

Abstract
Retinoic acid (RA) signaling is required to restrict heart size through limiting the posterior boundary of the vertebrate cardiac progenitor field within the anterior lateral plate mesoderm (ALPM). However, we still do not fully understand how different cardiac progenitor populations that contribute to the developing heart, including earlier-differentiating first heart field (FHF), later-differentiating second heart field (SHF), and neural crest-derived progenitors, are each affected in RA-deficient embryos. Here, we quantified the number of cardiac progenitors and differentiating cardiomyocytes (CMs) in RA-deficient zebrafish embryos. While Nkx2.5+ cells were increased overall in the nascent hearts of RA-deficient embryos, unexpectedly, we found that the major effect within this population was a significant expansion in the number of differentiating FHF CMs. In contrast to the expansion of the FHF, there was a progressive decrease in SHF progenitors at the arterial pole as the heart tube elongated. Temporal differentiation assays and immunostaining in RA-deficient embryos showed that the outflow tracts (OFTs) of the hearts were significantly smaller, containing fewer differentiated SHF-derived ventricular CMs and a complete absence of SHF-derived smooth muscle at later stages. At the venous pole of the heart, pacemaker cells of the sinoatrial node also failed to differentiate in RA-deficient embryos. Interestingly, genetic lineage tracing showed that the number of neural-crest derived CMs was not altered within the enlarged hearts of RA-deficient zebrafish embryos. Altogether, our data show that the enlarged hearts in RA-deficient zebrafish embryos are comprised of an expansion in earlier differentiating FHF-derived CMs coupled with a progressive depletion of the SHF, suggesting RA signaling determines the relative ratios of earlier- and later-differentiation cardiac progenitors within an expanded cardiac progenitor pool.
Graphical Abstract
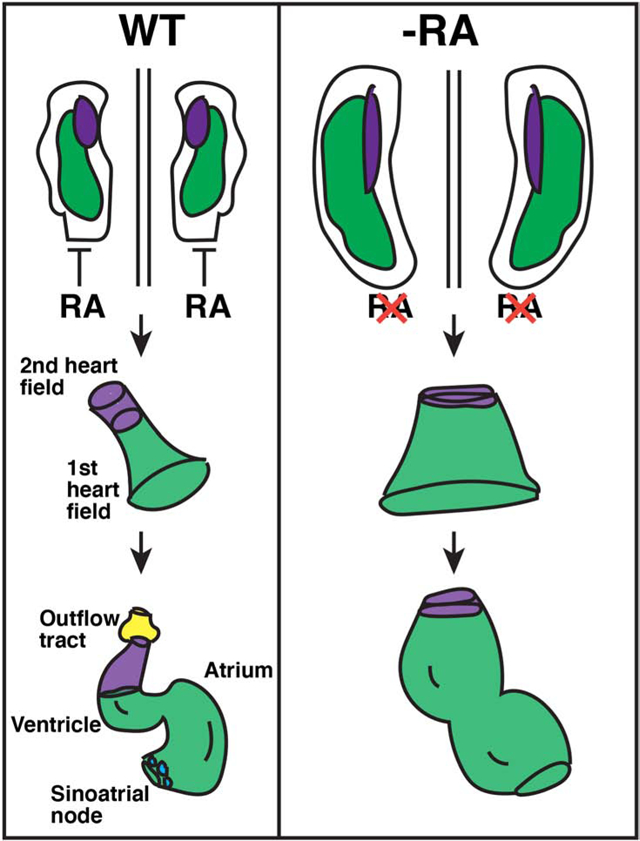
Introduction
Insufficient retinoic acid (RA) signaling results in congenital heart defects (CHDs) within all vertebrate embryos (Perl and Waxman, 2019; Stefanovic and Zaffran, 2017). Although numerous requirements for RA signaling in heart development have been defined, the earliest known role of RA signaling in vertebrate heart development is restriction of the cardiac progenitor pool within the anterior lateral plate mesoderm (ALPM) (Keegan et al., 2005; Ryckebusch et al., 2010; Sirbu et al., 2008; Waxman et al., 2008). Pharmacological inhibitors of RA signaling and neckless (nls) mutants (Begemann et al., 2001), which have a loss-of-function mutation in the major embryonic RA-producing enzyme Aldehyde dehydrogenase 1a2 (Aldh1a2 - formerly called Retinaldehyde dehydrogenase 2 (Raldh2)), have demonstrated that RA-deficiency in zebrafish embryos results in a posterior expansion of the cardiac progenitor field and enlarged hearts with a surplus number of differentiated atrial and ventricular CMs (Keegan et al., 2005; Waxman et al., 2008). Aldh1a2 knock-out (KO) mice also have a posterior expansion of heart field markers (Ryckebusch et al., 2008; Sirbu et al., 2008), supporting the conserved early requirements of RA signaling in limiting the cardiac progenitor pool. Furthermore, fate maps of the ALPM in zebrafish have shown that the augmentation of the cardiac progenitor pool within the ALPM of RA signaling-deficient embryos predominantly results from a posterior expansion into territories that normally harbor pharyngeal vascular, muscle, and forelimb progenitors, consistent with its requirement in anterior-posterior patterning (Sorrell and Waxman, 2011; Waxman et al., 2008). While studies in mice and zebrafish have shown that RA signaling in vertebrates divides the cardiac and forelimb progenitor fields within the ALPM through repression of FGF signaling (Sorrell and Waxman, 2011; Zhao et al., 2009), how RA signaling affects different vertebrate cardiac progenitor populations is still not fully understood.
The vertebrate heart is generated through the progressive temporal differentiation of different progenitor populations (Zaffran and Kelly, 2012). Earlier differentiating cardiac progenitors from within the ALPM called the first heart field (FHF) contribute primarily to the left ventricle in chickens and mice and approximately half of the single ventricle in zebrafish. Later differentiating cardiac progenitors that lie adjacent to the FHF called the second heart field (SHF) grow the heart at its poles, contributing to the right ventricle, outflow tract (OFT), and atria in four-chambered hearts, but primarily the arterial pole of the ventricle in zebrafish (Buckingham et al., 2005; Liu and Stainier, 2012). Despite studies examining these populations in RA-deficient mice (Ryckebusch et al., 2008; Sirbu et al., 2008), the requirements of RA signaling in these different progenitor cell populations remains unclear. The hearts of Aldh1a2 KO mice are severely dysmorphic and fail to loop, with defects in both the venous and arterial poles of the hearts proposed to be due exclusively to improper patterning and disorganization of the SHF (Niederreither et al., 1999; Ryckebusch et al., 2008). The posterior expansion of SHF markers within the ALPM of Aldh1a2 KO mice was associated with defects in differentiation markers at the venous pole (Ryckebusch et al., 2008; Sirbu et al., 2008). At the arterial pole, the OFT does not develop in Aldh1a2 KO mice, with in vitro explant experiments showing that cultured SHF progenitors from the Aldh1a2 KO mice fail to differentiate (Ryckebusch et al., 2008). Although both RA-deficient zebrafish and mice show a posterior expansion of the cardiac progenitors within the ALPM, the cardiac defects observed in Aldh1a2 KO mice have remained difficult to reconcile with the enlarged hearts of RA-deficient zebrafish embryos (Waxman et al., 2008). Assuming the requirements of RA signaling in early cardiac patterning are conserved, an exclusive expansion of the SHF markers in mice would imply the enlarged hearts of RA-deficient zebrafish are potentially due to increased SHF progenitors. However, in RA-deficient zebrafish an expansion of cardiac differentiation markers is evident by cardiac fusion stages (Waxman et al., 2008), which contrasts with the model proposed from mice and implies there may be an increase in FHF-derived CMs. Thus, the origins of the surplus CMs within the enlarged hearts of RA-deficient zebrafish embryos and, consequently, the conservation of the effects of loss of RA signaling in early cardiac patterning remain unclear.
In addition to the cardiac progenitors that originate from the ALPM, RA signaling is required for the proper addition of neural crest cells to the developing heart (El Robrini et al., 2016; Jiang et al., 2002; Niederreither et al., 2001). It has long been recognized that neural crest cells contribute to the mesenchyme within the OFT that is required for proper septation in four-chambered hearts (Kirby et al., 1983; Waldo et al., 1998). Neural crest cells in Aldh1a2 KO mice do not migrate properly and fail to populate the more proximal locations within the OFT leading to septation defects (El Robrini et al., 2016). Furthermore, conditional KOs of RA receptors (RARs) in the neural crest indicate that RA signaling non-cell autonomously influences neural crest function within the OFT (Jiang et al., 2002). Importantly, in addition to contributing to the OFT, more recent studies in zebrafish, chickens, and mice have demonstrated that cardiac neural crest cells also have conserved requirements contributing to CMs within the chambers (Abdul-Wajid et al., 2018; Cavanaugh et al., 2015; Tang et al., 2019). In zebrafish, an initial wave of neural crest cells contribute to the CMs in both chambers of the heart, while a later posterior wave of neural crest cells contribute to smooth muscle within the OFT (Cavanaugh et al., 2015), reminiscent with what has predominantly been studied in mice (Jiang et al., 2002; Waldo et al., 1998). However, we currently do not understand how the neural crest-derived CM progenitors are affected by loss of RA and if their expansion contributes to the enlarged hearts of RA-deficient embryos.
Here, we examined the effects of RA deficiency on the different CM progenitor sources in zebrafish embryos. Unexpectedly, we find that the primary contribution to the enlarged hearts of RA-deficient embryos are FHF-derived CMs. While the number of SHF progenitors is not initially affected in RA-deficient embryos, there is a subsequent reduction in the number of SHF progenitors at the arterial pole as the tube elongates, supporting that there is a progressive expansion of the FHF, which occurs in part at the expense of these SHF progenitors. Photoconversion-mediated lineage tracing showed that fewer SHF-derived CMs differentiate at the arterial pole and that there is a complete lack of SHF-derived smooth muscle within the OFT at later stages within the enlarged hearts of RA-deficient embryos, consistent with a failure of anterior SHF progenitors to differentiate in RA-deficient mice. Furthermore, at the venous pole of the heart in RA-deficient zebrafish embryos, pacemaker CMs of the sinoatrial node (SAN) fail to differentiate, which is similar to differentiation defects in the posterior SHF found in RA-deficient mice. By examining neural crest-derived CMs with genetic lineage tracing, we also found that there was not an effect on the number of neural crest-derived CMs in RA deficient embryos, indicating that neural crest cells do not contribute to enlargement of the hearts. Altogether, our data show that early RA signaling predominantly restricts the size of the FHF within the enlarged progenitor field and has conserved requirements promoting SHF differentiation in OFT and SAN differentiation at the venous pole, which provides new insights into the mechanisms underlying CHDs in RA-deficient vertebrates.
Results
RA promotes posterior nkx2.5+ progenitor specification within the ALPM
Despite previous studies in mice and zebrafish showing that RA signaling has a conserved requirement restricting the size of the cardiac progenitor field within the ALPM (Keegan et al., 2005; Ryckebusch et al., 2008; Sirbu et al., 2008; Waxman et al., 2008), how RA signaling affects the populations of earlier-differentiating FHF and later-differentiating SHF progenitors remains poorly understood. Furthermore, while previous work demonstrated RA-deficient zebrafish embryos have an expanded pool of progenitors expressing nkx2.5 during somitogenesis (Keegan et al., 2005), how RA signaling affects this progenitor population has not been examined in light of the studies showing the broader contributions of nkx2.5+ progenitors to the pharyngeal vasculature and muscles (Paffett-Lugassy et al., 2017; Paffett-Lugassy et al., 2013). Employing the nkx2.5:Kaede transgenic line, which recapitulates nkx2.5 expression and permits photoconversion-mediated lineage tracing that was used in subsequent experiments, we first examined its expression during mid- to late-somitogenesis stages (14 through 20 somite (s) stages; 16–19 hours post-fertilization (hpf)). During these developmental stages, the bilateral CM progenitors migrate to the midline, fuse to generate the cardiac cone, and the first aggregates of the posterior-lateral pharyngeal arch artery (PAA) progenitors form (Paffett-Lugassy et al., 2017; Paffett-Lugassy et al., 2013). In situ hybridization (ISH) of embryos treated with DMSO (control) and diethyl-aminobenzoic acid (DEAB), which inhibits the RA-producing retinaldehyde dehydrogenases (Aldh1as), beginning at the shield stage showed an enlargement in the nkx2.5 expression domain at the 14s stage (16 hpf; Fig. 1A,B), consistent with previous results (Keegan et al., 2005). However, in addition to corroborating previous observations, we found that at the 18s and 20s stages (18 and 19 hpf), the posterior nkx2.5 PAA populations failed to segregate in the RA-deficient embryos (Fig. 1D,F). The loss of these more posterior nkx2.5+ aggregates is consistent with the loss of pharyngeal arches observed at later stages (Begemann et al., 2001) and is reminiscent of previous examination of tbx5+ progenitor cells that fail to segregate into more anterior cardiac and posterior forelimb populations in RA-deficient embryos (Waxman et al., 2008).
Figure 1. RA signaling promotes specification of posterior nkx2.5+ progenitors.
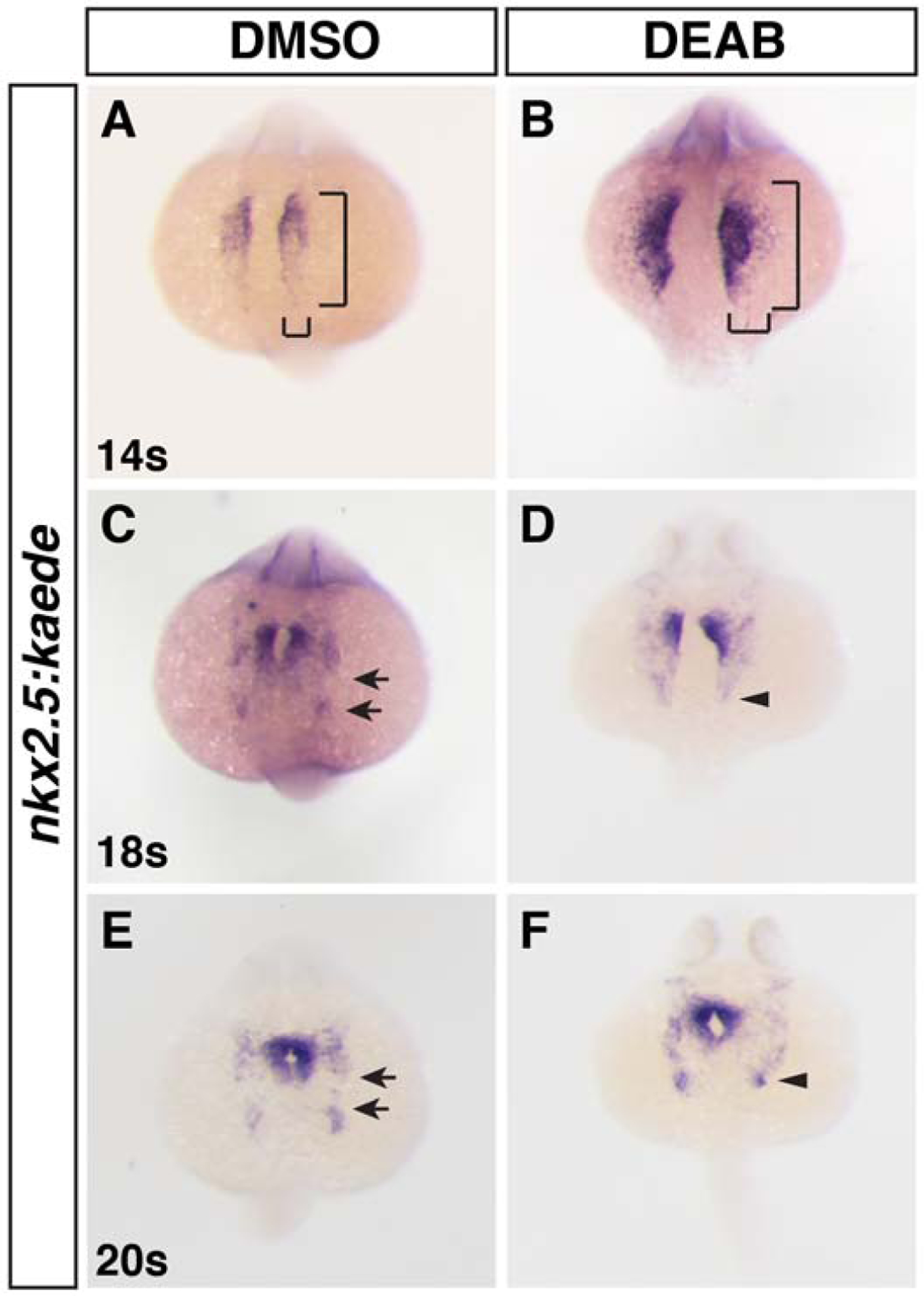
(A-F) ISH for nkx2.5:Kaede (Kaede) in DMSO-treated (control) and DEAB-treated embryos at the 14s, 18s, and 20s stages (16, 18, and 19 hpf). Brackets in A and B indicate the length and width of the nkx2.5:Kaede expression. Nkx2.5 expression is expanded laterally and posteriorly and the posterior pharyngeal artery progenitors are not a distinct aggregate in RA deficient embryos. Views are dorsal with anterior up. Arrows in C and E indicate the space between the anterior nkx2.5+ and posterior nkx2.5+ aggregates. Arrows in D and F indicate the posterior limit of nkx2.5 expression and lack of posterior nkx2.5 aggregates. n = 25 for all conditions examined.
The enlargement of the nkx2.5+ progenitor field and loss of the posterior nkx2.5+ PAA cell population is consistent with anteriorization shown previously for RA-deficient embryos (Ryckebusch et al., 2008; Waxman et al., 2008). Moreover, previous caged-fluorescein-mediated lineage tracing showed that in RA-deficient embryos cardiac progenitors extend more posteriorly within the ALPM into regions that normally harbor pharyngeal muscle, arch arteries, and forelimb progenitors (Waxman et al., 2008). Given that nkx2.5+ cells can contribute to multiple lineages (Guner-Ataman et al., 2013; Paffett-Lugassy et al., 2017), we wanted to confirm nkx2.5+ CM progenitors are expanded posteriorly within the ALPM of RA-deficient embryos. To assess fates of nkx2.5+ progenitors within the ALPM, we used the nkx2.5:Kaede transgenic line to perform lineage tracing via photoconversion in DMSO-treated (control) and DEAB-treated embryos. Small groups of nkx2.5:Kaede+ cells were photoconverted from green to red in anterior and posterior regions of the lateral expression domain at the 20s stage (19 hpf) and examined for their contribution to heart, OFT, ventral aorta, and PAAs at 48 hpf (Holowiecki et al., 2020; Rydeen and Waxman, 2016). Converting nkx2.5:Kaede+ cells more anterior and lateral to the forming cardiac cone in control embryos at the 20s stage (19 hpf) resulted in labeled cells that predominantly contribute to the OFT and ventral aorta (Fig. 2A–C). However, in RA signaling-deficient embryos, nkx2.5:Kaede+ cells that were converted in a similar region also contributed to CMs (Fig. 2D–F). Converting lateral nkx2.5:Kaede+ cells more posteriorly in control embryos showed that these cells predominantly contributed to the 3rd PAA (Fig. 2G–I), consistent with previous studies (Paffett-Lugassy et al., 2013). However, the posterior nkx2.5:Kaede+ progenitors in RA-deficient embryos contributed to the OFT, ventral aorta, and CMs in the arterial pole of the heart (Fig. 2J–L), similar to labeling cells more anteriorly. Therefore, our lineage tracing shows that within the enlarged nkx2.5+ field of RA-deficient embryos progenitors, nkx2.5+ progenitors that give rise to CMs are expanded posteriorly.
Figure 2. Cardiac progenitors are expanded posteriorly within the nkx2.5+ progenitor field.
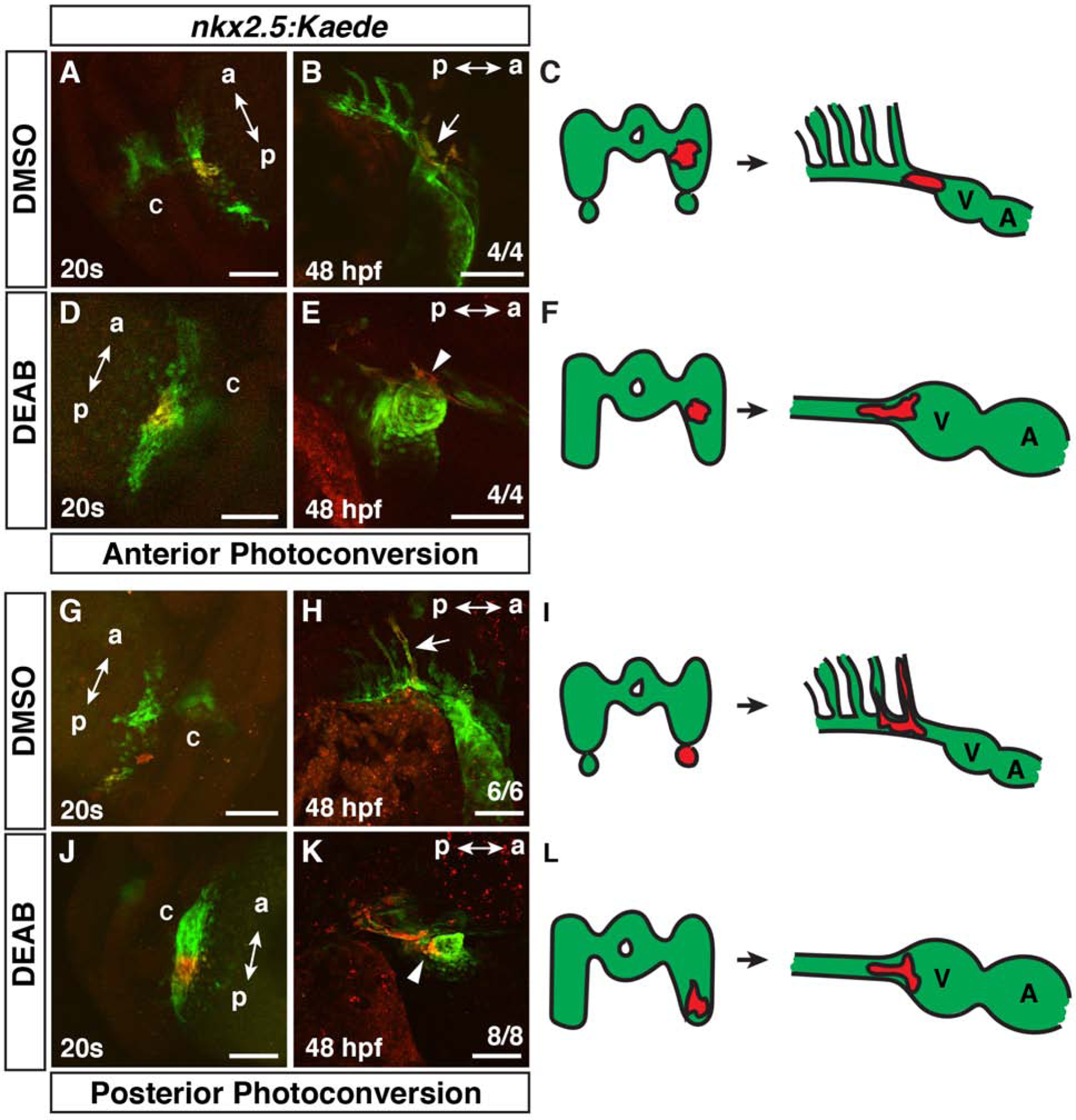
(A,B,D,E,G,H,J,K) Photoconversion-mediated lineage tracing of nkx2.5:Kaede+ progenitor cells in DMSO- and DEAB-treated embryos at the 20s stage (19 hpf) and at 48 hpf. Views in A,D,G, J are dorso-lateral with anterior up. Views in B,E,H,K are lateral with anterior right and dorsal up. a – anterior, p – posterior, c - cardiac cone. Embryos at 48 hpf (B,E,H,K) are the same embryos shown with photoconverted cells at the 20s stage (19 hpf) in (A,D,G,J). Arrow in B indicates photoconverted cells in the ventral aorta and OFT. (C,F,I,L) Schematics summarizing the lineage tracing in the anterior and posterior lateral nkx2.5:Kaede+ fields. V – ventricle, A – Atrium. In the anterior lateral nkx2.5+ region, 4 of 4 control embryos had labeled cells give rise to the ventral aorta/OFT, while 1 of the 4 also labeled the 3rd PAA. 4 of 4 DEAB-treated embryos with nkx2.5:Kaede+ cell clusters labeled in similar regions gave rise to the ventral aorta, OFT, and CMs. In the posterior lateral nkx2.5:Kaede+ field, 6 of 6 control embryos had photoconverted cells contribute to the 3rd PAA. 8 of 8 DEAB-treated embryos had photoconverted cells give rise to the ventral aorta, OFT, and CMs. Arrow in H indicates photoconverted cells in the 3rd PAA. Arrowheads in E and K indicated photoconverted CMs at the arterial pole. Scale bars – 100 μm.
RA-deficient zebrafish embryos have an increase in FHF-derived CMs and a reduction in SHF-derived CMs
The enlarged hearts of RA signaling-deficient zebrafish embryos are comprised of an increase in both atrial and ventricular CMs (Waxman et al., 2008). However, the origin of the progenitors that give rise to the surplus CMs has not been determined. Nkx2.5+ progenitors contribute to both the FHF and SHF (Guner-Ataman et al., 2013; Holowiecki et al., 2020; Paffett-Lugassy et al., 2017). Therefore, the expansion of Nkx2.5+ progenitors in the ALPM of RA-deficient embryos did not itself reveal the origin of the surplus CMs. Furthermore, although an expansion of CM differentiation markers can be seen as early as the 18–20s stages (18–19 hpf) in RA-deficient zebrafish embryos (Waxman et al., 2008), implying that their increase in CMs may in part be due to increased FHF progenitors, studies in mice have concluded there is only an expansion of the posterior SHF (Ryckebusch et al., 2008; Sirbu et al., 2008). To determine how loss of RA signaling affects these different cardiac progenitor populations, we quantified the number of earlier differentiating CMs (FHF) and cardiac progenitors (presumptive later-differentiating SHF) in WT and aldh1a2/nls zebrafish mutant embryos at the 20s stage (19 hpf) and 24 hpf. To distinguish between these fields, immunohistochemistry (IHC) was performed for Nkx2.5, which marks cardiac progenitors and differentiating CMs, and sarcomeric myosin (Mhc), which marks differentiating/differentiated CMs (Fig. 3A,B,E,F). Quantification of FHF, defined as Nkx2.5+/Mhc+ cells, and SHF, defined as Nkx2.5+/Mhc− cells, showed that the overall number of Nkx2.5+ cells was increased (Fig. 3C,G), consistent with the ISH (Fig. 1) and previous analysis (Keegan et al., 2005). However, within this cell population, the number of FHF cells and their proportion of the total Nkx2.5+ population was increased at both the 20s stage (19 hpf) and 24 hpf (Fig. 3C,D,G,H). In contrast to the earlier differentiating CMs, the number of SHF cells was not affected at the 20s stage (19 hpf) in RA-deficient embryos (Fig. 3C,D), but subsequently decreased at 24 hpf in nls embryos relative to WT embryos (Fig. 3G,H), implying that Nkx2.5+/Mhc− progenitors may be precociously differentiating and not maintaining the size of the SHF progenitor pool. Virtually equivalent results were found examining DEAB-treated embryos with the myl7:GFP transgene, which also marks differentiating CMs, although there was a more dramatic loss of SHF progenitors at 24 hpf (Fig. 4A–H). Together, these results show that the enlarged progenitor field of RA-deficient embryos within the ALPM is primarily the result of an expansion in FHF progenitors, along with a progressive loss of SHF progenitors.
Figure 3. RA restricts the size of the FHF and maintains the SHF progenitor population in nls mutant embryos.
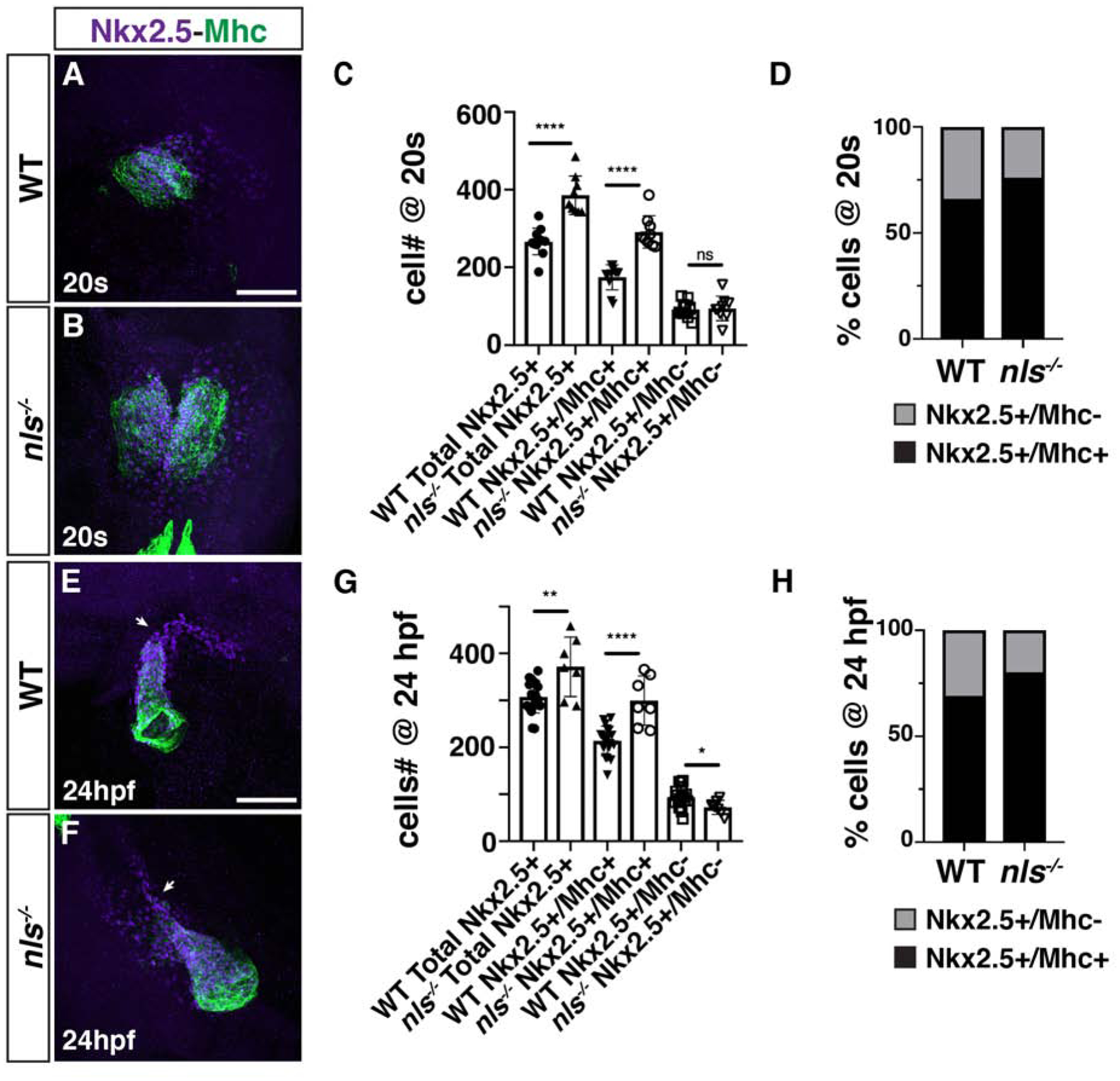
(A, B) IHC for Nkx2.5 (purple) and Mhc (green) in WT sibling and nls mutant embryos at the 20s stage (19 hpf). Nls mutant hearts were sometimes delayed in forming the cardiac cone relative to WT siblings. Views are dorsal with anterior up. Scale bar – 100 μm. (C) Quantification of the number of Nkx2.5+/Mhc+ and Nkx2.5+/Mhc− cells in WT sibling and nls mutant embryos at the 20s stage (19 hpf). **** indicates p<0.0001. ns – not significant. (D) Percentage of Nkx2.5+/Mhc+ and Nkx2.5+/Mhc− cells within the hearts of WT sibling and nls mutant embryos at the 20s stage (19 hpf). (E, F) IHC for Nkx2.5 (purple) and Mhc (green) in WT sibling and nls mutant embryos at 24 hpf. Frontal views with the arterial pole up. Arrows indicate the border between Nkx2.5+/Mhc+ and Nkx2.5+/Mhc− cells. Scale bar – 100 μm. (G) Quantification of the number of Nkx2.5+/Mhc+ and Nkx2.5+/Mhc− cells in WT sibling and nls mutant embryos at 24 hpf. * indicates p=0.0253, ** indicates p=0.0026, **** indicates p<0.0001. (H) Percentage of Nkx2.5+/Mhc+ and Nkx2.5+/Mhc− cells within the hearts of WT sibling and nls mutant embryos at 24 hpf.
Figure 4. RA restricts the size of the FHF and maintains the SHF progenitor population in DEAB-treated embryos.
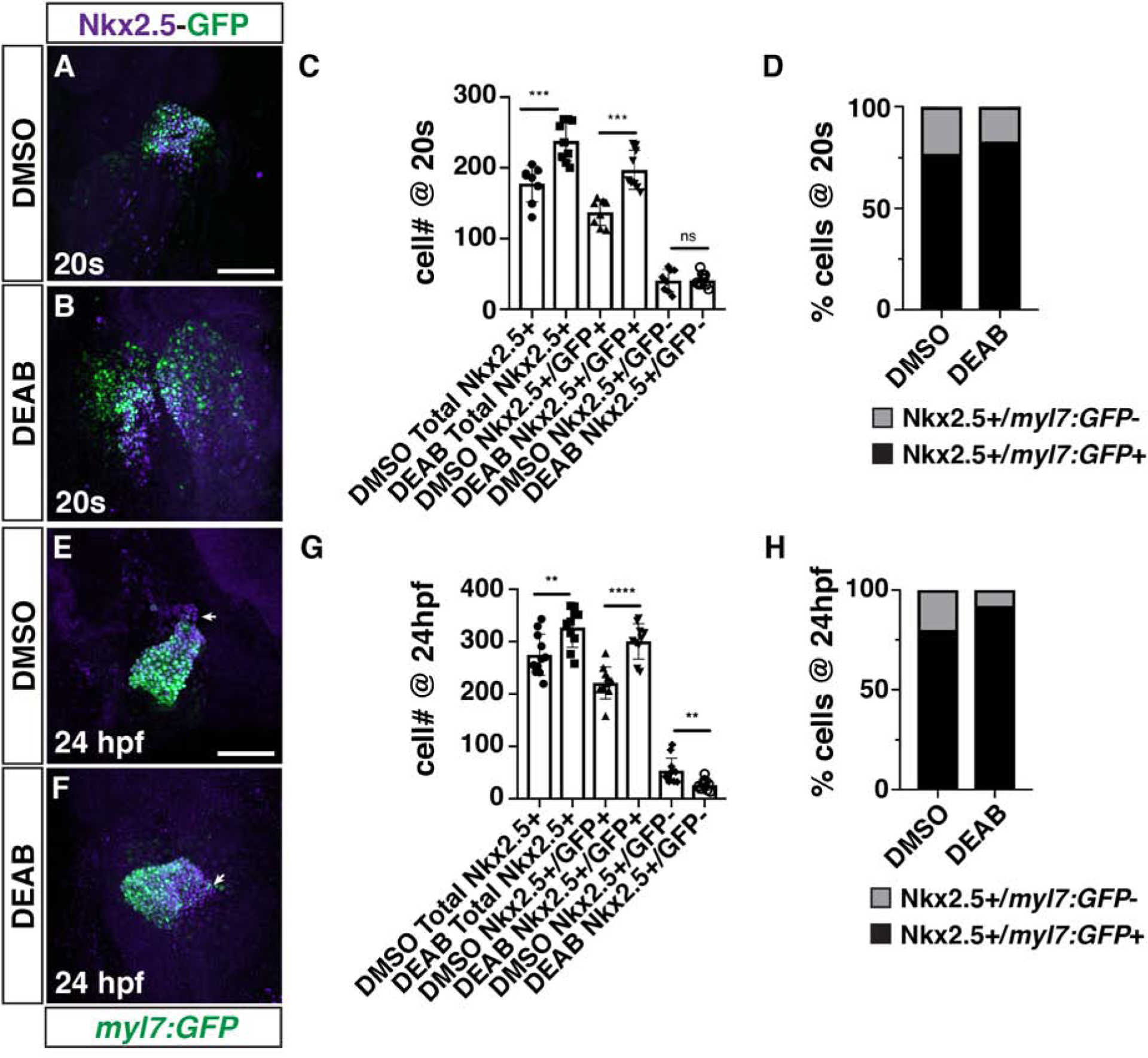
(A, B) IHC for Nkx2.5 (purple) and GFP (myl7:GFP - green) in DMSO- and DEAB-treated embryos at the 20s stage (19 hpf). The cardiac cone was sometimes delayed in forming in DEAB-treated embryos relative to DMSO-treated control embryos. Views are dorsal with anterior up. Scale bar – 100 μm. (C) Quantification of the number of Nkx2.5+/GFP+ and Nkx2.5+/GFP− cells in DMSO- and DEAB-treated embryos at the 20s stage (19 hpf). *** indicates p≤0.0004. (D) Percentage of Nkx2.5+/GFP+ and Nkx2.5+/GFP− cells within the hearts of DMSO- and DEAB-treated embryos at the 20s stage (19 hpf). (E, F) IHC for Nkx2.5 (purple) and GFP (myl7:GFP - green) in DMSO- and DEAB-treated embryos at 24 hpf. Frontal views with the arterial pole up. Arrows indicate the border between Nkx2.5+/GFP+ and Nkx2.5+/GFP− cells. Scale bar – 100 μm. (G) Quantification of the number of Nkx2.5+/GFP+ and Nkx2.5+/GFP− cells in DMSO- and DEAB-treated embryos at 24 hpf. ** indicates p≤0.0060, **** indicates p<0.0001. (H) Percentage of Nkx2.5+/GFP+ and Nkx2.5+/GFP− cells within the hearts of DMSO- and DEAB-treated embryos at 24 hpf.
Fewer SHF-derived cells differentiate at the OFT of RA-deficient zebrafish embryos
Given the progressive decrease in SHF (Nkx2.5+/Mhc−) cells in the absence of RA signaling at early stages of heart development, we were next interested to see if this trend resulted in decreased SHF-derived CMs at later stages. To examine the effect of RA-deficiency on the SHF-derived CMs in zebrafish embryos, we used a temporal differentiation assay with myl7:nls-KikGR transgenic embryos whereby earlier differentiating KikGR-expressing CMs are converted from green to red at 24 hpf and the number of later-differentiating green-only CMs are quantified at 48 hpf (Lazic and Scott, 2011). Consistent with the reduced number of SHF progenitors observed at 24 hpf, we found that there was a decrease in the number of green-only, later-differentiating SHF-derived ventricular CMs in the OFT of DEAB-treated embryos (Fig. 5A–C). To examine the effect of RA deficiency on differentiating SHF progenitors within the arterial pole of the developing heart tube, we performed ISH for mef2cb (Lazic and Scott, 2011) and ltbp3 (Zhou et al., 2011) in control and DEAB-treated embryos at 30 hpf. Similar to the appearance of SHF-derived KikGR-expressing ventricular CMs, both these markers were restricted to thin lines of cells at the arterial pole in DEAB-treated embryos, while in control embryos their expression covered a significant portion of the arterial pole (Fig. 6A–D). Since the SHF can also give rise to smooth muscle cells in the OFT, we performed IHC for Elastin b (Elnb) at 72 hpf, when these smooth muscle cells in the OFT starts to differentiate. Unlike WT sibling and DMSO-treated control embryos (Fig. 7A,C), nls mutant and DEAB-treated embryos lacked detectable Elnb in their OFTs (Fig. 7B,D). Thus, our results support that ventricular and smooth muscle-derived SHF-derived cells of the OFT are significantly reduced or absent in RA-signaling deficient embryos.
Figure 5. Reduced ventricular CMs differentiate at the arterial pole of RA-deficient embryos.
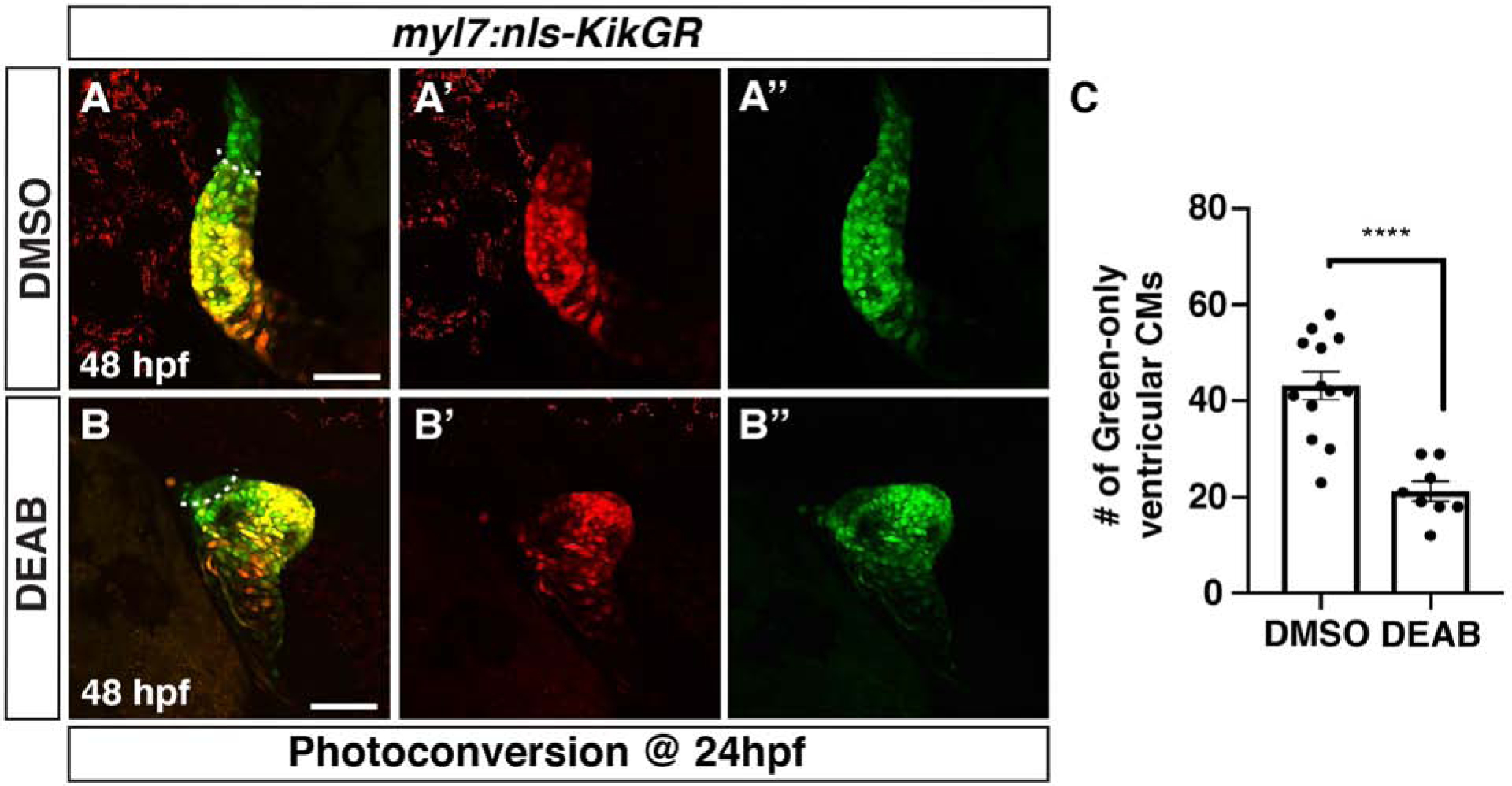
(AB”) DMSO- and DEAB-treated myl7:nls-KikGR embryos at 48 hpf. Embryos were photoconverted at 24 hpf. Dashed line demarcates the border between green-only and green+/red+ ventricular CMs. Views are lateral with the arterial pole up. Scale bars – 100 μm. (C) Quantification of the number of green-only cells within the ventricle. **** indicates p<0.0001.
Figure 6. SHF markers of the arterial pole are reduced in RA-deficient embryos.
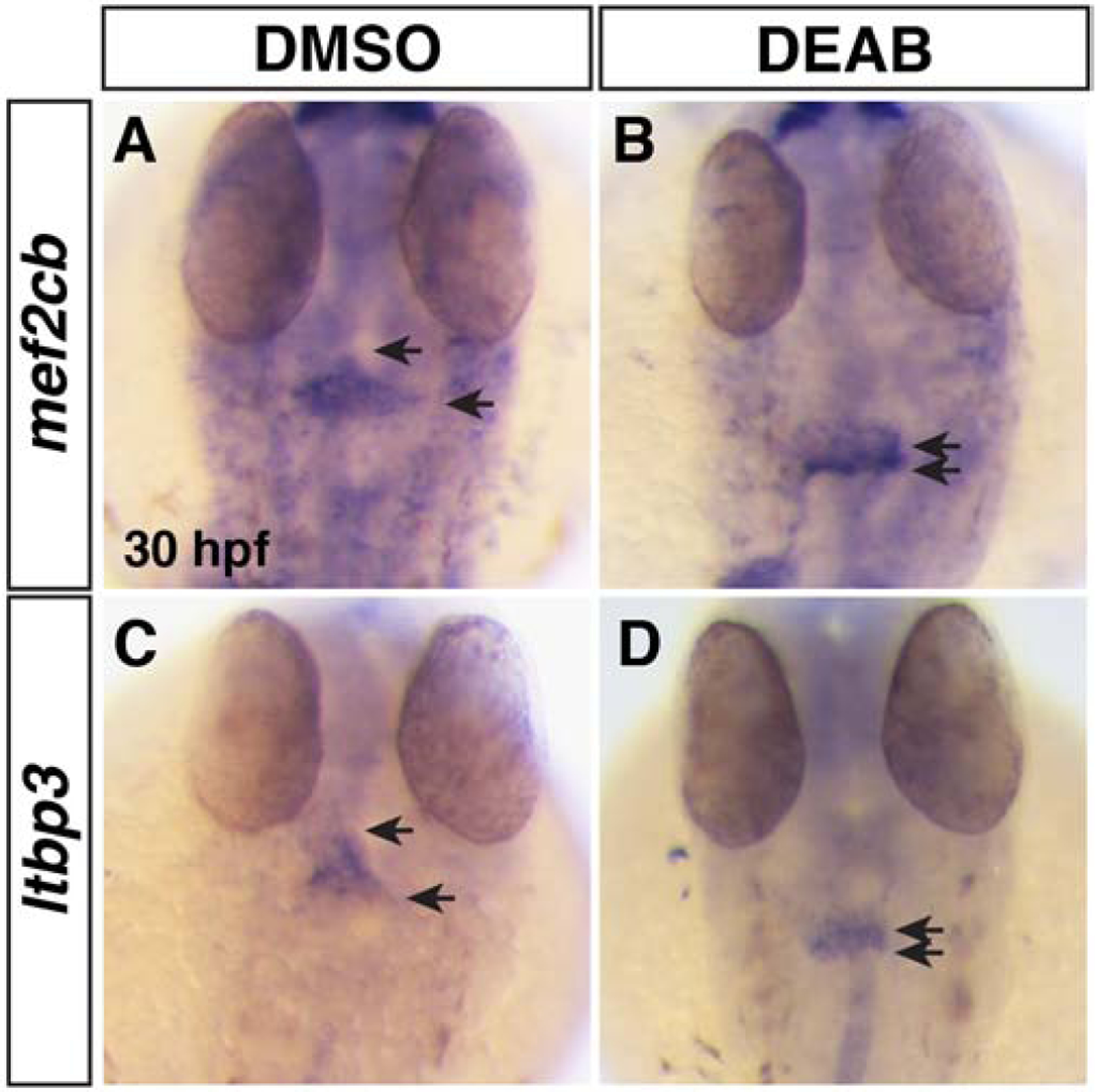
(A,B) ISH for mef2cb at 30 hpf in DMSO- and DEAB-treated embryos. (C,D) ISH for ltbp3 at 30 hpf in DMSO- and DEAB-treated embryos. Number of embryos examined per condition: mef2cb DMSO-treated n=31; DEAB-treated n=18; for ltbp3 DMSO-treated n=18; DEAB-treated n=16. Views are dorsal with anterior up. Arrows indicate the length of expression at the arterial poles.
Figure 7. Smooth muscle within the OFT is absent in RA-deficient embryos.
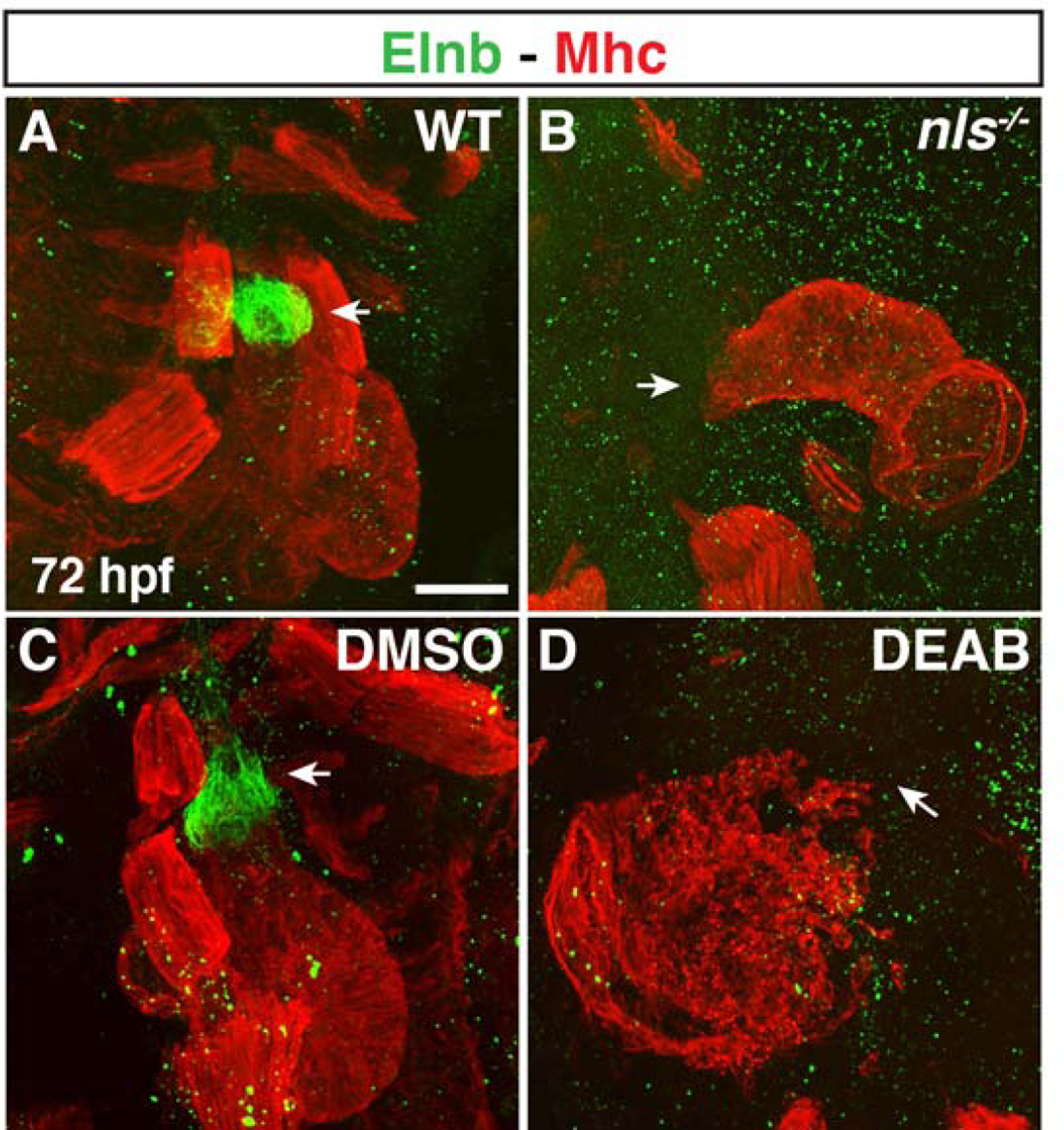
(A-D) IHC of Elnb (green) and Mhc (red) in hearts of (A,B) WT sibling and nls mutant embryos and (C,D) DMSO- and DEAB-treated embryos at 72 hpf. Number of embryos examined per condition: WT n=6, nls n=8, DMSO-treated n=12, DEAB-treated n=9. Views are lateral with arterial pole of the heart up. Arrows indicate the Elnb in WT sibling and DMSO-treated and the lack of Elnb in nls mutant and DEAB-treated at the arterial poles of the hearts. Scale bar – 50 μm.
The sinoatrial node (SAN) fails to differentiate in RA-deficient embryos
In mice, the posterior expansion of the SHF is accompanied by defects at the venous pole, including a loss of multiple BMPs and Tbx5 in the SAN, which harbors the specialized pacemaker CMs of the heart (Christoffels et al., 2010; Ryckebusch et al., 2008; Sirbu et al., 2008). Therefore, we wanted to determine if loss of RA signaling also affects the differentiation of the SAN at the venous pole in zebrafish. IHC for Isl1, a conserved marker of the SAN (de Pater et al., 2009; Tessadori et al., 2012), showed that Isl1+ cells are missing in DEAB-treated embryos (Fig. 8A,B). Moreover, using the enhancer trap line sqet33-mi59B, which reports fgf13a expression and marks the SAN with the atrium as well as additional cells adjacent to the venous pole of the heart (Poon et al., 2010), corroborated that the SAN is lost in RA-deficient embryos (Fig. 8C,D). Therefore, our results show that there are also conserved defects in the differentiation of cells at the venous pole in RA-deficient zebrafish embryos.
Figure 8. Pacemaker CMs do not differentiate in RA-deficient embryos.
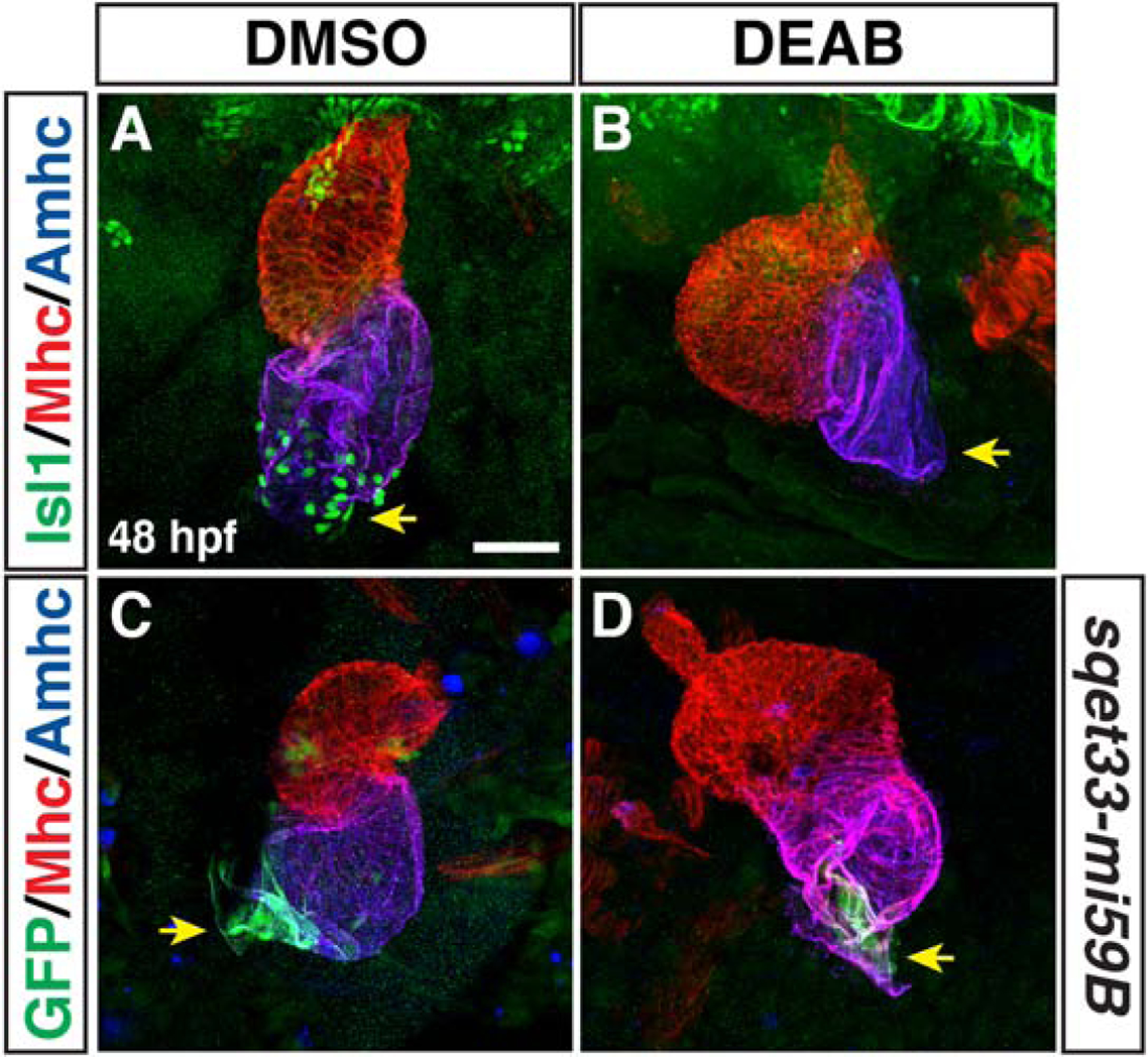
(A,B) IHC of Isl1 (green), Mhc (red), Amhc (blue) in hearts of DMSO- and DEAB-treated embryos at 48 hpf. Number of embryos examined: DMSO-treated n=25, DEAB-treated n=21. Arrows in A and B indicate Isl1 expression and the lack of Isl1 at the venous poles of the hearts in control and DEAB-treated embryos, respectively. (C,D) IHC of GFP (green), Mhc (red), Amhc (blue) in hearts of DMSO- and DEAB-treated sqet33-mi59B embryos at 48 hpf. Number of embryos examined: DMSO-treated n=11, DEAB-treated n=11. Arrows in C and D indicate the sqet33-mi59B GFP expression and its reduced expression at the venous poles of the hearts in control and DEAB-treated embryos, respectively. The remaining sqet33-mi59B expression in DEAB-treated embryos is adjacent to the atrial CMs. Views are ventral with anterior up. Scale bar – 50 μm.
Anterior pharyngeal muscles are not affected in RA-deficient embryos
Previous lineage tracing suggested that the SHF progenitors that contribute to the OFT in zebrafish embryos are located more anterior and medial to the FHF ventricular progenitors within the ALPM (Hami et al., 2011). Furthermore, nkx2.5+ progenitors that are located anterior and lateral to the forming heart tube in the 2nd arch will also give rise to later-differentiating cells in the OFT as well as dorsal-anterior pharyngeal muscles that are immediately posterior to the eye (Nagelberg et al., 2015; Paffett-Lugassy et al., 2017). The anterior pharyngeal muscles (dorsal mandibular and hyoid) are dysmorphic in nls mutants (Begemann et al., 2001). Therefore, we sought to understand if the contribution of anterior nkx2.5+ progenitors to the pharyngeal muscles was affected in RA-deficient embryos. To examine the contributions of nkx2.5+ cells to these pharyngeal muscles we used the nkx2.5:ZsYellow transgenic line. Although endogenous Nkx2.5 expression is not maintained in these cells, the nkx2.5:ZsYellow transgene labels the anterior pharyngeal muscles that are in part derived from the nkx2.5+ cells due to the perdurance of ZsYellow (Paffett-Lugassy et al., 2017). We found that while the anterior pharyngeal muscle had morphological defects, as has been previously reported (Begemann et al., 2001), the contribution of nkx2.5-derived cells was not overtly affected in DEAB-treated embryos (Fig. 9A–B”). As with DMSO-treated controls, the contribution of nkx2.5:ZsYellow+ cells increased from the anterior levator arcus palatini (lap) to the posterior adductor opercula (ao) muscles. Thus, our data suggest that despite a failure of the adjacent anterior SHF-derived cells to differentiate in RA-deficient embryos, loss of RA does not overtly affect the differentiation of anterior 2nd-arch derived pharyngeal muscle.
Figure 9. Anterior pharyngeal muscles are unaffected in RA-deficient embryos.
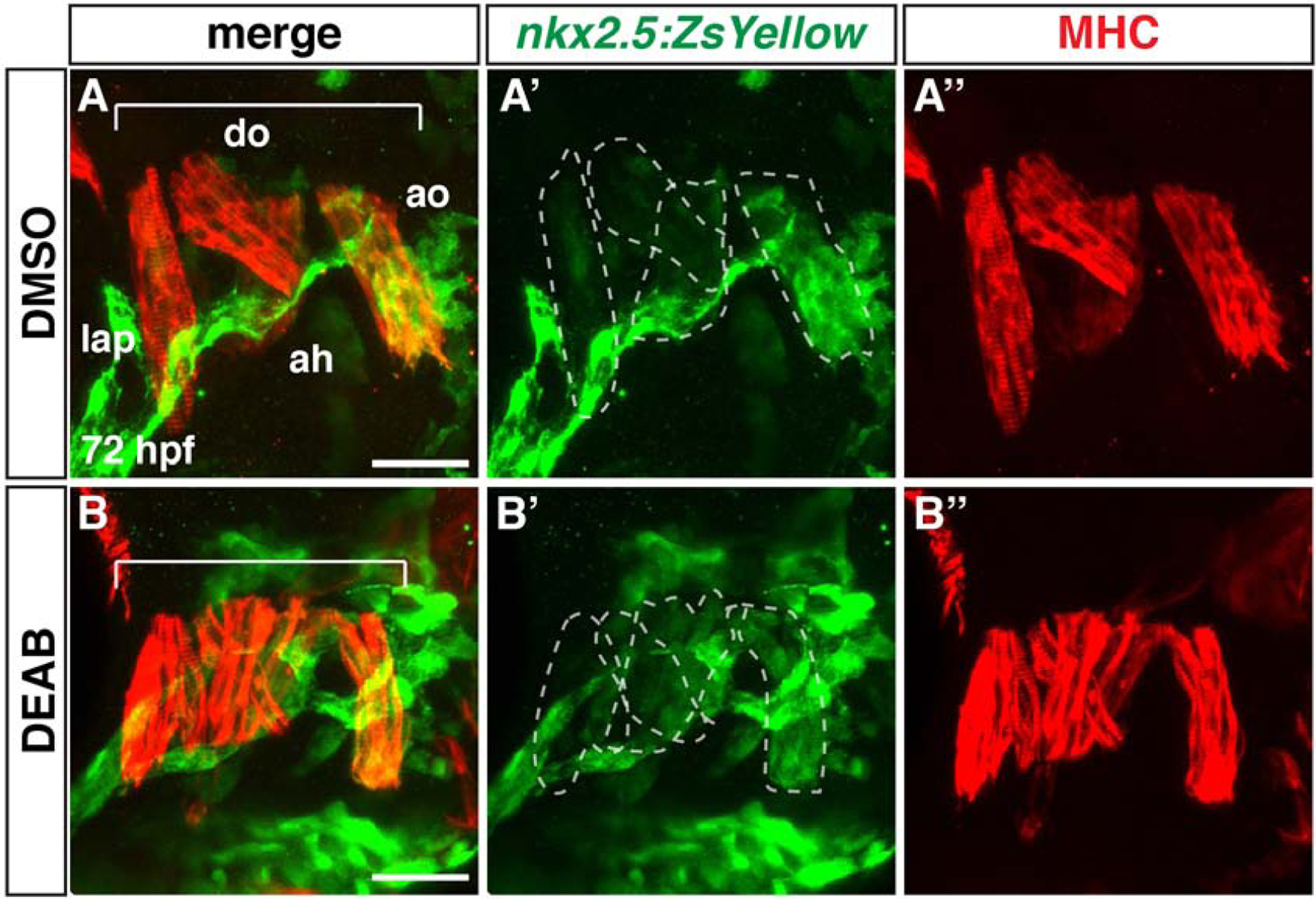
(A-B”) IHC for ZsYellow+ (green) and Mhc+ (red) pharyngeal muscles in DMSO- and DEAB-treated nkx2.5:ZsYellow embryos at 72 hpf. Views are frontal with anterior left and dorsal up. Brackets indicate the dorsal pharyngeal muscles. Dorsal mandibular arch: lap - levator arcus palatini, do - dilatator opercula. Dorsal hyoid arch: ah - adductor hyomandibulae, ao - adductor opercula. The dashed lines indicate the nkx2.5:ZsYellow+ muscles. The dorsal mandibular and hyoid arch muscles are dysmorphic in RA-deficient embryos. Number of embryos examined: DMSO-treated n=9, DEAB-treated n=6. Scale bar – 33 μm.
Neural crest derived CMs are not increased in RA signaling-deficient embryos
In mammals, RA-deficiency affects the migration of neural crest cells that contribute to smooth muscle within the OFT (El Robrini et al., 2016; Jiang et al., 2002). However, recent lineage-tracing studies in zebrafish have highlighted that distinct waves of neural crest contribute to CMs and smooth muscle: an earlier wave of anterior neural crest cells contribute to CMs within the heart, while a later posterior wave contributes to smooth muscle in the OFT and ventral aorta (Cavanaugh et al., 2015). Given the known effects of RA signaling on neural crest cells that contribute to the developing heart (El Robrini et al., 2016; Jiang et al., 2002; Niederreither et al., 2001), we sought to understand if the anterior neural crest-derived CMs were affected in RA-deficient embryos. To examine neural crest-derived CMs, we used the established neural crest lineage transgenic reporters sox10:gal4, UAS:Cre; ubb:loxP-EGFP-loxP-mCherry (referred to as NC:mCherry) (Cavanaugh et al., 2015). We found that there was not an effect on the number of neural crest-derived CMs in DEAB-treated embryos compared to control embryos (Fig. 10A–C). Thus, the surplus CMs in RA-deficient embryos appear to be solely due to an increase in FHF-derived CMs.
Figure 10. Neural crest-derived CMs are not affected in RA-deficient embryos.
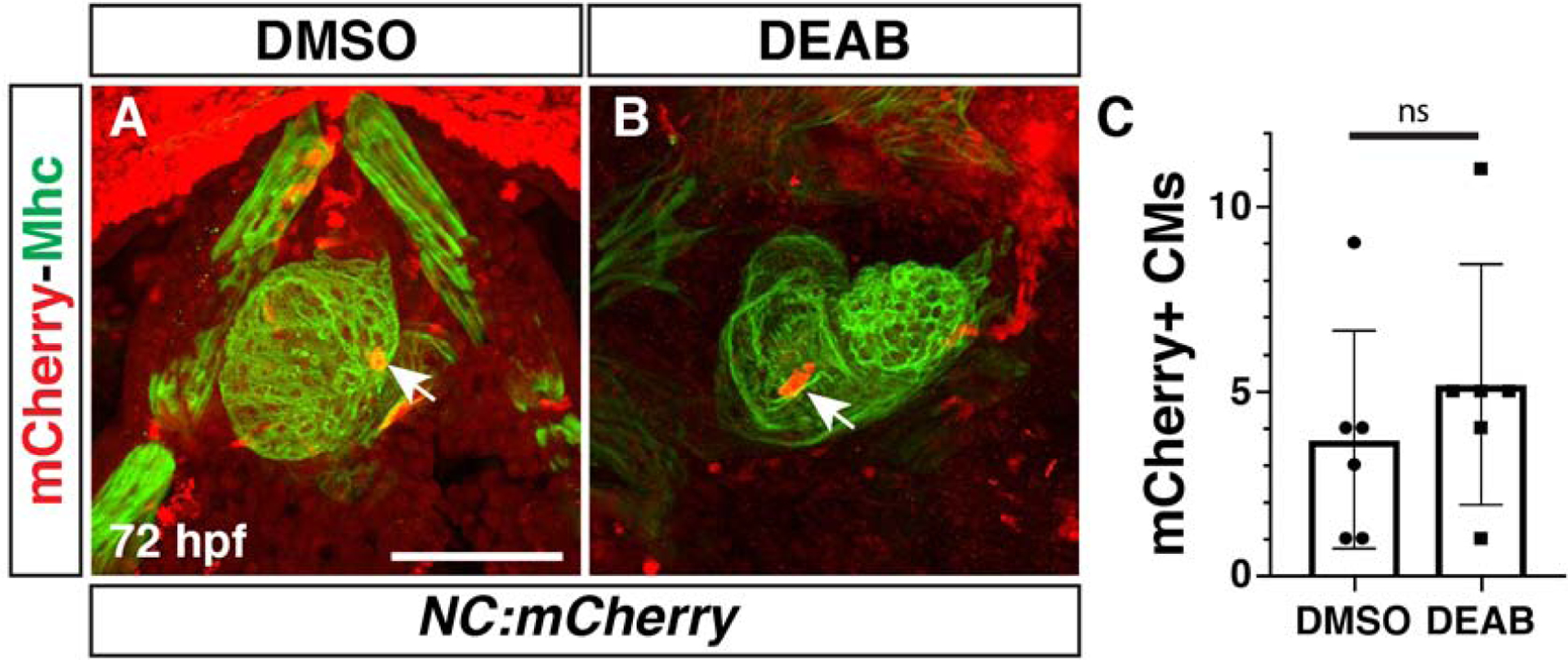
(A,B) IHC for mCherry (red) and Mhc (green) in DMSO- and DEAB-treated NC:mCherry embryos at 72 hpf. Views are ventral with anterior up. Arrows indicate labeled CMs in hearts. (C) Quantification of the NC:mCherry+ cells in the hearts of DMSO-treated and DEAB-treated embryos. Scale bar – 100 μm.
Discussion
Here, we have bridged gaps in our understanding of the requirements of RA signaling in patterning the vertebrate cardiac progenitor fields. Importantly, our data employing lineage tracing and cell quantification in zebrafish embryos support a model whereby early RA deficiency causes an expansion of FHF cardiac progenitors and not the SHF or neural-crest derived CMs (Fig. 11). Instead, SHF progenitors that generate the OFT at the arterial pole of the heart are progressively reduced and fail to differentiate, while pacemaker CMs at the venous pole of the atria also fail to differentiate. Hence, the consequences of early RA loss on the hearts of zebrafish embryos mirror defects in the anterior and posterior SHF-derived populations found in RA-deficient mice (Ryckebusch et al., 2008). An expansion in FHF-derived CMs shown here is consistent with the increase in early atrial and ventricular CM differentiation markers and differentiated CMs at later stages in RA-deficient zebrafish reported previously (Waxman et al., 2008). However, an increase in FHF-derived CMs from RA depletion contrasts with interpretations from work in Aldh1a2 KO mice, which concluded that the FHF was either not affected or possibly diminished relative to the SHF (Ryckebusch et al., 2008; Sirbu et al., 2008). While there could be species-specific differences with respect to the allocation of these progenitor populations in mice and zebrafish, we suspect one reason for the differences could be technical and due to the manner of CM quantification, which were determined from imaged whole hearts in zebrafish versus sections in mice (Ryckebusch et al., 2008). Given that our data further demonstrate highly conserved cardiac defects that result from early RA deficiency in zebrafish and mouse embryos, we anticipate that future analysis will show that an expansion of FHF-derived CMs occurs in the hearts of RA-deficient mice.
Figure 11. Model of early RA requirements in patterning the zebrafish heart.
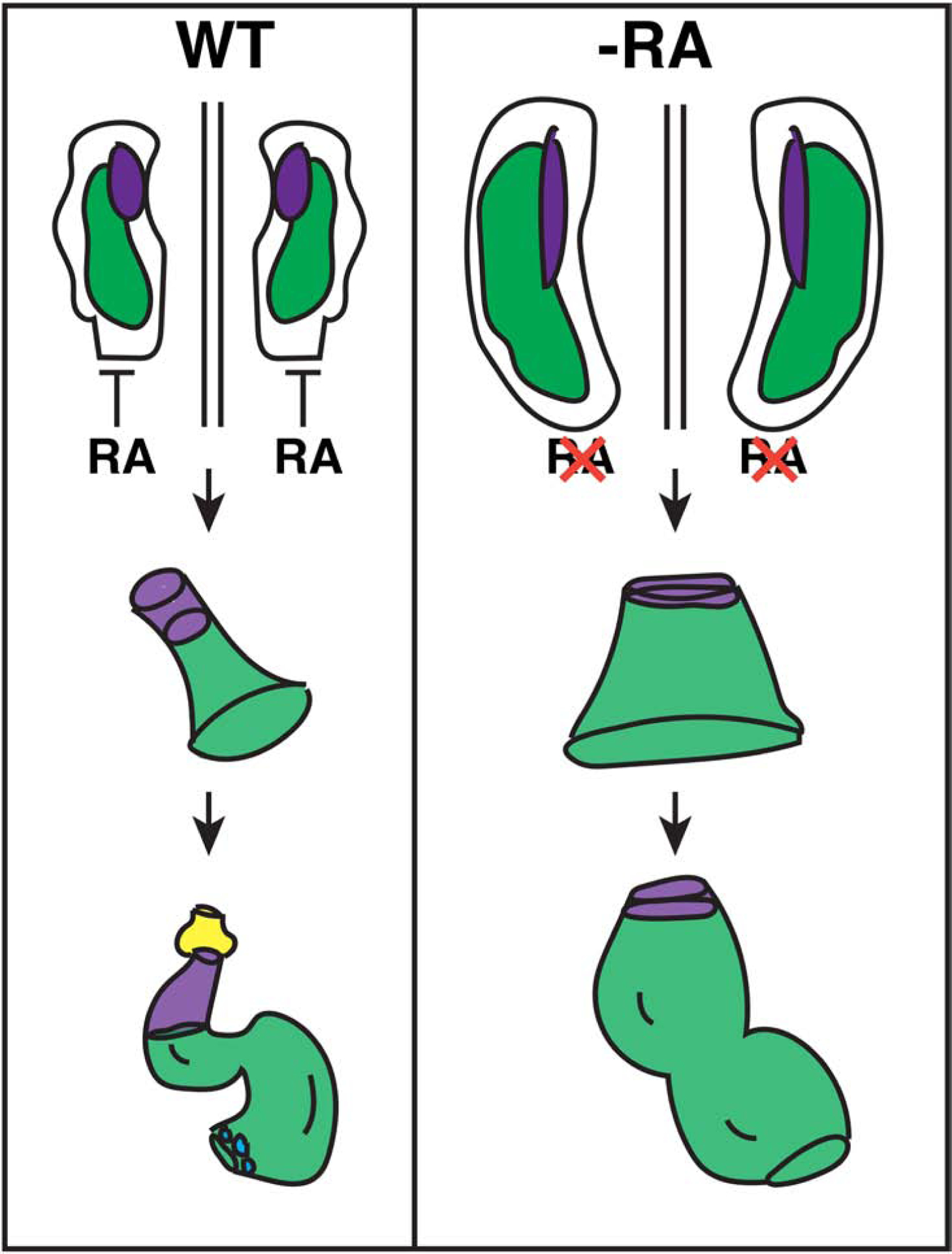
RA signaling restricts the cardiac progenitor populations within the ALPM. Normally, FHF (green) and anterior SHF (purple) lie adjacent to each other within bilateral populations. Posterior RA signaling limits the boundary of the cardiac progenitor field. As the tube forms, SHF-derived cells are added to the arterial pole of the heart tube, giving rise to ventricular CMs and smooth muscle in the bulbus arteriosus (yellow). At the venous pole, pacemaker CMs (blue) differentiate. In the absence of RA, the FHF progenitors are expanded, while the number of SHF progenitors are actually not initially affected, they may be expanded posteriorly within the ALPM. However, the SHF progenitors that will give rise to the OFT prematurely differentiate, leading to a loss of the SHF-derived-arterial pole and OFT in the enlarged hearts. Pacemaker CMs fail to differentiate at the venous pole of the enlarged hearts in RA-deficient embryos.
Vertebrate hearts are generated from the continuous differentiation of progenitors from the FHF and SHF. While progenitors can be designated FHF and SHF based on gene expression and developmental requirements in specific structures (Black, 2007; Kelly, 2012; Zaffran and Kelly, 2012), these designations are fundamentally defined by the timing of progenitor differentiation as they integrate into the heart. Our results indicate that the anterior-posterior patterning of the ALPM by RA signaling also effectively controls the differentiation rate of cardiac progenitors. The expansion of the anterior FHF progenitors at the expense of more posterior fates is reflected in increased number of early differentiating CMs. Moreover, one interpretation of the progressive loss of anterior SHF progenitors from the 20s stage (19 hpf) through 24 hpf, due to their premature differentiation, is effectively that there is a conversion of SHF to FHF in RA-deficient embryos. Our interpretation that premature differentiation is the most likely cause for the depletion of SHF progenitors is based on similar observations in RAR KO mice (Li et al., 2010). However, we recognize that we have not formally ruled out that decreases in proliferation rate or cell death could underlie the diminished SHF population at the arterial pole. Importantly, the increased number of earlier differentiating CMs is within an overall expansion in the size of the heart field, which occurs predominantly at the expense of the adjacent posterior progenitors and not more anterior non-cardiac fates, such as the pharyngeal muscle, or neural crest cells, as shown here. However, despite the lack of effect on these adjacent cell types in the anterior, our lineage-tracing shows that more posterior Nkx2.5+ cells in RA-deficient embryos contribute to the arterial pole of the heart, suggesting that SHF progenitors may also be expanded posteriorly within the ALPM despite their subsequent reduction. Thus, while restricting the overall progenitor field within the ALPM, RA also concurrently determines the rate of CM differentiation and consequently the relative ratio of FHF and SHF progenitors.
In comparing our data to previous studies of RA signaling in heart development at different developmental stages, we propose that defects in the inflow and outflow tracts of RA-deficient embryos from these different studies reflect progressive requirements in anterior-posterior patterning of the cardiac field. Mutations in RARs, their heterodimeric partners RXRs, or Vitamin-A deficient mouse embryos result in a spectrum of OFT and ventricular septal defects that are less severe than the profoundly dysmorphic hearts in Aldha1a2 KO mice (Kastner et al., 1994; Kastner et al., 1997; Li et al., 2010; Mendelsohn et al., 1994; Niederreither et al., 1999; Niederreither et al., 2001; Ryckebusch et al., 2008). Moreover, detailed work with RARα KOs in combination with RARβ or RXRα KOs showed that the OFT defects in these mice are due to a depletion of SHF progenitor cells after the early patterning of the progenitor field (Li et al., 2010). The precocious differentiation of SHF progenitors leading to a depletion of this cell population and loss of the OFT shown here from early RA deficiency in zebrafish embryos supports that there is a conserved requirement for RA signaling over multiple developmental stages to maintain the anterior SHF progenitor population in vertebrates. Furthermore, in addition to OFT defects, it is interesting that precocious differentiation of ventricular CMs was one of the first cellular defects reported in the hearts of RXRα, RXRβ, RARα KO and Vitamin-A deficient mice (Kastner et al., 1997) and is found in Aldh1a2 KO mice (Niederreither et al., 2001). While the genetic studies from mice, including tissue-specific KOs (Li et al., 2010), suggest there is a direct role for these receptors in these cardiac progenitors, it is not yet clear if these receptors are required specifically in the SHF progenitors of zebrafish at early stages. Assessing the tissue-specific requirements of RARs in zebrafish with engineered mutants may be tedious due to their broad expression and gene duplications (Waxman and Yelon, 2007). Taken together, these data suggest RA signaling is required over a prolonged period of heart development to maintain the anterior SHF progenitor population that generates the arterial pole of the heart through preventing their premature differentiation.
Early RA-deficiency in all vertebrates results in a posterior expansion of the cardiac progenitor field within the ALPM (Ryckebusch et al., 2008; Sirbu et al., 2008; Sorrell and Waxman, 2011; Waxman et al., 2008), which has been proposed to underlie defects in the posterior SHF and the venous pole of the heart (Ryckebusch et al., 2008). Within the ALPM, RA signaling initially promotes forelimb, posterior arch and muscle specification more posteriorly and represses the extent of the more anterior cardiac progenitor fields (Begemann et al., 2001; Waxman et al., 2008; Zhao et al., 2009). In mice, the transgenic RARE-lacZ reporter has shown that RA signaling overlaps with and occurs caudal to the posterior SHF within the ALPM (Sirbu et al., 2008). Early loss of RA signaling results in a concurrent posterior expansion of SHF markers and loss of posterior Tbx5 expression within the ALPM, as well as a subsequent reduction in Tbx5 at the venous pole in the dysmorphic heart tubes (Ryckebusch et al., 2008; Sirbu et al., 2008). While the specific effects on SAN differentiation have not been examined in RA-deficient mouse embryos, Tbx5 is required for SAN differentiation in mice and it has been inferred that RA signaling may lie upstream of Tbx5 in promoting the differentiation of pacemaker CMs (Christoffels et al., 2010).
In zebrafish, RA signaling determines the border between more anterior and posterior Tbx5+ progenitors that will contribute to the heart and forelimbs, respectively (Waxman et al., 2008). Furthermore, as shown here, early RA signaling also establishes the specification of anterior and posterior Nkx2.5+ progenitor populations. Whether or not RA signaling occurs directly in posterior CM progenitors in zebrafish embryos is less clear. RA signaling reporters have not shown expression in the ALPM (Mandal et al., 2013; Waxman and Yelon, 2011). While the lack of reporter expression does not rule out RA signaling occurs in these cells, RA responsive genes largely seem to be adjacent to CM specification markers (Dohn et al., 2019; Waxman et al., 2008). Nevertheless, our data demonstrate that early loss of RA signaling in zebrafish embryos results in a failure of the SAN to differentiate at the venous pole. However, it is not clear if the loss of the SAN in RA-deficient zebrafish embryos is due to diminished tbx5 expression. Previous studies examining tbx5 expression in RA-deficient embryos have not shown it is lost in CM progenitors or the nascent heart (Grandel and Brand, 2010; Waxman et al., 2008). Moreover, the posterior SHF in mammals contributes comparatively more cells to the atria than in zebrafish, where lineage-tracing has shown the contributions are minimal (de Pater et al., 2009; Lazic and Scott, 2011). In addition to the consequences of early loss of RA signaling, more recent data in mice has also shown that at comparatively later stages of cardiogenesis when the heart tube is elongating, RA signaling is still necessary to promote Tbx5 expression within the posterior SHF and limit the posterior expression domain of Tbx1 (De Bono et al., 2018). A failure to promote posterior Tbx5 and the maintenance of anterior Tbx1 expression results in improper specification of the posterior SHF and venous pole, and consequently atrioventricular septal defects in these mice that are similar to those found in patients with TBX5 mutations that cause Holt-Oram syndrome (De Bono et al., 2018). Thus, RA signaling has progressive requirements that first define the border between anterior and posterior progenitors within the ALPM and then between anterior and posterior cardiac progenitor fields as the venous pole is added. The proper delineation of these fields is necessary for normal SAN and atrial CM differentiation at the venous pole of the heart.
Conclusions
Overall, we have shown the early loss of RA signaling in zebrafish embryos results in enlarged hearts through an expansion of earlier differentiating FHF-derived CMs. Moreover, we show that early loss of RA signaling results in conserved defects at the arterial and venous poles of vertebrate hearts. At the arterial pole of the heart, OFT defects in RA-deficient embryos are due to the premature differentiation and depletion of anterior SHF progenitors, while at the venous pole of the heart the SAN fails to differentiate. Our work helps to resolve discrepancies in our understanding of the requirements of RA signaling within vertebrate heart development and provides new insights into the etiology of CHDs associated with RA deficiency in vertebrates. Future work will be dedicated to understanding the precise RA-dependent gene regulatory networks that pattern these cardiac progenitors within the ALPM and control the timing of their differentiation.
Material and Methods
Ethics statement
All zebrafish husbandry and experiments reported in the manuscript were done as outlined in approved IACUC protocols at the Cincinnati Children’s Hospital Medical Center.
Zebrafish lines used and developmental stages
The following zebrafish transgenic lines were used in described experiments: TgBAC(−36nkx2.5:ZsYellow)fb7 (Paffett-Lugassy et al., 2013), TgBAC(−36nkx2.5:Kaede)fb9 (Paffett-Lugassy et al., 2013), Tg(myl7:nls-KikGR)hsc6 (Lazic and Scott, 2011), Tg(myl7:EGFP)twu (Huang et al., 2003), Tg(−5sox10:GAL4, UAS:Cre)la2326, Tg(−3.5ubb:loxP-EGFP-loxP-mCherry)cz1701 (Cavanaugh et al., 2015), and sqet33-mi59B (Poon et al., 2010). The aldh1a2/nlsi26 allele was used (Begemann et al., 2001). Nls mutants were genotyped with PstI digest, which cuts the mutant allele, as reported previously (Begemann et al., 2001), following PCR using primers Nls-long F1 (5’- tgagtgggaggtacacttgaaagac) and nls-R (5’- gatcagagcgccgaggta). All embryos were raised at 28.5°C. Hpf indicated reflects developmental stages post-fertilization for embryos raised at 28.5°C.
DEAB treatments
DEAB (Sigma) treatments were performed as previously described (Waxman et al., 2008). Pools of ~30 zebrafish myl7:EGFP embryos were treated in 2 mls of embryo water in glass vials beginning at shield stage with DEAB (2.5 μM). An equivalent volume (0.25 μl) of DMSO (vehicle) was added for controls. Vials with embryos were placed on a nutator in the dark until the desired stage. Treated embryos show overt heart, hindbrain, and forelimb defects that are similar to the nls mutants (Begemann et al., 2001). Unless embryos were harvested for analysis at earlier stages, the DEAB was washed out at 24 hpf by transferring embryos to petri dishes with embryo water and the embryos were raised until the indicated stage for analysis.
Immunohistochemistry
Immunohistochemistry was performed similar to what was described previously (Waxman et al., 2008). Briefly, zebrafish embryos at the designated stages were fixed for 1 hr in 1% formaldehyde/1x phosphate-buffered saline (PBS). The fixed embryos were then repeatedly washed in 0.1% or 0.2% Saponin (Sigma) in 1X PBS. For earlier stages (20s through 24 hpf), 0.1% Saponin was used. For stages >24 hpf, 0.2% Saponin was used. Embryos were blocked for 1 hr with 1X PBS/10% sheep serum/2 mg/ml bovine serum albumin/0.1% or 0.2% Saponin. The embryos were incubated with primary antibodies diluted in the blocking solution overnight at 4°C. Following incubation, embryos were washed 3X in 0.1% or 0.2% Saponin/1xPBS and incubated for 2 hrs with secondary antibodies diluted in the blocking solution. Embryos were then washed multiple times in 0.1% or 0.2% Saponin/1xPBS. Following staining, embryos were post-fixed with 2% formaldehyde for 1 hr followed by additional washes with Saponin/1xPBS prior to mounting and imaging. All steps were performed at room temperature unless indicated. Primary antibodies used were: rabbit anti-Nkx2.5 (Gene Tex), chicken anti-GFP (Invitrogen), mouse anti-Mhc (DHSB), mouse anti-Amhc (DHSB), rabbit anti-Elnb (custom – reported in (Song et al., 2019)), rabbit anti-RCFP (Takara), rabbit anti-mCherry (Biovision), and rabbit anti-Isl1 (Gene Tex). Concentrations and secondary antibodies used are indicated in Supplemental Table 1. For imaging myl7:EGFP+, Mhc+ and Nkx2.5+ cells, embryos were flat-mounted between two coverslips ventral side down. For imaging Mhc, Elnb, ZsYellow, and mCherry, embryos were mounted on a coverslip with 0.6% low-melt agarose with the heart facing down or on their side. For imaging Isl1 and GFP (sqet33-mi59B), embryos were mounted on a coverslip with 1XPBS immediately prior to imaging with the heart facing down. All embryos were imaged using a Nikon A1 confocal microscope. Imaris software (Bitplane) was used to assist in the quantification of cells.
In situ hybridization
In situ hybridization was performed essentially as previously described (Oxtoby and Jowett, 1993). Probes for kaede (ZDB-EFG-080131-2), mef2cb (ZDB-GENE-040901-7), and ltbp3 (ZDB-GENE-060526-130) were used. Embryos were imaged using a Zeiss M2BioV12 stereomicroscope.
Lineage tracing and Temporal Differentiation Assays
Lineage tracing was performed on DEAB-treated embryos with the nkx2.5:Kaede transgene similar to what has been previously reported (Holowiecki et al., 2020; Rydeen and Waxman, 2016). Briefly, 2-well Ibidi μ-slides (cat# 80286) were covered in a thin layer of 2% agarose and wells for embryos were created by puncturing the agarose with a capillary tube. Embryos were then mounted with their dorsal side up and small clusters of nkx2.5:Kaede+ cells were photoconverted from green to red using the 20X objective with 10X zoom and the DAPI filter on a Nikon A1 confocal microscope. Following photoconversion, embryos were raised until 48 hpf when they were mounted on their side in embryo water, and imaged using a Nikon A1 confocal microscope. Contributions of converted red cells to the ventricle, ventral aorta, and pharyngeal arch arteries were scored following imaging.
To assess the temporal differentiation of CMs the myl7:nls-KikGR transgene was used similar to what has been previously reported (Rydeen and Waxman, 2016). DMSO (control) and DEAB-treated embryos were exposed to light from a DAPI filter for ~20 minutes at 24 hpf with a Zeiss M2BioV12 fluorescent stereomicroscope. At 48 hpf, embryos were sorted, anesthetized with Tricaine, mounted on a coverslip with a small spot of 3% methylcellulose in embryo water, and imaged with a Nikon A1 confocal microscope. Green-only cells were counted from images in Imaris (Bitplane).
Statistical analysis
Statistical significance for two conditions was determined using a two-tailed Student’s t-test. Statistical tests were performed using Graphpad Prism software. p<0.05 was considered statistically significant. Asterisks in all graphs indicate p<0.05. Error bars in all graphs indicate standard error of the mean (s.e.m).
Supplementary Material
Supplemental Table 1. Antibodies used.
Highlights.
Cell quantification reveals retinoic acid is required to restrict the amount of earlier-differentiating cardiac progenitors.
Temporal differentiation assays indicate retinoic acid is required to promote the differentiation of later-differentiation cells in the outflow tract.
Retinoic acid signaling is required to promote the differentiation of pacemaker cardiomyocytes at the venous pole of the heart.
Loss of retinoic acid does not affect the number of neural crest-derived cardiomyocytes that contribute to the heart.
Retinoic acid concurrently restricts the size of the heart field and proportion of earlier and later-differentiating cardiomyocytes.
Acknowledgements
We thank Jennifer Schumacher for reading and comments of the manuscript.
Funding
Work in the manuscript was supported by National Institutes of Health grants R01 HL141186 and R01 HL137766 to J.S.W and by National Institutes of Health grant T32 HL125204 and American Heart Association grant 18POST34050034 to A.H.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing interests
The authors declare no competing interests.
References
- Abdul-Wajid S, Demarest BL, Yost HJ, 2018. Loss of embryonic neural crest derived cardiomyocytes causes adult onset hypertrophic cardiomyopathy in zebrafish. Nat Commun 9, 4603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begemann G, Schilling TF, Rauch GJ, Geisler R, Ingham PW, 2001. The zebrafish neckless mutation reveals a requirement for raldh2 in mesodermal signals that pattern the hindbrain. Development 128, 3081–3094. [DOI] [PubMed] [Google Scholar]
- Black BL, 2007. Transcriptional pathways in second heart field development. Semin Cell Dev Biol 18, 67–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckingham M, Meilhac S, Zaffran S, 2005. Building the mammalian heart from two sources of myocardial cells. Nat. Rev. Genet 6, 826–835. [DOI] [PubMed] [Google Scholar]
- Cavanaugh AM, Huang J, Chen JN, 2015. Two developmentally distinct populations of neural crest cells contribute to the zebrafish heart. Dev. Biol 404, 103–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christoffels VM, Smits GJ, Kispert A, Moorman AF, 2010. Development of the pacemaker tissues of the heart. Circ. Res 106, 240–254. [DOI] [PubMed] [Google Scholar]
- De Bono C, Thellier C, Bertrand N, Sturny R, Jullian E, Cortes C, Stefanovic S, Zaffran S, Theveniau-Ruissy M, Kelly RG, 2018. T-box genes and retinoic acid signaling regulate the segregation of arterial and venous pole progenitor cells in the murine second heart field. Hum. Mol. Genet 27, 3747–3760. [DOI] [PubMed] [Google Scholar]
- de Pater E, Clijsters L, Marques SR, Lin YF, Garavito-Aguilar ZV, Yelon D, Bakkers J, 2009. Distinct phases of cardiomyocyte differentiation regulate growth of the zebrafish heart. Development 136, 1633–1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dohn TE, Ravisankar P, Tirera FT, Martin KE, Gafranek JT, Duong TB, VanDyke TL, Touvron M, Barske LA, Crump JG, Waxman JS, 2019. Nr2f-dependent allocation of ventricular cardiomyocyte and pharyngeal muscle progenitors. PLoS Genet 15, e1007962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Robrini N, Etchevers HC, Ryckebusch L, Faure E, Eudes N, Niederreither K, Zaffran S, Bertrand N, 2016. Cardiac outflow morphogenesis depends on effects of retinoic acid signaling on multiple cell lineages. Dev. Dyn 245, 388–401. [DOI] [PubMed] [Google Scholar]
- Grandel H, Brand M, 2010. Zebrafish limb development is triggered by a retinoic acid signal during gastrulation. Dev. Dyn [DOI] [PubMed] [Google Scholar]
- Guner-Ataman B, Paffett-Lugassy N, Adams MS, Nevis KR, Jahangiri L, Obregon P, Kikuchi K, Poss KD, Burns CE, Burns CG, 2013. Zebrafish second heart field development relies on progenitor specification in anterior lateral plate mesoderm and nkx2.5 function. Development 140, 1353–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hami D, Grimes AC, Tsai HJ, Kirby ML, 2011. Zebrafish cardiac development requires a conserved secondary heart field. Development 138, 2389–2398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holowiecki A, Linstrum K, Ravisankar P, Chetal K, Salomonis N, Waxman JS, 2020. Pbx4 limits heart size and fosters arch artery formation by partitioning second heart field progenitors and restricting proliferation. Development 147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CJ, Tu CT, Hsiao CD, Hsieh FJ, Tsai HJ, 2003. Germ-line transmission of a myocardium-specific GFP transgene reveals critical regulatory elements in the cardiac myosin light chain 2 promoter of zebrafish. Developmental dynamics : an official publication of the American Association of Anatomists 228, 30–40. [DOI] [PubMed] [Google Scholar]
- Jiang X, Choudhary B, Merki E, Chien KR, Maxson RE, Sucov HM, 2002. Normal fate and altered function of the cardiac neural crest cell lineage in retinoic acid receptor mutant embryos. Mech. Dev 117, 115–122. [DOI] [PubMed] [Google Scholar]
- Kastner P, Grondona JM, Mark M, Gansmuller A, LeMeur M, Decimo D, Vonesch JL, Dolle P, Chambon P, 1994. Genetic analysis of RXR alpha developmental function: convergence of RXR and RAR signaling pathways in heart and eye morphogenesis. Cell 78, 987–1003. [DOI] [PubMed] [Google Scholar]
- Kastner P, Messaddeq N, Mark M, Wendling O, Grondona JM, Ward S, Ghyselinck N, Chambon P, 1997. Vitamin A deficiency and mutations of RXRalpha, RXRbeta and RARalpha lead to early differentiation of embryonic ventricular cardiomyocytes. Development 124, 4749–4758. [DOI] [PubMed] [Google Scholar]
- Keegan BR, Feldman JL, Begemann G, Ingham PW, Yelon D, 2005. Retinoic acid signaling restricts the cardiac progenitor pool. Science 307, 247–249. [DOI] [PubMed] [Google Scholar]
- Kelly RG, 2012. The second heart field. Curr. Top. Dev. Biol 100, 33–65. [DOI] [PubMed] [Google Scholar]
- Kirby ML, Gale TF, Stewart DE, 1983. Neural crest cells contribute to normal aorticopulmonary septation. Science 220, 1059–1061. [DOI] [PubMed] [Google Scholar]
- Lazic S, Scott IC, 2011. Mef2cb regulates late myocardial cell addition from a second heart field-like population of progenitors in zebrafish. Dev. Biol 354, 123–133. [DOI] [PubMed] [Google Scholar]
- Li P, Pashmforoush M, Sucov HM, 2010. Retinoic acid regulates differentiation of the secondary heart field and TGFbeta-mediated outflow tract septation. Dev. Cell 18, 480–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Stainier DY, 2012. Zebrafish in the study of early cardiac development. Circ. Res 110, 870–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal A, Rydeen A, Anderson J, Sorrell MR, Zygmunt T, Torres-Vazquez J, Waxman JS, 2013. Transgenic retinoic acid sensor lines in zebrafish indicate regions of available embryonic retinoic acid. Developmental dynamics : an official publication of the American Association of Anatomists [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendelsohn C, Lohnes D, Decimo D, Lufkin T, LeMeur M, Chambon P, Mark M, 1994. Function of the retinoic acid receptors (RARs) during development (II). Multiple abnormalities at various stages of organogenesis in RAR double mutants. Development 120, 2749–2771. [DOI] [PubMed] [Google Scholar]
- Nagelberg D, Wang J, Su R, Torres-Vazquez J, Targoff KL, Poss KD, Knaut H, 2015. Origin, Specification, and Plasticity of the Great Vessels of the Heart. Curr. Biol 25, 2099–2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niederreither K, Subbarayan V, Dolle P, Chambon P, 1999. Embryonic retinoic acid synthesis is essential for early mouse post-implantation development. Nat. Genet 21, 444–448. [DOI] [PubMed] [Google Scholar]
- Niederreither K, Vermot J, Messaddeq N, Schuhbaur B, Chambon P, Dolle P, 2001. Embryonic retinoic acid synthesis is essential for heart morphogenesis in the mouse. Development 128, 1019–1031. [DOI] [PubMed] [Google Scholar]
- Oxtoby E, Jowett T, 1993. Cloning of the zebrafish krox-20 gene (krx-20) and its expression during hindbrain development. Nucleic Acids Res 21, 1087–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paffett-Lugassy N, Novikov N, Jeffrey S, Abrial M, Guner-Ataman B, Sakthivel S, Burns CE, Burns CG, 2017. Unique developmental trajectories and genetic regulation of ventricular and outflow tract progenitors in the zebrafish second heart field. Development 144, 4616–4624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paffett-Lugassy N, Singh R, Nevis KR, Guner-Ataman B, O’Loughlin E, Jahangiri L, Harvey RP, Burns CG, Burns CE, 2013. Heart field origin of great vessel precursors relies on nkx2.5-mediated vasculogenesis. Nat. Cell Biol 15, 1362–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perl E, Waxman JS, 2019. Reiterative Mechanisms of Retinoic Acid Signaling during Vertebrate Heart Development. J Dev Biol 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poon KL, Liebling M, Kondrychyn I, Garcia-Lecea M, Korzh V, 2010. Zebrafish cardiac enhancer trap lines: new tools for in vivo studies of cardiovascular development and disease. Dev. Dyn 239, 914–926. [DOI] [PubMed] [Google Scholar]
- Ryckebusch L, Bertrand N, Mesbah K, Bajolle F, Niederreither K, Kelly RG, Zaffran S, 2010. Decreased levels of embryonic retinoic acid synthesis accelerate recovery from arterial growth delay in a mouse model of DiGeorge syndrome. Circ. Res 106, 686–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryckebusch L, Wang Z, Bertrand N, Lin SC, Chi X, Schwartz R, Zaffran S, Niederreither K, 2008. Retinoic acid deficiency alters second heart field formation. Proc Natl Acad Sci U S A 105, 2913–2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rydeen AB, Waxman JS, 2016. Cyp26 Enzymes Facilitate Second Heart Field Progenitor Addition and Maintenance of Ventricular Integrity. PLoS Biol 14, e2000504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirbu IO, Zhao X, Duester G, 2008. Retinoic acid controls heart anteroposterior patterning by down-regulating Isl1 through the Fgf8 pathway. Dev. Dyn 237, 1627–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song YC, Dohn TE, Rydeen AB, Nechiporuk AV, Waxman JS, 2019. HDAC1-mediated repression of the retinoic acid-responsive gene ripply3 promotes second heart field development. PLoS Genet 15, e1008165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorrell MR, Waxman JS, 2011. Restraint of Fgf8 signaling by retinoic acid signaling is required for proper heart and forelimb formation. Developmental Biology 358, 44–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanovic S, Zaffran S, 2017. Mechanisms of retinoic acid signaling during cardiogenesis. Mech. Dev 143, 9–19. [DOI] [PubMed] [Google Scholar]
- Tang W, Martik ML, Li Y, Bronner ME, 2019. Cardiac neural crest contributes to cardiomyocytes in amniotes and heart regeneration in zebrafish. Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tessadori F, van Weerd JH, Burkhard SB, Verkerk AO, de Pater E, Boukens BJ, Vink A, Christoffels VM, Bakkers J, 2012. Identification and functional characterization of cardiac pacemaker cells in zebrafish. PLoS One 7, e47644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldo K, Miyagawa-Tomita S, Kumiski D, Kirby ML, 1998. Cardiac neural crest cells provide new insight into septation of the cardiac outflow tract: aortic sac to ventricular septal closure. Dev. Biol 196, 129–144. [DOI] [PubMed] [Google Scholar]
- Waxman JS, Keegan BR, Roberts RW, Poss KD, Yelon D, 2008. Hoxb5b acts downstream of retinoic acid signaling in the forelimb field to restrict heart field potential in zebrafish. Dev. Cell 15, 923–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waxman JS, Yelon D, 2007. Comparison of the expression patterns of newly identified zebrafish retinoic acid and retinoid X receptors. Developmental dynamics : an official publication of the American Association of Anatomists 236, 587–595. [DOI] [PubMed] [Google Scholar]
- Waxman JS, Yelon D, 2011. Zebrafish retinoic acid receptors function as context-dependent transcriptional activators. Developmental Biology 352, 128–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaffran S, Kelly RG, 2012. New developments in the second heart field. Differentiation 84, 17–24. [DOI] [PubMed] [Google Scholar]
- Zhao X, Sirbu IO, Mic FA, Molotkova N, Molotkov A, Kumar S, Duester G, 2009. Retinoic acid promotes limb induction through effects on body axis extension but is unnecessary for limb patterning. Curr. Biol 19, 1050–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Cashman TJ, Nevis KR, Obregon P, Carney SA, Liu Y, Gu A, Mosimann C, Sondalle S, Peterson RE, Heideman W, Burns CE, Burns CG, 2011. Latent TGF-beta binding protein 3 identifies a second heart field in zebrafish. Nature 474, 645–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table 1. Antibodies used.