

Abstract
IL-17A and IL-22 derived from Th17 cells play a significant role in mucosal immunity and inflammation. TGF-β and IL-6 promote Th17 differentiation, however, these cytokines have multiple targets. The identification and screening of additional molecules that regulate IL-17A and IL-22 responses in certain inflammatory conditions is of great clinical significance. Here, we show that CDDO-Im, a specific Nrf2 activator, promotes IL-17A and IL-22 responses in murine Th17 cells. In contrast, CDDO-Im inhibits IL-17A response in Multiple Sclerosis (MS) patient-derived PBMCs. However, Nrf2 specifically regulates IL-22 response in vivo. Nrf2 acts through the regulation of Antioxidant Response Element (ARE) binding motifs in target genes to induce or repress transcription. Promoter analysis revealed that Il17a, Rorc and Ahr genes have several ARE motifs. We showed that Nrf2 bind to ARE repressor (ARE-R2) of Rorc and inhibits Rorc-dependent IL-17A transactivation. The luciferase reporter assay data showed that CDDO-Im regulated Ahr promoter activity. ChIP-qPCR data showed that Nrf2 bind to ARE of AhR. Finally, we confirmed that the CDDO-Im-mediated induction of IL-22 production in CD4+ T cells was abrogated in CD4-specific Ahr knockout mice (AhrCD4). CH-223191, a specific AhR antagonist, inhibits CDDO-Im-induced IL-22 production in CD4+ T cells, which further confirmed the AhR-dependent regulation. Collectively, our data showed that Nrf2 via AhR pathways regulated IL-22 response in CD4+ T cells.
Introduction
IL-17A and IL-22 derived from Th17 (α,β CD4+ T cells producing IL-17A, IL-17F and IL-22) and Type 3 innate lymphoid cells (ILC3) have been shown to exert both protective and inflammatory functions. Exacerbated IL-17A response is associated with pathophysiology of many diseases including cystic fibrosis (CF), psoriasis, multiple sclerosis (MS), rheumatoid arthritis (RA) and allergic asthma (1–6). Neutralizing IL-17A or blocking Th17 cell differentiation has been shown to have a beneficial role in multiple mouse models of autoimmune inflammation and psoriasis (7–10). Additionally, preclinical neutralization of IL-17A in cystic fibrosis transmembrane receptor knockout (Cftr−/−) mice showed reduced neutrophil recruitment and airway inflammation (1). In contrast, IL-17A is critically important for mucosal immunity against Gram-negative bacteria and fungus (11–13). We also reported that IL-17A signaling in intestine is important for the maintenance of the gut microbiota including segmented filamentous bacteria (SFB) colonization (14). IL-22, another Th17 and ILC3-derived cytokine, has been shown to be critical for promoting tissue protective and regenerative responses in various organs (15–20). Thus, identification and evaluation of molecules that selectively regulate IL-17A and IL-22 responses in certain inflammatory conditions is of great clinical significance.
Murine Th17 cell differentiation (in vivo and in vitro) is influenced by cytokines including IL-6, TGF-β, IL-23 and IL-1β (21). Furthermore, IL-6 is reported to induce Th17 differentiation by upregulating the expression of RORγt, the master transcription factor of Th17 lineage, and IL-23R by inhibiting the expression of Foxp3 (22, 23). Additionally, Aryl hydrocarbon receptor (AhR) has been shown to regulate IL-22 and IL-17A expression in Th17 cells and ILC3 (24–27). However, Th17 cells can still differentiate in the absence of AhR (24). Moreover, targeting IL-17A or IL-23-mediated Th17 differentiation pathway is efficacious in several autoimmune diseases. However, blocking of IL-23 also has an impact on additional regulatory pathways mediated by IL-22. The inhibition of RORγt by small molecule inhibitors has been shown to be effective in limiting IL-17A responses in multiple inflammatory conditions (28–30). Thus, screening of additional molecules that regulate Th17 differentiation remains an attractive target for potential therapeutic intervention.
Drugs or Xenobiotics are metabolized by a series of metabolizing enzymes, categorized into phase I to III (31). AhR and Nuclear factor (erythroid-derived 2)-like 2 (Nrf2) regulate activity of phase 1 (cytochrome 450 monoxygenase, CYP, etc.) and phase II (GCLC, HO-1, NQO1, SOD3, etc.) enzymes, respectively (31). Nrf2 is a key regulator of the cellular redox state in mammalian cells (31, 32). Nrf2-mediated expression of GCLC, HO-1, NQO1, SOD3 exert cytoprotective, antioxidant, and anti-inflammatory effects in target tissues (32, 33). Furthermore, Nrf2 acts through the regulation of Antioxidant Response Element (ARE) binding motifs in target genes. The bindings between Nrf2 and AREs are predicted to induce transcription or repress transcription (ARE-R) of downstream proteins. Several studies showed that Nrf2 play an important role in suppressing Th17 immune responses in vivo (34, 35). Furthermore, T cell-specific Nrf2 activation protected mice from ischemia reperfusion-induced acute kidney injury which was correlated with reduced intracellular levels of IL-17A, IFNγ and TNFα (36). Despite these promising results, it remains unclear how Nrf2 suppresses Th17 cell-mediated inflammatory IL-17A responses. Furthermore, it is not known whether Nrf2 regulates IL-22 responses in Th17 cells. It is also possible that enhanced susceptibility of Nrf2−/− mice in multiple models of tissue inflammation/injury is due to dysregulation of Th17-dependent IL-17A and IL-22 responses.
We found that Nrf2 was highly expressed in Th17 cells. Il17a, Rorc and Ahr genes have several ARE motifs. Using Nrf2−/− and AhrCD4 knockout mice, we showed that Nrf2 activation induced both IL-17A and IL-22 responses in CD4+ T cell polarized to Th17 cells. However, Nrf2 activation inhibits IL-17A responses in RRMS patient-derived PBMC. In vivo, IL-22 but not IL-17A response was regulated by Nrf2 signaling. Nrf2 respectively binds to ARE-R2 motif of Rorc and ARE motif of Ahr in CD4+ T cells. Furthermore, our data suggested that Nrf2-induced IL-22 production was dependent on AhR. In summary, we showed that Nrf2 activation impacted IL-17A and IL-22 responses in CD4+ T cells.
Materials and Methods
Mice:
WT, IL-17A-GFP and Nrf2−/− mice (The Jackson Laboratory) in C57BL/6 background were housed in specific pathogen-free conditions at Children’s Hospital of Pittsburgh, Pittsburgh, PA and Stony Brook University, Stony Brook, NY. Ahrfl/fl and CD4-cre mice were purchased from The Jackson Laboratory to generate CD4+ T cell-specific Ahr conditional knockout mice. Ahrfl/fl and AhrCD4 mice were housed in specific pathogen-free conditions at Washington University School of Medicine in St. Louis. Spleens from Ahrfl/fl and AhrCD4 mice were shipped to Stony Brook University for Th17 differentiation assays. All of the animal studies were conducted with the approval of the University of Pittsburgh Institutional Animal Care and Use Committee and Stony Brook University Institutional Animal Care and Use Committee.
Ethics statement:
De-identified RRMS patient PBMCs were collected after obtaining informed consent and approved by an Institutional Review Board at Stony Brook University (IRB 1084248).
Animal treatments:
For Con A treatment, WT and Nrf2−/− mice were injected intravenously with Con A (Sigma Aldrich) at 10 μg/g body weight. Mice were euthanized at 2 hours after Con A administration. Splenocytes were isolated and processed for flow cytometry. Blood was harvested and centrifuged at 2000xg, 4 °C for 15 minutes. Then serum was used for ELISA.
For the adoptive transfer model, splenic CD4+ T cells from WT (CD45.1) and Nrf2−/− (CD45.2) mice were isolated using STEMCELL Technologies CD4+ T cells isolation kit, mixed (1:1), and transferred (106 cells/mice) intravenously into Rag1−/− mice. 50 μg Ovalbumin (Sigma Aldrich) and low dose (50 ng) LPS (Sigma Aldrich) were given on days 15, 21, 28 and 29 to isoflurane-anesthetized mice in sterile PBS (50 μl) by oropharyngeal aspiration-tongue pull technique as enumerated in Figure 3A. Lungs were harvested one day after last Ova+LPS administration and used for lymphocyte isolation and flow cytometry.
Figure 3: IL-17A and IL-22 responses in WT and Nrf2−/− mice in vivo:
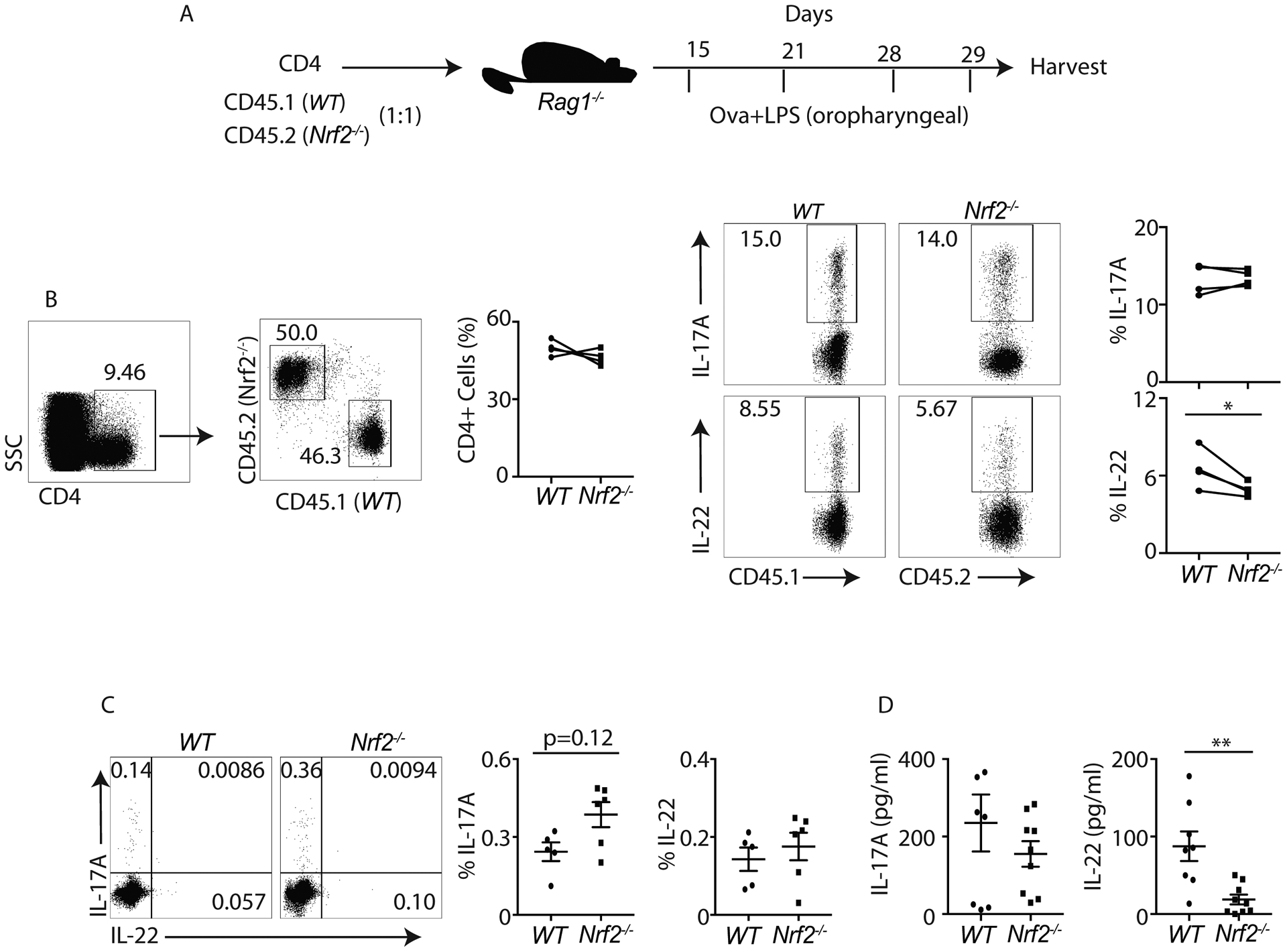
A) Schematic diagram shows OVA+LPS experimental strategy.
B) One day after last OVA+LPS administration, lung-derived lymphocytes from WT and Nrf2−/− mice were isolated and restimulated with PMA+Ionomycin. IL-17A or IL-22 producing CD4+ T cells were analyzed by flow cytometry. Data show the percentage of CD4+ T cells producing IL-17A or IL-22.
C and D) At 2 hours post ConA administration, WT and Nrf2−/− splenocytes were harvested and restimulated with BD leukocyte activation cocktail (1:200). Flow cytometry was utilized to examine the frequency of IL-17A or IL-22 producing CD4+ T cells (gated on CD3+CD4+) (C). Serum IL-17A and IL-22 concentrations were quantified by ELISA (D).
Figure 3C and 3D was generated from 2 independent experiments. Each symbol indicates experiments from a separate animal. Data presented as mean ± SEM on relevant graphs. *P ≤ 0.05; **P ≤ 0.01 (Mann-Whitney test, Two-tailed).
For Citrobacter rodentium (ATCC, 51459) infection, the bacteria were cultured overnight. Il17-GFP mice were fasted for 6 hours and orally administrated with 2.5x109 CFU of C. rodentium. At 24 h post infection, mice were injected with 5 μmol/kg body weight of CDDO-Im or DMSO intraperitoneally. After 4 days, distal colon was harvested for qPCR.
Cell Isolation:
Lung lymphocyte isolation is as described (15). For splenocyte and lymph node lymphocyte isolation, spleens were harvested and dissociated in 3 mL of 1xPBS through 70 μm strainer. Cells were pelleted at 500xg, 4 °C for 5 minutes. The pellet was resuspended in 1mL of 1xPBS containing 2% FBS and processed for sorting with EasySep™ Mouse Naïve CD4+ T cell Isolation Kit (STEMCELL Technologies, cat#:19765).
ROS estimation:
Naïve CD4+ (CD4+CD62L+) T cells from WT and Nrf2−/− mice were isolated from spleens using EasySep™ Mouse Naïve CD4+ T cell Isolation Kit (STEMCELL Technologies, cat#:19765), and stimulated with anti-CD3 (2.5 μg/ml), anti-CD28 (2.0 μg/ml) in the presence or absence of IL-6 (20 ng/ml) + TGF-β (1 ng/ml) for 24 hours. Cellular ROS was quantified using a DCFDA kits (Abcam) as per manufactured instructions. Stained cells were analyzed for MFI using a Becton Dickinson (BD) LSR II flow cytometer. FACS data were analyzed with the BD FACSDiva software.
Th17 differentiation:
Naïve CD4+ (CD4+CD62L+) cells from WT, Nrf2−/−, Ahrfl/fl and AhrCD4 mice were isolated from spleens and lymph nodes using EasySep™ Mouse Naïve CD4+ T cell Isolation Kit (STEMCELL Technologies, cat#:19765), and polarized in culture medium (IMDM+10% FBS+100 U/mL penicillin+100 μg/mL streptomycin) with CellXVivo Mouse Th17 Cell Differentiation Kit (R&D system, cat#: CDK017) in presence of CDDO-Im (Tocris Bioscience), TEMPOL (Sigma Aldrich), CH-223191 (STEMCELL Technologies) or vehicle control (DMSO). Cell culture medium was harvested on day 1 or day 4 for ELISA.
Flow cytometry:
Lung lymphocytes, splenic lymphocytes or differentiated Th17 cells were re-stimulated with Leukocyte Activation Cocktail, with BD GolgiPlug™ (BD, cat#:550583, 1:500) for 4 hours. After stimulation, cells were stained with Live-dead (Invitrogen, cat#: L34957) and eBiosciences antibodies against CD45 (30-F11), CD3 (17A2), CD4 (RMA4–5) at 4 °C for 20 minutes. Then cells were fixed with Foxp3/Transcription Factor Staining Buffer Set (eBiosciences, cat#: 00-5523-00) at 4 °C for 30 minutes and stained intracellularly with eBiosciences antibodies against IL-17A (ebio17b7), IL-22 (IL22JOP), AhR (4MEJJ), RORγt (B2D) overnight. Stained lymphocytes were acquired using a BD LSR II flow cytometer. Flow cytometric data were analyzed with BD FACSDiva or FlowJo.
qPCR:
RNA was isolated using RNeasy Plus Micro Kit (Qiagen, cat#: 74034) per manufacturer’s instructions. Isolated RNA was reversely transcribed into cDNA using iScript™ Reverse Transcription Supermix (Bio-rad, cat#: 1708840). 5 μL of SsoAdvanced Universal Probes Supermix (Bio-rad, cat#: 1725284) or SsoAdvanced Universal SYBR Green Supermix (Bio-rad, cat#: 1725274) was mixed with 0.5 μL of primers/probes and 4.5 μL of cDNA. Qiagen QuantiTech mouse primers Nrf2 (Mm_Nfe2I2_1_SG), Nrf1 (Mm_Nrf1_1_SG), Nrf3 (Mm_Nfe2I3_1_SG) and Applied Biosystem mouse primer-probe for Il17a (Mm00439618_m1), Il22 (Mm00444241_m1), Hprt (Mm00446968_m1), Ahr (Mm0047893_m1), Sod1 (Mm01344232_g1), Sod2 (Mm00449726_m1), Sod3 (Mm01213380_g1), Nqo1 (Mm001253561_m1), Cybb (Mm01287743), Il17f (Mm00521423), Il21 (Mm00517640), Csf2 (Mm01290062), human primer-probe for IL17A (Hs00174383), IDT mouse primer-probe for Gapdh (FP–TCATCAACGGGAAGCCCATCAC and RP–AGACTCCACGACATACTCAGCACCG) or IDT human primer-probe for HPRT (Hs.PT.58v.45621572), NQO1 (Hs.PT.58.2697277) were used for qPCR. Gene expression was quantified and normalized to Hprt or Gapdh.
CHIP-qPCR:
There are 5 transcriptional variants of murine Rorc listed in the NCBI database. 3 of them were excluded because they are predicted sequences by computational models. The other 2 including NM_001293734.1 and NM_011281.3 are sequenced and cited in many papers. NM_001293734.1 contains the sequence of NM_011281.3. So NM_001293734.1 was used for putative ARE analysis. 3 putative AREs and 3 putative ARE-Rs were predicted in NM_001293734.1 (Supplemental Figure 3A). Naïve CD4+ T cells were isolated with EasySep™ Mouse Naïve CD4+ T cell Isolation Kit (STEMCELL Technologies, cat#:19765) and polarized to Th17 cells with CellXVivo Mouse Th17 Cell Differentiation Kit (R&D system, cat#: CDK017). At 4 days post stimulation, cells were harvested and processed for CHIP with SimpleChIP® Enzymatic Chromatin IP Kit (Cell Signaling Technology, cat#: 9003S) as per manufacturer’s instructions. Anti-Nrf2 antibody (Cell Signaling Technology, cat#: 12721, 1:200) was used to pull down the Nrf2 binding DNA. Target DNA fragments were quantified by qPCR with primers listed in Supplemental Table 1.
ELISA:
Mouse serum and cell culture medium of mouse Th17 cells or human RRMS PBMCs were processed for IL-17A and IL-22 ELISA. ELISA was carried out with mouse or human IL-17A and IL-22 ELISA kit (Biolegend) as per manufacturer’s instructions.The optical density (OD) was read at 450 nm and 570 nm with the spectrometer.
Ahr Luciferase assays:
C10 cells were stably transduced with a reporter vector containing three copies of the xenobiotic response element (XRE) and a firefly luciferase reporter (Promega, Madison WI). 50,000 cells were seeded overnight at 37 °C in a 96 well white, clear bottom Corning Costar plates. Cells were treated for 8 hours with different concentrations of CDDO-Im (Dr. Thomas W. Kensler laboratory, University of Pittsburgh, PA). Cell lysis and luciferase activity were measured using the Luciferase Assay System (Promega) as per manufacturer’s instructions.
Transfections and IL-17A luciferase reporter assays:
Mycoplasma free HEK293T cells (ATCC, CRL-3216) were transiently transfected with IL-17 promoter construct tagged with firefly luciferase reporter (Addgene) and AAV-CMV-Nrf2 (Addgene) using X-tremeGENE 9 DNA transfection reagent. Renilla luciferase reporter vector was co-transfected as an internal control. Luminescence was measured after 48 hours of transfection by Dual-Glo Luciferase Assay system (Promega; E2940). Firefly luciferase activity was normalized with Renilla luciferase activity and the result was represented as relative light units (RLU).
RRMS patient-derived PBMC stimulation:
RRMS patient-derived PBMCs were thawed at 37 °C. Cells were added to 10 mL of Harvest Medium (IMDM+10% FBS+100 U/mL penicillin+100 μg/mL streptomycin) and centrifuged at 350xg for 5 min to pellet the cells. Cells were resuspended in Harvest Medium containing Dynabeads Human T-Activator CD3/CD28 (Gibco, cat#: 11161D) and cultured under CDDO-Im or vehicle control treatment for 5 days. On Day 5, culture medium was harvested and proceeded for ELISA analysis. Cells were harvested in RLT buffer (Qiagen) for further qPCR analysis.
Results
Nrf2-regulated antioxidant gene expression is suppressed during Th17 differentiation
We first confirmed Nrf2 expression in naïve CD4+ T cells and T helper cell subsets. Our data indicated that the expression of Nrf2 as compared to Nrf1 and Nrf3 was substantially enriched in naïve CD4+ T cells (Figure 1A). We further tested the expression of Nrf2 in Th1, Th2 and Th17 cells. Interestingly, Nrf2 expression was found to be the highest in differentiated Th17 cells in vitro in comparison to Th1 and Th2 cells (Figure 1B). We observed that Th17 condition medium inhibited antioxidant gene Nqo1 but induced Ahr expression compared to TCR-activated CD4+ T cells on day 1, suggesting that Nrf2 may be inhibited under Th17 differentiation condition (Figure 1C). In line with these observations cellular reactive oxygen species (ROS), the Nrf2-regulated molecules (37), were higher in IL-6 + TGF-β (Th17 condition medium) stimulated cells (Figure 1D). Furthermore, ROS levels were higher in Nrf2−/− mice at 1 day post TCR activation (Figure 1E). To further understand the role of Nrf2 in Th17 cell differentiation and functions, we used Nrf2−/− mice, and CDDO-Im, a potent activator of Nrf2 signaling (34, 38–41). Naïve CD4+ T cells were stimulated with plate bound anti-CD3 and anti-CD28 and treated with or without CDDO-Im (0M and 10−8M) under Th17 polarization condition for 24 hours. Consistent with our observation that Nqo1 was inhibited during Th17 differentiation (Figure 1C), no difference in Nqo1 expression was observed between WT and Nrf2−/− CD4+ T cells (Figure 1F). Indeed, CDDO-Im stimulation led to the Nrf2-dependent increase of Nqo1 expression in CD4+ T cells compared to vehicle control (Figure 1F and supplemental Figure 1A). Collectively, our data suggest that Nrf2-mediated antioxidant regulatory pathway in CD4+ T cells was suppressed while Ahr expression was promoted under Th17 polarization condition.
Figure 1: Nrf2-dependent antioxidant genes expression is suppressed in Th17 cells.
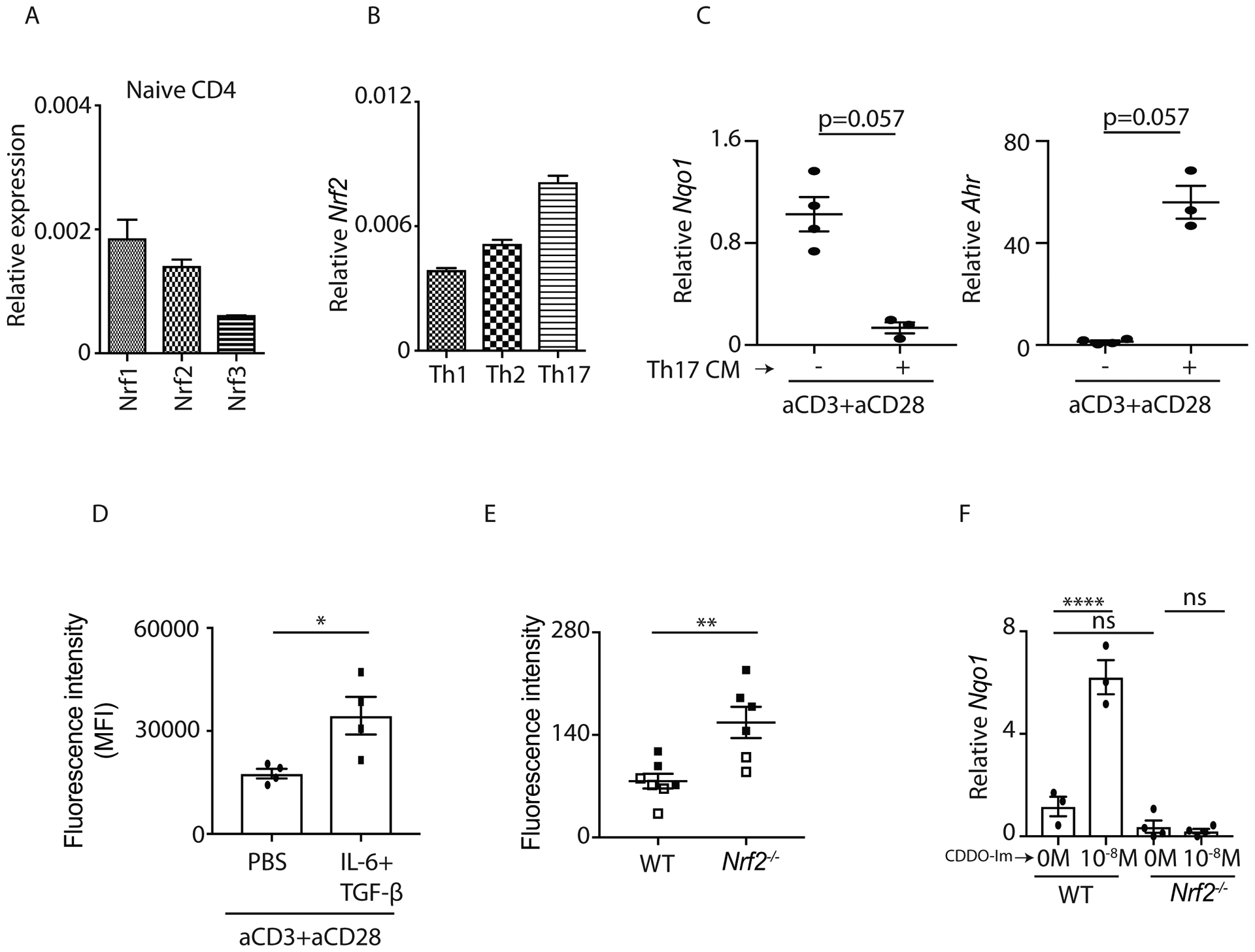
A and B) qPCR data (n=3) showed Nrf1, Nrf2 and Nrf3 expression in C57BL/6 mice-derived naïve CD4+ T cell (A) and different T helper subsets (B).
C) Naïve CD4+ T cells were isolated from the spleens of C57BL/6 mice and stimulated by anti-CD3+anti-CD28 with or without Th17 differentiation medium. RNA was extracted at 24 hours post stimulation and the transcripts (Nqo1 and Ahr) were analyzed by qPCR.
D) Mean Fluorescent Intensity (MFI) of cellular ROS in CD4+ T cells of C57BL/6 mice was quantified after 24-hours stimulation with indicated cytokines.
E) Fluorescence Intensity of cellular ROS in CD4+ T cells from WT and Nrf2−/− mice was measured at 24 hours post anti-CD3 and anti-CD28 stimulation.
F) Naïve CD4+ T cells were isolated from the spleens of C57BL/6 and Nrf2−/− mice and processed for Th17 differentiation. Cells were harvested at 24 hours post DMSO or CDDO-Im stimulation and the expression of Nqo1 was examined by qPCR.
Figure 1D, 1E and 1F were generated from 2 independent experiments. Each symbol indicates experiments from a separate animal. Data presented as mean ± SEM on relevant graphs. *P ≤ 0.05; **P ≤ 0.01; ****P ≤ 0.0001 (Mann Whitney test and One-Way ANOVA).
Nrf2 activation induced IL-22 responses in CD4+ T cells in vitro
We next investigated whether Nrf2 or enhancing Nrf2 activity with CDDO-Im has an impact on in vitro polarized Th17 cells. We polarized WT and Nrf2−/− mice-derived naïve CD4+ cells into Th17 cells with or without the presence of CDDO-Im (0M and 10−8M). On Day 4, we evaluated the frequency of IL-17A and IL-22 producing CD4+ T cells. There was no difference in IL-17A and IL-22 producing CD4+ T cells between WT and Nrf2−/− mice (Figure 2A), suggesting that elevated ROS level in the knockout mice does not have an impact on polarized Th17 cells. Interestingly, our flow cytometry data show that CDDO-Im modestly promotes IL-17A responses in WT but not in Nrf2−/− mice (Figure 2A). Consistent with prior literatures (42, 43), our intracellular flow cytometry staining for IL-22 is weak. Therefore, we used ELISA to measure IL-22 as well as IL-17A production. Our data showed that CDDO-Im promoted both IL-22 and IL-17A productions in WT but not in Nrf2−/− mice (Figure 2B). Furthermore, we found that other Th17 related cytokines were also regulated by Nrf2. An increased expression of Il17f and Il21 but not Csf2 transcript was observed in CDDO-Im stimulated Th17 cells (Figure 2C). Collectively, our data showed that Nrf2 activation enhanced IL-17A and IL-22 production in CD4+ T cells.
Figure 2: CDDO-Im regulates IL-17A and IL-22 responses in Th17 cells:
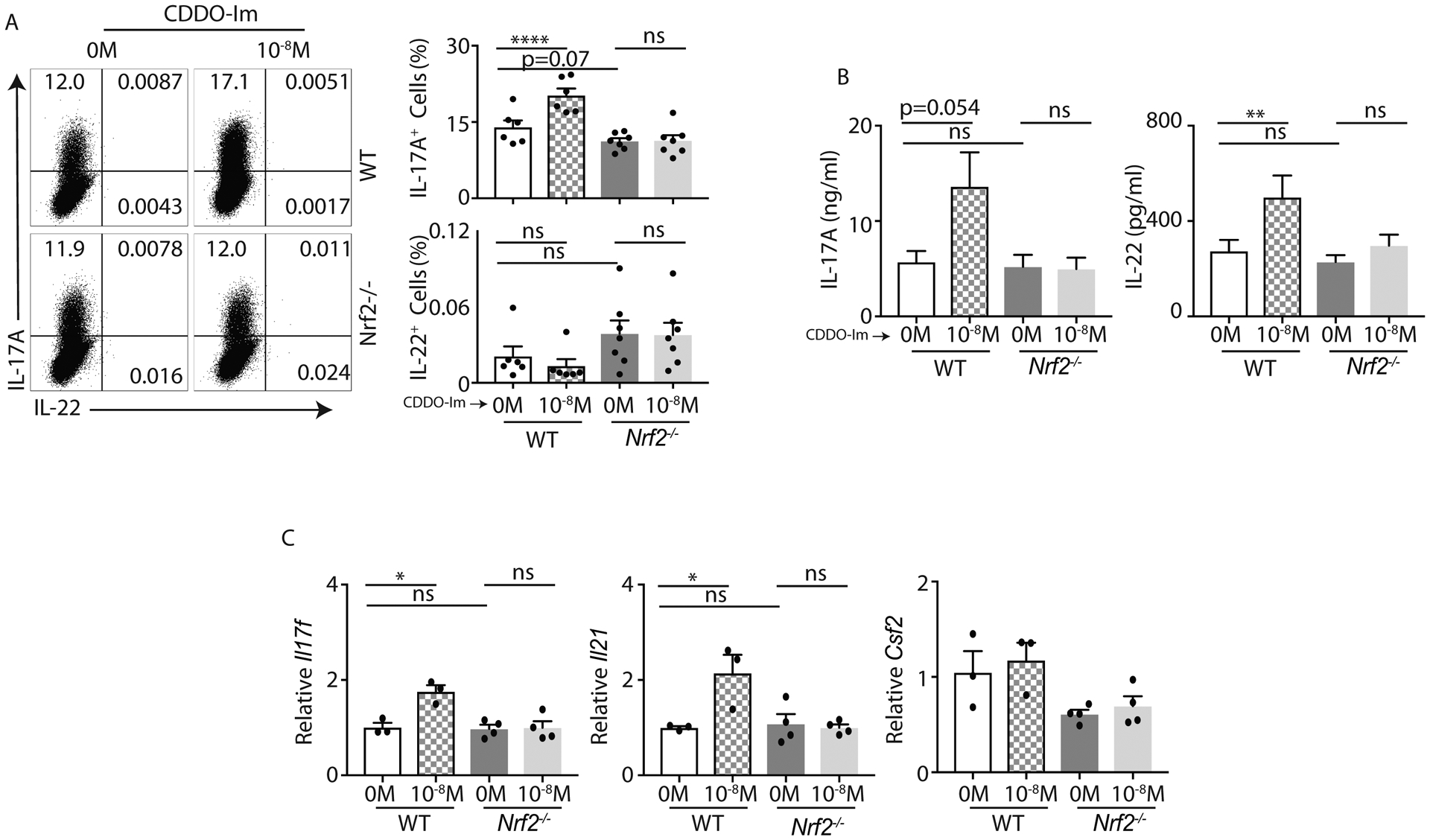
A and B) Naïve CD4+ T cells from C57BL/6 (WT) and Nrf2−/− mice were polarized to Th17 cells in presence of CDDO-Im or vehicle control. After 4-day culture, cells were restimulated with BD leukocyte activation cocktail for 4 hours and stained with anti-CD45, anti-CD3, anti-CD4, anti-IL-17A and anti-IL-22 antibodies. CD4+ T cells (gated on CD45+CD3+CD4+) producing IL-17A or IL-22 were analyzed by flow cytometry. Data show representative image (left panel) and percentage (right panel) of CD4+ T cells producing IL-17A and IL-22 in response to CDDO-Im or control vehicle (DMSO) stimulation. The concentration of IL-17A and IL-22 in the culture medium on day 4 was quantified by ELISA (B).
C) qPCR data show Il17f, Il21 and Csf2 expression on CDDO-Im or vehicle stimulated Th17 cells from WT and Nrf2−/− mice at 96 hours post stimulation.
Figure 2A and 2B were generated from 2 independent experiments. Each symbol indicates experiments from a separate animal. Data presented as mean ± SEM on relevant graphs. *P ≤ 0.05; **P ≤ 0.01; ****P ≤ 0.0001 (One-Way ANOVA).
Nrf2 regulates IL-22 responses in vivo
We have shown that the activation of Nrf2 by CDDO-Im promotes both IL-17A and IL-22 production in CD4+ T cells. Next, we evaluated whether Nrf2−/− mice had a defect in IL-17A and IL-22 response in vivo. We used Ovalbumin (Ova)+LPS and concanavalin A (Con A) mediated inflammation models (44, 45). Briefly, we adoptively transferred WT (CD45.1) and Nrf2−/− (CD45.2) CD4+ T cells (1:1) to Rag1−/− mice. OVA and LPS (oropharyngeal route) were administered on day 15, 21, 28 and 29 as shown in Figure 3A. CD4+ T cell frequency and IL-17A and IL-22 were analyzed one day after last Ova+LPS administration. Our data showed that CD4+ T cell percentage and IL-17A production in WT and Nrf2−/− mice were similar in the lungs of Rag1−/− mice (Figure 3B). Interestingly, CD4+ T cell-derived IL-22 response was significantly lower in Nrf2−/− mice (Figure 3B). We further validated our findings using a Con A-mediated immune cell activation model. We injected Con A into WT and Nrf2−/− mice. After 2 hours, CD4+ T cells were evaluated for IL-17A and IL-22 responses in the spleen (Figure 3C). We found a trend towards increased percentage of IL-17A producing CD4+ T cells. IL-22 intracellular staining in activated CD4+ T cell was weak; therefore, we performed ELISA to examine serum IL-22 concentration. Indeed, serum IL-22 concentration was significantly reduced in Nrf2−/− mice compared to WT mice (Figure 3D). Serum IL-17A concentration was similar in both groups (Figure 3D). Finally, we used Citrobacter rodentium infection model in which IL-22 was required for bacterial clearance and host protection (17). We showed that CDDO-Im administration induced around 1000-fold increase in Il22 transcript in the distal colon compared to DMSO treated group (Supplemental Figure 1B). These results suggest that Nrf2 activation regulates IL-22 but not IL-17A production in vivo.
Nrf2 activation inhibits IL-17A response in RRMS patient-derived PBMCs
Several studies have shown that Nrf2 plays an important role in suppressing IL-17A immune responses in vivo (34–36), which we have not seen in differentiated Th17 cells in vitro and two different in vivo models. CDDO-Im may act differently in murine and human derived CD4+ T cells to modulate IL-17A response. Nrf2-mediated immune regulation may be more important in autoimmune disorders where IL-17A plays a pathological role. It has been reported that multiple sclerosis (MS) patients have exacerbated IL-17A response (46). Therefore, we used Relapsing-Remitting Multiple Sclerosis (RRMS) patient-derived PBMCs to study IL-17A regulation by Nrf2 activation. We stimulated RRMS patient-derived PBMCs with anti-CD3 + anti-CD28 in presence of CDDO-Im (10−7M and 10−8M) or vehicle control. Consistent with the mice results, human data also showed that CDDO-Im induced NQO1 expression to a much greater extent at 10−7M compared to 10−8M (Supplemental Figure 2A). Additionally, CDDO-Im (10−7M and 10−8M) inhibited IL-17A at both transcript and protein level (Supplemental Figure 2B). Collectively, our data showed that Nrf2 activation inhibited IL-17A response in RRMS patient-derived PBMCs.
Superoxide dismutase 1 (SOD1) induction in CD4+ T cells is Nrf2-dependent and SOD mimetics regulates Th17 differentiation
To understand how Nrf2 regulates IL-17A and IL-22 response in CD4+ T cells, we next examined the expression patterns of critical cellular redox genes. Nrf2 has been shown to regulate superoxide dismutase Sod1, Sod3, and Cybb (Nox2) expression in non-immune cell types (32), and it has been reported that Sod3−/− and Cybb−/− mice have exacerbated Th17 responses (47–49). Therefore, we examined the expression level of these transcripts in Th17 cells at 24 hours and 4 days post differentiation. The expression of Sod1 is significantly induced by CDDO-Im in WT Th17 cells, whereas Sod2, Sod3, and Cybb were unaltered in both groups (Figure 4A and Supplemental Figure 2C). To further confirm the role of SOD in regulating IL-17A and IL-22 responses in CD4+ T cells, we utilized SOD mimetics. Interestingly, TEMPOL, a SOD mimetic agent, reduced the frequency of both IL-17A+ and IL-22+ CD4+ T cells polarized to Th17 cells (Figure 4B), which is inconsistent with the induction of IL-22 with CDDO-Im treatment at 10−8 M (Figure 2B). Collectively, our data suggested that Nrf2 activation regulated Sod1 expression in CD4+ T cells, however Nrf2-mediated IL-22 responses were independent of Nrf2-SOD1 axis.
Figure 4: Sod1 is induced by Nrf2 activation but may not be involved in Nrf2-mediated IL-17A and IL-22 responses in Th17 cells:
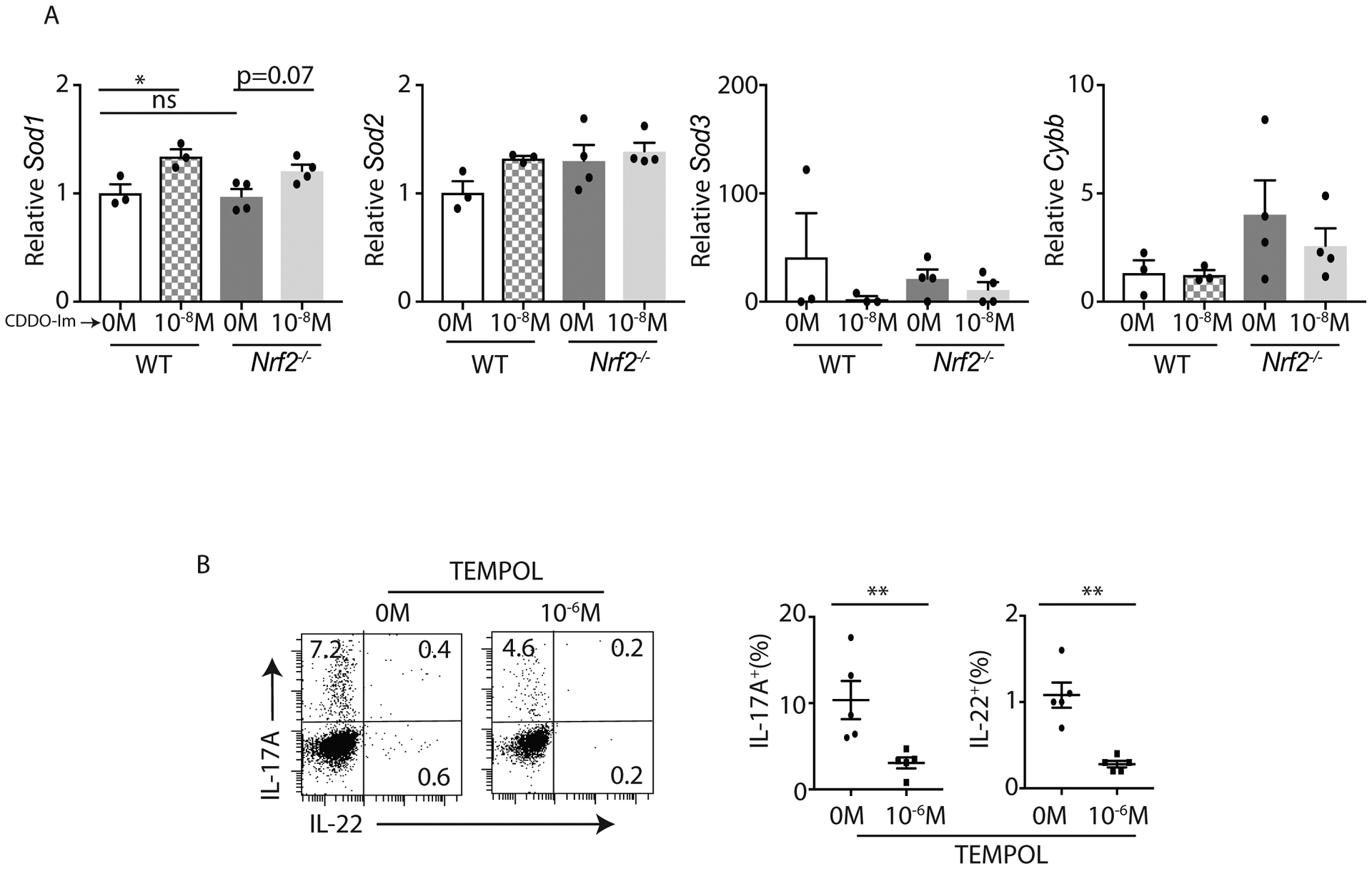
A) C57BL/6 (WT) and Nrf2−/− naïve CD4+ T cells were polarized to Th17 cells in presence of CDDO-Im or vehicle control. Differentiated Th17 cells were harvested on day 4 and processed for qPCR on Sod1, Sod2, Sod3 and Cybb.
B) Naïve CD4+ T cells from C57BL/6 mice were polarized to Th17 cells in presence of TEMPOL or vehicle control. After 4 days of culture, cells were stimulated with PMA+Ionomycin. CD4+ T cells (gated on CD3+CD4+) producing IL-17A and IL-22 were analyzed by flow cytometer. Data show the percentage of CD4+ T cells producing IL-17A and IL-22 in response to TEMPOL or control vehicle (DMSO) stimulation.
Figure 4B was generated from 2 independent experiments. Each symbol indicates experiments from a separate animal. Data presented as mean ± SEM on relevant graphs. *P ≤ 0.05; **P ≤ 0.01 (Mann-Whitney test, Two-tailed and One-Way ANOVA).
Nrf2 activation regulates Rorc and Ahr in CD4+ T cells
High Il22 expression in Th17 cells positively correlated with increased expression of RORγt (Rorc) and AhR (50). RORγt is a transcription factor that regulates IL-17A and IL-22 expression in Th17 cells (51). AhR is not a critical factor for Th17 differentiation. However, its activation is required for the production of IL-17A and IL-22 (24). Therefore, we firstly examined whether RORγt was involved in Nrf2-mediated regulation on Th17 cells. We analyzed the expression of RORγt by flow cytometry on day 4 of Th17 differentiation post CDDO-Im or vehicle stimulation. We found a reduced RORγt level in CDDO-Im treated WT but not Nrf2−/− CD4+ T cells (Figure 5A and 5B). We also identified and analyzed several putative AREs and ARE-Rs in Rorc promoter (Supplemental Figure 3A). We evaluated Nrf2 bindings to Rorc gene in Th17 cells. Our ChIP-qPCR data suggested that Nrf2 protein directly bind to ARE-R2 but not to other ARE or ARE-R motifs of Rorc promoter in CD4+ T cells (Supplemental Figure 3B and data not shown).
Figure 5: Nrf2 regulates RORγt expression as well as Ahr promoter activity:
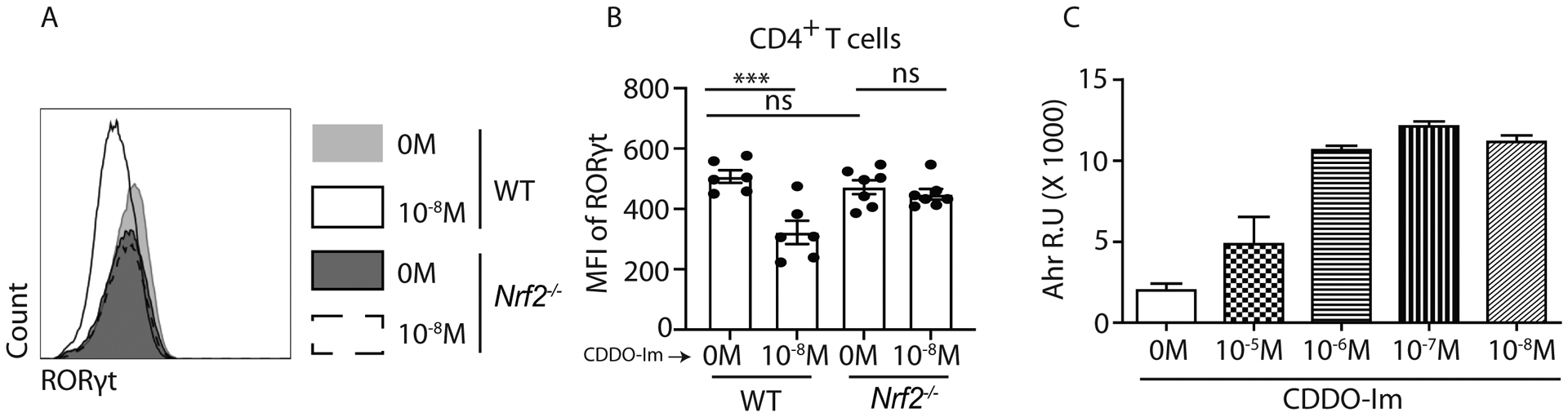
A and B) Naïve CD4+ T cells from WT and Nrf2−/− mice were polarized to Th17 cells in presence of CDDO-Im or vehicle control. After 4 days, cells were harvested and restimulated with BD leukocyte activation cocktail (1:200) for 4 hours. Then cells were stained with anti-CD45, anti-CD3, anti-CD4 and anti-RORγt antibodies. CD4+ T cells (gated on CD45+CD3+CD4+) positive were examined for RORγt expression by flow cytometry. Data show representative flow image (A) and mean fluorescent intensity (MFI) (B) of CD4+ T cells.
C) C10 cells stably transfected with AhR luciferase reporter vector were cultured in 24 wells plate and treated with indicated concentrations of CDDO-Im for 8 hours. Luciferase activity in response to CDDO-Im stimulation was quantified with Promega Luciferase Assay System.
Figure 5A, 5B and 5C were generated from 2 independent experiments. Figure 5A and 5C were representative of two independent experiments. Each symbol indicates experiments from a separate animal. Data presented as mean ± SEM on relevant graphs. ***P ≤ 0.001; (One-Way ANOVA).
The inhibition of RORγt by Nrf2 and the interaction between Nrf2 and Rorc surprisingly suggest a dual role of Nrf2 in regulating IL-17A responses in CD4+ T cells. It is possible that modest reduction in RORγt may not have an impact on IL-17A production. Using HEK293 cell line, we next investigated if Nrf2 inhibited or promoted Il17a transcription. HEK293T cells were transiently transfected with mouse Il17a promoter construct tagged with firefly luciferase reporter. Renilla luciferase reporter vector was co-transfected as an internal control. We found that Nrf2 inhibited RORγt-induced IL-17A activation (Supplemental Figure 3C).
To further study the role of AhR in Nrf2-regulated Th17 cells, we utilized an AhR reporter cell line which allowed the measurement of its transcriptional activity. Our data showed that CDDO-Im activated AhR in a dose dependent manner (Figure 5C). Consistent with the IL-17A and IL-22 induction by Nrf2, our results indicated a possible role of AhR in promoting Nrf2-mediated IL-17A and IL-22 responses in CD4+ T cells.
Nrf2-dependent regulation of IL-22 response in CD4+ T cells is dependent on AhR
AhR has been shown to be required for IL-22 generation in Th17 and ILC3 cells (24, 25). Therefore, we investigated if Nrf2 and AhR pathways were interconnected. We utilized CD4+ cell-specific Ahr knockout (AhrCD4) mice to investigate whether Nrf2-dependent regulation of IL-17A and IL-22 responses were modulated by AhR signaling. We firstly validated AhrCD4 mice by analyzing AhR expression in Ahrfl/fl and AhrCD4 mice with flow cytometry (Figure 6A). Naïve CD4+ T cells from Ahrfl/fl and AhrCD4 mice were oriented to Th17 cells in presence of CDDO-Im or vehicle control. On day 4, we analyzed the Th17 cells by flow cytometry and ELISA. CDDO-Im stimulation modestly induced IL-17A response in CD4+ T cells from both Ahrfl/fl and AhrCD4 mice (Figure 6B and 6C). This suggested that Nrf2-mediated IL-17A response was independent of AhR. Our intracellular flow cytometry staining for IL-22 was weak, therefore we used ELISA to measure IL-22. The results showed that CDDO-Im did not induce IL-22 production in AhrCD4 mice (Figure 6D), suggesting that Nrf2-regulated IL-22 response in CD4+ T cells is dependent on AhR. We further confirmed the results by using CH-223191, a cytosolic AhR antagonist. CH-223191 can abrogate the activation of AhR signaling by blocking the binding of AhR to its ligands (52). Our data showed that at the presence of CH-223191, CDDO-Im failed to induce IL-22 production in CD4+ T cells (Figure 6E). Collectively, the results suggested that Nrf2-induced IL-22 but not IL-17A production in CD4+ T cells were dependent on AhR.
Figure 6: Nrf2-mediated IL-22 induction in Th17 cells is dependent on AhR:
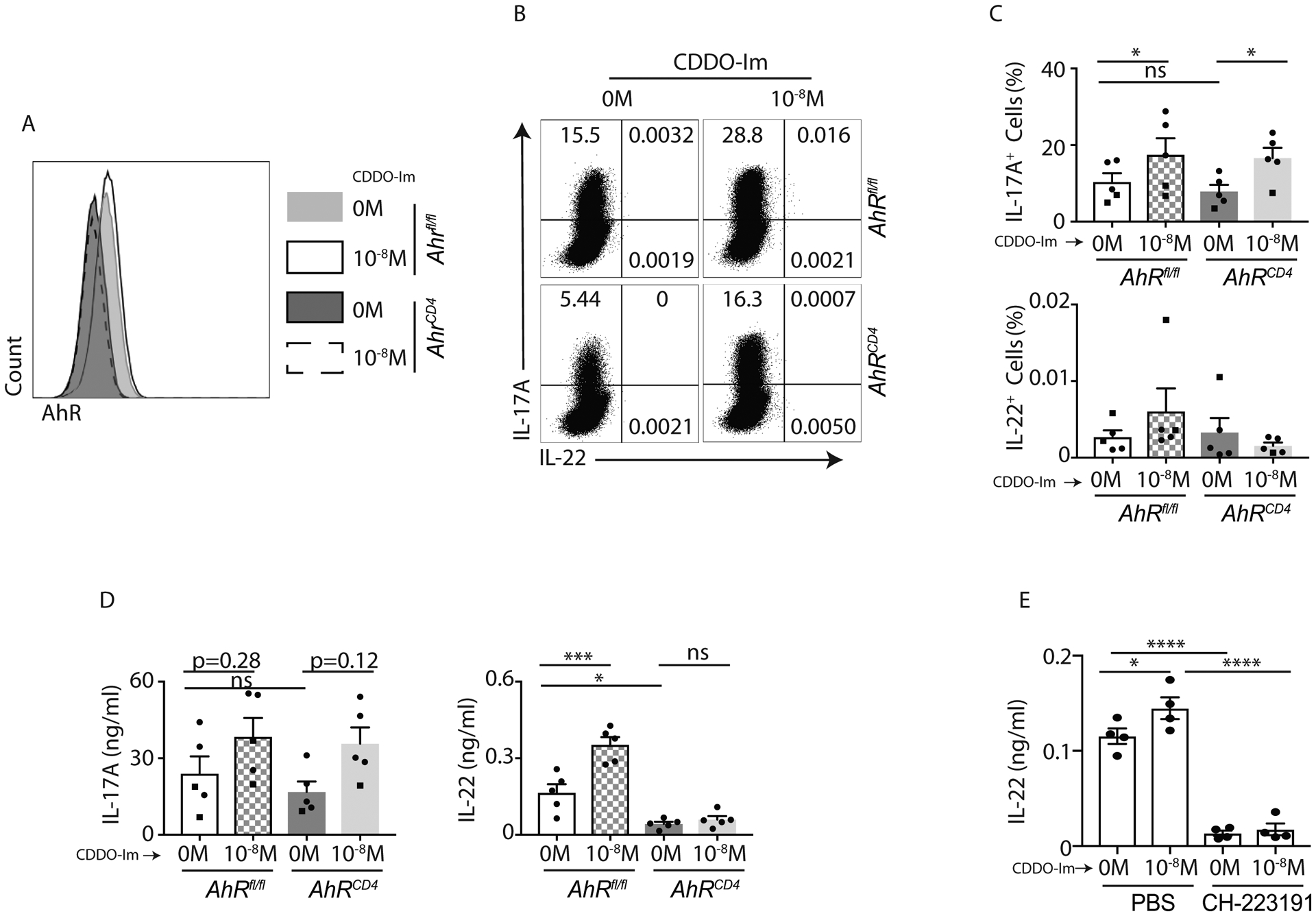
A-D) Naïve CD4+ T cells from AhRfl/fl and AhRCD4 mice were polarized to Th17 cells in presence of CDDO-Im or vehicle control for 4 days. Differentiated cells were restimulated with BD leukocyte cocktail (1:200). CD4+ T cells (gated on CD45+CD3+CD4+) were examined for AhR expression (A). IL-17A and IL-22 production were also analyzed by flow cytometry. Data show representative image (B) and percentage (C) of CD4+ T cells producing IL-17A and IL-22 in response to CDDO-Im or vehicle control (DMSO) stimulation. Culture medium on day 4 was harvested and analyzed for IL-17A and IL-22 concentrations by ELISA (D).
E) Naïve CD4+ T cells from C57BL/6 mice were polarized to Th17 cells in presence of CDDO-Im, CH-223191 or vehicle control for 4 days. On day 4, the culture medium was analyzed for IL-22 concentrations by ELISA.
Figure 6A and 6B was a representative image of two independent experiments. Figure 6B and 6D were generated from 2 independent experiments. Each symbol indicates experiments from a separate animal. Data presented as mean ± SEM on relevant graphs. *P ≤ 0.05; ***P ≤ 0.001; ****P ≤ 0.0001 (One-way ANOVA).
Nrf2 binds to the ARE motif of Ahr promoter in CD4+ T cells.
In order to test how AhR regulated Nrf2-mediated IL-22 production in CD4+ T cells, we firstly examined the expression of Nqo1, the Nrf2 targeted gene, in AhrCD4 mice. Interestingly, Nqo1 expression was reduced in CDDO-Im stimulated AhrCD4 CD4+ T cells compared to Ahrfl/fl CD4+ T cells, indicating that AhR may be a possible downstream target of Nrf2 signaling (Figure 7A). We next evaluated whether Nrf2 binds to Ahr promoter in Th17 cells. We analyzed mouse Ahr sequence and an ARE motif was found in mouse Ahr promoter (Figure 7B). Therefore, we perform CHIP-qPCR in differentiated Th17 cells by pulling down the Nrf2 binding sequences. As expected, our CHIP-qPCR data suggested that Nrf2 protein directly binded to the ARE motif of Ahr (Figure 7C). Collectively, our data showed that Nrf2 induces IL-22 production in CD4+ T cells by promoting AhR transcription.
Figure 7: Nrf2 binds to ARE of mouse Ahr:
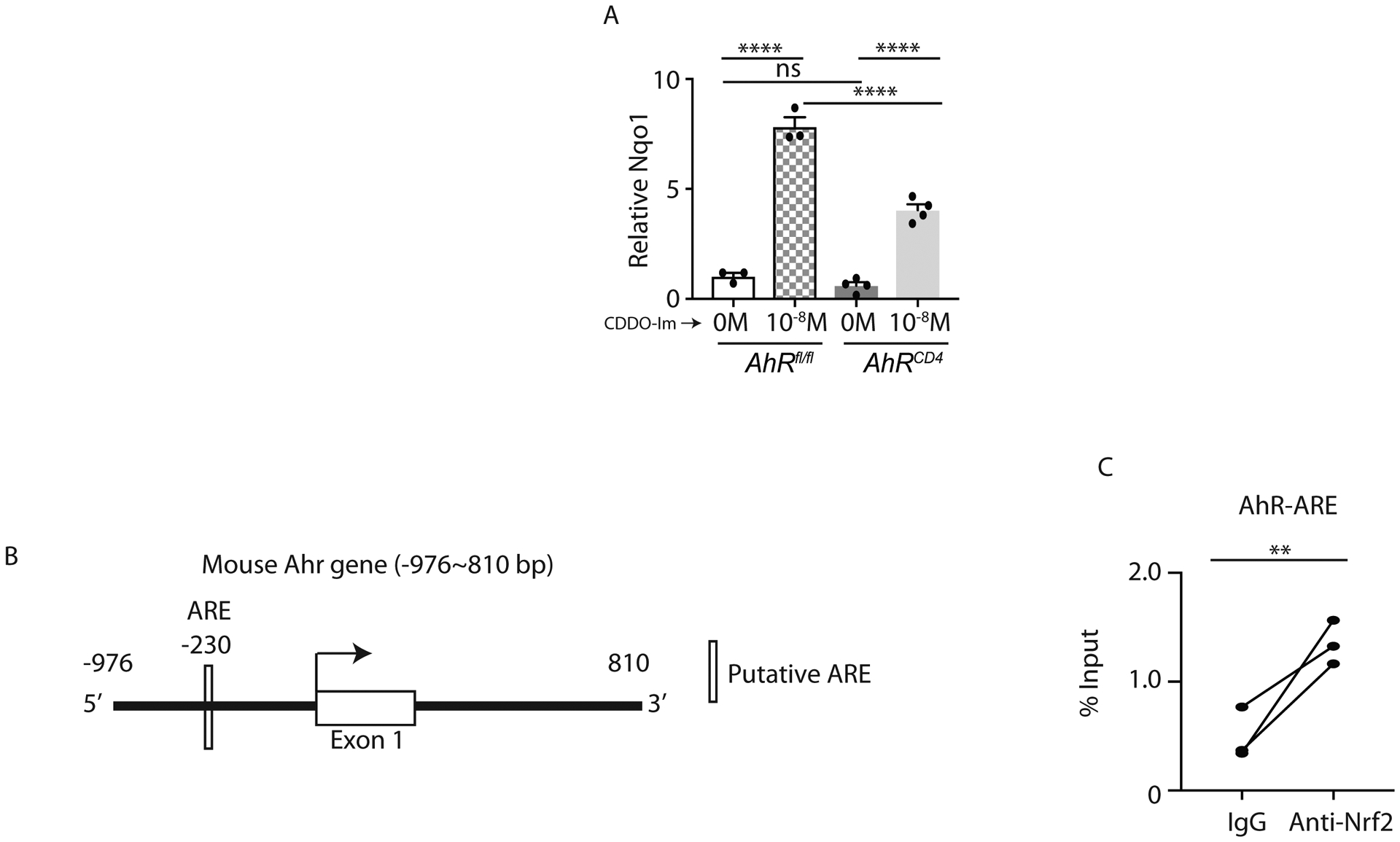
A) Naïve CD4+ T cells from AhRfl/fl and AhRCD4 mice were polarized to Th17 cells in presence of CDDO-Im or vehicle control for 4 days. Cells were harvested for qPCR analysis on Nqo1 expression.
B) Schematic diagram shows the putative ARE binding motifs in mouse Ahr gene.
C) Naïve CD4+ T cells isolated from the spleens and lymph nodes of C57BL/6 mice were cultured under the Th17 differentiation condition for 4 days. Cells were harvested and processed for CHIP with anti-Nrf2 antibody. The Nrf2-binding DNA was utilized for qPCR analysis to examine the interaction between the ARE motif of Ahr gene and Nrf2.
Data presented as mean ± SEM on relevant graphs. **P ≤ 0.01; ****P ≤ 0.0001 (Paired student t test and One-way ANOVA).
Discussion
Th17 cell-derived IL-17A and IL-22 are critical for mounting both inflammatory and tissue protective responses. Differentiated or activated CD4+ T cells co-produce IL-17A and IL-22. It remains unclear whether these cytokine responses can be modified differently to achieve tissue-specific or targeted functional outcome. A therapeutic strategy by promoting IL-22-dependent tissue protective or regenerative response is of great clinical significance. Our study provides evidence that modulating Nrf2 activity in CD4+ T cells specifically promotes IL-22 responses.
The importance of Nrf2 in inhibiting Tbet-dependent IFNγ and concurrently promoting GATA3-dependent Th2 responses has been reported (53). Additionally, the involvement of Nrf2 signaling in inhibiting murine and human IL-17A responses has been documented in several studies (34, 35, 38). However, how Nrf2 regulates IL-17A immune responses remains poorly studied. Additionally, the role of Nrf2 or Nrf2 activator in regulating IL-22 responses has not been thoroughly characterized. TGF-β and IL-6 are key cytokines required for Th17 cell-derived IL-17A and IL-22 responses. Several other mediators such as high salt, cellular redox and lipid metabolites also influence Th17 differentiation and/or its pathogenic potential (54–57). We observed that Th17 conditioned medium induced ROS. The immunomodulatory role of ROS has been documented in many studies (58, 59). ROS signaling implicated in T cell activation, proliferation, effector function and apoptosis (58, 59). It was obvious that Th17 condition medium inhibited Nrf2 regulated antioxidant machinery to become activated. Consistently, Nrf2−/− mice displayed an increased ROS levels suggesting an increased potential of knockout-derived CD4+ T cell to differentiate to Th17 cells. However, IL-17A and IL-22 responses were similar in Nrf2−/− mice and WT mice. Enhanced IL-17A and IL-22 responses in CD4+ T cells were only observed after Nrf2 activation (with CDDO-Im) suggesting a ROS independent regulation of Th17 differentiation. Indeed, TEMPOL inhibits both IL-17A and IL-22 response in CD4+ T cells. The impact of cellular ROS levels on Th17 differentiation is controversial. Studies have shown that ROS can inhibit or promote Th17 cell survival and induction (57, 60, 61). Additional work is required to understand the role of Nrf2-dependent ROS regulation in CD4+ T cells proliferation, maintenance and effector functions.
In contrast to in vitro observations, our in vivo murine results with Con A and OVA+LPS administration suggest that Nrf2 has modest or no impact on IL-17A induction but is important for the regulation of IL-22 response in CD4+ T cells. Interestingly, the in vivo IL-22 immune responses in Nrf2−/− mice are not fully reduced, suggesting the involvement of other factors. It is possible that AhR reciprocally regulates Nrf2 expression as suggested by findings in murine fibroblast (62). Alternatively, it is possible that intact IL-23 signaling in Nrf2−/− mice may still regulate IL-22 responses as we did not find a role of CDDO-Im in regulating IL-23R expression (data not shown). We showed that CDDO-Im-dependent IL-22 response was positively correlated with increased AhR activity and reduced RORγt expression. Furthermore, we showed that CDDO-Im regulated AhR activity and AhR-dependent IL-22 responses. Nrf2-dependent regulation of AhR ARE in CD4+ T cells is in line with a previous study in murine fibroblast (62). In this study, authors showed that CDDO-Im induces AhR, Cypa1a and Cypb1b transcripts in WT mice but not in Nrf2−/− mice.
We also found that CDDO-Im reduced RORγt protein levels and this result was inversely correlated with increased IL-17A responses in CD4+ T cells. We showed that Nrf2 bind to ARE-R2 of Rorc further confirmed negative regulation of RORγt. It is possible that modest changes of RORγt do not significantly affect IL-17/IL-22 response in CD4+ T cells during Th17 differentiation. Moreover, we observed that CDDO-Im induced IL-17A in AhRCD4 knockout mice. CDDO-Im is a potent inhibitor of ROS. The importance of ROS in regulating T cells proliferation and IL-2 generation has been well established. Additionally, a recent study showed that Nrf2 play an important role in modulating IL-2 responses in T cells (40). Thus, CDDO-Im-mediated early T cell activation during Th17 cell differentiation may have an impact on IL-17A responses independent of RORγt and AhR.
Th17 cells are plastic in nature capable of converting to other lineages in response to specific stimulation. It is possible that Nrf2 acts differently on differentiated/committed Th17 cells to inhibit inflammatory IL-17A response or skews them to Th2 or Th1 lineage that we have not investigated in present study. We showed that CDDO-Im act differently in murine and human-derived CD4+ T cells to modulate IL-17A response. This could be due to the presence of other cell types including antigen presenting cells in PBMCs. Additional work is required to determine the role of APC-dependent regulation of Th17 response in our model.
In conclusion, our work showed that Nrf2 through AhR promotes IL-22 production in CD4+ T cells. IL-17A regulation by Nrf2 needs further investigation. Finally, Nrf2 activator can be used in vivo to initiate IL-22-dependent tissue regenerative responses.
Supplementary Material
Key Points:
Nrf2 regulates IL-22 response in CD4+ T cells.
Nrf2 activation inhibits IL-17A response in multiple sclerosis patient-derived PBMCs.
Acknowledgments
We would like to acknowledge Dr. Thomas Kensler, Pittsburgh for critically reading the manuscript. We would like to acknowledge the Flow Cytometry Core at Stony Brook University. We thank Shannon Kanidinc, Artan Berisha, Makheni Jean-Pierre, and Ankita Singh for their help.
PK was supported by the Crohn’s and Colitis Foundation (476637), National Multiple Sclerosis Society (PP-1709-29192), NIH R01 DK121798-01, NIH R21 AI146696, NIH R21 AI149257 and the SUNY Research Foundation, MG supported by R01DK118568. AA was supported by Wellcome Trust/DBT India alliance intermediate fellowship (IA/I/12/1/500524) and DST-SERB from Department of Science and Technology, Govt of India.
References:
- 1.Hsu D, Taylor P, Fletcher D, van Heeckeren R, Eastman J, van Heeckeren A, Davis P, Chmiel JF, Pearlman E, and Bonfield TL. 2016. Interleukin-17 Pathophysiology and Therapeutic Intervention in Cystic Fibrosis Lung Infection and Inflammation. Infect Immun 84: 2410–2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rizzo HL, Kagami S, Phillips KG, Kurtz SE, Jacques SL, and Blauvelt A. 2011. IL-23-Mediated Psoriasis-Like Epidermal Hyperplasia Is Dependent on IL-17A. J Immunol 186: 1495–1502. [DOI] [PubMed] [Google Scholar]
- 3.Nakae S, Saijo S, Horai R, Sudo K, Mori S, and Iwakura Y. 2003. IL-17 production from activated T cells is required for the spontaneous development of destructive arthritis in mice deficient in IL-1 receptor antagonist. Proceedings of the National Academy of Sciences of the United States of America 100: 5986–5990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dutta R, and Trapp BD. 2011. Mechanisms of neuronal dysfunction and degeneration in multiple sclerosis. Prog Neurobiol 93: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ivanov S, Bozinovski S, Bossios A, Valadi H, Vlahos R, Malmhall C, Sjostrand M, Kolls JK, Anderson GP, and Linden A. 2007. Functional relevance of the IL-23-IL-17 axis in lungs in vivo. Am J Respir Cell Mol Biol 36: 442–451. [DOI] [PubMed] [Google Scholar]
- 6.Christenson SA, van den Berge M, Faiz A, Inkamp K, Bhakta N, Bonser LR, Zlock LT, Barjaktarevic IZ, Barr RG, Bleecker ER, Boucher RC, Bowler RP, Comellas AP, Curtis JL, Han MK, Hansel NN, Hiemstra PS, Kaner RJ, Krishnanm JA, Martinez FJ, O’Neal WK, Paine R 3rd, Timens W, Wells JM, Spira A, Erle DJ, and Woodruff PG. 2019. An airway epithelial IL-17A response signature identifies a steroid-unresponsive COPD patient subgroup. J Clin Invest 129: 169–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hueber W, Patel DD, Dryja T, Wright AM, Koroleva I, Bruin G, Antoni C, Draelos Z, Gold MH, Psoriasis Study G, Durez P, Tak PP, Gomez-Reino JJ, Rheumatoid Arthritis Study G, Foster CS, Kim RY, Samson CM, Falk NS, Chu DS, Callanan D, Nguyen QD, Uveitis Study G, Rose K, Haider A, and Di Padova F. 2010. Effects of AIN457, a fully human antibody to interleukin-17A, on psoriasis, rheumatoid arthritis, and uveitis. Sci Transl Med 2: 52ra72. [DOI] [PubMed] [Google Scholar]
- 8.Genovese MC, Van den Bosch F, Roberson SA, Bojin S, Biagini IM, Ryan P, and Sloan-Lancaster J. 2010. LY2439821, a humanized anti-interleukin-17 monoclonal antibody, in the treatment of patients with rheumatoid arthritis: A phase I randomized, double-blind, placebo-controlled, proof-of-concept study. Arthritis Rheum 62: 929–939. [DOI] [PubMed] [Google Scholar]
- 9.Patel DD, Lee DM, Kolbinger F, and Antoni C. 2013. Effect of IL-17A blockade with secukinumab in autoimmune diseases. Ann Rheum Dis 72 Suppl 2: ii116–123. [DOI] [PubMed] [Google Scholar]
- 10.Havrdova E, Belova A, Goloborodko A, Tisserant A, Wright A, Wallstroem E, Garren H, Maguire RP, and Johns DR. 2016. Activity of secukinumab, an anti-IL-17A antibody, on brain lesions in RRMS: results from a randomized, proof-of-concept study. J Neurol 263: 1287–1295. [DOI] [PubMed] [Google Scholar]
- 11.Chen K, McAleer JP, Lin Y, Paterson DL, Zheng M, Alcorn JF, Weaver CT, and Kolls JK. 2011. Th17 cells mediate clade-specific, serotype-independent mucosal immunity. Immunity 35: 997–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McAleer JP, Nguyen NLH, Chen K, Kumar P, Ricks DM, Binnie M, Armentrout RA, Pociask DA, Hein A, Yu A, Vikram A, Bibby K, Umesaki Y, Rivera A, Sheppard D, Ouyang WJ, Hooper LV, and Kolls JK. 2016. Pulmonary Th17 Antifungal Immunity Is Regulated by the Gut Microbiome. J Immunol 197: 97–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen K, Eddens T, Trevejo-Nunez G, Way EE, Elsegeiny W, Ricks DM, Garg AV, Erb CJ, Bo MH, Wang T, Chen W, Lee JS, Gaffen SL, and Kolls JK. 2016. IL-17 Receptor Signaling in the Lung Epithelium Is Required for Mucosal Chemokine Gradients and Pulmonary Host Defense against K. pneumoniae. Cell Host Microbe 20: 596–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kumar P, Monin L, Castillo P, Elsegeiny W, Horne W, Eddens T, Vikram A, Good M, Alexi A Bibby Schoenborn, K., Montelaro Ronald C., Metzger Dennis W., Gulati Ajay S., and Kolls Jay K.. 2016. Intestinal Interleukin-17 Receptor Signaling Mediates Reciprocal Control of the Gut Microbiota and Autoimmune Inflammation. Immunity 44: 659–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kumar P, Thakar MS, Ouyang W, and Malarkannan S. 2013. IL-22 from conventional NK cells is epithelial regenerative and inflammation protective during influenza infection. Mucosal Immunol 6: 69–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pociask DA, Scheller EV, Mandalapu S, McHugh KJ, Enelow RI, Fattman CL, Kolls JK, and Alcorn JF. 2013. IL-22 Is Essential for Lung Epithelial Repair following Influenza Infection. Am J Pathol 182: 1286–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zheng Y, Valdez PA, Danilenko DM, Hu Y, Sa SM, Gong Q, Abbas AR, Modrusan Z, Ghilardi N, de Sauvage FJ, and Ouyang W. 2008. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat Med 14: 282–289. [DOI] [PubMed] [Google Scholar]
- 18.Radaeva S, Sun R, Pan HN, Hong F, and Gao B. 2004. Interleukin 22 (IL-22) plays a protective role in T cell-mediated murine hepatitis: IL-22 is a survival factor for hepatocytes via STAT3 activation. Hepatology 39: 1332–1342. [DOI] [PubMed] [Google Scholar]
- 19.Zheng M, Horne W, McAleer JP, Pociask D, Eddens T, Good M, Gao B, and Kolls JK. 2016. Therapeutic Role of Interleukin 22 in Experimental Intra-abdominal Klebsiella pneumoniae Infection in Mice. Infect Immun 84: 782–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aujla SJ, Chan YR, Zheng M, Fei M, Askew DJ, Pociask DA, Reinhart TA, McAllister F, Edeal J, Gaus K, Husain S, Kreindler JL, Dubin PJ, Pilewski JM, Myerburg MM, Mason CA, Iwakura Y, and Kolls JK. 2008. IL-22 mediates mucosal host defense against Gram-negative bacterial pneumonia. Nat Med 14: 275–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zuniga LA, Jain R, Haines C, and Cua DJ. 2013. Th17 cell development: from the cradle to the grave. Immunol Rev 252: 78–88. [DOI] [PubMed] [Google Scholar]
- 22.Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, and Kuchroo VK. 2006. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 441: 235–238. [DOI] [PubMed] [Google Scholar]
- 23.Korn T, Mitsdoerffer M, Croxford AL, Awasthi A, Dardalhon VA, Galileos G, Vollmar P, Stritesky GL, Kaplan MH, Waisman A, Kuchroo VK, and Oukka M. 2008. IL-6 controls Th17 immunity in vivo by inhibiting the conversion of conventional T cells into Foxp3(+) regulatory T cells. Proceedings of the National Academy of Sciences of the United States of America 105: 18460–18465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Veldhoen M, Hirota K, Westendorf AM, Buer J, Dumoutier L, Renauld J-C, and Stockinger B. 2008. The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature 453: 106–109. [DOI] [PubMed] [Google Scholar]
- 25.Qiu J, Heller JJ, Guo XH, Chen ZME, Fish K, Fu YX, and Zhou L. 2012. The Aryl Hydrocarbon Receptor Regulates Gut Immunity through Modulation of Innate Lymphoid Cells. Immunity 36: 92–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kiss EA, Vonarbourg C, Kopfmann S, Hobeika E, Finke D, Esser C, and Diefenbach A. 2011. Natural aryl hydrocarbon receptor ligands control organogenesis of intestinal lymphoid follicles. Science 334: 1561–1565. [DOI] [PubMed] [Google Scholar]
- 27.Lee JS, Cella M, McDonald KG, Garlanda C, Kennedy GD, Nukaya M, Mantovani A, Kopan R, Bradfield CA, Newberry RD, and Colonna M. 2011. AHR drives the development of gut ILC22 cells and postnatal lymphoid tissues via pathways dependent on and independent of Notch. Nat Immunol 13: 144–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huh JR, and Littman DR. 2012. Small molecule inhibitors of RORgammat: targeting Th17 cells and other applications. Eur J Immunol 42: 2232–2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sun N, Guo H, and Wang Y. 2019. Retinoic acid receptor-related orphan receptor gamma-t (RORgammat) inhibitors in clinical development for the treatment of autoimmune diseases: a patent review (2016-present). Expert Opin Ther Pat 29: 663–674. [DOI] [PubMed] [Google Scholar]
- 30.Venken K, Jacques P, Mortier C, Labadia ME, Decruy T, Coudenys J, Hoyt K, Wayne AL, Hughes R, Turner M, Van Gassen S, Martens L, Smith D, Harcken C, Wahle J, Wang CT, Verheugen E, Schryvers N, Varkas G, Cypers H, Wittoek R, Piette Y, Gyselbrecht L, Van Calenbergh S, Van den Bosch F, Saeys Y, Nabozny G, and Elewaut D. 2019. RORgammat inhibition selectively targets IL-17 producing iNKT and gammadelta-T cells enriched in Spondyloarthritis patients. Nat Commun 10: 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu C, Li CY, and Kong AN. 2005. Induction of phase I, II and III drug metabolism/transport by xenobiotics. Arch Pharm Res 28: 249–268. [DOI] [PubMed] [Google Scholar]
- 32.Ma Q 2013. Role of nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol 53: 401–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Espinosa-Diez C, Miguel V, Mennerich D, Kietzmann T, Sanchez-Perez P, Cadenas S, and Lamas S. 2015. Antioxidant responses and cellular adjustments to oxidative stress. Redox Biol 6: 183–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pareek TK, Belkadi A, Kesavapany S, Zaremba A, Loh SL, Bai L, Cohen ML, Meyer C, Liby KT, Miller RH, Sporn MB, and Letterio JJ. 2011. Triterpenoid modulation of IL-17 and Nrf-2 expression ameliorates neuroinflammation and promotes remyelination in autoimmune encephalomyelitis. Sci Rep 1: 201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li B, Cui W, Liu J, Li R, Liu Q, Xie XH, Ge XL, Zhang J, Song XJ, Wang Y, and Guo L. 2013. Sulforaphane ameliorates the development of experimental autoimmune encephalomyelitis by antagonizing oxidative stress and Th17-related inflammation in mice. Exp Neurol 250: 239–249. [DOI] [PubMed] [Google Scholar]
- 36.Noel S, Martina MN, Bandapalle S, Racusen LC, Potteti HR, Hamad AR, Reddy SP, and Rabb H. 2015. T Lymphocyte-Specific Activation of Nrf2 Protects from AKI. J Am Soc Nephrol 26: 2989–3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hirayama A, Yoh K, Nagase S, Ueda A, Itoh K, Morito N, Hirayama K, Takahashi S, Yamamoto M, and Koyama A. 2003. EPR imaging of reducing activity in Nrf2 transcriptional factor-deficient mice. Free Radic Biol Med 34: 1236–1242. [DOI] [PubMed] [Google Scholar]
- 38.Liu M, Reddy NM, Higbee EM, Potteti HR, Noel S, Racusen L, Kensler TW, Sporn MB, Reddy SP, and Rabb H. 2014. The Nrf2 triterpenoid activator, CDDO-imidazolide, protects kidneys from ischemia-reperfusion injury in mice. Kidney Int 85: 134–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reddy NM, Suryanaraya V, Yates MS, Kleeberger SR, Hassoun PM, Yamamoto M, Liby KT, Sporn MB, Kensler TW, and Reddy SP. 2009. The triterpenoid CDDO-imidazolide confers potent protection against hyperoxic acute lung injury in mice. Am J Respir Crit Care Med 180: 867–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zagorski JW, Turley AE, Freeborn RA, VanDenBerg KR, Dover HE, Kardell BR, Liby KT, and Rockwell CE. 2018. Differential effects of the Nrf2 activators tBHQ and CDDO-Im on the early events of T cell activation. Biochem Pharmacol 147: 67–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xu D, Chen L, Chen X, Wen Y, Yu C, Yao J, Wu H, Wang X, Xia Q, and Kong X. 2017. The triterpenoid CDDO-imidazolide ameliorates mouse liver ischemia-reperfusion injury through activating the Nrf2/HO-1 pathway enhanced autophagy. Cell Death Dis 8: e2983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Plank MW, Kaiko GE, Maltby S, Weaver J, Tay HL, Shen W, Wilson MS, Durum SK, and Foster PS. 2017. Th22 Cells Form a Distinct Th Lineage from Th17 Cells In Vitro with Unique Transcriptional Properties and Tbet-Dependent Th1 Plasticity. J Immunol 198: 2182–2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rutz S, Noubade R, Eidenschenk C, Ota N, Zeng W, Zheng Y, Hackney J, Ding J, Singh H, and Ouyang W. 2011. Transcription factor c-Maf mediates the TGF-beta-dependent suppression of IL-22 production in T(H)17 cells. Nat Immunol 12: 1238–1245. [DOI] [PubMed] [Google Scholar]
- 44.Chen K, Pociask DA, McAleer JP, Chan YR, Alcorn JF, Kreindler JL, Keyser MR, Shapiro SD, Houghton AM, Kolls JK, and Zheng M. 2011. IL-17RA is required for CCL2 expression, macrophage recruitment, and emphysema in response to cigarette smoke. PloS one 6: e20333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nagata T, Mckinley L, Peschon JJ, Alcorn JF, Aujla SJ, and Kolls JK. 2008. Requirement of IL-17RA in Con A Induced Hepatitis and Negative Regulation of IL-17 Production in Mouse T Cells. J Immunol 181: 7473–7479. [DOI] [PubMed] [Google Scholar]
- 46.Babaloo Z, Aliparasti MR, Babaiea F, Almasi S, Baradaran B, and Farhoudi M. 2015. The role of Th17 cells in patients with relapsing-remitting multiple sclerosis: interleukin-17A and interleukin-17F serum levels. Immunol Lett 164: 76–80. [DOI] [PubMed] [Google Scholar]
- 47.Kwon MJ, Jeon YJ, Lee KY, and Kim TY. 2012. Superoxide dismutase 3 controls adaptive immune responses and contributes to the inhibition of ovalbumin-induced allergic airway inflammation in mice. Antioxid Redox Signal 17: 1376–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee YS, Cheon IS, Kim BH, Kwon MJ, Lee HW, and Kim TY. 2013. Loss of extracellular superoxide dismutase induces severe IL-23-mediated skin inflammation in mice. J Invest Dermatol 133: 732–741. [DOI] [PubMed] [Google Scholar]
- 49.Segal BH, Han W, Bushey JJ, Joo M, Bhatti Z, Feminella J, Dennis CG, Vethanayagam RR, Yull FE, Capitano M, Wallace PK, Minderman H, Christman JW, Sporn MB, Chan J, Vinh DC, Holland SM, Romani LR, Gaffen SL, Freeman ML, and Blackwell TS. 2010. NADPH oxidase limits innate immune responses in the lungs in mice. PLoS One 5: e9631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Perez LG, Kempski J, McGee HM, Pelzcar P, Agalioti T, Giannou A, Konczalla L, Brockmann L, Wahib R, Xu H, Vesely MCA, Soukou S, Steglich B, Bedke T, Manthey C, Seiz O, Diercks BP, Gnafakis S, Guse AH, Perez D, Izbicki JR, Gagliani N, Flavell RA, and Huber S. 2020. TGF-beta signaling in Th17 cells promotes IL-22 production and colitis-associated colon cancer. Nat Commun 11: 2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, and Littman DR. 2006. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 126: 1121–1133. [DOI] [PubMed] [Google Scholar]
- 52.Zhao B, Degroot DE, Hayashi A, He G, and Denison MS. 2010. CH223191 is a ligand-selective antagonist of the Ah (Dioxin) receptor. Toxicol Sci 117: 393–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rockwell CE, Zhang M, Fields PE, and Klaassen CD. 2012. Th2 skewing by activation of Nrf2 in CD4(+) T cells. J Immunol 188: 1630–1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ravindran R, Loebbermann J, Nakaya HI, Khan N, Ma H, Gama L, Machiah DK, Lawson B, Hakimpour P, Wang YC, Li S, Sharma P, Kaufman RJ, Martinez J, and Pulendran B. 2016. The amino acid sensor GCN2 controls gut inflammation by inhibiting inflammasome activation. Nature 531: 523–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang C, Yosef N, Gaublomme J, Wu C, Lee Y, Clish CB, Kaminski J, Xiao S, Meyer Zu Horste G, Pawlak M, Kishi Y, Joller N, Karwacz K, Zhu C, Ordovas-Montanes M, Madi A, Wortman I, Miyazaki T, Sobel RA, Park H, Regev A, and Kuchroo VK. 2015. CD5L/AIM Regulates Lipid Biosynthesis and Restrains Th17 Cell Pathogenicity. Cell 163: 1413–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kleinewietfeld M, Manzel A, Titze J, Kvakan H, Yosef N, Linker RA, Muller DN, and Hafler DA. 2013. Sodium chloride drives autoimmune disease by the induction of pathogenic TH17 cells. Nature 496: 518–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fu G, Xu Q, Qiu Y, Jin X, Xu T, Dong S, Wang J, Ke Y, Hu H, Cao X, Wang D, Cantor H, Gao X, and Lu L. 2017. Suppression of Th17 cell differentiation by misshapen/NIK-related kinase MINK1. J Exp Med 214: 1453–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.George-Chandy A, Nordstrom I, Nygren E, Jonsson IM, Postigo J, Collins LV, and Eriksson K. 2008. Th17 development and autoimmune arthritis in the absence of reactive oxygen species. Eur J Immunol 38: 1118–1126. [DOI] [PubMed] [Google Scholar]
- 59.Sena LA, Li S, Jairaman A, Prakriya M, Ezponda T, Hildeman DA, Wang CR, Schumacker PT, Licht JD, Perlman H, Bryce PJ, and Chandel NS. 2013. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity 38: 225–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhi L, Ustyugova IV, Chen X, Zhang Q, and Wu MX. 2012. Enhanced Th17 differentiation and aggravated arthritis in IEX-1-deficient mice by mitochondrial reactive oxygen species-mediated signaling. J Immunol 189: 1639–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gerriets VA, Kishton RJ, Nichols AG, Macintyre AN, Inoue M, Ilkayeva O, Winter PS, Liu X, Priyadharshini B, Slawinska ME, Haeberli L, Huck C, Turka LA, Wood KC, Hale LP, Smith PA, Schneider MA, MacIver NJ, Locasale JW, Newgard CB, Shinohara ML, and Rathmell JC. 2015. Metabolic programming and PDHK1 control CD4+ T cell subsets and inflammation. J Clin Invest 125: 194–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shin S, Wakabayashi N, Misra V, Biswal S, Lee GH, Agoston ES, Yamamoto M, and Kensler TW. 2007. NRF2 modulates aryl hydrocarbon receptor signaling: influence on adipogenesis. Mol Cell Biol 27: 7188–7197. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.