

Abstract
Background:
A combination of central muscle relaxants, chlorzoxazone and baclofen (chlorzoxazone-baclofen), has been proposed for treatment of cerebellar symptoms in human spinocerebellar ataxia (SCA). However, central muscle relaxants can worsen balance. The optimal dose for target engagement without toxicity remains unknown.
Objectives:
Using the genetically precise Atxn1154Q/2Q model of SCA1, we determine the role of cerebellar dysfunction in motor impairment. We also aim to identify appropriate concentrations of chlorzoxazone-baclofen needed for target engagement without toxicity to plan for human clinical trials.
Methods:
We use patch clamp electrophysiology in acute cerebellar slices and immunostaining to identify the specific ion channels targeted by chlorzoxazone-baclofen. Behavioral assays for coordination and grip strength are used to determine specificity of chlorzoxazone-baclofen for improving cerebellar dysfunction without off-target effects in Atxn1154Q/2Q mice.
Results:
We identify irregular Purkinje neuron firing in association with reduced expression of the ion channels Kcnma1 and Cacna1g in Atxn1154Q/2Q mice. Using in vitro electrophysiology in brain slices, we identify concentrations of chlorzoxazone-baclofen that improve Purkinje neuron spike regularity without reducing firing frequency. At a disease stage in Atxn1154Q/2Q mice when motor impairment is due to cerebellar dysfunction, orally administered chlorzoxazone-baclofen improves motor performance without affecting muscle strength.
Conclusion:
We identify a tight relationship between baclofen-chlorzoxazone concentrations needed to engage target, and levels above which cerebellar function will be compromised. We propose to use this information for a novel clinical trial design, using sequential dose escalation within each subject, to identify dose levels that are likely to improve ataxia symptoms while minimizing toxicity.
Keywords: Ataxia, SCA1, chlorzoxazone, baclofen, neuronal dysfunction
Introduction
The CAG triplet repeat-associated spinocerebellar ataxias (SCA) are a group of neurodegenerative diseases that result in progressive loss of motor function and early death [1]. Distinct genetic mutations underlie each of the CAG triplet repeat-associated SCAs; nevertheless, recent work illustrates that shared underlying mechanisms of disease may contribute to neuronal dysfunction across these disorders [2–5]. SCA1, one of the most common and well-studied of the SCAs, is caused by a glutamine-encoding CAG triplet expansion in the ATXN1 gene. Cerebellar Purkinje neurons are prominently affected in SCA1 and are believed to underlie motor impairment, while dysfunction of brainstem nuclei and other non-cerebellar structures correlates more closely with early death [6–8]. However, the relative contribution of these structures to motor impairment at various stages of disease remains unclear.
Recent work demonstrates that Purkinje neuron dysfunction is a central cause of motor impairment in rodent models of SCA. In murine models, abnormalities in Purkinje neuron membrane excitability often manifest as irregular, slow, or absent spiking. Interestingly, specific ion-channels have been associated with firing dysfunction across several of these models [2–5], suggesting shared mechanisms of disease and potentially overlapping therapeutic targets. These channels include the large conductance calcium-activated potassium (BK) channel, along with calcium sources that can activate BK function [5]. In the transgenic ATXN1[82Q] mouse model of SCA1, targeting reduced BK channel function with the FDA-approved potassium channel activating compounds chlorzoxazone and baclofen (chlorzoxazone-baclofen) can restore Purkinje neuron spiking and improve motor dysfunction [9]. Due to Purkinje neuron-specific expression of the ATXN1[82Q] transgene in this model of SCA1 [10], the ability of chlorzoxazone-baclofen to improve cerebellar symptoms without worsening non-cerebellar symptoms is not currently known.
The benefit of chlorzoxazone-baclofen on Purkinje neuron spiking has previously been assessed at a single concentration in the transgenic ATXN1[82Q] model of SCA1 [9]. In these mice, Purkinje neurons undergo depolarization block due to profound loss of BK channel current early in disease, causing a majority of cells to cease firing at the onset of symptoms [3]. Since ATXN1[82Q] transgene expression is restricted to Purkinje neurons in this model, it is unclear whether chlorzoxazone-baclofen may have off-target effects outside of the cerebellum. It is therefore imperative to identify the concentration ranges of chlorzoxazone-baclofen that can optimally improve Purkinje neuron function while minimizing extra-cerebellar toxicity. This will enable the design of future clinical trials to assess the safety and efficacy of chlorzoxazone-baclofen in human SCA.
In the present work, we use a genetically precise knock-in mouse model of SCA1 to explore appropriate concentrations and target engagement of chlorzoxazone-baclofen. Our studies demonstrate that BK channels are the likely calcium-activated potassium channel target in SCA1. Importantly, chlorzoxazone-baclofen co-treatment improves motor function in SCA1 mice at an age when the motor phenotype is not confounded by changes in grip strength. In addition, chlorzoxazone-baclofen show no signs of toxicity in SCA1 mice and do not worsen deficits in grip strength at a later disease timepoint when motor weakness is present. These data indicate that chlorzoxazone-baclofen treatment specifically targets cerebellar dysfunction in SCA1, and supports the further development of chlorzoxazone-baclofen as a treatment for cerebellar symptoms in SCA1.
Methods
Animals
All animal studies were reviewed and approved by the University of Michigan Institutional Animal Care and Use Committee and were conducted in accordance with the United States Public Health Service’s Policy on Humane Care and Use of Laboratory Animals. Atxn1154Q/2Q mice, which express an expanded CAG triplet repeat in the endogenous Atxn1 locus [11], were maintained on a C57BL/6 background. Heterozygous Atxn1154Q/2Q mice and wild-type littermate controls were used for all studies. All studies were performed at 14 weeks of age or 20 weeks of age. Sexes were balanced for all animal studies.
Supplemental Methods
Please see the Supplemental Methods section of this manuscript for additional information.
Results
Purkinje neuron dysfunction is associated with motor impairment preceding neurodegeneration in Atxn1154Q/2Q mice
Purkinje neuron dysfunction is an early feature of disease in many models of spinocerebellar ataxia (SCA) [2–4, 12–17]. Changes in Purkinje neuron firing are directly relevant to disease, as improving Purkinje neuron firing also improves motor dysfunction in murine SCA models [4, 9, 13, 15–17]. Abnormalities in Purkinje neuron firing are particularly relevant in SCA1, as pharmacologic and genetic strategies to improve Purkinje neuron intrinsic excitability improves not just motor dysfunction but also degeneration in murine models of disease [3, 17, 18]. Prior studies in SCA1 have focused primarily on the transgenic ATXN1[82Q] model of SCA1, where polyglutamine expanded ATXN1 expression is restricted to Purkinje neurons. While the ATXN1[82Q] model of SCA1 accurately models cerebellar features of disease, the relevance of cerebellar dysfunction to the overall motor phenotype is unknown in the more genetically precise Atxn1154Q/2Q model of SCA1, in which a polyglutamine-expanded Atxn1 transgene is knocked into the endogenous Atxn1 locus, and therefore widely expressed in the nervous system and elsewhere. In this model of SCA1 with a hyperexpanded polyglutamine repeat motor dysfunction and premature death have been attributed, at least in part to spinal cord and brainstem motor neuron degeneration [7, 11]. We therefore sought to identify whether cerebellar dysfunction is present, and relevant to the motor phenotype in Atxn1154Q/2Q mice.
We examined Purkinje neuron membrane excitability in Atxn1154Q/2Q mice at 14 weeks of age, an age at which motor impairment is prominent [11]. No evidence of neurodegeneration is present in Atxn1154Q/2Q mice even at 20 weeks of age (Supplementary Figure 1A–B). When compared to Purkinje neurons from wild-type littermate controls, Atxn1154Q/2Q Purkinje neurons display irregular spiking (Figure 1A–C) with no change in firing frequency (Figure 1D). Since Purkinje neuron firing differs between the anterior and posterior cerebellum in SCA murine models [4, 5], we independently analyzed neurons from the anterior cerebellum (lobules IV-V) and nodular zone (lobule X) and found a similar increase in spike irregularity with no change in firing frequency across both cerebellar regions. Additionally, since alterations in Purkinje neuron physiology may reflect changes in either intrinsic pacemaking or synaptic activity, we performed recordings in Atxn1154Q/2Q and wild-type control Purkinje neurons before and after the addition of synaptic inhibitors (6,7-dinitroquinoxaline-2,3-dione [DNQX], 10 µM; picrotoxin, 50 µM). No significant difference in either spike frequency (Supplementary Figure 1C) or regularity (Supplementary Figure 1D) was noted after the addition of synaptic inhibitors, suggesting that altered Purkinje neuron spiking in Atxn1154Q/2Q mice is intrinsically driven. When neurons were held at −80 mV and injected with depolarizing current, we found that Atxn1154Q/2Q neurons require a significantly lower amplitude of injected current to elicit depolarization block than wild-type littermate control neurons (Figure 1E). Importantly, these alterations in spiking were accompanied by no significant change in input resistance between Atxn1154Q/2Q and wild-type littermate control neurons (Figure 1F).
Figure 1. Alterations in Purkinje neuron physiology in Atxn1154Q/2Q mice.
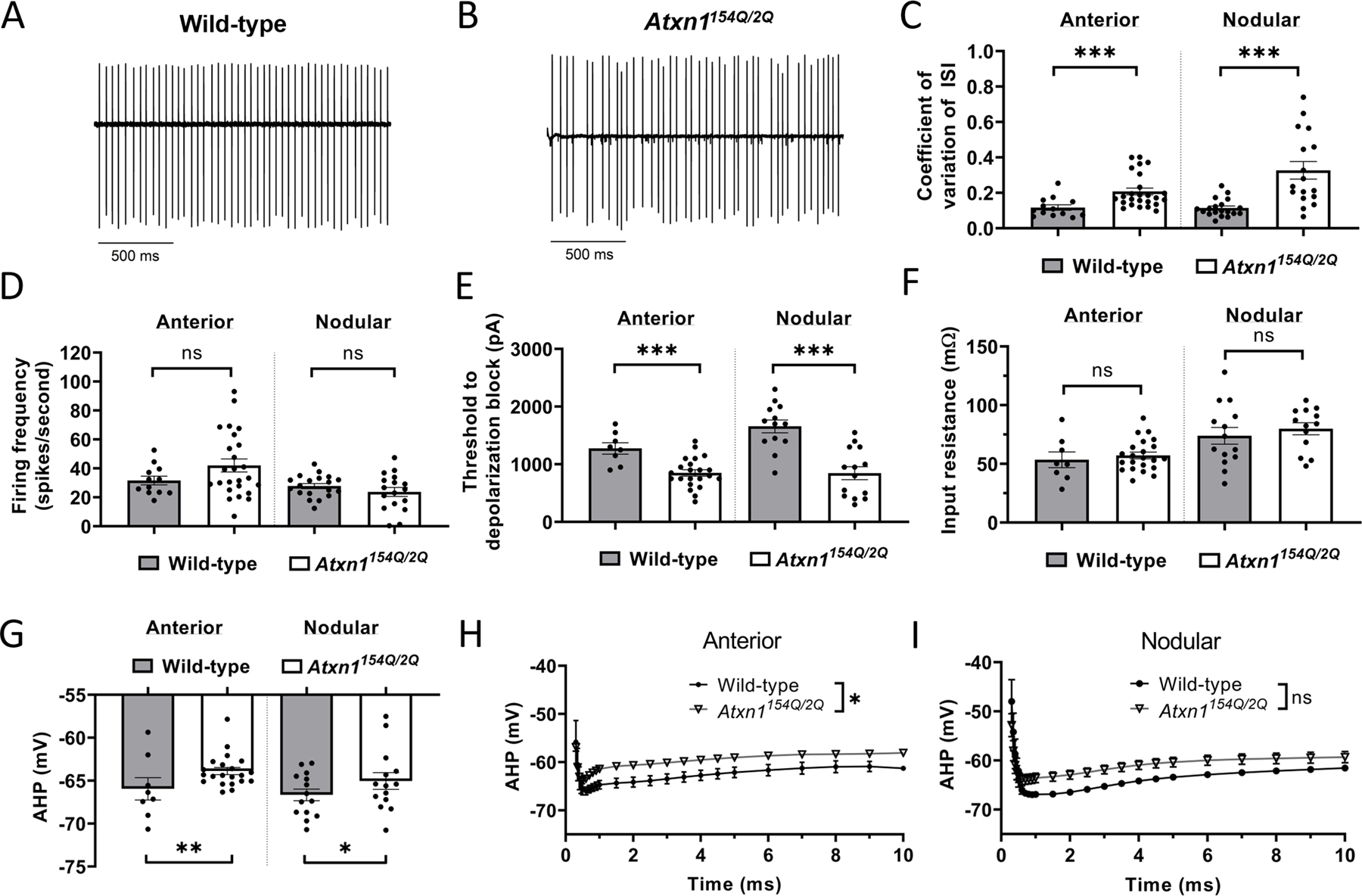
Representative spontaneous Purkinje neuron spiking is shown for 14 week-old (A) wild-type and (B) Atxn1154Q/2Q mice. (C) Regularity of Purkinje neuron spiking, as indicated by the coefficient of variation of the interspike interval, in wild-type and Atxn1154Q/2Q mice. (D) Firing frequency in wild-type and Atxn1154Q/2Q Purkinje neurons. (E) Threshold of injected current required to elicit depolarization block in wild-type and Atxn1154Q/2Q mice. (F) Input resistance in wild-type and Atxn1154Q/2Q Purkinje neurons. (G) Minimum spike amplitude, indicative of the fast afterhyperpolarization (AHP) in wild-type and Atxn1154Q/2Q Purkinje neurons. Quantification of the AHP and interspike interval of wild-type and Atxn1154Q/2Q Purkinje neurons in the (H) anterior cerebellar lobules and (I) posterior cerebellar lobules. * denotes p<0.05; ** denotes p<0.01, *** denotes p<0.001; ns denotes p>0.05. Two-tailed Student’s t-test (C-G); two-way repeated measures ANOVA with Holm-Sidak correction for multiple comparisons (H-I).
Since irregular Purkinje neuron spiking is associated with deficits in the afterhyperpolarization (AHP) in various mouse models of ataxia [2–4, 19], we examined differences in spike waveform between Atxn1154Q/2Q Purkinje neurons and wild-type littermate controls. We found that the AHP in Atxn1154Q/2Q mice was depolarized when compared to wild-type littermate control cells (Figure 1G–I). No other significant changes in spike waveform were noted in Atxn1154Q/2Q Purkinje neurons when compared to wild-type littermate controls (Supplementary Table 1). Together, these data indicate that a specific reduction in AHP amplitude accompanies irregular spiking and neuronal hyperexcitability in Atxn1154Q/2Q mice at a stage of disease with prominent motor impairment.
Changes in ion channel expression or function accompany changes in Purkinje neuron spiking in mouse models of SCA [12]. The observed irregular spiking and reduced AHP amplitude observed in Atxn1154Q/2Q Purkinje neurons is consistent with calcium-activated potassium (KCa) channel dysfunction or voltage-gated calcium (CaV) channel dysfunction [3, 15, 19–21]. We examined previously published microarray data to identify potential ion channel transcript dysregulation in Atxn1154Q/2Q and identified reduced transcripts for Kcnma1, Cacna1g, Trpc3 and Itpr1 [5, 22]. Reduced Kcnma1 and Cacna1g transcripts were observed at 14 weeks of age in Atxn1154Q/2Q mice when compared to wild-type littermate controls [5]. We therefore sought to quantify protein expression of large conductance KCa (BK) channels (encoded by Kcnma1) and putative calcium sources that may activate BK, including CaV3.1 (Cacna1g) [4], CaV2.1 (Cacna1a) [23], and the IP3 receptor (Itpr1) [24] in Atxn1154Q/2Q mice and wild-type littermate controls at 14 weeks of age. In both the anterior cerebellum (Figure 2A) and nodular zone (Figure 2B), fluorescence intensity of BK and CaV3.1 (Figure 2C) was reduced in the cerebellar molecular layer of Atxn1154Q/2Q mice compared to wild-type littermate controls, while the IP3 receptor and CaV2.1 (Figure 2C) showed no change in expression. Together, these data suggest that reduced cerebellar expression of BK and CaV3.1 underlies the altered physiology observed in Atxn1154Q/2Q mice.
Figure 2. Reduced expression of BK and CaV3.1 channels in Atxn1154Q/2Q cerebellum.
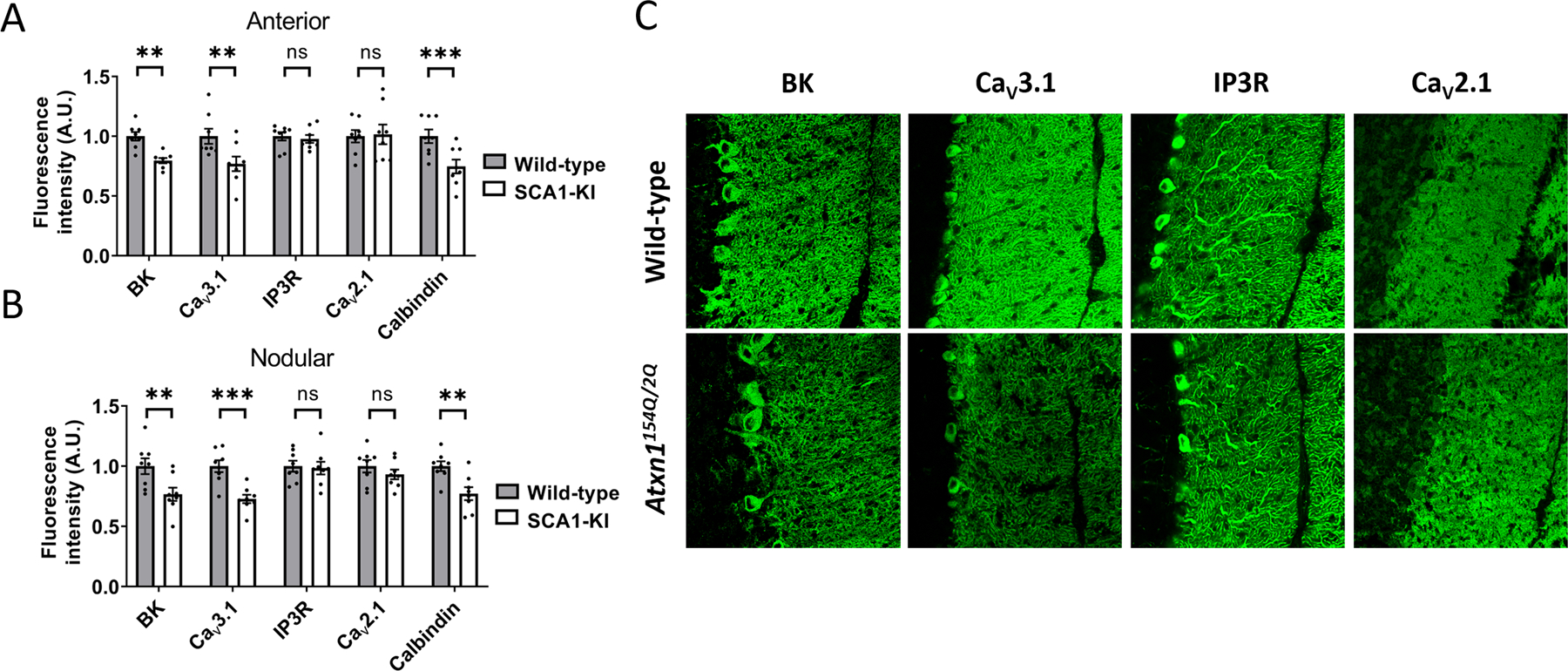
(A) Ion channel expression was measured in the anterior cerebellum (lobule 5) molecular layer of 14 week-old Atxn1154Q/2Q mice and wild-type littermate controls. (B) Ion channel expression was measured in the nodular zone (lobule 10) molecular layer of 14 week-old Atxn1154Q/2Q mice and wild-type littermate controls. (C) Representative 40x confocal microscopy images from the anterior cerebellum are shown for (C) BK, CaV3.1, IP3R, and CaV2.1 in Atxn1154Q/2Q mice and wild-type littermate controls. ** denotes p<0.01; *** denotes p<0.001; ns denotes p>0.05. Two-tailed Student’s t-test (F), with Holm-Sidak correction for multiple comparisons used in (A-B).
A combination of chlorzoxazone and baclofen improves Atxn1154Q/2Q Purkinje neuron firing
The altered Purkinje neuron spiking observed in symptomatic Atxn1154Q/2Q mice is similar to neuronal dysfunction seen in another mouse model of SCA1, the ATXN1[82Q] transgenic model [3]. More substantial loss of BK and Cav3.1 channel expression in ATXN1[82Q] mice results in a more prominent electrophysiologic phenotype, in which neurons undergo a complete loss of spiking [3]. This nevertheless represents a continuum with the irregular spiking observed in Atxn1154Q/2Q Purkinje neurons [5] (Figure 1). The shared underlying dysfunction of BK channels and reduced AHP amplitude suggests that BK channels may be an important target in SCA1.
In transgenic ATXN1[82Q] mice, combined treatment with chlorzoxazone and baclofen targets potassium channel dysfunction to improve Purkinje neuron spiking and motor impairment at both early- and mid-symptomatic ages [9]. Chlorzoxazone is a canonical KCa channel activator that can target BK channels [25]. Baclofen, a GABAB receptor agonist, is a known activator of subthreshold-activated potassium channels in Purkinje neurons [26]. Subthreshold-activated potassium channels can compensate for loss of the AHP and facilitate repetitive spiking in Purkinje neurons [2]. In ATXN1[82Q] mice, a combination of chlorzoxazone and baclofen (chlorzoxazone-baclofen) was necessary to achieve adequate KCa channel activation and restoration of function [9]. We wished to determine the necessity of a combination of these agents to improve the more modestly impaired firing in Atxn1154Q/2Q mice. Since SCA1-KI mice exhibit a gradation of Purkinje neuron firing abnormalities, this allows for investigation of dose-dependence of chlorzoxazone-baclofen to correct irregular spiking. It is important to determine the optimal concentrations of chlorzoxazone-baclofen as excessive activation of K+ channels results in reduction in Purkinje neuron firing frequency, which is independently correlated with motor dysfunction (reviewed in [27]).
The effect of different doses of chlorzoxazone and baclofen were assessed in acute cerebellar slices from 14-week Atxn1154Q/2Q mice by perfusing varying doses of either individual compounds or combinations of chlorzoxazone-baclofen. A high combined dose of chlorzoxazone (50 µM) and baclofen (2 µM) was able to improve spike regularity in Atxn1154Q/2Q Purkinje neurons, as indicated by a reduction in the coefficient of variation (CV) of the interspike interval (ISI) (Figure 3A). An intermediate combined dose of chlorzoxazone (10 µM) and baclofen (400 nM) similarly reduced the CV of the ISI in Atxn1154Q/2Q neurons (Figure 3A), while a lower combined dose of chlorzoxazone (5 µM) and baclofen (100 nM) had no effect on CV of the ISI (Figure 3A). Firing frequency was examined in these same recordings. Firing frequency was greatly suppressed by the high combined dose of chlorzoxazone (50 µM) and baclofen (2 µM), while no effect on firing frequency was observed at the intermediate combined dose of chlorzoxazone (10 µM) and baclofen (400 nM) (Figure 3B). A small reduction in firing frequency was observed in the low combined dose of chlorzoxazone (5 µM) and baclofen (100 nM) (Figure 3B). Interestingly, when treated with the intermediate dose of chlorzoxazone alone (10 µM; Figure 3C) or baclofen alone (400 nM; Figure 3D), the CV of the ISI was not changed from baseline. This is contrast to perfusion of higher dose chlorzoxazone alone (50 µM) or baclofen alone (2 µM), which both suppress firing frequency (Supplementary Figure 2A–B) while also reducing the CV of the ISI (Supplementary Figure 2C–D). Taken together, these data suggest that chlorzoxazone-baclofen improves Purkinje neuron spiking in a dose-responsive manner in Atxn1154Q/2Q mice, and that the intermediate dose of a combination of chlorzoxazone (10 µM) and baclofen (400 nM) has the optimal effect of improving Purkinje neuron spiking irregularity without affecting firing frequency in acute cerebellar slices. Additionally, these data clearly indicate the necessity of a combination of chlorzoxazone and baclofen to achieve the effect of improving spike regularity while not affecting firing frequency.
Figure 3. Chlorzoxazone-baclofen improves Atxn1154Q/2Q Purkinje neuron physiology in a dose-responsive manner.
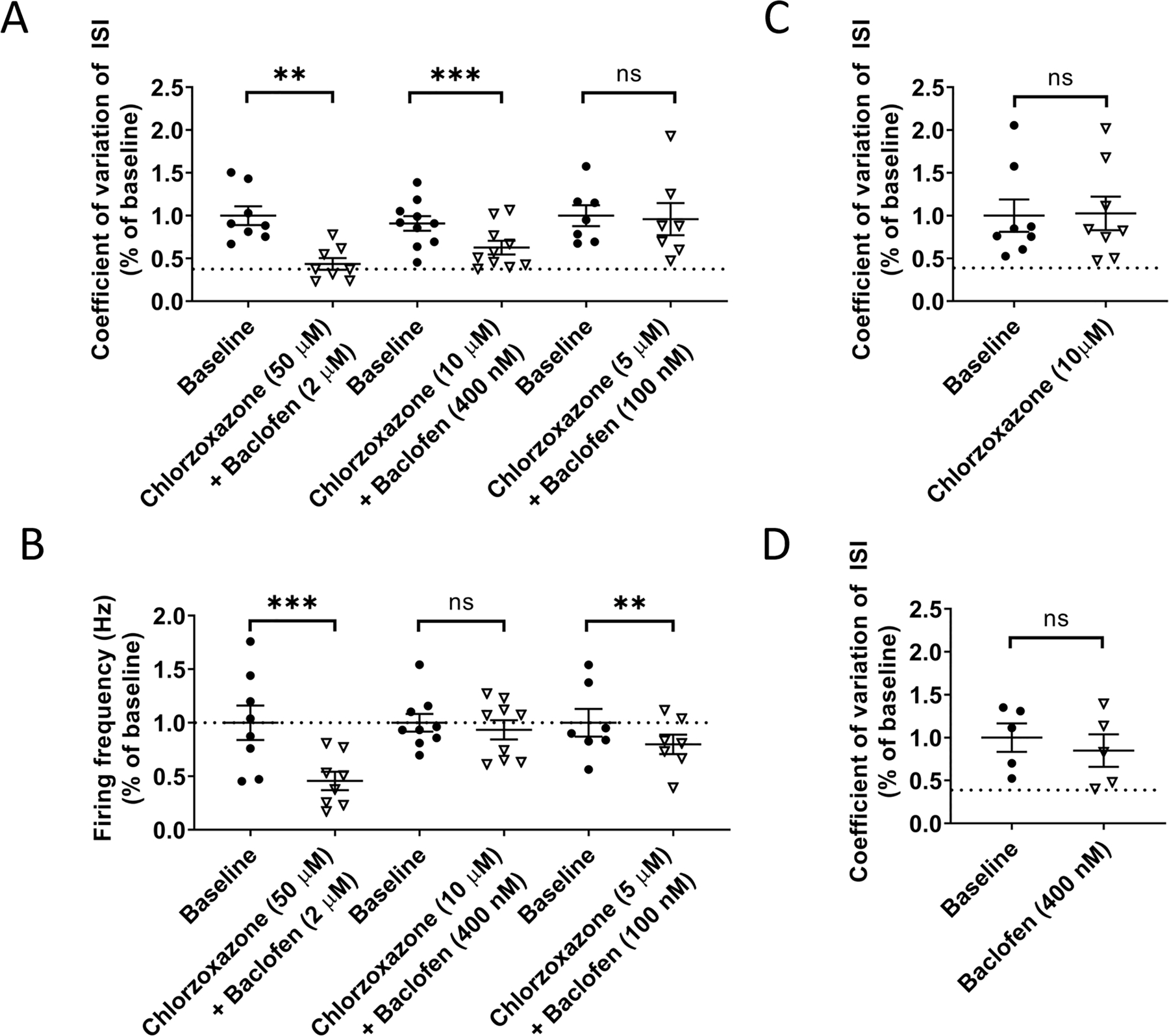
Acute cerebellar slices were prepared from 14 week-old Atxn1154Q/2Q mice for all recordings. (A) Regularity of Purkinje neuron spiking was recorded by measuring the coefficient of variation (CV) of the interspike interval (ISI). CV of the ISI was recorded at baseline and normalized to 1.0, and was then recorded after perfusion of differing concentrations of chlorzoxazone and baclofen into the bath. Dotted line represents normalized CV of the ISI from age-matched wild-type littermate controls. (B) Firing frequency was recorded at baseline and normalized to 1.0, and was then recorded after perfusion of differing concentrations of chlorzoxazone and baclofen into the bath. Dotted line represents normalized baseline firing frequency from age-matched wild-type littermate controls. (C) Similar to (A), CV of the ISI was recorded at baseline and after perfusion of an intermediate dose of chlorzoxazone (10 µM). Dotted line represents normalized CV of the ISI from age-matched wild-type littermate controls. (D) Similar to (C), CV of the ISI was recorded at baseline and after perfusion an intermediate dose of baclofen (400 nM). Dotted line represents normalized CV of the ISI from age-matched wild-type littermate controls. * denotes p<0.05; ** denotes p<0.01; *** denotes p<0.001; ns denotes p>0.05. Paired Student’s t-test.
Chlorzoxazone and baclofen specifically target cerebellar dysfunction in SCA1
Atxn1154Q/2Q mice have a hyperexpanded polyglutamine repeat. In human SCA1, large repeat expansions are associated with brainstem predominant disease and premature death in the second decade of life [28–30]. It is important to know whether cerebellar dysfunction contributes to motor impairment in Atxn1154Q/2Q mice and accurately serves as a model for testing therapy for cerebellar dysfunction, the major source of disability and quality of life in human SCA1. Atxn1154Q/2Q mice are described to have motor neuron involvement [7], which may confound interpretation of the rotarod task in mice. We therefore evaluated grip strength, a measure of motor neuron function, using the hanging wire task. At 14 weeks of age, no impairments in grip strength were noted in Atxn1154Q/2Q mice when compared to wild-type littermate controls (Figure 4A). Atxn1154Q/2Q mice were administered chlorzoxazone-baclofen in drinking water at a dose expected to achieve the intermediate concentrations (see Figure 4 of [9]) of chlorzoxazone (1–10 µM) and baclofen (100 nM-1 µM) in the brain. Importantly, chlorzoxazone-baclofen did not affect grip strength of 14 week-old Atxn1154Q/2Q mice (Figure 4A). The rotarod assay is commonly used to test balance and gait coordination, and reflects balance impairment when not confounded by impaired muscle strength [31]. At 14 weeks, Atxn1154Q/2Q mice display significantly impaired motor function on the rotarod assay when compared to wild-type littermate controls (Figure 4B). The absence of grip-strength impairment in the Atxn1154Q/2Q mice provides more confidence that the rotarod impairment at this stage of disease is due to cerebellar dysfunction. Treatment with chlorzoxazone-baclofen significantly improved motor performance in 14 week-old Atxn1154Q/2Q mice (Figure 4B).
Figure 4. Chlorzoxazone-baclofen improves cerebellar motor impairment in Atxn1154Q/2Q mice.
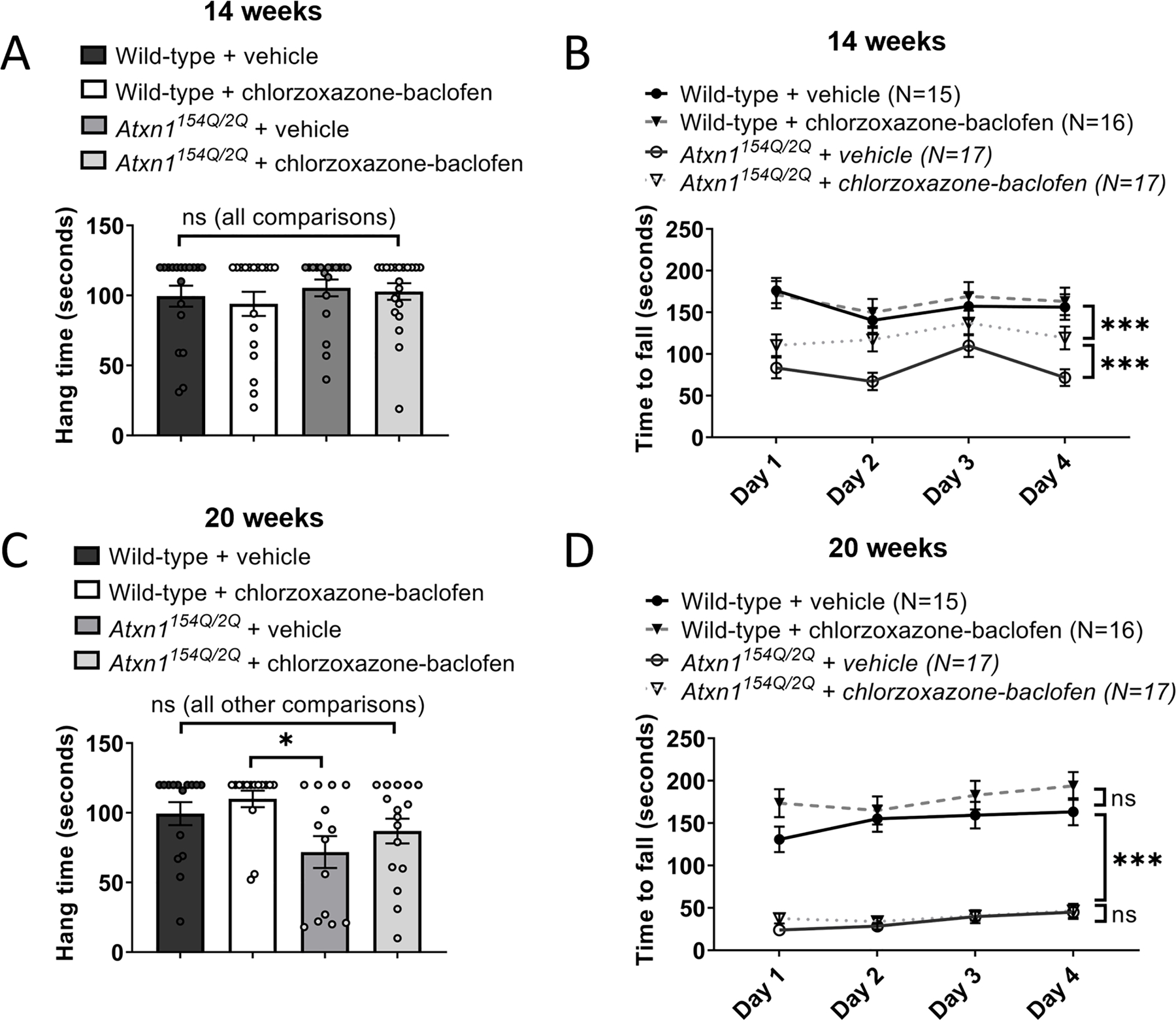
Atxn1154Q/2Q mice and wild-type littermate controls were treated with chlorzoxazone-baclofen in water bottles starting at 13 weeks of age. (A) Grip strength was measured in Atxn1154Q/2Q mice and wild-type littermate controls at 14 weeks of age using a hanging wire task. (B) The time to fall off of a rotarod at constant speed was recorded for Atxn1154Q/2Q mice and wild-type littermate controls at 14 weeks of age. (C) Similar to (A), at 20 weeks of age. (D) Similar to (B), at 20 weeks of age. * denotes p<0.05; ** denotes p<0.01; *** denotes p<0.001; ns denotes p>0.05. Two-way repeated measures ANOVA with Holm-Sidak correction for multiple comparisons (A, C); one-way ANOVA with Holm-Sidak correction for multiple comparisons (B, D).
We next assessed grip strength in the same cohorts of chlorzoxazone-baclofen treated Atxn1154Q/2Q mice and wild-type littermate controls at 20 weeks of age. Since there were no significant changes in grip strength due to treatment condition within each genotype (Figure 4C), we examined changes in grip strength when pooled by genotype at this later stage of disease. Atxn1154Q/2Q mice display a significant impairment in grip strength compared to wild-type littermate controls (Wild-type: 104.7 ± 5.112 seconds; Atxn1154Q/2Q: 79.90 ± 7.158 seconds; p = 0.0065, Student’s t-test; data presented as mean ± standard error of the mean). Reassuringly, chlorzoxazone-baclofen, which are FDA approved skeletal muscle relaxants, did not worsen the grip strength deficits in Atxn1154Q/2Q mice (Figure 4C). At this stage of disease, it would be expected that rotarod performance of Atxn1154Q/2Q mice would be further impaired, referable to the observed grip strength deficits. Consistent with this hypothesis, 20-week Atxn1154Q/2Q mice displayed significantly impaired motor performance on the rotarod compared to wild-type littermate controls to a larger degree than was observed at 14 weeks of age. Chlorzoxazone-baclofen did not rescue the rotarod impairment in Atxn1154Q/2Q mice at this stage of disease (Figure 4D), but, importantly, also did not worsen motor performance in 20 week-old Atxn1154Q/2Q mice, consistent with the lack of effect of chlorzoxazone-baclofen on grip strength at this age.
Since chlorzoxazone-baclofen is being considered for clinical use in patients with ataxia [9], we evaluated for potential toxicity of long-term use of this combination in Atxn1154Q/2Q mice. As reported previously, Atxn1154Q/2Q mice fail to gain weight compared to wild-type littermate controls, which is evident in both male and female mice at both 14 (Figure 5A–B, Supplementary Figure 3A) and 20 (Figure 5C–D, Supplementary Figure 3B) weeks of age [11]. Chlorzoxazone-baclofen did not accelerate this failure to gain weight. A slightly lower baseline weight was observed in chlorzoxazone-baclofen treated wild-type mice (Figure 5A), which was also noted in these same mice at 20 weeks of age (Figure 5C). However, this difference in weight was not accelerated in chlorzoxazone-baclofen treated female wild-type mice when compared to vehicle treated controls at the two recorded timepoints (Figure 5E), suggesting that these mice still gain weight normally and are not experiencing toxicity from chlorzoxazone-baclofen treatment. Together, these behavioral studies indicate that at 14 weeks, cerebellar dysfunction contributes meaningfully to motor impairment in Atxn1154Q/2Q mice and can be ameliorated by a treatment strategy targeting cerebellar dysfunction. At a later disease stage, Atxn1154Q/2Q mice display a motor phenotype that is driven by impairments of muscle strength. These findings reinforce the idea that chlorzoxazone-baclofen specifically targets motor impairment due to cerebellar dysfunction in SCA1.
Figure 5. Body weight is preserved in Atxn1154Q/2Q mice treated with chlorzoxazone-baclofen.
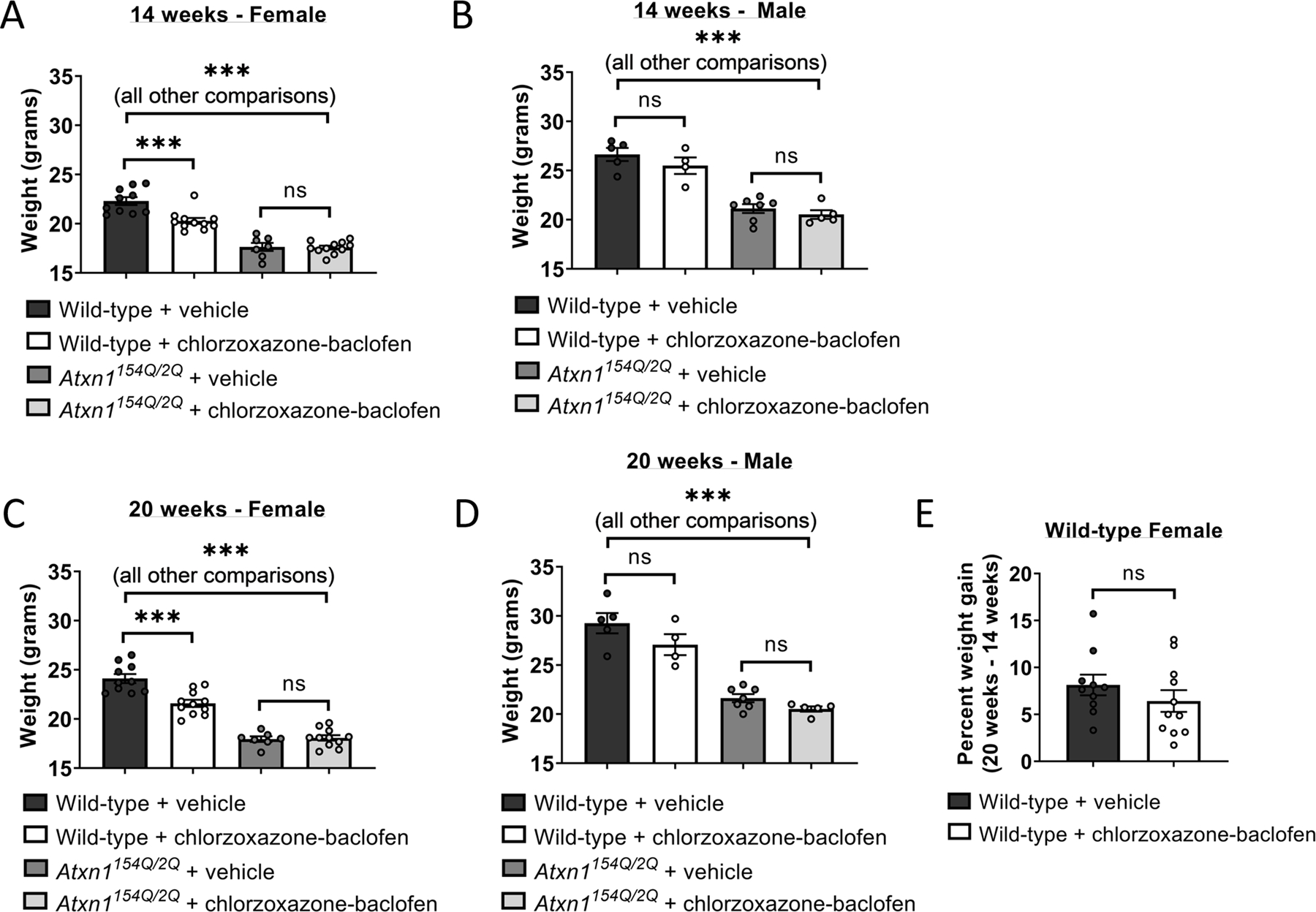
Body weight was measured in 14 week-old (A) male and (B) female Atxn1154Q/2Q mice and wild-type littermate controls after one week of water bottle treatment of chlorzoxazone-baclofen. Similar to (A-B), body weight was measured in 20 week-old (C) male and (D) female Atxn1154Q/2Q mice and wild-type littermate controls after 7 weeks of continuous chlorzoxazone-baclofen water bottle treatment. (E) Percent change in body weight from 14 to 20 weeks of age was calculated for wild-type female mice treated with either vehicle drinking water or water containing chlorzoxazone-baclofen. *** denotes p<0.001; ns denotes p>0.05. One-way ANOVA with Holm-Sidak correction for multiple comparisons (A-D); Two-tailed Student’s t-test (E).
Discussion
Chlorzoxazone-baclofen has been proposed for the treatment of cerebellar motor dysfunction in SCA [9]. However, the optimal dose of chlorzoxazone-baclofen for target engagement without off-target toxicity is not known. In the present study, we identify a dose range of chlorzoxazone-baclofen that improves Purkinje neuron spike regularity in the Atxn1154Q/2Q model of SCA1 without suppressing firing frequency. Importantly, chlorzoxazone-baclofen improves cerebellar motor dysfunction in Atxn1154Q/2Q mice at an age when motor impairment is cerebellar in origin, and does not negatively affect grip strength. At a later stage of disease, when motor impairment is worsened by impaired grip strength, chlorzoxazone-baclofen does not cause further impairment in motor function. Taken together, these studies suggest that chlorzoxazone-baclofen can be dosed to properly engage target in the cerebellum while simultaneously minimizing off-target toxicity. Human clinical trials of chlorzoxazone-baclofen will determine whether this drug combination may be effective to also treat cerebellar motor impairment in human SCA.
In the transgenic ATXN1[82Q] mouse model of SCA1, chlorzoxazone-baclofen improves cerebellar Purkinje neuron physiology and motor impairment by compensating for reduced potassium channel transcripts and function [9]. In the present study, chlorzoxazone-baclofen similarly improves motor function in Atxn1154Q/2Q mice at an age when motor dysfunction is cerebellar in origin. The less severe degree of potassium channel transcript reduction in Atxn1154Q/2Q mice compared to ATXN1[82Q] mice [5] explains the less severe Purkinje neuron firing abnormalities in Atxn1154Q/2Q mice. Despite a longer polyglutamine repeat in Atxn1154Q/2Q mice, the less severe cerebellar phenotype is likely due to more modest endogenous levels of mutant Atxn1 expression in Atxn1154Q/2Q mice as compared to the overexpression of mutant ATXN1 in ATXN1[82Q] Purkinje neurons [10, 11]. In ATXN1[82Q] mice, the benefit of chlorzoxazone-baclofen persists at later stage disease, despite prominent Purkinje neuron dendritic degeneration seen at this disease stage [9]. Chlorzoxazone-baclofen was, however, unable to sustain improved motor function in Atxn1154Q/2Q mice in later stage disease. This discrepancy may be explained by the genetic differences in these two models. The transgenic ATXN1[82Q] model of SCA1 overexpresses mutant ATXN1 specifically in cerebellar Purkinje neurons under control of the Pcp2 promoter [10]. Conversely, Atxn1154Q/2Q mice express glutamine-expanded Atxn1 at its endogenous locus, resulting in widespread expression throughout the central nervous system [11]. As a result, motor impairment in ATXN1[82Q] mice is cerebellar in origin, even in late stage disease, and largely reflects Purkinje neuron dysfunction. Spinal motor neuron involvement in Atxn1154Q/2Q mice likely contributes to motor impairment in this model of SCA1 [7, 11]. The current study suggests that motor dysfunction in mid-stage disease in Atxn1154Q/2Q mice is driven by cerebellar dysfunction, as evidenced by the improvement in motor function with chlorzoxazone-baclofen, whereas later stage motor impairment is a result of motor neuron dysfunction that is not improved by chlorzoxazone-baclofen. In this regard, Atxn1154Q/2Q mice likely recapitulate features of human SCA1, where morbidity is primarily cerebellar in origin in earlier stages of disease [32, 33], whereas end stage disease is due to cranial motor neuron dysfunction [34]. Chlorzoxazone-baclofen is likely therefore to have the maximal clinical benefit at stages of disease where motor dysfunction is cerebellar in origin. A combination of gene-specific disease-modifying therapy in combination with symptomatic therapy is likely to be most effective in improving quality of life in patients with ataxia.
Chlorzoxazone and baclofen are used clinically as muscle relaxants and are not recommended for simultaneous dosing in patients due to concerns about tolerability [35]. A major concern of using central muscle relaxants in SCA is the potential to worsen motor impairment [36]. However, the present data suggest that chlorzoxazone-baclofen can engage target in cerebellar Purkinje neurons while avoiding toxicity related to impaired muscle strength. An optimal effect of combined chlorzoxazone and baclofen treatment would be to improve the regularity of spiking in SCA1 Purkinje neurons without slowing firing frequency, as both irregular spiking and slow Purkinje neuron firing are associated with motor impairment in mouse models of ataxia [4, 12, 14–16, 19, 37]. In the current study, we identify a tight relationship between drug concentrations needed to engage target, and levels above which cerebellar function will be compromised. In planning for a human clinical trial with this drug combination to improve cerebellar ataxia, it will be important to identify the specific orally administered dose of chlorzoxazone-baclofen that achieves appropriate brain levels. We propose that a scheme of sequential dose escalation within each clinical trial-subject would be the most effective way to identify doses of chlorzoxazone-baclofen that engage target optimally without compromising cerebellar function, by allowing for monitoring of plasma and potentially cerebrospinal fluid levels of chlorzoxazone-baclofen along with effects on cerebellar motor function. In the current study, a high cerebellar concentration of chlorzoxazone-baclofen (50 µM and 2 µM, respectively) impairs cerebellar function, while a low concentration of chlorzoxazone-baclofen (5 µM and 0.2 µM, respectively) fails to engage target. An intermediate concentration of chlorzoxazone-baclofen (10 µM and 0.4 µM, respectively) at the site of action in the cerebellum is associated with optimal improvements in cerebellar physiology. Brain concentrations of baclofen are ~5 fold lower than plasma concentrations in both humans and mice [38]. Our studies in mice suggest that brain and plasma concentrations of chlorzoxazone reach near equilibrium in mice following chronic administration [9]. We therefore propose, based on the in vitro data obtained in the current study, that a plasma concentration range between 8–40 µM chlorzoxazone and 1–4 µM baclofen will be associated with improvement in cerebellar function in human SCA. It is likely that the relationship between administered drug dose, plasma/CSF concentrations of chlorzoxazone-baclofen, and changes in cerebellar function with each drug dose will need to be closely monitored for individual trial participants in a potential efficacy study.
Interestingly, the ion channels that underlie altered Purkinje neuron physiology in Atxn1154Q/2Q mice are also implicated in neuronal dysfunction in other mouse models of SCA. These channels, BK (encoded by Kcnma1) and CaV3.1 (encoded by Cacna1g), show reduced expression in another mouse model of SCA1 [5], several mouse models of SCA2 [5], and a mouse model of SCA7 [4]. BK channels rely upon intracellular calcium for activation, and recent evidence suggests that CaV3.1 may be an important calcium source for BK [4, 5]. Despite SCA1, SCA2, and SCA7 resulting from polyglutamine expansions in distinct genes, dysfunction of a BK channel functional module appears to contribute to altered Purkinje neuron spiking and cerebellar motor impairment in all of these disorders [5]. As illustrated in the present study, and in the ATXN1[82Q] model of SCA1 [9], chlorzoxazone-baclofen improves Purkinje neuron physiology through engagement of potassium channel targets. Therefore, it is possible that chlorzoxazone-baclofen may have relevance for treating motor impairment beyond SCA1. Notably, chlorzoxazone treatment has also been shown to improve aberrant Purkinje neuron spiking and motor performance in other murine models of cerebellar ataxia [37, 39–41]. A combination of chlorzoxazone-baclofen has not, however, been tested in rodent models of polyglutamine ataxia other than SCA1, although a related genetic strategy of viral BK channel replacement improves Purkinje neuron physiology in SCA7 [4]. It is tempting to speculate that a shared mechanism of dysfunction exists across multiple etiologies of SCA, and that a treatment strategy such as chlorzoxazone-baclofen may therefore have wider relevance for targeting cerebellar motor impairment.
Supplementary Material
Acknowledgments
Financial disclosure/conflict of interest: US Patent Application Number: 16/182965, titled: Therapeutic Combination for Treatment of Cerebellar Ataxia. Inventors: Vikram Shakkottai, David Bushart. Assignee: The Regents of the University of Michigan
Funding sources: NIH R01 NS085054 (VGS)
Footnotes
Financial Disclosure
VGS receives royalties from UpToDate Inc.
All other authors have no financial conflicts of interest to disclose.
References
- 1.Durr A, Autosomal dominant cerebellar ataxias: polyglutamine expansions and beyond. Lancet Neurol, 2010. 9(9): p. 885–94. [DOI] [PubMed] [Google Scholar]
- 2.Dell’Orco JM, Pulst SM, and Shakkottai VG, Potassium channel dysfunction underlies Purkinje neuron spiking abnormalities in spinocerebellar ataxia type 2. Human Molecular Genetics, 2017. 26(20): p. 3935–3945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dell’Orco JM, et al. , Neuronal Atrophy Early in Degenerative Ataxia Is a Compensatory Mechanism to Regulate Membrane Excitability. Journal of Neuroscience, 2015. 35(32): p. 11292–11307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stoyas CA, et al. , Nicotinamide Pathway-Dependent Sirt1 Activation Restores Calcium Homeostasis to Achieve Neuroprotection in Spinocerebellar Ataxia Type 7. Neuron, 2020. 105(4): p. 630-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chopra R, et al. , Altered Capicua expression drives regional Purkinje neuron vulnerability through ion channel gene dysregulation in spinocerebellar ataxia type 1. Hum Mol Genet, 2020. [DOI] [PMC free article] [PubMed]
- 6.Genis D, et al. , Clinical, neuropathologic, and genetic studies of a large spinocerebellar ataxia type 1 (SCA1) kindred: (CAG)n expansion and early premonitory signs and symptoms. Neurology, 1995. 45(1): p. 24–30. [DOI] [PubMed] [Google Scholar]
- 7.Orengo JP, et al. , Motor neuron degeneration correlates with respiratory dysfunction in SCA1. Dis Model Mech, 2018. 11(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rub U, et al. , Spinocerebellar ataxia type 1 (SCA1): new pathoanatomical and clinico-pathological insights. Neuropathol Appl Neurobiol, 2012. 38(7): p. 665–80. [DOI] [PubMed] [Google Scholar]
- 9.Bushart DD, et al. , Targeting potassium channels to treat cerebellar ataxia. Ann Clin Transl Neurol, 2018. 5(3): p. 297–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Burright EN, et al. , SCA1 transgenic mice: a model for neurodegeneration caused by an expanded CAG trinucleotide repeat. Cell, 1995. 82(6): p. 937–48. [DOI] [PubMed] [Google Scholar]
- 11.Watase K, et al. , A long CAG repeat in the mouse Sca1 locus replicates SCA1 features and reveals the impact of protein solubility on selective neurodegeneration. Neuron, 2002. 34(6): p. 905–19. [DOI] [PubMed] [Google Scholar]
- 12.Bushart DD and Shakkottai VG, Ion channel dysfunction in cerebellar ataxia. Neurosci Lett, 2019. 688: p. 41–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shakkottai VG, et al. , Early changes in cerebellar physiology accompany motor dysfunction in the polyglutamine disease spinocerebellar ataxia type 3. J Neurosci, 2011. 31(36): p. 13002–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hansen ST, et al. , Changes in Purkinje cell firing and gene expression precede behavioral pathology in a mouse model of SCA2. Human Molecular Genetics, 2013. 22(2): p. 271–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jayabal S, et al. , 4-aminopyridine reverses ataxia and cerebellar firing deficiency in a mouse model of spinocerebellar ataxia type 6. Sci Rep, 2016. 6: p. 29489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kasumu AW, et al. , Selective positive modulator of calcium-activated potassium channels exerts beneficial effects in a mouse model of spinocerebellar ataxia type 2. Chem Biol, 2012. 19(10): p. 1340–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hourez R, et al. , Aminopyridines correct early dysfunction and delay neurodegeneration in a mouse model of spinocerebellar ataxia type 1. J Neurosci, 2011. 31(33): p. 11795–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chopra R, et al. , Protein kinase C activity is a protective modifier of Purkinje neuron degeneration in cerebellar ataxia. Hum Mol Genet, 2018. 27(8): p. 1396–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Walter JT, et al. , Decreases in the precision of Purkinje cell pacemaking cause cerebellar dysfunction and ataxia. Nat Neurosci, 2006. 9(3): p. 389–97. [DOI] [PubMed] [Google Scholar]
- 20.Ly R, et al. , T-type channel blockade impairs long-term potentiation at the parallel fiber-Purkinje cell synapse and cerebellar learning. Proc Natl Acad Sci U S A, 2013. 110(50): p. 20302–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sausbier M, et al. , Cerebellar ataxia and Purkinje cell dysfunction caused by Ca2+-activated K+ channel deficiency. Proc Natl Acad Sci U S A, 2004. 101(25): p. 9474–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gatchel JR, et al. , The insulin-like growth factor pathway is altered in spinocerebellar ataxia type 1 and type 7. Proc Natl Acad Sci U S A, 2008. 105(4): p. 1291–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Womack MD, Chevez C, and Khodakhah K, Calcium-activated potassium channels are selectively coupled to P/Q-type calcium channels in cerebellar Purkinje neurons. J Neurosci, 2004. 24(40): p. 8818–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhao G, et al. , Type 1 IP3 receptors activate BKCa channels via local molecular coupling in arterial smooth muscle cells. J Gen Physiol, 2010. 136(3): p. 283–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu YC, Lo YK, and Wu SN, Stimulatory effects of chlorzoxazone, a centrally acting muscle relaxant, on large conductance calcium-activated potassium channels in pituitary GH3 cells. Brain Res, 2003. 959(1): p. 86–97. [DOI] [PubMed] [Google Scholar]
- 26.Tabata T, et al. , GABAergic activation of an inwardly rectifying K+ current in mouse cerebellar Purkinje cells. J Physiol, 2005. 563(Pt 2): p. 443–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chopra R and Shakkottai VG, Translating cerebellar Purkinje neuron physiology to progress in dominantly inherited ataxia. Future Neurol, 2014. 9(2): p. 187–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zoghbi HY, et al. , Spinocerebellar ataxia: variable age of onset and linkage to human leukocyte antigen in a large kindred. Ann Neurol, 1988. 23(6): p. 580–4. [DOI] [PubMed] [Google Scholar]
- 29.Ranum LP, et al. , Molecular and clinical correlations in spinocerebellar ataxia type I: evidence for familial effects on the age at onset. Am J Hum Genet, 1994. 55(2): p. 244–52. [PMC free article] [PubMed] [Google Scholar]
- 30.Haines JL, et al. , Spinocerebellar ataxia in a large kindred: age at onset, reproduction, and genetic linkage studies. Neurology, 1984. 34(12): p. 1542–8. [DOI] [PubMed] [Google Scholar]
- 31.Crawley JN, What’s wrong with my mouse? : behavioral phenotyping of transgenic and knockout mice. 2000, New York: Wiley-Liss. xiii, 329 p. [Google Scholar]
- 32.Ashizawa T, et al. , Clinical characteristics of patients with spinocerebellar ataxias 1, 2, 3 and 6 in the US; a prospective observational study. Orphanet J Rare Dis, 2013. 8: p. 177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schmitz-Hubsch T, et al. , Responsiveness of different rating instruments in spinocerebellar ataxia patients. Neurology, 2010. 74(8): p. 678–84. [DOI] [PubMed] [Google Scholar]
- 34.Diallo A, et al. , Survival in patients with spinocerebellar ataxia types 1, 2, 3, and 6 (EUROSCA): a longitudinal cohort study. Lancet Neurol, 2018. 17(4): p. 327–334. [DOI] [PubMed] [Google Scholar]
- 35.Expert P.b.t.A.G.S.B.C.U., American Geriatrics Society 2019 Updated AGS Beers Criteria(R) for Potentially Inappropriate Medication Use in Older Adults. J Am Geriatr Soc, 2019. 67(4): p. 674–694. [DOI] [PubMed] [Google Scholar]
- 36.Lexi-Comp Inc., Lexicomp online.
- 37.Egorova PA, et al. , In vivo analysis of cerebellar Purkinje cell activity in SCA2 transgenic mouse model. J Neurophysiol, 2016. 115(6): p. 2840–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Knutsson E, Lindblom U, and Martensson A, Plasma and cerebrospinal fluid levels of baclofen (Lioresal) at optimal therapeutic responses in spastic paresis. J Neurol Sci, 1974. 23(3): p. 473–84. [DOI] [PubMed] [Google Scholar]
- 39.Alvina K and Khodakhah K, KCa channels as therapeutic targets in episodic ataxia type-2. J Neurosci, 2010. 30(21): p. 7249–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gao Z, et al. , Cerebellar ataxia by enhanced Ca(V)2.1 currents is alleviated by Ca2+-dependent K+-channel activators in Cacna1a(S218L) mutant mice. J Neurosci, 2012. 32(44): p. 15533–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Egorova PA, Gavrilova AV, and Bezprozvanny IB, Ataxic Symptoms in Huntington’s Disease Transgenic Mouse Model Are Alleviated by Chlorzoxazone. Front Neurosci, 2020. 14: p. 279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ankri L, Yarom Y, and Uusisaari MY, Slice it hot: acute adult brain slicing in physiological temperature. J Vis Exp, 2014(92): p. e52068. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.