

Abstract
Type 1 diabetes (T1D) and autoimmune thyroid disease (AITD) are the most frequent chronic autoimmune diseases worldwide. Several autoimmune endocrine and non-endocrine disorders tend to occur together. T1D and AITD often cluster in individuals and families, seen in the formation of autoimmune polyendocrinopathy (AP). The close relationship between these two diseases is largely explained by sharing a common genetic background. The HLA antigens DQ2 (DQA1*0501-DQB1*0201) and DQ8 (DQA1*0301-DQB1*0302), tightly linked with DR3 and DR4, are the major common genetic predisposition. Moreover, functional single nucleotide polymorphisms (or rare variants) of various genes, such as the cytotoxic T-lymphocyte- associated antigen (CTLA4), the protein tyrosine phosphatase non-receptor type 22 (PTPN22), the interleukin-2 Receptor (IL2Ra), the Vitamin D receptor (VDR), and the tumor-necrosis-factor-α (TNF) that are involved in immune regulation have been identified to confer susceptibility to both T1D and AITD. Other genes including cluster of differentiation 40 (CD40), the forkhead box P3 (FOXP3), the MHC Class I Polypeptide-Related Sequence A (MICA), insulin variable number of tandem repeats (INS-VNTR), the C-Type Lectin Domain Containing 16A (CLEC16A), the Erb-B2 Receptor Tyrosine Kinase 3 (ERBB3) gene, the interferon-induced helicase C domain-containing protein 1 (IFIH1), and various cytokine genes are also under suspicion to increase susceptibility to T1D and AITD. Further, BTB domain and CNC homolog 2 (BACH2), C-C motif chemokine receptor 5 (CCR5), SH2B adaptor protein 3 (SH2B3), and Rac family small GTPase 2 (RAC2) are found to be associated with T1D and AITD by various independent genome wide association studies and overlap in our list, indicating a strong common genetic link for T1D and AITD. As several susceptibility genes and environmental factors contribute to the disease aetiology of both T1D and AITD and/or AP subtype III variant (T1D+AITD) simultaneously, all patients with T1D should be screened for AITD, and vice versa.
Keywords: type 1 diabetes, autoimmune thyroid disease, genetic link, susceptibility genes, HLA antigens, single nucleotide polymorphisms, autoimmune polyendocrinopathy
Epidemiology and Serology
Type 1 diabetes (T1D) and autoimmune thyroid disease (AITD) are two frequent autoimmune endocrine disorders. The prevalence of T1D is increasing worldwide and has nearly doubled in the past 40 years, with a prevalence of 9.5 per 10,000 people worldwide. The incidence worldwide is 15 per 100,000, ranging from 0.1 per 100,000 in China and Venezuela to 40.9 per 100,000 in Finland and in Arab countries between 2.4 in Oman and 29.0 in Saudia Arabia (1, 2). The highest incidence rates are at ages 5 to 9 years in girls and 10 to 14 in boys. The gender ratio is almost balanced with a slight preponderance of males (3–7). AITD, encompassing Graves’ disease (GD) and Hashimoto’s thyroiditis (HT), is defined as the presence of positive thyroid-related Ab. It is most commonly associated with other autoimmune glandular and non-glandular disorders. AITD peaks in the fourth decade for GD or fifth and sixth decade for HT. AITD is frequently combined with T1D in populations of various ancestries, occurring together more often than expected by the population prevalence of each disease GD is less prevalent than HT, only affecting approximately 1–1.5% of the general population, but is the underlying cause of 50–80% of cases of autoimmune hyperthyroidism. While subclinical hyperthyroidism can be diagnosed in only 0.12% of the non-diabetic population, it is prevalent in 6–10% of T1D patients (8–10).
The association of at least two autoimmune-induced glandular disorders is defined as autoimmune polyendocrinopathy (AP). Several non-endocrine autoimmune diseases can also be present (11, 12). AP is divided into a very rare, monogenic juvenile type I and one polygenic adult type II with three variants or subtypes, which are distinguished according to age of presentation, disease combinations, and modes of inheritance (13–17). The most prevalent AP type is the disease association of T1D and AITD in the same individual defined as subtype III variant. While the AP juvenile type has an annual incidence of 1–2:100,000, prevalence numbers are 1:600–1:900, 1:4,000, and 1:25,000 in Iranian Jews, Italians, and Finns, respectively. The adult AP form is far more common, with a worldwide incidence and prevalence of 1.4–4.5:100,000 and 14–45:1,000,000, respectively. Due to the high number of cases remaining unreported, the prevalence may approximate 1:20,000 (11, 18, 19). No accumulation in individual ethnic groups that deviate from the worldwide prevalence is known. The manifestation peak of adult AP is in the fourth-to-fifth decade depending on the combination of endocrine components with a female predominance of 75% (16, 20). T1D is the main cost driver in AP (21). The inheritance pattern seems to be autosomal dominant with incomplete penetrance, while several genetic loci may interact with environmental factors, such as deficiency of vitamin D and selenium, high iodine intake, and exposure to irradiation. The exact underlying pathogenic mechanisms are however not yet completely characterized. Also, chemical contaminants such as polybrominated diethyl ethers, polychlorinated biphenyls and their metabolites, binding to thyroid transport proteins and disrupting the thyroid function by displacing thyroxine, may be involved (22).
T1D is a T-cell mediated chronic disorder characterized by the loss of insulin-producing pancreatic β-cells and the appearance of insulitis. Biomarkers for T1D are autoantibodies (Ab) to islet cell antigens (ICA), tyrosine phosphatase (IA2), glutamic acid decarboxylase-65 (GAD), insulin (IAA), and zinc transporter ZnT8Solute carrier family 30 member 8 (SLC30A8). GD is defined by the presence of autoimmune-induced hyperthyroidism together with the presence of thyrotropin receptor autoantibodies (TSH-R-Ab) in general and stimulatory TSH-R-Ab (TSAb) in particular (23–29). HT is defined as primary hypothyroidism with an atrophic thyroid gland, an increased serum level of serum thyroid peroxidase (TPO) Ab and/or blocking TSH-R-Ab (TBAb) (24, 26, 30–32). As many as 25% of adolescents with T1D have thyroid-related Ab (15, 33–36). Long-term follow-up suggests that 30% of patients with T1D will develop AITD (10, 11, 15). TPO-Ab are present in 15–30% of adults and in 5–22% of children with T1D, while only 2–10 and 1–4%, respectively, are present in healthy controls (10, 19, 37). Up to 50% of TPO-Ab positive T1D patients develop an AITD. The diagnosis of AP III variant involves serological measurement of organ-specific Ab and subsequent functional testing of baseline thyrotropin (TSH), follicle-stimulating hormone (FSH), luteinizing hormone (LH), free triiodothyronine (fT3) and thyroxine (fT4), estradiol, cortisol, testosterone and fasting morning glucose as well as serum sodium (Na+), potassium (K+), and calcium (38).
Numerous reports on the individual susceptibility genes for T1D and/or AITD have been published, however scarce data are available pertaining to joint susceptibility. Therefore, in the present review we focus on the genetic link between these two clinically relevant endocrine autoimmune diseases and outline the underlying pathogenetic mechanisms causing AP.
Pathogenesis
In glandular autoimmunity, auto-aggression is considered multifactorial (39, 40). Antigen presenting cells (APC), such as ubiquitous dendritic cells, initiate the principal antigen-specific immune response (41). In non-lymphoid organs, immature dendritic cells pick up and fracture antigen molecules to subsequently migrate to the secondary lymphoid organs presenting their HLA class I or II associated antigen fragments. Together with chemokines/cytokines, antigen-specific T helper (Th) cells are activated via expansion of Th1 cells. They start to proliferate and exert tissue destructive activities. They further activate cytotoxic T lymphocytes, which stimulate the humoral immune response via expansion of Th2 cells and B-lymphocytes (42, 43). Activation of mononuclear phagocytes, another class of APC that also produce pro-inflammatory mediators accompanies the Th1 response. Auto-aggression occurs when immune tolerance, such as T suppressor cells that usually down regulate the overactive immune responses, is lost (41).
The regulatory T cell population (Treg) that inhibits the activation of CD4+ and CD25 T effector cells and regulates auto-aggressive T and B cell impacts the potential development of human autoimmune diseases (44). Naive CD4+ T cells, upon encountering their cognate, differentiate into effector cells, such as Th1, Th2, Th17, or Treg. These are further characterized by their cytokine production profiles and immune regulatory functions (44):
Th1 cells regulate antigen presentation and immunity against intracellular pathogens and produce tumor necrosis factor alpha (TNF) and interferon gamma (IFN-γ).
Th2 cells mediate humoral responses and immunity against parasites. They are important mediators of allergic diseases and produce interleukins (IL)-4, IL-5, and IL-13.
Th17 cells, which develop via an independent lineage from Th1 or Th2 cells, participate in inflammation and autoimmunity processes and express IL-17, IL-17F, IL-21, IL-22, and IL-26.
Treg mediate immune suppression by secretion of transforming growth factor-β (TGF-β) and IL-10 and express Forkhead box P3 (FOXP3) transcription factor (45).
The innate immune system steers the differentiation of Th cells by providing T cell receptor (TCR), and co-stimulatory signals as well as an appropriate cytokine microenvironment, which ultimately leads to the preferential induction of one specific cell lineage over the other. Of upmost importance for Th1, Th2, and Th17 cell differentiation are IFN-γ, IL-12 and IL-4, while IL-6 and TGF-β potently initiate Th17 differentiation (45).
Further possible mechanisms of autoimmunity include the hygiene hypotheses, which could play an important role in the worldwide increase of autoimmune diseases. It states that pathogens, parasites and commensal microorganisms protect against a variety of autoimmune conditions. Hence, the reduction in infection rates is likely to be one of several factors that has led to an increase in the frequency of certain autoimmune diseases (46). This is paired with changes in diet, especially the increase in food consumption of saponins, lectins, gliadin, and capsaicin, which can increase intestinal permeability, thus leading to increased uptake of endotoxin and inflammation. The increase in autoimmunity may enhance intake of epitopes cross-reactive with self. However, as the increase in autoimmunity did not occur directly after the switch from hunter-gatherer diet to the early agricultural diet, an underlying immunoregulatory deficit was necessary. The hygiene hypotheses does therefore not fully explain the rise in autoimmunity. It seems that genetic, molecular mimicry, and viral hypotheses are incoherent without a major simultaneous environmental change to weaken background immunoregulation, so that certain genotypes, in the presence of certain triggers, can develop these autoimmune diseases (47, 48).
Further mechanisms of co-occurrence other than the genetic-link could be shared, i.e., molecular amino acid signatures in the HLA-DR peptide-binding pocket predisposing to both T1D and AITD. A positively charged (Lys-71, Arg-74) HLA-DR pocket 4 was found to be critical for the development of both T1D and AITD, by accommodating autoantigenic peptides that may initiate both diseases or may facilitate the anchoring of the T-cell receptor to the peptide- MHC II complex (49). While differences exist in the pathogenesis of both diseases, epidemiological data revealed clustering of T1D and AITD in the same individual as well as in families, suggesting a common genetic basis for both diseases. Studies performing family-based association analyses in multiplex families demonstrated a joint genetic susceptibility that involves complex gene-gene and genetic-epigenetic interactions. As the molecular basis for the interactions between susceptibility genes in complex diseases remains unknown, the cumulative effect of increased statistical risk, as well as molecular interactions between susceptibility genes or their products, determine disease phenotype and severity (50–52). Even in countries with low disease incidence familial clustering has been observed (1).
The Genetic Link
Both T1D and AITD are multifactorial autoimmune endocrine diseases, with several susceptibility genes and environmental factors contributing to disease aetiology (15). Confirmed by genetic studies, a few genes confer risk for developing both AITD and T1D. These genes are denominated as joint susceptibility genes for AP III variant, suggesting that the gene product is implicated in the pathogenesis of both diseases (53, 54). Both whole-genome linkage screening and candidate gene studies have identified these genes (51, 55–57). Comprehensive analysis of gene-gene interactions in T1D showed that the more susceptibility risk alleles an individual carries, the higher the relative risk of developing disease, with a resulting odds ratio of 61. HLA remains the most important contributor to the overall risk, while additional gene interactions are likely to confer either protection or susceptibility (1, 58).
HLA Genes
Several genetic studies of patients with autoimmune diseases have shown specific contribution of HLA alleles, mostly HLA class II, to the genetic predisposition to autoimmune diseases. This is crucial for understanding their pathogenesis. There is a correlation between carriage of certain HLA class II alleles and an increased probability of developing the most common autoimmune diseases, including T1D and AITD. Several HLA class I and II alleles have been identified to influence both T1D and AITD, as well as AP (59–62). The HLA gene complex is located on chromosome 6p21 ( Figure 1 ) and encodes the major histocompatibility complex (MHC) proteins in humans. MHC class I proteins are heterodimers that consist of a long α-chain containing a transmembrane domain and a short universal β2-microglobulin chain while MHC class II proteins consist of long α- and β- chains carrying extracellular, transmembrane and short cytoplasmic domains. While HLA corresponding to MHC class I present peptides from inside the cell, HLA corresponding to MHC class II present antigens from outside the cell to T-lymphocytes. Class I genes encode for HLA antigens A, B, and C, class II genes encode for α- and β-chains of the heterodimeric HLA class II antigens DR, DP and DQ. Class III genes encode for complement components (63).
Figure 1.

T1D and AITD associated HLA alleles and haplotypes. Position of the HLA gene complex on the p arm of chromosome 6p21 (6p21.1-6p21.3) with around 3,500 kilo bases. The HLA class I region (blue) is located at the telomere side, while the HLA class II region (green) is located at the centromere side. DRB1, DQA1, and DQB1 alleles and haplotypes of the HLA class II (red) are associated with both T1D and AITD (AP III variant) in Caucasian subjects. Major susceptibility alleles in Caucasians are HLA-DRB1-03:01, HLA-DRB1-03:02, HLA-DRB1-04:01, HLA-DQA1-03:01, HLA-DQA1-05:01, HLA-DQB1-02:01, HLA-DQB1-03:01, and HLA-DQB1-03:02. The resulting haplotypes are HLA-DRB1-03:01-DQA1-05:01-DQB1-02:01, HLA-DRB1-04:01-DQA1-03:01-DQB1-03:01, and HLA-DRB1-04:01-DQA1-03:01-DQB1-03:02.
HLA class II molecules are synthesized in the endoplasmic reticulum. The immune response develops after the 13–18 amino-acid antigenic peptide is presented by APC, i.e., dendritic cells, B cells or macrophages, using the HLA class II molecule, and is recognized by the respective T-cell receptor on the CD4+ T-cell surface (64). HLA-DM, a nonconventional HLA class II molecule, is not polymorphic and cannot interact with antigenic peptides. However, it catalyzes the binding of antigenic peptide to HLA-DR. The HLA class II–peptide complexes are then delivered to the plasma membrane to present peptides to CD4+ T cells (65–67).
If an immune response is initiated, CD4+ T cells activate B cells for a subsequent production of specific autoantibodies and contribute to the recruitment of macrophages to the immune response. HLA molecules with point substitutions within the antigen-binding groove vary in their efficiency of binding and presentation of self-peptides followed by the initiation of an autoimmune response (68–70). Since HLA class II can present both exogenous and endogenous peptides to CD4+ T cells, many biomedical studies have focused on its role in the initiation of autoimmune responses. In patients with autoimmune diseases, such as T1D and AITD, autoantibodies are synthesized and lymphocytes often infiltrate into the target organ, leading to inflammation and even partial destruction. Because antigen presentation and further T cell activation are considered key components of the immune response, studying the peculiarities of antigen presentation as well as the structure and features of HLA proteins in particular is of utmost importance. Interestingly, the autoimmune diseases accompanied by Ab production are typically associated with HLA class II, while the diseases not accompanied by Ab production are more commonly associated with certain HLA class I alleles (71, 72). Although certain HLA Class II molecules may initiate the CD4+ T-cell mediated autoimmune response, HLA Class I molecules induce autoantigen-specific CD8+ T cell-mediated cytotoxicity, explaining with a plausible immunological rationale, and the genetic association of both HLA Class I and Class II molecules with glandular autoimmunity. This combination has synergistic and complementary effects on various stages of the autoimmune response (73).
HLA-DR3 is the major HLA class II allele contributing to the joint susceptibility for AITD and T1D. HLA haplotypes DR3-DQB1*02:01 and DR4-DQB1*03:02 contribute to AP III variant. The haplotypes HLA-DRB1*03:01-DQA1*05:01-DQB1*02:01 and HLA-DRB1*04:01-DQA1*03:01-DQB1*03:02 are significantly overrepresented in AP III compared to controls (72). The haplotypes for the AITD HT (DRB1*04:01-DQA1*03:01-DQB1*03:02/03:01) and GD (DRB1*03:01-DQA1*05:01-DQB1*02:01), as well as the two haplotypes identified for T1D (HLA-DQA1*03:01-DQB1*03:02 and HLA-DQA1*05:01-DQB1*02:01) are nearly identical. Hence, these HLA alleles and haplotypes confer susceptibility to both T1D and AITD (74–76). Interestingly, in Arabic populations, the haplotype DRB1*03:01-DQB1*02:01 was associated with T1D in individuals from Bahrain, Lebanon, and Tunisia, while the Japanese high-risk haplotypes DRB1*04:05-DQB1*04:01 and DRB1*09:01- DQB1*03:03 as well as the Caucasian haplotypes DRB1*03:01-DQB1*02:01 and DRB1*04:01-DQB1*03:02 were associated with T1D in individuals from Egypt, Morocco, Kuwait, Saudi Arabia, and Algeria (2).
The amino acid at position 57 of the DQ-β chain confers resistance or susceptibility to disease. An aspartic acid on position 57 in both alleles has a protective effect on T1D, while its absence provides susceptibility. In individuals in whom both alleles are non-Asp, the relative risk of T1D has been estimated to be 30 to 107 (1). The specific class II HLA-DR variant containing arginine at position 74 (DR-1-Arg74) increases the risk for AITD by blocking thyroglobulin peptides, especially Tg.2098. While bound to DR-1-Arg74, it could block continuous T cell activation and the autoimmune response in AITD. Of note, the position 74 in the β chain is exceptionally important, since this amino acid residue is located where the peptide-binding motif of HLA overlaps with the T-cell receptor docking site (77). The HLA-DR binding pocket has a unique amino acid signature. Four islet and thyroid peptides (Tg.1571, GAD.492, TPO.758, and TPO.338) were identified with the ability to bind to the HLA-DR binding pocket, being presented by antigen-presenting cells and elicited a T cell response. Both thyroid and islet peptides can bind to this flexible binding pocket and induce thyroid and islet specific T cell responses, thus triggering T1D and AITD in the same individual (78). Potential mechanisms how HLA genes activate endocrine autoimmunity and predispose to both T1D and AITD, even if the autoantigenic peptides are distinct, are shown in Figure 2 . Either one APC expresses both pancreas (islet) as well as thyroid autoantigens (peptides) which are embedded in pockets with HLA class II molecules and presented to the T cell within the immunological synapse, or two APC express either a pancreas or thyroid antigen, however both APC share a common amino acid which facilitates the anchoring of T cells (1, 79, 80).
Figure 2.
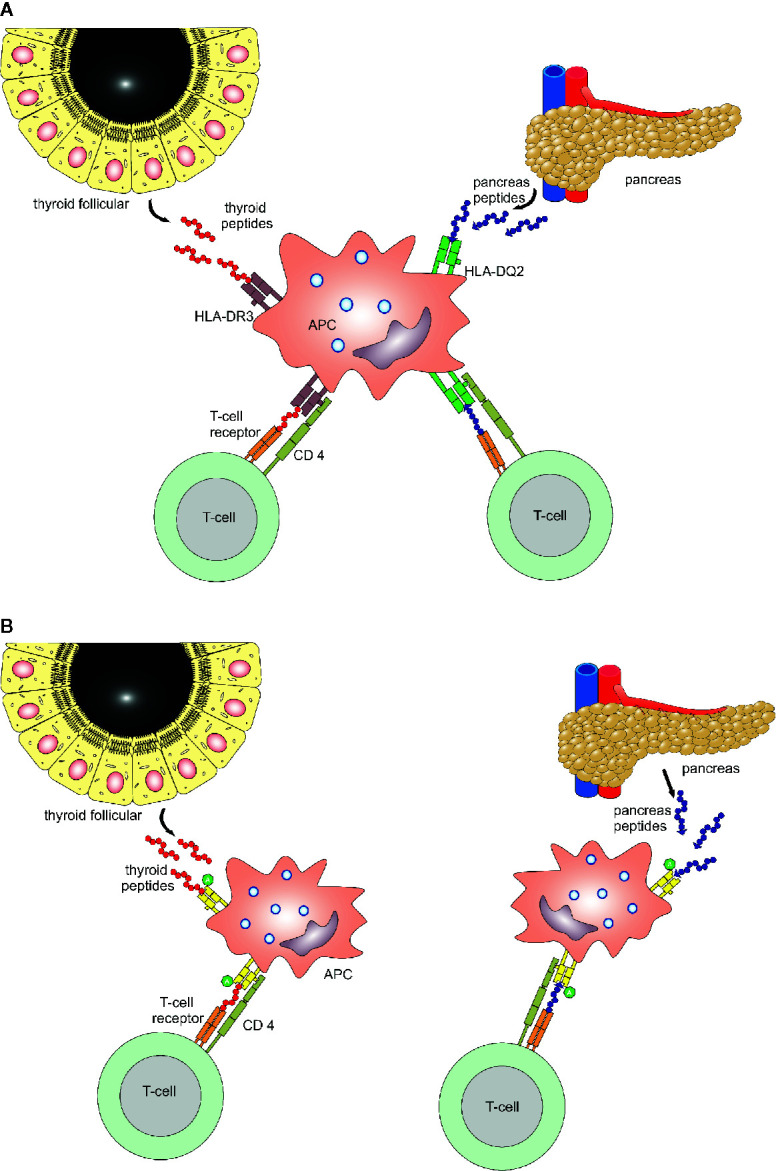
Binding mechanisms of islet and thyroid peptides. Two potential mechanisms for immunologically targeting several glands in patients with polyglandular autoimmunity. Panel (A) One APC expresses both pancreas as well as thyroid autoantigens (peptides) which are embedded in pockets with HLA class II molecules and presented to the T cell within the immunological synapse. Panel (B) Two APC express either a pancreas or thyroid antigen, however both APC share a common amino acid, which facilitates the anchoring of T cells.
Another risk allele for both T1D and AITD is HLA-DQB1 with Alanine at position 57 (Ala57), whereas Aspartic acid (Asp57) is protective. HLA-DQB1 confers strong susceptibility by Ala57 homozygosity in monoglandular and polyglandular autoimmunity (81). HLA-DQB1 Ala57 heterozygous women have an increased risk for AITD, whereas males have an increased risk for T1D, revealing sex-dependent increased susceptibility (81). The amino acid residue 57 is responsible for the formation of the DQA1– DQB1 heterodimer. If the aspartic acid is substituted by a neutral amino acid at this position, the HLA molecule will be able to present insulin fragments (82). HLA haplotypes DR3-DQ2 and DR4-DQ8 are associated with the presence of GAD and insulin Ab in patients with recent onset T1D (83). The specific HLA-DR9/DQ9 subtype might be associated to both T1D and AITD, as it was found in Japanese patients exhibiting both diseases (84). However, the most universal haplotypes positively associated with these diseases are DR3-DQ2 and DR4-DQ8 (85). In the Japanese population, the DRB1-DQB1 haplotypes DR4, DR8 and DR9 confer susceptibility to T1D and AITD (86). Moreover, the major susceptible HLA class II haplotype in Japanese associated with AP III was DRB1*04:05-DQB1*04:01 (87), and the haplotype DRB1∗09:01 was associated with the co-occurrence of T1D and AITD in Japan (88).
Sex alters the class I HLA-A association with T1D and AITD (60). In addition, patients with T1D and AITD share a common genetic background of HLA class II antigens DQ2 (DQA1*05:01- DQB1*02:01) and DQ8 (DQA1*03:01-DQB1*03:02) and overlapping functional single-nucleotide polymorphisms of various susceptibility genes involved in the immune regulation (10).
Population based differences are found in the association of HLA with T1D and AITD. Linkage analysis showed that the HLA class II alleles DQ and DR are strongly associated with both diseases. While DR3 is a major risk allele in German, Belgian, Filipino and Chinese Han populations, it does not confer susceptibility in the Japanese population. DR4 is major risk allele in Western Europe, America, South Asia and Africa but is not found in the Chinese Han population. DQA1*03:01, *05:01, and BQB1*02:01 confer risk in Caucasians, while DQB1*03:02 additionally confers risk in USA and DQB1*03:03 in Japan. DQB1*04:01 occurs in lower frequency in Caucasians but is a major risk allele in the Chinese Han population, probably due to its linkage disequilibrium with DRB1*04:05, which with DQB1*03:02 are further major risk alleles in the Chinese Han population (89). In the Northern Indian population, HLA-DRB1*03, DQA1*05, and DQB1*02 were associated with T1D, while worldwide in Caucasians HLA-DR4 (DRB1*04:01/04/05) and DQ8 (DQA1*03:01-DQB1*03:02) are associated with T1D. While in Northern India HLA-DRB1*04 and DRB1*03 were increased in patients with T1D, only the DRB1*03 allele was strongly associated with T1D and DRB1*04 was only increased in combination with DRB1*03. In Southern India however, both DRB1*03 and DRB1*04 contributed equally towards disease predisposition (73).
Single Nucleotide Polymorphisms in Non-HLA Genes
Additionally to HLA genes, several other genes, including the cytotoxic T-lymphocyte associated protein 4 (CTLA4, c.+6230G>A, rs3087243) and (CTLA4, c.49A>G, rs231775), the protein tyrosine phosphatase non-receptor type 22 (PTPN22, c.+1858 C>T, rs2476601), the interleukin 2 receptor subunit alpha (IL2Ra, c.A>G rs10795791), the vitamin D receptor (VDR, Bsm I rs1544410; Apa I rs7975232; Taq I rs731236), as well as the tumor necrosis factor (TNF, c.-863G>A, rs1800630) have been reported as susceptibility genes for T1D and AITD ( Table 1 ). Many other genes, such as cluster of differentiation 40 (CD40), forkhead box P3 (FOXP3), MHC class I polypeptide-related sequence A (MICA), insulin variable number of tandem repeats (INS-VNTR), C-type lectin domain containing 16 (CLEC16A), erb-B2 receptor tyrosine kinase 3 (ERBB3) gene, the interferon induced with helicase C domain 1 (IFIH1) gene and various cytokine genes, are assumed to be involved (90, 91).
Table 1.
Mutual non-HLA susceptibility genes in Type 1 diabetes and autoimmune thyroid disease.
| Gene | Location | Function | Mutations/Polymorphisms | Mutation/Polymorphism Phenotype |
|---|---|---|---|---|
| CONFIRMED | ||||
| CTLA4 | 2q33 | Encodes a receptor that is expressed on T cells and is a negative regulator of T-cell activation. | +6230G>A (rs3087243) 49A>G (rs231775) |
CTLA4 function or expression is affected by the threonine-to alanine substitution in the signal peptide of the CTLA4 protein, which leads to less efficient glycosylation in endoplasmic reticulum and reduced surface expression of CTLA4 protein. Increases susceptibility to T1D, AITD, and AP. |
| PTPN22 | 1p13 | Encodes lymphoid tyrosine phosphatase, a strong inhibitor of T-cell activation, expressed in B and T lymphocytes. Inhibits T lymphocyte antigen receptor signaling pathway | +1858C>T (rs2476601) | Polymorphisms lead to increased T-cell activation and enhanced susceptibility to T1D, AITD, and AP. |
| IL2Ra | 10p15 | Differentiation factor actively suppresses auto-reactive T cells via CD25 and regulates function of natural killer cells, B cells and Treg. | CD25 (rs10795791) | Polymorphisms in the CD25 gene region might affect function of Treg. Increased susceptibility to T1D and AITD, especially GD |
| VDR | 12q13.11 | Expressed on immune cells and directly inhibits activated T cells. Reduces production of pro-inflammatory cytokines (IFNγ). | Bsm I (rs1544410) Aps I (rs7975232) Taq I (rs731236) |
Polymorphisms lead to increased T-cell activation and enhanced susceptibility to T1D. |
| TNF | 6p21, within HLA class III region of MHC. | Located on chromosome 6p21, within HLA class III region of MHC. | -308 (rs1800629) | Increased transcription and production of TNF protein through polymorphisms in promoter region, higher levels of TNFα transcription facilitate inflammatory response in autoimmunity. Increases susceptibility to T1D and AITD. |
|
||||
| CD40 Gene | 20q13.12 | Influences both humoral and cell-mediated immune responses. | −1T>C (rs1883832) | Polymorphisms in the Kozak sequence are associated with autoimmunity, especially AITD. |
| FOXP3 | p arm of X chromosome (Xp11.23) |
FOXP3 transcription factor occupies the promoters for genes involved in regulatory T-cell function, controls regulatory T-cell differentiation, and is considered the master regulator of Treg development and function. | -2383C>T (rs3761549) | Polymorphisms might affect function of Treg hence increasing susceptibility to T1D and AITD. |
| MICA | p arm of chromosome 6 (6p21.33) | Encodes highly polymorphic cell surface glycoprotein. Protein expressed in two isoforms (MICA1 and MICA2), by alternative splicing. MICA protein is a ligand for receptors on natural killer cells and the stress-induced antigen is broadly recognized by intestinal epithelial T cells. |
MICA*A5 variant | Polymorphisms increase susceptibility for organ-specific autoimmune diseases. |
| INS-VNTR | 11p15 | Translation of pre-proinsulin, precursor of mature insulin. VNTR region of 14 to 15 bp consensus sequence upstream of insulin gene in insulin promoter. |
Short class I: (26–63 repeats), Intermediate class II: (64–139 repeats) Larger class III: (140–210 repeats) |
Polymorphisms might affect translation of pre-proinsulin, and production mature insulin. Increased susceptibility for T1D in AP type III. |
| CLEC16A | 16p13 | Encodes a member of the C-type lectin domain containing family. | rs12708716 | Polymorphisms increase susceptibility for organ-specific autoimmune diseases, T1D and AITD |
| ERBB3 | 12q13 | Encodes a member of the epidermal growth factor receptor (EGFR) family of receptor tyrosine kinases. | rs2292399 | Polymorphisms increase risk for organ-specific autoimmunity, especially T1D and AITD. |
| IFIH1 | 2q24.3 | Belongs to the pattern recognition receptor (PPR) family and is a sensor of double-stranded RNA, initiating antiviral activity through IFN production. | A946T (rs1990760) | Polymorphisms are associated with both T1D and AITD. |
| IL1R1 | 2q14.2 | Encodes a competitive inhibitor of interleukin-1–induced proinflammatory activity | VNTR | Polymorphisms are associated with autoimmunity, especially AITD. |
| IL4 Genes | 5q31.1 | Is involved in the regulation of immune and inflammatory responses. | -590C>T | Polymorphisms are associated with autoimmunity, especially AITD. |
CTLA4, cytotoxic T-lymphocyte associated protein 4; PTPN22, protein tyrosine phosphatase non-receptor type 22; IL2Ra, interleukin 2 receptor subunit alpha; VDR, Vitamin D receptor; TNF, Tumor necrosis factor; CD40, cluster of differentiation 40; FOXP3, forkhead box P3; MICA, MHC Class I Polypeptide-Related Sequence A; INS-VNTR, insulin variable number of tandem repeats; CLEC16A, C-Type Lectin Domain Containing 16; ERBB3, Erb-B2 Receptor Tyrosine Kinase 3 gene; IFIH1, interferon induced with helicase C domain 1; IL1R1, interleukin-1 receptor antagonist; IL4, interleukin 4; GD, Graves’ disease.
A major and confirmed susceptibility gene across different ethnic groups, for both T1D and AITD, is the CTLA4 gene. Being located on chromosome 2q33, it encodes a receptor expressed on T cells and serves as a negative regulator of T-cell activation. CTLA4 is involved in the interaction between T lymphocytes and APC. APC activate T lymphocytes by presenting an antigenic peptide bound to a HLA class II protein on the cell surface to the T lymphocyte receptor (92). On the surface of CD4+ T lymphocytes, co-stimulatory signals on the APC surface interact with receptors, such as CTLA4 during antigen presentation. CTLA4 downregulates T lymphocyte activation (93). Also related to autoimmunity is a 3’ untranslated region (3’ UTR), (AT)n microsatellite polymorphism with longer and shorter repeats of AT. Longer repeats are associated with decreased inhibitory function of CTLA4 and a reduced control of T cell proliferation by correlating with a shorter half-life of the CTLA4 mRNA than shorter repeats (94). Both single-nucleotide polymorphisms (SNP) or rare variants +6230G>A (rs3087243) and 49A>G (rs231775) promote development of autoimmunity, e.g., T1D and AITD by decreasing CTLA4 function. In patients with T1D and AITD (AP III), the CTLA4 G/G genotype of the +6230 G>A SNP is increased significantly (38, 95). In families with both T1D and AITD, a preferential transmission of the G allele of the CTLA4 49A>G SNP in exon 1 can be seen. The 49A>G SNP in the signal peptide of the CTLA4 protein results in a threonine-to-alanine substitution, leading to a less efficient glycosylation in the endoplasmic reticulum and reduced surface expression of the CTLA4 protein. This results in increased T-cell activation by affecting CTLA4 function and/or expression (37, 38).
Located on chromosome 1p13, the PTPN22 gene encodes the lymphoid tyrosine phosphatase (LYP), one of the strongest inhibitors of T cell activation. It inhibits the T lymphocyte antigen receptor-signaling pathway by binding to protein kinase Csk and limiting the response to antigens. It is expressed in immature and mature B and T lymphocytes. LYP, associated with the molecular adaptor protein CBL, regulates CBL function in the T cell antigen receptor signaling pathway, as it also binds to Csk, limiting the response to antigens (10, 37, 38). The minor T allele of the +1858 (rs2476601) C>T transition was observed to be associated with T1D, AITD, and AP III (96). The SNP causes a tryptophan for arginine substitution in the LYP protein at codon 620. Distinct isoforms of the protein are encoded through two transcript variants by alternative splicing of this gene (97–99). In addition, patients carrying the minor T allele of the PTPN22 +1858C>T SNP, had a twofold increased frequency of the HLA-DRB1*03 allele (57). While the minor T allele is known to be involved in altered T lymphocyte activation, a novel SNP in the promoter region of the PTPN22 gene G1123C, has been found to be associated with both T1D and AITD in Asian patients (98). Additional polymorphisms may also be causative (54).
The IL2Ra gene encoding CD25 is located on chromosome 10p15 and impacts production and function of regulatory T cells, actively suppressing autoreactive T cells in the periphery as a differentiation factor and via CD25. It regulates the function of natural killer cells and B cells and plays an important role in the development and function of Treg. A polymorphism in the CD25 gene region (rs10795791) thereby influences the development of the autoimmune diseases and is known to be associated with both T1D and AITD, especially GD (55, 100).
The VDR gene is expressed on immune cells and reduces the production of pro-inflammatory cytokines by directly inhibiting activated T cells. Vitamin D3 exerts its immune modulatory function through suppression of activated T cells, resulting in improvement of phagocytosis and suppression of gamma-interferon (IFNγ) production. It therefore may reduce the occurrence of T1D in humans. Three SNPs, BsmI (rs1544410), ApaI (rs7975232), and TaqI (rs731236), have been associated with T1D (101–103), while the BsmI and TaqI polymorphisms are also significantly associated with an increased AITD risk (104). Also, the VDR polymorphism Fokl has been associated with both T1D and AITD in the Brazilian population (105, 106).
The TNF gene is located within the HLA class III region of the MHC between HLA-B loci of class I and HLA-D loci of class II on chromosome 6p21. It encodes the pro-inflammatory cytokine TNF. Both the uncommon A allele of the G>A genotype of the -308 (rs1800629) SNP and the C/C genotype in the promoter region of the gene are associated with increased transcription and production of the TNF protein, which has been both implicated in the pathogenesis of autoimmune diseases as well as confers susceptibility to both T1D and AITD. The endogenous production of TNF is influenced by TNF promoter polymorphisms, thereby affecting messenger RNA (mRNA) and protein expression levels. Higher levels of TNF transcription may facilitate the inflammatory response in autoimmunity (91, 107). These SNP show a strong association with the occurrence of T1D and AITD in Asian and Caucasian populations (54, 108). Finally, HLA-DRB1*03 and TNF -308*A alleles were strongly associated in patients with AP III. These findings indicate similar immunogenetics of T1D and AITD (109–111).
Further genes are still a matter of discussion regarding their contribution to T1D and AITD. The CD40 gene-encoded protein also belongs to the TNF receptor superfamily and is mainly expressed in B-lymphocytes, monocytes, thyrocytes, orbital connective tissue, macrophages and dendritic cells, while the CD40 cell surface receptor is expressed on the surface of mature B cells, but not on plasma cells. CD40 influences both humoral and cell-mediated immune responses by interacting with the CD40 ligand on T cells (112). The CD40 ligand (CD40L) binds to the CD40 receptor and is predominantly expressed by activated CD4+ T cells, thus activating B-cells and other APC (113). The interaction of CD40-CD40L is vital for the activation of humoral immunity through triggering B cells and production of Ab. CD40 was associated with uncontrolled HLA class II expression and intercellular adhesion molecule 1 (ICAM-1) overexpression in the thyroid follicular cells of patients with AITD (114). Thyroid follicular cells might be able to function as APC under special circumstances (115). The CD40 −1 T>C SNP (rs1883832) in the Kozak sequence is associated with AITD, especially GD (116–118).
CD247 codes for CD3-zeta, a component of the TCR-CD3 signaling complex on T cells. CD3-zeta functions as an amplifier of TCR signaling and the CD3-zeta tyrosine phosphorylation is one of the first events occurring after TCR engagement (119).
The FOXP3 gene, located on the p arm of the X chromosome (Xp11.23), modulates the differentiation of regulatory T cells. It contains 11 coding exons and belongs to the forkhead/winged-helix family of transcriptional regulators. The FOXP3 transcription factor controls Treg differentiation and is considered the master regulator of Treg development by occupying the promoters for genes involved in Treg function. Genetic variants through mutations of the FOXP3 regulatory pathway reduces function and affects thymocytes developing within the thymus that during thymopoiesis are transformed into mature Tregs, promoting the development of autoimmunity (54, 90, 120). Both a haplotype consisting of 25 repeats of a microsatellite on allele 10 and the T allele of a C>T SNP at position -283 (rs1883832) were related with AP III. Because the microsatellite is located past the zinc finger domain of the FOXP3 gene, it affects downstream splicing, thereby impeding the function of the gene (121).
MICA is located on the p arm of chromosome 6 (6p21.33) and an associated locus within the MHC region. It encodes a highly polymorphic cell surface glycoprotein expressed in two isoforms formed by alternative splicing, MICA1 and MICA2. MICA2 is lacking exon 3. The MICA protein is expressed in epithelial and intestinal cells and is a ligand for receptors on the surface of natural killer cells. It acts as a possibly stress-induced antigen that is broadly recognized by intestinal epithelial T cells, binding to CD8 T cells carrying the integral membrane protein receptor natural killer group 2, member D (NKG2D) as well as natural killer cells. When engaged, the NKG2D–MICA complex results in the activation of T cell responses and natural killer cells against epithelial stressor cells expressing MICA on their surface. It is therefore also connected to organ-specific autoimmune diseases such as T1D and AITD (54, 90, 122, 123).
The INS gene region, encoding Insulin, is located on chromosome 11p5. Based on the number of the VNTRs, three classes are differentiated. An association of the insulin VNTR class I alleles with AP III has been found (51). A short class I VNTR penta-allelic 86-bp tandem repeat in the regulatory 5ʹ-UTR and a longer class III VNTR 600 bp tandem repeat polymorphism are related to lower and higher INS expression in the thymus, respectively (54, 90, 124). As insulin binds to insulin- like growth factor 1 receptors and exert its functions and the insulin-like, growth factor 1 receptor overlaps with TSH-R signaling in AITD, DNA alternations affecting the insulin expression influence the TSH-R signaling in AITD.
The CLEC16A gene, located on chromosome 16p13, contains a C-type lectin domain. The encoded protein is detected in immune cells, is implicated in pathogen recognition, and might predispose for immune mediated diseases e.g. T1D and AITD (125). The SNP rs2903692 shows a G>A transition significantly associated with both T1D and AITD, with the G allele increasing the risk for AP III (56).
The ERBB3 gene is located on chromosome 12q13. The SNP rs2292399 in intron 7 of ERBB3 has been shown to be associated with AITD and T1D with the A allele increasing the risk for AP III (56).
The IFIH1 gene, located at 2q24.3 chromosome, belongs to the pattern recognition receptor (PPR) family and is a cytosolic RNA sensor. The gene product, as a sensor of double-stranded RNA, initiates antiviral activity through the induction of IFN regulatory factors 3 and 7, leading to IFN production and apoptosis of virally infected cells, also promoting innate and adaptive immune responses. The SNP rs1990760 is a non-synonymous polymorphism, an alanine to threonine amino acid change at codon 946A>T, within the IFIH1 coding region located in the HNF-3b binding site. The polymorphism is associated with both T1D and AITD, especially GD. The correlation between viral infections and development of autoimmune diseases such as T1D and AITD makes IFIH1 a good susceptibility gene candidate (126–129).
Cytokine genes are also potential candidate genes for development of autoimmunity, especially AITD, since they are involved in the regulation of immune and inflammatory responses (130). The interleukin-1 receptor antagonist (IL-1RA) gene, located on chromosome 2q14.2, encodes a competitive inhibitor of interleukin-1–induced pro-inflammatory activity. IL1R1 modulates several interleukin-1–related immune and inflammatory responses and inhibits the activities of interleukin 1, interleukin 1-α and interleukin 1-β. A VNTR in intron 2 within the IL1R1 gene has been associated with AITD (131, 132). Genes encoding interleukin 4 (IL4), located on chromosome 5q31.1, are also involved in the regulation of immune and inflammatory responses. IL4 is produced by activated T cells and acts as a pleiotropic cytokine with immunomodulatory functions and is a ligand for the IL-4 receptor. A polymorphism at position −590C>T in the IL4 gene is associated with AITD (54, 133).
A list of susceptibility genes from several international publications obtained by a NCBI PubMed (https://pubmed.ncbi.nlm.nih.gov/) search for T1D, AITD, and the co-occurrence of both diseases from various populations worldwide is shown in Table 2 . In detail, in Japan, CTLA4 is strongly associated with T1D or AITD but not with AP III (87, 136). Furthermore, ERBB3, CLEC16A, and CTLA4 are associated with the co-occurrence of thyroid autoimmunity and T1D in the Japanese population (56, 87, 88). CTLA4, PTPN22, IFIH1, INS, CD247, and NAA25, are associated with both T1D and AITD in Sweden (119). The VDR polymorphism FokI is associated with both T1D and AITD in the Brazilian Hispanic population (105). The chemokine receptor type (CCR)5 - 32 bp deletion (Δ32) is associated with both diseases and with celiac disease in Poland (141). Finally, the signal transducer and activator of transcription (STAT)-4 is associated with both T1D and AITD in Korea (140), SESN3 with AITD in the Chinese Han population (139) and SLC26A4 is associated with AITD in the Tunisian population (138). Non-HLA genes like CTLA4 and PTPN22, which have been associated with both T1D and AITD in many Caucasian populations could only be associated with either T1D or AITD in certain Asian and Arab populations (2, 135, 137). Differences of association could also be found in IFIH1 and INS. Interestingly, population differences of IFIH1 susceptibility were also found within Caucasians populations in Europe (134, 135). This raises the question of how strong the population-based differences actually are and if they only occur between populations of different races or even within populations of the race.
Table 2.
List of population based susceptibility genes for T1D, AITD, and AP subtype III variant.
| Gene | Diseases | Country | Ethnecity | Reference |
|---|---|---|---|---|
| CTLA4 | T1D | Middle East & North Africa | Arab | Hatem Zayed, 2016 (2) |
| T1D | Poland | Caucasoid | Hanna Borysewicz-Sanczyk, 2020 (134) | |
| T1D | Great Britain | Caucasoid | Suna Onengut-Gumuscu 2015 (135) | |
| AITD | Great Britain, China | Caucasoid, Asian | Yul Hwangbo, 2018 (136) | |
| T1D AITD | Japan | Asian | Takuya Awata, 2008 (56) | |
| T1D AITD | Japan | Asian | Hisakuni Yamashita, 2011 (88) | |
| T1D AITD | Japan | Asian | Ichiro Horie, 2012 (87) | |
| T1D AITD | Sweden | Caucasoid | Dan Holmberg, 2016 (119) | |
| PTPN22 | T1D | Tunisia | Arab | Hatem Zayed, 2016 (2) |
| T1D | Great Britain | Caucasoid | Suna Onengut-Gumuscu 2015 (135) | |
| AITD | Jordan | Arab | Asem Alkhateeb, 2013 (137) | |
| AITD | Great Britain | Caucasoid | Yul Hwangbo, 2018 (136) | |
| T1D AITD | Sweden | Caucasoid | Dan Holmberg, 2016 (119) | |
| IL2RA | T1D | Japan | Asian | Hisakuni Yamashita, 2011 (88) |
| T1D | Great Britain | Caucasoid | Suna Onengut-Gumuscu 2015 (135) | |
| T1D | Poland | Caucasoid | Hanna Borysewicz-Sanczyk, 2020 (134) | |
| AITD | Great Britain | Caucasoid | Yul Hwangbo, 2018 (136) | |
| IFIH1 | T1D | Japan | Asian | Hisakuni Yamashita, 2011 (88) |
| T1D | Poland | Caucasoid | Hanna Borysewicz-Sanczyk, 2020 (134) | |
| T1D | Great Britain | Caucasoid | Suna Onengut-Gumuscu 2015 (135) | |
| T1D AITD | Sweden | Caucasoid | Dan Holmberg, 2016 (119) | |
| INS | T1D | Japan | Asian | Hisakuni Yamashita, 2011 (88) |
| T1D | Great Britain | Caucasoid | Suna Onengut-Gumuscu 2015 (135) | |
| T1D AITD | Sweden | Caucasoid | Dan Holmberg, 2016 (119) | |
| CLEC16A | T1D AITD | Japan | Asian | Takuya Awata, 2008 (56) |
| T1D AITD | Japan | Asian | Hisakuni Yamashita, 2011 (88) | |
| ERBB3 | T1D AITD | Japan | Asian | Takuya Awata, 2008 (56) |
| T1D AITD | Japan | Asian | Hisakuni Yamashita, 2011 (88) | |
| CD28 | T1D | Tunisia | Arab | Hatem Zayed, 2016 (2) |
| CD40 | GD | Japan | Asian | Naoya Inoue, 2012 (118) |
| CD69 | T1D | Great Britain | Caucasoid | Suna Onengut-Gumuscu 2015 (135) |
| CD226 | T1D | Great Britain | Caucasoid | Suna Onengut-Gumuscu 2015 (135) |
| CD247 | T1D | Tunisia | Arab | Hatem Zayed, 2016 (2) |
| T1D AITD | Sweden | Caucasoid | Dan Holmberg, 2016 (119) | |
| TNF | T1D | Great Britain | Caucasoid | Suna Onengut-Gumuscu 2015 (135) |
| IL2 | T1D | Great Britain | Caucasoid | Suna Onengut-Gumuscu 2015 (135) |
| IL10 | T1D | Great Britain | Caucasoid | Suna Onengut-Gumuscu 2015 (135) |
| IL21 | T1D | Great Britain | Caucasoid | Suna Onengut-Gumuscu 2015 (135) |
| IL27 | T1D | Great Britain | Caucasoid | Suna Onengut-Gumuscu 2015 (135) |
| IL7R | T1D | Japan | Asian | Hisakuni Yamashita, 2011 (88) |
| T1D | Great Britain | Caucasoid | Suna Onengut-Gumuscu 2015 (135) | |
| IL15 | T1D | Tunisia | Arab | Hatem Zayed, 2016 (2) |
| VDR | T1D AITD | Brazil | Hispanic | Denise Barreto Mory et al., 2016 (105) |
| NAA25 | T1D AITD | Sweden | Caucasoid | Dan Holmberg, 2016 (119) |
| TRB | T1D | Tunisia | Arab | Hatem Zayed, 2016 (2) |
| SLC26A4 | AITD | Tunisia | Arab | Rihab Kallel-Bouattour, 2017 (138) |
| SESN3 | AITD | Chinese Han | Asian | Wei Liu, 2018 (139) |
| FCRL3 | GD | Japan | Asian | Naoya Inoue, 2012 (118) |
| GD | China | Asian | Yul Hwangbo, 2018 (136) | |
| ZFAT | HT | Japan | Asian | Naoya Inoue, 2012 (118) |
| STAT4 | T1D AITD | Korea | Asian | Yongsoo Park, 2011 (140) |
| BANK1 | T1D | Tunisia | Arab | Hatem Zayed, 2016 (2) |
| ZAP70 | T1D | Tunisia | Arab | Hatem Zayed, 2016 (2) |
| SMOC2 | AITD | Jordan | Arab | Asem Alkhateeb, 2013 (137) |
| BACH2 | T1D | Great Britain | Caucasoid | Suna Onengut-Gumuscu 2015 (135) |
| AITD | Great Britain | Caucasoid | Yul Hwangbo, 2018 (136) | |
| IKZF1 | T1D | Great Britain | Caucasoid | Suna Onengut-Gumuscu 2015 (135) |
| IKZF3 | T1D | Great Britain | Caucasoid | Suna Onengut-Gumuscu 2015 (135) |
| IKZF4 | T1D | Great Britain | Caucasoid | Suna Onengut-Gumuscu 2015 (135) |
| AFF3 | T1D | Great Britain | Caucasoid | Suna Onengut-Gumuscu 2015 (135) |
| CCR5 | T1D | Great Britain | Caucasoid | Suna Onengut-Gumuscu 2015 (135) |
| T1D AITD CD | Poland | Caucasoid | Bartosz Słomiński, 2017 (141) | |
| CCR7 | T1D | Great Britain | Caucasoid | Suna Onengut-Gumuscu 2015 (135) |
| GLIS3 | T1D | Great Britain | Caucasoid | Suna Onengut-Gumuscu 2015 (135) |
| SH2B3 | T1D | Great Britain | Caucasoid | Suna Onengut-Gumuscu 2015 (135) |
| HT | United States | Caucasoid | Yul Hwangbo, 2018 (136) | |
| GRP183 | T1D | Great Britain | Caucasoid | Suna Onengut-Gumuscu 2015 (135) |
| RASGRP1 | T1D | Great Britain | Caucasoid | Suna Onengut-Gumuscu 2015 (135) |
| CTSH | T1D | Great Britain | Caucasoid | Suna Onengut-Gumuscu 2015 (135) |
| DEXI | T1D | Great Britain | Caucasoid | Suna Onengut-Gumuscu 2015 (135) |
| BCAR1 | T1D | Great Britain | Caucasoid | Suna Onengut-Gumuscu 2015 (135) |
| FUT2 | T1D | Great Britain | Caucasoid | Suna Onengut-Gumuscu 2015 (135) |
| UBASH3A | T1D | Great Britain | Caucasoid | Suna Onengut-Gumuscu 2015 (135) |
| ICOSLG | T1D | Great Britain | Caucasoid | Suna Onengut-Gumuscu 2015 (135) |
| RAC2 | T1D | Great Britain | Caucasoid | Suna Onengut-Gumuscu 2015 (135) |
| GD | China | Asian | Yul Hwangbo, 2018 (136) | |
| TSHR | GD | Great Britain, China | Caucasoid, Asian | Yul Hwangbo, 2018 (136) |
| RNASET2 | AITD | Great Britain, China | Caucasoid, Asian | Yul Hwangbo, 2018 (136) |
| CHRNA9 | GD | China | Asian | Yul Hwangbo, 2018 (136) |
| MMEL1 | GD | Great Britain | Caucasoid | Yul Hwangbo, 2018 (136) |
| PRICKLE1 | GD | Great Britain | Caucasoid | Yul Hwangbo, 2018 (136) |
| ITGAM | GD | Great Britain | Caucasoid | Yul Hwangbo, 2018 (136) |
| GPR174 | GD | China | Asian | Yul Hwangbo, 2018 (136) |
| SLAMF6 | GD | China | Asian | Yul Hwangbo, 2018 (136) |
| ABO | GD | China | Asian | Yul Hwangbo, 2018 (136) |
| LINC01550 | GD | China | Asian | Yul Hwangbo, 2018 (136) |
| TG | GD | China | Asian | Yul Hwangbo, 2018 (136) |
| FOXE1 | HT | United States | Caucasoid | Yul Hwangbo, 2018 (136) |
| VAV3 | HT | United States, Japan | Caucasoid, Asian | Yul Hwangbo, 2018 (136) |
| CAPZB | HT | United States | Caucasoid | Yul Hwangbo, 2018 (136) |
| PDE8B | HT | United States | Caucasoid | Yul Hwangbo, 2018 (136) |
| TRIB2 | AITD | Great Britain | Caucasoid | Yul Hwangbo, 2018 (136) |
| LPP | AITD | Great Britain | Caucasoid | Yul Hwangbo, 2018 (136) |
| FAM76B | AITD | Great Britain | Caucasoid | Yul Hwangbo, 2018 (136) |
T1D, Type 1 diabetes; AITD, Autoimmune thyroid disease; GD, Graves’ disease; HT, Hashimoto’s thyroiditis; CD, Celiac disease; CTLA4, cytotoxic T-lymphocyte associated protein 4; PTPN22, protein tyrosine phosphatase non-receptor type 22; IL2RA, interleukin 2 receptor subunit alpha; IFIH1, interferon-induced with helicase C domain 1; INS, insulin gene; CLEC16A, C-Type Lectin Domain Containing 16; ERBB3, Erb-B2 Receptor Tyrosine Kinase 3; CD28, cluster of differentiation 28; CD40, cluster of differentiation 40; CD3z, cluster of differentiation 3z; CD247, cluster of differentiation 247; IL7R, interleukin-7 receptor; IL15, interleukin-15; VDR, Vitamin D receptor; NAA25, N-Alpha-Acetyltransferase 25; TCRβ, T-cell receptor beta; SLC26A4, solute carrier family 26 member 4; CCR)5-(Δ32), Chemokine receptor type (CCR)5 - 32-bp deletion (Δ32); SESN3, Sestrin3; FCRL3, Fc receptor-like protein 3; ZFAT, Zinc Finger And AT-Hook Domain Containing; STAT4, Signal transducer and activator of transcription 4; BANK1, B Cell Scaffold Protein With Ankyrin Repeats 1; ZAP70, Zeta-chain-associated protein kinase 70; SMOC2, SPARC-related modular calcium-binding protein 2; BACH2, BTB domain and CNC homolog 2; IKZF1, IKAROS family zinc finger 1; IKZF3, IKAROS family zinc finger 3; IKZF4, IKAROS family zinc finger 4; AFF3, AF4/FMR2 family member 3; CCR5, C-C motif chemokine receptor 5; CCR7, C-C motif chemokine receptor 7; GLIS3, GLIS family zinc finger 3; SH2B3, SH2B adaptor protein 3; GRP183, G protein-coupled receptor 183; RASGRP1, RAS guanyl releasing protein 1; CTSH, cathepsin H; DEXI, Dexi homolog; BCAR1, BCAR1 scaffold protein, Cas family member; FUT2, fucosyltransferase 2; UBASH3A, ubiquitin associated and SH3 domain containing A; ICOSLG, inducible T cell costimulator ligand; RAC2, Rac family small GTPase 2; TSHR, thyroid stimulating hormone receptor; RNASET2, ribonuclease T2; CHRNA9, cholinergic receptor nicotinic alpha 9 subunit; MMEL1, membrane metalloendopeptidase like 1; PRICKLE1, prickle planar cell polarity protein 1; ITGAM, integrin subunit alpha M; GPR174, G protein-coupled receptor 174; SLAMF6, SLAM family member 6; ABO: ABO, alpha 1-3-N-acetylgalactosaminyltransferase and alpha 1-3-galactosyltransferase; LINC01550, long intergenic non-protein coding RNA 1550; TG, thyroglobulin; FOXE1, forkhead box E1; VAV3, vav guanine nucleotide exchange factor 3; CAPZB, capping actin protein of muscle Z-line subunit beta; PDE8B, phosphodiesterase 8B; TRIB2, tribbles pseudokinase 2; LPP, LIM domain containing preferred translocation partner in lipoma; FAM76B, family with sequence similarity 76 member B.
Further, CTLA4, PTPN22, IL2RA, BACH2, CCR5, SH2B3, and RAC2 are found to be associated with T1D and AITD by various independent genome wide association studies and overlap in our list, indicating a strong common genetic link for T1D and AITD.
Conclusion
The coexistence of different organ-specific and non- organ-specific autoimmune diseases in the same individual or family could be explained by sharing a common genetic background as well as a defective immune regulation. HLA regions, including DR3, DR4, in association with DQ2 and DQ8 are strongly associated with T1D, AITD, and AP III in Caucasians. HLA haplotypes in patients with AITD (HT and GD), as well as T1D were found to be nearly identical. However, in Arab and Asian populations, HLA susceptibility alleles and haplotypes differ from the ones found in European and American Caucasian populations. Also, population based differences are detected in the association between glandular autoimmunity with both HLA and non-HLA genes. Certain non-HLA susceptibility genes, such as CTLA4, PTPN22, IL2Ra, VDR, and TNF are involved in the activation of Treg that can react or cross-react with autoantigens. Therefore, polymorphisms in these genes confer further susceptibility to T1D, AITD, and AP III in Caucasians. The contribution of further genes e.g., CD40, FOXP3, MICA, INS-VNTR, CLEC16A, ERBB3, IFIH1, and various cytokine genes has not been definitely and/or fully clarified. In conclusion, the combined influence of genetic, epigenetic and environmental factors may lead to the onset of autoimmune disorders in different organs of the same subject or within families. Therefore, genetic screening is useful in patients with monoglandular autoimmunity e.g., T1D and Addison’s disease, as well as their first-degree relatives. In view of the possible long interval between the first manifestation of AP and the subsequent development of further autoimmune endocrinopathies, regular and long-term observation of patients is warranted. Furthermore, screening for autoimmune endocrine diseases is recommended regularly, especially for the offspring of patients with T1D and AITD, and AP III variant.
Author Contributions
LF conceptualized and designed the study, acquired and analyzed the data as well as drafted the article. GK Project initiation, conception and design, analysis and interpretation of data, drafting and critical revision of the article, as well as approval of the final version to be published. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The editorial assistance of Mr. Jan Längericht, pharmacist, Endocrine and Molecular Thyroid Laboratory, Department of Medicine I, Johannes Gutenberg University (JGU) Medical Center, Mainz, Germany, is much appreciated.
References
- 1. Huber A, Menconi F, Corathers S, Jacobson EM, Tomer Y. Joint genetic susceptibility to type 1 diabetes and autoimmune thyroiditis: from epidemiology to mechanisms. Endocr Rev (2008) 29(6):697–725. 10.1210/er.2008-0015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zayed H. Genetic Epidemiology of Type 1 Diabetes in the 22 Arab Countries. Curr Diabetes Rep (2016) 16(5):37. 10.1007/s11892-016-0736-4 [DOI] [PubMed] [Google Scholar]
- 3. Pundziute-Lycka A, Dahlquist G, Nystrom L, Arnqvist H, Bjork E, Blohme G, et al. The incidence of Type I diabetes has not increased but shifted to a younger age at diagnosis in the 0-34 years group in Sweden 1983-1998. Diabetologia (2002) 45(6):783–91. 10.1007/s00125-002-0845-2 [DOI] [PubMed] [Google Scholar]
- 4. Harjutsalo V, Sjoberg L, Tuomilehto J. Time trends in the incidence of type 1 diabetes in Finnish children: a cohort study. Lancet (2008) 371(9626):1777–82. 10.1016/S0140-6736(08)60765-5 [DOI] [PubMed] [Google Scholar]
- 5. Tuomilehto J. The emerging global epidemic of type 1 diabetes. Curr Diabetes Rep (2013) 13(6):795–804. 10.1007/s11892-013-0433-5 [DOI] [PubMed] [Google Scholar]
- 6. Patterson CC, Dahlquist GG, Gyurus E, Green A, Soltesz G, Group ES. Incidence trends for childhood type 1 diabetes in Europe during 1989-2003 and predicted new cases 2005-20: a multicentre prospective registration study. Lancet (2009) 373(9680):2027–33. 10.1016/S0140-6736(09)60568-7 [DOI] [PubMed] [Google Scholar]
- 7. Mobasseri M, Shirmohammadi M, Amiri T, Vahed N, Hosseini Fard H, Ghojazadeh M. Prevalence and incidence of type 1 diabetes in the world: a systematic review and meta-analysis. Health Promot Perspect (2020) 10(2):98–115. 10.34172/hpp.2020.18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Diana T, Wuster C, Olivo PD, Unterrainer A, Konig J, Kanitz M, et al. Performance and Specificity of 6 Immunoassays for TSH Receptor Antibodies: A Multicenter Study. Eur Thyroid J (2017) 6(5):243–9. 10.1159/000478522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Diana T, Wuster C, Kanitz M, Kahaly GJ. Highly variable sensitivity of five binding and two bio-assays for TSH-receptor antibodies. J Endocrinol Invest (2016) 39(10):1159–65. 10.1007/s40618-016-0478-9 [DOI] [PubMed] [Google Scholar]
- 10. Kahaly GJ, Frommer L, Schuppan D. Celiac disease and endocrine autoimmunity - the genetic link. Autoimmun Rev (2018) 17(12):1169–75. 10.1016/j.autrev.2018.05.013 [DOI] [PubMed] [Google Scholar]
- 11. Frommer L, Kahaly GJ. Autoimmune Polyendocrinopathy. J Clin Endocrinol Metab (2019) 104(10):4769–82. 10.1210/jc.2019-00602 [DOI] [PubMed] [Google Scholar]
- 12. Dittmar M, Libich C, Brenzel T, Kahaly GJ. Increased familial clustering of autoimmune thyroid diseases. Horm Metab Res (2011) 43(3):200–4. 10.1055/s-0031-1271619 [DOI] [PubMed] [Google Scholar]
- 13. Anaya JM. The diagnosis and clinical significance of polyautoimmunity. Autoimmun Rev (2014) 13(4-5):423–6. 10.1016/j.autrev.2014.01.049 [DOI] [PubMed] [Google Scholar]
- 14. Kahaly GJ. Polyglandular autoimmune syndromes. Eur J Endocrinol (2009) 161(1):11–20. 10.1530/EJE-09-0044 [DOI] [PubMed] [Google Scholar]
- 15. Dittmar M, Kahaly GJ. Polyglandular autoimmune syndromes: immunogenetics and long-term follow-up. J Clin Endocrinol Metab (2003) 88(7):2983–92. 10.1210/jc.2002-021845 [DOI] [PubMed] [Google Scholar]
- 16. Cutolo M. Autoimmune polyendocrine syndromes. Autoimmun Rev (2014) 13(2):85–9. 10.1016/j.autrev.2013.07.006 [DOI] [PubMed] [Google Scholar]
- 17. Kahaly GJ, Frommer L. Polyglandular autoimmune syndromes. J Endocrinol Invest (2018) 41(1):91–8. 10.1007/s40618-017-0740-9 [DOI] [PubMed] [Google Scholar]
- 18. Hansen MP, Kahaly GJ. Autoimmune polyglandular syndromes. Dtsch Med Wochenschr (2013) 138(7):319–26; quiz 27-8. 10.1055/s-0032-1327355 [DOI] [PubMed] [Google Scholar]
- 19. Hansen MP, Matheis N, Kahaly GJ. Type 1 diabetes and polyglandular autoimmune syndrome: A review. World J Diabetes (2015) 6(1):67–79. 10.4239/wjd.v6.i1.67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Betterle C, Zanchetta R. Update on autoimmune polyendocrine syndromes (APS). Acta Bio-Medica Atenei Parmensis (2003) 74(1):9–33. [PubMed] [Google Scholar]
- 21. Radermacher LK, Ponto K, Merkesdal S, Pomart V, Frommer L, Pfeiffer N, et al. Type I Diabetes is the Main Cost Driver in Autoimmune Polyendocrinopathy. J Clin Endocrinol Metab (2020) 105(4):e1307–15. 10.1210/clinem/dgz021 [DOI] [PubMed] [Google Scholar]
- 22. Ferrari SM, Fallahi P, Antonelli A, Benvenga S. Environmental Issues in Thyroid Diseases. Front Endocrinol (Lausanne) (2017) 8:50. 10.3389/fendo.2017.00050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Diana T, Daiber A, Oelze M, Neumann S, Olivo PD, Kanitz M, et al. Stimulatory TSH-Receptor Antibodies and Oxidative Stress in Graves Disease. J Clin Endocrinol Metab (2018) 103(10):3668–77. 10.1210/jc.2018-00509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li Y, Kim J, Diana T, Klasen R, Olivo PD, Kahaly GJ. A novel bioassay for anti-thyrotrophin receptor autoantibodies detects both thyroid-blocking and stimulating activity. Clin Exp Immunol (2013) 173(3):390–7. 10.1111/cei.12129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Diana T, Kanitz M, Lehmann M, Li Y, Olivo PD, Kahaly GJ. Standardization of a bioassay for thyrotropin receptor stimulating autoantibodies. Thyroid (2015) 25(2):169–75. 10.1089/thy.2014.0346 [DOI] [PubMed] [Google Scholar]
- 26. Kahaly GJ, Diana T. TSH Receptor Antibody Functionality and Nomenclature. Front Endocrinol (Lausanne) (2017) 8:28. 10.3389/fendo.2017.00028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Diana T, Brown RS, Bossowski A, Segni M, Niedziela M, Konig J, et al. Clinical relevance of thyroid-stimulating autoantibodies in pediatric graves’ disease-a multicenter study. J Clin Endocrinol Metab (2014) 99(5):1648–55. 10.1210/jc.2013-4026 [DOI] [PubMed] [Google Scholar]
- 28. Lytton SD, Kahaly GJ. Bioassays for TSH-receptor autoantibodies: an update. Autoimmun Rev (2010) 10(2):116–22. 10.1016/j.autrev.2010.08.018 [DOI] [PubMed] [Google Scholar]
- 29. Kahaly GJ. Bioassays for TSH Receptor Antibodies: Quo Vadis? Eur Thyroid J (2015) 4(1):3–5. 10.1159/000375445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Diana T, Olivo PD, Kahaly GJ. Thyrotropin Receptor Blocking Antibodies. Horm Metab Res (2018) 50(12):853–62. 10.1055/a-0723-9023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Diana T, Li Y, Olivo PD, Lackner KJ, Kim H, Kanitz M, et al. Analytical Performance and Validation of a Bioassay for Thyroid-Blocking Antibodies. Thyroid (2016) 26(5):734–40. 10.1089/thy.2015.0447 [DOI] [PubMed] [Google Scholar]
- 32. Kahaly GJ, Diana T, Glang J, Kanitz M, Pitz S, Konig J. Thyroid Stimulating Antibodies Are Highly Prevalent in Hashimoto’s Thyroiditis and Associated Orbitopathy. J Clin Endocrinol Metab (2016) 101(5):1998–2004. 10.1210/jc.2016-1220 [DOI] [PubMed] [Google Scholar]
- 33. Kordonouri O, Klinghammer A, Lang EB, Gruters-Kieslich A, Grabert M, Holl RW. Thyroid autoimmunity in children and adolescents with type 1 diabetes: a multicenter survey. Diabetes Care (2002) 25(8):1346–50. 10.2337/diacare.25.8.1346 [DOI] [PubMed] [Google Scholar]
- 34. Barker JM. Clinical review: Type 1 diabetes-associated autoimmunity: natural history, genetic associations, and screening. J Clin Endocrinol Metab (2006) 91(4):1210–7. 10.1210/jc.2005-1679 [DOI] [PubMed] [Google Scholar]
- 35. Tait KF, Marshall T, Berman J, Carr-Smith J, Rowe B, Todd JA, et al. Clustering of autoimmune disease in parents of siblings from the Type 1 diabetes Warren repository. Diabetes Med (2004) 21(4):358–62. 10.1111/j.1464-5491.2004.01162.x [DOI] [PubMed] [Google Scholar]
- 36. Holl RW, Bohm B, Loos U, Grabert M, Heinze E, Homoki J. Thyroid autoimmunity in children and adolescents with type 1 diabetes mellitus. Effect of age, gender and HLA type. Horm Res (1999) 52(3):113–8. 10.1159/000023446 [DOI] [PubMed] [Google Scholar]
- 37. Kahaly GJ, Hansen MP. Type 1 diabetes associated autoimmunity. Autoimmun Rev (2016) 15(7):644–8. 10.1016/j.autrev.2016.02.017 [DOI] [PubMed] [Google Scholar]
- 38. Biondi B, Kahaly GJ, Robertson RP. Thyroid Dysfunction and Diabetes Mellitus: Two Closely Associated Disorders. Endocr Rev (2019) 40(3):789–824. 10.1210/er.2018-00163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Baker JR, Jr. Autoimmune endocrine disease. JAMA (1997) 278(22):1931–7. 10.1001/jama.278.22.1931 [DOI] [PubMed] [Google Scholar]
- 40. Michels AW, Eisenbarth GS. Immunologic endocrine disorders. J Allergy Clin Immunol (2010) 125(2 Suppl 2):S226–37. 10.1016/j.jaci.2009.09.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Marrack P, Kappler J, Kotzin BL. Autoimmune disease: why and where it occurs. Nat Med (2001) 7(8):899–905. 10.1038/90935 [DOI] [PubMed] [Google Scholar]
- 42. Forster G, Otto E, Hansen C, Ochs K, Kahaly G. Analysis of orbital T cells in thyroid-associated ophthalmopathy. Clin Exp Immunol (1998) 112(3):427–34. 10.1046/j.1365-2249.1998.00613.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Otto EA, Ochs K, Hansen C, Wall JR, Kahaly GJ. Orbital tissue-derived T lymphocytes from patients with Graves’ ophthalmopathy recognize autologous orbital antigens. J Clin Endocrinol Metab (1996) 81(8):3045–50. 10.1210/jcem.81.8.8768872 [DOI] [PubMed] [Google Scholar]
- 44. Jonuleit H, Schmitt E, Stassen M, Tuettenberg A, Knop J, Enk AH. Identification and functional characterization of human CD4(+)CD25(+) T cells with regulatory properties isolated from peripheral blood. J Exp Med (2001) 193(11):1285–94. 10.1084/jem.193.11.1285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Onodera T, Toniolo A, Ray UR, Jenson AB, Knazek RA, Notkins AL. Virus-induced diabetes mellitus. XX. Polyendocrinopathy and autoimmunity. J Exp Med (1981) 153(6):1457–73. 10.1084/jem.153.6.1457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bach JF. The hygiene hypothesis in autoimmunity: the role of pathogens and commensals. Nat Rev Immunol (2018) 18(2):105–20. 10.1038/nri.2017.111 [DOI] [PubMed] [Google Scholar]
- 47. Rook GA. Hygiene hypothesis and autoimmune diseases. Clin Rev Allergy Immunol (2012) 42(1):5–15. 10.1007/s12016-011-8285-8 [DOI] [PubMed] [Google Scholar]
- 48. Okada H, Kuhn C, Feillet H, Bach JF. The ‘hygiene hypothesis’ for autoimmune and allergic diseases: an update. Clin Exp Immunol (2010) 160(1):1–9. 10.1111/j.1365-2249.2010.04139.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Menconi F, Osman R, Monti MC, Greenberg DA, Concepcion ES, Tomer Y. Shared molecular amino acid signature in the HLA-DR peptide binding pocket predisposes to both autoimmune diabetes and thyroiditis. Proc Natl Acad Sci USA (2010) 107(39):16899–903. 10.1073/pnas.1009511107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Golden B, Levin L, Ban Y, Concepcion E, Greenberg DA, Tomer Y. Genetic analysis of families with autoimmune diabetes and thyroiditis: evidence for common and unique genes. J Clin Endocrinol Metab (2005) 90(8):4904–11. 10.1210/jc.2004-2236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Villano MJ, Huber AK, Greenberg DA, Golden BK, Concepcion E, Tomer Y. Autoimmune thyroiditis and diabetes: dissecting the joint genetic susceptibility in a large cohort of multiplex families. J Clin Endocrinol Metab (2009) 94(4):1458–66. 10.1210/jc.2008-2193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Levin L, Ban Y, Concepcion E, Davies TF, Greenberg DA, Tomer Y. Analysis of HLA genes in families with autoimmune diabetes and thyroiditis. Hum Immunol (2004) 65(6):640–7. 10.1016/j.humimm.2004.02.026 [DOI] [PubMed] [Google Scholar]
- 53. Levin L, Tomer Y. The etiology of autoimmune diabetes and thyroiditis: evidence for common genetic susceptibility. Autoimmun Rev (2003) 2(6):377–86. 10.1016/S1568-9972(03)00080-6 [DOI] [PubMed] [Google Scholar]
- 54. Dittmar M, Kahaly GJ. Immunoregulatory and susceptibility genes in thyroid and polyglandular autoimmunity. Thyroid (2005) 15(3):239–50. 10.1089/thy.2005.15.239 [DOI] [PubMed] [Google Scholar]
- 55. Brand OJ, Lowe CE, Heward JM, Franklyn JA, Cooper JD, Todd JA, et al. Association of the interleukin-2 receptor alpha (IL-2Ralpha)/CD25 gene region with Graves’ disease using a multilocus test and tag SNPs. Clin Endocrinol (Oxf) (2007) 66(4):508–12. 10.1111/j.1365-2265.2007.02762.x [DOI] [PubMed] [Google Scholar]
- 56. Awata T, Kawasaki E, Tanaka S, Ikegami H, Maruyama T, Shimada A, et al. Association of type 1 diabetes with two Loci on 12q13 and 16p13 and the influence coexisting thyroid autoimmunity in Japanese. J Clin Endocrinol Metab (2009) 94(1):231–5. 10.1210/jc.2008-0718 [DOI] [PubMed] [Google Scholar]
- 57. Tomer Y, Dolan LM, Kahaly G, Divers J, D’Agostino RB, Jr, Imperatore G, et al. Genome wide identification of new genes and pathways in patients with both autoimmune thyroiditis and type 1 diabetes. J Autoimmun (2015) 60:32–9. 10.1016/j.jaut.2015.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bjornvold M, Undlien DE, Joner G, Dahl-Jorgensen K, Njolstad PR, Akselsen HE, et al. Joint effects of HLA, INS, PTPN22 and CTLA4 genes on the risk of type 1 diabetes. Diabetologia (2008) 51(4):589–96. 10.1007/s00125-008-0932-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Vriezinga SL, Auricchio R, Bravi E, Castillejo G, Chmielewska A, Crespo Escobar P, et al. Randomized feeding intervention in infants at high risk for celiac disease. N Engl J Med (2014) 371(14):1304–15. 10.1056/NEJMoa1404172 [DOI] [PubMed] [Google Scholar]
- 60. Flesch BK, Konig J, Frommer L, Hansen MP, Kahaly GJ. Sex Alters the MHC Class I HLA-A Association With Polyglandular Autoimmunity. J Clin Endocrinol Metab (2019) 104(5):1680–6. 10.1210/jc.2018-01974 [DOI] [PubMed] [Google Scholar]
- 61. Frommer L, Flesch BK, Konig J, Kahaly GJ. Amino Acid Polymorphisms in Hla Class II Differentiate Between Thyroid and Polyglandular Autoimmunity. J Clin Endocrinol Metab (2020) 105(6):1737–47. 10.1210/clinem/dgz164 [DOI] [PubMed] [Google Scholar]
- 62. Weinstock C, Matheis N, Barkia S, Haager MC, Janson A, Markovic A, et al. Autoimmune polyglandular syndrome type 2 shows the same HLA class II pattern as type 1 diabetes. Tissue Antigens (2011) 77(4):317–24. 10.1111/j.1399-0039.2011.01634.x [DOI] [PubMed] [Google Scholar]
- 63. Bernecker C, Ostapczuk M, Vordenbaumen S, Ehlers M, Thiel A, Schinner S, et al. HLA-A2 phenotype may be protective against Graves’ disease but not against Hashimoto’s thyroiditis in Caucasians. Horm Metab Res (2013) 45(1):74–7. 10.1055/s-0032-1323704 [DOI] [PubMed] [Google Scholar]
- 64. Neefjes J, Jongsma ML, Paul P, Bakke O. Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat Rev Immunol (2011) 11(12):823–36. 10.1038/nri3084 [DOI] [PubMed] [Google Scholar]
- 65. Kropshofer H, Vogt AB, Moldenhauer G, Hammer J, Blum JS, Hammerling GJ. Editing of the HLA-DR-peptide repertoire by HLA-DM. EMBO J (1996) 15(22):6144–54. 10.1002/j.1460-2075.1996.tb01002.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Pos W, Sethi DK, Wucherpfennig KW. Mechanisms of peptide repertoire selection by HLA-DM. Trends Immunol (2013) 34(10):495–501. 10.1016/j.it.2013.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Schulze MS, Wucherpfennig KW. The mechanism of HLA-DM induced peptide exchange in the MHC class II antigen presentation pathway. Curr Opin Immunol (2012) 24(1):105–11. 10.1016/j.coi.2011.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Gromme M, Neefjes J. Antigen degradation or presentation by MHC class I molecules via classical and non-classical pathways. Mol Immunol (2002) 39(3-4):181–202. 10.1016/S0161-5890(02)00101-3 [DOI] [PubMed] [Google Scholar]
- 69. Lang HL, Jacobsen H, Ikemizu S, Andersson C, Harlos K, Madsen L, et al. A functional and structural basis for TCR cross-reactivity in multiple sclerosis. Nat Immunol (2002) 3(10):940–3. 10.1038/ni835 [DOI] [PubMed] [Google Scholar]
- 70. Parham P. Presentation of HLA class I-derived peptides: potential involvement in allorecognition and HLA-B27-associated arthritis. Immunol Rev (1996) 154:137–54. 10.1111/j.1600-065X.1996.tb00932.x [DOI] [PubMed] [Google Scholar]
- 71. Sollid LM, Pos W, Wucherpfennig KW. Molecular mechanisms for contribution of MHC molecules to autoimmune diseases. Curr Opin Immunol (2014) 31:24–30. 10.1016/j.coi.2014.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Flesch BK, Matheis N, Alt T, Weinstock C, Bux J, Kahaly GJ. HLA class II haplotypes differentiate between the adult autoimmune polyglandular syndrome types II and III. J Clin Endocrinol Metab (2014) 99(1):E177–82. 10.1210/jc.2013-2852 [DOI] [PubMed] [Google Scholar]
- 73. Kumar N, Mehra NK, Kanga U, Kaur G, Tandon N, Chuzho N, et al. Diverse human leukocyte antigen association of type 1 diabetes in north India. J Diabetes (2019) 11(9):719–28. 10.1111/1753-0407.12898 [DOI] [PubMed] [Google Scholar]
- 74. Jacobson EM, Huber A, Tomer Y. The HLA gene complex in thyroid autoimmunity: from epidemiology to etiology. J Autoimmun (2008) 30(1–2):58–62. 10.1016/j.jaut.2007.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Zeitlin AA, Heward JM, Newby PR, Carr-Smith JD, Franklyn JA, Gough SC, et al. Analysis of HLA class II genes in Hashimoto’s thyroiditis reveals differences compared to Graves’ disease. Genes Immun (2008) 9(4):358–63. 10.1038/gene.2008.26 [DOI] [PubMed] [Google Scholar]
- 76. Dittmar M, Ide M, Wurm M, Kahaly GJ. Early onset of polyglandular failure is associated with HLA-DRB1*03. Eur J Endocrinol (2008) 159(1):55–60. 10.1530/EJE-08-0082 [DOI] [PubMed] [Google Scholar]
- 77. Chelvanayagam G. A roadmap for HLA-DR peptide binding specificities. Hum Immunol (1997) 58(2):61–9. 10.1016/S0198-8859(97)00185-7 [DOI] [PubMed] [Google Scholar]
- 78. Li CW, Osman R, Menconi F, Concepcion ES, Tomer Y. Flexible peptide recognition by HLA-DR triggers specific autoimmune T-cell responses in autoimmune thyroiditis and diabetes. J Autoimmun (2017) 76:1–9. 10.1016/j.jaut.2016.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Nicholson MJ, Hahn M, Wucherpfennig KW. Unusual features of self-peptide/MHC binding by autoimmune T cell receptors. Immunity (2005) 23(4):351–60. 10.1016/j.immuni.2005.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Deng L, Mariuzza RA. Recognition of self-peptide-MHC complexes by autoimmune T-cell receptors. Trends Biochem Sci (2007) 32(11):500–8. 10.1016/j.tibs.2007.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Kraus AU, Penna-Martinez M, Shoghi F, Seidl C, Meyer G, Badenhoop K. HLA-DQB1 pos. 57 defines susceptibility to isolated and polyglandular autoimmunity in adults: interaction with gender. J Clin Endocrinol Metab (2018) 104(6):1907–16. 10.1210/jc.2018-01621 [DOI] [PubMed] [Google Scholar]
- 82. Todd JA, Bell JI, McDevitt HO. HLA-DQ beta gene contributes to susceptibility and resistance to insulin-dependent diabetes mellitus. Nature (1987) 329(6140):599–604. 10.1038/329599a0 [DOI] [PubMed] [Google Scholar]
- 83. Hannelius U, Beam CA, Ludvigsson J. Efficacy of GAD-alum immunotherapy associated with HLA-DR3-DQ2 in recently diagnosed type 1 diabetes. Diabetologia (2020) 63(10):2177–81. 10.1007/s00125-020-05227-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Gobaru M, Ashida K, Yoshinobu S, Nagayama A, Kabashima M, Iwata S, et al. Human Leukocyte Antigen (HLA) Subtype-Dependent Development of Myasthenia Gravis, Type-1 Diabetes Mellitus, and Hashimoto Disease: A Case Report of Autoimmune Polyendocrine Syndrome Type 3. Am J Case Rep (2019) 20:1709–14. 10.12659/AJCR.918996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Zakharova MY, Belyanina TA, Sokolov AV, Kiselev IS, Mamedov AE. The Contribution of Major Histocompatibility Complex Class II Genes to an Association with Autoimmune Diseases. Acta Naturae (2019) 11(4):4–12. 10.32607/20758251-2019-11-4-4-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Katahira M, Maeda H, Tosaki T, Segawa S. The human leukocyte antigen class II gene has different contributions to autoimmune type 1 diabetes with or without autoimmune thyroid disease in the Japanese population. Diabetes Res Clin Pract (2009) 85(3):293–7. 10.1016/j.diabres.2009.06.007 [DOI] [PubMed] [Google Scholar]
- 87. Horie I, Kawasaki E, Ando T, Kuwahara H, Abiru N, Usa T, et al. Clinical and genetic characteristics of autoimmune polyglandular syndrome type 3 variant in the Japanese population. J Clin Endocrinol Metab (2012) 97(6):E1043–50. 10.1210/jc.2011-3109 [DOI] [PubMed] [Google Scholar]
- 88. Yamashita H, Awata T, Kawasaki E, Ikegami H, Tanaka S, Maruyama T, et al. Analysis of the HLA and non-HLA susceptibility loci in Japanese type 1 diabetes. Diabetes Metab Res Rev (2011) 27(8):844–8. 10.1002/dmrr.1234 [DOI] [PubMed] [Google Scholar]
- 89. Zhang XM, Wang HY, Luo YY, Ji LN. HLA-DQ, DR allele polymorphism of type 1 diabetes in the Chinese population: a meta-analysis. Chin Med J (Engl) (2009) 122(8):980–6. 10.3760/cma.j.issn.0366-6999.2009.08.019 [DOI] [PubMed] [Google Scholar]
- 90. Tomer Y, Menconi F. Type 1 diabetes and autoimmune thyroiditis: the genetic connection. Thyroid (2009) 19(2):99–102. 10.1089/thy.2008.1565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Tavares NA, Santos MM, Moura R, Araujo J, Guimaraes RL, Crovella S, et al. Association of TNF-alpha, CTLA4, and PTPN22 polymorphisms with type 1 diabetes and other autoimmune diseases in Brazil. Genet Mol Res (2015) 14(4):18936–44. 10.4238/2015.December.28.42 [DOI] [PubMed] [Google Scholar]
- 92. Teft WA, Kirchhof MG, Madrenas J. A molecular perspective of CTLA-4 function. Annu Rev Immunol (2006) 24:65–97. 10.1146/annurev.immunol.24.021605.090535 [DOI] [PubMed] [Google Scholar]
- 93. Brunner MC, Chambers CA, Chan FK, Hanke J, Winoto A, Allison JP. CTLA-4-Mediated inhibition of early events of T cell proliferation. J Immunol (1999) 162(10):5813–20. [PubMed] [Google Scholar]
- 94. Takara M, Kouki T, DeGroot LJ. CTLA-4 AT-repeat polymorphism reduces the inhibitory function of CTLA-4 in Graves’ disease. Thyroid (2003) 13(12):1083–9. 10.1089/10507250360731479 [DOI] [PubMed] [Google Scholar]
- 95. Dultz G, Matheis N, Dittmar M, Bender K, Kahaly GJ. CTLA-4 CT60 polymorphism in thyroid and polyglandular autoimmunity. Horm Metab Res (2009) 41(6):426–9. 10.1055/s-0029-1214414 [DOI] [PubMed] [Google Scholar]
- 96. Dultz G, Matheis N, Dittmar M, Rohrig B, Bender K, Kahaly GJ. The protein tyrosine phosphatase non-receptor type 22 C1858T polymorphism is a joint susceptibility locus for immunthyroiditis and autoimmune diabetes. Thyroid (2009) 19(2):143–8. 10.1089/thy.2008.0301 [DOI] [PubMed] [Google Scholar]
- 97. Smyth D, Cooper JD, Collins JE, Heward JM, Franklyn JA, Howson JM, et al. Replication of an association between the lymphoid tyrosine phosphatase locus (LYP/PTPN22) with type 1 diabetes, and evidence for its role as a general autoimmunity locus. Diabetes (2004) 53(11):3020–3. 10.2337/diabetes.53.11.3020 [DOI] [PubMed] [Google Scholar]
- 98. Kawasaki E, Awata T, Ikegami H, Kobayashi T, Maruyama T, Nakanishi K, et al. Systematic search for single nucleotide polymorphisms in a lymphoid tyrosine phosphatase gene (PTPN22): association between a promoter polymorphism and type 1 diabetes in Asian populations. Am J Med Genet A (2006) 140(6):586–93. 10.1002/ajmg.a.31124 [DOI] [PubMed] [Google Scholar]
- 99. Bottini N, Vang T, Cucca F, Mustelin T. Role of PTPN22 in type 1 diabetes and other autoimmune diseases. Semin Immunol (2006) 18(4):207–13. 10.1016/j.smim.2006.03.008 [DOI] [PubMed] [Google Scholar]
- 100. Bayer AL, Yu A, Adeegbe D, Malek TR. Essential role for interleukin-2 for CD4(+)CD25(+) T regulatory cell development during the neonatal period. J Exp Med (2005) 201(5):769–77. 10.1084/jem.20041179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Sahin OA, Goksen D, Ozpinar A, Serdar M, Onay H. Association of vitamin D receptor polymorphisms and type 1 diabetes susceptibility in children: a meta-analysis. Endocr Connect (2017) 6(3):159–71. 10.1530/EC-16-0110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Mukhtar M, Batool A, Wajid A, Qayyum I. Vitamin D Receptor Gene Polymorphisms Influence T1D Susceptibility among Pakistanis. Int J Genomics (2017) 2017:4171254. 10.1155/2017/4171254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. San-Pedro JI, Bilbao JR, Perez de Nanclares G, Vitoria JC, Martul P, Castano L. Heterogeneity of vitamin D receptor gene association with celiac disease and type 1 diabetes mellitus. Autoimmunity (2005) 38(6):439–44. 10.1080/08916930500288455 [DOI] [PubMed] [Google Scholar]
- 104. Feng M, Li H, Chen SF, Li WF, Zhang FB. Polymorphisms in the vitamin D receptor gene and risk of autoimmune thyroid diseases: a meta-analysis. Endocrine (2013) 43(2):318–26. 10.1007/s12020-012-9812-y [DOI] [PubMed] [Google Scholar]
- 105. Mory DB, Gabbay MA, Rocco ER, Kasamatsu T, Crispim F, Miranda WL, et al. High frequency of vitamin D receptor gene polymorphism FokI in Brazilian Type 1 diabetes mellitus patients with clinical autoimmune thyroid disease. Diabetol Metab Syndr (2016) 8:29. 10.1186/s13098-016-0145-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Houcken J, Degenhart C, Bender K, Konig J, Frommer L, Kahaly GJ. PTPN22 and CTLA-4 Polymorphisms Are Associated With Polyglandular Autoimmunity. J Clin Endocrinol Metab (2018) 103(5):1977–84. 10.1210/jc.2017-02577 [DOI] [PubMed] [Google Scholar]
- 107. Khan S, Mandal RK, Jawed A, Dar SA, Wahid M, Panda AK, et al. TNF-alpha -308 G > A (rs1800629) Polymorphism is Associated with Celiac Disease: A Meta-analysis of 11 Case-Control Studies. Sci Rep (2016) 6:32677. 10.1038/srep32677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Dittmar M, Kahaly GJ. Genetics of the autoimmune polyglandular syndrome type 3 variant. Thyroid (2010) 20(7):737–43. 10.1089/thy.2010.1639 [DOI] [PubMed] [Google Scholar]
- 109. Wilson AG, Symons JA, McDowell TL, McDevitt HO, Duff GW. Effects of a polymorphism in the human tumor necrosis factor alpha promoter on transcriptional activation. Proc Natl Acad Sci USA (1997) 94(7):3195–9. 10.1073/pnas.94.7.3195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Louis E, Franchimont D, Piron A, Gevaert Y, Schaaf-Lafontaine N, Roland S, et al. Tumour necrosis factor (TNF) gene polymorphism influences TNF-alpha production in lipopolysaccharide (LPS)-stimulated whole blood cell culture in healthy humans. Clin Exp Immunol (1998) 113(3):401–6. 10.1046/j.1365-2249.1998.00662.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Dittmar M, Kaczmarczyk A, Bischofs C, Kahaly GJ. The proinflammatory cytokine TNF-alpha -308 AA genotype is associated with polyglandular autoimmunity. Immunol Invest (2009) 38(3-4):255–67. 10.1080/08820130902766092 [DOI] [PubMed] [Google Scholar]
- 112. Tomer Y, Concepcion E, Greenberg DA. A C/T single-nucleotide polymorphism in the region of the CD40 gene is associated with Graves’ disease. Thyroid (2002) 12(12):1129–35. 10.1089/105072502321085234 [DOI] [PubMed] [Google Scholar]
- 113. Ferrari S, Giliani S, Insalaco A, Al-Ghonaium A, Soresina AR, Loubser M, et al. Mutations of CD40 gene cause an autosomal recessive form of immunodeficiency with hyper IgM. Proc Natl Acad Sci USA (2001) 98(22):12614–9. 10.1073/pnas.221456898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Tolosa E, Roura C, Marti M, Belfiore A, Pujol-Borrell R. Induction of intercellular adhesion molecule-1 but not of lymphocyte function-associated antigen-3 in thyroid follicular cells. J Autoimmun (1992) 5(1):119–35. 10.1016/S0896-8411(05)80056-3 [DOI] [PubMed] [Google Scholar]
- 115. Jacobson EM, Huber AK, Akeno N, Sivak M, Li CW, Concepcion E, et al. A CD40 Kozak sequence polymorphism and susceptibility to antibody-mediated autoimmune conditions: the role of CD40 tissue-specific expression. Genes Immun (2007) 8(3):205–14. 10.1038/sj.gene.6364375 [DOI] [PubMed] [Google Scholar]
- 116. Houston FA, Wilson V, Jennings CE, Owen CJ, Donaldson P, Perros P, et al. Role of the CD40 locus in Graves’ disease. Thyroid (2004) 14(7):506–9. 10.1089/1050725041517039 [DOI] [PubMed] [Google Scholar]
- 117. Wang XX, Wang XX, Chen T. Association between the CD40 rs1883832 polymorphism and Graves’ disease risk: a meta-analysis. EXCLI J (2019) 18:10–20. 10.17179/excli2017-916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Inoue N, Watanabe M, Yamada H, Takemura K, Hayashi F, Yamakawa N, et al. Associations between autoimmune thyroid disease prognosis and functional polymorphisms of susceptibility genes, CTLA4, PTPN22, CD40, FCRL3, and ZFAT, previously revealed in genome-wide association studies. J Clin Immunol (2012) 32(6):1243–52. 10.1007/s10875-012-9721-0 [DOI] [PubMed] [Google Scholar]
- 119. Holmberg D, Ruikka K, Lindgren P, Eliasson M, Mayans S. Association of CD247 (CD3zeta) gene polymorphisms with T1D and AITD in the population of northern Sweden. BMC Med Genet (2016) 17(1):70. 10.1186/s12881-016-0333-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Hori S, Nomura T, Sakaguchi S. Pillars Article: Control of Regulatory T Cell Development by the Transcription Factor Foxp3. J Immunol (2017) 198(3):981–5. 10.1126/science.1079490 [DOI] [PubMed] [Google Scholar]
- 121. Zhang L, Zhao Y. The regulation of Foxp3 expression in regulatory CD4(+)CD25(+)T cells: multiple pathways on the road. J Cell Physiol (2007) 211(3):590–7. 10.1002/jcp.21001 [DOI] [PubMed] [Google Scholar]
- 122. Bauer S, Groh V, Wu J, Steinle A, Phillips JH, Lanier LL, et al. Pillars Article: Activation of NK Cells and T Cells by NKG2D, a Receptor for Stress-Inducible MICA. J Immunol (2018) 200(7):2231–3. 10.1126/science.285.5428.727 [DOI] [PubMed] [Google Scholar]
- 123. Ide M, Dittmar M, Wurm M, Kanitz M, Kahaly GJ. [Polymorphisms of MICA microsatellites in thyroidal autoimmunity]. Med Klin (Munich) (2007) 102(1):11–5. 10.1007/s00063-007-1001-z [DOI] [PubMed] [Google Scholar]
- 124. Vafiadis P, Bennett ST, Todd JA, Nadeau J, Grabs R, Goodyer CG, et al. Insulin expression in human thymus is modulated by INS VNTR alleles at the IDDM2 locus. Nat Genet (1997) 15(3):289–92. 10.1038/ng0397-289 [DOI] [PubMed] [Google Scholar]
- 125. Robinson MJ, Sancho D, Slack EC, LeibundGut-Landmann S, Reis e Sousa C. Myeloid C-type lectins in innate immunity. Nat Immunol (2006) 7(12):1258–65. 10.1038/ni1417 [DOI] [PubMed] [Google Scholar]
- 126. Wawrusiewicz-Kurylonek N, Goscik J, Chorazy M, Siewko K, Posmyk R, Zajkowska A, et al. The interferon-induced helicase C domain-containing protein 1 gene variant (rs1990760) as an autoimmune-based pathology susceptibility factor. Immunobiology (2020) 225(1):151864. 10.1016/j.imbio.2019.10.013 [DOI] [PubMed] [Google Scholar]
- 127. Kang DC, Gopalkrishnan RV, Wu Q, Jankowsky E, Pyle AM, Fisher PB. mda-5: An interferon-inducible putative RNA helicase with double-stranded RNA-dependent ATPase activity and melanoma growth-suppressive properties. Proc Natl Acad Sci USA (2002) 99(2):637–42. 10.1073/pnas.022637199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature (2006) 441(7089):101–5. 10.1038/nature04734 [DOI] [PubMed] [Google Scholar]
- 129. Fujinami RS, von Herrath MG, Christen U, Whitton JL. Molecular mimicry, bystander activation, or viral persistence: infections and autoimmune disease. Clin Microbiol Rev (2006) 19(1):80–94. 10.1128/CMR.19.1.80-94.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Kemp EH, Kahaly GJ, Porter JA, Frommer L, Weetman AP. Autoantibodies against the calcium-sensing receptor and cytokines in autoimmune polyglandular syndromes types 2, 3 and 4. Clin Endocrinol (Oxf) (2018) 88(1):139–45. 10.1111/cen.13482 [DOI] [PubMed] [Google Scholar]
- 131. Tarlow JK, Blakemore AI, Lennard A, Solari R, Hughes HN, Steinkasserer A, et al. Polymorphism in human IL-1 receptor antagonist gene intron 2 is caused by variable numbers of an 86-bp tandem repeat. Hum Genet (1993) 91(4):403–4. 10.1007/BF00217368 [DOI] [PubMed] [Google Scholar]
- 132. Blakemore AI, Watson PF, Weetman AP, Duff GW. Association of Graves’ disease with an allele of the interleukin-1 receptor antagonist gene. J Clin Endocrinol Metab (1995) 80(1):111–5. 10.1210/jcem.80.1.7530255 [DOI] [PubMed] [Google Scholar]
- 133. Hunt PJ, Marshall SE, Weetman AP, Bell JI, Wass JA, Welsh KI. Cytokine gene polymorphisms in autoimmune thyroid disease. J Clin Endocrinol Metab (2000) 85(5):1984–8. 10.1210/jcem.85.5.6588 [DOI] [PubMed] [Google Scholar]
- 134. Borysewicz-Sanczyk H, Sawicka B, Wawrusiewicz-Kurylonek N, Glowinska-Olszewska B, Kadlubiska A, Goscik J, et al. Genetic Association Study of IL2RA, IFIH1, and CTLA-4 Polymorphisms With Autoimmune Thyroid Diseases and Type 1 Diabetes. Front Pediatr (2020) 8:481. 10.3389/fped.2020.00481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Onengut-Gumuscu S, Chen WM, Burren O, Cooper NJ, Quinlan AR, Mychaleckyj JC, et al. Fine mapping of type 1 diabetes susceptibility loci and evidence for colocalization of causal variants with lymphoid gene enhancers. Nat Genet (2015) 47(4):381–6. 10.1038/ng.3245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Hwangbo Y, Park YJ. Genome-Wide Association Studies of Autoimmune Thyroid Diseases, Thyroid Function, and Thyroid Cancer. Endocrinol Metab (Seoul) (2018) 33(2):175–84. 10.3803/EnM.2018.33.2.175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Alkhateeb A, Marzouka NA, Tashtoush R. Variants in PTPN22 and SMOC2 genes and the risk of thyroid disease in the Jordanian Arab population. Endocrine (2013) 44(3):702–9. 10.1007/s12020-013-9908-z [DOI] [PubMed] [Google Scholar]
- 138. Kallel-Bouattour R, Belguith-Maalej S, Zouari-Bradai E, Mnif M, Abid M, Hadj Kacem H. Intronic variants of SLC26A4 gene enhance splicing efficiency in hybrid minigene assay. Gene (2017) 620:10–4. 10.1016/j.gene.2017.03.043 [DOI] [PubMed] [Google Scholar]
- 139. Liu W, Zhang QY, Yuan FF, Wang HN, Zhang LL, Ma YR, et al. A dense mapping study of six European AITD susceptibility regions in a large Chinese Han Cohort of Graves’ disease. Clin Endocrinol (Oxf) (2018) 89(6):840–8. 10.1111/cen.13847 [DOI] [PubMed] [Google Scholar]
- 140. Park Y, Lee HS, Park Y, Min D, Yang S, Kim D, et al. Evidence for the role of STAT4 as a general autoimmunity locus in the Korean population. Diabetes Metab Res Rev (2011) 27(8):867–71. 10.1002/dmrr.1263 [DOI] [PubMed] [Google Scholar]
- 141. Slominski B, Lawrynowicz U, Mysliwska J, Ryba-Stanislawowska M, Skrzypkowska M, Mysliwiec M, et al. CCR5-Delta32 gene polymorphism is related to celiac disease and autoimmune thyroiditis coincidence in patients with type 1 diabetes. J Diabetes Complications (2017) 31(3):615–8. 10.1016/j.jdiacomp.2016.10.031 [DOI] [PubMed] [Google Scholar]