

Abstract
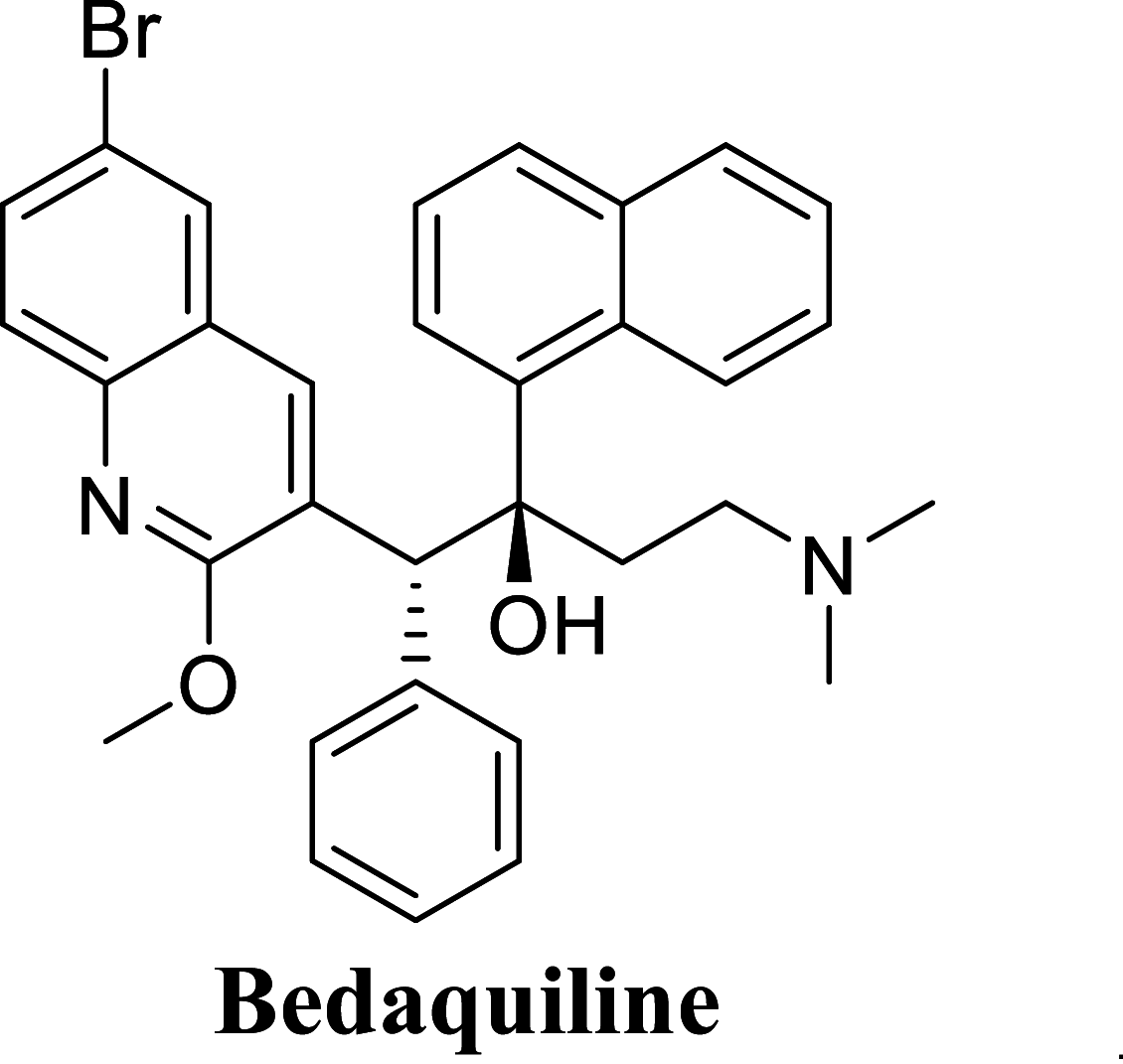
Since the last 4 decades, Bedaquiline has been the first drug discovered as a new kind of anti-tubercular agent and received FDA approval in December 2012 to treat pulmonary multi-drug resistance tuberculosis (MDR-TB). It demonstrates excellent efficacy against MDR-TB by effectively inhibiting mycobacterial ATP synthase. In addition to these apparent assets of Bedaquiline, potential disadvantages of Bedaquiline include inhibition of the hERG (human Ether-à-go-related gene; KCNH2), potassium channel (concurrent risk of cardiac toxicity), and risk of phospholipidosis due to its more lipophilic nature. To assist the effective treatment of MDR-TB, highly active Bedaquiline analogs that display a better safety profile are urgently needed. A structure-based virtual screening approach was used to address the toxicity problems associated with Bedaquiline. Among the virtually screened compound, CID 15947587 had significant docking affinity (− 5.636 kcal/mol) and highest binding free energy (ΔG bind − 85.2703 kcal/mol) towards the Mycobacterial ATP synthase enzyme with insignificant cardiotoxicity and lipophilicity. During MD simulation studies (50 ns), the molecule optimizes its conformation to fit better the active receptor site justifying the binding affinity. The obtained results showed that CID15947587 could be a useful template for further optimizing the MDR-TB inhibitor.
Supplementary Information
The online version contains supplementary material available at 10.1007/s40203-021-00086-x.
Keywords: Bedaquiline, HERG, Docking, MMGBSA, MD simulation
Introduction
Mycobacterium tuberculosis (Mtb, M. tuberculosis), the etiological agent of Tuberculosis (TB), is the world's deadliest infectious disease (Maitra et al. 2016). It is estimated that around 33% of the total population is affected by tuberculosis bacillus, and every year, eight million people develop tuberculosis, which kills around 1.8 million worldwide each year (Shah et al. 2007; Migliori et al. 2007). About 80% of TB cases have been reported in 23 countries, with the highest in Africa and Southeast Asia (Diacon et al. 2012; Mitnick et al. 2008). Additionally, the spread of HIV-infection has made a significant contribution to the re-escalation of TB disease. As the World Health Organization (WHO) indicates, about 33% of the 40 million HIV/AIDS patients are co-infected with TB (https://www.who.int/tb/publications/global_report/en/). The major problem with TB treatment is the occurrence of drug-resistant strains of Mtb (Controlled clinical trial of four short-course (6-month) regimens of chemotherapy for treatment of pulmonary tuberculosis 1974). On top of this, various clinical isolates of Mtb have been reported to be resistant to the first (Isoniazid, Rifampicin) and the several second-line anti-TB drugs (Amikacin, Kanamycin), responsible for multidrug-resistant (MDR) and extensively drug-resistant (XDR) strains (Dooley et al. 2013). Worldwide the number of MDR and XDR-TB cases is more prevalent in different underdeveloped and developing countries. However, few cases of MDR-TB have also been reported recently in developed countries, including several European countries (https://www.who.int/tb/areas-of-work/drug-resistant-tb/global-situation/en/). According to the WHO global tuberculosis report 2020, a total 465, 000 MDR-TB had been reported worldwide in 2019, and the three countries with the highest share of the global burden were India (27%), China (14%), and the Russian Federation (8%) (https://www.who.int/docs/default-source/hq-tuberculosis/global-tuberculosis-report2020/factsheet.pdf?sfvrsn=86820282_2; https://apps.who.int/iris/bitstream/handle/10665/336069/9789240013131-eng.pdf?ua=1). Therefore, to avert an unpreventable threat in the future, the WHO has been terrified to develop novel therapeutic compounds to strengthen drug-resistant TB treatment. A four-drug regimen (isoniazid (INH), rifampin (RIF), pyrazinamide (PZA), and either ethambutol (ETB) or streptomycin (SM)) is the present treatment for drug-susceptible TB, which is administered for a duration of six months (Nath and Ryoo 2013; Saxena et al. 2014). These drugs are generally given in combination for 4–6 months, followed by INH and RIF for the remaining treatment period to eradicate any persistent tubercle bacilli. The actual problem is the occurrence of MDR-TB, which shows resistance towards INH and RIF, and the XDR-TB that exhibits resistance against second-line injectable drugs along with INH and RIF (Seung et al. 2015; Jagadeb et al. 2019). Thus, there is a rising need to discover new effective anti-TB drugs to improve these strains’ treatment. Over the last several years, progress was made in the research and development of new drugs for TB, but only ten anti-TB drugs are in clinical trials so far (Jagadeb et al. 2019). After 40 years period, in December 2012, Bedaquiline (initially known as R207910, then TMC207, SIRTURO®, Janssen pharmaceuticals) is the only drug approved by FDA to treat MDR-TB (Mahajan 2013). The novel drug, Bedaquiline, escapes regular antibiotic’s mode of action of cell membrane penetration and targets the F-ATP synthase of the Mycobacteria tuberculosis on the cellular membrane preventing the production of ATP, enhancing the acidity of the cell, and stopping cellular function (Guillemont et al. 2011). Despite the therapeutic successes of Bedaquiline, it also has several limitations. Bedaquiline shows inhibition of the hERG potassium channels (human Ether-à-go-go-Related Gene; KCNH2), essential for normal cardiac repolarization. Inhibition of the hERG current causes QT interval prolongation resulting in potentially fatal ventricular tachyarrhythmia called Torsade de Pointes (concurrent risk of cardiac toxicity) (Treatment of Patients with MDR-TB 2014; Sanguinetti and Tristani-Firouzi 2006). Bedaquiline also shows a very lipophilic nature (measured log P 7.25), which may contribute to its chance of phospholipidosis, seen at high doses in preclinical models (Mesens et al. 2007). Their high lipophilicity may also contribute to long terminal elimination half-life, leading to tissue over-proportional accumulation at high doses or with daily dosing (Svensson et al. 2015; Patel et al. 2019). Therefore, these are compelling reasons for optimizing the Bedaquiline structure to reduce the toxicity burden of the drug-resistant TB treatment regimen. To address this, we explored the available drug repositories by virtual screening to identify drugs for MDR-TB that have low hERG inhibition and lipophilicity. In this article, we have collected similar analogs of Bedaquiline from the PubChem database and virtually screened for hERG inhibition and lipophilicity, then subjected to molecular docking, molecular dynamics (MD), and Density functional theory (DFT) study to find out the effective drug for MDR-TB.
Structure based virtual screening protocol
Virtual screening is rapidly becoming the primary application of computational docking methods, with many successes in discovering new lead compounds for pharmaceutical development (Cosconati et al. 2010). The idea is to cover an extensive library of available ligands to identify a small subset for purchase and experimental testing. In the present study, we have tried to optimize the Bedaquiline to overcome the toxicity problems associated with the structure-based virtual screening approach. The workflow of the virtual screening campaign is outlined in Fig. 1.
Fig. 1.

Virtual screening protocol
Computer hardware and software
Structure-based virtual screening was performed on HP Z2 G2 TOWER workstation (8-core processor) using the Glide and QuickPro module of Schrodinger on the Linux Ubuntu enterprise version 20.4 as an operating system. The crystal structure of Mycobacterial ATP Synthase was retrieved from the Protein Data Bank (PDB) with the accession code 4V1F. The binding free energies of the protein–ligand complex are processed by the MM-GBSA approach utilizing the Prime, while MD Simulation was performed using the academic Desmond. Single point energy calculations using density functional theory were performed using the Jaguar module of Schrödinger.
Database (ligand) building and toxicity prediction
272 similar analogs of Bedaquiline were acquired from the PubChem database by performing a similarity search option in the database (Library of ligands are shown in supplementary Fig. S1). The structures of the ligands were generated in the CDX format using the tool ChemBio Draw ultra-version 14. These ligands were then converted to the mol format and subjected to ligand preparation by the LigPrep module. LigPrep was performed to desalt and make all possible tautomers and states at pH 7.0 using Epik, definite chiralities were retained, and ligands were minimized using the OPLS_2005 force field. The toxicity of these prepared ligands was predicted by QikProp module. The QikProp is a fast, accurate, easy-to-use absorption, distribution, metabolism, and excretion (ADME) prediction program designed by Professor William L. Jorgensen. QikProp predicts physically noteworthy descriptors and pharmaceutically relevant properties of organic molecules, either individually or in batches (Patel et al. 2018; Vistoli et al. 2008).
Molecular docking study
The protein crystal structure of the Mycobacterial ATP synthase co-crystallized with Bedaquiline (PDB: 4V1F) was obtained from the Protein Data Bank (https://www.rcsb.org/structure/4V1F). Protein preparation was carried out with the Protein Preparation Wizard in Maestro. The protein was prepared after ensuring chemical correctness, assigning bond orders, eliminating water molecules, and adding hydrogens for pH 7.0 using Epik. Prime was used to complete missing side chains and loops, and termini were capped. Using the default constraint of 0.30 Å RMSD and the OPLS 2005 force field, a restrained minimization of the protein structure was performed to complete protein preparation (Schrödinger Release 2008). The binding site was defined around the co-crystallized Bedaquiline, and the receptor grid was prepared around the Bedaquiline. Molecular docking was performed using the Glide ligand docking module in SP (Standard precision) mode, where the receptor grid defined in the receptor grid generation folder was selected for the docking of ligands, which were prepared by LigPrep. The binding conformations were examined to identify critical interactions (Halgren et al. 2004; Friesner et al. 2004).
Prime MM-GBSA binding free energy analysis
The MM-GBSA approach was used to assess binding free energies (ΔGBind) of the selected protein–ligand complexes using the default parameters of the Prime MM-GBSA modules in the Schrödinger software. The Prime MM-GBSA analysis was carried out by the Glide pose viewer file. The MM-GBSA calculations are used to evaluate the relative binding affinity of ligands to the receptor (reported in kcal/mol). The MM-GBSA binding energies are estimates of the free binding energies; the more negative value shows stronger binding (Vijayakumar et al. 2014; Patel et al. 2020a; Casalvieri et al. 2020).
Molecular dynamics simulations
Molecular dynamics (MD) simulations for the top dock protein–ligand complex was carried out using the Desmond program, an explicit solvent MD package along with a fixed OPLS 2005 force field (Desmond Molecular Dynamics System, D. E. Shaw Research, New York, NY, 2018-4 2017). The protein–ligand complex was prepared in protein preparation wizard with the predefined SPC (simple point charge) water model and orthorhombic box shape (size of the box as 10 Å × 10 Å × 10 Å distance). The sodium chloride with the approximately physiological concentration of 0.15 M was placed in 10 Å buffer regions between the protein atoms and the simulation box to set the ionic strength using the system-built option. Minimization jobs were performed to relax the system into a local energy minimization; afterward this model system was submitted to 50 ns MD simulation steps with the OPLS_2005 force field. Noose-Hover chain thermostat algorithm at 300 K, Martyna-Tobias-Klein barostat algorithm at 1.01325 bar, isotropic coupling, Coulombic cutoff at 0.9 nm. The rest of the parameters were default (Patel et al. 2020b; Jin et al. 2020). The trajectories of MD simulations evaluated for ligand-receptor interactions were identified using the Simulation Interaction Diagram (SID) tool.
Density functional theory (DFT) calculations
After completion of the structure-based virtual screening, it is needed to determine the structural behavior of the active compound and to explore how structural orientation, any biological influence part in the structure, and what parameters may force the biological activeness of the molecule. For this reason, single-point energy calculations using DFT were performed to explore the detailed aspects in terms of structure, electronics, and energy states of every atom of compound. Both the virtual screened compound and Bedaquiline were taken into the Jaguar platform in Schrodinger to calculate the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) by using Lee–Yang–Parr correlation functional theory (B3LYP) incorporation of basis set 6-31G* level and hybrid DFT with Beckes 3-parameter exchange potential (Bochevarov et al. 2013; Panwar and Singh 2020). These parameters play a significant role in explaining the magnitude of compounds’ interaction in the binding pocket of Mycobacterial ATP Synthase.
Result and discussion
This study's virtual screening protocol is based on the application of sequential filters to find out a selective Mycobacterial ATP Synthase inhibitor. The workflow of the virtual screening campaign is outlined in Fig. 1.
Very first, 272 analogs of Bedaquiline were acquired from the PubChem database by performing a similarity search option in the database. Cardiotoxicity is one of the adverse effects of Bedaquiline reported in clinical trials because of an increased incidence of QT segment prolongation on electrocardiogram (Treatment of Patients with MDR-TB 2014; Sanguinetti and Tristani-Firouzi 2006).
Therefore, in the beginning, in silico prediction of the hERG inhibition using QikProp has been calculated, and the analogs which were showing less hERG inhibition (QPloghERG < − 7.806) than Bedaquiline has been considered for the next filter (Supplementary Table S1). Phospholipidosis is another problem associated with Bedaquiline due to lipophilicity, leading to tissue over-proportional accumulation at high doses (Guillemont et al. 2011). The potential for excessively proportional tissue accumulation has limited the full exploration of its possible dose range. In general, highly lipophilic drugs are also prone to liver toxicity (Guillemont et al. 2011). Considering the lipophilic characteristic of Bedaquiline, we employed the next filter, Predicted octanol/water partition coefficient (QPlogPo/w). The Bedaquiline analogs, which are showing less QPlogPo/w (Supplementary Table S2) than Bedaquiline (QPlogPo/w < 7.832) subjected to docking study, leaving a total of 80 analogs (Supplementary Table S3).
Molecular docking studies
The molecular docking tool, GLIDE, was used for ligand docking studies into the Mycobacterial ATP synthase enzyme binding pocket. Docking methodology was validated by measuring RMSD of the co-crystalized (internal) ligand and extracted internal ligand of the docked target protein–ligand complex structure, which served as a control docking model as shown in Fig. 2. The docking result showed that glide SP docking evaluated the optimal orientation of the co-crystallized ligand. RMSD value of 1.1254 suggested that the methodology was perfect for predicting the binding affinity for unknown ligands. Top scoring compounds’ docking results are given in Table 1, and the remaining is shown in Supplementary Table S3.
Fig. 2.
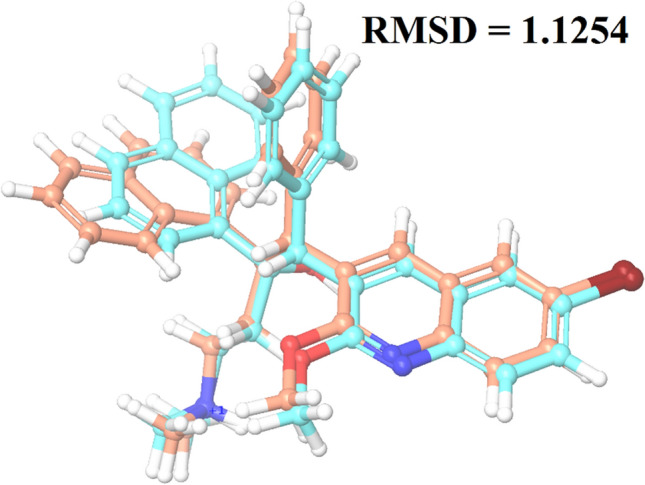
Data obtained in the validation of the molecular docking protocol, the impeccably overlapped conformation of the docked ligand Bedaquiline with respect to its crystallized conformation obtained from the bioactive complex structure; cyan ligand: native ligand; sky blue ligand: docked pose
Table 1.
Glide SP docking score of the top Bedaquiline analogs against Mycobacterial ATP synthase
| Compound code | Docking score | Glide gscore | Glide emodel |
|---|---|---|---|
|
− 5.636 | − 5.64 | − 65.052 |
|
− 5.593 | − 5.597 | − 65.642 |
|
− 5.576 | − 5.577 | − 66.983 |
|
− 5.503 | − 5.497 | − 64.74 |
|
− 5.501 | − 5.505 | − 65.332 |
|
− 5.488 | − 5.491 | − 62.115 |
|
− 5.404 | − 5.475 | − 56.017 |
|
− 5.377 | − 5.379 | − 60.10 |
|
− 5.375 | − 5.378 | − 63.21 |
|
− 5.357 | − 5.361 | − 63.928 |
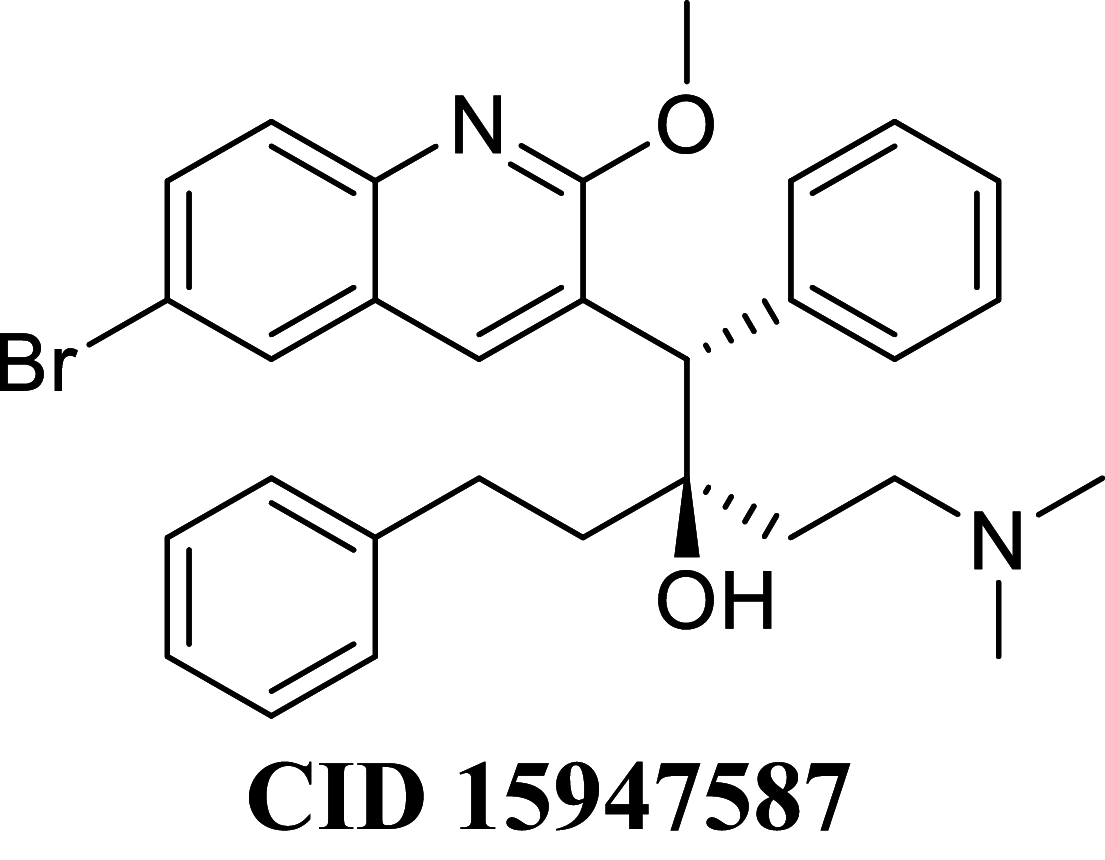
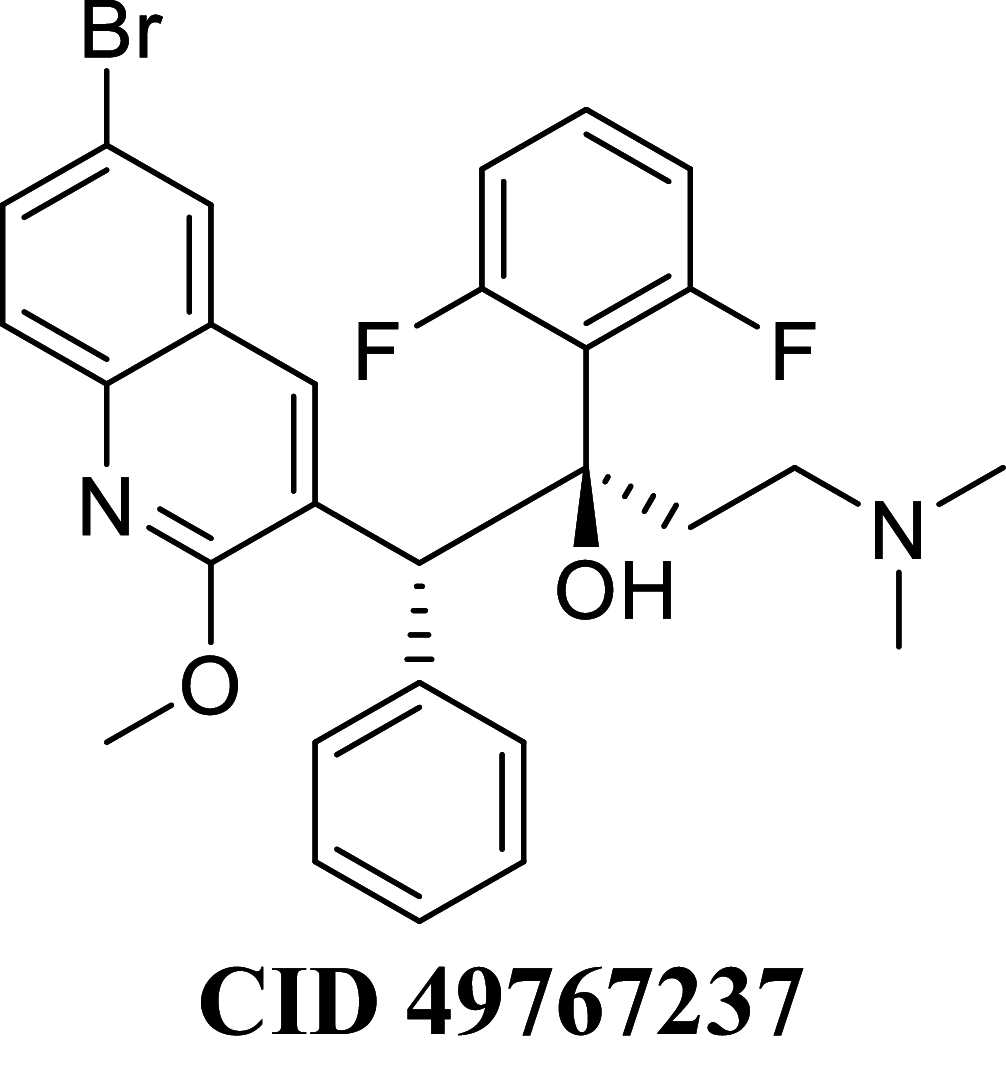
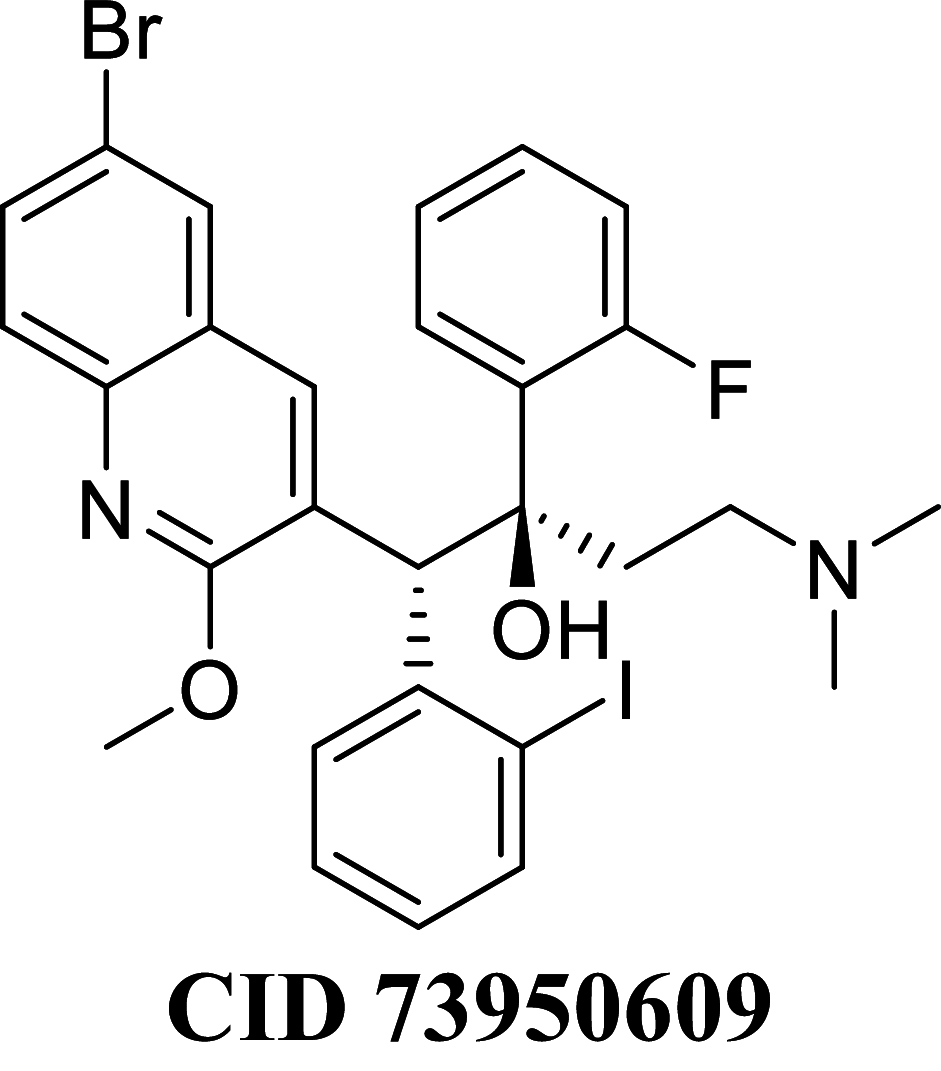
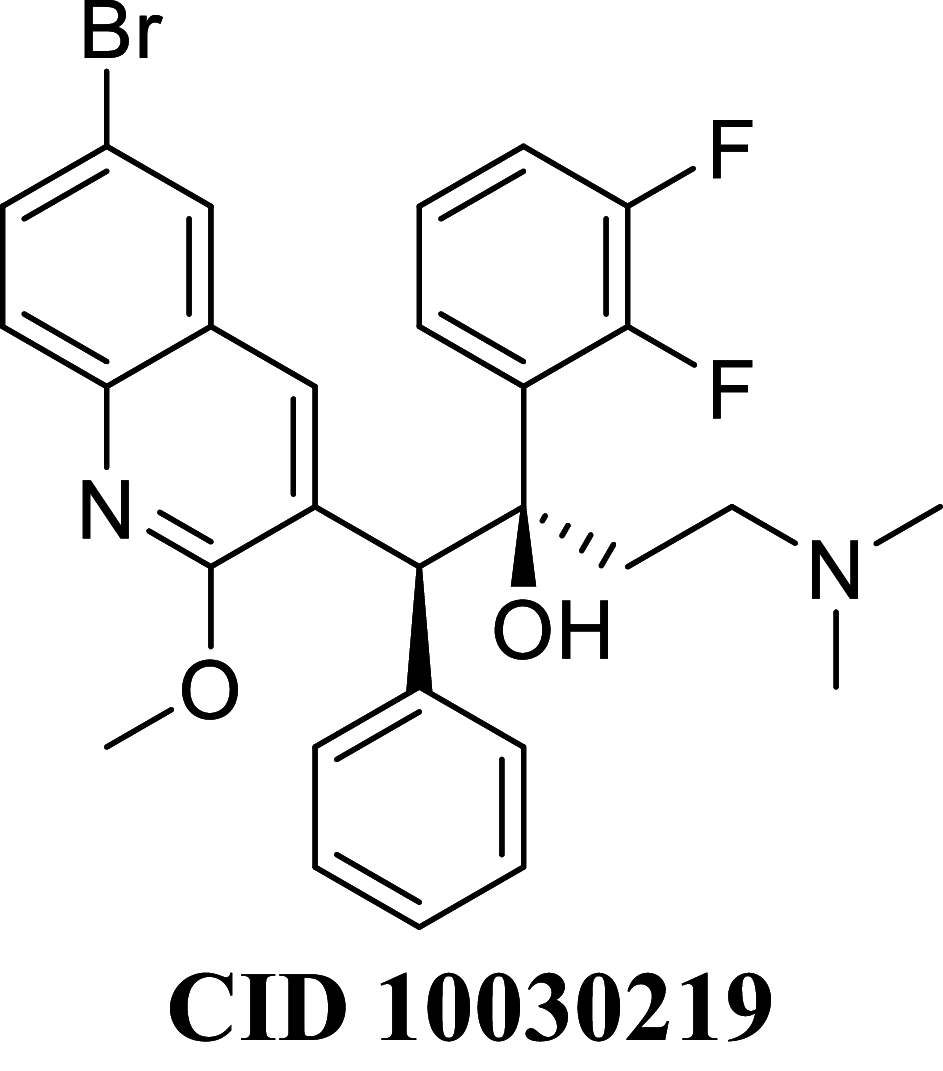
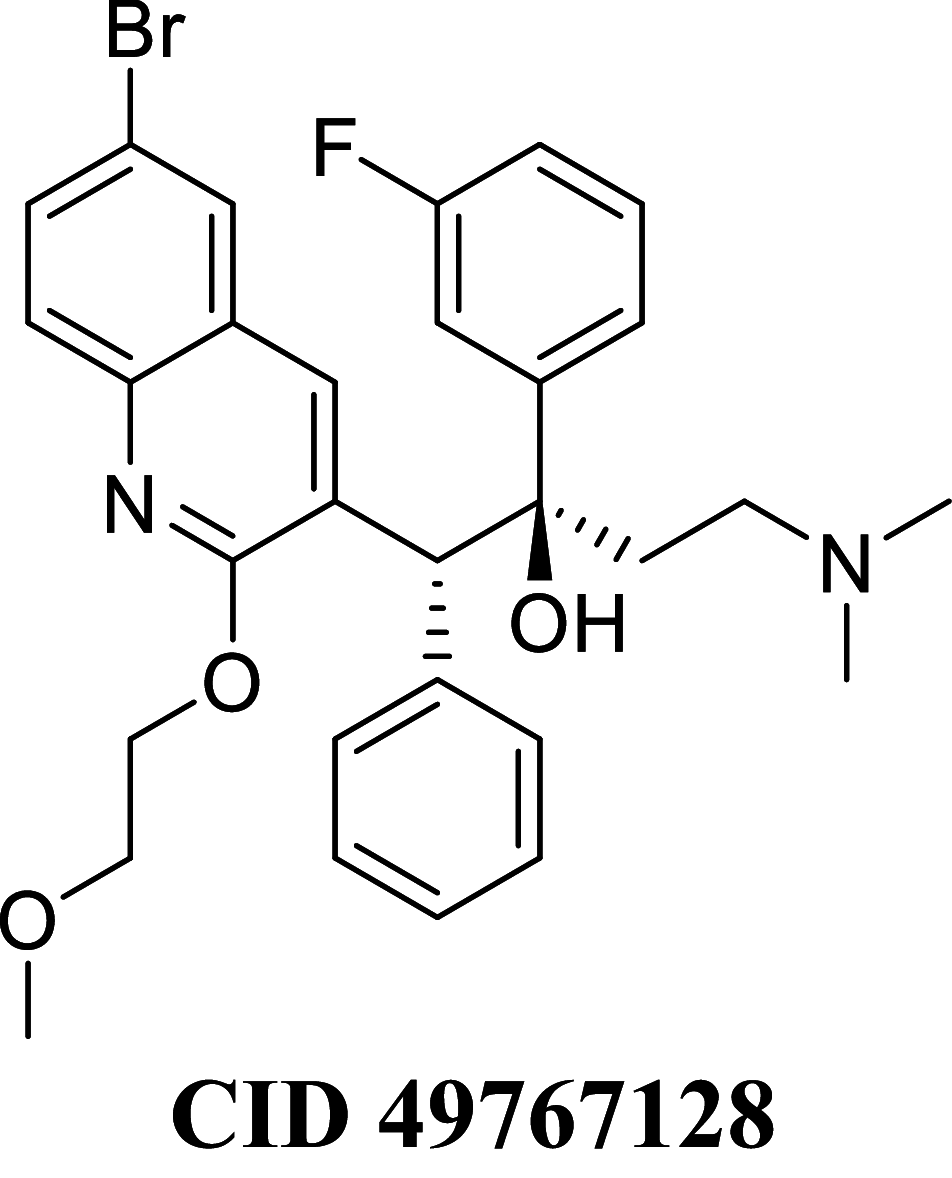
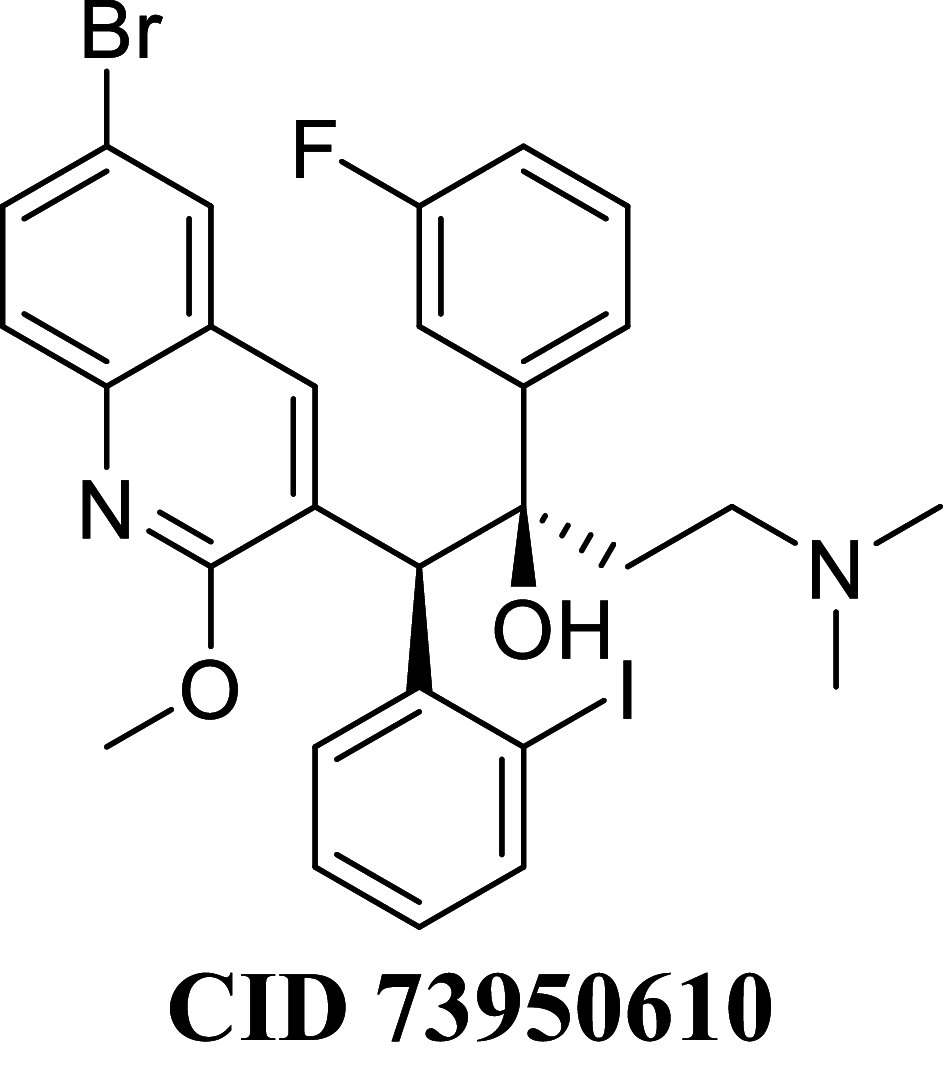
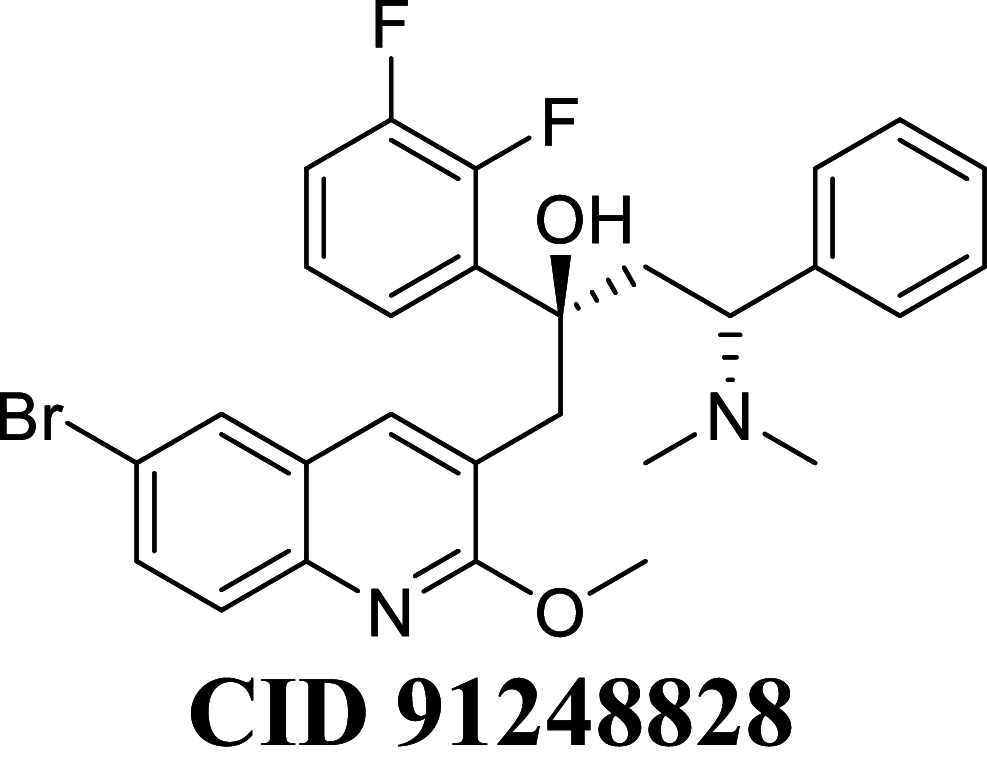
Docking result demonstrated that among the screened Bedaquiline analogs, 09 analogs namely: CID15947587, CID49767237, CID73950609, CID10030219, CID49767128, CID73950610, CID91248828, CID74223217 and CID73950613 were having higher binding potency compared to Bedaquiline (− 5.357 kcal/mol). All these 09 analogs have a similar binding mode of interaction to Bedaquiline. The docking interaction showed that Bedaquiline form pi-pi stacking with Phe69, pi cation with Tyr68, and salt bridge (between terminal tertiary amine and Glu65) (Fig. 3a). The top dock score analogs CID15947587 (Fig. 3b) and CID49767237 (Fig. 3c) were shown similar binding approaches. Docking study revealed that acidic amino acid, i.e., Glutamic acid (Glu65), is crucial for forming hydrogen bonds and salt bridges.
Fig. 3.

Binding-interaction analysis of a Bedaquiline; b CID15947587; c CID49767237 with Mycobacterial ATP synthase (PDB: 41VF) domain
MMGBSA binding free energy analysis
MM-GBSA binding free energy analysis was carried out of the nine potent protein–ligand complexes along with Bedaquiline to assess the affinity of ligands to the target protein receptors. The binding free energies (ΔGBind) evaluated by this method are more efficient than the Glide score values for the assortment of protein–ligand complexes. The primary energy components, such as Coulomb or Electrostatics Interaction energy (ΔGBind Coulomb), Lipophilic Interaction energy (ΔGBind Lipo), Generalized Born electrostatic solvation energy (ΔGSolv-GB) and van der Waals interaction energy (ΔGBind vdW) altogether contribute to the analysis of MM-GBSA-based relative binding affinity. The binding energies and the contributing factors calculated for the protein dock complexes are mentioned in Table 2. Among the nine complexes studied, two complexes showed high binding free energies, namely CID 15947587 (ΔGBind = − 85.2703 kcal/mol) and 49767237(ΔGBind = − 80.9703 kcal/mol) than Bedaquiline. The identified top nine Bedaquiline analogs presented comparatively better docking and binding free energy for the control molecules and therefore represent excellent candidates for further in-vitro investigation (Fig. 4).
Table 2.
Binding free energy components for the protein ligand complexes calculated by MM-GBSA analysis
| Compound code |
MMGBSA (Kcal/mol) | Prime energy | ||||
|---|---|---|---|---|---|---|
| ΔGBind | ΔGCoulomb | ΔGLipo | ΔGSolv_GB | ΔGvdW | ||
| CID 15947587 | − 85.2703 | − 76.5889 | − 47.4356 | 81.2595 | − 43.9773 | − 10,046.1 |
| CID 49767237 | − 80.9703 | − 80.1919 | − 38.356 | 76.3010 | − 37.3592 | − 10,036.4 |
| Bedaquiline | − 80.8048 | − 77.8186 | − 40.9149 | 79.3202 | − 40.7356 | − 10,033.2 |
| CID 49767128 | − 79.8811 | − 79.0659 | − 38.3894 | 78.1032 | − 34.7068 | − 10,053.2 |
| CID 73950609 | − 79.7602 | − 80.8131 | − 38.2541 | 77.9185 | − 37.3034 | − 10,040 |
| CID 10030219 | − 79.2284 | − 80.4851 | − 37.8998 | 78.2522 | − 37.7738 | − 10,035.9 |
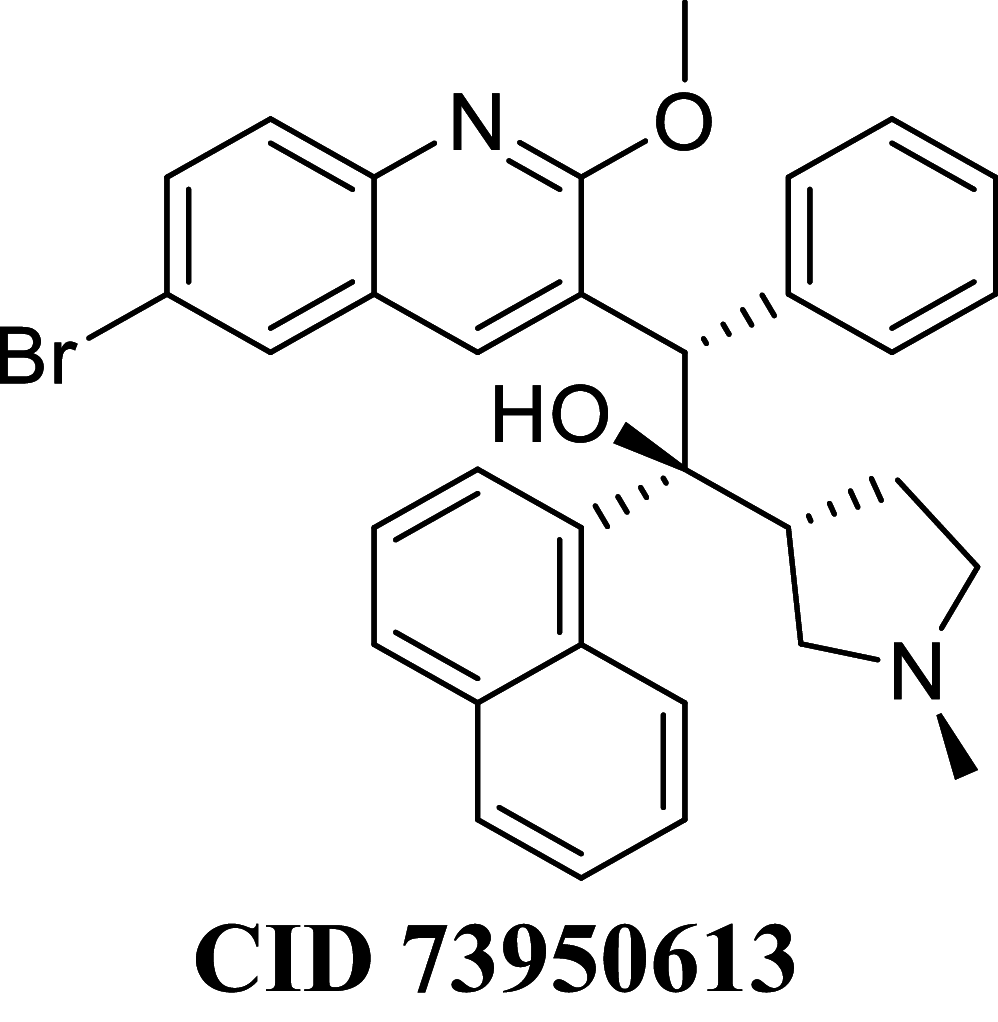
| CID 73950613 | − 78.7324 | − 81.4297 | − 38.8476 | 78.4303 | − 37.7080 | − 10,048 |
| CID 73950610 | − 77.8839 | − 78.2756 | − 38.9479 | 77.4190 | − 38.7299 | − 10,040.4 |
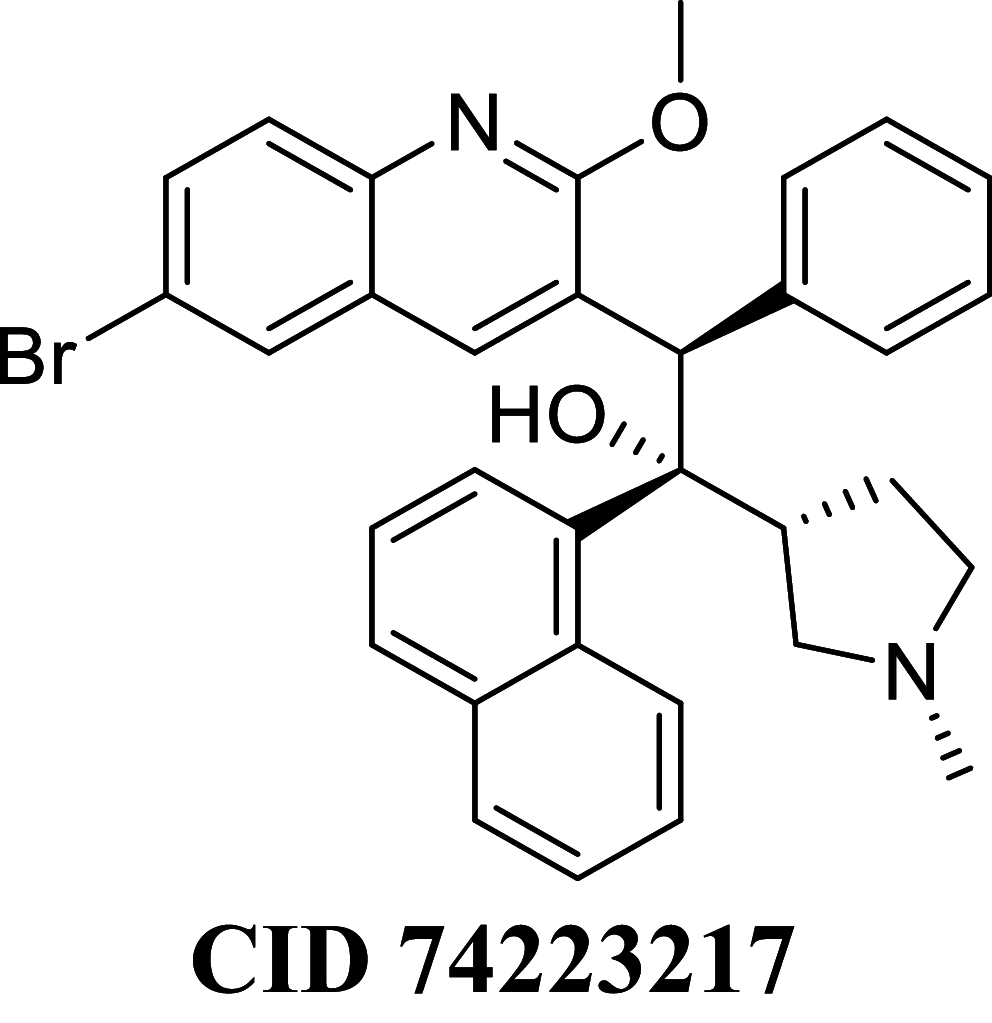
| CID 74223217 | − 71.1490 | − 85.8053 | − 39.2107 | 81.4227 | − 36.0980 | − 9994.4 |
| CID 91248828 | − 70.8466 | − 75.343 | − 39.464 | 76.2293 | − 31.0807 | − 10,037.5 |
ΔGBind: binding free energy; ΔGCoulomb: Coulomb or electrostatics interaction energy; ΔG Lipo: lipophilic interaction energy; ΔGSolv_GB: Generalized born electrostatic solvation energy, ΔGvdW: Van der Waals interaction energy
Fig. 4.

The correlation plot between MM-GBSA values (primary y-axis) and docking score (secondary y-axis) for the control molecule (Bedaquiline) and 9 finalized potential hits
Molecular dynamic simulation study
A molecular docking study was performed using the rigid crystal structure of Mycobacterial ATP synthase. Hence, we have evaluated target receptor and lead compounds interactions in the dynamic behavior using molecular dynamic simulation to obtain the stable binding conformation. As observed from Table 1, CID15947587 was found to have a higher docking score than among screened compounds; hence CID15947587 in complex with Mycobacterial ATP synthase were considered for the molecular dynamic simulation for 50 ns, using simple point charge (SPC) water mode. The root mean square deviation (RMSD), root mean square fluctuation (RMSF), and protein–ligand contacts were analyzed from the MD simulation trajectories to study thermodynamic conformational stability during 50 ns period.
MD simulation trajectories’ RMSD analysis denotes the protein backbone’s stability, when bound with the specific ligand within the dynamic condition. It also provides brief insights into its structural conformation during the MD simulation. The lower RMSD value throughout the MD simulation suggests the higher stability of the protein–ligand complex, whereas a higher RMSD value shows comparatively low stability of the protein–ligand complex (Patel et al. 2020b; Bhowmick et al. 2020). The RMSD graph result of CID15947587 is shown in Fig. 5. Initially, the graph line showed an increasing trend from 0 to 10 ns with RMSD value ranging from 1.2 to 3.6 Å, then showed little stability from 10 to 28 ns. After 30 ns, a promising result was observed, and the graph line was stable till 50 ns. The overall RMSD analysis revealed that fluctuations in a graph during 50 ns simulation are within the standard range of RMSD. Ligand RMSD of 1–3 Å indicates that the reported lead compound bound tightly within the cavity of Mycobacterial ATP synthase enzyme binding pocket.
Fig. 5.

Time-dependent protein–ligand RMSD plot (Angstrom) of the CID15947587 with Mycobacterial ATP synthase enzyme binding pocket
The RMSF value represents the mobility and flexibility of each protein residue during the entire simulation. Greater RMSF values indicate more flexibility during the MD simulation, while the lower value of RMSF reflects the stability of the system (Patel et al. 2020b). CID15947587-mycobacterial ATP synthase complex yielded little fluctuations at Glu65, Tyr50, and Phe51 residue, which is perfectly acceptable. The RMSF plot (Fig. 6) confirmed that the ligand contacted residues had less fluctuation.
Fig. 6.

Time-dependent protein RMSF plot (Angstrom) of the CID 15947587 with mycobacterial ATP synthase enzyme binding pocket
Protein–ligand interactions can be monitored throughout the simulation. Residue Glu65 played a crucial role in forming a hydrogen bond with CID15947587, Tyr68 and Phe74 formed pi cation and pi-pi stacking with CID15947587, respectively. Leu70, Phe74, Pro83, Met21, Ala66, Tyr68, Phe69, and Leu72 were involved in hydrophobic interactions with the ligand. The ionic interaction has also been observed between the tertiary amine of the CID15947587 and Glu65 residue, as shown in Fig. 7. Comparing Fig. 2 with Fig. 7, the important hydrogen bond, which has been shown in the initial docking models did not change during the MD simulations.
Fig. 7.

Simulation interactions diagram, 2D binding interaction of CID 15947587 with mycobacterial ATP synthase enzyme binding pocket along with bar diagram
Additionally, Fig. 8 shows the total number of specific contacts protein makes with ligand throughout the trajectory. The contribution of amino acids in each trajectory frame of 50 ns MD simulation as shown in the bottom panel of Fig. 8, which represent the number of contacts and their density (the darker shade of orange shows more than one contact in that frame). Key interaction seen during each frame was with Glu65, which was consistent during the complete simulation process. Other interactions were also found with Ile70, Phe74, Tyr68, Phe69, and Leu72, which were not consistent during the simulation.
Fig. 8.

Protein–ligand contacts showing good contacts (darker shades) with the amino acid residues over 50 ns time period of simulation of the CID 15947587 with mycobacterial ATP synthase enzyme binding pocket
The five molecular properties of ligand (ligand RMSD, the radius of gyration [rGyr], Molecular surface area [MolSA], solvent accessible surface area [SASA], and polar surface area [PSA]) were also studied to check the stability of the CID5947587 in the Mycobacterial ATP synthase enzyme binding pocket as shown in Fig. 9. Ligand RMSD suggests root mean square deviation of a ligand with respect to the reference conformation (usually, the initial frame is used as the reference, and it is regarded as time t = 0).
Fig. 9.

Ligand properties during 50 ns simulations for CID15947587: a ligand RMSD, root mean square deviation, b radius of gyration (rGyr), c molecular surface area (MolSA), d solvent accessible surface area (SASA), and e polar surface area (PSA)
The RMSD values of CID 15947587 were below 1.5 Å. The radius of gyration is used to evaluate the ‘extendedness’ of a ligand and is equivalent to its principal moment of inertia; the radius of gyration throughout the 50 ns simulation remained constant and ranged from 4.65 to 4.95 Å for CID15947587. MolSA (440–470 Å2), SASA (350–450 Å2), and PSA (32–48 Å2) plot also suggested the stability of CID15947587 during the simulation process.
DFT analysis
Frontier molecular orbitals of compounds specify a crucial role of charge-transfer interactions with the binding site of mycobacterial ATP synthase. Both the active compound (CID15947587) and Bedaquiline were examined to understand the electronic and energetic states. The HOMO is the orbital of the highest energy (electron-rich), so energetically, it is the easiest to remove the electron from the orbital, in contrast, LUMO is the lowest-lying orbital that is empty (lack of the electron), so energetically it is the easiest to add more electrons into these orbital (Jordaan et al. 2020; Amala et al. 2019; Ganesan et al. 2020).
For CID15947587 and Bedaquiline, in which HOMO’s outcome ranges from − 0.25837 to − 0.30843 and LUMO − 0.14441 to − 0.14673 and HOMO and LUMO’s energy gap (HOMO–LUMO gap/HLG) was 0.11164 and 0.16402 for CID15947587 and Bedaquiline as tabularized in Table 3. The position of HOMO–LUMO orbitals of CID 15947587 (Fig. 10a) suggests that the hydrophobic region of the molecule alters the topology of HOMO–LUMO orbitals, and also energies are localized. HOMO and LUMO cover the quinoline ring of the hit molecule. Bedaquiline was found to have HOMO orbital mainly covering naphthalene ring and LUMO orbital covering quinoline ring (Fig. 10b). The presence of negative values of HOMO–LUMO for both compounds implies good stability and important to form stable ligand–protein complex. Further, the energy gap represents an effective tool to determine the most active compounds and their way of mechanism. Lesser energy gap between the HOMO and LUMO energies has a considerable impact on the intermolecular charge transfer and bioactivity of compounds (Amala et al. 2019; Murray and Politzer 2011). Consequently, a more energy gap observed in the compounds negatively affects the electron to move from the HOMO to the LUMO, which subsequently led to a weak affinity of the inhibitor for Mycobacterial ATP synthase. Compared with Bedaquiline (0.16402 eV), the HLG value of CID 15947587 (0.11164 eV) is lower. Thus, CID15947587 is most than Bedaquiline.
Table 3.
Single point energy (Jaguar) output value of frontier orbital energies
| Compounds | HOMO (eV) | LUMO (eV) | HLG (eV) |
|---|---|---|---|
| CID 15947587 | − 0.25837 | − 0.14673 | 0.11164 |
| Bedaquiline | − 0.30843 | − 0.14441 | 0.16402 |
HLG HOMO–LUMO gap
Fig. 10.

Plots of the HOMO, LUMO and HLG of CID 15947587 (a) and Bedaquiline (b)
Conclusion
In summary, we performed a successful structure-based virtual screening on 272 structurally similar analogs of Bedaquiline collected from the PubChem database. Bedaquiline is the first MDR-TB inhibitor since 1971, having selective Mycobacterial synthase inhibitory activity with the adverse effect of cardiotoxicity and phospholipidosis. In order to assist the effective treatment of MDR-TB, highly active Bedaquiline analogs that display a better safety profile are urgently needed. Among 272 structurally similar analogs of Bedaquiline, 78 analogs were found to have insignificant cardiotoxicity and lipophilicity, which were subjected to molecular docking, binding free energy calculation, molecular dynamics simulation, and DFT analysis. When we examined the affinity of 78 compounds against Mycobacterial ATP synthase, we observed that nine compounds with CID15947587, CID49767237, CID49767128, CID73950609, CID10030219, CID73950613, CID73950610, CID74223217, and CID91248828 showed significant docking score and binding free energy. Among the virtually screened compounds CID 73950609, CID 73950610 and CID 91248828 were novel for the anti-mycobacterial activity. From identified hits, CID15947587 were selected for MD simulation and DFT calculations. MD simulation suggested that the complex was stable for 50 ns. The HOMO–LUMO orbital energy gap showed that CID 15947587 might be considered as a lead compound for further development in this field as MDR-TB Mycobacterial ATP synthase inhibitor.
Supplementary Information
Below is the link to the electronic supplementary material.
Author contributions
Author IA and HP was involved in the idea generation and performing the computational chemistry work. HJ, YS have contributed in the molecular dynamic simulation study. RG and VJ contributed for the manuscript writing and grammatical check.
Funding
The authors would like to thank ‘Indian Council of Medical Research (ICMR) Ministry of Health and Family Welfare, Department of Health Research Govt. of India’ (Grant no. ISRM/12(11)/2019) for funding the project.
Declarations
Conflict of interest
All authors declare no actual or potential conflict of interest including any financial, personal, or other relationships with other people or organizations.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- (1974) Controlled clinical trial of four short-course (6-month) regimens of chemotherapy for treatment of pulmonary tuberculosis. Lancet 2(7889):1100–1106 [PubMed]
- (2017) Desmond Molecular Dynamics System, D. E. Shaw Research, New York, NY, 2018-4. Maestro-Desmond Interoperability Tools, Schrödinger, New York, NY, 2018-4
- Amala M, Rajamanikandan S, Prabhu D, Surekha K, Jeyakanthan J. Identification of anti-filarial leads against aspartate semialdehyde dehydrogenase of Wolbachia endosymbiont of Brugia malayi: combined molecular docking and molecular dynamics approaches. J Biomol Struct Dyn. 2019;37(2):394–410. doi: 10.1080/07391102.2018.1427633. [DOI] [PubMed] [Google Scholar]
- Bhowmick S, AlFaris NA, AlTamimi JZ, AlOthman ZA, Aldayel TS, Wabaidur SM, Islam MA. Screening and analysis of bioactive food compounds for modulating the CDK2 protein for cell cycle arrest: multi-cheminformatics approaches for anticancer therapeutics. J Mol Struct. 2020 doi: 10.1016/j.molstruc.2020.128316. [DOI] [Google Scholar]
- Bochevarov AD, Harder E, Hughes TF, Greenwood JR, Braden DA, Philipp DM, Rinaldo D, Hall MD, Zhang J, Friesner RA. Jaguar: a high-performance quantum chemistry software program with strengths in life and materials sciences. Int J Quantum Chem. 2013;113(18):2110–2142. doi: 10.1002/qua.24481. [DOI] [Google Scholar]
- Casalvieri KA, Matheson CJ, Backos DS, Reigan P. Molecular docking of substituted pteridinones and pyrimidines to the ATP-binding site of the N-terminal domain of RSK2 and associated MM/GBSA and molecular field datasets. Data Brief. 2020;29:105347. doi: 10.1016/j.dib.2020.105347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosconati S, Forli S, Perryman AL, Harris R, Goodsell DS, Olson AJ. Virtual screening with AutoDock: theory and practice. Expert Opin Drug Discov. 2010;5(6):597–607. doi: 10.1517/17460441.2010.484460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diacon AH, Donald PR, Pym A, Grobusch M, Patientia RF, Mahanyele R, Bantubani N, Narasimooloo R, De Marez T, van Heeswijk R, Lounis N, Meyvisch P, Andries K, McNeeley DF. Randomized pilot trial of eight weeks of bedaquiline (TMC207) treatment for multidrug-resistant tuberculosis: long-term outcome, tolerability, and effect on emergence of drug resistance. Antimicrob Agents Chemother. 2012;56(6):3271–3276. doi: 10.1128/AAC.06126-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dooley KE, Nuermberger EL, Diacon AH. Pipeline of drugs for related diseases: tuberculosis. Curr Opin HIV AIDS. 2013;6:579–585. doi: 10.1097/COH.0000000000000009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE, Francis P, Shenkin PS. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem. 2004;47(7):1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- Ganesan MS, Raja KK, Murugesan S, Kumar BK, Rajagopal G, Thirunavukkarasu S. Synthesis, biological evaluation, molecular docking, molecular dynamics and DFT studies of quinolone–fluoroproline amide hybrids. J Mol Struct. 2020 doi: 10.1016/j.molstruc.2020.128360. [DOI] [Google Scholar]
- Guillemont J, Meyer C, Poncelet A, Bourdrez X, Andries K. Diarylquinolines, synthesis pathways and quantitative structure–activity relationship studies leading to the discovery of TMC207. Future Med Chem. 2011;3(11):1345–1360. doi: 10.4155/fmc.11.79. [DOI] [PubMed] [Google Scholar]
- Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT, Banks JL. Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J Med Chem. 2004;47(7):1750–1759. doi: 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- https://apps.who.int/iris/bitstream/handle/10665/336069/9789240013131-eng.pdf?ua=1. Assessed on 7/11/2020
- https://www.rcsb.org/structure/4V1F
- https://www.who.int/docs/default-source/hq-tuberculosis/global-tuberculosis-report2020/factsheet.pdf?sfvrsn=86820282_2
- https://www.who.int/tb/areas-of-work/drug-resistant-tb/global-situation/en/
- https://www.who.int/tb/publications/global_report/en/
- Jagadeb M, Rath SN, Sonawane A. In silico discovery of potential drug molecules to improve the treatment of isoniazid-resistant Mycobacterium tuberculosis. J Biomol Struct Dyn. 2019;37(13):3388–3398. doi: 10.1080/07391102.2018.1515116. [DOI] [PubMed] [Google Scholar]
- Jin Z, Wang Y, Yu XF, Tan QQ, Liang SS, Li T, Zhang H, Shaw PC, Wang J, Hu C. Structure-based virtual screening of influenza virus RNA polymerase inhibitors from natural compounds: molecular dynamics simulation and MM-GBSA calculation. Comput Biol Chem. 2020;85:107241. doi: 10.1016/j.compbiolchem.2020.107241. [DOI] [PubMed] [Google Scholar]
- Jordaan MA, Ebenezer O, Damoyi N, Shapi M. Virtual screening, molecular docking studies and DFT calculations of FDA approved compounds similar to the non-nucleoside reverse transcriptase inhibitor (NNRTI) efavirenz. Heliyon. 2020;6(8):e04642. doi: 10.1016/j.heliyon.2020.e04642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahajan R. Bedaquiline: first FDA-approved tuberculosis drug in 40 years. Int J Appl Basic Med Res. 2013;3(1):1–2. doi: 10.4103/2229-516X.112228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maitra A, Bates S, Shaik M, Evangelopoulos D, Abubakar I, McHugh TD, Lipman M, Bhakta S. Repurposing drugs for treatment of tuberculosis: a role for non-steroidal anti-inflammatory drugs. Br Med Bull. 2016;118(1):138–148. doi: 10.1093/bmb/ldw019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesens N, Verbeeck J, Rouan M, Vanparys P (2007) Elucidating the role of M2 in the preclinical safety profile of TMC207. Abstract on the 38th Union World Conference on Lung Health, Cape Town South Africa
- Migliori GB, Besozzi G, Girardi E, Kliiman K, Lange C, Toungoussova OS, Ferrara G, Cirillo DM, Gori A, Matteelli A, Spanevello A, Codecasa LR, Raviglione MC, SMIRA/TBNET Study Group Clinical and operational value of the extensively drug-resistant tuberculosis definition. Eur Respir J. 2007;30(4):623–626. doi: 10.1183/09031936.00077307. [DOI] [PubMed] [Google Scholar]
- Mitnick CD, Shin SS, Seung KJ, Rich ML, Atwood SS, Furin JJ, Fitzmaurice GM, Alcantara Viru FA, Appleton SC, Bayona JN, Bonilla CA, Chalco K, Choi S, Franke MF, Fraser HS, Guerra D, Hurtado RM, Jazayeri D, Joseph K, Llaro K, Mestanza L, Mukherjee JS, Muñoz M, Palacios E, Sanchez E, Sloutsky A, Becerra MC. Comprehensive treatment of extensively drug-resistant tuberculosis. N Engl J Med. 2008;359(6):563–574. doi: 10.1056/NEJMoa0800106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray JS, Politzer P. The electrostatic potential: an overview. Wiley Interdiscip Rev Comput Mol Sci. 2011;1(2):153–163. doi: 10.1002/wcms.19. [DOI] [Google Scholar]
- Nath H, Ryoo S. First- and second-line drugs and drug resistance. Tuberc Curr Issues Diagn Manag. 2013 doi: 10.5772/54960. [DOI] [Google Scholar]
- Panwar U, Singh SK. Atom-based 3D-QSAR, molecular docking, DFT, and simulation studies of acylhydrazone, hydrazine, and diazene derivatives as IN-LEDGF/p75 inhibitors. Struct Chem. 2020 doi: 10.1007/s11224-020-01628-3. [DOI] [Google Scholar]
- Patel H, Dhangar K, Sonawane Y, Surana S, Karpoormath R, Thapliyal N, Jagtap R. In search of selective 11β-HSD type 1 inhibitors without nephrotoxicity: an approach to resolve the metabolic syndrome by virtual based screening. Arab J Chem. 2018;11(2):221–232. doi: 10.1016/j.arabjc.2015.08.003. [DOI] [Google Scholar]
- Patel H, Pawara R, Pawara K, Ahmed F, Shirkhedkar A, Surana S. A structural insight of bedaquiline for the cardiotoxicity and hepatotoxicity. Tuberculosis. 2019;117:79–84. doi: 10.1016/j.tube.2019.06.005. [DOI] [PubMed] [Google Scholar]
- Patel HM, Shaikh M, Ahmad I, Lokwani D, Surana SJ. BREED based de novo hybridization approach: generating novel T790M/C797S-EGFR tyrosine kinase inhibitors to overcome the problem of mutation and resistance in non-small cell lung cancer (NSCLC) J Biomol Struct Dyn. 2020 doi: 10.1080/07391102.2020.1754918. [DOI] [PubMed] [Google Scholar]
- Patel HM, Ahmad I, Pawara R, Shaikh M, Surana SJ. J Biomol Struct Dyn. 2020 doi: 10.1080/07391102.2020.1734092. [DOI] [PubMed] [Google Scholar]
- Schrödinger Release . Protein preparation wizard. New York: Epik Schrödinger, LLC; 2008. [Google Scholar]
- Sanguinetti MC, Tristani-Firouzi M. hERG potassium channels and cardiac arrhythmia. Nature. 2006;440(7083):463–469. doi: 10.1038/nature04710. [DOI] [PubMed] [Google Scholar]
- Saxena S, Renuka J, Yogeeswari P, Sriram D. Discovery of novel mycobacterial DNA gyrase B inhibitors: in silico and in vitro biological evaluation. Mol Inform. 2014;33(9):597–609. doi: 10.1002/minf.201400058. [DOI] [PubMed] [Google Scholar]
- Seung KJ, Keshavjee S, Rich ML. Multidrug-resistant tuberculosis and extensively drug-resistant tuberculosis. Cold Spring Harb Perspect Med. 2015;5(9):a017863. doi: 10.1101/cshperspect.a017863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah NS, Wright A, Bai GH, Barrera L, Boulahbal F, Martín-Casabona N, Drobniewski F, Gilpin C, Havelková M, Lepe R, Lumb R, Metchock B, Portaels F, Rodrigues M, Rüsch-Gerdes S, Van Deun A, Vincent V, Laserson K, Wells C, Cegielski JP. Worldwide emergence of extensively drug-resistant tuberculosis. Emerg Infect Dis. 2007;13(3):380–387. doi: 10.3201/eid1303.061400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svensson EM, Murray S, Karlsson MO, Dooley KE. Rifampicin and rifapentine significantly reduce concentrations of bedaquiline, a new anti-TB drug. J Antimicrob Chemother. 2015;70(4):1106–1114. doi: 10.1093/jac/dku504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treatment of Patients with MDR-TB (2014) Briefing document, TMC 207 (bedaquiline). Anti-infective Drugs Advisory Committee, NDA pp 204–384
- Vijayakumar B, Parasuraman S, Raveendran R, Velmurugan D. Identification of natural inhibitors against angiotensin I converting enzyme for cardiac safety using induced fit docking and MM-GBSA studies. Pharmacogn Mag. 2014;10(Suppl 3):S639–S644. doi: 10.4103/0973-1296.139809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vistoli G, Pedretti A, Testa B. Assessing drug-likeness—what are we missing? Drug Discov Today. 2008;13(7–8):285–294. doi: 10.1016/j.drudis.2007.11.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.