

Abstract
Early acute kidney injury (AKI) contributes to severe morbidity and mortality in critically burned patients. Renal inflammation plays a vital role in the progression of early AKI, acting as a therapeutic target. Astaxanthin (ATX) is a strong antioxidant widely distributed in marine organisms that exerts many biological effects in trauma and disease. ATX is also suggested to have anti-inflammatory activity. Hence, we attempted to explore the role of ATX in protecting against early postburn AKI via its anti-inflammatory effects and the related mechanisms. A severely burned model was established for histological and biochemical assessments based on adult male rats. We found that oxidative stress-induced tissue inflammation participated in the development of early AKI after burn injury and that the MyD88-dependent TLR4/NF-κB pathway was activated to regulate renal inflammation. The TLR4 and NF-κB inhibitors TAK242 and PDTC showed similar effects in attenuating burn-induced renal inflammation and early AKI. Upon ATX treatment, the release of inflammatory mediators in the kidneys was downregulated, while the TLR4/MyD88/NF-κB axis was inhibited in a dose-related manner. TAK242 and PDTC could enhance the anti-inflammatory effect of high-dose ATX, whereas lipopolysaccharide (LPS) reversed its action. Furthermore, the expression of heme oxygenase (HO)-1 was upregulated by ATX in a dose-related manner. Collectively, the above data suggest that ATX protects against renal inflammation in a dose-related manner by regulating the TLR4/MyD88/NF-κB axis and HO-1 and ultimately prevents early AKI following severe burns.
Subject terms: Acute kidney injury, Trauma
Introduction
Severely burned patients (total body surface area (TBSA) ≥ 20%) must face a series of devastating circulatory or organ issues after the initial thermal insult, and these issues can largely be attributed to burn-induced ischemia/hyporeperfusion, oxidative damage, and secondary inflammation or cell death1–3. As an organ with a rich blood supply and polyunsaturated fatty acids, the kidney is vulnerable to attack from reactive oxygen species (ROS) and the subsequent inflammatory response under ischemic conditions, leading to development of AKI4,5. In terms of burn injury, early acute kidney injury (AKI) usually occurs within the first 3 days after trauma, contributes to a poor prognosis and even high mortality (80%) and morbidity, is apt to give rise to a series of rapid pathological changes in tubular structures and predisposes patients to sequential functional deterioration of the kidney2,6. The incidence of acute renal dysfunction in burn patients receiving critical care can be up to 40%2. We previously provided several lines of evidence to support the key roles of ROS-related oxidative stress and secondary local mitochondria-related apoptosis in pathophysiological issues during the progression of burn-induced early AKI7. Furthermore, we found clues indicating that renal tissue inflammation should be taken into account as another important issue following ischemia or ROS burst8. Multiple studies investigating severe trauma (including burns), large surgery and sepsis have suggested the effectiveness of anti-inflammatory strategies to protect against AKI or other acute organ injuries following the above conditions8–10. However, early and timely interventions for burn-induced organ inflammation are still worthy of further exploration.
Nuclear factor (NF)-κB is regarded as a potential and crucial intermediate regulator of local inflammatory conditions in early AKI after burn injury8. We observed that the selective antioxidant molecular hydrogen may inhibit burn-induced release of inflammatory cytokines in rat kidneys through an NF-κB-mediated signaling pathway and ultimately attenuate early AKI and reduce apoptosis8. Furthermore, the Toll-like receptor (TLR)4/NF-κB signaling pathway has been suggested to be a classic cascade that is able to regulate development of the inflammatory response in diverse organ injuries following ischemia–reperfusion (IR)11. Moreover, the effects of nephroprotectants and preconditioning treatment on different types of kidney injury are closely related to downregulation of signals included in the TLR4/NF-κB pathway5,12. Recently, several researchers have observed a regulatory role of the TLR4 pathway in burn-induced sepsis and lung or cerebral injury, indicating that it is a promising therapeutic target. In addition to inflammation, a reactive oxygen species (ROS) burst is also accompanied by the expression of endogenous antioxidant proteins, such as heme oxygenase-1 (HO-1) and Nrf2, in some ischemic or LPS-induced organ injuries as a self-defense response13,14. HO-1 has been reported to have a nephroprotective role in renal injury caused by diverse stimuli due to its antagonism against oxidative damage and its regulation of several biological processes13,15,16. Considering burn-induced hypodynamic circulation and increased oxidative stress, the TLR4/NF-κB pathway and HO-1 are potential players in the development of burn-induced early AKI.
Astaxanthin (3,3′-dihydroxy-b,b'-carotene-4,4′-dione, ATX) is a natural carotenoid easily obtained from marine organisms and exhibits more robust and powerful antioxidative effects than other carotenoids17. Previously, researchers demonstrated that pretreatment or immediate administration of ATX can attenuate oxidative stress-induced toxicity in tubular epithelial cells and I/R-induced or diabetes-related renal injury in mice by reducing oxidative stress, inflammation and tubular apoptosis18,19. In terms of burn injury, we observed that ATX exerted a protective effect against burn-induced early AKI by ameliorating oxidative damage and downregulating mitochondria-related apoptosis via the PI3K/Akt/Bad pathway7. Combined with similar reports of its effects on oxidative damage and inflammation in other organ injuries, ATX seems to be a promising and nontoxic therapeutic reagent to prevent early AKI after burn injury20,21.
Given the important roles of oxidative stress and secondary renal inflammation in severe burn-induced early AKI, we hypothesized a possible role of ATX in protection via its anti-inflammatory effects and the potential mechanisms of action in early postburn AKI through the regulation of the TLR4/NF-κB pathway and HO-1, we and aimed to explore more details to support clinical treatment.
Results
Severe burn induced acute kidney injury in the early stage after injury, accompanied by increased oxidative stress and renal inflammation, whereas TAK242 or PDTC attenuated most of the above changes
Similar to a previous report, under a microscope, hematoxylin and eosin (HE)-stained sections showed typical histological changes in the kidney after burn injury, mainly including the absence of the proximal tubular brush border, blebbing of apical membranes, separation of tubular epithelial cells from the basement membrane, or aggregation of cells and proteins in the luminal region (Fig. 1a). Interestingly, unlike vehicle 1, application of both the selective TLR4 inhibitor TAK242 and the NF-κB activation inhibitor PDTC seemed to attenuate burn-induced histological damage in the kidneys (Fig. 1a). Furthermore, the tubular damage scores showed a gradual and significant increase from 6 h until 48 h post burn injury (Fig. 1b). The scores of the TAK242 and PDTC groups were significantly lower than those of the 48 h group after burn injury, while there was no difference between the scores of the vehicle 1 and burn groups at 48 h post burn injury (Fig. 1b).
Figure 1.

Histological, functional and biological assessments of kidney after burn. (a) Representative HE-stained images of renal tissues at a magnification of 200 × ; (b) Tubular damage score according to HE-stained slices; (c) Blood-based detection of creatinine and NGAL; (d) Oxidative stress evaluation based on commercial kits of ROS, MDA and SOD; (e) The release levels of inflammatory mediators after insults. n = 8 for each group. The results are expressed as the mean ± SD. *p < 0.05, **p < 0.01, v.s. sham; #p < 0.05, ##p < 0.01, v.s. vehicle1; @p < 0.05, nsp > 0.05.
In terms of renal function, similarly, we observed that burn injury increased the levels of sCr and NGAL at 6 h post burn injury, and levels peaked at 48 h after burn injury, whereas TAK242 had a stronger effect on attenuating the burn-induced increase in sCr and NGAL than PDTC. Vehicle1 had no effect on the levels of sCr and NGAL in rats at 48 h after burn injury (Fig. 1c).
In terms of oxidative status and secondary inflammation, we observed an obvious ROS burst in rat kidneys beginning in the early stage (6 h) after burn injury that persisted until 48 h post burn injury (Fig. 1d). Similarly, renal MDA levels exhibited a significant increase from 6 to 48 h after burn injury (Fig. 1d). Conversely, the representative endogenous antioxidant enzyme SOD showed a marked decrease after burn injury (Fig. 1d). The increased renal oxidative stress after burn injury conformed to the results of our previous study; nevertheless, neither vehicle 1 nor PDTC affected the burn-induced changes in ROS generation or the levels of MDA and SOD (Fig. 1d). However, interestingly, TAK242 significantly decreased the ROS burst and the MDA level in burned rat kidneys 48 h after burn injury (Fig. 1d). Furthermore, the renal mRNA levels of typical inflammatory mediators, such as MPO, IL-1β and IL-6, in burned rats increased significantly and gradually after insult and peaked 48 h after burn injury, although a slight decrease occurred 24 h post burn injury compared to the obvious elevation observed 6 h and 12 h post burn injury (Fig. 1e). Vehicles showed no effect on the mRNA transcription of MPO, IL-1β and IL-6, while both TAK242 and PDTC markedly lowered the burn-induced increase in MPO, IL-1β and IL-6 48 h after burn insult (Fig. 1e).
Activation of the TLR4/MyD88/NF-κB signaling pathway contributed to regulation of renal inflammation following burn injury
We also investigated potential signaling pathways related to renal inflammation induced by burn injury. According to the immunofluorescence results and subsequent tubule count, typical TLR4-positive (FITC-labeled, green) renal tubules appeared 12 h post burn injury, representing an increased distribution of TLR4 (Fig. 2a,b). The increases continued to 48 h post burn (Fig. 2a and b). In contrast to the slight effect of vehicle, both TAK242 and PDTC were able to reduce the burn-induced increase in TLR4-positive tubules 48 h after insult, and the effect of TAK242 seemed to be more obvious (Fig. 2a,b). Further immunoblot detection supported the observation based on immunofluorescence staining, and the expression of TLR4 showed a similar tendency (Fig. 2c). Specifically, TLR4 expression was significantly upregulated from 6 h post burn injury until 48 h, while there was only a slight increase at 6 h after burn injury. TAK242 exhibited a stronger effect in reversing the marked increase in TLR4 expression than PDTC. Vehicle exerted no regulation on TLR4 expression. In view of the potential importance of MyD88 in the TLR4 signaling pathway, we investigated the expression of MyD88 in rat kidneys after burn injury (Fig. 2c). The results showed that the expression of MyD88 significantly increased beginning at 6 h post burn injury and showed a gradual increasing tendency over time, similar to TLR4. TAK242 and PDTC obviously downregulated MyD88 expression 48 h post burn injury, while there were no differences between the 48 h burn and vehicle groups.
Figure 2.

Immunofluorescent and immunoblotting detection of TLR4 pathway. (a) Immunofluorescence double staining of TLR4; (b) The double-blind counting of TLR4 positively-stained tubules (green-labeled), n = 12 per group; (c) Representative bands of Western blot on TLR4 and MyD88, n = 6 for each group. The results are expressed as the mean ± SD. *p < 0.05, **p < 0.01, v.s. sham; #p < 0.05, ##p < 0.01, v.s. vehicle1; @@p < 0.01, nsp > 0.05.
Further immunoblot detection was performed to determine the possible activation of signals downstream of TLR4/MyD88 (Fig. 3). Finally, we observed increased phosphorylation of inhibitor of nuclear factor kappa-B kinase subunit (IKK)α + β, NF-kappa-B inhibitor (IκB)α, and NF-κB p65 beginning at 6 h after burn injury. All three peaks appeared at 48 h. The inhibitory effect of TAK242 on the burn–induced phosphorylation of IKKα + β and IκBα in kidneys was significant, whereas PDTC showed a more powerful ability to downregulate NF-κB p65 phosphorylation than TAK242. However, both were able to reverse the burn-induced phosphorylation of IKKα + β, IκBα, and NF-κB p65 at 48 h after insult, while vehicle showed no effect on phosphorylation of these proteins.
Figure 3.

Immunoblotting detection on downstream signals of MyD88 dependent TLR4 pathway. n = 6 for each group. The results are expressed as the mean ± SD. *p < 0.05, **p < 0.01, v.s. sham; #p < 0.05, ##p < 0.01, v.s. vehicle1; @p < 0.05, @@p < 0.01, nsp > 0.05.
All the above results indicate that typical activation of the MyD88-dependent TLR4/NF-κB signaling pathway might be involved in the induction of early renal inflammation after burn injury.
ATX dose-relatedly attenuated burn-induced tissue inflammation in rat kidneys
In our previous study, we observed the typical distribution of inflammatory mediators in burned rat renal tissue based on the results of immunohistochemical (IHC) staining. In this study, according to the results of IHC staining, we detected increased MPO, IL-1β and IL-6 immunoreactivity in the renal tubules of burned rats at the selected time window, similar to the groups treated with the classic selective TLR4 activator lipopolysaccharide (LPS) or the ATX solvent (vehicle 2) (Fig. 4). However, ATX seemed to dose-relatedly decrease the number of MPO-, IL-1β- or IL-6-positive tubular cells in rats 48 h after burn injury (Fig. 4). The results of IHC assessment were further supported by qRT-PCR detection (Fig. 4). The burn 48 h and vehicle groups exhibited similar expression of different inflammatory mediators, such as MPO, IL-1β and IL-6, although both showed slightly weaker effects than the LPS group. The levels of MPO, IL-1β and IL-6 in all three groups were significantly higher than in the sham group. All doses of ATX downregulated burn-increased MPO, IL-1β and IL-6 mRNA expression at 48 h after burn injury and displayed a dose-related effect. The effect peaked at the dose of 20 mg/kg. All these results suggest that ATX might dose-relatedly attenuate burn-induced inflammation in rat kidneys.
Figure 4.

Astaxanthin dose-relatively decreases the burn-induced release of inflammatory mediators. (a) Representative immunohistochemical images and tissue mRNA levels of MPO with ATX treatment; (b) Representative immunohistochemical images and tissue mRNA levels of IL-1β with ATX treatment; (c) Representative immunohistochemical images and tissue mRNA levels of IL-6 with ATX treatment. n = 6 for each group. The results are expressed as the mean ± SD. *p < 0.05, **p < 0.01, v.s. sham; #p < 0.05, ##p < 0.01, v.s. vehicle2; @p < 0.05, @@p < 0.01, nsp > 0.05.
ATX downregulated activation of the TLR4/MyD88/NF-κB signaling pathway in kidneys after burn injury in a dose-related manner
According to the aforementioned role of the TLR4/MyD88/NF-κB signaling pathway in regulating burn-induced renal tissue inflammation, we investigated the potential effect of ATX on the pathway (Fig. 5). Western blotting results showed that both LPS and burn insult induced significant elevation of TLR4 and MyD88 and secondary phosphorylation of IKKα + β, IκBα, and NF-κB p65 compared to the sham group. All three doses of ATX were able to remarkably downregulate TLR4 and MyD88 expression, and the strongest effect appeared in the 20 mg/kg ATX group. In addition, downstream downregulation of IKKα + β, IκBα, and NF-κB p65 phosphorylation was detected in all three ATX treatment groups. The peak of regulation was dose-dependent. Overall, ATX seemed to inhibit activation of the TLR4/MyD88/NF-κB signaling cascade.
Figure 5.

Astaxanthin dose-relatively downregulates the activation of TLR4/myD88/NF-κB signaling pathway. n = 6 for each group. The results are expressed as the mean ± SD. * p < 0.05, ** p < 0.01, v.s. sham; # p < 0.05, ## p < 0.01, v.s. vehicle2; @ p < 0.05, @@ p < 0.01, ns p > 0.05.
ATX ameliorated burn-induced renal inflammation through the MyD88-dependent TLR4/NF-κB pathway
Combined application of the selective TLR4 activator LPS and TAK242 or PDTC was introduced to determine the anti-inflammatory role of ATX based on the TLR4/MyD88/NF-κB pathway in the progression of burn-induced AKI. The pretreatment with TAK242 assisted high-dose ATX in enhancing the downregulation of TLR4, MyD88, phosphorylated IKKα + β, phosphorylated IκBα, and phosphorylated NF-κB p65 expression, whereas PDTC pretreatment showed a similar but weaker effect on this signaling pathway, with the exception of NF-κB p65 phosphorylation (Fig. 6a). The renal release of selected inflammatory mediators (MPO, IL-1β and IL-6) was also attenuated further with the help of TAK242 and PDTC in the ATX treatment groups (Fig. 6b). On the other hand, compared to the high dose of ATX, simultaneous LPS injection remarkably abolished the downregulation of TLR4 and MyD88 expression and phosphorylation of IKKα + β, IκBα, and NF-κB p65 caused by ATX treatment in the kidneys of burned rats (Fig. 6a). Furthermore, we observed an obvious increase in the mRNA expression of selected inflammatory mediators (MPO, IL-1β and IL-6) in burned rat kidneys treated with the combined application of high-dose ATX and LPS compared to the single application of ATX (Fig. 6b). All these results support an intermediate role of the MyD88-dependent TLR4/NF-κB pathway in mediating the anti-inflammatory effect of ATX.
Figure 6.

Astaxanthin attenuates burn-induced inflammation through regulating TLR4 /MyD88/NF-κB pathway. (a) Representative bands of western blot are shown after introduction of LPS, TAK242 and PDTC. LPS reversed the effect of high-dose ATX on expression of TLR4, MyD88 and downstream phosphorylated activation of IKKα/β, Iκ, BαNF-κB, while both TAK242 and PDTC could help to enhance the effect of ATX; (b) The inhibition of ATX on release of selected inflammatory mediators could be abolished by LPS, and enhanceb by using TAK242 or PDTC. n = 6 for each group. The results are expressed as the mean ± SD. *p < 0.05, **p < 0.01, v.s. ATX 20 mg/kg; #p < 0.05, ##p < 0.01.
ATX dose-relatedly increased the distribution and expression of HO-1 in rat kidneys after burn injury
Based on immunofluorescence staining, we observed increased red-labeled renal tubules 48 h after burn injury, representing HO-1 distribution. According to the count under a microscope, the number of HO-1-positive tubules increased significantly in the 48 h burn group. The administration of medium- and high-dose ATX in burned rats resulted in more red-labeled tubules than observed in the burn group in the selected visual fields, while low-dose ATX showed a weaker ability to further increase HO-1 distribution (Fig. 7a,b). The vehicle of the ATX solution (vehicle 2) showed no effect on burn-induced tubular HO-1 expression (Fig. 7a,b). Furthermore, immunoblot results showed that burn insult induced a significant upregulation of HO-1 expression in the kidneys of burned rats compared to expression in the sham group, while medium- and high-dose ATX administration further increased renal HO-1 expression after burn injury (Fig. 7c). Both vehicle and low-dose ATX showed a slight effect on HO-1 expression upregulation compared to the burn group 48 h after injury (Fig. 7c). Collectively, ATX seemed to further upregulate the renal HO-1 protein level in burned rats in a dose-related manner.
Figure 7.

Astaxanthin treatment dose-related increased the distribution and expression of HO-1 in kidneys of burned rats. (a) Representative images of Immunofluorescence staining of HO-1 in Kidneys; (b) The double-blind counting of HO-1 positively-stained tubules (red-labeled) n = 12 per group; (c) Representative bands of HO-1 protein expression, n = 6 per group. . The results are expressed as the mean ± SD. *p < 0.05, **p < 0.01, v.s. sham; ##p < 0.01, v.s. vehicle2; @p < 0.05, @@p < 0.01, nsp > 0.05.
Discussion
In the present study, we are the first to initiate a systematic investigation of the role of local inflammation and potentially related regulators in the progression of early AKI after burn injury, and to assess the potential protective effect of ATX via its anti-inflammatory effects as a further study following our previous work. Herein, our findings showed the following: (1) burn-induced renal inflammation contributes to the development of early AKI, with a concurrent increase in oxidative stress; (2) the TLR4/NF-κB pathway is involved in regulation of renal inflammation after burn insult and is dependent on MyD88; (3) ATX dose-relatedly attenuates activation of the TLR4/MyD88/NF-κB cascade and the subsequent release of inflammatory mediators; and (4) ATX further upregulates renal expression of HO-1 in a dose-related manner to assist in its regulation of oxidative stress and the TLR4 pathway.
In the early stage after burn injury, the risk of shock accompanied by a reduction in generalized renal blood flow (RBF) causes a series of endothelial and tubular changes, such as endothelial cell swelling and disruption of the endothelial monolayer, as a result of activation of the coagulation system and impaired vascular reactivity, which contributes to early tubular damage post burn injury1. In the current study, we observed typical regional changes in the kidneys of a severe burn model over time, including tubular epithelial necrosis and tubular dilation. Moreover, progression of histological damage in the kidneys is accompanied by deterioration of renal function and increases in the classic functional indices sCr and NGAL22. A subsequent investigation revealed that burn injury induced a remarkable increase in ROS generation and a decrease in antioxidant enzymes, representing increased oxidative stress 6 h post burn injury, which persisted during the early stage after burn injury, similar to our previous report8. The release of inflammatory mediators in kidneys exhibited a time-dependent increase after burn injury and showed a late peak compared with that of oxidative stress, suggesting that it was a result of a continuous ROS burst. After burn injury, leukocytes, endothelial cells and the renal tubular epithelium contribute to generation of several proinflammatory cytokines, such as MPO, IL-6, and IL-1β8. Increased release of proinflammatory mediators leads to enhanced vasoconstriction, vessel occlusion and a decreased number of microvessels, which results in edema of the outer medulla, increased tubular injury, and possible tubulointerstitial fibrosis23. Previously, we observed clues that suggested a connection between ROS-related oxidative stress and local inflammation in burned rat kidneys, and NF-κB seemed to act as a potential regulator to mediate burn-induced renal inflammation8. As a classic signaling cascade, other researchers have exposed the importance of TLR4 and NF-κB in regulating innate and adaptive immune responses to renal ischemic injury5,23,24, and the role of the TLR4/NF-κB cascade in ischemic injuries of other organs by regulating immune and subsequent inflammatory responses has been reported25,26. Interestingly, we observed that both TAK242 and PDTC seemed to attenuate the histological and functional deterioration of rat kidneys 48 h after burn injury, which provided clues about the potential value of the TLR4/NF-κB cascade in burn-induced AKI. As a specific inhibitor of TLR4, the benefit of TAK242 against histological kidney damage indicates a potential role of TLR4 in burn-induced AKI, while the effect of the NF-κB inhibitor PDTC suggests a pivotal role of NF-κB. Furthermore, administration of either TAK242 or PDTC markedly attenuated the elevated release of four proinflammatory mediators in burn-stimulated kidneys, which suggests a possible effect of the TLR4/NF-κB signaling pathway on regulation of the inflammation related to the development of burn-induced AKI. However, only TAK242 decreased the increased ROS level in kidneys and enhanced the activities of antioxidant enzymes after burn injury, indicating a potential relationship between TLR4 and ROS-mediated oxidative stress.
In terms of the typical TLR4/NF-κB signaling cascade, activated TLR4 can induce downstream phosphorylation of IKKs through a MyD88-dependent or MyD88-independent route and further lead to disintegration of the IKK complex27,28. Subsequently, phosphorylation of IκBα favors degradation of IκBα and subsequent dissociation of the NF-κB p65 subunit from the IκBα/NF-κB/Rel complex27,28. The dissociated p65 subunit is activated via phosphorylation and then translocates into the nucleus, which ultimately mediates the transcription of multiple genes27,28. Several in vivo results from other researchers illustrated that many types of agents display a nephroprotective effect in ischemia–reperfusion-induced AKI by regulating TLR4/NF-κB pathway-mediated inflammation in the kidneys11,29–31. In this study, we further investigated the detailed role of the TLR4/NF-κB signaling cascade in burned rats via immunofluorescence staining and western blotting. The time-dependent increase in the distribution and expression of TLR4 and MyD88 suggests that MyD88 is involved in regulating upstream activation of the NF-κB pathway. Subsequently, the renal tissue of rats showed sequentially increased phosphorylation of signals, including IKKα + β, IκBα, and NF-κB p65, along with increased release of proinflammatory cytokines. Combined with the reverse effect of TAK242 and PDTC blockade on renal structure, function and proinflammatory mediators, our results indicate that the MyD88-dependent TLR4/NF-κB signaling pathway is involved in regulation of burn-induced inflammation and related injury in kidneys and might be a pivotal target to prevent or treat burn-induced early AKI.
Astaxanthin (ATX) is a natural and strong antioxidant that can be extracted from multiple crustaceans32. ATX is considered to be a safe and hopeful therapeutic agent in several diseases, as well as a safe feed additive and dietary supplement21,33–35. Our previous study primarily demonstrated the protective value of ATX in burn-induced early AKI, and the mechanism of action was found to be related to its ability to attenuate oxidative stress and secondary renal cell apoptosis7. Inflammation is another crucial result of circulatory or tissue ROS bursts following surgery or severe trauma. ATX has been suggested to have an anti-inflammatory role based on several in vivo and in vitro studies on neurologic injury/dysfunction/disease, dermatitis, lung injury, and diabetes-related disorders, among others36–42. In terms of burn injury, we observed a beneficial effect of ATX in preventing deterioration of burn wounds by ameliorating tissue inflammation in wounds43. The results of prior studies have suggested that the anti-inflammatory property of ATX might be attributed to regulation of certain inflammation-related signaling molecules, such as NF-κB, JAK/STATS, PI3K/Akt, MAPK, ERK, and MSK, all of which might lead to secondary release of inflammatory cytokines42,44–48. In particular, NF-κB has elicited more attention from researchers due to its crucial role in regulating the inflammatory cascade under diverse pathophysiological statuses of multiple organs27. Furthermore, based on in vivo and in vitro subarachnoid hemorrhage (SAH) models, Zhang et al. demonstrated that TLR4 plays a crucial role in mediating neuroinflammation caused by insults resulting from SAH, while MyD88 and NF-κB act downstream, and the neuroprotective effect of ATX primarily relies on inhibiting the TLR4 signaling pathway and the subsequent proinflammatory response36. In the current study, our observations support the importance of the TLR4/MyD88/NF-κB signaling axis in regulating renal inflammation after burn injury, and ATX exhibited a dose-dependent inhibitory effect on activation of the TLR4 pathway and the secondary increase in the release of inflammatory mediators. Single or combined application of the signal inhibitor TAK242 or PDTC and the classic TLR4 agonist LPS led to enhancement or reversal of the anti-inflammatory effect of high-dose ATX, which further indicates that ATX might ameliorate burn-induced renal inflammation by influencing the TLR4/MyD88/NF-κB signaling pathway.
Similar to apoptosis, inflammation is another event that follows an ROS burst caused by multiple factors. Under trauma or stress, certain inner biological defenses exist to protect against possible organ or tissue injury. The kidney is able to exert adaptive and protective mechanisms to limit harmful effects5. As a heat shock protein, HO-1 can be induced rapidly to address both oxidative and cellular stress, presenting powerful antioxidant or anti-inflammatory characteristics49,50. Furthermore, previous researchers observed that quick induction of HO-1 expression can be initiated in the kidneys of an AKI model even 3 to 6 h after both ischemia/reperfusion and nephrotoxin insult13,16. In the prior and current study, we also detected early upregulation of HO-1 in the kidneys of burned rats, suggesting its potential value in regulating burn-induced AKI. TLR4 is closely related to HO-1 expression, whereas HO-1 can negatively regulate TLR4 by influencing ROS generation or certain signals51,52. The antioxidant and anti-inflammatory properties of ATX also rely on activation of HO-153,54. In a streptozotocin-induced diabetic rat model, Yeh et al. found that ATX administration attenuated diabetes-related ocular nerve injury by upregulating HO-1 to decrease oxidative stress and secondary inflammation and that NF-κB acted downstream following HO-139. The beneficial potential of ATX against airway inflammation has also been observed with in vivo and in vitro models and was reported to be closely related to the participation of HO-155. In the present study, burn injury led to a remarkable increase in HO-1 in renal tubules in response to thermal insult, whereas medium- and high-dose ATX further upregulated HO-1. Considering the effect of ATX on ROS production and inflammation in kidneys after burn injury, the obtained data support our proposed role of HO-1 in mediating the ability of ATX to modulate renal inflammation by influencing oxidative stress and TLR4 in the development of early AKI after burn injury.
In summary, the current study demonstrated that local inflammation plays an important role in burn-induced early AKI and that the MyD88-dependent TLR4/NF-κB pathway is involved in regulation of renal inflammation (Fig. 8). ATX exhibited anti-inflammation-based protection against early AKI after burn injury, and the beneficial effect can be attributed to its relief of burn-induced oxidative stress and secondary inflammation by influencing the TLR4/MyD88/NF-κB axis. Furthermore, HO-1 acts as a mediator that links the antioxidant and anti-inflammatory effects of ATX. Our study still has some limitations: more work is still required to determine the direct action of ATX on TLR4 or the existence of a potential mediator between ATX and TLR4 signaling; gene-editing technique may help to define the regulation of ATX on signals with less bias; some other signals relating to inflammatory regulation also act downstream of ATX possibly. Nevertheless, our current findings further verify the potential value of ATX as a promising and safe therapeutic agent to improve the outcome of burn patients during the early stage and enrich our knowledge of its mechanism of action.
Figure 8.
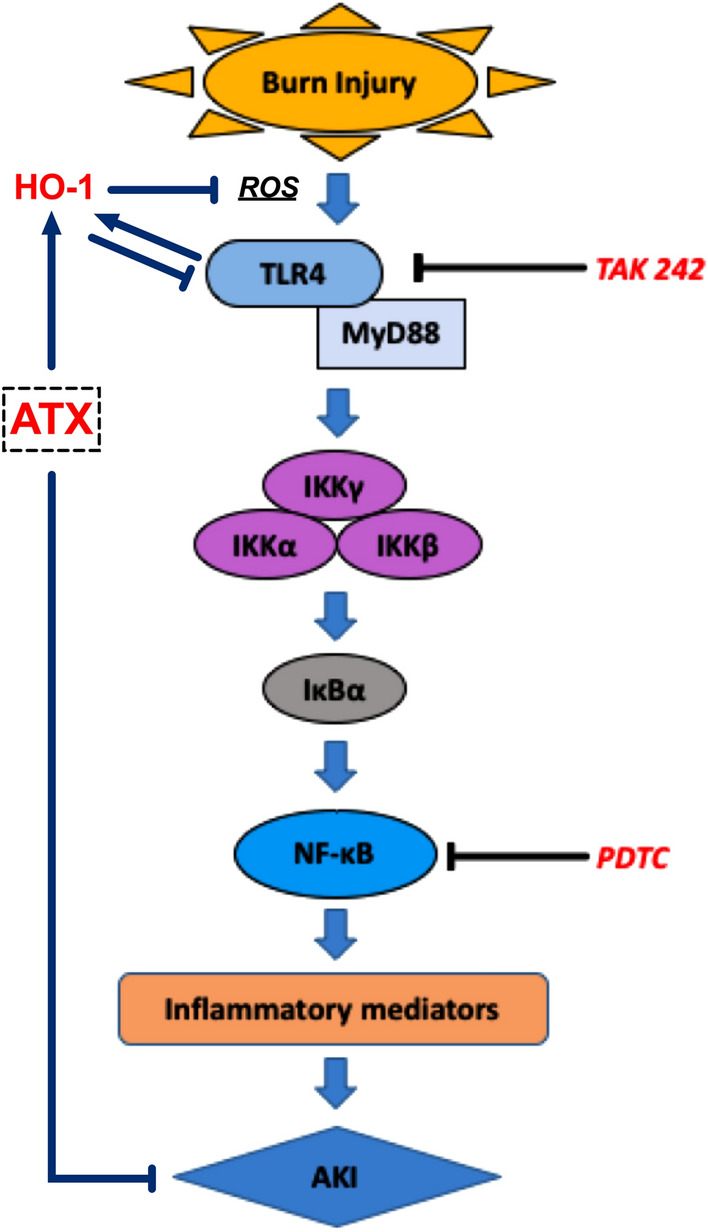
The schematic diagram of this study.
Methods
Experimental animals and severe burn model establishment
The current study was performed according to protocols approved by The Second Affiliated Hospital Zhejiang University School of Medicine Institutional Animal Care and Use Committee (2016-144) and strictly followed the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals. The present study was also carried out in compliance with the ARRIVE guidelines. Adult male 6–7-week Sprague–Dawley (SD) rats weighing approximately 220–250 g were purchased from the Shanghai Slac Laboratory Animal Company (Shanghai, China) and housed with a 12-h light/dark cycle in a filtered-air unit at a constant temperature and humidity. All animals were given free access to food and water. Every animal was housed in one single cage after operation.
The severely burned rat model was established as previously described7,8. Specifically, all animals received sodium pentobarbital (50 mg/kg) by intraperitoneal (ip) injection as anesthesia before sham or burn operation. The burn model was established by exposing the rats to a 15 s immersion in 100 °C hot water to generate a large-scale full-thickness dermal burn. The thermally damaged area occupied approximately 40% of the TBSA. The rats in the sham group were treated with 25 °C water on the shaved dorsum after anesthesia. During the operation, the breath and heart rate of burn rats were carefully monitored to ensure that all rats were anesthetized and free of pain before postanesthesia recovery. Liquid resuscitation was preceded by ip injection of lactated Ringer’s solution (LRS) at 4 ml/kg/TBSA immediately and 6 h after the operation. In addition, all rat models were housed in individual cages and administered 0.25 mg/kg buprenorphine via subcutaneous injection immediately and every 12 h postburn for analgesia. A pain and distress scale reported previously was introduced for guiding the analgesia therapy7.
Animal grouping and drug administration
The study can be divided into two parts, as shown in Fig. 9. Specifically, in part I, sixty-four SD rats were randomly assigned to eight groups with the help of the random-number table: the sham group, the four burn groups (6 h, 12 h, 24 h, 48 h), the vehicle treatment (burn plus vehicle 1) groups, and the burn plus TAK242 or PDTC groups (n = 8 per group). Both TAK242 (MCE, USA) and PDTC (Beyotime, China) were dissolved in a vehicle solution (vehicle 1, 1% dimethyl sulfoxide in distilled water). Animals in the TAK242 or PDTC group were given TAK242 at a dose of 3 mg/kg or PDTC at a dose of 100 mg/kg by intravenous tail (tail iv) injection 0.5 h preburn56,57. Both the sham and burn groups received equal volumes of distilled water, while the burn plus vehicle 1 group was administered an equal volume of vehicle 1 solution. The time-window of observation and sample collection (48 h after sham operation or burn) in controls of sham, TAK242 and PDTC groups was selected according to the results suggesting the most obvious renal injury and renal inflammation found in the 48 h postburn group.
Figure 9.

The experimental design and grouping.
In part II, eighty SD rats were randomly assigned to ten groups with the help of the random-number table: the sham group, the LPS treatment group, the three ATX-treatment burn groups, the burn plus ATX plus TAK242 or PDTC groups and the burn plus ATX plus LPS group (n = 8 per group). ATX (Sigma-Aldrich, St. Louis, MO, USA) solution was prepared in vehicle 2 (polyethylene glycol 400-N,N-dimethylacetamide (PEG400), purchased from Sigma-Aldrich, St. Louis, MO, USA (50:50, v/v)) at concentrations of 5 mg/ml, 10 mg/ml and 20 mg/ml. The rats in the ATX treatment groups received ATX at 5 mg/kg, 10 mg/kg or 20 mg/ml via tail iv injection7,43. In the sham group, rats were given equal volumes (1 ml/kg) of distilled water via tail iv injection. The animals in the vehicle 2 groups were given 1 ml/kg PEG400 (50:50, v/v) without any drugs via tail iv injection. All the above treatments were administered 30 min after surgery7,43. The LPS control group received 4 mg/kg lipopolysaccharide (LPS) immediately after sham operation via the ip routine58. The ATX plus LPS group was given 20 mg/kg ATX 30 min postburn and 4 mg/kg LPS 1 h after burn induction via ip injection58,59. TAK242 and PDTC were administered via tail iv injection or intraperitoneally 0.5 h preburn at 3 mg/kg and 100 mg/kg in the selected burn plus ATX groups as mentioned above.
All rats were sacrificed through overdose of sodium pentobarbital. Except for those in the three burn groups (sacrificed at 6 h, 12 h, and 24 h postburn), the animals in the other groups were sacrificed at 48 h post sham operation or burn insult.
Histological preparation
Following euthanasia, both kidneys were dissected after cardiac perfusion with phosphate-buffered saline (PBS) (pH = 7.2) and were maintained in 4% paraformaldehyde at 4 °C or in a − 80 °C freezer for subsequent assessment7,8. Frozen or paraffin-embedded kidney samples were sectioned for further staining.
Histological examination
Hematoxylin and eosin (HE) staining was performed for histological examination, and the stained slices were observed and imaged under a microscope (Leica, Solms, Germany). Twelve high-magnification files were randomly selected for observation from six slices per group in a blind manner by two pathologists. The tubular damage score was evaluated based on the percentage of injured renal cortical tubules and ranked as 0: normal, 1: less than 10%, 2: 11 to 25%, 3: 26 to 75%, and 4: greater than 75%7,8.
Renal function evaluation
Blood samples were collected to detect the levels of serum creatinine (sCr) by using a clinical chemistry analyzer system and kits as previously reported (Au5800; Beckman Coulter, CA, USA)7,8. The levels of serum neutrophil gelatinase-associated lipocalin (NGAL) were determined using a commercial NGAL ELISA kit for RAT (EK0855; Boster, Wuhan, China).
Immunofluorescence staining
After rewarming, washing and antigen retrieval, the 7-μm-thick frozen slices were separately incubated with anti-TLR4 (1:100; ab22048; Abcam, Cambridge, UK) or anti-HO-1 (1:200; ab13243; Abcam, Cambridge, UK) antibody overnight at 4 °C. After being rinsed with PBS, the sections were incubated with a FITC-labeled goat anti-mouse (1:50; BA1101; Boster, Wuhan, China) or Cy3-labeled goat anti-rabbit (1:50; BA1032; Boster, Wuhan, China) secondary antibody for 2 h at 37 °C in the dark43. The sections were rinsed and stained with DAPI (100 ng/ml; Boster, Wuhan, China) for 8 min at room temperature and then mounted with VECTASHIELD mounting medium (H-1000; Vector, CA, USA). All the slices were observed and imaged under a fluorescence microscope (Leica, Solms, Germany). The TLR4-positive (green stained) or HO-1-positive (red stained) tubules were counted under double-blinded conditions. Two independent investigators who were blinded to the group assignments calculated the number of immunostained tubules. At least two visual fields per slide and six slides per group were evaluated by the two investigators.
Immunohistochemistry (IHC) staining
After deparaffinization, rehydration and antigen retrieval, the sections were incubated with anti-MPO antibody (1:100; ab9535; Abcam, Cambridge, UK), anti-IL-1β antibody (1:100; SC-7884; Santa Cruz Biotechnology, CA, USA), and anti-IL-6 antibody (1:100; SC-1265-R; Santa Cruz, CA, USA) overnight at 4 °C7,8. After incubation with goat anti-rabbit secondary antibody (Boster, Wuhan, China), the samples were visualized with a 3,3-diaminobenzidine (DAB) kit (Boster, Wuhan, China). The mounted sections were observed and photographed under a microscope at 200 × magnification (DM2500; Leica, Solms, Germany).
Oxidative stress assessment
Kidney tissue homogenate was prepared as previously reported7,8. ROS generation was detected with a DCFH-DA-based Reactive Oxygen Species Assay Kit (KGT010-1; KeyGEN Biotech, Nanjing, China) and expressed as n-fold compared to that of the sham group. An indirect index of oxidative stress, the malondialdehyde (MDA) level, was measured with a thiobarbituric acid reactive species (TBARS) assay kit (KGT003-1; KeyGEN Biotech, Nanjing, China) and is expressed in nmol/mg protein. SOD activity in the skin tissues of burn wounds was evaluated using a commercial assay kit from KeyGEN Biotech (KGT00150; Nanjing, China). The results are expressed in U/mg protein. Absorbance values were measured using a microplate reader (Model 680 Microplate Reader; BIO-RAD, CA, USA).
Quantitative real-time polymerase chain reaction (qRT-PCR) analysis
qRT-PCR was performed to determine the mRNA expression levels of myeloperoxidase (MPO), interleukin (IL)-1β, and IL-6. Briefly, total RNA was extracted from frozen kidney tissues with TRIzol Reagent (Invitrogen, Carlsbad, CA, USA) and RNase-Free DNase I (Qiagen, Duesseldorf, Germany)8. A SuperScript First-Strand Synthesis System for reverse transcription PCR (Invitrogen, Carlsbad, CA, USA) was introduced to synthesize cDNAs, and RNA and cDNA concentrations, and then, purities were measured via BIO-RAD spectrophotometry (SMARTSPEC Plus, BIO-RAD, CA, USA)8. The primers (Table 1) were designed using Primer Premier 6.0 (PREMIER Biosoft, CA, USA) and synthesized by Shanghai Biological Engineering Co., Ltd. (Shanghai, China)8. PCR amplifications were conducted with Power SYBR Master Mix (Invitrogen, Carlsbad, CA, USA) in an IQ 5 Real-time PCR system (BIO-RAD, CA, USA)8. β-Actin was used as an internal standard to show relative expression levels. Relative quantification of the target gene expression levels was conducted using the 2−∆∆Ct method.
Table 1.
The oligonucleotide primers used for PCR amplification.
| Gene | Genbank accession | Primer sequences (5′ to 3′) | Size (bp) | Annealing (°C) |
|---|---|---|---|---|
| Rat MPO | NM_001107036.1 | GCTACGGGATGGCGATAGGTTT | 126 | 63 |
| GACACGGTAGTGATGCCAGTGTT | ||||
| Rat IL-1β | NM_031512.2 | CCTAGGAAACAGCAATGGTCGGGAC | 126 | 63 |
| GTCAGAGGCAGGGAGGGAAACAC | ||||
| Rat IL-6 | NM_012589.2 | CAATCAGAATTGCCATTGCACAA | 169 | 63 |
| CGTCTTCGCAAGAGGAAGAGCAGT | ||||
| Rat β-actin | EF156276.1 | CCAACCGTGAAAAGATGACCCAGAT | 97 | 63 |
| CCAGAGGCATACAGGGACAACA |
Western blot analysis
Generally, the lysed protein samples were subjected to SDS-PAGE and transferred onto nitrocellulose membranes via electrophoresis, while aliquots of samples were used to determine the protein concentration of each sample using a bicinchoninic acid (BCA) kit (KGPBCA, KeyGEN Biotech, Nanjing, China)7,8. Subsequently, membranes were incubated in blocking buffer for 2 h and incubated overnight at 4 °C with the following primary antibodies: anti-TLR4 (1:200; SC-10741; Santa Cruz, CA, USA), anti-p-IKKα + β (1:500; ab2064; Abcam, Cambridge, UK), anti-IKKα + β (1:1000; ab178870; Abcam, Cambridge, UK), anti-p-IκBα (1:1000; ab12135; Abcam, Cambridge, UK), anti-IκBα (1:500; ab32518; Abcam, Cambridge, UK), anti-p-NF-κB p65 (1:500; #3033; Cell Signaling Technology, Boston, USA), anti-NF-κB p65 (1:1000; ab16502; Abcam, Cambridge, UK), and anti-HO-1 (1:2000; ab13243; Abcam, Cambridge, UK). GAPDH (1:15,000; ab8245; Abcam, Cambridge, UK) was used as a control on the same membranes. Secondary antibodies were applied, and the bands were detected with West Dura Extended Duration Substrate (Pierce, USA) and X-ray film (Kodak, USA). Then relative density of bands (based on gray value) was analyzed using Bandscan 5.0 software (Glyko, USA) via comparison with GAPDH expression7,8.
Statistical analysis
All data are presented as the means ± standard deviation (SD). GraphPad Prism version 7 (San Diego, CA, USA) and SPSS 19 (SPSS, Chicago, IL, USA) were used for statistical analyses. Multiple comparisons were analyzed using one-way analysis of variance (ANOVA) followed by a Bonferroni post hoc test. A value of p < 0.05 was regarded as statistically significant.
Supplementary Information
Acknowledgements
This research was supported by the following grants: National Natural Science Foundation of China (NSFC) No. 81671909 and 81901958, Zhejiang Provincial Natural Science Foundation of China under Grants No. LY18H150004, LY19H150004 and LY20H150010, and Key Research and Development Project of Sichuan Science and Technology Department No. 2018SZ0380.
Abbreviations
- AKI
Acute kidney injury
- IHC
Immunochemistry
- IKK
Inhibitor of nuclear factor kappa-B kinase
- IκBα
Nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, alpha
- IL
Interleukin
- IR
Ischemia–reperfusion
- IP
Intraperitoneal injection
- MyD88
Myeloid differentiation primary response gene 88
- NGAL
Neutrophil gelatinaseassociated lipocalin
- NF
Nuclear factor
- qRT-PCR
Quantitative real time polymerase chain reaction
- sCr
Serum creatinine
- TLR
Toll like receptor
Author contributions
Conceptualization, S.G, Q.F. and M.Y.; methodology, S.G. and C.H.; validation, S.G., M.Y. and L.Z.; formal analysis, L.G. and C.Y.; investigation, S.G., L.G. and M.Y.; resources, M.Y. and C.Y.; data curation, Q.F.; writing—original draft preparation, S.G., L.G., Q.F., Y.L. and X.G. writing—review and editing, S.G., Q.F. and X.G.; visualization, S.G.; supervision, C.H.; project administration, S.G. and C.H.. All authors have read and agreed to the published version of the manuscript.
Competing interests
The authors declare no competing interests.
Footnotes
The original online version of this Article was revised: The original version of this Article contained an error in the Acknowledgments section, which now reads: “This research was supported by the following grants: National Natural Science Foundation of China (NSFC) No. 81671909 and 81901958, Zhejiang Provincial Natural Science Foundation of China under Grants No. LY18H150004, LY19H150004 and LY20H150010, and Key Research and Development Project of Sichuan Science and Technology Department No. 2018SZ0380.”
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Songxue Guo and Linsen Guo.
Change history
1/12/2022
A Correction to this paper has been published: 10.1038/s41598-022-05213-y
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-021-86146-w.
References
- 1.Colpaert K, Hoste EA. Acute kidney injury in burns: a story of volume and inflammation. Crit. Care. 2008;12:192. doi: 10.1186/cc7106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Clark A, et al. Acute kidney injury after burn. Burns. 2017;43:898–908. doi: 10.1016/j.burns.2017.01.023. [DOI] [PubMed] [Google Scholar]
- 3.Thalji SZ, Kothari AN, Kuo PC, Mosier MJ. Acute kidney injury in burn patients. Ann. Surg. 2017;266:376–382. doi: 10.1097/SLA.0000000000001979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Elks CM, et al. A blueberry-enriched diet attenuates nephropathy in a rat model of hypertension via reduction in oxidative stress. PLoS ONE. 2011;6:e24028. doi: 10.1371/journal.pone.0024028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zuk A, Bonventre JV. Acute kidney injury. Annu. Rev. Med. 2016;67:293–307. doi: 10.1146/annurev-med-050214-013407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mosier MJ, et al. Early acute kidney injury predicts progressive renal dysfunction and higher mortality in severely burned adults. J. Burn Care Res. 2010;31:83–92. doi: 10.1097/BCR.0b013e3181cb8c87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guo SX, et al. Astaxanthin attenuates early acute kidney injury following severe burns in rats by ameliorating oxidative stress and mitochondrial-related apoptosis. Mar. Drugs. 2015;13:2105–2123. doi: 10.3390/md13042105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guo SX, et al. Effects of hydrogen-rich saline on early acute kidney injury in severely burned rats by suppressing oxidative stress induced apoptosis and inflammation. J. Transl. Med. 2015;13:183. doi: 10.1186/s12967-015-0548-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.He L, et al. Mangiferin attenuate sepsis-induced acute kidney injury via antioxidant and anti-inflammatory effects. Am. J. Nephrol. 2014;40:441–450. doi: 10.1159/000369220. [DOI] [PubMed] [Google Scholar]
- 10.Nozaki Y, et al. Signaling through the interleukin-18 receptor α attenuates inflammation in cisplatin-induced acute kidney injury. Kidney Int. 2012;82:892–902. doi: 10.1038/ki.2012.226. [DOI] [PubMed] [Google Scholar]
- 11.O'Neill S, et al. Heat shock protein 90 inhibition abrogates TLR4-mediated NF-κB activity and reduces renal ischemia-reperfusion injury. Sci. Rep. 2015;5:12958. doi: 10.1038/srep12958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nair AR, Masson GS, Ebenezer PJ, Del Piero F, Francis J. Role of TLR4 in lipopolysaccharide-induced acute kidney injury_Protection by blueberry. Free Radic. Biol. Med. 2014;71:16–25. doi: 10.1016/j.freeradbiomed.2014.03.012. [DOI] [PubMed] [Google Scholar]
- 13.Nath KA. Heme oxygenase-1 and acute kidney injury. Curr. Opin. Nephrol. Hypertens. 2014;23:17–24. doi: 10.1097/01.mnh.0000437613.88158.d3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carchman EH, Rao J, Loughran PA, Rosengart MR, Zuckerbraun BS. Heme oxygenase-1-mediated autophagy protects against hepatocyte cell death and hepatic injury from infection/sepsis in mice. Hepatology. 2011;53:2053–2062. doi: 10.1002/hep.24324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nath KA. Heme oxygenase-1: a provenance for cytoprotective pathways in the kidney and other tissues. Kidney Int. 2006;70:432–443. doi: 10.1038/sj.ki.5001565. [DOI] [PubMed] [Google Scholar]
- 16.Bolisetty S, et al. Heme oxygenase-1 inhibits renal tubular macroautophagy in acute kidney injury. J. Am. Soc. Nephrol. 2010;21:1702–1712. doi: 10.1681/ASN.2010030238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ambati R, Moi P, Ravi S, Aswathanarayana R. Astaxanthin: sources, extraction, stability, biological activities and its commercial applications—a review. Mar. Drugs. 2014;12:128–152. doi: 10.3390/md12010128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Qiu X, et al. Protective effects of astaxanthin against ischemia/reperfusion induced renal injury in mice. J. Transl. Med. 2015;13:28. doi: 10.1186/s12967-015-0388-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim YJ, Kim YA, Yokozawa T. Protection against oxidative stress, inflammation, and apoptosis of high-glucose-exposed proximal tubular epithelial cells by astaxanthin. J. Agric. Food Chem. 2009;57:8793–8797. doi: 10.1021/jf9019745. [DOI] [PubMed] [Google Scholar]
- 20.Landon R, et al. Impact of astaxanthin on diabetes pathogenesis and chronic complications. Mar. Drugs. 2020;18:357–420. doi: 10.3390/md18070357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li J, Guo C, Wu J. Astaxanthin in liver health and disease: a potential therapeutic agent. Drug Des. Dev. Ther. 2020;14:2275–2285. doi: 10.2147/DDDT.S230749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ronco C. Acute kidney injury: from clinical to molecular diagnosis. Crit. Care. 2016;20:201. doi: 10.1186/s13054-016-1373-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bonventre JV, Yang L. Cellular pathophysiology of ischemic acute kidney injury. J. Clin. Invest. 2011;121:4210–4221. doi: 10.1172/JCI45161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kanagasundaram NS. Pathophysiology of ischaemic acute kidney injury. Ann. Clin. Biochem. 2015;52:193–205. doi: 10.1177/0004563214556820. [DOI] [PubMed] [Google Scholar]
- 25.Zhu L, et al. Exercise preconditioning regulates the toll-like receptor 4/nuclear factor-κB signaling pathway and reduces cerebral ischemia/reperfusion inflammatory injury: a study in rats. J. Stroke Cerebrovasc. Dis. 2016 doi: 10.1016/j.jstrokecerebrovasdis.2016.07.033. [DOI] [PubMed] [Google Scholar]
- 26.Li J, et al. Resveratrol attenuates inflammation in the rat heart subjected to ischemia-reperfusion: Role of the TLR4/NF-κB signaling pathway. Mol Med Rep. 2015;11:1120–1126. doi: 10.3892/mmr.2014.2955. [DOI] [PubMed] [Google Scholar]
- 27.Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;132:344–362. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- 28.Campbell KJ, Perkins ND. Regulation of NF-kappaB function. Biochem. Soc. Symp. 2006;73:165–180. doi: 10.1042/bss0730165. [DOI] [PubMed] [Google Scholar]
- 29.Yang Y, et al. Renoprotective approaches and strategies in acute kidney injury. Pharmacol. Ther. 2016;163:58–73. doi: 10.1016/j.pharmthera.2016.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li Y-W, et al. Protective effect of tea polyphenols on renal ischemia/reperfusion injury via suppressing the activation of TLR4/NF-κB p65 signal pathway. Gene. 2014;542:46–51. doi: 10.1016/j.gene.2014.03.021. [DOI] [PubMed] [Google Scholar]
- 31.Wang L, et al. Picroside II protects rat kidney against ischemia/reperfusion-induced oxidative stress and inflammation by the TLR4/NF-κB pathway. Exp. Therap. Med. 2015;9:1253–1258. doi: 10.3892/etm.2015.2225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ambati RR, Phang SM, Ravi S, Aswathanarayana RG. Astaxanthin: sources, extraction, stability, biological activities and its commercial applications—a review. Mar. Drugs. 2014;12:128–152. doi: 10.3390/md12010128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Higuera-Ciapara I, Félix-Valenzuela L, Goycoolea FM. Astaxanthin: a review of its chemistry and applications. Crit. Rev. Food Sci. Nutr. 2006;46:185–196. doi: 10.1080/10408690590957188. [DOI] [PubMed] [Google Scholar]
- 34.Valenti MT, Perduca M, Romanelli MG, Mottes M, Dalle Carbonare L. A potential role for astaxanthin in the treatment of bone diseases (review) Mol. Med. Rep. 2020;22:1695–1701. doi: 10.3892/mmr.2020.11284. [DOI] [PubMed] [Google Scholar]
- 35.Brotosudarmo THP, Limantara L, Setiyono E. Structures of astaxanthin and their consequences for therapeutic application. Int. J. Food Sci. 2020;2020:2156582-16. doi: 10.1155/2020/2156582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang X, et al. Astaxanthin mitigates subarachnoid hemorrhage injury primarily by increasing sirtuin 1 and inhibiting the Toll-like receptor 4 signaling pathway. FASEB J. 2019;33:722–737. doi: 10.1096/fj.201800642RR. [DOI] [PubMed] [Google Scholar]
- 37.Kumar A, Dhaliwal N, Dhaliwal J, Dharavath RN, Chopra K. Astaxanthin attenuates oxidative stress and inflammatory responses in complete Freund-adjuvant-induced arthritis in rats. Pharmacol. Rep. 2020;72:104–114. doi: 10.1007/s43440-019-00022-z. [DOI] [PubMed] [Google Scholar]
- 38.Bi J, Cui R, Li Z, Liu C, Zhang J. Astaxanthin alleviated acute lung injury by inhibiting oxidative/nitrative stress and the inflammatory response in mice. Biomed. Pharmacother. 2017;95:974–982. doi: 10.1016/j.biopha.2017.09.012. [DOI] [PubMed] [Google Scholar]
- 39.Yeh P-T, Huang H-W, Yang C-M, Yang W-S, Yang C-H. Astaxanthin inhibits expression of retinal oxidative stress and inflammatory mediators in streptozotocin-induced diabetic rats. PLoS ONE. 2016;11:e0146438. doi: 10.1371/journal.pone.0146438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ying C-J, et al. Anti-inflammatory effect of astaxanthin on the sickness behavior induced by diabetes mellitus. Cell Mol. Neurobiol. 2015;35:1027–1037. doi: 10.1007/s10571-015-0197-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Park JH, et al. Anti-inflammatory effect of astaxanthin in phthalic anhydride-induced atopic dermatitis animal model. Exp. Dermatol. 2018;27:378–385. doi: 10.1111/exd.13437. [DOI] [PubMed] [Google Scholar]
- 42.Sun S-Q, Zhao Y-X, Li S-Y, Qiang J-W, Ji Y-Z. Anti-tumor effects of astaxanthin by inhibition of the expression of STAT3 in prostate cancer. Mar. Drugs. 2020;18:415. doi: 10.3390/md18080415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fang Q, et al. Astaxanthin protects against early burn-wound progression in rats by attenuating oxidative stress-induced inflammation and mitochondria-related apoptosis. Sci. Rep. 2017;7:41440–41513. doi: 10.1038/srep41440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Speranza L, et al. Astaxanthin treatment reduced oxidative induced pro-inflammatory cytokines secretion in U937: SHP-1 as a novel biological target. Mar. Drugs. 2012;10:890–899. doi: 10.3390/md10040890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fu J, et al. Astaxanthin alleviates spinal cord ischemia-reperfusion injury via activation of PI3K/ Akt/GSK-3β pathway in rats. J. Orthop. Surg. Res. 2020 doi: 10.1186/s13018-020-01790-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cai X, et al. Astaxanthin prevents against lipopolysaccharide-induced acute lung injury and sepsis via inhibiting activation of MAPK/NF-κB. Am. J. Transl. Res. 2019;11:1884–1894. [PMC free article] [PubMed] [Google Scholar]
- 47.Kim YH, Koh H-K, Kim D-S. Down-regulation of IL-6 production by astaxanthin via ERK-, MSK-, and NF-κB-mediated signals in activated microglia. Int. Immunopharmacol. 2010;10:1560–1572. doi: 10.1016/j.intimp.2010.09.007. [DOI] [PubMed] [Google Scholar]
- 48.Han J, et al. Astaxanthin ameliorates lipopolysaccharide-induced neuroinflammation, oxidative stress and memory dysfunction through inactivation of the signal transducer and activator of transcription 3 pathway. Mar. Drugs. 2019;17:123–218. doi: 10.3390/md17020123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bak S-U, et al. Heme oxygenase-1 (HO-1)/carbon monoxide (CO) axis suppresses RANKL-induced osteoclastic differentiation by inhibiting redox-sensitive NF-κB activation. BMB Rep. 2017;50:103–108. doi: 10.5483/BMBRep.2017.50.2.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sudan K, et al. TLR4 activation alters labile heme levels to regulate BACH1 and heme oxygenase-1 expression in macrophages. Free Radic. Biol. Med. 2019;137:131–142. doi: 10.1016/j.freeradbiomed.2019.04.024. [DOI] [PubMed] [Google Scholar]
- 51.Mandal P, et al. Adiponectin and heme oxygenase-1 suppress TLR4/MyD88-independent signaling in rat Kupffer cells and in mice after chronic ethanol exposure. J. Immunol. 2010;185:4928–4937. doi: 10.4049/jimmunol.1002060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chillappagari S, et al. Impaired TLR4 and HIF expression in cystic fibrosis bronchial epithelial cells downregulates hemeoxygenase-1 and alters iron homeostasis in vitro. Am. J. Physiol. Lung Cell Mol. Physiol. 2014;307:L791–L799. doi: 10.1152/ajplung.00167.2014. [DOI] [PubMed] [Google Scholar]
- 53.Wang Y, et al. Remote ischemic preconditioning protects against liver ischemia-reperfusion injury via heme oxygenase-1-induced autophagy. PLoS ONE. 2014;9:e98834–e98912. doi: 10.1371/journal.pone.0098834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wu H, et al. Astaxanthin as a potential neuroprotective agent for neurological diseases. Mar. Drugs. 2015;13:5750–5766. doi: 10.3390/md13095750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nian L, et al. Astaxanthin suppresses cigarette smoke and lipopolysaccharide-induced airway inflammation through induction of heme oxygenase-1. Cell. Mol. Biol. (Noisy-le-grand) 2019;65:94–99. [PubMed] [Google Scholar]
- 56.Montegudo AE, et al. Pyrrolidine dithiocarbamate improves mortality in a rat model of severe hemorrhage. J. Surg. Res. 2013;179:e149–e155. doi: 10.1016/j.jss.2012.01.006. [DOI] [PubMed] [Google Scholar]
- 57.Zhong X, et al. TAK242 suppresses the TLR4 signaling pathway and ameliorates DCD liver IRI in rats. Mol. Med. Rep. 2019;20:1–10. doi: 10.3892/mmr.2019.10439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Plotnikov EY, et al. Mechanisms of LPS-induced acute kidney injury in neonatal and adult rats. Antioxidants (Basel) 2018;7:105. doi: 10.3390/antiox7080105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Islam MS, Miao L, Yu H, Han Z, Sun H. Ethanol extract of illicium henryi attenuates LPS-induced acute kidney injury in mice via regulating inflammation and oxidative stress. Nutrients. 2019;11:1412–1418. doi: 10.3390/nu11061412. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.