

Abstract
Although autophagy is a type of programmed cell death, it is also essential for cell survival upon tolerable level of various stress events. For the cell to respond adequately to an external and/or internal stimulus induced by cellular stress, autophagy must be controlled in a highly regulated manner. By using systems biology techniques, here we explore the dynamical features of autophagy induction. We propose that the switch-like characteristic of autophagy induction is achieved by a control network, containing essential feedback loops of four components, so-called autophagy inducer, autophagy controller, mTORC1 and autophagy executor, respectively. We show how an autophagy inducer is capable to turn on autophagy in a cellular stress-specific way. The autophagy controller acts as a molecular switch and not only promotes autophagy but also blocks the permanent hyperactivation of the process via downregulating the autophagy inducer. In this theoretical analysis, we explore in detail the properties of all four proposed controlling elements and their connections. Here we also prove that the kinetic features of this control network can be considered accurate in various stress processes (such as starvation, endoplasmic reticulum stress and oxidative stress), even if the exact components may be different. The robust response of the resulting control network is essential during cellular stress.
Subject terms: Biochemistry, Molecular biology
Introduction
The Greek word ‘autophagy’ means ‘self-eating’, referring to the ability of the cells to digest their own components with respect to various external and internal signals. Basal autophagy is observed even under physiological conditions; however, this process gets more efficient upon increasing cellular stress level1–3. Traditionally, autophagy was classified as a cell death mechanism4; however, many scientific results have been revealed that autophagy also has an essential role in cellular survival upon various stress events (such as starvation or endoplasmic reticulum (ER) stress)5,6. These data clearly suggest that the crucial function of autophagy is to maintain cellular homoeostasis, while excessive level of permanent autophagy can result in cell death5,7–9.
Due to the essential role of autophagy in regulating cellular homoeostasis and stress response, both the induction and the downregulation of the process are tightly controlled10. One of the most important elements of the process is unc51-like autophagy activating kinase 1/2 (ULK1/2), the mammalian homologue of yeast Atg111,12. ULK1/2 controls the early stage of autophagy via forming a so-called autophagy induction complex with ATG13, ATG101 and FIP20013–16. This complex can phosphorylate Beclin1, the mammalian homologue of yeast Atg617. Beclin1 forms a multiprotein complex with other molecules (such as VPS34, ATG14 and AMBRA1), to enhance the formation of the double-membrane structure (so-called isolation membrane) to engulf cytoplasmic material for autophagosome formation10,18. Although the molecular mechanism of autophagy induction seems to be universal, the process also has stress-specific regulators upon various stress events (i.e., starvation, oxidative exposure, and ER stress).
Aminoacid- or glucose-deprivation-induced cellular stress is tightly controlled by both mammalian target of rapamycin (mTOR) and AMP-protein kinase (AMPK)19–21. mTOR, when in a complex with other proteins (such as Raptor, MLST8, PRAS40 and Deptor), called mTORC1 is the master regulator of cellular growth and metabolism22. AMPK is a heterotrimeric protein complex and it has an essential role in maintaining energy homoeostasis by sensing the change of cellular AMP/ATP ratio19. mTORC1 inhibits autophagy under nutrient-rich conditions, meanwhile AMPK promotes the autophagy upon starvation23. The precise crosstalk between mTORC1 and AMPK is achieved via a double-negative feedback loop24,25. In addition, both kinases regulate ULK1/2 directly. mTORC1-dependent phosphorylation of ULK1/2 results in its inactivation, whereas AMPK is able to induce ULK1/223,26. Interestingly, ULK1/2 kinase inhibits both AMPK and mTORC1 via phosphorylation, generating negative and double-negative feedback loops in the control network23,27–30.
The nuclear factor erythroid 2-related factor 2 (NRF2) has a key role to enable cell adaptation to oxidative stress by promoting the transcription of more than 2000, mainly cytoprotective genes31–33. NRF2 is bound to KEAP1 into an inactive complex under physiological conditions; however, p62 (also known as SQSMT1; sequestosome) quickly gets activated upon oxidative stress34,35. Active p62 has a high binding affinity to KEAP1, therefore enhancing the dissociation of active NRF2 from KEAP136–38. Besides, p62 targets proteins to be transferred to autophagosome and induces their autophagy-dependent degradation34,39. Moreover, NRF2 promotes the expression of many autophagy genes, such as ATG3, ATG5, ATG7, p62 and GABARAPL1 upon oxidative stress40. Recently, we have also shown the regulatory connection between AMPK and NRF2 upon oxidative stress41. Although AMPK has a transient activation followed by NRF2 induction during oxidative stress, we found that NRF2 deficiency resulted in a permanent activation of AMPK. Our results show that NRF2 is essential to downregulate autophagy via repressing AMPK transcription upon prolonged oxidative stress41.
Interestingly, ER stress induced by harmful external and internal effects (such as oxidative agents, accumulation of not properly folded proteins) also immediately induces the formation of autophagosomes42. The ER stress response mechanism turns on a complex network of signalling pathways, called unfolded protein response (UPR). UPR has three well-defined branches controlled by ER membrane-associated proteins, called IRE1 (inositol requiring 1), PERK (PKR-like ER kinase) and ATF6 (activating transcription factor 6)43. Although both IRE1 and ATF6 mainly induce UPR target genes (such as chaperones), the key role of PERK pathway is to block the protein translation43. According to the level of ER stress, each branches of UPR are able to enhance autophagy44. It has shown that tolerable ER stress results in autophagy induction to promote cellular survival, but excessive level of ER stress leads to transient autophagy followed by apoptotic cell death45. With systems biology methods, we have also claimed that the feedback loops between the branches of UPR are crucial to the proper cellular life-and-death decision upon ER stress46,47.
Although many biologists are focusing on the mechanism of autophagy induced by various cellular stress events, the dynamical features of the regulatory network of cellular stress-specific response mechanism have not been explored yet. By using systems biology techniques, here we present a general model of autophagy induction by focusing on the key elements and feedback loops. To give a qualitative description about the control network, we studied both the induction and the downregulation of autophagy upon cellular stress. This approach is able to analyse the dynamical characteristic of the control network; therefore, it can result in medically relevant observations and results (e.g., disease-specific drug targets or biomarkers for autophagy malfunction).
Materials and methods
A control network with feedback loops can describe a dynamic autophagy regulation system
Mathematical models are useful to understand the precise molecular mechanisms that control important aspects of cell physiology, such as cell growth and division or cellular life-and-death decision48,49. The theoretical modelling of a biological system can give a proper directionality to molecular biological experiments by giving a qualitative description about the dynamical characteristic of the cellular regulatory networks.
Our analysis mainly focuses on the kinetic features of autophagy induction upon various cellular stress events, such as starvation, ER stress or oxidative exposure. We have thoroughly studied more than 100 scientific papers from the life science and medical fields to build a control network model of autophagy-dependent survival. For the references of the publications used to build the model, see Supplementary Table 1. According to the already published scientific data, we propose that regulated autophagy induction can be described by a wiring diagram of four different regulators, called autophagy inducer (AUIN), autophagy controller (AUCO), mTORC1 and autophagy executors (AUEX), respectively (Fig. 1). The biological evidence suggests that AUIN is able to promote autophagy via indirect upregulation of AUEX by both upregulating AUCO and downregulating mTORC1. Corresponding to the already published data, we claim that mTORC1 prevents the induction of AUIN and AUCO. The common features of AUCO are as follows: (1) inhibits mTORC1, (2) inhibits AUIN and (3) promotes AUEX. Based on experimental data, we propose when AUEX is active, the autophagy-dependent cell survival is turned on. As AUCO can also inhibit both mTORC1 and AUIN, two negative feedback loops (i.e., AUCO ┤ AUIN − > AUCO and AUCO ┤ mTORC1 ┤ AUIN − > AUCO) are generated. Both AUIN and AUCO promote AUEX referring to their importance in autophagy induction.
Fig. 1. The comprehensive wiring diagram of autophagy induction upon cellular stress.
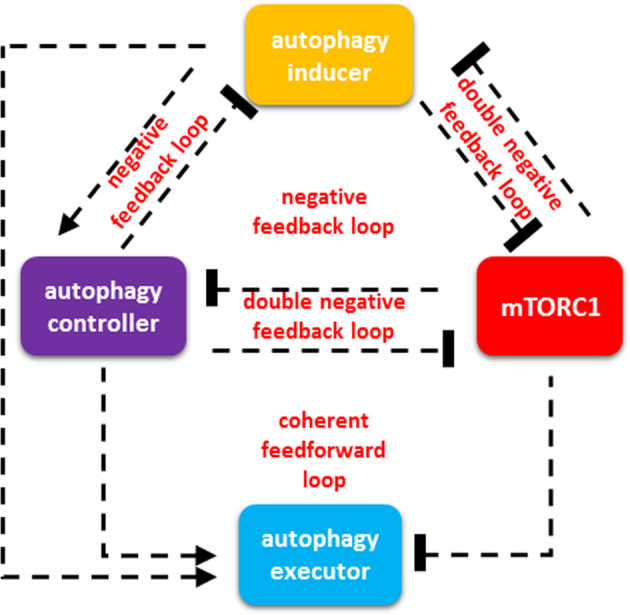
A general wiring diagram to describe the precise mechanism of autophagy induction. The regulatory elements and their connections of life-and-death decision when the autophagy inducers (AUIN), the autophagy controllers (AUCO), the autophagy executors (AUEX) and the elements of mTORC1 pathway are grouped together in isolated orange, purple, blue and red boxes, respectively. Dashed lines show how the molecules can influence each other. Blocked end lines denote inhibition.
Each component has an active and an inactive form in the model. The cellular stress is used as an input parameter in the control network. For details about the codes and software used for simulations, see the Supplementary Information.
The question immediately arises, which components of the control network are named as AUIN, AUCO and AUEX. Based on data obtained from the literature, we created a table containing the potential regulators of autophagy induction. With the already published scientific data, we propose which components of autophagy regulation could be AUIN, AUCO and AUCO, and details about them are collected in Supplementary Table 1. For example, AMPK and GADD34 act like AUIN; NRF2, CHOP and ULK1/2 are considered to be AUCO; whereas ATG5, ATG7 and Beclin1 can work as AUEX. We propose that this control network can be regulated in a stress-type-specific manner. The three columns of the Supplementary Table 1 contain all the possible AUIN, AUCO and AUEX at mTOR inhibition, upon ER stress or during oxidative stress. Besides, all the experimentally proved regulators of the network diagram with the possible regulatory connections are shown in Supplementary Fig. 1. All the relevant literature about the sign of the regulatory connections (i.e., the control molecules activate or inhibit eachother) are collected in Supplementary Table 2.
Results
The proper behaviour of the model is proved upon three various stress events
To confirm the accuracy of our autophagy regulation model, here we investigate the kinetic features of stress-type-specific autophagy controlling networks. To support our observations with relevant and existing biological evidence, we analyse here three, well-defined stress events as follows: starvation, ER stress and oxidative exposure.
Upon starvation stress
Biologists have already shown that cellular food supply is controlled by the AMPK-ULK1/2-mTORC1 regulatory triangle23. AMPK is able to promote autophagy (i.e., increasing the relative activity of ATG genes) by phosphorylating ULK1/2, the key regulator of autophagosome formation23,50. Besides, AMPK directly inhibits mTORC1 via phosphorylation upon nutrient depletion23,51–53. mTORC1 inhibits autophagy under nutrient-rich condition by downregulating both ULK1/2 and AMPK23–26. Interestingly, ULK1/2 inhibits AMPK, generating a negative feedback loop in the control network of autophagy induction27,54 (Fig. 2A).
Fig. 2. System-level feedbacks guarantee a robust stress response mechanism upon starvation.

A The wiring diagram is plotted to describe the precise mechanism of autophagy induction under starvation. Dashed lines show how the molecules can influence each other. Blocked end lines denote inhibition. B Phase plane diagrams are plotted upon excessive levels of stress. The balance curves of AMPK (orange) and ULK1/2 (purple) are plotted. The phase plane is shown for stress = 0.75. Intersection of nullclines represents the unstable (unfilled circle) steady state. Trajectories are depicted with grey lines. The temporal dynamics (C) is simulated with stress = 0.75. For details about the codes and software used for simulations, see the Supplementary Information.
To study the role of experimentally proved regulatory motifs in the control network, the overall steady-state response of the system is computed, generating a so-called phase plane diagram. In this case, our ordinary differential equation system is simplified to a pair of differential equation for ULK1/2/dt and AMPK/dt, respectively. We assume that all the other components are in steady state. The coordinate system is spanned by ULK1/2 and AMPK, and then the so-called balance curves, namely ULK1/2/dt = 0 (purple) and AMPK/dt = 0 (orange) are plotted (Fig. 2B). Balance curve (or mathematically called as nullcline) means that the rate of activation of the given component is exactly balanced by the rate of its degradation. The nullcline of ULK1/2 is S-shaped due to the double-negative feedback loops in the control network. Where the nullclines intersect each other upon starvation stress, the control network has one unstable steady state.
In case of starvation, the negative feedback loop between ULK1/2 and AMPK results in a sustained oscillatory characteristic (Fig. 2B, C). AMPK ensures the activation of ULK1/2, which, after a certain time delay, promotes the inactivation of AMPK (Fig. 2B). The phase plane diagram of ULK1/2 and AMPK with one unstable intersection shows a limit cycle oscillation, where grey arrows indicate the direction of motion among the limit cycle. Corresponding to our previous experimental results55, time course of sustained oscillation of ULK1/2 and AMPK has also depicted and AUEX gets periodically active (Fig. 2C) too, suggesting that autophagy has an ON and OFF characteristic under starvation.
Upon ER stress
It has been already experimentally confirmed that autophagy-dependent survival is followed by apoptotic cell death upon excessive level of ER stress and this mechanism is under the control of UPR5,6,45. The biological evidence suppose that two signal transducers of UPR, namely GADD34 and CHOP, have essential roles in ER stress response56,57. According to the experimental data, GADD34 promotes autophagy upon ER stress via downregulating mTORC158. CHOP is a transcription factor that controls gene transcription involved in apoptosis59, but it also has a positive effect on transcription of various autophagy genes (p62, ATG3 and ATG12)60. Here we suggest that GADD34 acts like AUIN, whereas CHOP might be AUCO with respect to ER stress. To confirm our assumption, we compare several well-known experimental data about GADD34 and CHOP to the kinetic analysis of our control network (Figs. 3 and 4).
Fig. 3. Both GADD34 and CHOP have important functions at endoplasmic reticulum stress: upregulation of CHOP or GADD34.

The computational simulations are determined in the overexpression of A CHOP and B GADD34 upon excessive levels of cellular stress. The temporal dynamics is simulated with high stress (stress = 9) combined with A CHOP-T = 1.25 or B GADD34-T = 2. Grey background refers to possible cell death. For details about the codes and software used for simulations, see the Supplementary Information.
Fig. 4. Both GADD34 and CHOP have important functions at endoplasmic reticulum stress: downregulation of CHOP or GADD34.

The computational simulations are determined in the absence of A CHOP and B GADD34 upon excessive levels of cellular stress. The temporal dynamics is simulated with high stress (stress = 9) combined with A CHOP-T = 0.1 or B GADD34-T = 0.1. Grey background refers to possible cell death. For details about the codes and software used for simulations, see the Supplementary Information.
Upregulation of either CHOP or GADD34 results in a short and transient activation of autophagy followed by the quick re-activation of mTORC1 and a possible cell death (Fig. 3A, B). These results nicely refer to that experimental data when overexpression of either CHOP or GADD34 rapidly turns on apoptotic cell death59,61.
Besides, it has experimentally already shown that addition of catalytically inactive GADD34 (GADD34ΔC/ΔC) result in pre-mature cell death in the presence of ER stress62. It is also well-known that CHOP-deleted cells are much less sensitive to ER stress compared to wild-type strain63. Consistent with the experimental data, downregulation of CHOP causes hyperactivation of both GADD34 and AUEX (Fig. 4A). In contrast, depletion of GADD34 completely diminishes autophagy, i.e., neither CHOP nor AUEX gets activated in our time-course simulation (Fig. 4B).
Our analysis clearly points out that GADD34 belongs more likely to AUIN, whereas CHOP might carry the dynamical characteristic of AUCO, further confirming that our general model can be properly used to describe ER stress response mechanism.
Upon oxidative stress
In our previous biological study, we have shown that NRF2 has an essential role in downregulating AMPK upon oxidative stress41, suggesting NRF2 works as AUCO, whereas other data assume that AMPK might be AUIN21 in our model (Fig. 5A). Similar to starvation, upon oxidative exposure, AMPK is essential to induce autophagy, but alone is not sufficient to maintain autophagy. AMPK turns on NRF2, which later inhibits AMPK via a negative feedback loop.
Fig. 5. The precise level of autophagy controller is essential in stress response.

Different mutant phenotypes are simulated: A Keap1 depletion (NRF2 activation) is mimicked upon excessive level of cellular stress (stress = 7.5, kaac = 5). B Depletion of autophagy controller is achieved on various levels (stress = 7.5, AUCO-T = 0.01, see lines marked with ‘a’; stress = 0.03, see lines marked with ‘b’; stress = 0.05, see lines marked with ‘c’). Grey background refers to possible cell death. For details about the codes and software used for simulations, see the Supplementary Information.
Gonzales et al.64 has recently shown experimentally that depletion of KEAP1 results in a downregulation of autophagy upon oxidative stress, but the molecular mechanism was not confirmed. As the key role of KEAP1 is to keep NRF2 in an inactive complex, we mimic KEAP1 depletion by increasing the amount of NRF2 (Fig. 5A). Although NRF2 has a positive effect on autophagy, its high level immediately inhibits the activation of AMPK. AMPK is essential for the induction of autophagy; therefore, autophagy remains inactive and cells might enhance cell death.
These results clearly show that NRF2 is the key controller of autophagy by switching ON and OFF the process upon oxidative stress.
AUCOs are the main switch elements for autophagy regulation upon cellular stress
Our analysis suggests that AUIN is essential for autophagy induction, but the exact role of AUCO is still a bit vague, as both theoretical and biological data have shown that the inhibition of various AUCOs (such as ULK1/2, CHOP or NRF2) result in various response. Therefore, we study the dynamical characteristic of autophagy induction when the total level of AUCO is systematically decreased upon cellular stress (Fig. 5B).
In that case, when the amount of AUCO is reduced by not so heavily (AUCO-T = 0.03 or 0.05, stress = 7.5), AUIN together with the reduced amount of AUCO is able to enhance AUEX (see lines ‘a’ and ‘b’ on Fig. 5B). As AUCO is not strong enough to downregulate AUIN properly, therefore autophagy gets hyperactivated upon excessive level of cellular stress. This kinetic behaviour was experimentally observed when the relative activity of NRF2 was depleted41, whereas cellular survival was significantly increased in the absence of CHOP63,65.
When AUCO-T is fully inhibited (AUCO-T = 0.01, stress = 7.5), AUEX cannot be active (see lines ‘c’ on Fig. 5B). Although AUIN becomes high and tries to induce AUEX (and AUCO as well), it alone is not sufficient to maintain autophagy. However, the high level of AUIN is sufficient to keep mTORC1 inactive. This dynamical feature is completely consistent with that experimental data when ULK1/2 is inhibited during starvation24.
Our analysis shows that a proper level of AUCO is essential for both turning ON and OFF autophagy, acting like a switch upon cellular stress.
Discussion and conclusions
Inspired by both experimental data and our previous theoretical analysis, we have created a general control model of autophagy regulation to analyse the potential roles of elements and feedback loops describing the dynamical characteristic of the response mechanism upon various cellular stress events. In this work, we have explored the systems-level properties of the control network using a mathematical model.
It is well-known that mTORC1 level is high and inhibits autophagy at physiological conditions. Here we grouped the molecules controlling autophagy regulation into three groups, called as AUIN, AUCO and AUEX, respectively (Fig. 1). The double-negative feedback loops between AUIN and AUCO; AUCO and mTORC1 generate bistability in the system with one physiological state and one autophagy state. Interestingly, two negative feedback loops are also present between AUIN and AUCO, and between AUCO and mTORC1 in the control network, suggesting that AUIN-AUCO-mTORC1 regulatory triangle has a critical effect on stress response mechanism. The possible regulatory connections supported by biological evidence are collected in Supplementary Table 2.
The question immediately arises, who are the exact elements hiding under our group names (i.e., AUIN, AUCO and AUEX) with respect to various cellular stress mechanisms. The presence of some proteins of the control network has been already experimentally proved in case of rapamycin treatment or starvation, revealing that AUIN might be AMPK and AUCO might be ULK1/2 (Fig. 2); however, these elements are not properly studied upon ER stress. Here we confirm that GADD34 and CHOP are well-known regulators of autophagy upon ER stress (Figs. 3 and 4), but these results need further experimental clarification in the future (the possible regulators are collected in Supplementary Table 1 and Supplementary Fig. 1). We also claim that always more than one protein from the same group takes part in the reaction to guarantee a robust stress response mechanism in any circumstances.
Although NRF2, CHOP and ULK1/2 are all called AUCO in our model, diminishing one of them results in different outcomes upon cellular stress. Similar to the published experimental data, downregulation of ULK1/2 completely blocks proper autophagy13,23,24,66, whereas either CHOP or NRF2 depletion results in the hyperactivation of the autophagy41,63,65 (Fig. 5B). In each case, AUIN (AMPK or GADD34) gets activated leading to the downregulation of mTORC1. AUIN promotes the activation of both AUCO and AUEX. As AUCO is essential for AUEX induction, our computer simulations confirm that in the total absence of AUCO, AUEX cannot be active. Meanwhile hyperactivation of AUEX can only be observed in that case if AUCO is not fully inactivated, assuming that neither NRF2 nor CHOP depletion results in complete inactivation of AUCO in the control network. These analyses also suggest that besides the cellular stress-specific AUCOs (i.e., NRF2 in oxidative stress and CHOP in ER stress), a non-stress-specific AUCO might be always present upon cellular stress and has an important role in regulating autophagy. We propose that this non-stress-specific AUCO is ULK1/2, as it is essential for autophagosome formation and is completely sufficient to block autophagy during starvation. However, these connections have to be clarified later experimentally.
Our computational model of the control network suggests that AUCO is the key switch controlling the jump between ON and OFF state of autophagy induction with respect to cellular stress level. Corresponding to our systems biological analysis, we propose that ULK1/2 is the main switch, whereas stress-specific side switches are also operating to make a precise answer upon cellular stress. For example, NRF2 could be an oxidative stress-dependent side switch. If this theory is valid, the main and side switches have to crosstalk to each other to generate an accurate cellular decision. To further clarify this assumption, we checked the possible connections between NRF2 and ULK1/2. As ULK1/2 is a kinase, first we identified potential Ser and Thr phosphorylation sites on NRF2 with Group-based Prediction System 5.067 and NetPhos 3.168. We found more than one consensus phosphorylation motifs of ULK1/2 on NRF2, suggesting that ULK1/2 might be able to control the NRF2 activity. By using the online available NRF2ome69, we also found that NRF2 is able to bind the promoter region of ULK2, suggesting that NRF2 might be a potential transcription factor of the kinase. It has recently been proved experimentally that NRF2 controls autophagosome genes, including ULKl/240. These data assume potential feedback loops between the main and side switches upon oxidative exposure; however, these connections later must be proven experimentally.
To highlight the medical relevance of the presented model, we note that the most commonly occurring complex diseases of the society (i.e., neurodegenerative diseases, metabolic diseases and carcinogenesis) are connected to the malfunction of autophagy. Therefore, we explored the dynamical characteristic of the network controlling autophagy induction. For example, acute lung injury induced by bacterial lipopolysaccharide (LPS) is a common critical illness characterized by inflammatory cytokine expression and cell death, although its molecular mechanism is poorly understood. Ito et al has recently revealed that GADD34 attenuates LPS-induced sepsis and acute tissue injury through suppressing macrophage activation, while GADD34 deficiency drastically increased lethality in LPS induction70. Interestingly, Wang et al has recently shown that addition of EGCG (green tee flavonoid), a natural compound, is able to protect pro-inflammatory cytokine induced injuries in insulin-producing cells through the mitochondrial pathway and therefore ameliorates LPS-induced acute lung injury71. Our model supposes that GADD34 is an AUIN (see Figs. 3 and 4). Since EGCG is a well-known AMPK activator (AUIN activator in our model) / mTORC1 inhibitor, question immediately arises, what if EGCG can protect cells in GADD34 deficiency in LPS-induced acute injury? To test this assumption, we simulate EGCG treatment combining with/without GADD34 depletion upon cellular stress (Fig. 6A, B). In case of EGCG treatment (kaai = 50, kimtor =50, stress = 5) the autophagy-dependent survival becomes hyper-active. By depleting GADD34 (AUIN-T = 0.1) during EGCG treatment GADD34 gets decreased, but AUCO (this might be CHOP) remains high resulting in active autophagy-dependent survival. These results need experimental confirmation, but illustrate the way how our systems biology approach can be applied to predict better various treatment and disease scenarios, and the design of the experiments to increase our knowledge on these medically relevant systems. Such better understanding may help us to modulate autophagy-dependent cellular decision with a long-term aim of a possible therapeutical intervention.
Fig. 6. Exploring the mechanism of the lipopolysaccharide (LPS)-induced acute tissue injury with the model system.

EGCG ameliorates GADD34 deficiency in LPS-induced acute tissue injury are simulated. The computational simulations determined the EGCG pretreatment (kaai = 50, kimtor = 50) combined with excessive level of cellular stress (stress = 5) in the A presence or B absence of GADD34 (GADD34-T = 0.1, kaai = 50, kimtor = 50). For details about the codes and software used for simulations, see the Supplementary Information.
Here we introduced a general, controlling network for autophagy that unifies our existing knowledge on how autophagy is regulated in various stress events. We pointed out that the robust response of this control network is essential during cellular stress and the key components, such as switches, could maintain a proper autophagy regulation to enable efficient stress response but inhibit overactivation. Applying this network model concept to other stress events and to disease settings, such as neurodegenerative diseases, cancer and Crohn’s disease, will be a helpful approach to understand the kinetic properties of autophagy regulation in these complex diseases.
Supplementary information
Acknowledgements
This work was supported by the ÚNKP-19-3-I-SE-81, New National Excellence Program of the Ministry for Innovation and Technology, by the ÚNKP-20-4-I-SE-32 New National Excellence Program of the Ministry for Innovation and Technology from the source of the National Research, Development And Innovation Fund, by a STIA-20-KF of Semmelweis University. O.K., M.H. and M.M. were supported by NKFIH FK-134267 (National Research, Development and Innovation Office, Hungary). T.K. was supported by the Biotechnological and Biosciences Research Council, UK Core Strategic Programme Grant (BB/CSP17270/1) and Institute Strategic Programme Grant for Gut Microbes and Health (BB/R012490/1 and its constituent projects, BBS/E/F/000PR10353 and BBS/E/F/000PR10355).
Author contributions
O.K., M.H., M.M. and T.K. designed the theroetical analysis. M.H., M.M. and O.K. generated the mathematical models and created the computer simulations. All authors discussed the results and wrote the manuscript together. M.H. and O.K. prepared the related figures and tables.
Conflict of interest
The authors declare no competing interests.
Ethics statement
The authors declare that this study did not require ethical approval.
Footnotes
Edited by B. Zhivotovsky
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s41419-021-03599-7.
References
- 1.Yin Z, Pascual C, Klionsky DJ. Autophagy: machinery and regulation. Micro. Cell. 2016;3:588–96. doi: 10.15698/mic2016.12.546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wirawan E, Vanden Berghe T, Lippens S, Agostinis P, Vandenabeele P. Autophagy: for better or for worse. Cell Res. 2012;22:43–61. doi: 10.1038/cr.2011.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jin S. Autophagy, mitochondrial quality control, and oncogenesis. Autophagy. 2006;2:80–4. doi: 10.4161/auto.2.2.2460. [DOI] [PubMed] [Google Scholar]
- 4.Clarke PG. Developmental cell death: morphological diversity and multiple mechanisms. Anat. Embryol. (Berl.) 1990;181:195–213. doi: 10.1007/BF00174615. [DOI] [PubMed] [Google Scholar]
- 5.Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2007;8:741–52. doi: 10.1038/nrm2239. [DOI] [PubMed] [Google Scholar]
- 6.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu Y, Levine B. Autosis and autophagic cell death: the dark side of autophagy. Cell Death Differ. 2015;22:367–76. doi: 10.1038/cdd.2014.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Green DR, Levine B. To be or not to be? How selective autophagy and cell death govern cell fate. Cell. 2014;157:65–75. doi: 10.1016/j.cell.2014.02.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kroemer G, Levine B. Autophagic cell death: the story of a misnomer. Nat. Rev. Mol. Cell Biol. 2008;9:1004–10. doi: 10.1038/nrm2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Parzych KR, Klionsky DJ. An overview of autophagy: morphology, mechanism, and regulation. Antioxid. Redox Signal. 2014;20:460–73. doi: 10.1089/ars.2013.5371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dikic I, Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018;19:349–64. doi: 10.1038/s41580-018-0003-4. [DOI] [PubMed] [Google Scholar]
- 12.Eskelinen EL. Autophagy: supporting cellular and organismal homeostasis by self-eating. Int. J. Biochem. Cell Biol. 2019;111:1–10. doi: 10.1016/j.biocel.2019.03.010. [DOI] [PubMed] [Google Scholar]
- 13.Ganley IG, Lam du H, Wang J, Ding X, Chen S, Jiang XULK1. ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J. Biol. Chem. 2009;284:12297–305. doi: 10.1074/jbc.M900573200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hosokawa N, et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol. Biol. Cell. 2009;20:1981–91. doi: 10.1091/mbc.e08-12-1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jung CH, et al. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol. Biol. Cell. 2009;20:1992–2003. doi: 10.1091/mbc.e08-12-1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hosokawa N, Sasaki T, Iemura S, Natsume T, Hara T, Mizushima N. Atg101, a novel mammalian autophagy protein interacting with Atg13. Autophagy. 2009;5:973–9. doi: 10.4161/auto.5.7.9296. [DOI] [PubMed] [Google Scholar]
- 17.Russell RC, et al. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat. Cell Biol. 2013;15:741–50. doi: 10.1038/ncb2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bento CF, et al. Mammalian autophagy: how does it work? Annu. Rev. Biochem. 2016;85:685–713. doi: 10.1146/annurev-biochem-060815-014556. [DOI] [PubMed] [Google Scholar]
- 19.Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012;13:251–62. doi: 10.1038/nrm3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sarbassov DD, Ali SM, Sabatini DM. Growing roles for the mTOR pathway. Curr. Opin. Cell Biol. 2005;17:596–603. doi: 10.1016/j.ceb.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 21.Tamargo-Gomez, I. & Marino, G. AMPK: regulation of metabolic dynamics in the context of autophagy. Int. J. Mol. Sci. 19, 3812 (2018). [DOI] [PMC free article] [PubMed]
- 22.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–93. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alers S, Loffler AS, Wesselborg S, Stork B. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks. Mol. Cell Biol. 2012;32:2–11. doi: 10.1128/MCB.06159-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Holczer M, Hajdu B, Lorincz T, Szarka A, Banhegyi G, Kapuy O. A double negative feedback loop between mTORC1 and AMPK kinases guarantees precise autophagy induction upon cellular stress. Int. J. Mol. Sci. 2019;20:22. doi: 10.3390/ijms20225543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ling NXY, et al. mTORC1 directly inhibits AMPK to promote cell proliferation under nutrient stress. Nat. Metab. 2020;2:41–9. doi: 10.1038/s42255-019-0157-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim J, Kundu M, Viollet B, Guan KLAMPK. and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011;13:132–U71. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Loffler AS, et al. Ulk1-mediated phosphorylation of AMPK constitutes a negative regulatory feedback loop. Autophagy. 2011;7:696–706. doi: 10.4161/auto.7.7.15451. [DOI] [PubMed] [Google Scholar]
- 28.Jung CH, Seo M, Otto NM, Kim DH. ULK1 inhibits the kinase activity of mTORC1 and cell proliferation. Autophagy. 2011;7:1212–21. doi: 10.4161/auto.7.10.16660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dunlop EA, Hunt DK, Acosta-Jaquez HA, Fingar DC, Tee AR. ULK1 inhibits mTORC1 signaling, promotes multisite Raptor phosphorylation and hinders substrate binding. Autophagy. 2011;7:737–47. doi: 10.4161/auto.7.7.15491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee JW, Park S, Takahashi Y, Wang HG. The association of AMPK with ULK1 regulates autophagy. PLoS ONE. 2010;5:e15394. doi: 10.1371/journal.pone.0015394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ma Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharm. Toxicol. 2013;53:401–26. doi: 10.1146/annurev-pharmtox-011112-140320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nguyen T, Nioi P, Pickett CB. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J. Biol. Chem. 2009;284:13291–5. doi: 10.1074/jbc.R900010200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stewart D, Killeen E, Naquin R, Alam S, Alam J. Degradation of transcription factor Nrf2 via the ubiquitin-proteasome pathway and stabilization by cadmium. J. Biol. Chem. 2003;278:2396–402. doi: 10.1074/jbc.M209195200. [DOI] [PubMed] [Google Scholar]
- 34.Jain A, et al. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J. Biol. Chem. 2010;285:22576–91. doi: 10.1074/jbc.M110.118976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McMahon M, Itoh K, Yamamoto M, Hayes JD. Keap1-dependent proteasomal degradation of transcription factor Nrf2 contributes to the negative regulation of antioxidant response element-driven gene expression. J. Biol. Chem. 2003;278:21592–600. doi: 10.1074/jbc.M300931200. [DOI] [PubMed] [Google Scholar]
- 36.Komatsu M, et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010;12:213–23. doi: 10.1038/ncb2021. [DOI] [PubMed] [Google Scholar]
- 37.Komatsu M, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007;131:1149–63. doi: 10.1016/j.cell.2007.10.035. [DOI] [PubMed] [Google Scholar]
- 38.Lau A, et al. A noncanonical mechanism of Nrf2 activation by autophagy deficiency: direct interaction between Keap1 and p62. Mol. Cell Biol. 2010;30:3275–85. doi: 10.1128/MCB.00248-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stolz A, Ernst A, Dikic I. Cargo recognition and trafficking in selective autophagy. Nat. Cell Biol. 2014;16:495–501. doi: 10.1038/ncb2979. [DOI] [PubMed] [Google Scholar]
- 40.Pajares M, et al. Transcription factor NFE2L2/NRF2 is a regulator of macroautophagy genes. Autophagy. 2016;12:1902–16. doi: 10.1080/15548627.2016.1208889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kosztelnik M, et al. Suppression of AMPK/aak-2 by NRF2/SKN-1 down-regulates autophagy during prolonged oxidative stress. FASEB J. 2019;33:2372–87. doi: 10.1096/fj.201800565RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ogata M, et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol. Cell Biol. 2006;26:9220–31. doi: 10.1128/MCB.01453-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–6. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- 44.Rashid HO, Yadav RK, Kim HR, Chae HJ. ER stress: autophagy induction, inhibition and selection. Autophagy. 2015;11:1956–77. doi: 10.1080/15548627.2015.1091141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Holczer M, Marton M, Kurucz A, Banhegyi G, Kapuy O. A comprehensive systems biological study of autophagy-apoptosis crosstalk during endoplasmic reticulum stress. Biomed. Res. Int. 2015;2015:319589. doi: 10.1155/2015/319589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kapuy O, Marton M, Banhegyi G, Vinod PK. Multiple system-level feedback loops control life-and-death decisions in endoplasmic reticulum stress. FEBS Lett. 2020;594:1112–23. doi: 10.1002/1873-3468.13689. [DOI] [PubMed] [Google Scholar]
- 47.Marton, M., Kurucz, A., Lizak, B., Margittai, E., Banhegyi, G. & Kapuy, O. A systems biological view of life-and-death decision with respect to endoplasmic reticulum stress-the role of PERK pathway. Int. J. Mol. Sci. 18, 58 (2017). [DOI] [PMC free article] [PubMed]
- 48.Tyson JJ, Chen KC, Novak B. Sniffers, buzzers, toggles and blinkers: dynamics of regulatory and signaling pathways in the cell. Curr. Opin. Cell Biol. 2003;15:221–31. doi: 10.1016/S0955-0674(03)00017-6. [DOI] [PubMed] [Google Scholar]
- 49.Strogatz, S. H. Nonlinear Dynamics and Chaos (Addison-Wesley, 1994).
- 50.Egan DF, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331:456–61. doi: 10.1126/science.1196371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gwinn DM, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell. 2008;30:214–26. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115:577–90. doi: 10.1016/S0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- 53.Meley D, et al. AMP-activated protein kinase and the regulation of autophagic proteolysis. J. Biol. Chem. 2006;281:34870–9. doi: 10.1074/jbc.M605488200. [DOI] [PubMed] [Google Scholar]
- 54.Dite TA, et al. The autophagy initiator ULK1 sensitizes AMPK to allosteric drugs. Nat. Commun. 2017;8:571. doi: 10.1038/s41467-017-00628-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Holczer M, Hajdu B, Lorincz T, Szarka A, Banhegyi G, Kapuy O. Fine-tuning of AMPK-ULK1-mTORC1 regulatory triangle is crucial for autophagy oscillation. Sci. Rep. 2020;10:17803. doi: 10.1038/s41598-020-75030-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ma Y, Hendershot LM. Delineation of a negative feedback regulatory loop that controls protein translation during endoplasmic reticulum stress. J. Biol. Chem. 2003;278:34864–73. doi: 10.1074/jbc.M301107200. [DOI] [PubMed] [Google Scholar]
- 57.Harding HP, et al. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell. 2000;6:1099–108. doi: 10.1016/S1097-2765(00)00108-8. [DOI] [PubMed] [Google Scholar]
- 58.Holczer M, Banhegyi G, Kapuy O. GADD34 keeps the mTOR pathway inactivated in endoplasmic reticulum stress related autophagy. PLoS ONE. 2016;11:e0168359. doi: 10.1371/journal.pone.0168359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.McCullough KD, Martindale JL, Klotz LO, Aw TY, Holbrook NJ. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol. Cell Biol. 2001;21:1249–59. doi: 10.1128/MCB.21.4.1249-1259.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.B’Chir W, et al. The eIF2alpha/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res. 2013;41:7683–99. doi: 10.1093/nar/gkt563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Harding HP, Zhang Y, Bertolotti A, Zeng H, Ron D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol. Cell. 2000;5:897–904. doi: 10.1016/S1097-2765(00)80330-5. [DOI] [PubMed] [Google Scholar]
- 62.Adler HT, et al. Leukemic HRX fusion proteins inhibit GADD34-induced apoptosis and associate with the GADD34 and hSNF5/INI1 proteins. Mol. Cell Biol. 1999;19:7050–60. doi: 10.1128/MCB.19.10.7050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zinszner H, et al. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 1998;12:982–95. doi: 10.1101/gad.12.7.982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gonzalez Y, Aryal B, Chehab L, Rao VA. Atg7- and Keap1-dependent autophagy protects breast cancer cell lines against mitoquinone-induced oxidative stress. Oncotarget. 2014;5:1526–37. doi: 10.18632/oncotarget.1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.B’Chir W, et al. Dual role for CHOP in the crosstalk between autophagy and apoptosis to determine cell fate in response to amino acid deprivation. Cell Signal. 2014;26:1385–91. doi: 10.1016/j.cellsig.2014.03.009. [DOI] [PubMed] [Google Scholar]
- 66.Chan EY, Kir S, Tooze SA. siRNA screening of the kinome identifies ULK1 as a multidomain modulator of autophagy. J. Biol. Chem. 2007;282:25464–74. doi: 10.1074/jbc.M703663200. [DOI] [PubMed] [Google Scholar]
- 67.Wang C., et al. GPS 5.0: an update on the prediction of kinase-specific phosphorylation sites in proteins. Genomics Proteomics Bioinformatics. 18, 72–80 2020. [DOI] [PMC free article] [PubMed]
- 68.Blom N, Sicheritz-Ponten T, Gupta R, Gammeltoft S, Brunak S. Prediction of post-translational glycosylation and phosphorylation of proteins from the amino acid sequence. Proteomics. 2004;4:1633–49. doi: 10.1002/pmic.200300771. [DOI] [PubMed] [Google Scholar]
- 69.Turei D, et al. NRF2-ome: an integrated web resource to discover protein interaction and regulatory networks of NRF2. Oxid. Med. Cell Longev. 2013;2013:737591. doi: 10.1155/2013/737591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ito S, Tanaka Y, Oshino R, Okado S, Hori M, Isobe KI. GADD34 suppresses lipopolysaccharide-induced sepsis and tissue injury through the regulation of macrophage activation. Cell Death Dis. 2016;7:e2219. doi: 10.1038/cddis.2016.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang J, Fan SM, Zhang J. Epigallocatechin-3-gallate ameliorates lipopolysaccharide-induced acute lung injury by suppression of TLR4/NF-kappaB signaling activation. Braz. J. Med. Biol. Res. 2019;52:e8092. doi: 10.1590/1414-431x20198092. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.