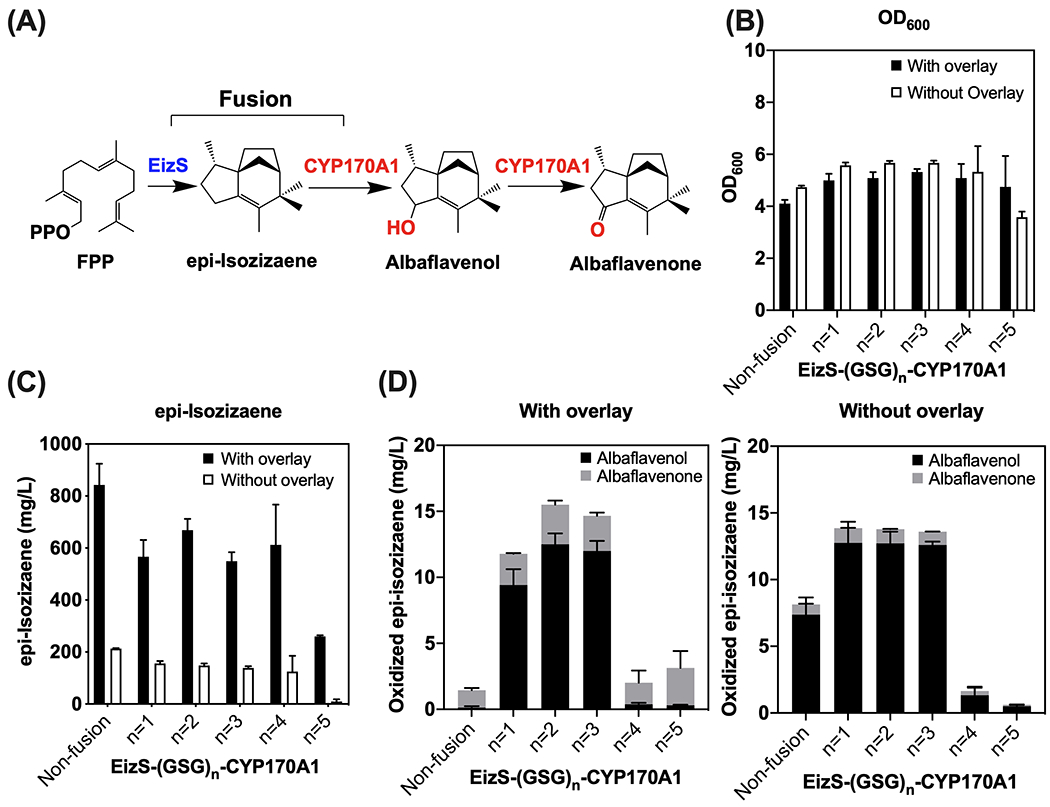
Production of oxidized epi-isozizaene with engineered enzyme fusions in E. coli DH1. (A) Metabolic pathway of oxidized epi-isozizaene production from glucose using enzyme fusions of EizS and CYP170A1, EizS-(GSG)n-CYP170A1 (n = 1–5); FPP, farnesyl pyrophosphate. (B) OD600 of production strains after 72 hours. (C) Production of epi-isozizaene with or without the solvent overlay. (D) Production of oxidized epi-isozizaene (albaflavenol, albaflavenone) with or without the solvent overlay. The concentrations of epi-isozizaene, albaflavenol, and albaflavenone are reported based on the equivalent concentration of (−)-trans caryophyllene. Error bars indicate one standard deviation of triplicates.
Compared with the non-fusion control, the epi-isozizaene production level was a little lower in the strains engineered with fusion enzymes (Figure 6C). On the other hand, the total oxidized products (albaflavenol and albaflavonone) were notably increased in the fusions with shorter linkers (GSG)1-3 for both conditions, with and without added solvent overlay (Figure 6D). As expected, the improvement of oxidized products by enzyme fusions was more significant when the overlay was used. The highest oxidized epi-isozizaene level (13 mg/L albaflavenol, 3 mg/L albaflavenone) was observed by the fusion EizS-(GSG)2-CYP170A1 with a (GSG)2 linker, which achieved 90- and 2.3-fold increase in albaflavenol and albaflavenone production, respectively. This result demonstrated the viability of engineering a fusion protein between terpene synthase and P450 for sesquiterpene oxidation, suggesting possible applications of this enzyme fusion strategy to functionalize diverse terpenoid compounds.
4. Discussion
In this study, we used an enzyme fusion strategy by directly linking terpene synthase and cytochrome P450 to facilitate the heterologous production of oxidized terpenoids in engineered microbial systems. Unlike the natural biosynthesis process in plants (Cheng et al., 2007; Pateraki et al., 2015), engineered microbial systems usually lack cellular compartments and spatial regulation to contain the volatile molecules. By taking advantage of the proximal environment created by the fusion enzyme, we expected the fusion can facilitate the capture of volatile terpene molecules for downstream P450s and improve the terpene oxidation. Using the monoterpene 1,8-cineole hydroxylation as a model system, fusion proteins of 1,8-cineole synthase (CS) and P450cin (CYP176A1) were constructed with different lengths of peptide linkers. The engineered CS-P450cin fusions showed improved hydroxylation of 1,8-cineole at both in vitro and in vivo conditions. During the in vitro hydroxycineole production, all CS-P450cin fusions showed a faster accumulation of 1,8-cineole than the non-fused individual enzyme case (Figure 3C). This might be a result from a reduced loss of 1,8-cineole in the proximal environment created by the fusion enzyme that facilitates the capture of 1,8-cineole and reduces the diffusion loss in solution. The increased availability of the terpene substrate could be a key factor that leads to higher production rates observed for P450cin in fusion proteins (Supplementary Table S3), which indicated that the non-fused P450cin could remain in a suboptimal activity in a two-step terpene oxidation. As many terpenes are usually hydrophobic and volatile compounds, the availability of terpene substrates is critical to reaching the maximum activity for a consequent P450 enzyme in the multi-step enzymatic oxidation of terpenoids. Additionally, the nature of CS dimerization (Karuppiah et al., 2017) could play an important role in enhancing the entire activity of a CS-P450cin fusion protein. Similar results were reported in a fusion of E. coli beta-galactosidase (LacZ) and the dimeric galactose dehydrogenase (GalDH) from Pseudomonas fluorescens, which showed improved enzyme activities when they are linked to each other (Ljungcrantz et al., 1989).
While spatial proximity explains the most advantages of a fusion protein relative to the corresponding non-fused enzymes, we observed fusion proteins with different linker lengths showed distinct activities. Surprisingly, the best fusion protein (G4) with 4-repeats of Gly-Ser-Gly linker from the in vitro conditions did not show the best production level under the in vivo conditions (Figure 3C, Figure 4C). This indicated that the optimal linker length for a fusion protein may vary from the in vitro and in vivo conditions because of their inconsistent reaction conditions, such as the ratio of P450cin and redox partners. This may also suggest that an optimal linker length plays a key role in selecting the best activity of a fusion enzyme. Similar observations have also been reported previously. For example, a fusion of Marinobacter aquaeolei P450 (CYP153) and a CYP116B reductase showed 67% improvement of activity by adding two extra amino acids in the linker (Hoffmann et al., 2016), and in another example, a ten amino-acid linker was found to present the best activity in the fusion of P450cin with its native flavodoxin (CinC) (Belsare et al., 2014).
To experimentally reveal the difference caused by the linker length, we pursued the structural analysis for G3 and G4 fusions which are selected with the best activities at the in vitro conditions. The SEC-SAXS data and modeling results suggested they showed different structural rigidities for the G4 fusion protein with a 12-aa linker and is less constrained, relative to the G3 fusion containing a 9-aa linker (Table 2, Figure 5D). This difference brought by the linker length indicated its importance in maintaining the rigidity of a fusion complex as well as reaching the best activity of a fusion protein. In addition to the linker length, the different ratios of P450 and redox partners could change the P450 conversion in a wide range (Hawkes et al., 2010; Khatri et al., 2017). Therefore, precision modulation of the in vivo P450-redox ratio presents a promising strategy to improve the P450 conversion yield for microbial production systems (Li et al., 2020; Schiffer et al., 2015).
Besides, the orientation of active sites in a fusion protein is another critical factor to achieve improved catalysis over the non-fused enzymes. In the assessment of the enzyme orientation in a fusion protein, we found reversing the order of CS and P450cin in the fusion protein dramatically decreased the catalytic efficiency (Table 3), and even did not show significant improvement of the activity over the non-fusion control. Given that the key interface residues R102 and R346 (Madrona et al., 2014) of P450cin for the constitution of P450cin-Cdx were intact for both CS-P450cin and P450cin-CS fusions (Supplementary Figure S3), we think this inefficient fusion construction might be more likely attributed to their relative position of the active sites in two domains. As the structural modeling predicted, the two active sites of CS and P450cin showed an unfavorable orientation in the P450cin-CS fusion protein, which may increase the difficulty for 1,8-cineole to access the binding sites of P450cin (Supplementary Figure S3). We also observed similar results in another negative example that implemented the enzyme fusion strategy in perillyl alcohol production by the hydroxylation of limonene (Supplementary Information). In this case, structural modeling predicted unfavorable orientation of two active sites for limonene synthase (LS) and CYP153A6 in the built fusion protein (Supplementary Figure S5B), and the experimental results also did not show any improvement in perillyl alcohol production in five fusion proteins engineered with different linker lengths (Supplementary Figure S5C).
The SAXS structural analysis suggested that the linker length can change the structural flexibility of domains in a fusion protein, but it may not easily change the orientation of enzyme active sites in a fusion complex due to the associated rigidity. Thus, for those fusion proteins showing an unfavorable orientation between two active sites, the loop structure at the terminus of the first enzyme might be a key to overcome the native rigidity of two domains and to facilitate the favorable orientations in the fusion. In a comparison of positive and negative examples we studied, we observed that each of two negative examples (LS-CYP153A6 and P450cin-CS fusions) shows a relatively short C-terminal loop of the first enzyme (i.e. LS has a 4-aa loop and P450cin has a 7-aa loop), while the first enzymes of positive examples (CS-P450cin and EizS-CYP170A1 fusions) have long C-terminal loops (i.e. CS has a 19-aa loop and EizS has a 21-aa loop) (Supplementary Figure S6). Therefore, the engineering of terminal loops could be a potential target to design fusion protein more adequately.
While engineering of fusion proteins shows a feasible approach for desirable enzymatic characteristics, it is still challenging to achieve the optimal activities. For example, it is difficult to precisely control the distance or orientation of enzymatic modules in a fusion protein. Also, the correct folding of a large multidomain protein could be difficult. Many P450 enzymes from plants are membrane-bound and have shown poor soluble expression in the bacterial hosts such as E. coli (Moser and Pichler, 2019). Thus, more consideration would be needed when engineering a fusion for eukaryotic terpene synthases and the membrane bound P450s in heterologous hosts. For example, truncating plastidial targeting sequence was found to improve the heterologous expression and catalytic efficiency of (4S)-limonene synthase from Mentha spicata (spearmint) in E. coli (Hyatt et al., 2007; Williams et al., 1998). Also, modifying N-terminal sequences of cytochrome P450 (CYP71D18, M. spicata) increased P450 expression as well as limonene hydroxylation rate in E. coli and yeast (Haudenschild et al., 2000).
The successful construction of a fusion enzyme relies on many factors, including domain order, inter-domain distance, domain orientation, and linker properties (Yu et al., 2015). Although several modeling programs have been reported to facilitate the designing of linkers for fusion protein engineering (Crasto and Feng, 2000; Liu et al., 2015), an in-depth structural analysis could be more important to design the optimal linker and the enzyme orientations to build an enzyme fusion with improved catalysis. Based on our experimental results and structural modeling analysis for fusion enzymes with both positive and negative results, we found the key factors in designing an efficient fusion protein include linker length, enzyme orientation, and potentially the terminal loops of each domain. Therefore, the combination of these factors guided by structural modeling would be an effective strategy to design and optimize the activity of a fusion enzyme.
On the other hand, the P450 bioconversion of terpenoids to oxidized products observed in our studies is still low. This could result from the relatively low percentage of active P450 as observed in the P450cin example (Supplementary Table S4). In this case, it is noted that the engineered fusion proteins could change and lower the functional expression of the active portion of P450cin. Compared with the non-fusion P450cin, most fusions (G2-G5) showed a lower percentage of the active P450cin portion despite higher productivities for hydroxycineole. Further optimization of the P450 enzymes and the in vivo microbial production system is necessary to improve the conversion yield, such as increasing the active P450 portion, identifying more efficient redox partners and the ratio of P450 and redox partners (Hawkes et al., 2010; Kimmich et al., 2007), engineering fusions of redox partners for enhanced electron transfer (Bakkes et al., 2017, 2015), optimizing the metabolic pathway for terpene substrate overproduction and co-factor balance, developing in situ extraction reagent (e.g. resins) to facilitate the collection of oxidized products (Alonso-Gutierrez et al., 2013).
5. Conclusions
The functionalization of terpene molecules using cytochrome P450 enzymes presents opportunities to produce various bioproducts that are frequently more value-added than the original terpene itself. In this study, we developed a strategy to improve the terpene hydroxylation efficiency by linking terpene synthase and P450 enzyme and facilitating the accessibility of terpene molecules to P450 enzymes in heterologous microbial systems. We demonstrated this strategy for monoterpene 1,8-cineole hydroxylation and achieved more than a 5-fold increase in hydroxycineole production using fusion proteins in vitro and 3-fold in vivo over the control using individual enzymes. Structural characterization of these enzyme fusions revealed that the length of the linker affects the flexibility, which eventually affects the catalytic activity, of the fusion enzymes. We also applied the enzyme fusion strategy to the oxidation of a sesquiterpene epi-isozizaene, in which up to 90-fold improvement in albaflavenol production was achieved by the fusion enzyme when the overlay was used. Our results suggested that engineering of fusion enzymes between terpene synthase and P450 shows a simple and efficient strategy toward the heterologous production of oxidized terpenes. Additionally, the structure-based prediction and evaluation would guide the design of fusion proteins with improved catalysis.
Supplementary Material
Acknowledgments
This work was part of the DOE Joint BioEnergy Institute (http://www.jbei.org) supported by the US Department of Energy, Office of Science, Office of Biological and Environmental Research, through Contract DE-AC0205CH11231 between Lawrence Berkeley National Laboratory and the US Department of Energy. SAXS data was collected at the SIBYLS beamline in the Advanced Light Source, which is supported by DOE BES, by the DOE OBER IDAT program, by NIH ALS-ENABLE (P30 GM124169) and by NIH S10OD018483. We would also like to acknowledge the help of beamline staff, Daniel Rosenberg and Michal Hammel.
Footnotes
Competing interests
TSL has a financial interest in Maple Bio.
References
- Aaron JA, Lin X, Cane DE, Christianson DW, 2010. Structure of epi-isozizaene synthase from Streptomyces coelicolor A3(2), a platform for new terpenoid cyclization templates,. Biochemistry 49, 1787–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aharoni A, Jongsma MA, Bouwmeester HJ, 2005. Volatile science? Metabolic engineering of terpenoids in plants. Trends Plant Sci. 10, 594–602. [DOI] [PubMed] [Google Scholar]
- Alonso-Gutierrez J, Chan R, Batth TS, Adams PD, Keasling JD, Petzold CJ, Lee TS, 2013. Metabolic engineering of Escherichia coli for limonene and perillyl alcohol production. Metab. Eng 19, 33–41. [DOI] [PubMed] [Google Scholar]
- Avalos JL, Fink GR, Stephanopoulos G, 2013. Compartmentalization of metabolic pathways in yeast mitochondria improves the production of branched-chain alcohols. Nat. Biotechnol 31, 335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakkes PJ, Biemann S, Bokel A, Eickholt M, Girhard M, Urlacher VB, 2015. Design and improvement of artificial redox modules by molecular fusion of flavodoxin and flavodoxin reductase from Escherichia coli. Sci. Rep 5, 12158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakkes PJ, Riehm JL, Sagadin T, Rühlmann A, Schubert P, Biemann S, Girhard M, Hutter MC, Bernhardt R, Urlacher VB, 2017. Engineering of versatile redox partner fusions that support monooxygenase activity of functionally diverse cytochrome P450s. Sci. Rep 7, 9570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belsare KD, Ruff AJ, Martinez R, Shivange AV, Mundhada H, Holtmann D, Schrader J, Schwaneberg U, 2014. P-LinK: A method for generating multicomponent cytochrome P450 fusions with variable linker length. Biotechniques 57, 13–20. [DOI] [PubMed] [Google Scholar]
- Bergman A, Siewers V, 2016. Chapter 7 - Metabolic engineering strategies to convert carbohydrates to aviation range hydrocarbons, in: Chuck CJBT-B. for A. (Ed.), . Academic Press, pp. 151–190. [Google Scholar]
- Bernhardt R, 2006. Cytochromes P450 as versatile biocatalysts. J. Biotechnol 124, 128–145. [DOI] [PubMed] [Google Scholar]
- Brosey CA, Yan C, Tsutakawa SE, Heller WT, Rambo RP, Tainer JA, Ivanov I, Chazin WJ, 2013. A new structural framework for integrating replication protein A into DNA processing machinery. Nucleic Acids Res. 41, 2313–2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang MCY, Eachus RA, Trieu W, Ro D-K, Keasling JD, 2007. Engineering Escherichia coli for production of functionalized terpenoids using plant P450s. Nat. Chem. Biol 3, 274. [DOI] [PubMed] [Google Scholar]
- Cheng A-X, Lou Y-G, Mao Y-B, Lu S, Wang L-J, Chen X-Y, 2007. Plant terpenoids: biosynthesis and ecological functions. J. Integr. Plant Biol 49, 179–186. [Google Scholar]
- Classen S, Hura GL, Holton JM, Rambo RP, Rodic I, McGuire PJ, Dyer K, Hammel M, Meigs G, Frankel KA, Tainer JA, 2013. Implementation and performance of SIBYLS: a dual endstation small-angle X-ray scattering and macromolecular crystallography beamline at the Advanced Light Source. J. Appl. Crystallogr 46, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrado RJ, Varner JD, DeLisa MP, 2008. Engineering the spatial organization of metabolic enzymes: mimicking nature’s synergy. Curr. Opin. Biotechnol 19, 492–499. [DOI] [PubMed] [Google Scholar]
- Crasto CJ, Feng J, 2000. LINKER: a program to generate linker sequences for fusion proteins. Protein Eng. Des. Sel 13, 309–312. [DOI] [PubMed] [Google Scholar]
- Dueber JE, Wu GC, Malmirchegini GR, Moon TS, Petzold CJ, Ullal AV, Prather KLJ, Keasling JD, 2009. Synthetic protein scaffolds provide modular control over metabolic flux. Nat. Biotechnol 27, 753–759. [DOI] [PubMed] [Google Scholar]
- Dyer KN, Hammel M, Rambo RP, Tsutakawa SE, Rodic I, Classen S, Tainer JA, Hura GL, 2014. High-Throughput SAXS for the Characterization of Biomolecules in Solution: A Practical Approach, in: Chen YW. (Ed.), Structural Genomics: General Applications. Humana Press, Totowa, NJ, pp. 245–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiser A, Do RKG, Šali A, 2000. Modeling of loops in protein structures. Protein Sci. 9, 1753–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke D, Petoukhov MV, Konarev PV, Panjkovich A, Tuukkanen A, Mertens HDT, Kikhney AG, Hajizadeh NR, Franklin JM, Jeffries CM, Svergun DI, 2017. {\it ATSAS 2.8}: a comprehensive data analysis suite for small-angle scattering from macromolecular solutions. J. Appl. Crystallogr 50, 1212–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Gao F, Liu D, Zhang H, Nie X, Yang C, 2016. Engineering the methylerythritol phosphate pathway in cyanobacteria for photosynthetic isoprene production from CO2. Energy Environ. Sci 9, 1400–1411. [Google Scholar]
- Guo H, Yang Y, Xue F, Zhang H, Huang T, Liu W, Liu H, Zhang F, Yang M, Liu C, Lu H, Zhang Y, Ma L, 2017. Effect of flexible linker length on the activity of fusion protein 4-coumaroyl-CoA ligase::stilbene synthase. Mol. Biosyst 13, 598–606. [DOI] [PubMed] [Google Scholar]
- Haudenschild C, Schalk MFAU - Karp F, Karp FFAU - Croteau R, Croteau R, 2000. Functional expression of regiospecific cytochrome P450 limonene hydroxylases from mint (Mentha spp.) in Escherichia coli and saccharomyces cerevisiae. Archi. Biochem. Biophys 379, 127–136. [DOI] [PubMed] [Google Scholar]
- Hawkes DB, Adams GW, Burlingame AL, Ortiz de Montellano PR, De Voss JJ, 2002. Cytochrome P450cin (CYP176A), isolation, expression, and characterization . J. Biol. Chem 277, 27725–27732. [DOI] [PubMed] [Google Scholar]
- Hawkes DB, Slessor KE, Bernhardt PV, De Voss JJ, 2010. Cloning, expression and purification of cindoxin, an unusual Fmn-containing cytochrome P450 redox partner. ChemBioChem 11, 1107–1114. [DOI] [PubMed] [Google Scholar]
- Hoffmann SM, Weissenborn MJ, Gricman Ł, Notonier S, Pleiss J, Hauer B, 2016. The impact of linker length on P450 fusion constructs: activity, stability and coupling. ChemCatChem 8, 1591–1597. [Google Scholar]
- Hura GL, Menon AL, Hammel M, Rambo RP, Poole II FL, Tsutakawa SE, Jenney FE Jr, Classen S, Frankel KA, Hopkins RC, Yang S, Scott JW, Dillard BD, Adams MWW, Tainer JA, 2009. Robust, high-throughput solution structural analyses by small angle X-ray scattering (SAXS). Nat. Methods 6, 606–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyatt DC, Youn B, Zhao Y, Santhamma B, Coates RM, Croteau RB, Kang C, 2007. Structure of limonene synthase, a simple model for terpenoid cyclase catalysis. Proc. Natl. Acad. Sci 104, 5360 LP–5365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins CM, Waterman MR, 1994. Flavodoxin and NADPH-flavodoxin reductase from Escherichia coli support bovine cytochrome P450c17 hydroxylase activities. J. Biol. Chem 269, 27401–27408. [PubMed] [Google Scholar]
- Kang A, George KW, Wang G, Baidoo E, Keasling JD, Lee TS, 2016. Isopentenyl diphosphate (IPP)-bypass mevalonate pathways for isopentenol production. Metab. Eng 34, 25–35. [DOI] [PubMed] [Google Scholar]
- Karuppiah V, Ranaghan KE, Leferink NGH, Johannissen LO, Shanmugam M, Ní Cheallaigh A, Bennett NJ, Kearsey LJ, Takano E, Gardiner JM, van der Kamp MW, Hay S, Mulholland AJ, Leys D, Scrutton NS, 2017. Structural basis of catalysis in the bacterial monoterpene synthases linalool synthase and 1,8-cineole synthase. ACS Catal. 7, 6268–6282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khatri Y, Schifrin A, Bernhardt R, 2017. Investigating the effect of available redox protein ratios for the conversion of a steroid by a myxobacterial CYP260A1. FEBS Lett. 591, 1126–1140. [DOI] [PubMed] [Google Scholar]
- Kimmich N, Das A, Sevrioukova I, Meharenna Y, Sligar SG, Poulos TL, 2007. Electron Transfer between Cytochrome P450cin and Its FMN-containing Redox Partner, Cindoxin. J. Biol. Chem 282, 27006–27011. [DOI] [PubMed] [Google Scholar]
- Klocke JA, Darlington MV, Balandrin MF, 1987. 1,8-Cineole (Eucalyptol), a mosquito feeding and ovipositional repellent from volatile oil ofHemizonia fitchii (Asteraceae). J. Chem. Ecol 13, 2131–2141. [DOI] [PubMed] [Google Scholar]
- Kourtz L, Dillon K, Daughtry S, Madison LL, Peoples O, Snell KD, 2005. A novel thiolase–reductase gene fusion promotes the production of polyhydroxybutyrate in Arabidopsis. Plant Biotechnol. J 3, 435–447. [DOI] [PubMed] [Google Scholar]
- Leita BA, Warden AC, Burke N, O’Shea MS, Trimm D, 2010. Production of p-cymene and hydrogen from a bio-renewable feedstock–1,8-cineole (eucalyptus oil). Green Chem. 12, 70–76. [Google Scholar]
- Li S, Du L, Bernhardt R, 2020. Redox Partners: Function Modulators of Bacterial P450 Enzymes. Trends Microbiol. 28, 445–454. [DOI] [PubMed] [Google Scholar]
- Liu C-L, Tian T, Alonso-Gutierrez J, Garabedian B, Wang S, Baidoo EEK, Benites V, Chen Y, Petzold CJ, Adams PD, Keasling JD, Tan T, Lee TS, 2018. Renewable production of high density jet fuel precursor sesquiterpenes from Escherichia coli. Biotechnol. Biofuels 11, 285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Chin JX, Lee D-Y, 2015. SynLinker: an integrated system for designing linkers and synthetic fusion proteins. Bioinformatics 31, 3700–3702. [DOI] [PubMed] [Google Scholar]
- Ljungcrantz P, Carlsson H, Mansson MO, Buckel P, Mosbach K, Buelow L, 1989. Construction of an artificial bifunctional enzyme, .beta.-galactosidase/galactose dehydrogenase, exhibiting efficient galactose channeling. Biochemistry 28, 8786–8792. [DOI] [PubMed] [Google Scholar]
- Madrona Y, Hollingsworth SA, Tripathi S, Fields JB, Rwigema J-CN, Tobias DJ, Poulos TL, 2014. Crystal structure of cindoxin, the P450cin redox partner. Biochemistry 53, 1435–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meharenna YT, Li H, Hawkes DB, Pearson AG, De Voss J, Poulos TL, 2004. Crystal structure of P450cin in a complex with its substrate, 1,8-cineole, a close structural homologue to d-camphor, the substrate for P450cam,. Biochemistry 43, 9487–9494. [DOI] [PubMed] [Google Scholar]
- Mendez-Perez D, Alonso-Gutierrez J, Hu Q, Molinas M, Baidoo EEK, Wang G, Chan LJG, Adams PD, Petzold CJ, Keasling JD, Lee TS, 2017. Production of jet fuel precursor monoterpenoids from engineered Escherichia coli. Biotechnol. Bioeng 114, 1703–1712. [DOI] [PubMed] [Google Scholar]
- Meynial Salles I, Forchhammer N, Croux C, Girbal L, Soucaille P, 2007. Evolution of a Saccharomyces cerevisiae metabolic pathway in Escherichia coli. Metab. Eng 9, 152–159. [DOI] [PubMed] [Google Scholar]
- Moser S, Pichler H, 2019. Identifying and engineering the ideal microbial terpenoid production host. Appl. Microbiol. Biotechnol 103, 5501–5516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pateraki I, Heskes AM, Hamberger B, 2015. Cytochromes P450 for terpene functionalisation and metabolic engineering BT - biotechnology of isoprenoids, in: Schrader J, Bohlmann J (Eds.), . Springer International Publishing, Cham, pp. 107–139. [DOI] [PubMed] [Google Scholar]
- Pelikan M, Hura GL, Hammel M, 2009. Structure and flexibility within proteins as identified through small angle X-ray scattering. Gen. Physiol. Biophys 28, 174–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham SQ, Pompidor G, Liu J, Li X-D, Li Z, 2012. Evolving P450pyr hydroxylase for highly enantioselective hydroxylation at non-activated carbon atom. Chem. Commun 48, 4618–4620. [DOI] [PubMed] [Google Scholar]
- Putnam CD, Hammel M, Hura GL, Tainer JA, 2007. X-ray solution scattering (SAXS) combined with crystallography and computation: defining accurate macromolecular structures, conformations and assemblies in solution. Q. Rev. Biophys 40, 191–285. [DOI] [PubMed] [Google Scholar]
- Rambo RP, Tainer JA, 2013. Accurate assessment of mass, models and resolution by small-angle scattering. Nature 496, 477–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambo RP, Tainer JA, 2011. Characterizing flexible and intrinsically unstructured biological macromolecules by SAS using the Porod-Debye law. Biopolymers 95, 559–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redding-Johanson AM, Batth TS, Chan R, Krupa R, Szmidt HL, Adams PD, Keasling JD, Soon Lee T, Mukhopadhyay A, Petzold CJ, 2011. Targeted proteomics for metabolic pathway optimization: Application to terpene production. Metab. Eng 13, 194–203. [DOI] [PubMed] [Google Scholar]
- Renault H, Bassard J-E, Hamberger B, Werck-Reichhart D, 2014. Cytochrome P450-mediated metabolic engineering: current progress and future challenges. Curr. Opin. Plant Biol 19, 27–34. [DOI] [PubMed] [Google Scholar]
- Roy A, Yang J, Zhang Y, 2012. COFACTOR: an accurate comparative algorithm for structure-based protein function annotation. Nucleic Acids Res. 40, W471–W477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarria S, Wong B, Martín HG, Keasling JD, Peralta-Yahya P, 2014. Microbial synthesis of pinene. ACS Synth. Biol 3, 466–475. [DOI] [PubMed] [Google Scholar]
- Schiffer L, Anderko S, Hobler A, Hannemann F, Kagawa N, Bernhardt R, 2015. A recombinant CYP11B1 dependent Escherichia coli biocatalyst for selective cortisol production and optimization towards a preparative scale. Microb. Cell Fact 14, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneidman-Duhovny D, Hammel M, Tainer JA, Sali A, 2016. FoXS, FoXSDock and MultiFoXS: Single-state and multi-state structural modeling of proteins and their complexes based on SAXS profiles. Nucleic Acids Res. 44, W424–W429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneidman-Duhovny D, Hammel M, Tainer JA, Sali A, 2013. Accurate SAXS profile computation and its assessment by contrast variation experiments. Biophys. J 105, 962–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw JJ, Berbasova T, Sasaki T, Jefferson-George K, Spakowicz DJ, Dunican BF, Portero CE, Narváez-Trujillo A, Strobel SA, 2015. Identification of a fungal 1,8-cineole synthase from hypoxylon sp. with specificity determinants in common with the plant synthases . J. Biol. Chem 290, 8511–8526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slessor KE, Hawkes DB, Farlow A, Pearson AG, Stok JE, De Voss JJ, 2012. An in vivo cytochrome P450cin (CYP176A1) catalytic system for metabolite production. J. Mol. Catal. B Enzym 79, 15–20. [Google Scholar]
- Urlacher VB, Girhard M, 2019. Cytochrome P450 monooxygenases in biotechnology and synthetic biology. Trends Biotechnol. 37, 882–897. [DOI] [PubMed] [Google Scholar]
- van Beilen JB, Holtackers R, Lüscher D, Bauer U, Witholt B, Duetz WA, 2005. Biocatalytic production of perillyl alcohol from limonene by using a novel Mycobacterium sp. cytochrome P450 alkane hydroxylase expressed in Pseudomonas putida. Appl. Environ. Microbiol 71, 1737 LP–1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams DC, McGarvey DJ, Katahira EJ, Croteau R, 1998. Truncation of limonene synthase preprotein provides a fully active ‘pseudomature’ form of this monoterpene cyclase and reveals the function of the amino-terminal arginine pair. Biochemistry 37, 12213–12220. [DOI] [PubMed] [Google Scholar]
- Yang J, Zhang Y, 2015. I-TASSER server: new development for protein structure and function predictions. Nucleic Acids Res. 43, W174–W181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Li T, Tang K, Zhou X, Lu M, Ounkham WL, Spain SM, Frost BJ, Lin H, 2017. Highly efficient conversion of terpenoid biomass to jet-fuel range cycloalkanes in a biphasic tandem catalytic process. Green Chem. 19, 3566–3573. [Google Scholar]
- Yu K, Liu C, Kim B-G, Lee D-Y, 2015. Synthetic fusion protein design and applications. Biotechnol. Adv 33, 155–164. [DOI] [PubMed] [Google Scholar]
- Zhang Y, 2009. I-TASSER: Fully automated protein structure prediction in CASP8. Proteins Struct. Funct. Bioinforma. 77, 100–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Lei L, Vassylyev DG, Lin X, Cane DE, Kelly SL, Yuan H, Lamb DC, Waterman MR, 2009. Crystal structure of albaflavenone monooxygenase containing a moonlighting terpene synthase active site. J. Biol. Chem 284, 36711–36719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Lin X, Lei L, Lamb DC, Kelly SL, Waterman MR, Cane DE, 2008. Biosynthesis of the sesquiterpene antibiotic albaflavenone in Streptomyces coelicolor A3(2). J. Biol. Chem 283, 8183–8189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo R, Zhang Y, Jiang C, Hackett JC, Loria R, Bruner SD, Ding Y, 2017. Engineered P450 biocatalysts show improved activity and regio-promiscuity in aromatic nitration. Sci. Rep 7, 842. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.