

Abstract
Human genetic studies established MET gene as a risk factor for autism spectrum disorders. We have previously shown that signaling mediated by MET receptor tyrosine kinase, expressed in early postnatal developing forebrain circuits, controls glutamatergic neuron morphological development, synapse maturation, and cortical critical period plasticity. Here we investigated how MET signaling affects synaptic plasticity, learning and memory behavior, and whether these effects are age-dependent. We found that in young adult (postnatal 2–3 months) Met conditional knockout (Metfx/fx:emx1cre, cKO) mice, the hippocampus exhibits elevated plasticity, measured by increased magnitude of long-term potentiation (LTP) and depression (LTD) in hippocampal slices. Surprisingly, in older adult cKO mice (10–12 months), LTP and LTD magnitudes were diminished. We further conducted a battery of behavioral tests to assess learning and memory function in cKO mice and littermate controls. Consistent with age-dependent LTP/LTD findings, we observed enhanced spatial memory learning in 2–3 months old young adult mice, assessed by hippocampus-dependent Morris water maze test, but impaired spatial learning in 10–12 months mice. Contextual and cued learning were further assessed using a Pavlovian fear conditioning test, which also revealed enhanced associative fear acquisition and extinction in young adult mice, but impaired fear learning in older adult mice. Lastly, young cKO mice also exhibited enhanced motor learning. Our results suggest that a shift in the window of synaptic plasticity and an age-dependent early cognitive decline may be novel circuit pathophysiology for a well-established autism genetic risk factor.
Keywords: autism, neurodevelopmental disorders, cortical circuits, synaptic plasticity, electrophysiology, learning and memory
1. Introduction
A putative pathophysiology of many neurodevelopmental and neuropsychiatric disorders, including autism spectrum disorders (ASD), is the disrupted timing synapse development, circuit connectivity, and activity-dependent plasticity changes (Aldinger et al., 2011; Berg and Geschwind, 2012; Zoghbi and Bear, 2012; Berger et al., 2013). The human MET gene is a replicated genetic risk factor for ASD. For instance, the rs1858830 ‘C’ allele in MET promoter reduces gene transcription and protein translation in cortical tissue of autism patients (Campbell et al., 2006; Campbell et al., 2007; Aldinger et al., 2015), and is nearly absent in individuals with Rett Syndrome (Plummer et al., 2013). Neuroimaging studies also revealed that the ‘C’ allele modulates functional and structural cortical connectivity in both typically developing persons and individuals with ASD (Rudie et al., 2012). MET encodes a receptor tyrosine kinase, MET, that controls multiple aspects of cortical circuit development, including morphological development of cortical projection neurons, dendritic spine genesis, maturation of excitatory synapse, and the critical period of cortical circuit plasticity (Judson et al., 2009; Qiu et al., 2011; Qiu et al., 2014; Chen et al., 2020).
A critical feature of MET signaling in cortical circuits is that it is largely limited to the early postnatal period. In mice, MET expression temporally coincides heightened period of synaptogenesis, extensive neurite growth, and formation of circuits, but is precipitously down-regulated prior to synapse maturation and activity-dependent circuit refinement (Lein et al., 2007; Judson et al., 2009; Judson et al., 2011; Chen et al., 2020). MET signaling also profoundly affects the molecular constituents of glutamatergic synapses, and by doing so, controls the timing of circuit connectivity, maturation, and plasticity. For instance, in hippocampus CA1 neurons from Met conditional knockout mice (i.e. Metfx/fx:emx1cre, referred hereafter as MetcKO or cKO) (Qiu et al., 2014), or from cultured hippocampus slices subjected to MET kinase activity inhibition (Ma et al., 2019), glutamatergic synapses mature earlier; there were decreased levels of silent synapses, increased synaptic content of glutamate receptor subunits GluA1, GluN2A, and a reduction of GluN2B. The altered glutamate receptor subunits may indicate disrupted timing of plasticity, as GluN2B-containing NMDA receptors exhibit high calcium permeability compared with GluN2A-containing receptors (Erreger et al., 2005; Shipton and Paulsen, 2014). Consistently, critical period plasticity in the visual cortex terminates early in MetcKO mice, but can be extended to beyond normal critical period with ectopic, sustained MET signaling in a controllable Met transgenic mouse line (Chen et al., 2020).
Given that MET signaling occurs throughout multiple cortical domains, it is conceivable that altered timing of synaptic plasticity may have broader implications on learning, memory and circuit plasticity throughout development. It would be interesting to investigate how the potentially altered synapse maturation and plasticity affects learning and memory behaviors. Insights gained from these studies may offer better understanding on neurobiology of a prominent autism risk gene. In this study, we investigated how loss of MET signaling in cKO mice affects hippocampal plasticity, measured by long-term potentiation (LTP, (Bliss and Gardner-Medwin, 1973; Bliss and Collingridge, 1993) and long-term depression (LTD), and assessed multiple hippocampus-dependent learning behaviors. We found an age-dependent, opposing effects of cKO on LTP/LTD and learning and memory; while young adult cKO (2–3 months) showed enhanced hippocampal plasticity and learning, older adult cKO (10–12 months) mice exhibited a decline in plasticity and learning. These findings further support the critical role of MET signaling in controlling the timing of synaptic plasticity, and in shaping the developmental trajectory of brain circuits and behavior. To our best knowledge, this is the first study that reveals an autism genetic risk factor in regulating the timing of plasticity and learning, which may be a novel pathophysiological substrate for autism risk.
2. Experimental Procedures
2.1. Animals
The forebrain-specific conditional knockout mice (cKO, Metfx/fx:Emx1cre) were generated by breeding hemizygote Metfx/+ male mice with an Emx1Cre knock-in allele (Gorski et al., 2002) to homozygous female Metfx/fx mice, as reported previously (Judson et al., 2009; Qiu et al., 2014). Littermate mice that do not contain the Emx1cre allele (Metfx/fx or Metfx/+) were used as controls. Metfx/fx line was provided by Dr. Snorri Thorgeirsson (NIH/Center for Cancer Research, Bethesda, MD), and Emx1Cre line was provided by Dr. Kevin Jones (University of Colorado, Boulder, CO). Both lines were back-crossed onto the C57Bl/6 background for more than ten generations. Offspring were genotyped by polymerase chain reaction (Judson et al., 2009; Judson et al., 2010). Because postnatal MET is largely selectively expressed in cortical excitatory neurons, this cKO approach abolishes MET protein in cortical tissues (Judson et al., 2009; Ma et al., 2019). cKO mice do not show overt phenotypes, and appear similar in size as same-sex littermate controls. Two age groups of mice were used for plasticity and behavior studies; mice of postnatal day (PD) 2–3 months old are designated as ‘young adult’, and mice of 10–12 months are designated as ‘older adult’. An approximately equal number of male and female mice were used. Mice of desired genotypes were assigned to experimental groups randomly. Where applicable, mice IDs and data were coded during experiments and analyses so that the experimenters were blinded to genotypes and grouping. All experimental procedures conforms to NIH guidelines and were approved by the Institutional Animal Care and Use Committee of the University of Arizona.
2.2. Electrophysiology
2.2.1. Hippocampal brain slice preparation
Mice were euthanized with 3–5% isoflurane, and brains were collected from either young adult or older adult mice of desired genotypes. To enhance brain slice viability, intra-cardiac perfusion of ice-cold choline solution (in mM: 110 choline chloride, 25 NaHCO3, 2.5 KCl, 1.25 NaH2PO4, 0.5 CaCl2, 7 MgSO4, 25 D-glucose, 11.6 sodium ascorbate, and 3.1 sodium pyruvate, saturated with 95% O2 / 5% CO2) was performed. Mice were decapitated and brains were quickly dissected out. Horizontal slices (300 μm thick) at the hippocampus level were made in ice-cold choline solution using a vibratome (VT-1200S, Leica). The hippocampus from each hemisphere was dissected out, and kept in artificial cerebrospinal fluid (ACSF, contains in mM: 126 NaCl, 2.5 KCl, 26 NaHCO3, 2 CaCl2, 2 MgCl2, 1.25 NaH2PO4, and 10 d-glucose; saturated with 95% O2 / 5% CO2) for 30 min at 35°C, and then maintained at 22°C RT until recording.
2.2.2. Field potential recording, LTP & LTD
The hippocampal slices were transferred to an interface chamber (Automate Scientific, Berkeley, CA), and superfused with ACSF. Field excitatory postsynaptic potentials (fEPSPs) were recorded in the CA1 stratum radiatum layer using a glass patch electrode at RT to facilitate long-term slice viability. The patch electrode had an electrical resistance of 1–2 MΩ at 1kHz when filled with ACSF. fEPSPs were evoked by an tungsten stimulating electrode (FHC, Bowdoin, ME) that was placed on the Schaffer collateral inputs ~200 μm away from the recording site. Biphasic stimulus (graded levels ranging from 10–250 μA, 100 μs duration, 0.05 Hz) was generated using a Digidata 1440A interface (Molecular Devices, San Jose, CA) and pClamp 10.2 software. The stimulus was delivered through an optic isolator (Iso-flex, A.M.P.I). fEPSP signals were amplified using a differential amplifier (model 1800, A–M Systems, Carlsborg, WA), low-pass filtered at 2 kHz and digitized at 10 kHz.
A stimulus – fEPSP input – output curve was first obtained by measuring fEPSP slope (first 1-ms response after fiber volley) as a function of the fiber volley amplitude. A stimulus intensity that produced a ~40–50% maximum fEPSP amplitude was then adopted and kept constant throughout the experiments.
After establishing stable baseline response of stimulus-evoked fEPSPs for at least 10 min, an LTP induction stimulation protocol was applied. It is well-established that synaptic stimulation pattern is critical for LTP/LTD induction and maintenance (Zhang et al., 2011; Kenney and Manahan-Vaughan, 2013). For LTP induction, we adopted a tetanus stimulation protocol that consisted of two, 1-sec trains of stimulation at 100Hz (Qiu et al., 2006). LTD was induced by low-frequency stimulation, which consists 1 Hz paired pulses (with 50 ms inter-pulse interval) for a 15-min duration (i.e., a total of 900 paired stimuli) (Qiu et al., 2006). fEPSP was continuously recorded for 60 min post LTP/LTD induction.
2.3. Behavior testing
2.3.1. Morris water maze test
A 72-cm diameter circular pool was filled with RT (~23 °C) water to a depth of 30 cm. Water is made opaque with white, non-toxic, Tempera paint. The pool was placed in a room with prominent visual cues. The circular arena was divided in four equal quadrants/zones. An additional zone close to wall (7.5cm) was defined to measure thigmotaxis. Mice were placed in one of four starting locations of the pool and allowed to swim until they reached a 7.5 × 7.5 cm submerged platform. On finding the platform, mice remained on the platform for 20 s before the next trial. If in a trial mice failed to find the platform within 90 s, they were guided to the platform by the experimenter and remained on the platform for 60 s before being removed. A video camera was used to record movement at 15 fps using Ethovision XT (Noldus Information Technology, Leesburg, VA). Mice were automatically detected using a weighed centroid machine vision algorithm, and tracking was implemented by Ethovision. During the training phase, mice had four trials per day with an inter-trial interval of one hour for eight consecutive days. A probe trial was conducted right after the training in Day 8, during which mice were allowed to freely swim while the submerged platform was removed. Time spent in the target quadrant, and number of platform crosses were quantified for the probe trial.
On Days 9 −11, mice were further tested for their reverse learning, during which the platform was moved to the opposite quadrant and mice were trained to re-learn the new position of the platform. For this reverse learning, the same training paradigms were used as those adopted in Day 1–8. Another probe test was performed on Day 11 following the completion of the reverse learning training session. Ethovision XT 14.0 and MATLAB was used to analyze and visualize the percent time spent in the each quadrant, the number of platform location crossings, and mice swimming speed.
2.3.2. Fear conditioning test
The Pavlovian fear conditioning learning test (Goode et al., 2019) was conducted in standard mouse conditioning chambers (17 × 17 × 25 (h) cm; Stoelting, Wood Dale, IL) housed in sound-attenuating cabinets. Testing protocol was programmed using AnyMaze video tracking system (Stoelting, Wood Dale, IL). Each chamber was identical in size and consisted of two removable Plexiglass sides, a rear wall, and a hinged Plexiglass door with a round opening (Context A). The grid floor contained 20 steel rods that spaced 0.8 cm apart that were connected to a current generator (shock source) to deliver foot shocks (unconditional stimulus – US). The chamber can be fitted with a new set of removable context to alter the enclosure’s color and texture (Context B), enabling associative learning with environmental cues. Locomotor activity was recorded on a digital camera at 25fps, which was analyzed offline to quantify freezing behavior using AnyMaze software. Mice immobility was considered freezing if they lasted at least 1 sec. A loudspeaker mounted on the inside wall of cabinet was used to deliver auditory tone (conditional stimulus – CS). The sound attenuating chamber was illuminated with ~50 lux ambient light during the experiment.
All mice underwent a 30-min habituation in the conditioning chamber the day prior to fear training. Fear conditioning training was conducted on the first day (Day 1) in the conditioning chamber with the grid floor (Context A), during which five tone – shock/CS - US pairings consisted of a 30-s (2 kHz, 80 dB) tone that co-ended with a 1-s (0.6 mA) foot shock. Tone-shock pairing was delivered with a 150 sec inter-trial interval (ITI). After the final CS - US pairing, mice remained in the conditioning chamber for 1 min before being returned to their home cages. Mice freezing behavior was quantified only during the tone period. Following Day 1 training, a contextual recall memory test was performed in Day 2, during which mice were introduced to the chamber (Context A) for 300 sec. Time spend in freezing was quantified. In Day 3, mice were introduced to the chamber with a new context (Context B, with new textured flat floor and wall inserts to change the appearance of the chamber). After a 120-sec habituation, the same tone (CS) was delivered for 30 sec, and repeated 5 times with 150 sec ITI. For extinction training in Day 4 (Context A), the tone was played for 30 sec, followed by a 30 sec interval. This is repeated for 45 cycles, which was divided into five 9-trial blocks for analysis of extinction learning (Goode et al., 2019). Following this extinction training, an extinction recall memory test was conducted on Day 5 (Context A), during which the tone was played for 30 sec, with 30 sec interval, and repeated for 10 times. Percent of freezing time was quantified during these ten trials. All freezing data presented were measured during CS presentation.
2.3.3. Open field
Open field test was used to assess basal locomotor activity, behavioral reactivity to a novel environment, and anxiety. Mice were placed in an open field (45 × 45 cm2) enclosure and were habituated for 10 min. The activity were recorded by Ethovision video-tracking system for 5 min. Total distance and percent of time spent in the center of arena (22.5 × 22.5 cm2) were quantified.
2.3.4. Rotarod test
Mice were placed on the beam of an accelerating rotarod (Ugo Basile, Italy). The latency to fall was recorded for each mouse, which provided a measurement of motor learning and coordination. Mice were tested in two sequential 6-day sessions, similar to that described previously (Ha et al., 2016). The first session (day 1–6) consisted of three trials each day, during each trial the rotarod is linearly accelerated from 4 to 40 rpm in 5 min. The inter-trial interval was 30 min. The second session included another 6-day (day 7–12) learning, three trials each day with each trial adopting an 8 to 80 rpm acceleration in 5 min.
2.3.5. Elevated plus maze test
The elevated plus maze (EPM) test was used to evaluate anxiety in mice by using their innate preference for dark and enclosed spaces. The EPM is 4 feet from the ground and consists of four runways (9 in x 2.5 in rectangle) arranged perpendicularly, two of which are enclosed with 5 inch high walls and two of which are open. The amount of time spent in the closed arms of the maze is used as a measure of anxiety-like behavior. Mice were put on the maze and their activity was recorded by Ethovision for a five minute session.
2.4. Histology/neuronal cell counting.
Mice were anesthetized with 3% isoflurane, the brains were fixed through intra-cardiac perfusion of 4% PFA in 0.1M PBS. The brains were quickly dissected out, post fixed in 4% PFA overnight, cryo-protected in sequential 15% and 30% sucrose. 40-μm sections were cut on a sliding microtome (Leica SM 2000R). Free floating sections were stained with Nissl reagents (toluidine blue), and destained with 70% ethanol. Sections were dehydrated in ethanol gradients (50–100%), cleared in xylene and mounted with DPX medium. Images were acquired on a microscope (Zeiss Imager M2) with 20X objectives. Cell counting was conducted using FIJI/ImageJ. Cells only with neuronal soma clearly identifiable and were located in upper layer 5 in the prelimbic region were counted. Cell density was expressed as number of neurons per mm2.
2.5. Statistical analysis
All results were reported as mean ± s.e.m. (standard error of the mean). To ensure rigor and transparency, the experimenters were unaware of mouse genotypes/grouping. Sample sizes and number of independent experiments were estimated by power analysis using an R script that takes pre-specified effect size, type I and II errors as input arguments. Score of some behavior data were independently analyzed by two experimenters. For all the behavior tests, we first analyzed sex-disaggregated data, and did not find any statistical significance. Data from both sexes were pooled together for further analysis. Statistical analyses and graphing were performed using GraphPad Prism 8.0, Microsoft Excel, MATLAB. Figures were prepared using Adobe Creative Cloud. Shapiro–Wilk test and F test were used to test normality and equal variance. Student t test or two-way analysis of variations with repeated measures (rmANOVA) was used when data passed normality and equal variance tests. Sidak’s multiple comparison (MCT) test was used for post hoc comparison following two-way ANOVA. A nonparametric Mann-Whitney test was used for non-normally distributed/ordinal type data (e.g. number of platform crosses). p < 0.05 was considered statistically significant for all tests.
3. Results
3.1. Enhanced hippocampus synaptic plasticity in young adult (2–3 months) mice
We have previously shown that in MetcKO mice, the hippocampal Schaffer collateral- CA1 circuit matures earlier, and there is altered glutamate receptor subunit composition at synaptic sites (Qiu et al., 2014). In addition, we recently found that cortical critical period plasticity closes early in MetcKO mice (Chen et al., 2020). Considering synaptic plasticity is a putative cellular substrate for learning and memory (Martin et al., 2000; Takeuchi et al., 2014; Asok et al., 2019), this raises the possibility that timing of synaptic plasticity may be altered in the hippocampus and other MET expressing cortical circuits, thus affects learning and memory behaviors.
We started by probing long term potentiation (LTP) plasticity of at the Schaffer collateral - CA1 synapses using acute hippocampus slice preparations from 2–3 months (‘young adult’) MetcKO (Metfx/fx:Emx1cre) mice and their littermate controls (Metfx/+ or Metfx/fx, Emx1WT). fEPSP responses were recorded in response to electric stimulation of the Schaffer collateral input pathway (Fig. 1A), with increasing stimulus intensity lead to larger fiber volley and fEPSP slope/amplitude. By adopting a paired stimuli pattern with varying inter-stimulus intervals (ISI), presynaptic function related to facilitation of synaptic transmission can be estimated. We first quantified the input-output responses by plotting fEPSP slope as a function of the fiber volley amplitude (Fig. 1A). Linear fitting of the response curve revealed no significant difference in the slopes of the fitted lines (Ctrl, N = 8 mice; cKO, N = 9 mice. F(1, 149) = 1.68, p = 0.19). Next, we tested paired pulse response, using stimulus pairs with ISI ranging from 20–100 ms. Two-way rmANOVA revealed no significant effects on groups (Fig. 1B. Ctrl, N = 10 mice; cKO, N = 10 mice. F(1, 90) = 1.69, p = 0.19).
Fig. 1. Young MetcKO mice show enhanced LTP at the Schaffer collateral-CA1 synapse.
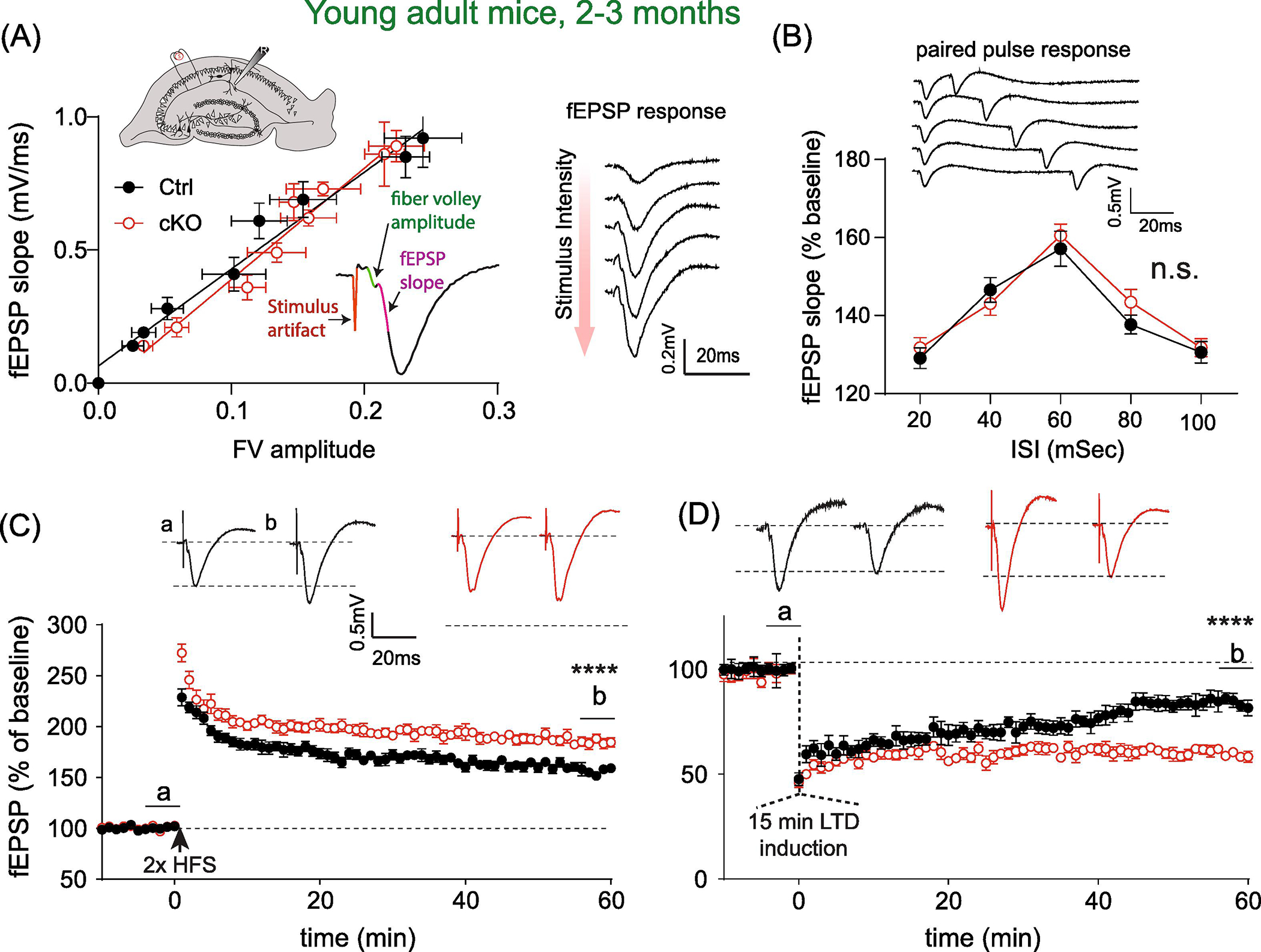
(A) Input-output curve of basal synaptic transmission measured as fEPSP slope as a function of fiber volley. Linear fitting of the response curves revealed no significant difference in the slopes of the fitted lines. Ctrl, N = 8 slices; cKO, n = 9 slices. F(1, 149) = 1.68, p = 0.19. Insets: top, illustration of fEPSP recording in the striatum radiatum layer in CA1, in response to electric stimulation. Bottom, a representative fEPSP trace is displayed, fiber volley amplitude and slope calculation are labeled. Example of recording traces showing increased fEPSP amplitude/slope in response to increased stimulus intensity is displayed to the right. (B) Quantification of paired pulse responses (PPR) from both groups. Main effects of genotype: F(1, 90) = 1.69, p = 0.19. Top, samples of PPR with inter-stimulus intervals (ISI) ranging from 20 to 100 msec. (C) Young adult cKO slices showed enhanced LTP induced by two trains of high frequency tetanus stimulation. fEPSP (percent baseline) at 56–60 min post tetanus: Ctrl, 156.3 ± 2.05; N = 11; cKO, 184.8 ± 3.04, N = 12. t(21) = 7.62, P < 0.0001. (D) Young cKO slices showed higher magnitude of LTD induced by paired low frequency stimulation. fEPSP percent baseline at 56–60 min post tetanus: Ctrl, 84.0 ± 1.03; N = 10 mice; cKO, 59.5 ± 0.69, N = 8 mice. t(16) = 18.8, P < 0.0001).
We next probed long-term synaptic plasticity by conducting both LTP and LTD recording at the Schaffer collateral - CA1 synapse. We found that LTP magnitude (fEPSP slope normalized as percent of baseline) was significantly increased when measured at 56–60 min post tetanus stimulation (Fig. 1C. Ctrl, 156.3 ± 2.05; N = 11 mice; cKO, 184.8 ± 3.04, N = 12 mice. t(21) = 7.62, p < 0.0001). In addition, LTD levels were also dramatically reduced at ~ 1h post LTD induction (Fig. 1E. Ctrl, 84.0 ± 1.03; N = 10 mice; cKO, 59.5 ± 0.69, N = 8 mice. t(16) = 18.8, p < 0.0001). Therefore, at this young adult age, basal levels of synaptic transmission, and presynaptic properties were not affected by the Met cKO, yet there is significantly increased synaptic plasticity, measured as enhanced LTP magnitude at the Schaffer collateral - CA1 synapse.
3.2. Reduced synaptic plasticity in the hippocampus in older adult (10–12 months) mice
We next investigated whether the enhanced synaptic plasticity in MetcKO mice persists and whether it is age-dependent. We conducted the same fEPSP/LTP recording to probe basal synaptic transmission and plasticity in hippocampal slices from older adult mice (10–12 months age). Again, no significant difference was found on the stimulus-fEPSP slope input-output curve (Fig. 2A. Ctrl, N = 8 mice; cKO, N = 7 mice. Comparison on the slopes: F(1, 131) = 0.38, p = 0.54). Paired pulse responses, at ISIs ranging from 20–100 ms, also showed no significant differences (Fig. 2B. Ctrl, N = 10 mice; cKO, N = 10 mice. F(1, 90) = 1.56, p = 0.22 for group effects. Two-way rmANOVA).
Fig. 2. Older adult (10–12 months) MetcKO mice show decreased LTP magnitude at the hippocampal Schaffer collateral- CA1 synapse.
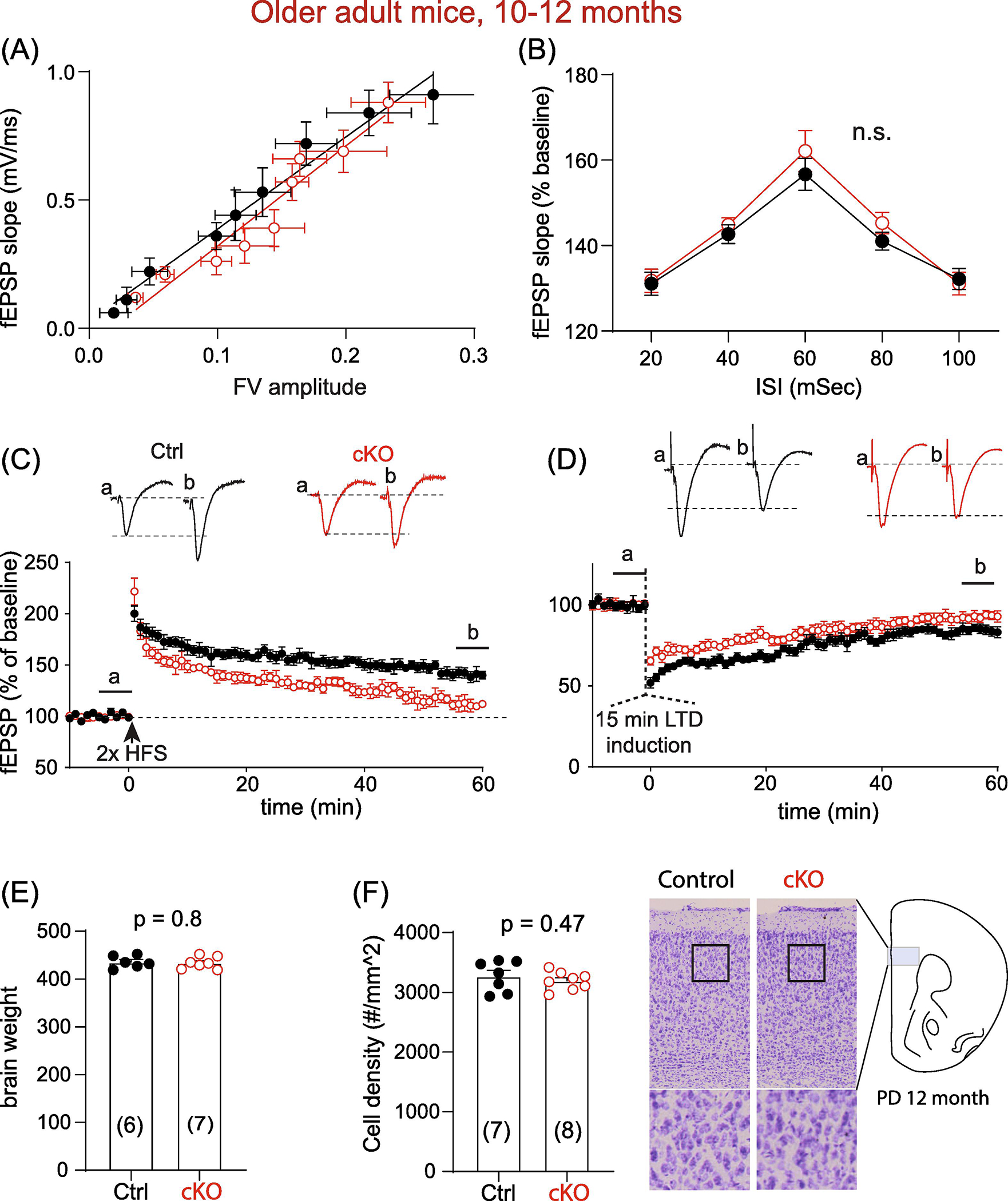
(A) Input-output curve of stimulus evoked fEPSP, measured by linear fitting of the fEPSP slope as a function of fiber volley amplitude. Comparison on the slopes: F(1, 131) = 0.38, p = 0.54. Ctrl, N = 8 mice; cKO, N = 7 mice). (B) Both groups did not differ in paired pulse responses at ISIs ranging from 20–100 ms. F(1, 90) = 1.56, p = 0.22 for main group effects. Ctrl, N = 10 mice; cKO, N = 10 mice. Two-way rmANOVA. (C) Old adult cKO slices showed reduced LTP. fEPSP measured at 56–60 min post tetanus: Ctrl, 140.9 ± 2.64; N = 10; cKO, 111.4 ± 2.67, N = 8. t(16) = 7.76, p < 0.0001. (D) Older adult cKO slices showed lower magnitude of LTD. fEPSP percent baseline at 56–60 min post tetanus: Ctrl, 84.5 ± 1.7; N = 11 mice; cKO, 93.1 ± 0.1.26, N = 8 mice. t(19) = 3.92, p = 0.0009). (E) Brain weight of older adult Ctrl and cKO mice do not differ (Ctrl, 435.7 ± 5.2g, N = 6; cKO, 433.9 ± 6.7g, N = 7. t(11) = 0.26, p = 0.80). (F) No significant difference on prefrontal neuronal density was observed for older adult Ctrl and cKO mice (number of cells/mm2, Ctrl, 3271 ± 97, N = 7; cKO, 3192 ± 53, N = 8. t(13) = 0.74, p = 0.47).
Surprisingly, we found that magnitude of high frequency stimulation-induced LTP was dramatically reduced in older adult slices (Fig. 2C, fEPSP slope measured as percent of baseline at 56–60 min post tetanus stimulation: Ctrl, 140.9 ± 2.64; N = 10 mice; cKO, 111.4 ± 2.67, N = 8 mice. t(16) = 7.76, p < 0.0001). In addition, LTD levels were also dramatically reduced at 56 – 60 min post LTD induction (Fig. 2D. Ctrl, 84.5 ± 1.7; N = 11 mice; cKO, 93.1 ± 0.1.26, N = 8 mice. t(19) = 3.92, p = 0.0009). This impaired LTP/LTD is not likely related to aging-dependent neuronal loss or degeneration in the cKO brain, because neither total brain weight (Fig. 2E. Ctrl, 435.7 ± 5.2g, N = 6 mice; cKO, 433.9 ± 6.7g, N = 7 mice. t(11) = 0.26, p = 0.80) nor neuronal density in cKO prefrontal cortex (Fig. 2F. Number of cells/mm2, Ctrl, 3271 ± 97, N = 7 mice; cKO, 3192 ± 53, N = 8 mice. t(13) = 0.74, p = 0.47) differ between control and cKO mice. Therefore, older adult MetcKO mice exhibited reduced levels of plasticity in the hippocampus, although basal synaptic transmission and presynaptic facilitation remained unaltered.
3.3. Young adult METcKO show enhanced MWM spatial learning
Given the age-dependent alteration in hippocampus LTP, a putative cellular mechanism for learning and memory (Martin et al., 2000; Takeuchi et al., 2014; Asok et al., 2019), we performed behavioral tests in MetcKO and their littermate controls to assess various forms of learning and memory. The Morris water maze (MWM) is a paradigm that tests spatial and associative learning, and known to be dependent on hippocampus function (Paul et al., 2009; Morellini, 2013). Mice were trained to find a hidden platform in a circular pool during an 8-day training (Fig. 3A). The pool was placed in a well-illuminated room to allow association of ambient visual cues with spatial learning. At the end of the training, a probe trial, during which the hidden platform was removed, was conducted to assess spatial memory retention. After that, mice were re-trained for three days to locate a new platform location in order to assess the reverse learning capability and cognitive flexibility.
Fig. 3. Young adult MetcKO mice show enhanced spatial learning and memory assessed by Morris water maze test.
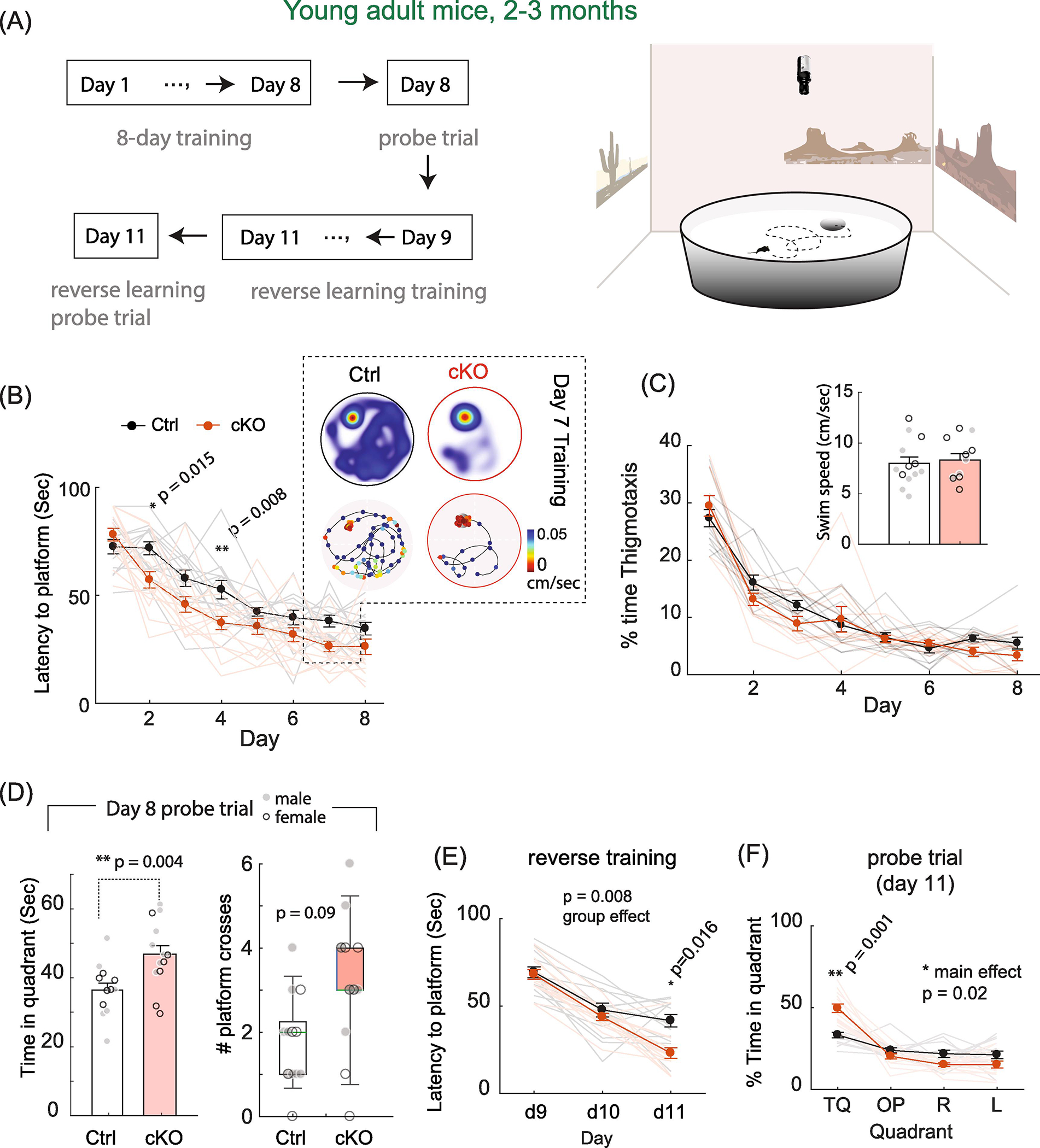
(A) Flowchart of an 11-day MWM protocol. Tests were conducted using digital recorded videos and Ethovision in a room with prominent visual cues to facilitate associative learning. (B) MetcKO mice showed faster learning during the acquisition period. Main effect on groups, F(1,25) = 20.7, p = 0.001. Ctrl, N = 13; cKO, N = 14. In addition, Day 2 and Day 4 showed significant differences. * p = 0.015, ** p = 0.008, Sidak’s MCT. Color inset indicate one representative training session in Day 7. Heatmap, time spent at location of arena; line graph, moving trajectory of mice. Marker color denotes swimming speed at the corresponding location. (C) MetcKO mice spent similar amount of time near the pool wall during the 8-day training, as indicated by the percent of thigmotaxis time. Main effect of group, F(1,25) = 1.25, p = 0.27). Inset, MetcKO mice also showed similar swim speed averaged across the 8-day training period. Averaged speed, Ctrl, 7.98 ± 0.63 cm/sec; cKO, 8.3 ± 0.62 cm/sec. t(22) = 0.37, p = 0.71. (D) MetcKO mice spent significantly more time in the target quadrant during probe trial. Ctrl, 36.5 ± 2.0 sec, N = 13; cKO, 46.8 ± 2.5 sec, N = 14. t(25) = 3.18, p = 0.004, unpaired t test. Grey filled circle, male data points; open circle, female data points. In addition, MetcKO mice showed increased number of platform crosses. p = 0.09. (E) MetcKO showed faster learning during the reverse training phase. Main effect on group, F(1,25) = 8.32, *p = 0.008. Two-way rmANOVA), and significantly shorter time in finding the new platform location at Day 11 (*p = 0.016. Sidak’s MCT). (F) MetcKO mice spent more time in the new target quadrant during the Day 11 probe trial. Ctrl, 48.1 ± 3.47 sec; cKO, 59.6 ± 2.67 sec. **p = 0.001. Two-way rmANOVA with Sidak’s MCT.
We found that young adult MetcKO mice of 2–3 months age demonstrated superb learning during the training phase, as they required significantly shorter time to locate the platform across the 8-day training period (Fig. 3B. Main effect of groups, F(1,25) = 20.7, p = 0.0001; Group × training interaction, F(7, 140) = 2.19, p = 0.038. Ctrl, N = 13 mice; cKO, N = 14 mice). There was a significantly shorter time on locating the platform on Day 2 (p = 0.015) and Day 4 (p = 0.008, Sidak’s MCT). cKO mice spent similar amount of time near the pool wall area, as measured by the percent of thigmotaxis time during the training (Fig. 3C. Main effect of groups, F(1,25) = 1.25, p = 0.27). These mice also demonstrated similar swim speed when swim speed was averaged across the 8-day training period (Fig. 3C, inset. Averaged speed, Ctrl, 7.98 ± 0.63 cm/sec; cKO, 8.3 ± 0.62 cm/sec. t(22) = 0.37, p = 0.71). In the Day 8 probe trial, during which the platform was removed, MetcKO mice spent significantly more time in the target quadrant (Fig. 3D. Ctrl, 36.5 ± 2.0 sec, N = 13; cKO, 46.8 ± 2.5 sec, N = 14; t(25) = 3.18, p = 0.004). MetcKO mice also showed increased number of platform crosses during probe trial, although this increase did not reaching significant level (Fig. 3D. median (range) of number of crosses: Ctrl, 2.0 (0 – 4); cKO, 3.0 (0 – 6). WT, N = 13; cKO, N = 14. p = 0.09, Mann-Whitney U test). Next, we assessed the capability of these mice to re-learn the new platform locations by training them to locate the platform in a three-day reverse learning trial (Day 9 – Day 11). MetcKO mice also showed overall faster reverse learning (Fig. 3E. Main effect on group, F(1,25) = 8.32, p = 0.008, two-way rmANOVA; training x group interaction, F(2,50) = 4.95, p = 0.011), with significantly shorter time in finding the new platform location on Day 11 (p = 0.016. Sidak’s MCT). During the Day 11 probe trial (with platform removed) conducted at the end of training, MetcKO mice spent significantly more time in the new target quadrant location (Fig. 3F. Ctrl, 48.1 ± 3.47; cKO, 59.6 ± 2.67. p = 0.001. Two-way rmANOVA with post hoc Sidak’s MCT).
3.4. Older adult MetcKO mice show impaired MWM spatial learning
We next conducted MWM test in older adult MetcKO mice (10–12 months) and their littermate controls (Fig. 4). MetcKO mice showed a significantly altered time course of learning during the 8-day training phase (Fig. 4A. Main group effect, F(1,22) = 71.2, p < 0.0001. Group × training interaction, F(7, 154) = 0.57, p = 0.78. Ctrl, N = 12 mice; cKO, N = 12 mice). MetcKO also differs from control at each day from Day 5– 8 (Day 5, p = 0.006; Day 6, p = 0.023; Day 7, p = 0.018; Day 8, p = 0.049; two-way rmANOVA with post hoc Sidak’s MCT). cKO mice spent similar time near the pool wall zone, as measured by the percent of thigmotaxis time during the training (Fig. 4B. Main effect of group, F(1,22) = 3.67, p = 0.07). Older adult cKO mice also showed similar swim speed (averaged across the 8-day training period. Fig. 4B, inset. Averaged speed, Ctrl, 8.60 ± 0.49 cm/sec; cKO, 8.09 ± 0.62 cm/sec. t(22) = 0.64, p = 0.53). During the Day 8 probe trial, MetcKO mice spent significantly less time in the target quadrant (Fig. 4C. Ctrl, 45.1 ± 1.87 sec, N = 12 mice; cKO, 33.2 ± 2.01 sec, N = 12 mice. t(22) = 4.33, p = 0.0003). cKO mice also exhibited a decreased number of platform crosses during the probe trial (Fig. 4C. Median (range) of number of crosses: Ctrl, 3.0 (1.0 – 6.0); cKO, 1.0 (0 – 3). N = 12 for both groups, p = 0.007. Mann-Whitney U test). MetcKO mice also demonstrated overall slower reverse learning in Day 9 –11 (Fig. 4D. Main effect of group, F(1,20) = 10.79, p = 0.0037; Group × training day interaction, F(2,40) = 3.93, p = 0.027. Two-way rmANOVA), with significantly higher latency in finding the new platform location on Day 11 (p = 0.017, post hoc Sidak’s MCT). During the probe trial conducted at the end of Day 11 training, MetcKO mice spent significantly less time in the new target quadrant location (Fig. 4E. Ctrl, 55.3 ± 3.28 sec; cKO, 44.2 ± 2.12 sec. p = 0.0005. Two-way rmANOVA with post hoc Sidak’s MCT) and more time in the opposite quadrant (Ctrl, 10.8 ± 3.3 sec; cKO, 21.5 ± 2.50 sec. p = 0.0025).
Fig. 4. Older adult (10–12 months age) MetcKO mice showed impaired MWM spatial memory learning.

(A) Older adult MetcKO mice showed faster learning during the 8-day acquisition phase (Ctrl, N = 12; cKO, N = 12. group factor, F(1,22) = 71.2, p < 0.0001), with Day 5 through Day 8 exhibited statistically significant differences (** p < 0.01, * p < 0.05, two way rmANOVA with post hoc Sidak’s MCT). (B) MetcKO mice spent similar amount of time near the pool wall area during the 8-day training, as indicated by the percent of thigmotaxis time. Main effect of group, F(1,22) = 3.67, p = 0.07). Inset, MetcKO mice also showed similar swim speed. Ctrl, 8.60 ± 0.49 cm/sec; cKO, 8.09 ± 0.62 cm/sec. t(22) = 0.64, p = 0.53. (C) MetcKO mice show significantly less time in the target quadrant during probe trial (Ctrl, 45.1 ± 1.87 sec, N = 12; cKO, 33.2 ± 2.01 sec, N = 12. t(22) = 4.33, ***p = 0.0003, unpaired t test); and significantly decreased number of platform crosses (Median (range) for Ctrl, 3.0 (1.0–6.0) crosses; cKO, 1.0 (0–3) crosses, N = 12 for both groups. p = 0.007, Mann-Whitney U test). (D) MetcKO mice exhibited faster learning during the reverse training phase. F(1,20) = 10.79, p = 0.0037 for the group effect, two-way rmANOVA. A significantly longer time was needed to locate the new platform at day 11. p = 0.017, post hoc Sidak’s MCT. (E) MetcKO mice spent less time in the new target quadrant location (Ctrl, 55.3 ± 3.28 sec; cKO, 44.2 ± 2.12 sec. p = 0.0005, two-way rmANOVA with post hoc Sidak’s MCT) and more time in the opposite quadrant (Ctrl, 10.8 ± 3.3 sec; cKO, 21.5 ± 2.5 sec. p = 0.0025).
3.5. Young adult MetcKO mice showed increased fear conditioning learning
The age-dependent disruption of hippocampus plasticity and MWM spatial learning prompted us to investigate whether impaired learning is generalizable to other associative learning tasks. We conducted a Pavlovian fear conditioning test to investigate associative fear memory. Fear conditioning learning and memory involve other MET-expressing limbic cortical structures (Judson et al., 2009; Judson et al., 2011), such as the amygdala and prefrontal cortex, which are critical for learning and storage of the Pavlovian CS-US memory, and generate fear reactions (Kim and Jung, 2006; Carrere and Alexandre, 2015; Ressler and Maren, 2019). Therefore, fear learning could be affected by Met genetic deletion. We adopted a 5-day fear conditioning learning protocol, which is detailed in Fig. 5. Five training sessions was conducted in Day 1, in which a mild foot shock (US) was paired with an auditory cue (CS) (see Methods) in Context A. Mice gradually learned to associate the CS with US (Fig. 5B). In Day 2, mice were placed in the same context, while no US or CS stimulus were presented. Freezing time reflected the contextual memory recall from previous learning (Fig. 5C). Day 3 tested cued memory, by placing mice in Context B and only presented with CS for five sessions (Fig. 5D). Fear extinction training was followed on Day 4 in Context A, during which mice were presented with only CS for 45 times, but no US was delivered (Fig. 5E). The extinction recall test, during which mice were presented with only the CS, was conducted in Day 5 in Context A to further assess the memory extinction (Fig. 5F).
Fig. 5. Fear conditioning learning protocol.
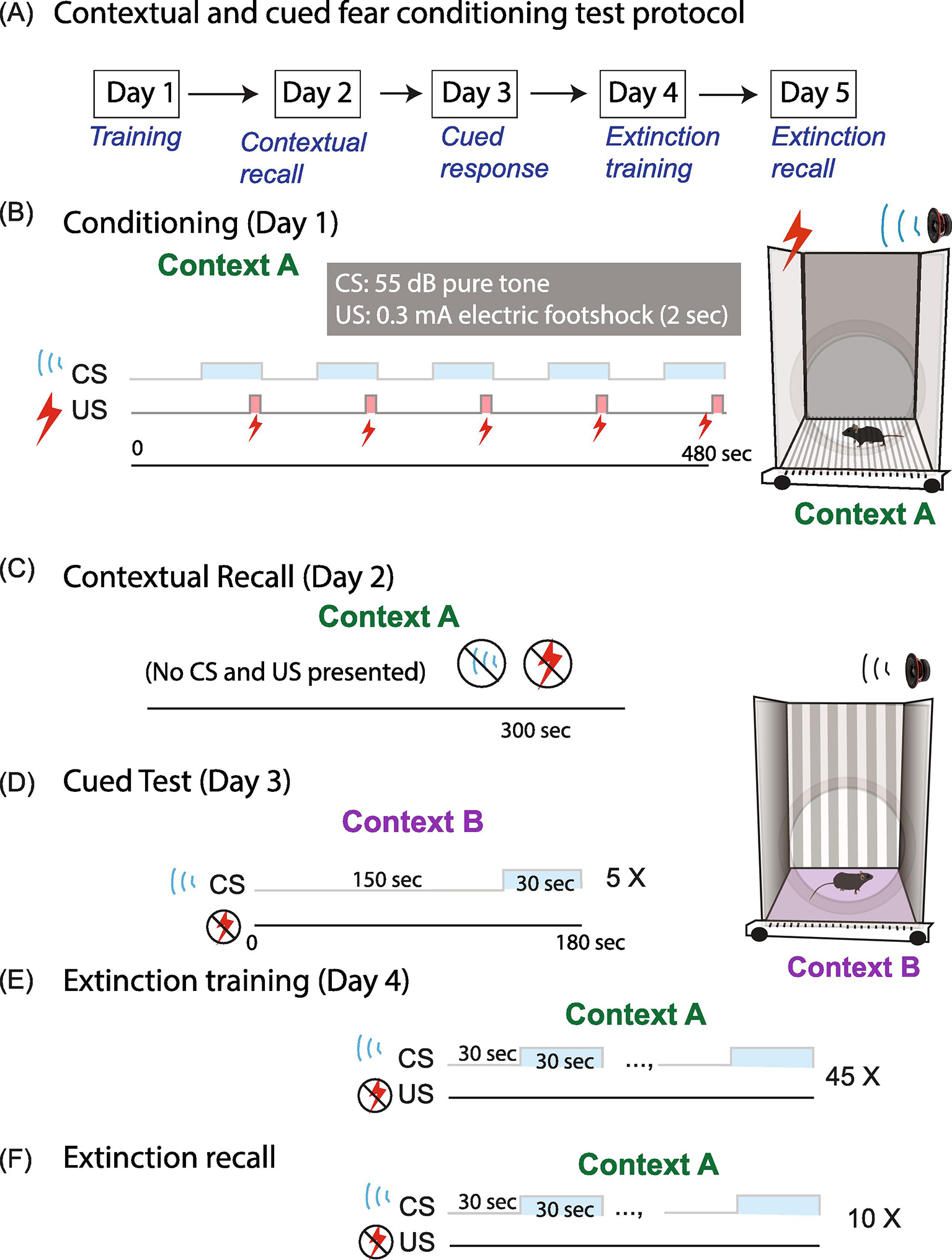
(A) Protocol outline. (B) Day 1 has five training sessions in which a mild foot shock (US) was paired with an auditory cue (CS) in Context A. (C) In day 2, mice are placed in the same context with no US or CS stimulus presented. Freezing time is measured. (D) Day 3 tests the cued memory, by placing the mice in Context B and presenting only with CS for five sessions. (E) Fear extinction training paradigm, conducted at Day 4 in Context A, during which mice are presented with only CS, but no US delivered for 45 trials. (F) Extinction recall test, during which mice are presented with only the CS for 10 trials, is conducted in Day 5 in Context A.
Both young adult MetcKO mice and controls showed increased freezing as the number of US-CS paring increases (Fig. 6A). In addition, MetcKO showed an overall faster learning during the US-CS conditioning phase (Main effect on group, F(1,27) = 31.3, p < 0.0001. Group × pairing sessions interaction, F(4,108) = 4.19, p = 0.003. WT, N = 14; cKO, N = 15 mice. Two-way rmANOVA). A significant increase in freezing was also found on session 2 (p = 0.005) and session 4 (p = 0.0004, post hoc Sidak’s MCT). MetcKO mice also seemed to learn better in associating the context cues with fear memory; in the contextual recall test conducted on Day 2, MetcKO mice showed significant enhanced contextual recall memory (Fig. 6B. t(27) = 4.11, p = 0.0005. unpaired t test). In the cued CS test on Day 3, MetcKO differed significantly from control mice in freezing (Fig. 6C. Main effect on group, F(1,27) = 7.00, p = 0.013. Group × tone sessions interaction, F(4, 108) = 0.72, p = 0.58. Ctrl, N = 14; cKO, N = 15 mice. Two way rmANOVA). When freezing was averaged in all five trials, MetcKO mice demonstrated a significant increase in cued fear response (Fig. 6D. t(27) = 2.62, p = 0.015, unpaired t test). Mice then underwent the fear extinction training in Day 4, during which 45 sessions were analyzed in five 9-trial blocks (see Methods). Both groups showed reduced freezing as the trials/sessions proceed, yet MetcKO mice learned fear extinction faster (Fig. 6E. Main effect on, F(1,27) = 6.05, p = 0.021. Group x training interaction, F(4,108) = 4.35, p = 0.0027, Two-way rmANOVA). There was also significantly less freezing during block 5 training (post hoc Sidak’s MCT, p = 0.0006). Lastly, mice were tested for their extinction recall on Day 5. MetcKO mice showed a significant difference in freezing across the ten trials (Fig. 6F. Main effects on group, F(1,23) = 6.86, p = 0.015. Group × training interaction, F(9, 207) = 0.69, p = 0.71. WT, N = 12; cKO, N = 13). When freezing was averaged from the ten trials, MetcKO showed a significant reduction in freezing during the extinction recall (Fig. 6G, Ctrl, 27.9 ± 0.82; cKO, 23.4 ± 1.46. t(23) = 2.62, p = 0.015). Therefore, young adult MetcKO mice exhibit enhanced Pavlovian fear conditioning US-CS learning as well as increased fear memory extinction.
Fig. 6. Young adult (2–3 months age) MetcKO mice show increased fear conditioning learning.
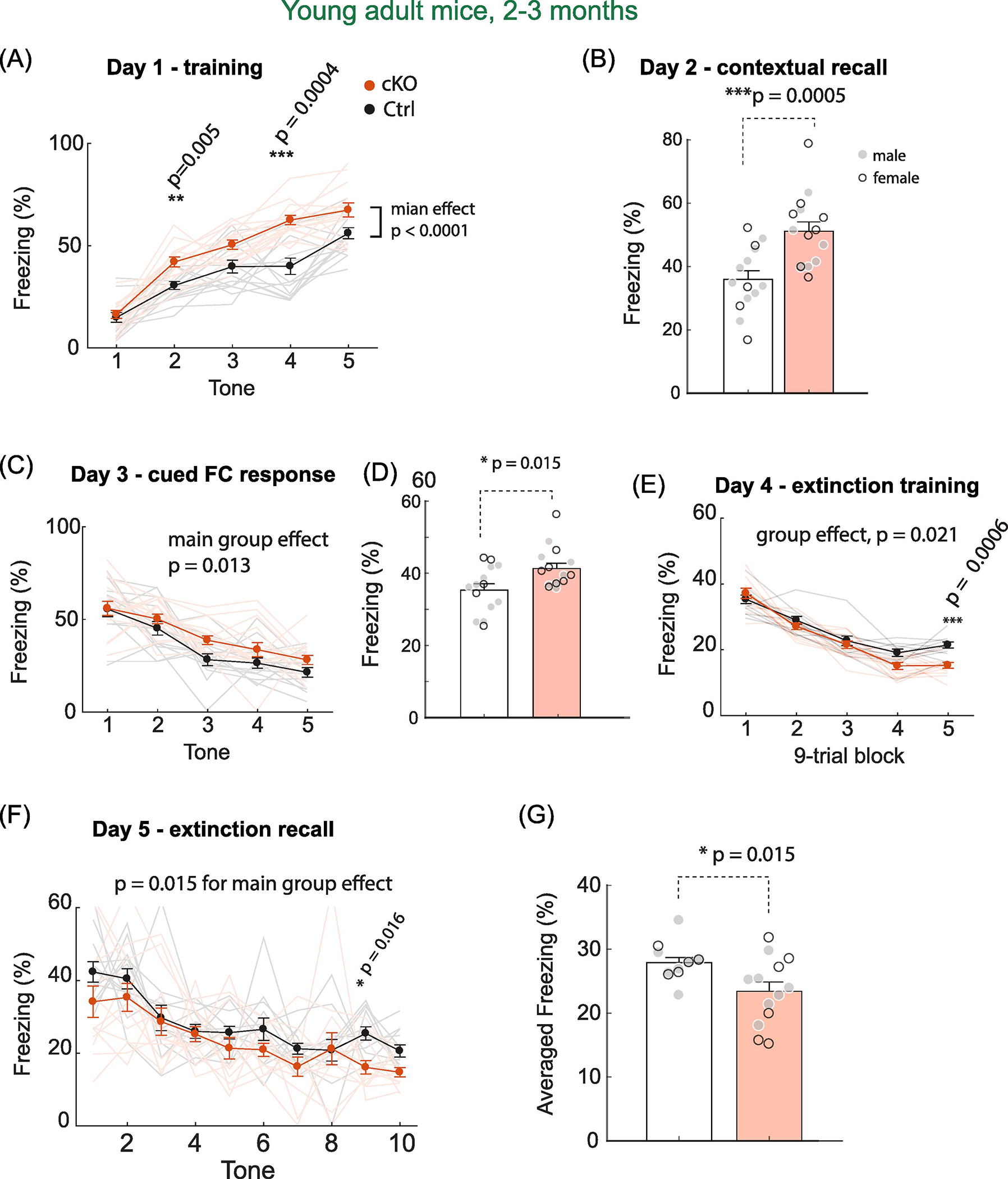
(A) Increased percent of freezing time with US-CS pairing in both group of mice. MetcKO mice showed a significantly increased freezing time compared with controls. Main genotype effect, F(1,27) = 31.3, p < 0.0001. Post hoc Sidak’s MCT revealed a significant increase in freezing on session 2 (p = 0.005) and session 4 (p = 0.0004). (B) MetcKO mice showed significantly increased contextual freezing. t(27) = 4.11, p < 0.0005. unpaired t test. (C) MetcKO showed overall increased freezing in the cued memory test. F(1,27) = 7.00, p = 0.013, two way rmANOVA. WT, N = 14; cKO, N = 15 mice. (D) Combined averaged cued freezing in five trials. MetcKO mice showed a significant increase in cued fear responses. t(27) = 2.62, p = 0.015. (E) Fear extinction in MetcKO mice was faster. Main group effect, F(1,27) = 6.05, *p = 0.021. Two-way rmANOVA. There was also significantly less freezing during block 5 training. Sidak’s MCT, p = 0.0006. (F) Fear extinction recall were tested with ten CS tones. MetcKO mice showed a significant reduction in freezing across the ten trials. Main group effects, F(1,23) = 6.86, p = 0.015. (G) Averaged freezing from the ten extinction recall trials. MetcKO showed a significant reduction in freezing. Ctrl, 27.9 ± 0.82 sec; cKO, 23.4 ± 1.46 sec. t(23) = 2.62, * p = 0.015.
3.6. Older adult MetcKO mice demonstrate declined fear learning and memory
We next asked whether the increased fear conditioning learning and extinction were age-dependent. 10–12 months older adult mice of MetcKO and their littermate controls underwent the same fear conditioning training on Day 1. Both groups demonstrated increased freezing as the number of US-CS pairing increased (Fig. 7A). MetcKO mice showed overall slower learning during the US-CS conditioning phase (Main effect for group, F(1, 20) = 37.7, p < 0.0001. Group × training interaction, F(4, 80) = 7.11, p < 0.0001. N = 11 mice for both groups. Two-way rmANOVA). A significant increase in freezing was found on session 4 (p = 0.0012) and session 5 (p = 0.0021, two-way rmANOVA with post hoc Sidak’s MCT). Contextual recall test on Day 2 revealed that MetcKO mice performed poorly in associating the contextual cues with fear memory (Fig. 7B. Averaged time freezing, Ctrl, 55.7 ± 4.65 sec; cKO, 33.6 ± 3.09 sec. N = 11 for both groups. t(20) = 3.96, p = 0.0008, unpaired t test). In the CS-cued test on Day 3, MetcKO showed overall reduced freezing in response to the CS auditory cue (Fig. 7C. N = 10 for both groups, F(1,18) = 11.2, p = 0.0035. Two-way rmANOVA). When freezing was averaged in all five trials, MetcKO mice showed a significant decrease in cued fear response (Fig. 7D. t(18) = 2.80, p = 0.012). Next, older adult mice were subjected to extinction training in Day 4. Both groups exhibited reduced freezing as the trials/sessions proceeded. MetcKO mice demonstrated overall slower learned fear extinction, but did not reach statistical significance (Fig. 7E. Main effect on group, F(1,18) = 2.94, p = 0.10. N = 10 for both groups. Two-way rmANOVA). Lastly, mice were tested for their extinction recall on Day 5. MetcKO mice showed a significant overall increase in freezing across the 10-trial extinction recall test (Fig. 7F. Main effect on group, F(1,18) = 14.2, p = 0.0014. Group x trial interaction, F(9, 162) = 1.86, p = 0.06. N = 10 for both groups). When averaged freezing was quantified from the 10 trials, MetcKO showed a significant increase in freezing during the extinction recall phase (Fig. 7G. Ctrl, 25.2 ± 1.32 sec; cKO, 32.1 ± 1.25 sec. t(18) = 3.77, p = 0.0014). Therefore, in contrast to young adult mice, older adult MetcKO mice at 10–12 months age exhibit impaired Pavlovian fear conditioning US-CS learning, as well as decreased fear memory extinction.
Fig. 7. Older adult (10–12 months) MetcKO mice show impaired learning fear conditioning learning.
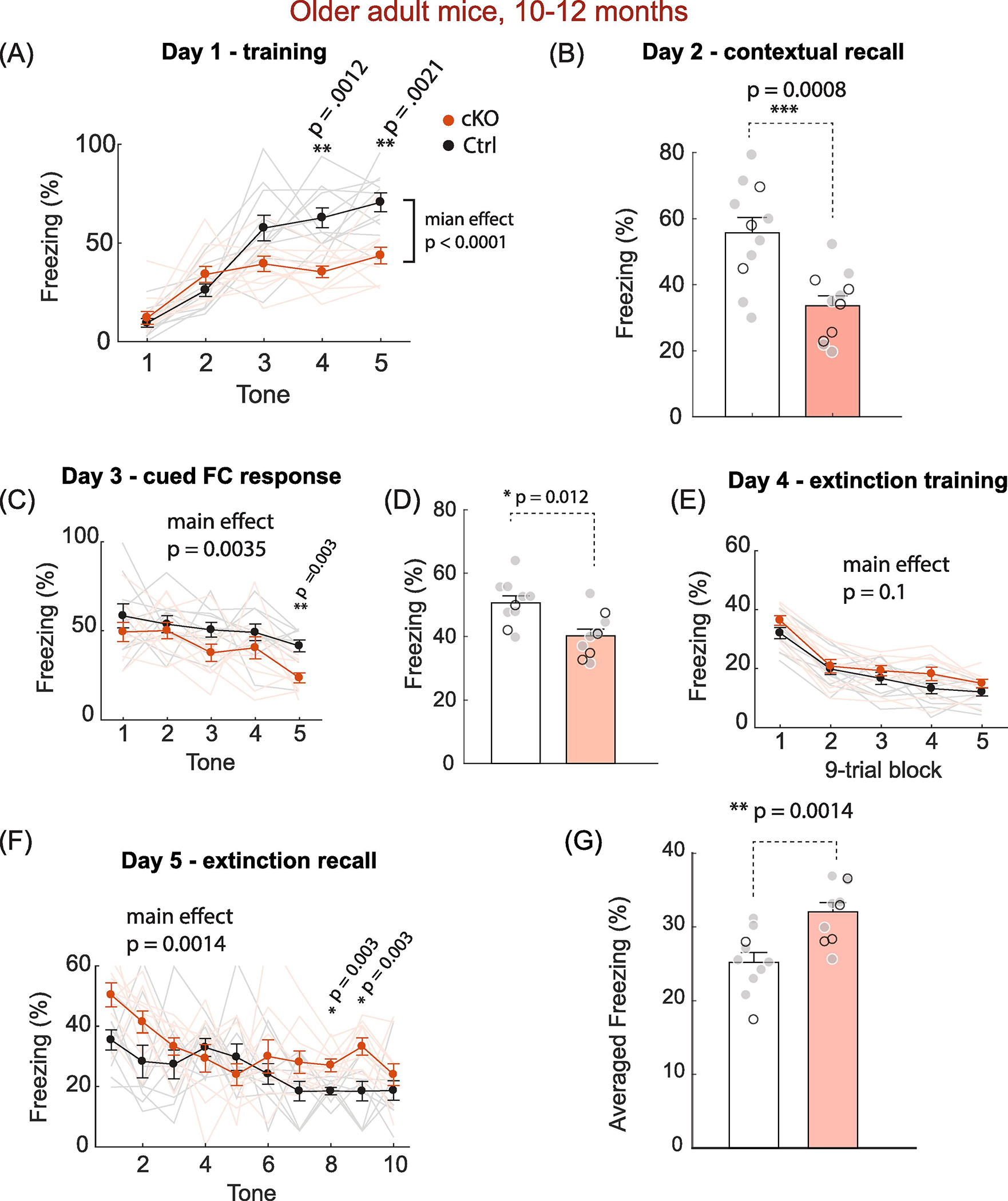
(A) Both groups showed increased percent of freezing with US-CS pairing. MetcKO mice showed an overall significantly decreased freezing time compared that from control. Effect for group, F(1, 20) = 37.7, p < 0.0001; Effect for interaction, F(4, 80) = 7.11, p < 0.0001. N = 11 mice for both groups. Two way rmANOVA. post hoc Sidak’s MCT also revealed a significant increase in freezing in session 4 (**p = 0.0012) and session 5 (**p = 0.0021). (B) MetcKO mice showed impaired contextual memory in the contextual recall test. Average freezing time, Ctrl, 55.7 ± 4.65 sec; cKO, 33.6 ± 3.09 sec. N = 11 for both groups. t(20) = 3.96, ***p = 0.0008, unpaired t test). (C) MetcKO showed overall reduced freezing in response to the CS cue across five trials. F(1,18) = 11.2, **p = 0.0035. N = 10 for both groups, two way rmANOVA). (D) Averaged freezing was measured across all five trials. MetcKO mice showed an overall increase in cued fear response. t(18) = 2.80, p = 0.012. unpaired t test. (E) Learned fear extinction in MetcKO mice was slower overall across the five sessions of 9-trial blocks, but did not reach statistical significance. F(1,18) = 2.94, p = 0.10. two-way rmANOVA. (F) MetcKO mice showed a significant overall increase in freezing across the 10-trial extinction recall test. Main group effect, F(1,18) = 14.2, p = 0.0014. (G) Overall averaged freezing, quantified from the ten Day 5 trials, was significantly increased in MetcKO mice during the extinction recall. Ctrl, 25.2 ± 1.32 sec; cKO, 32.1 ± 1.25 sec. t(18) = 3.77, p = 0.0014.
3.7. Normal locomotor activity, levels of anxiety, but enhanced motor learning in young adult MetcKO mice
The age-dependent alteration in synaptic plasticity, spatial and associative learning in MetcKO mice prompted us to ask how MetcKO may affect other types of learning. Because MET is transiently expressed in the developing mouse cortex, including the motor cortex (Judson et al., 2009), we conducted experiments aimed at assessing locomotor activity, anxiety-related behaviors. In addition, we employed a rotarod test to assess motor learning behavior. We first tested locomotor activity in MetcKO and control mice using an open field activity chamber, then measured anxiety-like behaviors using elevated plus maze (EPM) in another cohort of mice (Fig. 8A). Mice activity in the open field was tracked for 5 min. Both total distance traveled and time spent in the center were quantified. For young adult mice, neither total distance traveled (Fig. 8B. Ctrl, 2239 ± 118 cm, N = 14 mice; 2131 ± 133 cm, N = 13 mice. t(25) = 1.17, p = 0.25), nor the percent of time spent in center of arena (Fig. 8C. Ctrl, 29.6 ± 3.2; cKO, 24.3 ± 3.1, t(25) = 1.19, p = 0.24) was significantly different between the two groups. In addition, the two groups did not differ in their time spent in the open arm of the EPM (Fig. 8D. Ctrl, 15.7 ± 3.2 sec, N = 11; cKO, 12.5 ± 2.1 sec; N = 12. t(21) = 0.86, p = 0.39). For the 10–12 months older adult mice, there was also no difference in total distance traveled (Fig. 8E. Ctrl, 1547 ± 129.4 cm, N = 15 mice; cKO, 1509 ± 156.0 cm, N = 12 mice. t(25) = 0.19, p = 0.85), or the percent of time spent in center of arena (Fig. 8F. Ctrl, 25.2 ± 1.5; cKO, 23.7 ± 2.0. t(25) = 0.60, p = 0.55). Both groups also spent similar amount of time in the open arm in the EPM test (Fig. 8G. Ctrl, 12.9 ± 1.8 sec, N = 11; cKO, 16.6 ± 2.3 sec; N = 12. t(21) = 1.25, p = 0.23. These data indicate normal basal locomotor activity, and similar levels of anxiety or reaction to novelty in MetcKO mice at both ages.
Fig. 8. MetcKO mice show normal locomotor activity and anxiety, but increased motor learning at young adult (2–3 months) age.
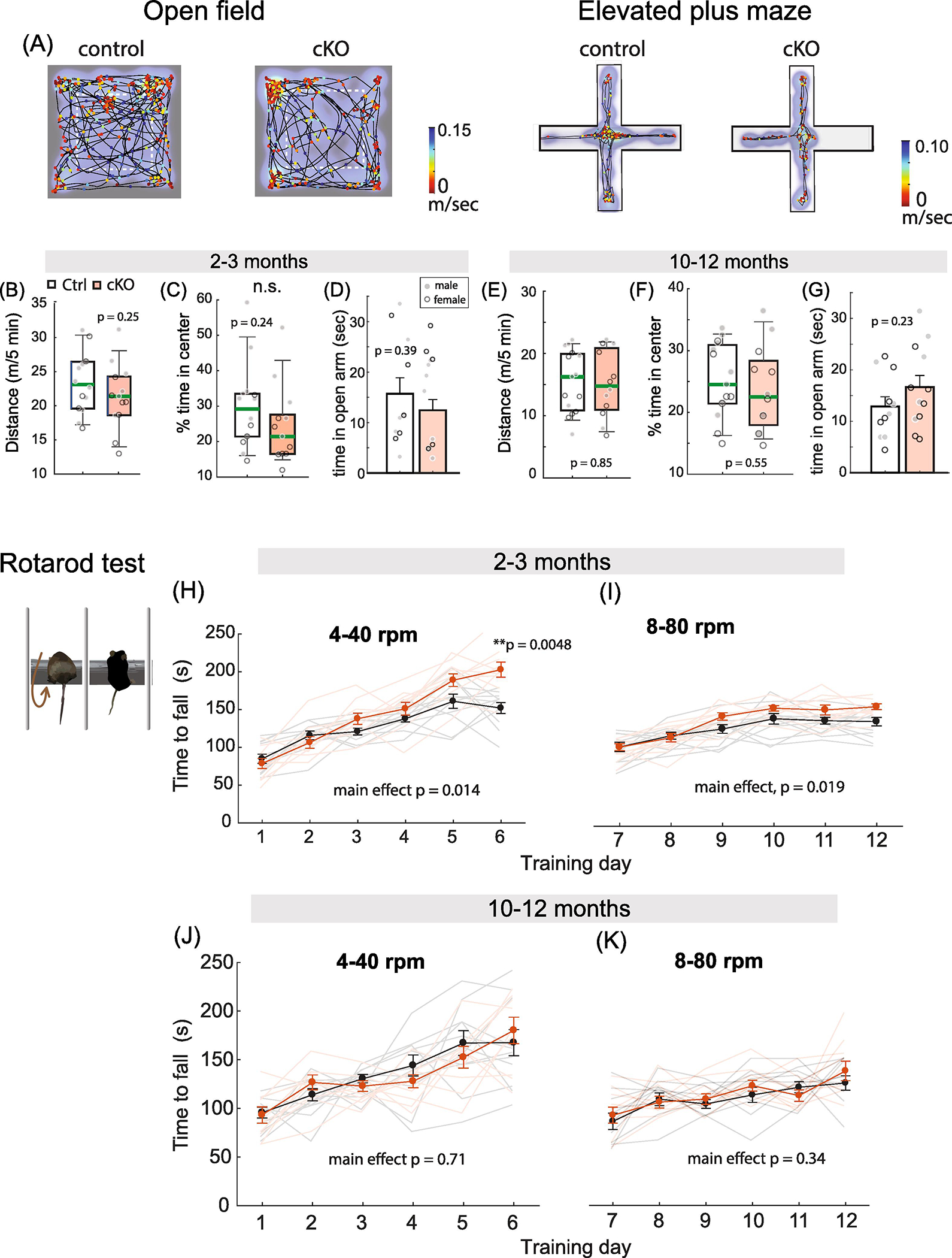
(A) A representative young adult control and MetcKO mice open field test and elevated plus maze test results. Heat map denotes time spent on the arena locations. Line graph indicates moving trajectories, marker color represents speed at the locomotion. (B) No significant difference in the total distance traveled between young adult control and cKO mice. Ctrl, 2239 ± 118 cm, N = 14 mice; 2131 ± 133 cm, N = 13 mice. t(25) = 1.17, p = 0.25. (C) No significant difference was found on the percent of time spent in center of arena in young control and cKO mice. Ctrl, 29.6 ± 3.2; cKO, 24.3 ± 3.1, t(25) = 1.19, p = 0.24. (D) Control and cKO mice spent similar amount of time in the open arm of the elevated plus maze (Ctrl, 15.7 ± 3.2 sec, N = 11; cKO, 12.5 ± 2.1 sec; N = 12. t(21) = 0.86, p = 0.39). (E) No significant difference on distance traveled between old adult control and cKO mice in open field test. Ctrl, 1547 ± 129.4 cm, N = 15 mice; cKO, 1509 ± 156.0 cm, N = 12 mice. t(25) = 0.19, p = 0.85. (F) Time spent in center of arena do not differ between older adult control and cKO mice. Ctrl, 25.2 ± 1.5; cKO, 23.7 ± 2.0. t(25) = 0.60, p = 0.55. (G) Both control and cKO mice spent similar amount of time in the open arm in the EPM test (Ctrl, 12.9 ± 1.8 sec, N = 11; cKO, 16.6 ± 2.3 sec; N = 12. t(21) = 1.25, p = 0.23). (H) cKO mice showed faster overall motor learning that was dependent on training sessions during the 4 −40 rpm training. Main group effect, F(1,19) =7.28, p = 0.014; Group x training sessions interaction, F(5,95) = 5.64. p = 0.0001. Two-way rmANOVA. cKO mice also exhibited a significant increase by Day 6 (p = 0.0048, Sidak’s MCT). (I) There was overall increased performance for the 8 – 80 rpm motor training as well. Main effect on groups, F(1,19) = 6.49, p = 0.019. Two-way rmANOVA. (J) There were no significant differences for the 4–40 rpm motor learning. Main group effect, F(1,17) = 0.14, p = 0.71. Two-way rmANOVA. (K) Older adult cKO mice perform similarly on the 8 – 80 rpm motor learning. Main effect on group, F(1,17) = 0.97, p = 0.34.
Next we examined motor learning ability of young adult mice on a rotarod (Ha et al., 2016). Mice were first tested on an accelerating rotarod (4 – 40 rpm linear acceleration in 5 min) for six consecutive days, with three training sessions performed per day. We found that both control and MetcKO mice showed motor learning with increased training days, while MetcKO mice showed faster overall learning curve (Fig. 8H. Main effect for group, F(1,19) =7.28, p = 0.014. Ctrl, N = 11, cKO, N = 10. Two-way rmANOVA) that is dependent on the training sessions (group × training session interaction, F(5,95) = 5.64, p = 0.0001). MetcKO mice motor performance also exhibited a significant increase by Day 6 (p = 0.0048, Sidak’s MCT). When tested on a more demanding condition (8–80 rpm linear acceleration in 5 min), young cKO mice also performed better, sustaining overall longer time before falling off the rotor beam (Fig. 8I. Main effect of group, F(1,19) = 6.49, p = 0.019. Two-way rmANOVA). Next, older adult mice were tested for their motor learning ability using the same paradigm. There was no difference between control and MetcKO mice in their overall performance during the six day training period when tested on the 4 – 40 rpm training (Fig. 8J. Main effect of group, F(1,17) = 0.14, p = 0.71. Ctrl, N = 10; cKO, N = 9. Two-way rmANOVA). There was also no difference when tested with the 8 – 80 rpm paradigm (Fig. 8K. Main effect of group, F(1,17) = 0.97, p = 0.34. Two-way rmANOVA). Therefore, young adult but not old adult MetcKO mice exhibit an enhanced motor learning, while basal locomotor activity and anxiety related behaviors do not seem affected by age.
DISCUSSION
In this study, we report the that genetic disruption of Met in developing mouse cortical circuits results in overall opposite learning and memory phenotypes that are depending on animal age. In addition, these behavior phenotypes are associated with age-dependent disrupted synaptic plasticity measured in the hippocampus. To the best of our knowledge, this study provides the first evidence that a neurodevelopmental risk gene bi-directionally controls age-dependent synaptic plasticity, memory and learning behavior.
The ASD-associated genetic variation in MET gene (e.g. the rs1858830 ‘C’ allele in human MET gene promotor), which increases autism risk by 2.27 fold, reduces MET mRNA and protein levels in the brains of autism patients, likely through altered interactions with transcription factors and synaptic signaling molecules (Campbell et al., 2006; Campbell et al., 2007; Plummer et al., 2013; Xie et al., 2016). How this lowered MET expression influences cognition, synaptic plasticity, learning and memory, social and language skills, and other behavioral phenotypes related to ASD remains unclear. In addition to MET, the developing brain is shaped by a myriad of molecular signaling mediated by growth factors that signal through RTKs (Park and Poo, 2013). This is consistent with the reported enrichment of ASD risk genes that are related to growth factor signaling (Wittkowski et al., 2014), and may account for the observed aberrant growth trajectories of ASD brains (Levitt and Campbell, 2009; Berg and Geschwind, 2012). Yet, MET signaling is unique in multiple ways; first, the signaling engages pleiotropic molecular pathways in the brain that are capable of activating PI3K, AKT, mTOR and small GTPases (Qiu et al., 2014; Xie et al., 2016; Chen et al., 2020). These molecular networks are known to control neuronal and spine morphogenesis, and synaptic transmission and plasticity (Kim and Lisman, 1999; Gipson and Johnston, 2012; Sanchez-Alegria et al., 2018); second, MET signaling crosstalks with other neurodevelopmental hub genes, including FOXP2 and MeCP2 (Mukamel et al., 2011; Plummer et al., 2013), which likely modify broad developmental milestones of cortex; third, MET signaling coincides a time window of peak synaptogenesis and neurite growth, but is precipitously down-regulated prior to synapse maturation and activity-dependent circuit refinement (Lein et al., 2007; Judson et al., 2009; Judson et al., 2011; Chen et al., 2020); As such, through the intrinsic control of its temporal expression, MET signaling is ideally positioned to regulate key developmental events, including neurogenesis, neurite outgrowth, and synaptic connectivity. Lastly, through cell type-specific expression (Wu and Levitt, 2013; Kast et al., 2017) and temporal control of synapse maturation (Qiu et al., 2014), MET signaling modifies cortical circuit connectivity (Qiu et al., 2011), critical period plasticity (Chen et al., 2020), and behavior (Okaty et al., 2015; Thompson and Levitt, 2015). These pleiotropic signaling effects may account for, in part, the broad biological effects when MET signaling was dysregulated (Judson et al., 2009; Judson et al., 2010; Qiu et al., 2011; Qiu et al., 2014; Peng et al., 2016; Heun-Johnson and Levitt, 2018; Chen et al., 2020).
The developing synapse is a critical target disrupted in many syndromic forms of neurodevelopmental disorders that affect cognition, including intellectual disability, and ASD (Harlow et al., 2010; Wang et al., 2011; Zoghbi and Bear, 2012; Wang et al., 2016). Disruptions of the molecular constituents of excitatory synapses are evident with deleterious, rare mutations in signaling proteins that alter synaptic structure and function (Durand et al., 1996; Gilman et al., 2011; Hamdan et al., 2011; Ebert and Greenberg, 2013; Phillips and Pozzo-Miller, 2015), and thus may explain the cognitive deficits associated with these rare mutations. We have previously reported that MetcKO mice show disrupted cortical circuit development. For example, MetcKO mice showed altered excitatory connectivity topology in frontal cortex circuits (Qiu et al., 2011), which were manifested in specific cortical projection neuron populations, potentially reflecting the mosaic cortical expression patterns of MET (Kast et al., 2019). MetcKO mice also exhibited an early termination of critical period plasticity in the visual cortex, measured by in vivo single unit recordings combined with monocular deprivation-induced ocular dominance plasticity (Chen et al., 2020). Strikingly, turning off MET signaling after a prolonged Met transgenic expression beyond normal cortical critical period also opens a temporal window of heightened plasticity (Chen et al., 2020), indicating MET signaling is a bi-directional determinant for cortical plasticity.
How does MetcKO potentially affects the timing of synaptic plasticity? The effects MetckO may be related to altered expression of synaptic glutamate receptor subunits across development, as we have found that at a very early developmental stage (PD 12–15), MetcKO mice show increased AMPAR subunit GluA1, GluN2A and decreased GluN2B at the synaptic sites in CA1 hippocampus. As a result, hippocampal circuit in MetcKO mice matures early, possesses less number of silent synapses (Qiu et al., 2014), and leads to strengthened basal synaptic transmission and elevated hippocampus synaptic plasticity (LTP, (Ma et al., 2019)). However, impaired hippocampus LTP was found in P56–70 mice when LTP was induced through a theta burst stimulation protocol (Ma et al., 2019). This study (Ma et al., 2019) contrasts with current finding that elevated LTP in MetcKO hippocampal slices when LTP was inducted using two trains of tetanus stimulation. It has been well established that different patterns of stimulation engages differential activation of NMDA receptors and voltage-gated calcium channels, and determines the plasticity outcomes (Larson and Munkacsy, 2015; Matt et al., 2018). Further corroborating the genetic results, pharmacological inhibition of MET kinase activity in cultured hippocampus slices also leads to similar changes on synaptic plasticity (Ma et al., 2019). The current study also found enhanced LTD in young adult MetcKO mice, which also contrasts with the Ma et al. (2019) findings with impaired LTD. This apparent discrepancy may be due to the slight differences in age or variability in experimental conditions. It has been previously reported that activation of MET signaling by its only known ligand, hepatocyte growth factor (HGF), in juvenile rat hippocampus slices enhanced LTP in CA1 region by augment NMDA receptor currents, but with no effect on LTD (Akimoto et al.). Therefore, it appears that MET signaling under ex vivo conditions can be complicated and further studies are warranted to determine its role in hippocampus plasticity and the underlying molecular mechanisms. Taken together, our work support the emerging view that autism is developmental disorder with disrupted timing of critical period plasticity (Berger et al., 2013).
Although intriguing, previous studies only went so far as to allude MET signaling as an intrinsic mechanism that control neuronal maturation and synaptic plasticity at an early developmental stage. Built on these findings, the current work reveals that disrupted MET signaling alters trajectory of synaptic plasticity and learning and memory behavior potentially across the life span. Long-term memory formation requires molecular signaling mechanisms governing synaptic plasticity and controlling experience-dependent establishment and maintenance of synaptic connections within the CNS (Takeuchi et al., 2014), both processes are known to be age-dependent (Baudry, 1998; Lynch et al., 2006; Oberman and Pascual-Leone, 2013) and are associated with synapse loss that is caused by aging or neurodegeneration (Barnes, 1994; Bach et al., 1999; Barnes et al., 2000; Barnes, 2003; Temido-Ferreira et al., 2019). Other LTP properties, such as input specificity, could be impaired as a result of aging in mice (Ris and Godaux, 2007). In addition to regulating synaptic plasticity, MET signaling has been shown to promote synaptogenesis, and its termination precedes the peak of synapse pruning. This indicates the potential to persistently regulate the circuit connectivity and enable the preservation of memory-subserving synaptic connections.
While MET is clearly an autism genetic risk factor (SFARI category 2 ‘strong candidate’ gene), it is extremely challenging to connect a specific gene or signaling system to behavior. We chose these learning and memory-related behavior tests based on our previous findings on the role of MET in neuron morphology, synaptic maturation and plasticity (Qiu et al., 2014; Peng et al., 2016; Chen et al., 2020). Even though learning and memory deficits are not a defining behavior domain for ASD, ASD risk genes frequently convert on synapse enriched molecules that affect plasticity outcomes, learning and memory (Chung et al., 2012; Gilbert and Man, 2017; Morimura et al., 2017; Heavner and Smith, 2020). Although the current study does not address the observed early cognitive decline and reduced synaptic plasticity in older adult mice, our work reveals novel, potential roles of MET signaling in the adult or possibly aging brain, in which MET protein is primarily of synaptic origin (Eagleson et al., 2013). Thus, MET signaling occupies both ends of the developmental spectrum. It has also been reported that MET protein levels are greatly reduced in Alzheimer’s disease (AD) patient brains (Hamasaki et al., 2014; Oswald et al., 2017). In addition, the only known ligand for MET, HGF, is also increased under neurodegenerative conditions (Machida et al., 2004; Wong et al., 2014; Wright and Harding, 2015). These findings coalesce with impaired synaptic plasticity changes associated with normal aging or neurodegeneration (Barnes, 1994; Bach et al., 1999; Barnes et al., 2000; Barnes, 2003; Temido-Ferreira et al., 2019). Whether reduction of MET expression and the resulting plasticity changes contribute to aging or neurodegeneration is an interesting point to address in future studies. It is possible that reduced MET signaling may crosstalk to AD molecular networks and contribute to the age- related cognitive decline in AD (Hullinger and Puglielli, 2017; Mostafavi et al., 2018). For these reasons, it would be informative to evaluate the whether MetckO modifies normal aging and AD related pathologies in future studies.
In addition to the role of MET signaling in cortical circuits subserving spatial and fear learning and memory, we also found that young MetcKO mice demonstrated enhanced motor learning. This indicates that MET signaling may be a generalized mechanism that controls maturation and plasticity in other MET-expressing brain regions. In addition, the considerable amount of time mice spent in the center of the open field maze may suggest elevated anxiety levels; this possibility, however, was negated by the findings that both control and MetcKO mice spent similar amount of time in the open arm of an EPM. These mice show similar performance in open field test, indicating normal basal locomotor activity and anxiety levels. A previous behavior study on MetcKO mice (Thompson and Levitt, 2015) found adult mice (P90–140) showed hypoactivity in an activity chamber, a reduction in spontaneous alternation in T maze, and increased performance on a steady-speed rotarod. Surprisingly, Metfx/fx/Nestincre but not Metfx/fx/emx1cre cKO mice exhibited deficits in contextual fear learning. Because Metfx/fx/Nestincre deletes Met in all the neurons, subcortical and interneuron circuits may contribute to the differences between these two cKO lines. This is particularly interesting considering that Met is expressed in hindbrain autonomic circuits prenatally (Wu and Levitt, 2013; Kast et al., 2017). Compared with the current study, potential differences in learned contextual fear may be due to different testing paradigms and/or animal ages. Nonetheless, these combined results suggest a complex contribution of Met in the development of cortical circuits mediating plasticity and cognitive behavior, and that the effects of disrupting developmental Met expression are dependent upon circuit-specific deletion patterns and perhaps more importantly, the developmental age. It would be also interesting to investigate other behavioral domains that are highly relevant to autism, including behavior stereotypy and the social cognitive domain in future studies.
In summary, the present study reveals a developmental pleiotropic signaling system that is relevant to autism controls the timing of synaptic plasticity development and cognitive function in an age-dependent manner. Our results support the view that autism and other neurodevelopmental disorders may be a disorder of disrupted critical period plasticity (LeBlanc and Fagiolini, 2011; Berg and Geschwind, 2012; Berger et al., 2013). Data presented here support the hypothesis that MET signaling is critical on the timing and experience-dependent circuit plasticity, and may shed light on the shared neurobiology of neurodegeneration and aging, both conditions featuring synaptic dysfunction and impaired plasticity. Our work also provides a rationale to further investigate the molecular link between MET signaling and synaptic plasticity/behavior by harnessing the power of high-throughput transcriptomics to achieve a comprehensive understanding on the relationship of the pleiotropic molecular signaling with behavior. The current finding raises a tantalizing possibility that synapse mature early may be subjected to an accelerated aging-related synaptic dysfunction and cognitive decline, an interesting issue that awaits future studies.
Acknowledgements
This study was supported by NIH/NIMH grant R01MH111619 (S.Q.), and institution startup fund from The University of Arizona (S.Q.).
Footnotes
Conflict of interest
The authors declare that they have no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference
- Akimoto M, Baba A, Ikeda-Matsuo Y, Yamada MK, Itamura R, Nishiyama N, Ikegaya Y, Matsuki N (2004) Hepatocyte growth factor as an enhancer of nmda currents and synaptic plasticity in the hippocampus. Neuroscience 128:155–162. [DOI] [PubMed] [Google Scholar]
- Aldinger KA, Plummer JT, Qiu S, Levitt P (2011) SnapShot: genetics of autism. Neuron 72:418–418 e411. [DOI] [PubMed] [Google Scholar]
- Aldinger KA, Lane CJ, Veenstra-VanderWeele J, Levitt P (2015) Patterns of Risk for Multiple Co-Occurring Medical Conditions Replicate Across Distinct Cohorts of Children with Autism Spectrum Disorder. Autism Res 8:771–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asok A, Leroy F, Rayman JB, Kandel ER (2019) Molecular Mechanisms of the Memory Trace. Trends Neurosci 42:14–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bach ME, Barad M, Son H, Zhuo M, Lu YF, Shih R, Mansuy I, Hawkins RD, Kandel ER (1999) Age-related defects in spatial memory are correlated with defects in the late phase of hippocampal long-term potentiation in vitro and are attenuated by drugs that enhance the cAMP signaling pathway. Proc Natl Acad Sci U S A 96:5280–5285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes CA (1994) Normal aging: regionally specific changes in hippocampal synaptic transmission. Trends Neurosci 17:13–18. [DOI] [PubMed] [Google Scholar]
- Barnes CA (2003) Long-term potentiation and the ageing brain. Philos Trans R Soc Lond B Biol Sci 358:765–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes CA, Rao G, Orr G (2000) Age-related decrease in the Schaffer collateral-evoked EPSP in awake, freely behaving rats. Neural Plast 7:167–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudry M (1998) Synaptic plasticity and learning and memory: 15 years of progress. Neurobiol Learn Mem 70:113–118. [DOI] [PubMed] [Google Scholar]
- Berg JM, Geschwind DH (2012) Autism genetics: searching for specificity and convergence. Genome Biol 13:247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger JM, Rohn TT, Oxford JT (2013) Autism as the Early Closure of a Neuroplastic Critical Period Normally Seen in Adolescence. Biol Syst Open Access 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliss TV, Gardner-Medwin AR (1973) Long-lasting potentiation of synaptic transmission in the dentate area of the unanaestetized rabbit following stimulation of the perforant path. J Physiol 232:357–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL (1993) A synaptic model of memory: long-term potentiation in the hippocampus. Nature 361:31–39. [DOI] [PubMed] [Google Scholar]
- Campbell DB, D’Oronzio R, Garbett K, Ebert PJ, Mirnics K, Levitt P, Persico AM (2007) Disruption of cerebral cortex MET signaling in autism spectrum disorder. Ann Neurol 62:243–250. [DOI] [PubMed] [Google Scholar]
- Campbell DB, Sutcliffe JS, Ebert PJ, Militerni R, Bravaccio C, Trillo S, Elia M, Schneider C, Melmed R, Sacco R, Persico AM, Levitt P (2006) A genetic variant that disrupts MET transcription is associated with autism. Proc Natl Acad Sci U S A 103:16834–16839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrere M, Alexandre F (2015) A pavlovian model of the amygdala and its influence within the medial temporal lobe. Front Syst Neurosci 9:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Ma X, Nehme A, Wei J, Cui Y, Cui Y, Yao D, Wu J, Anderson T, Ferguson D, Levitt P, Qiu S (2020) Time-delimited signaling of MET receptor tyrosine kinase regulates cortical circuit development and critical period plasticity. Mol Psychiatry. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung L, Bey AL, Jiang YH (2012) Synaptic plasticity in mouse models of autism spectrum disorders. Korean J Physiol Pharmacol 16:369–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand GM, Kovalchuk Y, Konnerth A (1996) Long-term potentiation and functional synapse induction in developing hippocampus. Nature 381:71–75. [DOI] [PubMed] [Google Scholar]
- Eagleson KL, Milner TA, Xie Z, Levitt P (2013) Synaptic and extrasynaptic location of the receptor tyrosine kinase met during postnatal development in the mouse neocortex and hippocampus. J Comp Neurol 521:3241–3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert DH, Greenberg ME (2013) Activity-dependent neuronal signalling and autism spectrum disorder. Nature 493:327–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erreger K, Dravid SM, Banke TG, Wyllie DJ, Traynelis SF (2005) Subunit-specific gating controls rat NR1/NR2A and NR1/NR2B NMDA channel kinetics and synaptic signalling profiles. J Physiol 563:345–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert J, Man HY (2017) Fundamental Elements in Autism: From Neurogenesis and Neurite Growth to Synaptic Plasticity. Front Cell Neurosci 11:359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilman SR, Iossifov I, Levy D, Ronemus M, Wigler M, Vitkup D (2011) Rare de novo variants associated with autism implicate a large functional network of genes involved in formation and function of synapses. Neuron 70:898–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gipson TT, Johnston MV (2012) Plasticity and mTOR: towards restoration of impaired synaptic plasticity in mTOR-related neurogenetic disorders. Neural Plast 2012:486402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goode TD, Ressler RL, Acca GM, Miles OW, Maren S (2019) Bed nucleus of the stria terminalis regulates fear to unpredictable threat signals. Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorski JA, Talley T, Qiu M, Puelles L, Rubenstein JL, Jones KR (2002) Cortical excitatory neurons and glia, but not GABAergic neurons, are produced in the Emx1-expressing lineage. J Neurosci 22:6309–6314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha S, Lee D, Cho YS, Chung C, Yoo YE, Kim J, Lee J, Kim W, Kim H, Bae YC, Tanaka-Yamamoto K, Kim E (2016) Cerebellar Shank2 Regulates Excitatory Synapse Density, Motor Coordination, and Specific Repetitive and Anxiety-Like Behaviors. J Neurosci 36:12129–12143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamasaki H, Honda H, Suzuki SO, Hokama M, Kiyohara Y, Nakabeppu Y, Iwaki T (2014) Down-regulation of MET in hippocampal neurons of Alzheimer’s disease brains. Neuropathology 34:284–290. [DOI] [PubMed] [Google Scholar]
- Hamdan FF, Daoud H, Piton A, Gauthier J, Dobrzeniecka S, Krebs MO, Joober R, Lacaille JC, Nadeau A, Milunsky JM, Wang Z, Carmant L, Mottron L, Beauchamp MH, Rouleau GA, Michaud JL (2011) De novo SYNGAP1 mutations in nonsyndromic intellectual disability and autism. Biol Psychiatry 69:898–901. [DOI] [PubMed] [Google Scholar]
- Harlow EG, Till SM, Russell TA, Wijetunge LS, Kind P, Contractor A (2010) Critical period plasticity is disrupted in the barrel cortex of FMR1 knockout mice. Neuron 65:385–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heavner WE, Smith SEP (2020) Resolving the Synaptic versus Developmental Dichotomy of Autism Risk Genes. Trends Neurosci 43:227–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heun-Johnson H, Levitt P (2018) Differential impact of Met receptor gene interaction with early-life stress on neuronal morphology and behavior in mice. Neurobiol Stress 8:10–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hullinger R, Puglielli L (2017) Molecular and cellular aspects of age-related cognitive decline and Alzheimer’s disease. Behav Brain Res 322:191–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judson MC, Amaral DG, Levitt P (2011) Conserved subcortical and divergent cortical expression of proteins encoded by orthologs of the autism risk gene MET. Cereb Cortex 21:1613–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judson MC, Eagleson KL, Wang L, Levitt P (2010) Evidence of cell-nonautonomous changes in dendrite and dendritic spine morphology in the met-signaling-deficient mouse forebrain. J Comp Neurol 518:4463–4478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judson MC, Bergman MY, Campbell DB, Eagleson KL, Levitt P (2009) Dynamic gene and protein expression patterns of the autism-associated met receptor tyrosine kinase in the developing mouse forebrain. J Comp Neurol 513:511–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kast RJ, Wu HH, Levitt P (2019) Developmental Connectivity and Molecular Phenotypes of Unique Cortical Projection Neurons that Express a Synapse-Associated Receptor Tyrosine Kinase. Cereb Cortex 29:189–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kast RJ, Wu HH, Williams P, Gaspar P, Levitt P (2017) Specific Connectivity and Unique Molecular Identity of MET Receptor Tyrosine Kinase Expressing Serotonergic Neurons in the Caudal Dorsal Raphe Nuclei. ACS Chem Neurosci 8:1053–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenney J, Manahan-Vaughan D (2013) Learning-facilitated synaptic plasticity occurs in the intermediate hippocampus in association with spatial learning. Front Synaptic Neurosci 5:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim CH, Lisman JE (1999) A role of actin filament in synaptic transmission and long-term potentiation. J Neurosci 19:4314–4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JJ, Jung MW (2006) Neural circuits and mechanisms involved in Pavlovian fear conditioning: a critical review. Neurosci Biobehav Rev 30:188–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson J, Munkacsy E (2015) Theta-burst LTP. Brain Res 1621:38–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBlanc JJ, Fagiolini M (2011) Autism: a “critical period” disorder? Neural Plast 2011:921680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lein ES et al. (2007) Genome-wide atlas of gene expression in the adult mouse brain. Nature 445:168–176. [DOI] [PubMed] [Google Scholar]
- Levitt P, Campbell DB (2009) The genetic and neurobiologic compass points toward common signaling dysfunctions in autism spectrum disorders. J Clin Invest 119:747–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch G, Rex CS, Gall CM (2006) Synaptic plasticity in early aging. Ageing Res Rev 5:255–280. [DOI] [PubMed] [Google Scholar]
- Ma X, Chen K, Lu Z, Piechowicz M, Liu Q, Wu J, Qiu S (2019) Disruption of MET Receptor Tyrosine Kinase, an Autism Risk Factor, Impairs Developmental Synaptic Plasticity in the Hippocampus. Dev Neurobiol 79:36–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machida S, Tanaka M, Ishii T, Ohtaka K, Takahashi T, Tazawa Y (2004) Neuroprotective effect of hepatocyte growth factor against photoreceptor degeneration in rats. Invest Ophthalmol Vis Sci 45:4174–4182. [DOI] [PubMed] [Google Scholar]
- Martin SJ, Grimwood PD, Morris RG (2000) Synaptic plasticity and memory: an evaluation of the hypothesis. Annu Rev Neurosci 23:649–711. [DOI] [PubMed] [Google Scholar]
- Matt L, Kirk LM, Chenaux G, Speca DJ, Puhger KR, Pride MC, Qneibi M, Haham T, Plambeck KE, Stern-Bach Y, Silverman JL, Crawley JN, Hell JW, Diaz E (2018) SynDIG4/Prrt1 Is Required for Excitatory Synapse Development and Plasticity Underlying Cognitive Function. Cell Rep 22:2246–2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morellini F (2013) Spatial memory tasks in rodents: what do they model? Cell Tissue Res 354:273–286. [DOI] [PubMed] [Google Scholar]
- Morimura N, Yasuda H, Yamaguchi K, Katayama KI, Hatayama M, Tomioka NH, Odagawa M, Kamiya A, Iwayama Y, Maekawa M, Nakamura K, Matsuzaki H, Tsujii M, Yamada K, Yoshikawa T, Aruga J (2017) Autism-like behaviours and enhanced memory formation and synaptic plasticity in Lrfn2/SALM1-deficient mice. Nat Commun 8:15800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostafavi S et al. (2018) A molecular network of the aging human brain provides insights into the pathology and cognitive decline of Alzheimer’s disease. Nat Neurosci 21:811–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukamel Z, Konopka G, Wexler E, Osborn GE, Dong H, Bergman MY, Levitt P, Geschwind DH (2011) Regulation of MET by FOXP2, genes implicated in higher cognitive dysfunction and autism risk. J Neurosci 31:11437–11442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberman L, Pascual-Leone A (2013) Changes in plasticity across the lifespan: cause of disease and target for intervention. Prog Brain Res 207:91–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okaty BW, Freret ME, Rood BD, Brust RD, Hennessy ML, deBairos D, Kim JC, Cook MN, Dymecki SM (2015) Multi-Scale Molecular Deconstruction of the Serotonin Neuron System. Neuron 88:774–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oswald F, Kloble P, Ruland A, Rosenkranz D, Hinz B, Butter F, Ramljak S, Zechner U, Herlyn H (2017) The FOXP2-Driven Network in Developmental Disorders and Neurodegeneration. Front Cell Neurosci 11:212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H, Poo MM (2013) Neurotrophin regulation of neural circuit development and function. Nat Rev Neurosci 14:7–23. [DOI] [PubMed] [Google Scholar]
- Paul CM, Magda G, Abel S (2009) Spatial memory: Theoretical basis and comparative review on experimental methods in rodents. Behav Brain Res 203:151–164. [DOI] [PubMed] [Google Scholar]
- Peng Y, Lu Z, Li G, Piechowicz M, Anderson MA, Uddin Y, Wu J, Qiu S (2016) The autism associated MET receptor tyrosine kinase engages early neuronal growth mechanism and controls glutamatergic circuits development in the forebrain. Mol Psychiatry. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips M, Pozzo-Miller L (2015) Dendritic spine dysgenesis in autism related disorders. Neurosci Lett 601:30–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plummer JT, Evgrafov OV, Bergman MY, Friez M, Haiman CA, Levitt P, Aldinger KA (2013) Transcriptional regulation of the MET receptor tyrosine kinase gene by MeCP2 and sex-specific expression in autism and Rett syndrome. Transl Psychiatry 3:e316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu S, Lu Z, Levitt P (2014) MET receptor tyrosine kinase controls dendritic complexity, spine morphogenesis, and glutamatergic synapse maturation in the hippocampus. J Neurosci 34:16166–16179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu S, Zhao LF, Korwek KM, Weeber EJ (2006) Differential reelin-induced enhancement of NMDA and AMPA receptor activity in the adult hippocampus. J Neurosci 26:12943–12955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu S, Anderson CT, Levitt P, Shepherd GM (2011) Circuit-specific intracortical hyperconnectivity in mice with deletion of the autism-associated Met receptor tyrosine kinase. J Neurosci 31:5855–5864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ressler RL, Maren S (2019) Synaptic encoding of fear memories in the amygdala. Curr Opin Neurobiol 54:54–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ris L, Godaux E (2007) Synapse specificity of long-term potentiation breaks down with aging. Learn Mem 14:185–189. [DOI] [PubMed] [Google Scholar]
- Rudie JD, Hernandez LM, Brown JA, Beck-Pancer D, Colich NL, Gorrindo P, Thompson PM, Geschwind DH, Bookheimer SY, Levitt P, Dapretto M (2012) Autism-associated promoter variant in MET impacts functional and structural brain networks. Neuron 75:904–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Alegria K, Flores-Leon M, Avila-Munoz E, Rodriguez-Corona N, Arias C (2018) PI3K Signaling in Neurons: A Central Node for the Control of Multiple Functions. Int J Mol Sci 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shipton OA, Paulsen O (2014) GluN2A and GluN2B subunit-containing NMDA receptors in hippocampal plasticity. Philos Trans R Soc Lond B Biol Sci 369:20130163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi T, Duszkiewicz AJ, Morris RG (2014) The synaptic plasticity and memory hypothesis: encoding, storage and persistence. Philos Trans R Soc Lond B Biol Sci 369:20130288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temido-Ferreira M, Coelho JE, Pousinha PA, Lopes LV (2019) Novel Players in the Aging Synapse: Impact on Cognition. J Caffeine Adenosine Res 9:104–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson BL, Levitt P (2015) Complete or partial reduction of the Met receptor tyrosine kinase in distinct circuits differentially impacts mouse behavior. J Neurodev Disord 7:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, McCoy PA, Rodriguiz RM, Pan Y, Je HS, Roberts AC, Kim CJ, Berrios J, Colvin JS, Bousquet-Moore D, Lorenzo I, Wu G, Weinberg RJ, Ehlers MD, Philpot BD, Beaudet AL, Wetsel WC, Jiang YH (2011) Synaptic dysfunction and abnormal behaviors in mice lacking major isoforms of Shank3. Hum Mol Genet 20:3093–3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X et al. (2016) Altered mGluR5-Homer scaffolds and corticostriatal connectivity in a Shank3 complete knockout model of autism. Nat Commun 7:11459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittkowski KM, Sonakya V, Bigio B, Tonn MK, Shic F, Ascano M, Nasca C, Gold-Von Simson G (2014) A novel computational biostatistics approach implies impaired dephosphorylation of growth factor receptors as associated with severity of autism. Transl Psychiatry 4:e354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong WK, Cheung AW, Yu SW, Sha O, Cho EY (2014) Hepatocyte growth factor promotes long-term survival and axonal regeneration of retinal ganglion cells after optic nerve injury: comparison with CNTF and BDNF. CNS Neurosci Ther 20:916–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright JW, Harding JW (2015) The Brain Hepatocyte Growth Factor/c-Met Receptor System: A New Target for the Treatment of Alzheimer’s Disease. J Alzheimers Dis 45:985–1000. [DOI] [PubMed] [Google Scholar]
- Wu HH, Levitt P (2013) Prenatal Expression of MET Receptor Tyrosine Kinase in the Fetal Mouse Dorsal Raphe Nuclei and the Visceral Motor/Sensory Brainstem. Dev Neurosci 35:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Li J, Baker J, Eagleson KL, Coba MP, Levitt P (2016) Receptor Tyrosine Kinase MET Interactome and Neurodevelopmental Disorder Partners at the Developing Synapse. Biol Psychiatry. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Storm DR, Wang H (2011) Bidirectional synaptic plasticity and spatial memory flexibility require Ca2+-stimulated adenylyl cyclases. J Neurosci 31:10174–10183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoghbi HY, Bear MF (2012) Synaptic dysfunction in neurodevelopmental disorders associated with autism and intellectual disabilities. Cold Spring Harb Perspect Biol 4. [DOI] [PMC free article] [PubMed] [Google Scholar]