

Abstract
Recurrent breast cancer presents significant challenges with aggressive phenotypes and treatment resistance. Therefore, novel therapeutics are urgently needed. Here, we report that murine recurrent breast tumor cells, when compared with primary tumor cells, are highly sensitive to ferroptosis. Discoidin Domain Receptor Tyrosine Kinase 2 (DDR2), the receptor for collagen I, is highly expressed in ferroptosis-sensitive recurrent tumor cells and human mesenchymal breast cancer cells. EMT regulators, TWIST and SNAIL, significantly induce DDR2 expression and sensitize ferroptosis in a DDR2-dependent manner. Erastin treatment induces DDR2 upregulation and phosphorylation independent of collagen I. Furthermore, DDR2 knockdown in recurrent tumor cells reduces clonogenic proliferation. Importantly, both the ferroptosis protection and reduced clonogenic growth may be compatible with the compromised YAP/TAZ upon DDR2 inhibition. Collectively, these findings identify the important role of EMT-driven DDR2 upregulation in recurrent tumors in maintaining growth advantage but activating YAP/TAZ-mediated ferroptosis susceptibility, providing potential strategies to eradicate recurrent breast cancer cells with mesenchymal features.
Keywords: DDR2, collagen, breast cancer, recurrence, ferroptosis, Hippo, YAP, TAZ
Introduction:
Breast cancer is typically treated by resection of the primary tumor followed by some combinations of adjuvant radiation, chemotherapy, and hormonal therapy. Most patients show responses to these treatments targeting the primary tumors and may enter remission. However, a fraction of patients will develop recurrent breast cancer months to years after initial treatments(1). While the somatic mutations and oncogenic pathways leading to the initial growth of primary breast cancers have been extensively studied, far less is known in these aggressive and generally incurable recurrent breast tumors. These recurrent tumors are often resistant to the therapies effective for primary tumors and account for the majority of morbidity and mortality in patients with breast cancers.
Our poor understanding of tumor recurrence is due, in large part, to the lack of appropriate animal models. To overcome this limitation, several genetically engineered mouse (GEM) models of recurrent breast cancers have been developed based on the doxycycline-inducible expression of oncogenes in the mammary gland. Expression of an activated form of oncogenes, such as Her2/Neu (2), Pik3ca (3), c-Myc (4), or Wnt (5) via doxycycline consistently leads to the development of primary breast cancer. Afterward, these oncogenes can be withdrawn by the removal of doxycycline, resulting in tumor regression below detectable levels, similar to the tumor remission in patients with breast cancer. Remarkably, tumors will eventually recur at the sites of primary tumors to form “recurrent tumors” after varying latency periods. Such tumor recurrence also bears significant similarities to breast cancer recurrence in patients. For example, tumors may recur over a long and variable timeframe, similar to the varying timing of recurrences in human breast cancer. Also, during the latency period between primary and recurrent tumors, residual tumor cells are expected to exist in the primary tumor sites, analogous to minimal residual disease in patients with breast cancer. Finally, the growth of the recurrent tumors is often independent of the initiating oncogenes, reminiscent of the finding that recurrent breast cancers have often lost the initial oncogenes and become insensitive to the treatments initially effective for the primary tumors. Recent studies have found that recurrent tumors undergo widespread epigenomic and transcriptional alterations associated with elevated RIPK3(6), G9a histone methyltransferase (7), and NRF2 activation(8). All these features of recurrent tumors are required for tumor recurrence.
Another shared feature of the mouse recurrent tumors(7, 9) and human recurrent breast tumors(10) is their mesenchymal features from epithelial-mesenchymal transition (EMT). EMT encompasses various dynamic changes in cellular organization from epithelial to mesenchymal phenotypes, with significant changes in migration and invasion. Tumor cells adopt EMT during tumor progression, invasion, chemo-resistance, and metastasis(10). EMT is characterized by loss of the epithelial cell markers (such as E-cadherin, cytokeratin) as well as the acquisition of mesenchymal markers (e.g., vimentin)(11).
This EMT also re-wires the metabolic process and renders tumor cells addicted to the uptake of extracellular cystine and susceptible to ferroptosis(12–15). External cystine is imported into human mammalian cells via xCT, a cystine-glutamate antiporter, for the synthesis of the tri-peptide glutathione (GSH)(16). GSH is an essential cofactor for GPX4 that attenuates lipid peroxidation, membrane damage, and ferroptosis(17). Therefore, the pharmacological inhibition of xCT transport by erastin increases lipid peroxidation and induces ferroptosis(18). The process of ferroptosis is highly regulated and many genetic determinants have been identified by chemical or genetic approaches (19, 20). Recently, we and other group has found the dysregulation of Hippo effectors YAP (Yes-associated protein 1) and TAZ (transcriptional coactivator with PDZ-binding motif) in determining ferroptosis(15, 21–24). One receptor kinase associated with EMT is Discoidin Domain Receptor Tyrosine Kinase 2 (encoded by DDR2), which become activated and phosphorylated when engaged by its ligand collagen I(25). Also, the activation of DDR2 regulates the levels of SNAIL protein through its intracellular binding partner Src kinase to maintain mesenchymal fate and invasive phenotypes (26, 27).
In this study, we found that murine recurrent breast tumor cells are highly sensitive to ferroptosis. Through the analysis of differential gene expression of the ferroptosis essential kinases identified in a previous screen, we found DDR2 expression is grossly exaggerated in the recurrent breast cancer cells and contributes to their susceptibility to ferroptosis. DDR2 expression is triggered by EMT regulators and contributes to the EMT-enhanced ferroptosis. Interestingly, erastin can activate the phosphorylation of DDR2 in the absence of its ligand collagen I. Unexpectedly, this DDR2 upregulation is also essential for the clonogenic growth of the recurrent cells. Mechanistic study shows that DDR2 promotes the ferroptosis of recurrent tumor cells by activating YAP/TAZ-mediated expression through Src. Therefore, the mesenchymal-associated DDR2 upregulation during breast cancer recurrence provides a growth advantage but also leads to the collateral vulnerability to ferroptosis through the Hippo pathway.
Results:
Recurrent tumor cells are sensitive to ferroptosis and expressed high levels of DDR2
To compare the ferroptosis sensitivity of primary and recurrent breast tumor cells, we isolated and expanded tumor cells from Her2-driven murine MTB/TAN model for primary breast tumors (before the oncogenic withdrawal) and recurrent breast tumors (after the recurrence)(6). Compared with primary tumor cells which were only sensitive to high doses of erastin, we found that recurrent tumor cells exhibited a much higher sensitivity to erastin treatment (Fig. 1A). When we examined the levels of reduced glutathione (GSH), that recurrent tumor cells have ~2 fold higher in basal GSH levels (Supplementary Fig. 1A), consistent with reported NRF2 in recurrent tumor cells (8). However, erastin depleted GSH to <2% in recurrent tumor cell lines while primary tumor cells could still maintain ~8% of GSH (Supplementary Fig. 1B). These data suggest that recurrent tumor cells require more on xCT and external cystine to maintain high GSH levels and survival. Consistently, when the RNA expression of Slc7a11 (encoding xCT), Gpx4, and Acsl4 was measured, the erastin-induced Slc7a11 expression was more dramatically induced in recurrent tumor cells (Supplementary Fig. 1C–E).
Fig. 1. Recurrent tumor cells are sensitive to ferroptosis and show high DDR2 expression.
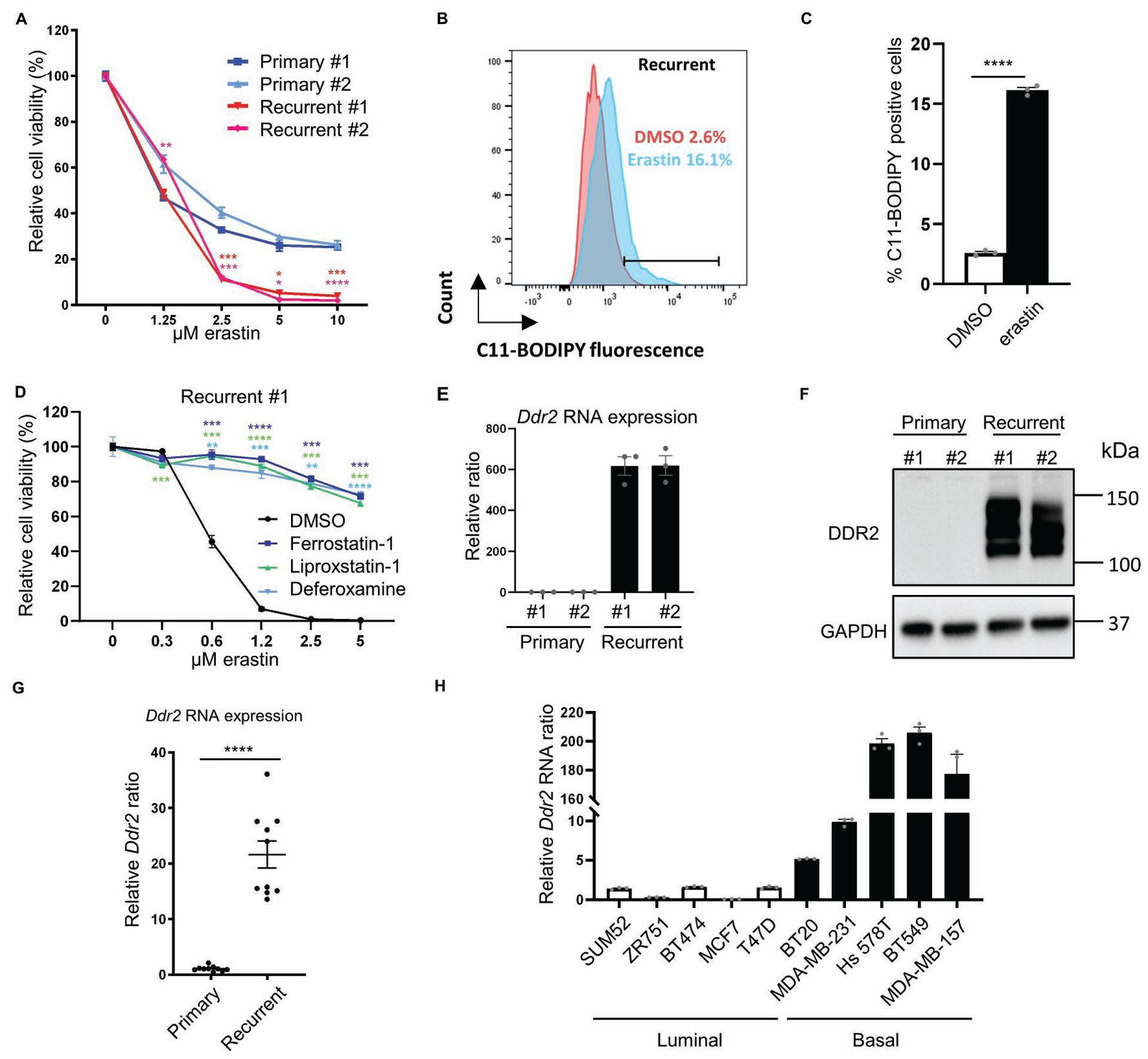
(A) Recurrent tumor cells were more sensitive to erastin treatment. Primary and recurrent murine tumor cells were treated with increasing indicated doses of erastin for 18 hours and the viability was measured by Celltiter Glo assay. n=3 biological replicates. (B-C) Erastin treatment (0.5μM, 18 hours) in recurrent tumor cells increased lipid peroxidation as determined by C11-BODIPY staining (B) and the quantification of % positive cells (C). (D) The erastin-induced cell death in recurrent tumor cells was rescued by ferroptosis inhibitors (ferrostatin-1, 10 μM; liproxstatin-1, 2 μM) and iron chelator (deferoxamine, 100 μM) as determined by Celltiter Glo assay after 19 hours of incubation. n=3 biological replicates. (E) Ddr2 expression was dramatically elevated in recurrent tumor cells by qRT-PCR. (F) Western blot showed a robust DDR2 protein expression only in recurrent tumor cell lines. (G) Comparison of Ddr2 RNA expression by qRT-PCR between 10 primary and 10 recurrent mouse tumors showed an overall increase in recurrent tumors. (H) DDR2 RNA is highly expressed in human basal breast cancer cell lines. Comparison of DDR2 RNA expression between 5 luminal (empty bars) and 5 basal (solid bars) established human breast cell lines expressed higher levels of DDR2 mRNA. (A, D) Two-way ANOVA, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 Dunnett’s multiple comparisons. (C, G) ****p<0.0001, two-tailed Student’s t-test. n = 10 samples for each group. Bars show standard error of the mean.
We also found that erastin treatment significantly increased lipid peroxidation in recurrent tumor cells (Fig. 1B–C). Also, the erastin-induced cell deaths could be rescued by ferroptosis inhibitors (ferrostatin-1, liproxstatin-1) and iron chelator (deferoxamine) in both recurrent tumor cells (Fig. 1D and Supplementary Fig. 1F). Therefore, recurrent tumor cells, when compared with primary tumor cells, were highly sensitive to erastin-induced ferroptosis.
To elucidate the genetic determinants of recurrent-specific ferroptosis sensitivity in breast cancers, we compared the expression of 34 ferroptosis essential genes identified in our previous kinome screen of ferroptosis(28) in primary and recurrent tumor cells(6). DDR2 emerged as a gene of interest since it was essential for ferroptosis and significantly upregulated in recurrent tumor cells(6). Next, we validated the expression of the Ddr2 mRNA and found recurrent tumor cells have dramatically higher (~600 folds) expression (Fig 1E). Consistently, while undetectable in the primary tumor cells, DDR2 protein was robustly expressed in the recurrent tumor cells (Fig 1F). Mouse recurrent tumors had an average of ~20- fold higher expression of Ddr2 mRNA than primary tumors (Fig 1G). Therefore, the recurrent tumor and cells, compared with the primary tumor and cells, have exaggerated Ddr2 expression.
Given DDR2 is a potential regulator of EMT(25), we examined the EMT markers in primary and recurrent tumor cells (Supplementary Fig. 1G–I). We found the upregulation of Snail mRNA in both recurrent tumor cells and upregulation of Twist mRNA in one of the recurrent tumor cells (Supplementary Fig. 1G–H). E-cadherin was lost in both recurrent tumor cells (Supplementary Fig. 1I). Next, we further tested whether DDR2 expression levels correlate to luminal or basal (mesenchymal-enriched) features of established human breast cell lines (Figure 1H). The sensitivity of these breast cancer cells to cystine-deprived death was previously determined and revealed a higher cystine addiction of basal-type cells (12). Among the tested cell lines, DDR2 was significantly up-regulated in cystine-addicted basal cell lines when compared with five cystine-independent luminal human breast cancer cell lines (Figure 1H). We further found DDR2 was significantly elevated in basal-like breast tumors when compared with luminal B type breast tumor in both TCGA(29) and METABRIC(30). Moreover, in METABRIC, we found the Claudin-low subtype, the most mesenchymal type of breast cancer (31), has significantly higher DDR2 expression when comparing with other subtypes. (Supplementary Fig. 1J–K). Collectively, these data suggest that DDR2 is significantly over-expressed in the murine recurrent breast tumors and human basal-type breast cancer tumors, both with strong ferroptosis susceptibility and mesenchymal features(9, 12).
DDR2 elevation in recurrent tumor cells contributes to its ferroptosis sensitivity
Next, we further validated the essential role of DDR2 in ferroptosis as predicted from our screen(28). We transduced recurrent tumor cells with non-targeting control shRNA or two independent Ddr2 shRNAs and placed under increasing doses of erastin. Ddr2 knockdown significantly conferred resistance to the erastin-induced cell death based on CellTiter Glo assay (Fig. 2A) and crystal violet staining (Fig. 2B, Supplementary Fig. 2A). We further used CellTox Glo assay to measure the released protease upon cell death and found both Ddr2 shRNAs almost completely abolished erastin-induced protease release (Fig. 2C). Next, C11-BODIPY staining revealed that Ddr2 knockdown decreased lipid peroxidation under erastin treatment (Fig 2D, E). Collectively, these data showed that Ddr2 knockdown protected recurrent tumor cells from erastin-induced ferroptosis. Consistently, DDR2 knockdown also protected human MDA-MB-231 cells from erastin-induced ferroptosis as indicated by CellTiter Glo assay (Supplementary Fig. 2B) and CellTox Glo assay (Supplementary Fig. 2C). Moreover, cystine deprivation also induces ferroptosis(12, 15). Consistently, we found that recurrent tumor cells were highly sensitive to cystine deprivation, and Ddr2 knockdown significantly rescued the cell death upon cystine deprivation using CellTiter Glo assay (Figure 2F) and crystal violet staining (Figure 2G, Supplementary Fig. 2D). Furthermore, we found that Ddr2 knockdown significantly mitigated the GSH depletion during cystine deprivation in recurrent tumor cells (Supplementary Fig. 2E), suggesting a role of GSH preservation in the ferroptosis protection phenotypes
Fig. 2. Genetic and chemical inhibition of DDR2 rescued ferroptosis in recurrent tumor cells.
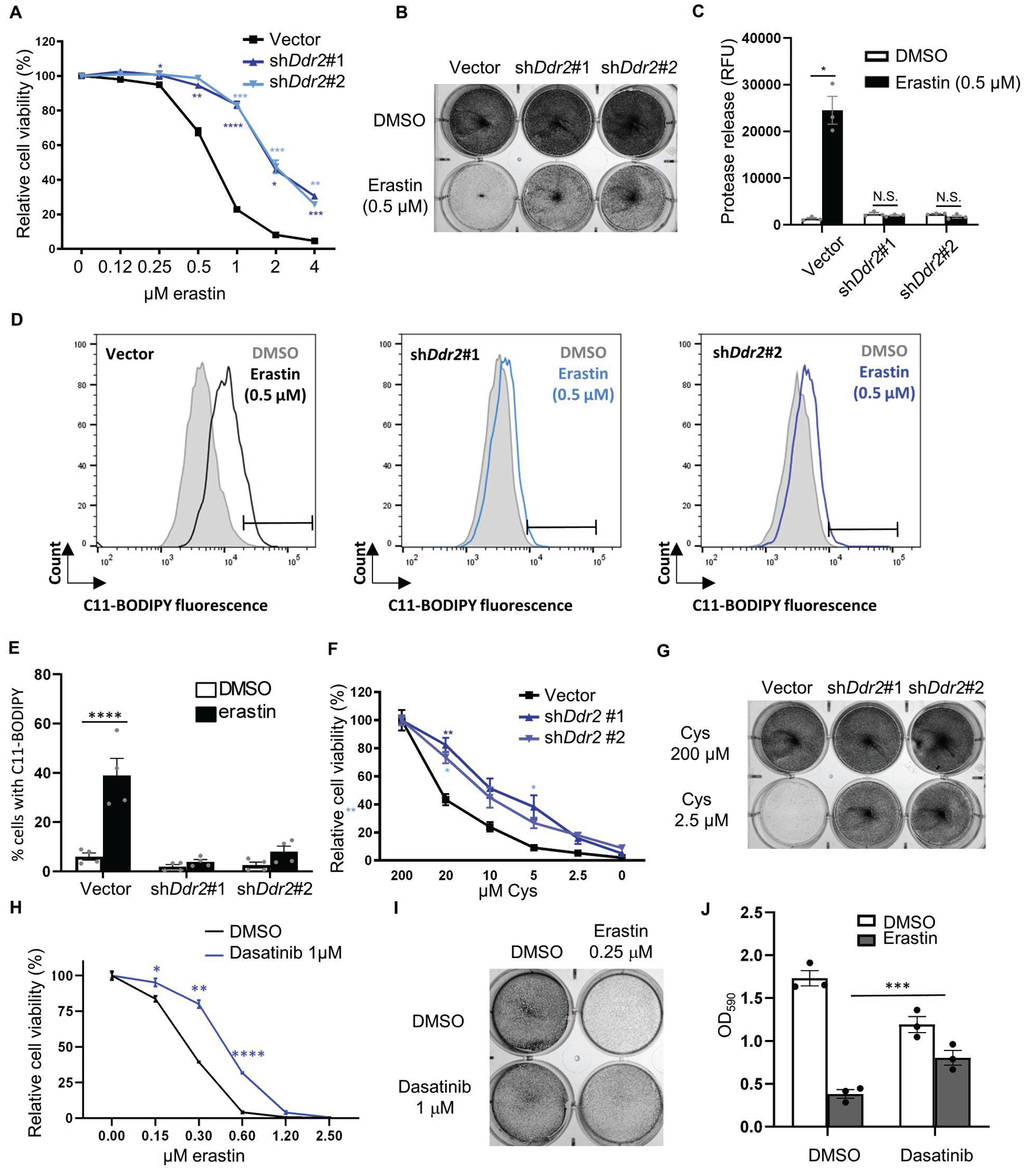
(A-E) Ddr2 knockdown rescued ferroptosis triggered by erastin. Recurrent tumor cells transduced with control or two Ddr2 shRNAs were treated with erastin for 19 hours and the cellular viability was determined by CellTiter Glo (A) and crystal violet staining (B). The effects on cell death were determined by CellTox Glo (C) and lipid peroxidation by C11-BODIPY staining (D, E). Representative data from one out of four independent experiments are shown (D). Quantification of lipid peroxidation in (E). (F-G) DDR2 knockdown rescued the reduction in cell viability triggered by cystine deprivation. Recurrent tumor cells transduced with control or Ddr2 shRNAs were treated with cystine deprivation and the cell numbers were determined by CellTiter Glo after 18 hours of incubation (F) or crystal violet staining after 16 hours of incubation (G). (H-J) DDR2 inhibitor, dasatinib (1 μM), rescued cell death triggered by erastin. Recurrent tumor cells were treated with dasatinib, erastin alone or in combination, and rescue effects of dasatinib were determined by CellTiter Glo (H), crystal violet staining (I), quantification of crystal violet (J). (A, F, H) n=3 biological replicates. (A, C, E, F, H, J) Two-way ANOVA, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 Dunnett’s multiple comparisons. Bars show S.E.M.
To prevent the off-target effect of shRNA-mediated knockdown, we reconstituted the human DDR2 (hDDR2) mRNA in mouse recurrent tumor cells transduced with shRNA targeting mouse but not human DDR2 cDNA (Supplementary Fig. 2F, G). First, we found that the ferroptosis protection phenotypes by Ddr2 shRNA were abolished by human DDR2 cDNA (Supplementary Fig. 2H). Next, we examined GSH levels (Supplementary Fig. 2I). Consistent with previous results of cystine deprivation (Supplementary Fig. 2E), Ddr2 knockdown also preserved GSH levels under erastin (Supplementary Fig. 2I). hDDR2 significantly mitigated the GSH preservation by Ddr2 knockdown (Supplementary Fig. 2I). Previously, we found that erastin dramatically increased xCT expression in recurrent tumor cells (Supplementary Fig. 1C), which was suppressed by Ddr2 knockdown but restored by human DDR2 cDNA (Supplementary Fig. 2J). These data further confirmed the specificity of DDR2 in modulating ferroptosis.
DDR2 is a receptor kinase that can be potently inhibited by DDR2 inhibitors, including dasatinib(32). We found the inhibition of DDR2 by a low dose of dasatinib (1 μM) rescued erastin-induced ferroptosis as determined by CellTiter Glo assay (Fig 2H), crystal violet staining (Fig. 2I), and its quantification (Fig. 2J). These data indicate that pharmacological inhibition of DDR2 can also prevent ferroptosis.
To understand whether the ferroptosis protection of Ddr2 knockdown was restricted to erastin, we tested the recurrent tumor cells with two GPX4 inhibitors (ML162 and RSL3). Both genetic or pharmacological inhibition of DDR2 significantly protected against death induced by ML162 and RSL3 (Supplementary Fig. 2M, N), indicating a broader ferroptosis protection of DDR2 inhibition.
Next, we expressed the DDR2 cDNA in primary tumor cells and found significantly ferroptosis-enhancing effects using CellTox Green assay (Supplementary Fig. 2O). Collectively, these data indicate that the elevated Ddr2 expression in the murine recurrent breast tumor cells is both sufficient and essential for their ferroptosis sensitivities.
EMT regulators promote DDR2 expression
Given the reported connection between EMT and DDR2(26, 27), we transduced EMT regulators Twist (Fig. 3A) and Snail (Fig. 3B) in the mouse primary tumor cells. Consistent with the previous finding (33), the transduction of either Twist or Snail significantly increased the expression of the Ddr2 mRNA and protein (Fig 3C and Supplementary Fig. 3A). These results indicate that either EMT regulator was sufficient to induce the expression of Ddr2 mRNA. In human T47D cells, the transduction with TWIST or SNAIL also increased the protein expression of DDR2 (Fig. 3D). Therefore, these two EMT regulators can induce the expression of DDR2 and may contribute to the high DDR2 expression in the murine and human breast tumor cells.
Fig. 3. EMT regulators upregulate DDR2 expression.
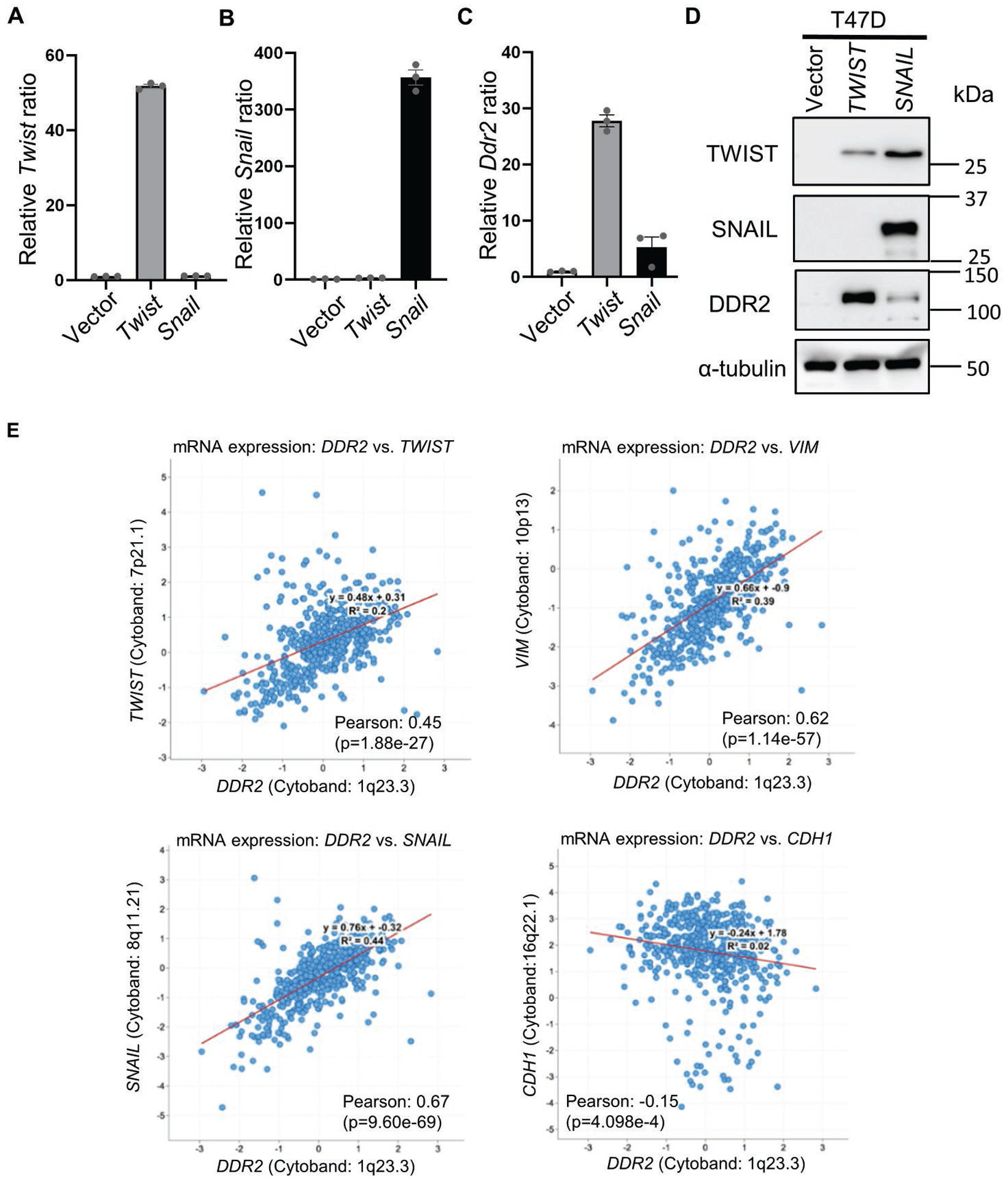
(A-C) Twist and Snail increased Ddr2 RNA expression. Primary tumor cells overexpressing Twist (A) or Snail (B) showed an increase in Ddr2 RNA expression as determined by RT-PCR (C). (D) T47D cells transduced with TWIST or SNAIL overexpression showed DDR2 upregulation by Western blots. (E) The correlation of DDR2 with mesenchymal markers TWIST, SNAIL and VIM, and epithelial marker, CDH1 in human breast cancer dataset from TCGA. Bars show S.E.M..
Next, we examined the relationship between the expression levels of DDR2 and various EMT-related genes in TCGA. DDR2 mRNA expression in the TCGA breast tumors showed a significant positive correlation with known mesenchymal markers, including TWIST, SNAIL, and VIM, and negative correlation with epithelial marker, CDH1 (Figure 3E). These in vivo correlation data further support the regulatory relationship between DDR2 and EMT process as a genetic determinant of EMT-associated ferroptosis.
Given EMT markers has been associated with poor patient prognosis in breast cancer(34–37), we analyzed whether DDR2 expression in primary tumors was correlated with different clinical outcome in breast cancer cohorts by prognostic database PROGgene V2(38). Indeed, we found that a high level of DDR2 in primary breast tumors was correlated with poor overall survival in the Huang dataset (GSE48390)(39) and the Enerly dataset (GSE19783)(40) (Supplementary Fig. 3B, C). These data suggest the strong correlation of high DDR2 expression level with poor clinical outcome of breast cancer patients.
EMT-driven DDR2 upregulation determines sensitivity to ferroptosis
Given our previous findings that EMT is associated with cystine addiction(12), we wished to examine the role of the EMT-upregulated DDR2 in determining ferroptosis sensitivity. Since antibiotic selection on murine primary tumor cells to establish stably transduced lines is technically challenging, we test this hypothesis using human breast cancer T47D cells. First, we established T47D cell lines overexpressing TWIST or SNAIL and found it more susceptible to erastin-induced ferroptosis by Celltiter Glo (Fig. 4A), CytoTox Glo (Fig. 4B), and lipid peroxidation (Supplementary Fig. 4A, B). Reciprocally, the knockdown of Twist or Snail shRNA in recurrent tumor cells conferred ferroptosis resistance (Supplementary Fig. 4C, D). Similarly, knockdown DDR2 by shRNAs in TWIST or SNAIL-expressing T47D significantly mitigated the enhanced ferroptosis as measured in cell viability (Fig. 4C, E) and cell death-associated protease release (Fig. 4D, F). Taken together, these data suggest that the DDR2 upregulation by EMT contributes to the enhanced ferroptosis sensitivity in breast cancer cells.
Fig. 4. EMT-mediated DDR2 upregulation determines its sensitivity to ferroptosis.
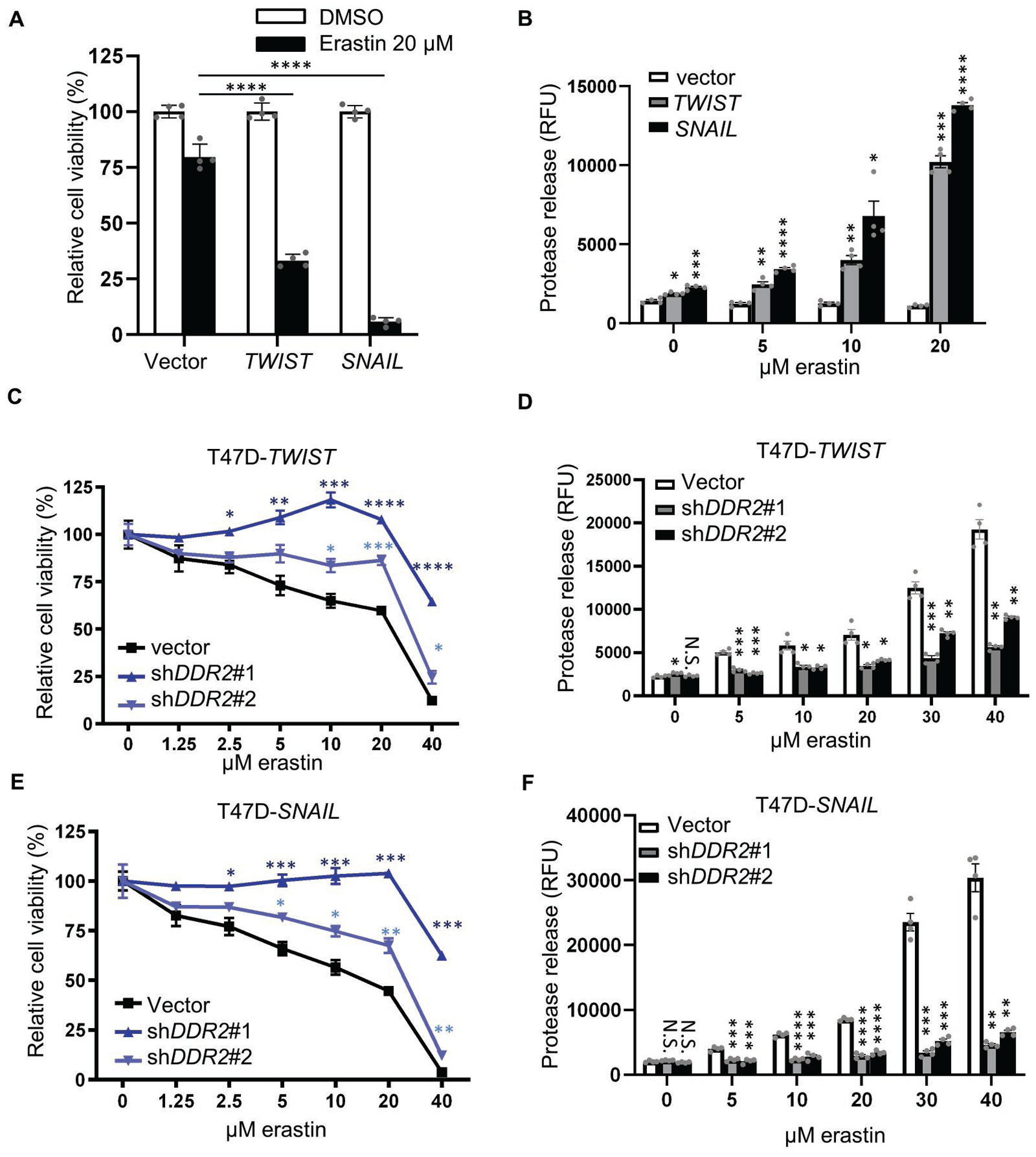
(A-B) T47D cells overexpressing TWIST or SNAIL were more sensitive to ferroptosis as determined by CellTiter Glo assay (viability) (A) and CellTox Glo (death-associated protease release) (B) after 24 hours of incubation. (C-F) DDR2 knockdown abolished the sensitivity to ferroptosis induced by TWIST or SNAIL overexpression. The DDR2 was knocked down by two shRNAs in T47D cells overexpressing TWIST or SNAIL. The viability of indicated cells, when treated with an increasing dose of erastin for 24 hours, was determined by CellTiter Glo assay(viability) (C, E) or CellTox Glo assay (death-associated protease release) (D, F). (C, E) n=4 biological replicates. (A-F) Two-way ANOVA, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 Dunnett’s multiple comparisons. Bars show S.E.M..
DDR2 knockdown reduced cell proliferation and YAP/TAZ activity
Given the unexpected and dramatic upregulation of Ddr2 in the recurrent tumors, we investigated its functional role. Interestingly, we noticed a cell density-dependent effect of Ddr2 knockdown on cell proliferation. When grown on higher density, Ddr2 knockdown only had modest effects on cell proliferation. However, when seeded a low cell density, Ddr2 knockdown led to a noticeable decrease in cell proliferation. Thus, we employed clonogenic assay to determine the effects of Ddr2 knockdown in recurrent tumor cells and found a significantly reduced colony formation (Fig. 5A, quantified in Fig. 5B). Consistently, when DDR2 was knocked down in MDA-MB-231, the colony formation was dramatically reduced (Supplementary Fig. 5A, quantified in Supplementary Fig. 5B). To validate this proliferation phenotype over time, we labeled each nuclei with histone H2B-mCherry for the continuous quantification of cell numbers. These cells were then transduced by control or two Ddr2 shRNAs and monitored by Incucyte S3 for 4 days (Fig. 5C). Indeed, Ddr2 knockdown in recurrent tumor cells led to a significant and consistent decrease in cell numbers (Fig. 5C). Similar reduced proliferation was also noted upon the knockdown of DDR2 in MDA-MB-231 cells (Supplementary Fig. 5C). To test the possibility of whether the ferroptosis resistance upon Ddr2 knockdown is due to proliferation arrest, we induced proliferation arrest in recurrent tumor cells with thymidine and found no effects on the ferroptosis resistance upon Ddr2 knockdown (Supplementary Fig. 5D, E). Collectively, these data showed that the exaggerated expression of DDR2 is essential for cellular proliferation of both murine recurrent breast cancer and human basal-type breast cancer cells.
Fig. 5. DDR2 regulates cell proliferation and YAP/TAZ activity.
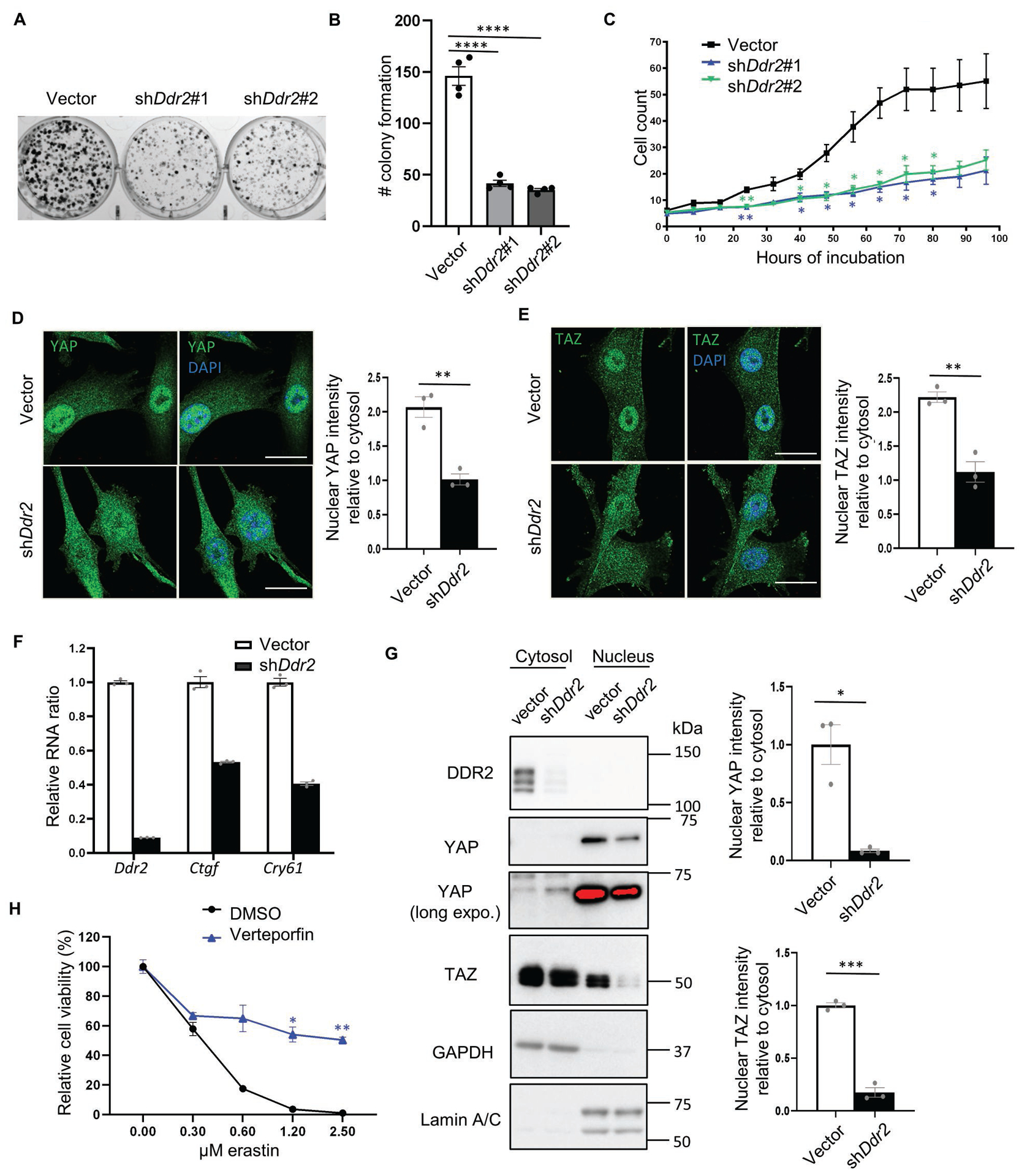
(A-B) The clonogenic assay showed Ddr2 knockdown reduced the number of colony formation. Recurrent tumor cells transduced with control or Ddr2 shRNAs were plated on 6 well plates (250 cells/well) and incubated for 10 days. The cells were then fixed by paraformaldehyde and stained with crystal violet for direct counting of colony numbers as quantified in (B). (C) Recurrent tumor cells with Ddr2 knockdown showed a reduction in cell proliferation under image-based monitoring of cell numbers. Recurrent tumor cells stably expressing histone H2B-mCherry were knocked down by control or Ddr2 shRNAs and plated in a 96-well plate. The image of each well was taken every 8 hours using Incucyte S3 for the quantification of cell numbers. (D-E) Ddr2 knockdown decreased the nuclear localization of YAP(D)/TAZ(E). Recurrent tumor cells were labeled by YAP(D) and TAZ(E) specific antibodies with a counterstain of DAPI for nuclei. Quantification was performed using line analysis by Image J. Nuclear (DAPI positive) and nuclear (DAPI negative) intensity were determined by the average of 5 cells for each sample and 3 independent repeats. Scale bar, 10μm. (F) RT-PCR validated the downregulation of Ctgf and Cyr61, two canonical YAP/TAZ target genes, upon Ddr2 knockdown in recurrent tumor cells. (G) Nuclear/cytosol fractionation showed reduced nuclear YAP/TAZ upon Ddr2 knockdown in recurrent tumor cells. GAPDH: cytosolic marker; Lamin A/C: nuclear marker. Relative nuclear/cytosolic YAP and TAZ ratio was determined by ImageJ. (H) Pharmacological suppression YAP by verteporfin (2 μM) mitigated erastin induced ferroptosis in recurrent tumor cells as determined by CellTiter Glo assay. (B) One-way ANOVA, ****p < 0.0001 Tukey’s multiple comparisons. (G) *p<0.05, ***p<0.001, two-tailed Student’s t-test. (C, H) n=3 biological replicates. Two-way ANOVA, *p < 0.05, **p < 0.01, Dunnett’s multiple comparisons. Bars show S.E.M..
The Hippo pathway is recognized as an important cell density-dependent signaling regulator(41). YAP and TAZ are the two downstream effectors of the Hippo pathway which serve as coactivators of TEAD transcription factors to promote cell proliferation and oncogenesis(41). When the Hippo pathway is activated (such as high cell density), YAP/TAZ is phosphorylated for proteasomal degradation and retained cytosolically. When the Hippo pathway is inactivated (such as low cell density), YAP/TAZ is nuclearly translocated and interacts with TEAD to trigger gene expression(41). Thus, we first tested whether Ddr2 knockdown affects the localization of YAP/TAZ. Using immunofluorescent labeling of YAP- and TAZ- specific antibodies with DAPI for nuclei, we found that Ddr2 knockdown decreased nuclear localization of both YAP and TAZ protein using line analysis (Fig. 5D, E).
Next, we measured the RNA expression of Ctgf and Cyr61, two canonical YAP/TAZ target genes(41). We found Ddr2 knockdown in recurrent tumor cells significantly reduced the expression of both Ctgf and Cyr61 (Fig. 5F), consistent with the reduced YAP/TAZ activities. Furthermore, cellular fractionation revealed that Ddr2 knockdown decreased nuclear/cytosolic ratio of YAP and TAZ proteins to 8.3% and 17.4%, respectively (Figure 5G). Next, we examined whether nuclear YAP/TAZ was differentially expressed between primary and recurrent tumor cells and found that recurrent tumor cell lines have more nuclear YAP/TAZ and upregulation of Cyr61 expression (Supplementary Fig. 5F, G). Taken together, these data suggest that Ddr2 knockdown significantly reduced the nuclear YAP/TAZ and activities in the recurrent tumor cells.
YAP/TAZ activity is well recognized to drive the proliferation of cancer cells(41, 42). Furthermore, several reports have revealed the critical role of YAP/TAZ activity in the ferroptosis susceptibility(15, 21–23). To confirm the essential role of YAP/TAZ in ferroptosis, we treated recurrent tumor cells with verteporfin (YAP/TAZ inhibitors) that significantly protected erastin-induced ferroptosis (Fig. 5H). These data suggest that downregulated YAP/TAZ activity upon Ddr2 knockdown may contribute to both ferroptosis protection and reduced clonogenic phenotypes.
Erastin-induced DDR2 phosphorylation regulates ferroptosis
To further investigate the connection between DDR2 and ferroptosis, we determined how erastin affects the levels of DDR2 protein in primary and recurrent tumor cells. The primary tumor cells did not have a detectable DDR2 protein before and after erastin treatment (Fig. 6A). However, the robust DDR2 expression in the recurrent tumor cells was further enhanced by erastin (Fig. 6A). To validate the specificity of these enhanced DDR2 protein signals, we knocked down DDR2 by shRNA and noted a reduction in both basal and enhanced levels of DDR2 expression, indicating the specificity of this increase in DDR2 protein (Fig. 6B). RT-PCR also revealed an upregulation of DDR2 RNA expression under erastin treatment (Fig. 6C). These data suggest that erastin treatment elevates the mRNA and protein levels of DDR2.
Figure 6. DDR2 phosphorylation sensitizes YAP/TAZ mediated ferroptosis through Src activity.
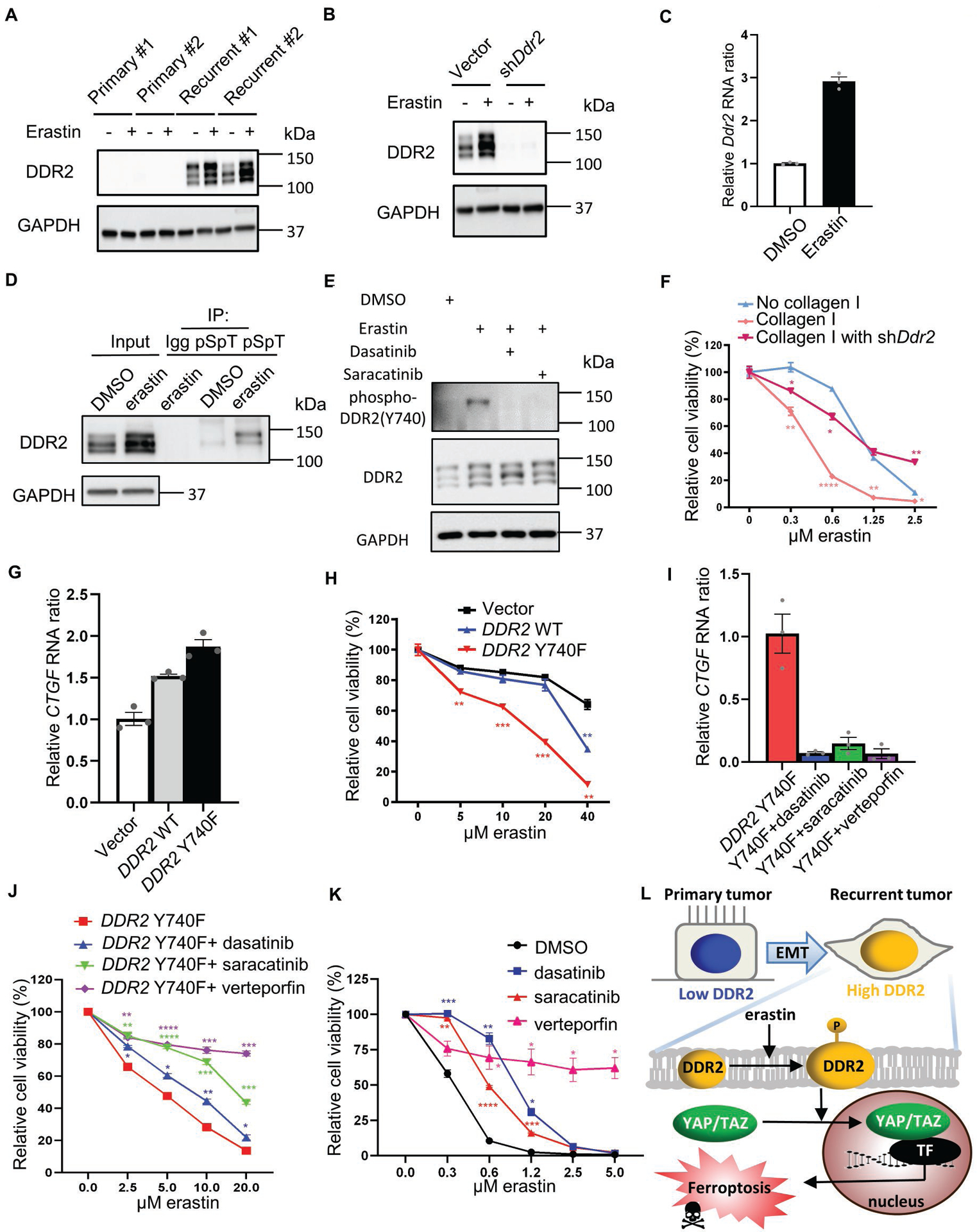
(A) DDR2 upregulation in recurrent tumor cells was further enhanced by erastin. Western blots measured DDR2 levels in two primary and two recurrent tumor cell lines before or after erastin treatment (0.5 μM, 16 hours). (B) DDR2 knockdown abolished basal and enhanced DDR2 protein expression. Recurrent tumor cells transduced with Ddr2 shRNA were treated with erastin (0.5 μM, 16 hours) for Western blots. (C) RT-PCR showed the upregulation of Ddr2 mRNA by erastin treatment (0.5 μM, 16 hours) in recurrent tumor cells. (D) Erastin treatment increased DDR2 phosphorylation. The protein lysate of recurrent tumor cells treated with DMSO or erastin (0.5 μM, 16 hours) were immunoprecipitated by the pan-phospho-Ser/Thr (pSpT) antibody and blotted with DDR2 antibody for phosphorylated DDR2. (E) Erastin triggered phosphorylation of DDR2 protein at Y740. Recurrent tumor cells treated with erastin (0.5 μM) and DMSO, dasatinib (1 μM), or saracatinib (1 μM) for 16 hours were stained with phospho-DDR2(Y740) antibody for DDR2 phosphorylation and activation. (F) Collagen I coating increased ferroptosis sensitivity by DDR2. Recurrent tumor cells transduced with Ddr2 shRNA were plated on a regular or collagen I coating plate for 19 hours of erastin treatment and CellTiter Glo assay. (G) The constitutively active mutant of DDR2 (Y740F) increased the expression of YAP/TAZ targeted gene, CTGF. The CTGF RNA expression in T47D cells overexpressing empty vector, DDR2 wild type cDNA, or constitutively active mutant of DDR2 (Y740F) was determined by RT-PCR. (H) DDR2 Y740F mutant increased ferroptosis sensitivity. The erastin sensitivity of T47D cells in (G) were tested using CellTiter Glo assay. (I-J) Pharmacological suppression of DDR2, Src, and YAP decreased the CTGF upregulation and ferroptosis sensitivity promoted by DDR2 Y740F cDNA. T47D cells overexpressing DDR2 Y740F cDNA were treated with DMSO, dasatinib (1 μM, DDR2 inhibitor), saracatinib (1 μM, Src inhibitor), or verteporfin (2 μM, YAP inhibitor). CTGF expression was determined after 18 hours of incubation by RT-PCR in (I). Ferroptosis sensitivity was determined by co-treatment with erastin for 24 hours and CellTiter Glo assay in (J). (K) Pharmacological suppression of DDR2, Src, and YAP prevented recurrent tumor cells from erastin-induced ferroptosis. Recurrent tumor cells under erastin treatment were co-treated with DMSO, dasatinib (1 μM), saracatinib (1 μM), or verteporfin (2 μM) for 18 hours for CellTiter Glo assay. (L) Schematic illustration of EMT-driven DDR2 upregulation in determining ferroptosis through the regulation of YAP/TAZ. (F, H, J, K) n=3 biological replicates. Two-way ANOVA, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 Dunnett’s multiple comparisons. Bars show S.E.M.
Given the DDR2 phosphorylation determines its kinase and signaling activity(43), we examined the effects of erastin treatments. Owing to the unavailability of mouse site-specific phosphorylated DDR2 antibody, the protein lysates of mouse recurrent tumor cells treated with either DMSO or erastin were pulled down by Phospho-(Ser/Thr) (pSpT) antibody and blotted with DDR2 antibody for phosphorylated DDR2 (Fig. 6D). We found that erastin significantly enhanced phosphorylated DDR2, suggesting the activation of DDR2 under ferroptosis-inducing conditions (Fig. 6D). Next, mouse and human DDR2 protein sequences are evolutionarily conserved especially around its well-known phosphorylation site at Y740. Thus, we used human site-specific phosphorylated DDR2 (phospho-Y740) antibody to validate the increase in phosphorylated DDR2 in mouse recurrent tumor cells under erastin treatment (Fig. 6E). Indeed, we observed a phosphorylated DDR2 (phospho-Y740) signal under erastin treatment that was abolished by dasatinib (DDR2 inhibitor) (Fig. 6E). Given Src, the binding partner of DDR2 was reported to phosphorylate Y740 on DDR2(43), we confirmed that saracatinib (Src inhibitor) abolished DDR2 phosphorylation under erastin treatment (Fig. 6E). To further characterize the erastin-induced DDR2 phosphorylation, we co-treated with N-acetylcysteine (NAC), liproxstatin-1 (Lip-1), 2-mercaptoethanol (Beta-ME), or ferrostatin-1 (Fer-1) with erastin (Supplementary Fig. 6A). These antioxidants and ferroptosis inhibitors significantly reduced erastin-induced DDR2 phosphorylation (Supplementary Fig. 6A). These data suggest that ferroptosis inducer, erastin, triggers the upregulation and phosphorylation/activation of DDR2 protein that is associated with ferroptosis signaling.
Given that DDR2 is a receptor tyrosine kinase for collagen I(44), we thus tested whether collagen I coating can further enhance the sensitivity to erastin. Indeed, we found that recurrent tumors plated on collagen I-coated slides were more sensitive to erastin (Fig. 6F), and this increase in sensitivity was abolished by DDR2 knockdown (Fig. 6F).
Src kinase has been reported to regulate YAP/TAZ activity by direct phosphorylation in several studies(45–47). Given the binding of DDR2 regulates Src activity(48), we tested whether DDR2 phosphorylation also regulates YAP/TAZ activity through Src kinase. Fig. 3D showed that T47D has no detectable level of endogenous DDR2 expression. We thus transduced vector, DDR2 wild type cDNA or constitutively active mutant of DDR2 (Y740F) (43) in T47D cell line. The overexpression of wild-type DDR2, when compared with empty vector, increased the expression of CTGF (Fig. 6G). Importantly, DDR2 Y740F mimicking DDR2 phosphorylation increased CTGF expression at an even higher level than wild-type DDR2 (Fig. 6G).
Given YAP/TAZ activity in determining ferroptosis has been recently reported(15, 21–23), we further treated T47D cells overexpressing empty vector, DDR2 wild-type, or Y740F cDNA with erastin. Consistently, wild-type DDR2 sensitized the cells to erastin (Fig. 6H), which was further enhanced by DDR2 Y740F mutant (Fig. 6H). These data support that the phosphorylation on DDR2 regulates YAP/TAZ activities and ferroptosis sensitivity.
To investigate the role of DDR2 phosphorylation in regulating ferroptosis sensitivity through Src-YAP/TAZ, we inhibited DDR2-Y740F with dasatinib (DDR2 inhibitor), saracatinib (Src inhibitor), or verteporfin (YAP/TAZ inhibitor) (Fig. 6I–J). Indeed, we found these inhibitors repressed the expression level of CTGF mRNA (Fig. 6I). Consistently, these inhibitors also significantly reduced ferroptosis sensitivity (Fig. 6J). These data suggest that Src and YAP/TAZ are indeed the downstream regulators of DDR2 in determining ferroptosis sensitivity. Furthermore, we validated the protective effect of dasatinib, saracatinib, and verteporfin in mitigating the erastin-induced cell death (Fig. 6K) and induction of ferroptosis markers (CHAC1 and PTGS2) (18)(Supplementary Fig. 6B, C) in recurrent tumor cells. Taken together, these results suggested that DDR2 phosphorylation activates YAP/TAZ mediated transcription by Src kinase to trigger ferroptosis. Together, we proposed a model by which the EMT-mediated DDR2 over-expression during breast tumor recurrence contributes significantly to ferroptosis susceptibility via regulation of YAP/TAZ pathways (Fig. 6L).
Discussion
Many genetic determinants have been identified by forward genetic screens to regulate ferroptosis through the metabolisms of cystine and lipid as well as iron biology(49, 50). For example, a whole-genome siRNA screen in HT-1080 cells treated with erastin discovered that cysteine-tRNA synthetase regulates ferroptosis by trans-sulfation(51). Another CRISPR whole-genome screen found that acyl-CoA synthetase long-chain family member 4 (ACSL4) regulates ferroptosis by lipid reprogramming(52). Recently, ATM(53), MESH1(20), and KEAP1 glycosylation(54, 55) were identified as relevant determinants of ferroptosis. Furthermore, the relevance of cell density and contact in the ferroptosis revealed Hippo effectors YAP/TAZ as determinants of ferroptosis(15, 21–23). However, much remains unknown about the cell-type-specific determinants of ferroptosis, including the connection between EMT and ferroptosis. Here, we provide compelling evidence that the over-expression of DDR2 in breast tumor cells with mesenchymal features are responsible for their exquisite sensitivity to ferroptosis (Fig. 6L). DDR2 is intimately connected with the mesenchymal differentiation and induced by EMT regulators SNAIL or TWIST(26, 48, 56–58). Therefore, the elevated DDR2 may provide the molecular underpinning by which EMT and mesenchymal features are associated with ferroptosis sensitivity. Unexpectedly, DDR2 is also essential for the proliferation and viability of the recurrent tumor cells. Both the ferroptosis protection and reduced proliferation phenotypes caused by DDR2 knockdown can be explained by the repression of YAP/TAZ proteins and activities. Therefore, EMT-regulated DDR2-YAP/TAZ provides the molecular connection between the proliferative state and ferroptosis susceptibility in the recurrent breast tumor cells with prominent mesenchymal features.
EMT is associated with induction of DDR2, which becomes phosphorylated when activated by collagen I, an abundant and critical protein in the extracellular matrix. Also, the activation of DDR2 regulates SNAIL protein in an Src-dependent manner to maintain mesenchymal fate and invasive phenotypes (26, 27). Furthermore, DDR2 expression and activation in breast and other tumors are associated with poor outcome and more invasive cancer(57, 59–61). Therefore, DDR2 plays a critical role in the signaling alterations during EMT to integrate extracellular signals (collagens and hypoxia) to invasive phenotypes, mobility, and chemo-resistance(62, 63). Therefore, inhibition of DDR2 has shown encouraging therapeutic efficacy(64, 65). Here, we discover that elevated DDR2 in the mouse recurrent tumors and human basal-type breast cancer cells also sensitizes these breast cancer cells to ferroptosis-inducing stimuli. Therefore, triggering ferroptosis may have significant therapeutic potential to target these cell populations that resist to most therapeutic agents. Also, our results suggest that a high level of DDR2 may predict the tumors which are likely to respond to these ferroptosis-inducing therapeutics.
In breast cancer, several elegant studies have shown that DDR2 plays a critical role in both tumor cells and stromal cells during breast cancer progression and metastasis. Consistent with the DDR2 expression in the basal-type breast cancer cells, DDR2 is expressed in the human triple-negative breast tumors and the tumor stroma(56). DDR2 is involved in the intimate and reciprocal signaling exchanges between breast tumor cells and mesenchymal stromal cells(63). DDR2 is also used by breast tumor leader cells as the biomechanical cue for directed collective migration and invasion (66). In the stromal cells, DDR2 regulates collagen fiber remodeling at the tumor-stromal boundary to shape the tumor microenvironment(62). Therefore, it will be important to determine whether the ferroptosis may also affect the interaction of the tumor-stromal cells.
While collagen I was found to enhance the ferroptosis sensitivity (Fig. 6F), most of our experiments did not involve the addition of collagen I to activate DDR2 either in the ferroptosis screen(53) or subsequent experiments. We speculate that the ferroptosis-inducing conditions, including extreme oxidative stresses, may achieve enough DDR2 activation to trigger ferroptosis. Another contributing factor is that the DDR2 is grossly exaggerated in the recurrent tumor cells that may further sensitize these cells to ferroptosis-inducing signals. Such collagen-independent DDR2 signaling is also mediated by Pregnancy-associated plasma protein A during pregnancy-associated breast cancer (67). Therefore, the enhanced oxidative stress in the tumor microenvironment may enhance the collagen I-activation of DDR2 to promote tumor invasion and metastasis.
The somatic mutations in DDR2 are found in 4% of non-small cell lung cell carcinoma and other tumor types(68). In these tumors bearing mutated DDR2, the dysregulated DDR2 signaling contribute to oncogenesis and tumor progression. Therefore, DDR2 inhibitor, dasatinib, has been approved to target tumors with oncogenic DDR2 mutations(68). However, our results may suggest that the DDR2 inhibitors may interfere with the ferroptosis which will be induced by oxidative stresses or ferroptosis-inducing therapeutics. The in vivo relevance of such effects on ferroptosis remains to be determined. One obvious implication is the combining DDR2 inhibitors with ferroptosis-inducing agents may mitigate the therapeutic efficacy.
Our study also has significant therapeutic implications for many non-cancer human diseases. Ferroptosis has been involved in the neurodegenerations and other human diseases, such as neurotoxicity(69), acute renal failure(70), hepatic(71), and cardiac(72) injuries and ischemia-reperfusion injury(73). Therefore, modulating ferroptosis may have therapeutic potentials among those diseases. Our results suggest that DDR2 inhibitors, many of which have been approved by the FDA for cancer treatments, may interfere with ferroptosis and improve the disease progression and clinical outcomes of these devastating diseases involving dysregulated ferroptosis.
Methods
Cell culture
SUM52, ZR751, BT474, MCF7 T47D, BT20, MDA-MB-231, Hs 578T, BT549 cells, and MDA-MB-157cells were obtained from the Cell Culture Facility at Duke University (Durham, NC, USA). These cell lines have been authenticated by STR DNA profiling and tested to be mycoplasma-free before being frozen by the Cell Culture Facility. These cell lines were maintained for less than 6 months. Primary and recurrent breast MTB/TAN tumor cells were described previously(6). All cell lines were cultured in DMEM (GIBCO-11995) supplemented with 10% heat-inactivated fetal bovine serum (#10082147, ThermoFisher) and antibiotics (streptomycin, 10,000 UI/ml and penicillin, 10,000 UI/ml, #15140122, ThermoFisher) in a humidified incubator at 37°C and 5% CO2. For primary tumor cells, 5 μg/ml insulin, 10 ng/ml EGF, 1 μg/ml hydrocortisone, 5 μg/ml prolactin, 1 μM progesterone, and 2 μg/ml doxycycline were supplemented to the culture media to maintain Her2/neu expression. For recurrent tumor cells, 5 μg/ml insulin and 10 ng/ml EGF were supplemented to the culture media. To prepare media for cystine deprivation, DMEM without glutamine, methionine, and cystine (#21013024, ThermoFisher) supplement with 10% heat-inactivated fetal bovine serum, 1 × antibiotic, L-glutamine (4 mM, Sigma), and L-methionine (0.2 mM, Sigma) were further added with the indicated concentration of cystine (from 0–200 μM, Sigma). Cells were first plated and cultured in complete DMEM media. The cells were then washed twice with PBS and replaced with media with a low concentration of cystine. After 18 to 26 hours of incubation, the cells were further analyzed by indicated assays.
Constructs and lentivirus viral infections
shRNAs targeting mouse DDR2 RNA were purchase from Sigma (TRCN0000023594, TRCN0000361395). shRNA targeting human DDR2 RNA were purchase from Sigma (TRCN00000121117, TRCN0000121262, and TRCN0000121172). DDR2 cDNA from pDONR223-DDR2 (#23897, Addgene) was subcloned to PLX302 (#25896, Addgene) using Gateway cloning for lentiviral expression. Point mutants (K608A, Y740F) were generated using QuikChange II XL Site-Directed Mutagenesis Kit (#200521, Agilent). pWZL (control vector), pWZL Blast Twist ER (#18799, Addgene), pWZL Blast Snail ER (#18798, Addgene). Lentivirus expressing specific constructs was generated by transfecting HEK-293T cells in 6 well plates with a 1: 1: 0.1 ratio of lentiviral vector: pMD2.G: psPAX2 with TransIT-LT1 transfection reagent (Mirus). For retrovirus, a 1: 0.1: 1 ratio of retroviral vector: pCMV-VSV-G: pUMVC was used. After filtering through cellulose acetate membrane (0.45 μm, #28145–481, VWR), 250 ul of media with lentivirus were added to a 60mm dish of indicated cells with polybrene (8ug/ml) and selected with puromycin.
Cell viability and cytotoxicity
Cell viability assay was performed and determined by CellTiter-Glo luminescent cell viability assay (Promega) following the manufacturer’s protocol. CellTiter-Glo substrate (15 μl) was added to the cells cultured in a 96-well plate with 100 μl media for 10 min of shaking and quantification of signal intensity using chemiluminescence plate reader. Cytotoxicity assay was measured by the rupture of the cell membrane and released protease using CellTox Glo cytotoxicity assay (Promega) by following the manufacturer’s protocol. CellTox Glo substrate (20 μl) was added to 20 μl of culture media in a 96-well plate for 30 min of incubation at 37 degrees and quantification of signal intensity using a fluorescence plate reader. CellTox Green assay (Promega) was performed by adding dye (1:1000) to the media for quantification of cell death by a fluorescence plate reader. For crystal violet staining, cells were first fixed with 4% paraformaldehyde for 20min at RT. After washed with PBS, the cells were incubated with filtered 0.2% crystal violet in methanol for 30 min at RT. GSH level was determined by GSH/GSSG-Glo Assay (Promega) following the manufacturer’s protocol.
Western blots
Western blotting was performed as previously described(74). Nuclear and cytoplasmic fractions of cell lysates were isolated following manufacturer’s protocol (#78835, ThermoFisher). Protein concentrations were quantified using BCA assay (#23227, ThermoFisher). Around 20 μg of protein was loaded on 8% SDS-PAGE gels, transferred to PDVF membrane, blocked with 5% non-fat milk in 1xTBST, incubated with primary antibodies overnight at 4°C. Primary antibodies: DDR2 (1:1000, PA1879, Boster); phospho-DDR2 Y740 (1:1000, MAB25382, R&D systems); α-tubulin (1:1000, sc-32293, Santa Cruz); β-tubulin (1:1000, #2128, Cell Signaling); GAPDH (1:2000, sc-25778, Santa Cruz); Twist1/2 (1:1000, GTX127310, GeneTex); Snail (1:1000, #3895, Cell signaling; Lamin A/C (1:1000, #4777T, Cell signaling); TAZ (1:1000, 560235, BD biosciences); YAP (1:1000, sc376830, Santa Cruz); YAP (1:1000, #14074, Cell Signaling); E-cadherin (1:1000, 610404, BD biosciences). For immunoprecipitation, Phospho-(Ser/Thr) Phe Antibody (1:100, #9631, Cell Signaling).
Quantitative real-time PCR
RNA was extracted and purified using the RNeasy Mini Kit (Qiagen) following the manufacturer’s protocol. Reverse transcription to cDNA was performed using random hexamers and reverse transcriptase SuperScript III (Invitrogen). Quantitative real-time PCR was performed by adding cDNA, primers, and Power SYBR Green PCR Mix (Applied Biosystems) and analyzed on StepOnePlus Real-time PCR system (Applied Biosystems). Samples were technically triplicated for mean+/− SEM. Data were representative of at least two independent repeats. Mouse β-actin (reference gene) primers: sense, 5’- GGC TGT ATT CCC CTC CAT CG −3’, antisense, 5’- CCA GTT GGT AAC AAT GCC ATG T-3’; Mouse DDR2 primers: sense, 5’ ATC ACA GCC TCA AGT CAG TGG-3’, antisense, 5’- TTC AGG TCA TCG GGT TGC AC-3’. Mouse CTGF primers: sense, 5’- GCC TAC CGA CTG GAA GAC AC-3’, antisense, 5’- GGA TGC ACT TTT TGC CCT TCT TA-3’. Mouse CYR61 primers: sense, 5’- CTG CGC TAA ACA ACT CAA CGA-3’, antisense, 5’- GCA GAT CCC TTT CAG AGC GG-3’. Mouse CHAC1 primers: sense, 5’- CTG TGG ATT TTC GGG TAC GG-3’, antisense, 5’- CCC CTA TGG AAG GTG TCT CC-3’. Mouse PTGS2 primers: sense, 5’- TTC AAC ACA CTC TAT CAC TGG C-3’, antisense, 5’- AGA AGC GTT TGC GGT ACT CAT-3’. Mouse GPX4 primers: sense, 5’- GAT GGA GCC CAT TCC TGA ACC-3’, antisense, 5’- CCC TGT ACT TAT CCA GGC AGA-3’. Mouse xCT primers: sense, 5’- GGC ACC GTC ATC GGA TCA G-3’, antisense, 5’- CTC CAC AGG CAG ACC AGA AAA-3’ Mouse ACSL4 primers: sense, 5’- CTC ACC ATT ATA TTG CTG CCT GT-3’, antisense, 5’- TCT CTT TGC CAT AGC GTT TTT CT-3’ Human DDR2 primers: sense, 5’-CCA GTC AGT GGT CAG AGT CCA-3’, antisense, 5’- GGG TCC CCA CCA GAG TGA TAA-3’. Human CTGF primers: sense, 5’- CAG CAT GGA CGT TCG TCT G-3’, antisense, 5’- AAC CAC GGT TTG GTC CTT GG-3’. Human CYR61 primers: sense, 5’- ACC GCT CTG AAG GGG ATC T-3’, antisense, 5’- ACT GAT GTT TAC AGT TGG GCT G-3’ Human GAPDH primers: sense, 5’- GAG TCA ACG GAT TTG GTC GT-3’, antisense, 5’- TTG ATT TTG GAG GGA TCT CG-3’.
Lipid peroxidation C11-BODIPY assay
Lipid peroxidation was determined by C11-BODIPY staining according to the manufacturer’s protocol (D3861, ThermoFisher Scientific). In short, control or DDR2 knockdown cells were treated with vehicle control or 0.5 μM erastin for 16 hours. The medium was then replaced by a 10μM C11-BODIPY-containing medium for 1h. After harvested, washed, and resuspended in PBS plus 1% BSA, the levels of lipid peroxidation were determined by flow cytometry (FACSCanto ™ II, BD Biosciences).
Quantification of cell growth
For the clonogenic assay, two hundred and fifty cells/well were plated in 6 well plates. Depending on the cell types, the samples were fixed by 4% paraformaldehyde after 6–10 days of incubation and stained with crystal violet for counting of colony number. For image-based quantification of cell count, mouse recurrent tumor cells or MDA-MB-231 cells were first transduced with stably expressed histone H2B-mCherry (#89766, Addgene) for fluorescent labeling of the nucleus. For 96 well plate, each well was seeded with 250 cells/well of control or DDR2 shRNA expressing cells. The image of each well was taken every 8 hours using Incucyte S3 (Essenbio). The number of cells of each image was then quantified by the software provided by Incucyte S3.
Immunofluorescence microscopy
Mouse recurrent tumor cells were washed twice with PBS and fixed in 3.7% paraformaldehyde for 15 min. The cells were then permeabilized with 0.2% Triton X-100 and blocked using 2% BSA for 20 min. Fixed samples were incubated with primary antibodies overnight and secondary antibodies for 30 min. Immunofluorescence microscopy was performed using a confocal microscope with Airyscan for super-resolution quality (880, Zeiss). Antibody: YAP (1:100, #14074, Cell Signaling); TAZ (1:100, #560235, BD biosciences); Goat anti-rabbit IgG Alexa Fluor 488 (1:500, A11034, ThermoFisher).
Statistical analysis
Individual data points representing the number of biological replicates have been provided in each bar graph. For the line graph, the numbers of biological replicates have been provided in the figure legends. Data represent the mean +/− the standard error of the mean. p-values were determined by one way ANOVA with Tukey’s multiple comparisons, two way ANOVA test with Dunnett’s multiple comparisons, or a two-tailed Student’s t-test in Graphpad. Error bars represent SEM, and significance between samples is denoted as *p < 0.05; **p < 0.01; ***p < 0.001; and ****p < 0.0001.
Supplementary Material
Acknowledgments
We are grateful for technical support from the members of the Chi lab. We also appreciate the generosity of Dr. Everardo Macias for allowing access to the Incucyte S3. We acknowledge the financial support in part by DOD grants (W81XWH-17-1-0143, W81XWH-15-1-0486, W81XWH-19-1-0842, W81XWH-20-1-0907) and NIH grants (R01GM124062, 1R01NS111588-01A1, 1R21-AI149205).
Footnotes
Data availability
All data and reagents supporting the findings of this study are available from the authors upon reasonable request.
Conflict of interest statement
The authors have declared that no conflict of interest exists.
References
- 1.Kimbung S, Loman N, Hedenfalk I. Clinical and molecular complexity of breast cancer metastases. Semin Cancer Biol. 2015;35:85–95. [DOI] [PubMed] [Google Scholar]
- 2.Moody SE, Sarkisian CJ, Hahn KT, Gunther EJ, Pickup S, Dugan KD, et al. Conditional activation of Neu in the mammary epithelium of transgenic mice results in reversible pulmonary metastasis. Cancer Cell. 2002;2(6):451–61. [DOI] [PubMed] [Google Scholar]
- 3.Liu P, Cheng H, Santiago S, Raeder M, Zhang F, Isabella A, et al. Oncogenic PIK3CA-driven mammary tumors frequently recur via PI3K pathway-dependent and PI3K pathway-independent mechanisms. Nat Med. 2011;17(9):1116–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boxer RB, Jang JW, Sintasath L, Chodosh LA. Lack of sustained regression of c-MYC-induced mammary adenocarcinomas following brief or prolonged MYC inactivation. Cancer Cell. 2004;6(6):577–86. [DOI] [PubMed] [Google Scholar]
- 5.Gunther EJ, Moody SE, Belka GK, Hahn KT, Innocent N, Dugan KD, et al. Impact of p53 loss on reversal and recurrence of conditional Wnt-induced tumorigenesis. Genes Dev. 2003;17(4):488–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lin CC, Mabe NW, Lin YT, Yang WH, Tang X, Hong L, et al. RIPK3 upregulation confers robust proliferation and collateral cystine-dependence on breast cancer recurrence. Cell Death Differ. 2020;27(27):2234–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mabe NW, Fox DB, Lupo R, Decker AE, Phelps SN, Thompson JW, et al. Epigenetic silencing of tumor suppressor Par-4 promotes chemoresistance in recurrent breast cancer. J Clin Invest. 2018;128(10):4413–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fox DB, Garcia NMG, McKinney BJ, Lupo R, Noteware LC, Newcomb R, et al. NRF2 activation promotes the recurrence of dormant tumour cells through regulation of redox and nucleotide metabolism. Nat Metab. 2020;2(4):318–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moody SE, Perez D, Pan TC, Sarkisian CJ, Portocarrero CP, Sterner CJ, et al. The transcriptional repressor Snail promotes mammary tumor recurrence. Cancer Cell. 2005;8(3):197–209. [DOI] [PubMed] [Google Scholar]
- 10.Tsai JH, Yang J. Epithelial-mesenchymal plasticity in carcinoma metastasis. Genes Dev. 2013;27(20):2192–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119(6):1420–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tang X, Ding CK, Wu J, Sjol J, Wardell S, Spasojevic I, et al. Cystine addiction of triple-negative breast cancer associated with EMT augmented death signaling. Oncogene. 2017;36(30):4235–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tang X, Wu J, Ding CK, Lu M, Keenan MM, Lin CC, et al. Cystine Deprivation Triggers Programmed Necrosis in VHL-Deficient Renal Cell Carcinomas. Cancer Res. 2016;76(7):1892–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tang X, Keenan MM, Wu J, Lin CA, Dubois L, Thompson JW, et al. Comprehensive profiling of amino acid response uncovers unique methionine-deprived response dependent on intact creatine biosynthesis. PLoS Genet. 2015;11(4):e1005158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang WH, Huang Z, Wu J, Ding C-KC, Murphy SK, Chi J-T. A TAZ-ANGPTL4-NOX2 axis regulates ferroptotic cell death and chemoresistance in epithelial ovarian cancer. Molecular Cancer Research. 2019;18(1):79–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Burdo J, Dargusch R, Schubert D. Distribution of the cystine/glutamate antiporter system xc- in the brain, kidney, and duodenum. J Histochem Cytochem. 2006;54(5):549–57. [DOI] [PubMed] [Google Scholar]
- 17.Timmerman LA, Holton T, Yuneva M, Louie RJ, Padro M, Daemen A, et al. Glutamine Sensitivity Analysis Identifies the xCT Antiporter as a Common Triple-Negative Breast Tumor Therapeutic Target. Cancer Cell. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dixon SJ, Patel DN, Welsch M, Skouta R, Lee ED, Hayano M, et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife. 2014;3:e02523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ding C-KC, Rose J, Sun T, Wu J, Chen P-H, Lin C-C, et al. MESH1 is a cytosolic NADPH phosphatase that regulates ferroptosis. Nature Metabolism. 2020;2:270–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang WH, Ding CKC, Sun T, Hsu DS, Chi JT. The Hippo Pathway Effector TAZ Regulates Ferroptosis in Renal Cell Carcinoma Cell Reports. 2019;28(10):2501–8.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu J, Minikes AM, Gao M, Bian H, Li Y, Stockwell BR, et al. Intercellular interaction dictates cancer cell ferroptosis via NF2-YAP signalling. Nature. 2019;572:402–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang W-H, Chi J-T. Hippo pathway effectors YAP/TAZ as novel determinants of ferroptosis. Molecular & Cellular Oncology. 2019;7(1):1699375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lin CC, Chi JT. Ferroptosis of epithelial ovarian cancer: genetic determinants and therapeutic potential. Oncotarget. 2020;11(39):3562–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang K, Corsa CA, Ponik SM, Prior JL, Piwnica-Worms D, Eliceiri KW, et al. The collagen receptor discoidin domain receptor 2 stabilizes SNAIL1 to facilitate breast cancer metastasis. Nat Cell Biol. 2013;15(6):677–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim D, You E, Jeong J, Ko P, Kim JW, Rhee S. DDR2 controls the epithelial-mesenchymal-transition-related gene expression via c-Myb acetylation upon matrix stiffening. Sci Rep. 2017;7(1):6847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gonzalez ME, Martin EE, Anwar T, Arellano-Garcia C, Medhora N, Lama A, et al. Mesenchymal Stem Cell-Induced DDR2 Mediates Stromal-Breast Cancer Interactions and Metastasis Growth. Cell Rep. 2017;18(5):1215–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen P-H, Wu J, Ding C-KC, Lin C-C, Pan S, Bossa N, et al. Kinome screen of ferroptosis reveals a novel role of ATM in regulating iron metabolism. Cell Death & Differentiation. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cancer Genome Atlas N. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486(7403):346–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Prat A, Parker JS, Karginova O, Fan C, Livasy C, Herschkowitz JI, et al. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res. 2010;12(5):R68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Terai H, Tan L, Beauchamp EM, Hatcher JM, Liu Q, Meyerson M, et al. Characterization of DDR2 Inhibitors for the Treatment of DDR2 Mutated Nonsmall Cell Lung Cancer. ACS Chem Biol. 2015;10(12):2687–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grither WR, Divine LM, Meller EH, Wilke DJ, Desai RA, Loza AJ, et al. TWIST1 induces expression of discoidin domain receptor 2 to promote ovarian cancer metastasis. Oncogene. 2018;37(13):1714–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Blanco MJ, Moreno-Bueno G, Sarrio D, Locascio A, Cano A, Palacios J, et al. Correlation of Snail expression with histological grade and lymph node status in breast carcinomas. Oncogene. 2002;21(20):3241–6. [DOI] [PubMed] [Google Scholar]
- 35.Come C, Magnino F, Bibeau F, De Santa Barbara P, Becker KF, Theillet C, et al. Snail and slug play distinct roles during breast carcinoma progression. Clin Cancer Res. 2006;12(18):5395–402. [DOI] [PubMed] [Google Scholar]
- 36.Elloul S, Elstrand MB, Nesland JM, Trope CG, Kvalheim G, Goldberg I, et al. Snail, Slug, and Smad-interacting protein 1 as novel parameters of disease aggressiveness in metastatic ovarian and breast carcinoma. Cancer. 2005;103(8):1631–43. [DOI] [PubMed] [Google Scholar]
- 37.Martin TA, Goyal A, Watkins G, Jiang WG. Expression of the transcription factors snail, slug, and twist and their clinical significance in human breast cancer. Ann Surg Oncol. 2005;12(6):488–96. [DOI] [PubMed] [Google Scholar]
- 38.Goswami CP, Nakshatri H. PROGgeneV2: enhancements on the existing database. BMC Cancer. 2014;14:970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huang CC, Tu SH, Lien HH, Jeng JY, Huang CS, Huang CJ, et al. Concurrent gene signatures for han chinese breast cancers. PLoS One. 2013;8(10):e76421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Enerly E, Steinfeld I, Kleivi K, Leivonen SK, Aure MR, Russnes HG, et al. miRNA-mRNA integrated analysis reveals roles for miRNAs in primary breast tumors. PLoS One. 2011;6(2):e16915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zanconato F, Cordenonsi M, Piccolo S. YAP/TAZ at the Roots of Cancer. Cancer Cell. 2016;29(6):783–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Koo JH, Plouffe SW, Meng Z, Lee DH, Yang D, Lim DS, et al. Induction of AP-1 by YAP/TAZ contributes to cell proliferation and organ growth. Genes Dev. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang K, Kim JH, Kim HJ, Park IS, Kim IY, Yang BS. Tyrosine 740 phosphorylation of discoidin domain receptor 2 by Src stimulates intramolecular autophosphorylation and Shc signaling complex formation. J Biol Chem. 2005;280(47):39058–66. [DOI] [PubMed] [Google Scholar]
- 44.Vogel W, Gish GD, Alves F, Pawson T. The discoidin domain receptor tyrosine kinases are activated by collagen. Mol Cell. 1997;1(1):13–23. [DOI] [PubMed] [Google Scholar]
- 45.Li P, Silvis MR, Honaker Y, Lien WH, Arron ST, Vasioukhin V. alphaE-catenin inhibits a Src-YAP1 oncogenic module that couples tyrosine kinases and the effector of Hippo signaling pathway. Genes Dev. 2016;30(7):798–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lamar JM, Xiao Y, Norton E, Jiang ZG, Gerhard GM, Kooner S, et al. SRC tyrosine kinase activates the YAP/TAZ axis and thereby drives tumor growth and metastasis. J Biol Chem. 2019;294(7):2302–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Si Y, Ji X, Cao X, Dai X, Xu L, Zhao H, et al. Src Inhibits the Hippo Tumor Suppressor Pathway through Tyrosine Phosphorylation of Lats1. Cancer Res. 2017;77(18):4868–80. [DOI] [PubMed] [Google Scholar]
- 48.Ikeda K, Wang LH, Torres R, Zhao H, Olaso E, Eng FJ, et al. Discoidin domain receptor 2 interacts with Src and Shc following its activation by type I collagen. J Biol Chem. 2002;277(21):19206–12. [DOI] [PubMed] [Google Scholar]
- 49.Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell. 2017;171(2):273–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, et al. Ferroptosis: process and function. Cell Death Differ. 2016;23(3):369–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hayano M, Yang WS, Corn CK, Pagano NC, Stockwell BR. Loss of cysteinyl-tRNA synthetase (CARS) induces the transsulfuration pathway and inhibits ferroptosis induced by cystine deprivation. Cell Death Differ. 2016;23(2):270–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. 2017;13(1):91–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen PH, Wu J, Ding CC, Lin CC, Pan S, Bossa N, et al. Kinome screen of ferroptosis reveals a novel role of ATM in regulating iron metabolism. Cell Death Differ. 2019;27(27):1008–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen PH, Smith TJ, Wu J, Siesser PF, Bisnett BJ, Khan F, et al. Glycosylation of KEAP1 links nutrient sensing to redox stress signaling. EMBO J. 2017;36(15):2233–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen P-H, Chi J-T, Boyce M. KEAP1 has a sweet spot: A new connection between intracellular glycosylation and redox stress signaling in cancer cells. Molecular & Cellular Oncology. 2017;4(6):e1361501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Toy KA, Valiathan RR, Nunez F, Kidwell KM, Gonzalez ME, Fridman R, et al. Tyrosine kinase discoidin domain receptors DDR1 and DDR2 are coordinately deregulated in triple-negative breast cancer. Breast Cancer Res Treat. 2015;150(1):9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Corsa CA, Brenot A, Grither WR, Van Hove S, Loza AJ, Zhang K, et al. The Action of Discoidin Domain Receptor 2 in Basal Tumor Cells and Stromal Cancer-Associated Fibroblasts Is Critical for Breast Cancer Metastasis. Cell Rep. 2016;15(11):2510–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Walsh LA, Nawshad A, Medici D. Discoidin domain receptor 2 is a critical regulator of epithelial–mesenchymal transition. Matrix Biology. 2011;30(4):243–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tsai M-C, Li W-M, Huang C-N, Ke H-L, Li C-C, Yeh H-C, et al. DDR2 overexpression in urothelial carcinoma indicates an unfavorable prognosis: a large cohort study. Oncotarget. 2016;7(48). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.SASAKI S, UEDA M, IGUCHI T, KANEKO M, NAKAYAMA H, WATANABE T, et al. DDR2 Expression Is Associated with a High Frequency of Peritoneal Dissemination and Poor Prognosis in Colorectal Cancer. Anticancer Research. 2017;37(5):2587–91. [DOI] [PubMed] [Google Scholar]
- 61.Fan Y, Xu Z, Fan J, Huang L, Ye M, Shi K, et al. Prognostic significance of discoidin domain receptor 2 (DDR2) expression in ovarian cancer. Am J Transl Res. 2016;8(6):2845–50. [PMC free article] [PubMed] [Google Scholar]
- 62.Bayer SV, Grither WR, Brenot A, Hwang PY, Barcus CE, Ernst M, et al. DDR2 controls breast tumor stiffness and metastasis by regulating integrin mediated mechanotransduction in CAFs. Elife. 2019;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gonzalez ME, Martin EE, Anwar T, Arellano-Garcia C, Medhora N, Lama A, et al. Mesenchymal Stem Cell-Induced DDR2 Mediates Stromal-Breast Cancer Interactions and Metastasis Growth. Cell Reports. 2017;18(5):1215–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Grither WR, Longmore GD. Inhibition of tumor-microenvironment interaction and tumor invasion by small-molecule allosteric inhibitor of DDR2 extracellular domain. Proc Natl Acad Sci U S A. 2018;115(33):E7786–E94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tu MM, Lee FYF, Jones RT, Kimball AK, Saravia E, Graziano RF, et al. Targeting DDR2 enhances tumor response to anti-PD-1 immunotherapy. Sci Adv. 2019;5(2):eaav2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hwang PY, Brenot A, King AC, Longmore GD, George SC. Randomly Distributed K14(+) Breast Tumor Cells Polarize to the Leading Edge and Guide Collective Migration in Response to Chemical and Mechanical Environmental Cues. Cancer Res. 2019;79(8):1899–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Slocum E, Craig A, Villanueva A, Germain D. Parity predisposes breasts to the oncogenic action of PAPP-A and activation of the collagen receptor DDR2. Breast Cancer Res. 2019;21(1):56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hammerman PS, Sos ML, Ramos AH, Xu C, Dutt A, Zhou W, et al. Mutations in the DDR2 kinase gene identify a novel therapeutic target in squamous cell lung cancer. Cancer Discov. 2011;1(1):78–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Skouta R, Dixon SJ, Wang J, Dunn DE, Orman M, Shimada K, et al. Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. Journal of the American Chemical Society. 2014;136(12):4551–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Angeli JPF, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ, et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nature cell biology. 2014;16(12):1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Du K, Oh SH, Sun T, Yang W-H, Chi J-TA, Diehl AM. Inhibiting xCT/SLC7A11 induces ferroptosis of myofibroblastic hepatic stellate cells and protects against liver fibrosis. bioRxiv. 2019:2019.12.23.886259. [Google Scholar]
- 72.Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. Glutaminolysis and transferrin regulate ferroptosis. Molecular cell. 2015;59(2):298–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Linkermann A, Skouta R, Himmerkus N, Mulay SR, Dewitz C, De Zen F, et al. Synchronized renal tubular cell death involves ferroptosis. Proc Natl Acad Sci U S A. 2014;111(47):16836–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lin CC, Kitagawa M, Tang X, Hou MH, Wu J, Qu DC, et al. CoA synthase regulates mitotic fidelity via CBP-mediated acetylation. Nat Commun. 2018;9(1):1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.