

Abstract
Background:
Congenital fibrosis of the extraocular muscles(CFEOM) is characterized by ptosis and non-progressive restrictive ophthalmoplegia. CFEOM1 is a stereotypical phenotype with isolated bilateral ptosis, bilateral ophthalmoplegia, absent upgaze, and globe infraduction. CFEOM3 is a more variable phenotype that can include unilateral disease, absent ptosis, residual upgaze, and/or orthotropia. Most cases of CFEOM1 result from recurrent heterozygous KIF21A missense mutations and less commonly from recurrent heterozygous TUBB3 missense mutations. While most cases of CFEOM3 result from recurrent heterozygous TUBB3 missense mutations, several pedigrees harbored pathogenic variants in KIF21A. Here, we asked if Lebanese pedigrees with CFEOM3 harbor pathogenic variants in TUBB3 or KIF21A.
Materials and Methods
Families affected with congenital cranial dysinnervation disorders were prospectively recruited from the American University of Beirut pediatric ophthalmology clinic and included two probands with CFEOM. KIF21A hotspot exons and TUBB3 coding sequence were sequenced. Available family members were sequenced for co-segregation analysis.
Results
Both families were found to have CFEOM3 and to harbor pathogenic variants in KIF21A(OMIM 608283). A simplex proband with CFEOM3 from a consanguineous Iraqi family harbored a de novo heterozygous KIF21A c.2860C>T variant(p.R954W); this variant accounts for the majority of reported KIF21A mutations but is typically implicated in CFEOM1. A Lebanese father with CFEOM3 and his son with CFEOM1 segregated a heterozygous KIF21A c.2830G>C variant (p.E944Q), previously reported in an individual with CFEOM1.
Conclusions
These results support prior reports of KIF21A mutations as a rare cause of CFEOM3. These families are Middle Eastern or Chinese, supporting a genetic modifier in these populations.
Keywords: CCDD, CFEOM, congenital fibrosis of extraocular muscles, KIF21A gene, pathogenic variants
INTRODUCTION
Congenital cranial dysinnervation disorders (CCDD) was coined in 2002 as an umbrella term to encompass congenital and non-progressive neurodevelopmental disorders characterized by abnormal eye, eyelid, and/or facial movement (1). Mutations in multiple different genes have been incriminated in the pathogenesis of the CCDDs (1–3), and studies of these mutant genes has led to the consensus that CCDDs are neurogenic and result from altered development of specific cranial motor neurons or from altered cranial axon growth and guidance.
Congenital fibrosis of the extraocular muscles (CFEOM) is a CCDD characterized by variable congenital ptosis and non-progressive ophthalmoplegia affecting the function of muscles primarily in the oculomotor distribution, and includes two dominant subtypes (CFEOM1, CFEOM3), and one recessive subtype (CFEOM2). CFEOM1 is defined as a autosomal dominant fully penetrant disorder in which affected individuals have isolated congenital bilateral ophthalmoplegia and bilateral ptosis, globe infraduction, and the inability to raise either eye above the horizontal midline (4). The extent of horizontal eye movement can vary and many affected individuals have aberrant residual eye movements. Recurrent heterozygous missense variants in KIF21A are the primary genetic cause of CFEOM1; these variants can arise de novo or be inherited. The resulting amino acid substitutions cluster in two portions of the encoded anterograde kinesin motor protein: a small stretch of a coiled-coil domain of the stalk and the lateral aspect of the motor domain (5–7).
CFEOM3 is defined as an autosomal dominant form of CFEOM in which affected individuals do not meet CFEOM1 criteria; they may have unilateral or absence of ptosis, unilateral ophthalmoplegia, orthotropic or hypertropic globes, and/or the ability to elevate one or both eyes above the midline. Genetic analysis of families segregating CFEOM3 identified recurrent missense variants in TUBB3, which encodes a beta-tubulin isoform that is a component of the microtubule cytoskeleton (8). Specific TUBB3 amino acid substitutions cause either isolated or syndromic CFEOM (8, 9).
While KIF21A mutations are virtually all fully penetrant and result in a stereotypical CFEOM1 phenotype, five pedigrees have been reported that met CFEOM3 criteria yet harbored KIF21A variants (4, 10–12). We now report a Lebanese and an Iraqi pedigree who also have CFEOM3 secondary to mutations in KIF21A.
MATERIALS and METHODS
This research was approved by the Institutional Review Board at the American University of Beirut. Written informed consent was obtained from participants or legal guardians with assent forms provided to children according to the Declaration of Helsinki for research involving human subjects.
Prospective recruitment of 26 probands and family members, totaling 129 participants, segregating CCDDs was conducted at the American University of Beirut Medical Center between April 2013 and April 2018. Using the classification summarized above, 2 pedigrees diagnosed with CFEOM were identified from the patient database. The proband and family members were seen at the Pediatric Ophthalmology and Neurology clinics and underwent general physical, neurological and ophthalmological examinations, including cranial nerve assessment, sensorimotor assessment, and dilated fundoscopy.
Saliva samples (Oragene, DNA Genotek Inc., Ottawa, Ontario, Canada) were obtained from participating family members and salivary DNA was extracted. All coding exons and intron-exon boundaries of TUBB3 and exons 8, 20, and 21 of KIF21A were sequenced as previously reported (5, 8). Primer sequences are available on request.
CLINICAL PRESENTATIONS
Family 1:
The proband was a 3-year-old Lebanese boy who presented to the clinic with severe chin-up posture and bilateral ptosis since infancy and was previously described (13). Medical history was otherwise negative and there was no reported parental consanguinity. Family history was significant for a father who reported a history of ptosis in the right eye for which he underwent eyelid surgery. On examination, the child had severe chin-up posture of 60° and bilateral ptosis with absent upper eyelid creases (Figure 1A). Severe bilateral dryness of the inferior corneas was noted on slit-lamp examination. Motility assessment confirmed severe elevation deficiency (−5.0) with normal horizontal ductions; Bell’s phenomenon was not present. He was orthotropic on Krimsky test with chin-up posture. Objective fundus excyclotorsion was noted (+2) bilaterally. Additional cranial nerve and general neurological testing was normal. Magnetic resonance imaging (MRI) of the brain and orbits revealed hypoplastic superior recti/levator complexes bilaterally (Figure 1B). Brain imaging was normal. Examination of the father revealed right eye exotropia with limited adduction and depression of the right eye and full motility of the left eye (Figure 1C). He had right-sided upper eyelid scars suggesting previous unilateral frontalis sling surgery. Additional cranial nerve and general neurological testing was normal. The child and his father were diagnosed with CFEOM1 and CFEOM3, respectively.
Figure 1. Family 1 clinical presentation.
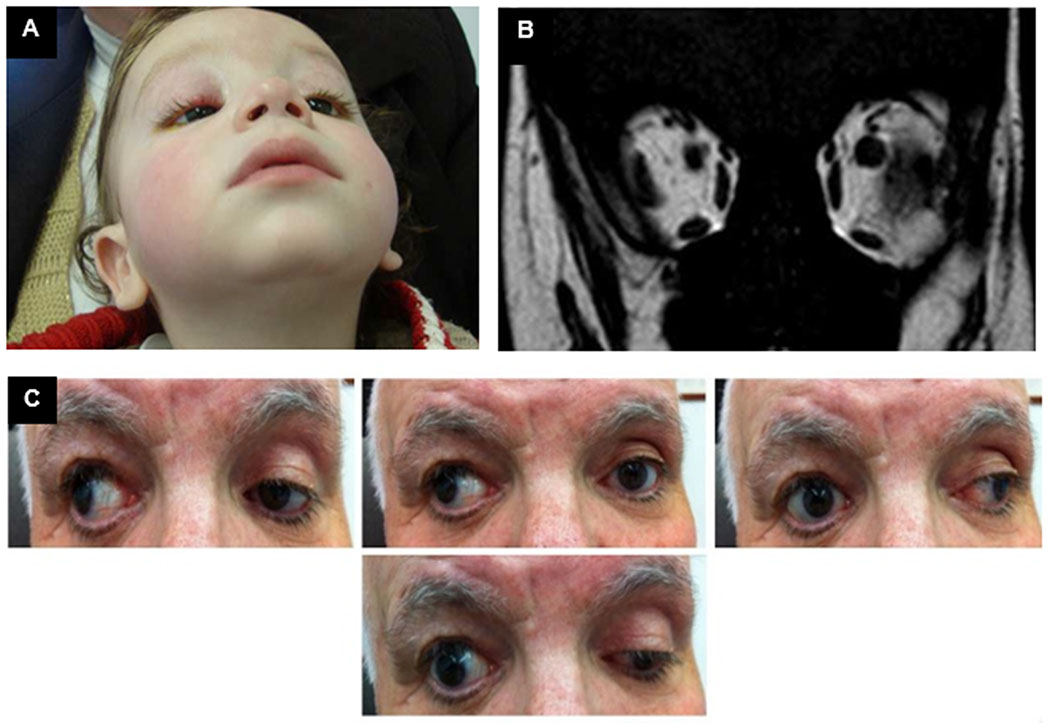
A. Proband 1 at presentation with severe chin-up posture. B. Proband 1 magnetic resonance imaging of orbits shows bilateral superior rectus muscle hypoplasia. C. Father of Proband 1 extraocular motility testing shows exotropia on primary gaze, with limited adduction and depression of the right eye. Left eye movements are full.
The proband underwent forced duction testing and strabismus surgery under general anesthesia. Neither eye could be supraducted on forced duction testing and there was bilateral inferior rectus restriction (−5.0). Both inferior recti were recessed (5.5 mm) with subsequent decrease in the chin-up posture to 25°. Transposition of both horizontal recti toward the superior rectus muscle was performed 5 months later with posterior augmentation and further reduction achieved in the chin-up posture to 5°. Subsequent bilateral frontalis sling suspension corrected the ptosis. Postoperatively, only minimal chin-up posture was noted, while motility examination still showed limited supraduction (−4.0) with orthotropia on the Krimsky test.
Family 2:
The proband was a 9-year-old Iraqi girl who presented with right upper lid ptosis. Her parents were first cousins and unaffected, and she had 2 unaffected sisters. At the time of presentation, she had already undergone right lateral rectus recession and medial rectus resection for exotropia and frontalis sling surgery for right upper eyelid ptosis. She had moderate esotropia in primary gaze and right hypotropia with superior oblique overaction and an A pattern. Motility examination confirmed limited elevation of the right eye in adduction, but the eye could elevate above midline in abduction (Figure 2A). Bell’s phenomenon was present. Left eye motility was full. Additional cranial nerve and general neurological assessment was normal. Brain and orbit CT scan revealed a hypoplastic right superior rectus complex (Figure 2B). Proband 2 was diagnosed with CFEOM3 of the right eye with consecutive esotropia. She underwent subsequent bilateral superior oblique tenectomy and left medial rectus recession with marked improvement in her strabismus. She was then lost to follow-up and no further eyelid surgery took place.
Figure 2. Family 2 clinical presentation.
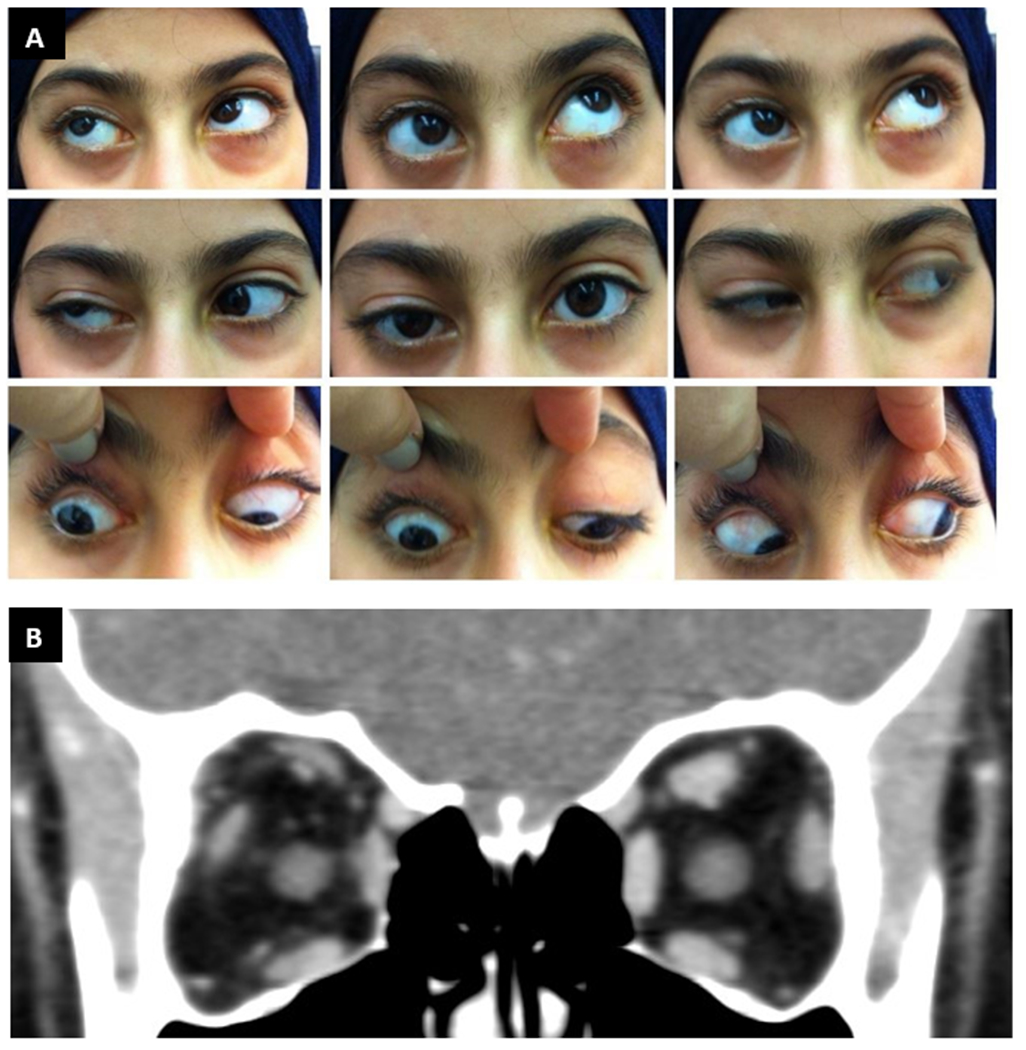
A. Proband 2 extraocular motility testing reveals ptosis of right upper lid, esotropia in primary gaze and right hypotropia, but retained ability to elevate the right eye above midline in abduction. Left eye movements are full. B. Proband 2 orbital CT scan reveals hypoplastic right superior rectus complex.
GENETIC EVALUATION
Sequencing of DNA from family 1 revealed that the proband and his father harbored a KIF21A heterozygous 2830G>C (hg38:chr12:39,332,617) variant predicted to result in a p.E944Q non-conservative amino acid substitution (Figure 3A, B). E944 is located within the cluster of CFEOM1 variants in the third coiled-coil region of the KIF21A stalk (Figure 3C).
Figure 3. Family 1 and Family 2 genetics.
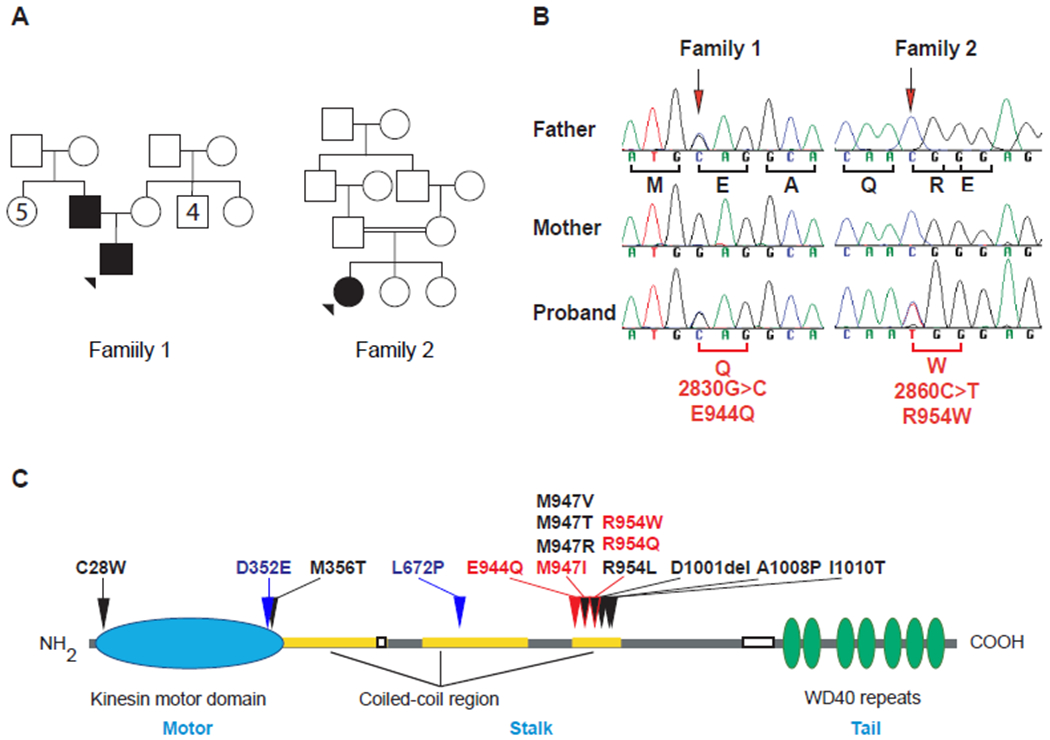
A. Pedigree schematics of Family 1 and Family 2. Squares male, circles females, filled squares or circles denote affected individual. B. Sequencing chromatograms reveal heterozygous KIF21A mutations in the father and proband of family 1 (2830G>C) and the proband of family 2 (2860C>T). The position of the mutation is marked by an arrow and the nucleotide change and predicted amino acid substitution indicated under the Proband sequence. C. Schematic presentation of the KIF21A kinesin protein structure with the motor domain denoted in blue, stalk in grey with coiled-coil domains in yellow, and the tail domain in green. The relative locations of reported KIF21A amino acid substitutions are represented over the protein. The amino acid substitutions that have been reported to cause CFEOM3 (and CFEOM1 as well) are denoted in red, including E944Q and R954W reported in this paper. The two amino acid substitutions reported to cause syndromic CFEOM are denoted in blue.
Sequencing of DNA from family 2 revealed that the proband harbored a KIF21A heterozygous c.2860C>T (hg38:chr12:39,332,405) variant that was absent in her parents and predicted to result in a p.R954W amino acid substitution (Figure 3A, B). R954 is located within the cluster of CFEOM1 variants in the third coiled-coil region of the KIF21A stalk (Figure 3C).
DISCUSSION
We report two pedigrees with CFEOM3 who harbor KIF21A mutations, bringing the number of such pedigrees to at least seven. The first pedigree is of Lebanese descent, is not known to be consanguineous, and includes a son diagnosed with CFEOM1 and his father diagnosed with CFEOM3 secondary to unilateral CFEOM. Both harbored a KIF21A c.2830G>C variant (p.E944Q). This same variant was previously reported de novo in a white patient of European descent with CFEOM1 (6). Notably, a KIF21A M947I substitution, three amino acids downstream, was reported to result in CFEOM3 in a small consanguineous Saudi Arabian pedigree and included mutation-positive individuals with residual upgaze. By contrast, M947R, M947T, and M947V substitutions have been reported to result in CFEOM1 (4, 5, 14).
The second pedigree we report is a simplex case in a consanguineous Iraqi family. She was also diagnosed with CFEOM3 secondary to unilateral CFEOM. She harbored a de novo KIF21A c.2860C>T variant (p.R954W). R954W KIF21A amino acid substitution is the most common cause of CFEOM and is present in >85% of CFEOM1 cases, both simplex and inherited (5). This individual, however, is at least the fifth report of a R954 substitution resulting in CFEOM3 rather than CFEOM1. The first report was in a large consanguineous Turkish pedigree that included mutation-positive individuals with absent or unilateral ptosis, orthotropic primary globe position, and/or ability to elevate the globes above the midline, as well as one with a normal exam (4). The second and third reports were from Shandong and Guangdong provinces in China. The first described a daughter diagnosed with CFEOM1 and mother diagnosed with CFEOM3 based on the absence of ptosis (10). The second described a simplex de novo case with unilateral ptosis and ability to elevate the eye above midline (11). Affected members of these three pedigrees all harbored the R954W substitution. Interestingly, there is a third report from China of a large pedigree in which five affected members were diagnosed with CFEOM1, while three affected members were diagnosed with CFEOM3 because of residual upgaze or absence of ptosis (12). This pedigree segregated the KIF21A R954Q substitution, which alters the same amino acid residue as R954W and is the second most common substitution reported in CFEOM1.
The Iraqi and Lebanese pedigrees reported here bring to seven the number of pedigrees reported with CFEOM3 resulting from KIF21A variants. Remarkably, four of these seven pedigrees are Middle Eastern (Turkish, Saudi Arabian, Iraqi, and Lebanese descent) of which three report consanguinity, while the remaining three pedigrees are Chinese. The mutations harbored by the seven pedigrees alter KIF21A amino acid residues E944, M947, and R954, all of which are located in the third coiled-coil domain of the KIF21A stalk. The stalk interacts with the motor domain to autoinhibit the KIF21A protein, and pathogenic mutations in the stalk have been shown to attenuate autoinhibition and cause stalling of developing axons in the superior division of the oculomotor nerve. This, in turn, results in failure of innervation of the superior division target muscles, the superior rectus and levator palpebrae superioris, and the CFEOM phenotype in mice and humans (4). The restriction, to date, of KIF21A-CFEOM3 mutations to Middle Eastern and Chinese pedigrees suggests that there may be genetic modifiers of the CFEOM phenotype present in these two populations.
KIF21A is an anterograde kinesin that uses energy to ‘walk’ along microtubules, and TUBB3 is a component of the neuronal microtubule along which KIF21A ‘walks’. This functional relationship and the increasing number of cases that describe further overlap between KIF21A and TUBB3 phenotypes support a shared developmental mechanism. This is highlighted further by two additional case reports. The first is of a Hispanic child with CFEOM1, facial weakness, and developmental delay who harbored a KIF21A D352E substitution (15). While facial weakness and developmental delay accompany multiple TUBB3 variants, this was the first report of these findings with a KIF21A variant. D352 is located on the lateral aspect of the motor domain adjacent to M356; M356 interacts with the stalk third coiled-coil domain to autoinhibit KIF21A, and M356T was previously reported to cause isolated CFEOM1 by attenuating this autoinhibition (5, 7). The second case report is of a white child of European descent who harbored a KIF21A L672P substitution (16). He had CFEOM, facial weakness, moderate intellectual disability, and a progressive peripheral sensory-motor neuropathy, again phenotypes that co-segregate with specific TUBB3 variants and had not been previously reported with KIF21A variants. Brain MRI findings were also similar to those reported with TUBB3 variants. L672 is in the middle of the second coiled-coil domain and the effect of this variant on function has not been explored. Thus, future work dissecting the etiology of these overlapping phenotypes may help to identify a shared mechanism between the KIF21A- and TUBB3-CFEOM.
Acknowledgments:
We thank Ms. Lama Chaar for her assistance in recruitment of families and data collection
Funding: ECE is a Howard Hughes Medical Institute Investigator.
Footnotes
Disclosure of interests: The authors report no conflicts of interest.
Written informed consent is documented for all participants.
Ethical approval: All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional review board in compliance with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
REFERENCES
- 1.Gutowski NJ, Chilton JK. The congenital cranial dysinnervation disorders. Arch Dis Child. 2015;100(7):678–81. [DOI] [PubMed] [Google Scholar]
- 2.Bosley TM, Abu-Amero KK, Oystreck DT. Congenital cranial dysinnervation disorders: a concept in evolution. Curr Opin Ophthalmol. 2013;24(5):398–406. [DOI] [PubMed] [Google Scholar]
- 3.Whitman M, Hunter DG, Engle EC. Congenital Fibrosis of the Extraocular Muscles. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, et al. , editors. GeneReviews((R)). Seattle (WA)1993. [PubMed] [Google Scholar]
- 4.Yamada K, Chan WM, Andrews C, Bosley TM, Sener EC, Zwaan JT, Mullaney PB, Ozturk BT, Akarsu AN, Sabol LJ, et al. Identification of KIF21A mutations as a rare cause of congenital fibrosis of the extraocular muscles type 3 (CFEOM3). Invest Ophthalmol Vis Sci. 2004;45(7):2218–23. [DOI] [PubMed] [Google Scholar]
- 5.Yamada K, Andrews C, Chan WM, McKeown CA, Magli A, de Berardinis T, Loewenstein A, Lazar M, O’Keefe M, Letson R, et al. Heterozygous mutations of the kinesin KIF21A in congenital fibrosis of the extraocular muscles type 1 (CFEOM1). Nat Genet. 2003;35(4):318–21. [DOI] [PubMed] [Google Scholar]
- 6.Chan WM, Andrews C, Dragan L, Fredrick D, Armstrong L, Lyons C, Geraghty MT, Hunter DG, Yazdani A, Traboulsi EI, et al. Three novel mutations in KIF21A highlight the importance of the third coiled-coil stalk domain in the etiology of CFEOM1. BMC Genet. 2007;8:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cheng L, Desai J, Miranda CJ, Duncan JS, Qiu W, Nugent AA, Kolpak AL, Wu CC, Drokhlyansky E, Delisle MM, et al. Human CFEOM1 mutations attenuate KIF21A autoinhibition and cause oculomotor axon stalling. Neuron. 2014. Apr 16;82(2):334–49. doi: 10.1016/j.neuron.2014.02.038. Epub 2014 Mar 20. PMID: 24656932; PMCID: PMC4002761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tischfield MA, Baris HN, Wu C, et al. Human TUBB3 mutations perturb microtubule dynamics, kinesin interactions, and axon guidance. Cell 2010;74–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chew S, Balasubramanian R, Chan WM, Kang PB, Andrews C, Webb BD et al. A novel syndrome caused by the E410K amino acid substitution in the neuronal β-tubulin isotype 3. Brain. 2013. February;136(2):522–535. 10.1093/brain/aws345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang X, Yamada K, Katz B, Guan H, Wang L, Andrews C, Zhao G, Engle EC, Chen H, Tong Z, et al. KIF21A mutations in two Chinese families with congenital fibrosis of the extraocular muscles (CFEOM). Mol Vis. 2010. Oct 13;16:2062–70. PMID: 21042561; PMCID: PMC2965570. [PMC free article] [PubMed] [Google Scholar]
- 11.Chen J, Ye Q, Deng D, Yan J, Lin H, Shen T, Lin Y. KIF21A mutation in two Chinese families with congenital fibrosis of the extraocular muscles type 1 and 3. Mol Med Rep. 2016. October;14(4):3145–51. doi: 10.3892/mmr.2016.5624. Epub 2016 Aug 11. PMID: 27513105; PMCID: PMC5042766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lu S, Zhao C, Zhao K, Li N, Larsson C. Novel and Recurrent KIF21A Mutations in Congenital Fibrosis of the Extraocular Muscles Type 1 and 3. Arch Ophthalmol. 2008;126(3):388–394. doi: 10.1001/archopht.126.3.388 [DOI] [PubMed] [Google Scholar]
- 13.Al-Haddad CE, Abdulaal M. Transposition surgery for inferior rectus fibrosis. J Pediatr Ophthalmol Strabismus. 2015. March 4;52 Online:e1–3. doi: 10.3928/01913913-20150224-01. PMID: 25735010. [DOI] [PubMed] [Google Scholar]
- 14.Yamada K, Hunter DG, Andrews C, Engle EC. A novel KIF21A mutation in a patient with congenital fibrosis of the extraocular muscles and Marcus Gunn jaw-winking phenomenon. Arch Ophthalmol. 2005;123(9):1254–1259. [DOI] [PubMed] [Google Scholar]
- 15.Ali Z, Xing C, Anwar D, Itani K, Weakley D, Gong X, Pascual JM, Mootha VV. A novel de novo KIF21A mutation in a patient with congenital fibrosis of the extraocular muscles and Möbius syndrome. Mol Vis. 2014. March 28;20:368–75. PMID: 24715754; PMCID: PMC3976685. [PMC free article] [PubMed] [Google Scholar]
- 16.Soliani L, Spagnoli C, Salerno GG, Mehine M, Rizzi S, Frattini D, Koskenvuo J, Fusco C. A Novel De Novo KIF21A Variant in a Patient With Congenital Fibrosis of the Extraocular Muscles With a Syndromic CFEOM Phenotype. J Neuroophthalmol. 2020. March 3. doi: 10.1097/WNO.0000000000000921. Epub ahead of print. PMID: 32141982. [DOI] [PubMed] [Google Scholar]