

Abstract
Pannexin 1 (Panx1) channels export ATP and may contribute to increased concentration of the vasodilator ATP in plasma during hypoxia in vivo. We hypothesized that Panx1 channels and associated ATP export contribute to hypoxic vasodilation, a mechanism that facilitates the matching of oxygen delivery to metabolic demand of tissue. Male and female mice devoid of Panx1 (Panx1−/−) and wild-type controls (WT) were anesthetized, mechanically ventilated, and instrumented with a carotid artery catheter or femoral artery flow transducer for hemodynamic and plasma ATP monitoring during inhalation of 21% (normoxia) or 10% oxygen (hypoxia). ATP export from WT vs. Panx1−/−erythrocytes (RBC) was determined ex vivo via tonometer experimentation across progressive deoxygenation. Mean arterial pressure (MAP) was similar in Panx1−/− (n = 6) and WT (n = 6) mice in normoxia, but the decrease in MAP in hypoxia seen in WT was attenuated in Panx1−/− mice (−16 ± 9% vs. −2 ± 8%; P < 0.05). Hindlimb blood flow (HBF) was significantly lower in Panx1−/− (n = 6) vs. WT (n = 6) basally, and increased in WT but not Panx1−/− mice during hypoxia (8 ± 6% vs. −10 ± 13%; P < 0.05). Estimation of hindlimb vascular conductance using data from the MAP and HBF experiments showed an average response of 28% for WT vs. −9% for Panx1−/− mice. Mean venous plasma ATP during hypoxia was 57% lower in Panx1−/− (n = 6) vs. WT mice (n = 6; P < 0.05). Mean hypoxia-induced ATP export from RBCs from Panx1−/− mice (n = 8) was 82% lower than that from WT (n = 8; P < 0.05). Panx1 channels participate in hemodynamic responses consistent with hypoxic vasodilation by regulating hypoxia-sensitive extracellular ATP levels in blood.
NEW & NOTEWORTHY Export of vasodilator ATP from red blood cells requires pannexin 1. Blood plasma ATP elevations in response to hypoxia in mice require pannexin 1. Hemodynamic responses to hypoxia are accompanied by increased plasma ATP in mice in vivo and require pannexin 1.
Keywords: blood flow, erythrocyte, hypoxia, pannexin, vasodilation
INTRODUCTION
Appropriate matching of oxygen (O2) delivery to metabolic demand of tissue during acute systemic hypoxia is predominantly mediated through augmented tissue blood flow (1). The net local vasodilation of peripheral tissue beds (e.g., skeletal muscle and the integument) during hypoxia is regularly observed in healthy humans (2–4), is highly phylogenetically conserved (5), and has been shown to be mediated by multiple mechanisms (4, 6, 7). Despite robust sympathetic activation and elevated α-adrenergic vasoconstrictor tone during hypoxia (8, 9), intraluminal stimulation of type 2 purinergic (P2) receptors along the endothelium by extracellular ATP uniquely elicits both pronounced vasodilation and attenuation of sympathetic vasoconstriction in humans (6, 10, 11).
ATP is a primary ligand for P2 receptors and plasma concentrations appear to increase during hemoglobin deoxygenation conditions, presumably via nonlytic, cellular ATP export (12–14). One well documented conduit for ATP export is pannexin1 (Panx1), a large, nonjunctional nonselective membrane channel allowing small molecule passage between the intracellular compartment and the extracellular milieu (15, 16). Panx1 channels are sensitive to hypoxia (17), and importantly, are present in various cell types (i.e., red blood cells (RBCs), vascular endothelium, and vascular smooth muscle cells) that are plausible regulators of both vascular tone and the plasma ATP concentration (18–20). Indeed, vasodilation develops in a concentration-dependent manner in congruence with increases in plasma ATP during simultaneous hemoglobin (Hb) deoxygenation in humans (3), and ex vivo pharmacological evidence suggests a role for Panx1 in the regulated export of ATP by RBCs in hypoxia (20, 21).
Presently, there is no mechanistic evidence elucidating the contribution of Panx1 channels to ATP-regulated hypoxic vasodilation in vivo. Although investigations in vitro point toward an active role for RBCs in hypoxia-induced ATP export through Panx1 channels (17, 20), the role of Panx1 channels in response to systemic hypoxia in vivo has not been defined. Therefore, we tested the hypothesis that Panx1 channel-mediated increases in plasma ATP are mechanistically linked to hypoxic vasodilation in vivo. To do so, we examined systemic (mean arterial blood pressure) and regional hemodynamics (hindlimb blood flow) in wild-type (WT) and Panx1 null (22) (Panx1−/−) mice and changes in plasma ATP in response to hypoxia. RBC-mediated ATP export from Panx1-deficient mice was examined to further elucidate the role of Panx1 to plasma ATP and hypoxic vasodilation.
METHODS
Transparency and Openness
The data supporting the findings of this study are available from the corresponding author upon reasonable request.
Wild-Type and Panx1−/− Mice
Animal procedures and protocols were approved by the Duke University and Durham VA Medical Center Animal Care and Use Committees and conformed to APS and Federal guidelines. Wild-type (WT) (C57/BL6, strain control; n = 26) and Panx1−/− (n = 26) mice, 3–6 mo of age and of both sexes, were used. Panx1 genotyping was confirmed as described in 2011 in Seminario-Vidal et al. (22) At baseline, the mice do not differ from respective WT controls (including littermates) with respect to fertility, viability of offspring, litter sizes, birth weight, or gross organ appearance. No morphological or cell volume differences were observed between WT and Panx1−/− RBCs using microscopy, hematocrit (HCT), and cell counting techniques (23).
Animal Preparation and Induction of Systemic Hypoxia
We used mechanical ventilation (and general anesthesia) to control minute ventilation while exposing mice to hypoxia, thereby preventing compensatory (and potentially confounding) changes in minute ventilation. Mice were initially anesthetized with 4% isoflurane (100% O2; 0.5 mL/min flow rate) in an induction chamber. Following induction, mice were transferred onto a 37°C temperature-controlled heating pad, anesthetized with 2% isoflurane via nose-cone inhalation, and monitored for breathing rate and also response to toe pinch to assess the plane of anesthesia. Once adequately anesthetized, tracheostomy surgery was performed and mice were mechanically ventilated at a rate of 140 breaths/min and a tidal volume of 180 µL (Harvard Apparatus (Holliston, MA) MiniVent ventilator; Model 845). During tracheostomy placement and for at least 10 min thereafter, 100% O2 was delivered. Gases for inhalation were mixed via a gas blender (Vyaire Bird Blender 3800 Series, Yorba Linda, CA) and tubing was used to connect the gas blend to the air inflow port on the ventilator. Upon stabilization, a normoxic period, breathing 21% O2 (balance N2) for 10 min, was followed by hypoxia at 10% FiO2 (balance N2) for 30 min. Gas switching in the blender was confirmed using an in-line O2 analyzer (MiniOx Oxygen Analyzer), and venous blood gases were sampled from the inferior vena cava at experiment termination and measured (Siemens RapidPoint 405). All mice underwent these same procedures regardless of Protocol assignment. Mice were humanely euthanized by rapid exsanguination and bilateral thoracotomy following all hemodynamic experiments.
Protocol 1: Systemic Hemodynamics
To gain broad insight into whether cardiovascular regulation (i.e., vasodilation) during hypoxia is modulated by Panx1 channels, we first determined the mean arterial pressure (MAP) and heart rate (HR) during systemic hypoxia (10% O2) in WT (n = 6; male = 4, female = 2) and Panx1−/− mice (n = 6; male = 6). To do so, a small incision was made midline in the ventral neck and the left carotid artery exposed, isolated, and cannulated with polyethylene tubing for MAP monitoring via a pressure transducer (AD Instruments, MLT844). Systemic blood pressure (MAP) was measured continuously, recorded with Windaq or LabChart data acquisition software, binned into 30 s averages across the experiment, and partitioned into 5-min segments throughout the 30-min exposure to hypoxia. Heart rate was computed from the beat-to-beat blood pressure waveform over 60 s for each 5-min segment.
Protocol 2: Local Hemodynamics (Hindlimb Blood Flow)
Because changes in MAP can result from central (cardiac output) and/or peripheral (vascular resistance) changes, the purpose of this protocol was to determine regional (hindlimb, femoral artery) blood flow (HBF) during systemic hypoxia in a second group of WT (n = 6; male = 5, female = 1) and Panx1−/− mice (n = 6; male = 5, female = 1). To do so, a small incision was made distal to the inguinal ligament. The left femoral artery was exposed and dissected free of the femoral vein and nerve, and a flow probe (Transonic 0.5PSB) was applied to the vessel segment. Mean blood flow was measured continuously, recorded with LabChart acquisition software, binned into 30 s averages across the experiment, and partitioned into 5-min segments throughout the 30-min exposure to hypoxia. Technical limitations precluded the blood flow and blood pressure measurements from being made in the same mice.
Venous Plasma ATP and Hb Measurement
Similar to previous reports (3, 24, 25), a venous whole blood sample of 200 µL was drawn into a EDTA-precoated syringe from the inferior vena cava after 30 min of hypoxic (10% O2) exposure and immediately mixed into 270 µL of ATP stop-solution as described by Gorman and colleagues (13, 25) for subsequent measurement of plasma ATP and Hb. In this setting, EDTA acts to inhibit ATPases and also serves as the anticoagulant. Samples were centrifuged at 10,000 g at room temperature for 2 min to thoroughly remove from the plasma any cells that might otherwise artifactually contribute to the measured plasma ATP, and the supernatant was removed and diluted twofold with PBS. The sample was again centrifuged to remove any remaining cells. Plasma ATP was assayed by the luciferin-luciferase technique with use of a tube luminometer (Promega 20/20) within 10 min from blood acquisition. Plasma Hb in the same sample was assessed using a FluoStar (BMG LABTECH, Cary, NC) spectrophotometer and used to estimate the %hemolysis and therefore the extent to which ATP could be due to hemolysis (3, 25). Hemolysis correction formulas were established by lysing RBCs from WT (1.95 µmol/g Hb, r2 = 0.98) and Panx1−/− (2.39 µmol/g Hb, r2 = 0.98) mice and performing linear regression relating intracellular ATP and Hb concentrations as outlined in the guidelines by Gorman and Feigl (13, 25). The hematocrit was measured in three animals from each group, averaged, and used in the computation of total plasma ATP.
Ex Vivo: RBC Deoxygenation, Pharmacological Stimulation, and Measurement of Extracellular ATP
The overall purpose of these procedures was to identify the O2-sensitive ATP release capacity from WT and Panx1−/− mice through examination of RBCs, a cell type well recognized for its ability to export ATP and suggested to be directly involved in hypoxic vasodilation (3, 26, 27). Fresh whole blood (0.5 mL) was collected into a heparinized vial via submandibular cheek bleeding method from WT (n = 14) and Panx1−/− (n = 14) nonanesthetized mice. Mice serving as blood donors were not used in other experiments. RBCs were isolated by centrifugation (500 g at 4°C for 10 min, to separate the majority of plasma and leukocytes) and the plasma and buffy coat were removed. Packed RBCs were resuspended and washed three times in excess PSS (in mM: 4.7 KCl, 2.0 CaCl2, 1.2 MgSO4, 140.5 NaCl, 21.0 Tris; 5.5 dextrose; with 0.5% BSA; pH 7.4) to remove remaining plasma and any cells remaining in the supernatant. Since RBCs have little or no surface ATPase activity of their own (21), inhibiting ATPases was not required here, and therefore heparin sufficed as the anticoagulant choice in this series. This method of isolation yields a RBC suspension devoid of platelets and less than one leukocyte per 50 high-power fields (3, 28). All studies on isolated RBCs were performed immediately after processing given the known behavioral changes in nonfresh (3 or more h postacquisition) RBCs (21, 29, 30). No attempt was made to preserve the low PO2 of the venous blood obtained during initial sample processing.
A 10% RBC suspension (in Krebs buffer) was placed in a rotating bulb tonometer and warmed to 37°C (Eschweiler GmbH & Co. KG, Germany). To produce a normoxic isocapnic environment, RBCs were exposed to a blend of 21% O2 and 5.8% CO2 (balance N2) gases. Gases were blended via a custom gas blender (MCQ Gas Blender Series 100, Italy), humidified, and introduced into the closed tonometer system. Progressive RBC deoxygenation was induced by lowering O2 in step-wise fashion to 10%, 5%, and 2.5% for 10 min each. Across all conditions, CO2 was 5.8% and the balance of gas was N2. Oxygenation was confirmed by blood gas analysis (Siemens Rapid Point 405 Series Automatic Blood Gas System, Los Angeles, CA) (3). The signal transduction linking hypoxia to Panx1-mediated ATP export from RBCs appears to involve a Gi (inhibitory) protein (31). In order to test whether ATP export from Panx1−/− RBCs was intact downstream of the hypoxic stimulus (but upstream of Panx1), we measured responses to either incremental Hb deoxygenation or mastoparan 7, a highly selective activator of the inhibitory G-protein (Gi) that has been implicated in O2-sensitive ATP export from RBCs (31).
ATP was measured via the luciferin-luciferase technique, with light emission during the reaction detected by luminometry. A sample of 10% HCT was diluted 250-fold and a 200 μL RBC suspension (0.04% HCT) was injected into a cuvette containing 100 μL of 10 mg/mL crude firefly tail extract (Sigma) and 100 μL of 0.5 mg/mL D-luciferin (RPI) mixed in PBS. Extracellular ATP was normalized to a cell count of 4 × 108 cells/mL. Intracellular ATP was determined by lysing a 50 μL sample of RBC suspension (10% HCT) with water and analyzing for ATP and Hb. A standard curve for ATP (Calbiochem) was obtained in RBC suspension for each individual experiment. To confirm that ATP export was not due to hemolysis, RBC suspension aliquots acquired for ATP analysis were analyzed for cell-free Hb and samples in which cell-free Hb was significantly high were excluded from the study (3, 28, 30). We measured absorbance at wavelengths of 380, 415, and 450 nm in order to calculate cell-free Hb as previously described (3, 13, 30, 32, 33).
Statistics
All values are reported as means ± SD. Specific hypothesis testing across groups over time was performed using two-way repeated measures ANOVA. All pairwise multiple comparison procedures were via the Student–Newman–Keuls method. Paired t tests were used where appropriate and indicated, and intergroup comparisons were made with unpaired t tests. Significance was set at P < 0.05 and SigmaPlot was used for statistical analysis.
RESULTS
The Hypotensive Response to Systemic Hypoxia is Blunted in Panx1−/− Mice
Body weights of Panx1−/− mice did not differ significantly from WT littermates, as previously described (means ± SD: 27.75 ± 3.55 g vs. 25.83 ± 4.32 g, respectively) (22). Baseline MAP did not differ significantly between WT and Panx1−/− mice in normoxia (85 ± 3 mmHg vs. 82 ± 2 mmHg, respectively; P > 0.05), nor at the end of catheterization surgery while mice breathed 100% O2 (86 ± 3 mmHg vs. 82 ± 2 mmHg, respectively; P > 0.05). Figure 1 displays the values averaged over 30 s and obtained every 5 min. Upon exposure to hypoxia (10% O2 FiO2), MAP was significantly lower than during the normoxic baseline period at each subsequent time point in WT mice (P < 0.05) but no decrease was observed in Panx1−/− mice (Fig. 1A). In addition, upon transition from normoxia to hypoxia, the change in MAP differed significantly between WT and Panx1−/− mice at all postbaseline time-points (Fig. 1B). The percent changes from baseline MAP in response to hypoxia were (values are at minute 5 and after each 5-min interval through minute 30): −5.3 ± 4.1%, −6.3 ± 3.0%, −11.0 ± 7.4%, −13.7 ± 6.1%, −14.7 ± 8.0%, and −16.2 ± 9.7% in WT mice; and 1.5 ± 5.2%, 0.4 ± 5.7%, 0.6 ± 4.3%, −0.8 ± 4.0%, −1.7 ± 5.8%, and −1.8 ± 8.6% in Panx1−/− mice, progressing from minute 5 to minute 30, respectively. Basal heart rate did not differ significantly between WT and Panx1−/− mice (646 ± 28 bpm vs. 598 ± 58 bpm; P > 0.05). Heart rate tended to increase progressively in WT mice from the normoxic baseline to the hypoxia exposure (values are at minute 5 and after each 5-min interval through minute 30): (%Δ = −0.5 ± 6.0%, 1.1 ± 3.4%, 0.7 ± 4.6%, 2.6 ± 8.1%, 5.2 ± 10.4%, 5.8 ± 12.1%, (P > 0.05; Fig. 2, A and B), but the change was not statistically significant, and no change (or trend) was observed in Panx1−/− mice during hypoxia (values are at minute 5 and after each 5-min interval through minute 30): %Δ = −0.8 ± 6.4%, −0.3 ± 7.1%, −0.1 ± 7.9%, −0.2 ± 8.1%, −0.6 ± 9.3%, −3.8 ± 8.8%, (P > 0.05; Fig. 3, A and B).
Figure 1.
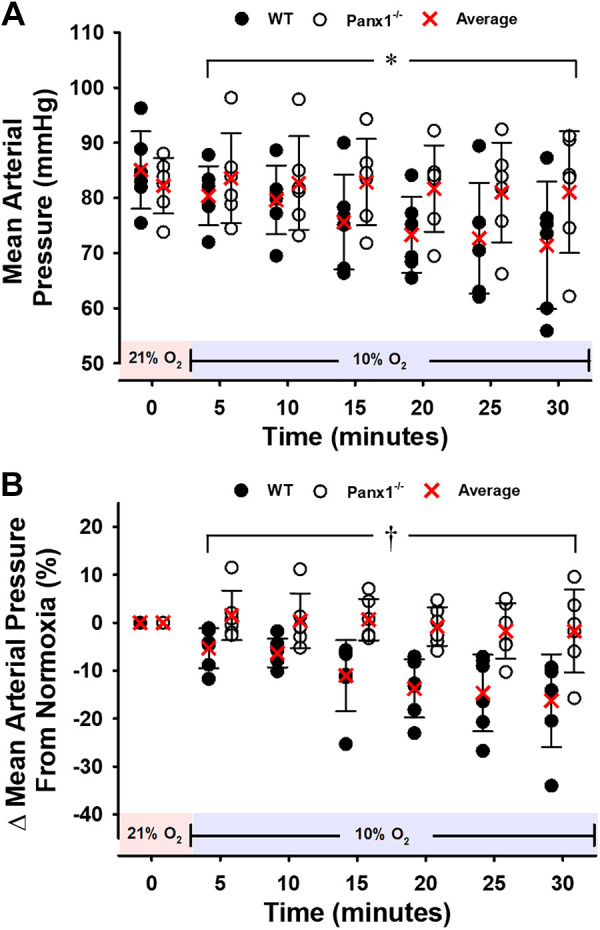
Mean arterial blood pressure (MAP) for WT or Panx1−/− mice during a 30-min hypoxic challenge. Individual values and means ± SD are shown. A and B absolute values and percent change from baseline. *P < 0.05 vs. baseline for WT but not for Panx1−/− mice. †P < 0.05 compared to WT as determined by two-way repeated-measures ANOVA with Student–Newman–Keuls post hoc analysis. n = 6 for WT mice (4 males, 2 females) and n = 6 for Panx1−/− mice (6 males). Red shading denotes normoxia (21% FiO2) and blue shading denotes hypoxia (10% FiO2). Panx1, pannexin 1; WT, wild-type.
Figure 2.
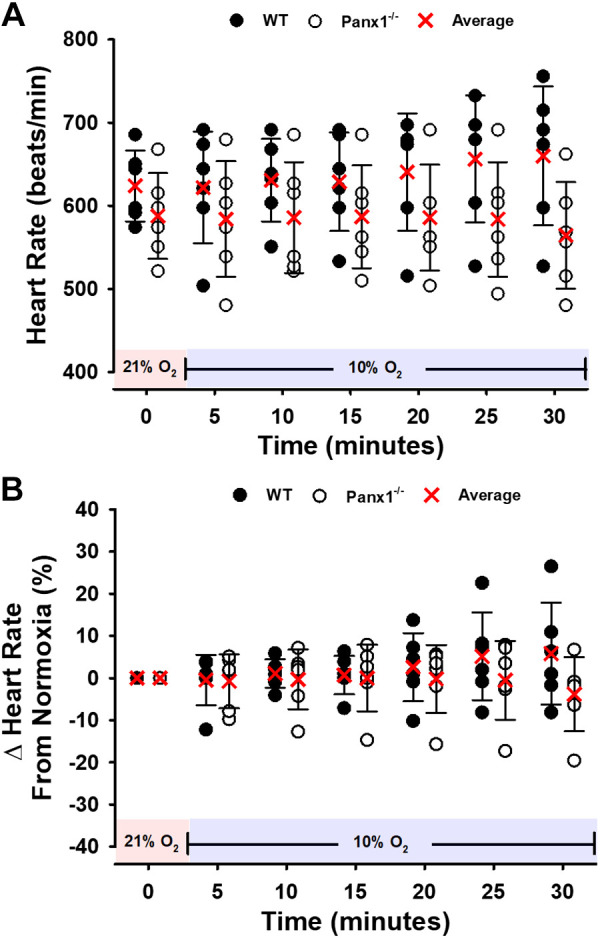
Heart rate (HR) for WT or Panx1−/− mice during a 30-min hypoxic challenge. Individual values and means ± SD are shown. A and B: absolute values and percent change from baseline. n = 6 for WT mice (4 males, 2 females) and n = 6 for Panx1−/− mice (6 males). Red shading denotes normoxia (21% FiO2) and blue shading denotes hypoxia (10% FiO2). Panx1, pannexin 1; WT, wild-type.
Figure 3.
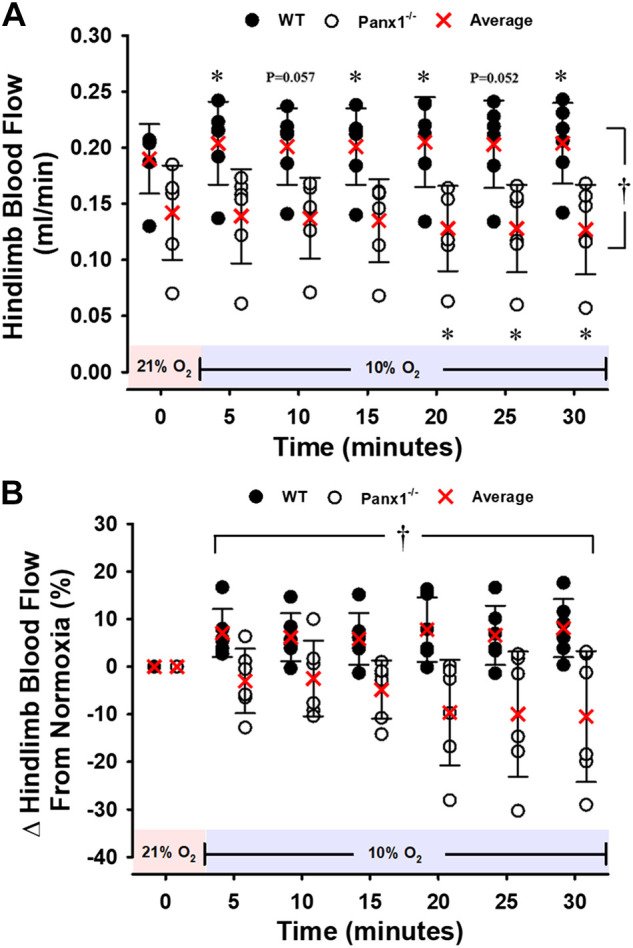
Hindlimb blood flow (HBF) for WT or Panx1−/− mice during a 30-min hypoxic challenge. Individual values and means ± SD are shown. A and B: absolute values and percent change from baseline. *P < 0.05 vs. respective baseline. †P < 0.05 compared to WT as determined by two-way repeated-measures ANOVA with Student–Newman–Keuls post hoc analysis. n = 6 for WT mice (5 males, 1 females) and n = 6 for Panx1−/− mice (5 males, 1 females). Red shading denotes normoxia (21% FiO2) and blue shading denotes hypoxia (10% FiO2). Panx1, pannexin 1; WT, wild-type.
The Hyperemic Response to Systemic Hypoxia is Blunted in Panx1−/− Mice
At baseline, hindlimb blood flow (HBF) measured in a second group of mice was significantly lower in Panx1−/− vs. WT mice during normoxia (0.14 ± 0.04 mL/min vs. 0.19 ± 0.03 mL/min; P < 0.05), and remained lower throughout hypoxia (Fig. 3A). In WT mice, hypoxia evoked increases in HBF from normoxic baseline 0.20 ± 0.04 mL/min at minute 5; (P < 0.05), 0.20 ± 0.03 mL/min at minute 10 (P = 0.057), (0.20 ± 0.03 mL/min at minute 15 (P < 0.05), 0.21 ± 0.04 mL/min at minute 20 (P < 0.05), 0.20 ± 0.04 mL/min at minute 25 (P = 0.052), and 0.20 ± 0.04 mL/min at minute 30 (P < 0.05, Fig. 3A). In contrast, Panx1−/− mice had a reduction in HBF from baseline to hypoxia, reaching statistical significance (P < 0.05) at minutes 20, 25, and 30 (0.14 ± 0.04 mL/min, 0.14 ± 0.03 mL/min, 0.14 ± 0.04 mL/min, 0.13 ± 0.04 mL/min, 0.13 ± 0.04 mL/min, and 0.13 ± 0.04 mL/min, from minute 5 to minute 30, respectively). Furthermore, the effect of hypoxia (%Δ from normoxia) on HBF was significantly different between WT and Panx1−/− mice across the entire trial (Fig. 3B). In a second series of experiments in Panx1−/− and littermate controls (N = 5 Panx1+/+ and 6 Panx1−/−; all male), the hypotensive response to hypoxia was again blunted in Panx1−/− mice, relative to WT littermates (not shown). In order to further probe the hypoxia-mediated influence on vascular tone, we used the MAP and HBF data from the two mouse groups to estimate hindlimb vascular conductance (HVC; i.e., estimation of vasodilation or vasoconstriction) in response to hypoxia. Figure 4 depicts increases in HVC reaching over 25% in WT mice, yet a contrasting mild decrease in HVC in Panx1−/− mice.
Figure 4.
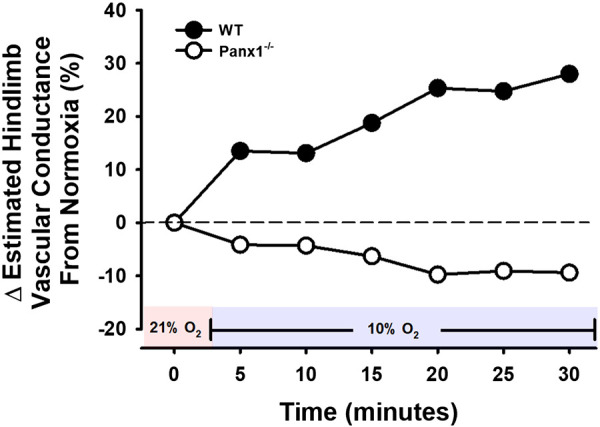
Calculated, estimated hindlimb vascular conductance during a 30-min hypoxic challenge using MAP from mouse group 1 and HBF from mouse group 2. Hindlimb vascular conductance = (group average HBF/group average MAP) × 100. Values are % change from baseline. n = 12 for WT mice (9 males, 3 females) and n = 12 for Panx1−/− mice (11 males, 1 females). Red shading denotes normoxia (21% FiO2) and blue shading denotes hypoxia (10% FiO2). HBF, hindlimb blood flow; MAP, mean arterial pressure; Panx1, pannexin 1; WT, wild-type.
Increase of Venous Plasma ATP during Systemic Hypoxia is Suppressed in Panx1−/− Mice
Plasma levels of ATP were similar in WT (n = 4; 95.1 ± 34.2 nM) and Panx1−/− mice (n = 4; 88.4 ± 30.8 nM; P > 0.05) under normoxic conditions. Blood was sampled after 30 min of hypoxia in groups of mice separate from those sampled for normoxia exposure only (n = 6 WT and n = 6 Panx1−/−). After 30 min of systemic hypoxia (10% FiO2; Pao2 = 47 ± 6 mmHg in WT vs. 44 ± 6 mmHg in Panx1−/−), venous plasma ATP was higher in hypoxic WT mice than at baseline, whereas in Panx1−/− mice plasma ATP was not higher during hypoxia (Fig. 5). Plasma ATP in hypoxia was greater in WT (n = 6) than Panx1−/− mice (n = 6) (166 ± 23 nM vs. 71 ± 6 nM; P < 0.05; Fig. 5). The difference in the mean plasma ATP between normoxia and hypoxia was +70.4 nM in WT and −17.1 nM in Panx1−/− mice (note: data are unpaired). To determine whether this observation could be simply the result of differences in levels of circulating ATP during the 35-min experiment, plasma ATP was measured during 30 min of normoxia in a separate group of mice from those exposed to hypoxic gases, and plasma ATP did not differ significantly between WT vs. Panx1−/− mice (values noted above under normoxic conditions).
Figure 5.
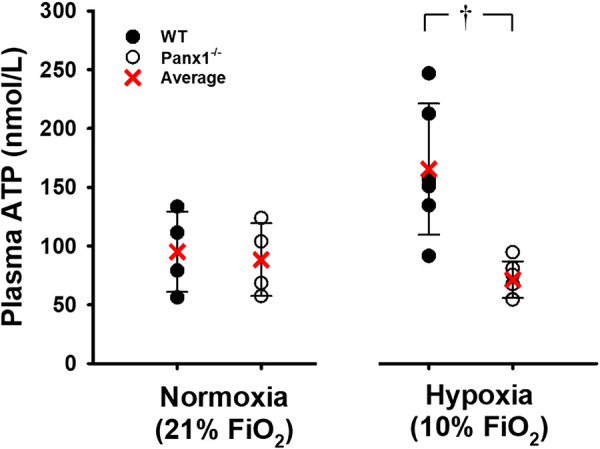
Plasma ATP concentration (nM) for WT or Panx1−/− mice after 30 min of breathing either 21% or 10% O2. Individual values and means ± SD are shown. †P < 0.05 compared to WT as determined by unpaired t test. For normoxia time control, n = 4 for WT mice (4 males) and n = 4 for Panx1−/− mice (4 males). For hypoxia, n = 6 for WT mice (4 males, 2 females) and n = 6 for Panx1−/− mice (6 males). Panx1, pannexin 1; WT, wild-type.
Regulated Export of ATP from RBCs of Panx1−/− Mice is Impaired; P50 is Unaltered
Exported ATP from isolated RBCs of WT mice during graded hypoxia was significantly greater than in normoxia (P < 0.05). In contrast, no hypoxia-induced increase in ATP was observed from RBCs from Panx1−/− mice (Fig. 6A). Moreover, despite a nonsignificantly lower basal ATP from Panx1−/− RBCs, the ATP export response to hypoxia in Panx1−/− mice was impaired at each level of deoxygenation (Fig. 6B). RBC intracellular ATP was similar between WT (n = 8) and Panx1 null mice (n = 8) (2.03 ± 0.51 vs. 2.31 ± 0.56 μmoles/g Hb; P > 0.05). Congruent with responses to the hypoxic stimulus, ATP export from WT RBCs in response to the Gi-protein activator Mastoparan 7 increased by 31%, whereas ATP release from Panx1−/− RBCs did not significantly increase in response to Mastoparan 7 (P < 0.05) (Fig. 7).
Figure 6.
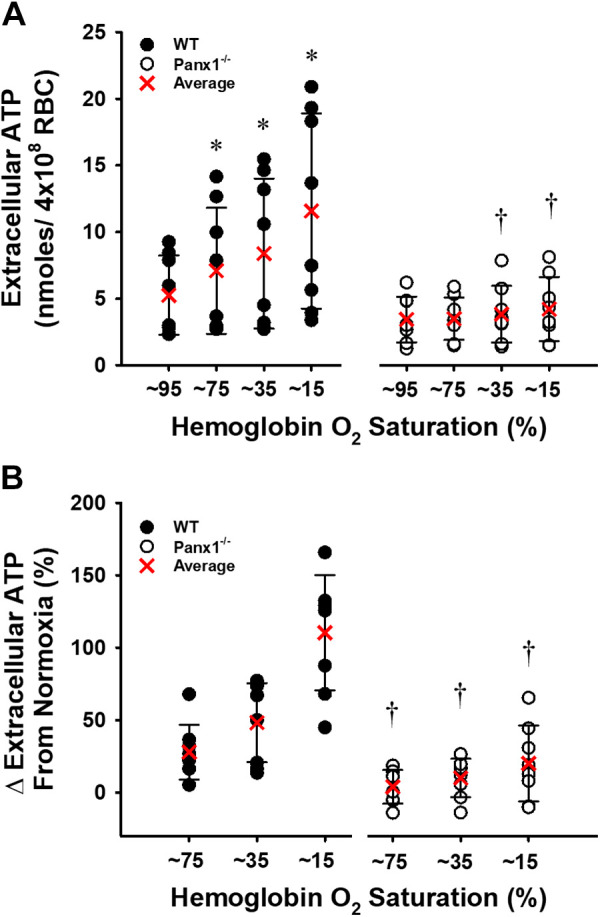
Extracellular ATP increased progressively with the deoxygenation of WT and, to a lesser extent, Panx1−/− RBCs. Individual values and means ± SD are shown. Absolute values are summarized in (A) and the percent change from baseline in (B). *P < 0.05 vs. respective baseline and †P < 0.05 compared to WT as determined by two-way repeated-measures ANOVA with Student–Newman–Keuls post hoc analysis. n = 8 for WT mice (6 males, 2 females) and n = 8 for Panx1−/− mice (6 males, 2 females). Panx1, pannexin 1; WT, wild-type.
Figure 7.
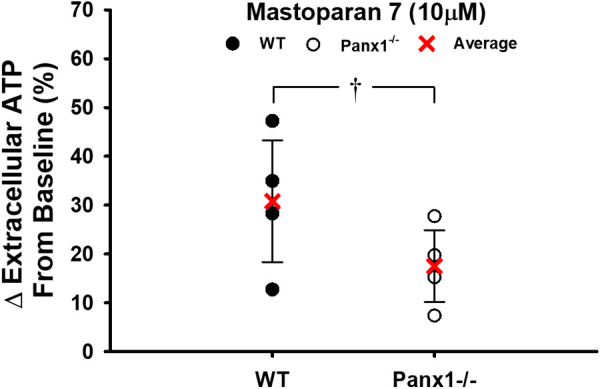
Percent change in extracellular ATP following RBC incubation of RBCs from WT or Panx1−/− mice with mastoparan 7 (10 µM). Individual values and means ± SD are shown. †P < 0.05 compared to WT as determined by unpaired t test. n = 4 for WT mice (4 males) and n = 4 for Panx1−/− mice (4 males). Panx1, pannexin 1; WT, wild-type.
In the present experiments, RBC lysis rarely exceeded 0.4%, and the predicted [ATP] that would result from this amount of RBC lysis was smaller by an order of magnitude than the differences in supernatant ATP we measured when comparing WT vs. Panx1−/− RBCs in hypoxia. Furthermore, the extent of RBC lysis, as calculated based on the measured cell-free [Hb] detected in the supernatant in response to hypoxia did not differ between WT RBCs (0.250 ± 0.067% RBC lysis at 5% O2) vs. Panx1−/− RBCs (which displayed weaker ATP export, 0.213 ± 0.046% RBC lysis at 5% O2). In addition, cell-free [ATP] and cell-free [Hb] in supernatant in response to deliberate RBC lysis were strongly correlated (r2 = 0.98), as expected, but cell-free [ATP] correlated very weakly (r2 = 0.04) with cell-free [Hb] in supernatants from RBC suspensions exposed to hypoxia. Collectively, these observations show a dissociation between cell-free ATP and cell-free Hb during hypoxia and indicate that hemolysis is not the driving factor in the hypoxia-induced appearance of extra-RBC ATP. Hb-O2 affinity of RBCs from WT and Panx1−/− was equivalent (P50 = 45.1 vs. 45.3 mmHg; Fig. 8).
Figure 8.
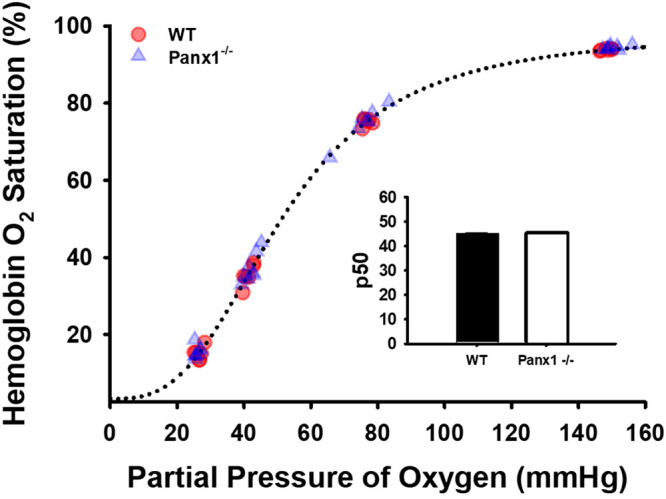
Oxygen binding curves of RBCs from WT (n = 6 males) or Panx1−/− mice (n = 7 males). The insert shows mean and standard error of P50 values. Mean P50 values (inset) did not differ significantly as determined by unpaired t test. Panx1, pannexin 1; WT, wild-type.
DISCUSSION
The present investigation suggests that Panx1-deficient mice have an impaired vasodilatory response to hypoxia in vivo, a finding associated with lower plasma ATP concentrations in vivo and depressed ATP export from RBCs during hypoxia ex vivo. The blunted hemodynamic response to hypoxia in Panx1−/− mice was evidenced via the inability to either effectively lower systemic MAP or augment regional HBF. Although examined independently, coupling MAP and HBF observations (and computing an estimated hindlimb vascular conductance) supports the notion for poor modulation of O2-sensitive regulation of vascular tone in Panx1 null mice. Furthermore, attenuated hemodynamic changes in Panx1-deficient mice during hypoxia could not be explained by the differences in heart rate, which was essentially unchanged in both groups of mice. In response to hypoxia, the change in MAP was accompanied by concordant changes in plasma ATP concentrations across WT and Panx1−/− mice, with Panx1 deficient mice demonstrating less change in plasma ATP in hypoxia as compared to WT mice. Reflecting one potential explanation and cellular source for plasma ATP, increases in extracellular ATP generated by Panx1−/− RBCs in hypoxia were significantly lower than those by WT RBCs. The decreased ability of Panx1−/− RBCs to export ATP in hypoxia could not be explained by differences in Hb O2 affinity, intracellular ATP content, or RBC susceptibility to hemolysis in hypoxia (which was low in both WT and Panx1−/− RBCs in these experiments), as these did not differ from those of WT RBCs. Indeed, RBC lysis is an important potential confounder when interpreting experiments addressing the determinants of regulated ATP export from RBCs, as we and others have discussed (34–37). Furthermore, the Panx1-dependent differences (Panx1−/− vs. WT) in the accumulation of extracellular ATP in response to hypoxia were greater by an order of magnitude than the predicted [ATP] that would result from the small degree of RBC lysis we observed. Taken together, these data point to an important role for intravascular ATP in the control of vascular tone during hypoxemia and for Panx1 channels in physiologically relevant, hemolysis-independent extracellular ATP export.
Hypoxia Elicits Panx1-Sensitive Vasodilation and Plasma ATP Elevation In Vivo
In humans and certain animal species (e.g., canines and rats) hypoxemia elicits regional vasodilation in multiple vascular beds, with associated increases in plasma ATP concentration (3). Intravascular ATP administration evokes dose-sensitive and robust vasodilation in humans (2, 10). Accordingly, we sought to test the hypothesis that Panx1 channel-mediated increases in plasma ATP are mechanistically linked to hypoxic vasodilation in vivo. In support of this hypothesis, we observe that Panx1-deficient mice exposed to hypoxia fail to: 1) increase plasma ATP concentration, 2) exhibit the anticipated hypotensive and regional hyperemic responses suggestive of vasodilation, and 3) increase ATP export from RBCs.
In the present study, WT mice exhibited a drop in blood pressure during systemic hypoxia, whereas blood pressure in Panx1 null mice did not change significantly. Unlike humans, blood pressure reduction to hypoxia is the expected response in mice (38). In support of the notion that the hypotension during hypoxia in WT mice was in fact due to peripheral vasodilation, we also observed increases in hindlimb hyperemia in response to hypoxia. Given that reductions in perfusion pressure in the face of increased blood flow equate to an elevated vascular conductance, peripheral vasodilation as indicated by the estimated increases in hindlimb vascular conductance is apparent (Figs. 1A, 3A, and 4). The absolute values of femoral blood flow were similar to values previously reported in anesthetized, resting mice (39, 40). Interestingly, and in striking contrast to WT mice, a ∼10% reduction in baseline hindlimb flow was observed in Panx1−/− mice, raising the possibility that basal (nonhypoxic) Panx1-mediated ATP export may contribute to basal control of peripheral vascular resistance. Whether the differences in baseline HBF might complicate the interpretation of the subsequent hypoxia-induced HBF changes is not known definitively, but we would not expect the baseline differences to weaken or qualify the conclusion that the differences in hypoxia are significant. Notably, the differences in BP and HBF remained significant when expressed as percent change. The time courses of the changes in blood flow and pressure were roughly similar. Isolated RBC microfluidic chamber studies suggest that the release dynamics are quite fast (on the order of milliseconds), therefore ATP export is unlikely to be rate-limiting in the hemodynamic response to hypoxia (41, 42). Differences in the flow and pressure responses between Panx1−/− vs. WT mice were apparent by ∼5 min after the onset of hypoxia. In addition, there was a nonsignificant trend toward a compensatory increase in heart rate in the WT mice that was not seen in the Panx1−/− mice (Fig. 2, A and B). Therefore, differences in the (direct, reflexive, or mediator-driven) chronotropic response to systemic hypoxia could not account for the differences in blood pressure to hypoxia in Panx1−/− vs. WT mice. Taken together, these data indicate that the hemodynamic responses during hypoxia consistent with hypoxic vasodilation are in part mediated by Panx1 channels. Whether Panx1 deletion may alter other determinants of blood flow, such as microvascular density, has not been investigated to our knowledge and was not examined in our studies. Panx1 may contribute to hemodynamic responses to other metabolic and pathologic stimuli (metabolic change, hemorrhage), but we focused exclusively on one stimulus, hypoxia, and future work is needed to determine the role of Panx1 more broadly in adaptive vasoregulation.
In response to hypoxia, the change in MAP correlated significantly with changes in plasma ATP concentrations across WT and Panx1−/− mice (r2 = 0.39, P < 0.05), with Panx1-deficient mice demonstrating lower hypoxia-induced changes in plasma ATP as compared to WT littermate mice. These data collectively show for the first time that Panx1 channels mediate an increase in plasma ATP during hypoxia and are mechanistically linked to hypoxic vasodilation in vivo. Panx1 channels do conduct ATP (43), in contrast to the cystic fibrosis transmembrane conductance regulator (CFTR) channel, which was once thought to be a candidate as an “ATP channel” (44). Although our animal preparation included mechanical ventilation as a means to regulate respiration during hypoxia, future studies are needed to determine whether Panx1 may contribute to such hemodynamic responses to hypoxia in awake mice, as in our anesthetized mice. In addition, future investigations examining whether the role of Panx1 and ATP in vascular tone might vary by the anatomic region (e.g., cerebral blood flow) is warranted. Mouse models and receptor-pharmacological approaches could inform whether ATP itself or a derivative such as adenosine is ultimately responsible for stimulating vasodilation in response to hypoxia. Nevertheless, we observe clear impairments in systemic and regional hemodynamics and attenuated plasma ATP during hypoxia in whole body Pan1 deficient mice.
RBC Export of ATP is Mediated via Panx1 Channels
Cellular export of ATP can variably increase or decrease vascular tone depending on the cell type, stimulus, and purinergic receptor involvement. For example, phenylephrine-induced vasoconstriction via the α-1D (α1D) adrenergic receptor on vascular smooth muscle cells (VSMCs) appears to involve the export of ATP via Panx1 (45). Alternatively, intravascular ATP can act as a sympatholytic agent, blunting vasoconstrictor (pressor) responses to adrenergic agonists (46). Luminal ATP can act as an endothelium-dependent vasodilator by stimulating the activity of endothelial nitric oxide synthase and endothelium-dependent hyperpolarization (47, 48). The cellular source(s) contributing to increased plasma ATP during hypoxia have been extensively debated (45). However, evidence has suggested against roles for sympathetic nerves or skeletal muscle as the primary sources for the release of vasodilator plasma ATP during hypoxemia (49–51), in part due to the physical barrier of the vascular smooth muscle coupled to a high density of membrane-bound ectonucleotidases (48, 52, 53). Nevertheless, tissue perfusion and the resupply of O2-carrying blood to the region of low oxygen tension is sufficient to increase intravascular (luminal) ATP (3, 24, 48, 54). Thus, the cellular source of the increased plasma ATP presumably lies within the blood itself or vascular lumen, with red blood cells and endothelial cells being candidates.
Hypoxia appears to promote ATP release from isolated endothelial cells or vascular smooth muscle cells only weakly (55). Therefore, given that RBCs can directly regulate vascular tone and export ATP in response to hypoxia, coupled to the fact that ATP elicits vasodilation capable of overriding sympathetic vasoconstrictor tone, RBCs are a strong candidate contributor to plasma ATP accumulation during hypoxia. Moreover, RBC export of ATP basally and in response to Hb deoxygenation has been previously linked to Panx1, which may serve as a conduit for hypoxic ATP export from RBCs (20). ATP export from RBCs can occur in response to various stimuli, and the underlying mechanisms and responsible signaling elements may vary (35). However, a line of evidence suggests a role for Panx1 as one mediator in the release of ATP from RBCs. For example, we have shown previously that pharmacological inhibitors of Panx1 inhibit hypoxic ATP export from human RBCs (21). Under nonhypoxic conditions, Panx1 inhibitors also decrease the release of ATP from malaria-infected RBCs basally and in response to a combination of stimuli promoting cAMP formation such as the beta-adrenergic agonist, isoproterenol, the adenylate cyclase activator, forskolin, and the phosphodiesterase inhibitor, papaverine (56). Shear-stress-induced RBC ATP export is also sensitive to Panx1 inhibitors (41). Leal-Denis (23) reported that cAMP agonists promoted a minor but significant release of ATP from WT (but not Panx1−/−) murine RBCs attached to poly-d-lysine-coated coverslips. Notably, pharmacological inhibitors of Panx1 can have off-target (Panx1-independent) actions, limiting the ability to link their effects with the Panx1 function. In this regard, Panx1−/− mice become an important tool to test the role of Panx1 in hypoxia-induced ATP export in vivo, as in our study. Consistent with this notion, the presented data indicate that RBCs from Panx1 deficient mice do not strongly export ATP in response to the physiological stimulus of hemoglobin deoxygenation. To this end, lack of export cannot be explained by hemolysis (see RESULTS) nor depletion of intracellular ATP, which is several orders of magnitude more concentrated than is plasma ATP (30) and is regenerated via glycolysis during hypoxia (30, 57, 58). Collectively, these findings support the assertion that hypoxia-induced ATP export via Panx1 channels from RBCs may be sufficient to trigger the in vivo vasodilatory response to hypoxia.
In hypoxia, the docking of deoxygenated Hb with the RBC membrane-resident Band 3 protein (anion exchanger 1 (AE1)) is favored, and cytoskeletal ankyrin-dependent RBC deformability increases in concert with increases in RBC glucose uptake and ATP export (59, 60). However, the specific mechanism linking hypoxia to Panx1-mediated ATP export is unknown. The increase in ATP export does not appear to drive the increase in RBC deformability, as we showed previously that the Panx1 inhibitors (and ATP-release inhibitors) carbenoxolone and glibenclamide did not attenuate human RBC deformability (21, 41). Finally, Leal Denis et al. (23) showed that the ATP release in response to mastoparan 7-induced cell swelling of RBCs was Panx1-dependent. We are not aware of published data on RBCs from Panx1−/− mice examining whether Band 3 binding and downstream effects such as glycolytic flux and glucose transport are modulated.
Plasma ATP in the Context of Other Vasodilatory Mediators
Hypoxic vasodilation is understood to be frequently endothelium-dependent as are pharmacologically-elicited ATP-mediated responses (49, 61, 62). That is, both systemic hypoxia and direct agonism by intraluminal ATP are mediated in part via endothelium-dependent hyperpolarization, NO, and vasodilating prostaglandins. When acting as an endothelium-dependent vasodilator, ATP exported by RBCs is postulated to elicit vasodilation principally via luminal P2Y (likely subtypes 1, 2, and/or 4) purinergic receptors (63). Moreover, ATP-induced vasodilation evokes an ascending vasodilatory response upstream from the longitudinal site of mediator release within the vasculature (64, 65). Because Panx1-mediated export of ATP occurs upstream of endothelial cell stimulation and consequential vasodilation, inhibition of Panx1 would limit ATP movement into the plasma and thus could presumably precede and account for contributions observed by specific downstream mediation from adenosine, nitric oxide, vasodilator prostaglandins, and endothelium-dependent hyperpolarization. RBCs are capable of exporting not only the vasodilator ATP, but also vasodilator S-nitrosothiols (SNOs; nitric oxide derivatives) during hypoxia, and both mediators have been implicated in hypoxic vasodilation and the regulation of blood flow distribution in various model systems. The relative roles of these two mediators remain to be fully parsed experimentally, including their differential roles in micro- vs. macrohemodynamic responses to hypoxia or other metabolic signals of tissue demand. RBC-derived SNO, alternatively, acts locally as an endothelium-independent vasodilator. ATP-dependent conducted vasodilatory activity may function in tandem with SNO-induced vasodilatory activity. Both mediators are exported by the RBC and exquisitely O2-sensitive via direct coupling to Hb saturation/desaturation (66) and may fine-tune regional blood flow redistribution in accordance with the local O2 demand.
Experimental Considerations
Although we aimed to first examine the hypoxic vasodilatory response to hypoxia in whole body Panx1 null mice, we recognize that in order to unequivocally link RBCs and the (lysis-independent) release of ATP via Panx1 channel to hypoxic vasodilation in vivo, cell-specific deletion is desired. Future studies using cell-specific Panx1−/− mice and/or cross-transfusion will aid in deeper understanding of the contribution of RBC ATP export to hypoxic vasodilation in vivo. Along these lines, once exported into the plasma, ATP may undergo hydrolysis leading to the formation of ADP or adenosine, which also act as vasodilators. Whether ATP itself or such a derivative/precursor is the responsible vasodilator agent to hypoxia in the present study cannot be determined from these studies and is worthy of future investigation. Nonetheless, the present study demonstrates that Panx1 coupled with higher plasma ATP are at minimum required to observe hypoxic vasodilation.
Lastly, the present investigation did not measure blood pressure and blood flow in the same mice, and as a result vasodilation can only be inferred. Direct measures of vasodilation in vivo in mice are challenging outside of mouse window model experiments and rely on vascular tone computations. The present experiment sought to prioritize mechanical ventilation and functional organ stability over the potentially confounding responses associated with ventilation, carotid cannulation, vessel excision, and blood sampling. To this end, the data presented minimize reflex changes in hemodynamics due to trauma and provide a more stable profile by which to observe singular unperturbed endpoints. Nonetheless, a computation of vascular tone by estimating hindlimb vascular conductance across the two groups of mice suggests modulation of peripheral vasodilation to hypoxia in Panx1 null mice (Fig. 4).
Conclusions
In this study, we show a key role for the Panx1 channel in O2-sensitive plasma ATP accumulation and concomitant hypoxic vasodilation in mice in vivo. RBCs from WT, but not Panx1−/−, mice exported ATP in proportion to the degree of RBC deoxygenation ex vivo. Collectively, these findings reveal the distinct contribution of Panx1 to hypoxic vasodilation, and highlight its role as an O2-sensitive ATP conduit and controller of vascular tone. These new findings suggest future work examining whether Panx1-mediated ATP export from RBCs may contribute to O2-sensitive hemodynamic regulation and disturbance, as in anemia or sepsis.
GRANTS
This study was supported by Grants from VA (BX-003478 to T.J.M) and NIH (R01 GM-113838 to T.J.M, T32 HL-007057 supporting B.S.K., and R56 HL-136909-A1 to E.R.L).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
B.S.K., M.A.S., and T.J.M. conceived and designed research; B.S.K., M.A.S., D.A.L.D., H.Z., and T.J.M. performed experiments; B.S.K., M.A.S., D.A.L.D., H.Z., and T.J.M. analyzed data; B.S.K., M.A.S., E.R.L., H.Z., and T.J.M. interpreted results of experiments; B.S.K., M.A.S., and T.J.M. prepared figures; B.S.K., M.A.S., D.A.L.D., and T.J.M. drafted manuscript; B.S.K., M.A.S., E.R.L., D.A.L.D., H.Z., and T.J.M. edited and revised manuscript; B.S.K., M.A.S., E.R.L., D.A.L.D., H.Z., and T.J.M. approved final version of manuscript.
REFERENCES
- 1.Ross JM, Fairchild HM, Weldy J, Guyton AC. Autoregulation of blood flow by oxygen lack. Am J Physiol 202: 21–24, 1962. doi: 10.1152/ajplegacy.1962.202.1.21. [DOI] [PubMed] [Google Scholar]
- 2.Gonzalez-Alonso J, Richardson RS, Saltin B. Exercising skeletal muscle blood flow in humans responds to reduction in arterial oxyhaemoglobin, but not to altered free oxygen. J Physiol 530: 331–341, 2001. doi: 10.1111/j.1469-7793.2001.0331l.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kirby BS, Crecelius AR, Voyles WF, Dinenno FA. Impaired skeletal muscle blood flow control with advancing age in humans: attenuated ATP release and local vasodilation during erythrocyte deoxygenation. Circ Res 111: 220–230, 2012. doi: 10.1161/CIRCRESAHA.112.269571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Simmons GH, Minson CT, Cracowski J-L, Halliwill JR. Systemic hypoxia causes cutaneous vasodilation in healthy humans. J Appl Physiol 103: 608–615, 2007. doi: 10.1152/japplphysiol.01443.2006. [DOI] [PubMed] [Google Scholar]
- 5.Olson KR, Donald JA, Dombkowski RA, Perry SF. Evolutionary and comparative aspects of nitric oxide, carbon monoxide and hydrogen sulfide. Respir Physiol Neurobiol 184: 117–129, 2012. doi: 10.1016/j.resp.2012.04.004. [DOI] [PubMed] [Google Scholar]
- 6.Kirby BS, Voyles WF, Carlson RE, Dinenno FA. Graded sympatholytic effect of exogenous ATP on postjunctional alpha-adrenergic vasoconstriction in the human forearm: implications for vascular control in contracting muscle. J Physiol 586: 4305–4316, 2008. doi: 10.1113/jphysiol.2008.154252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Umbrello M, Dyson A, Feelisch M, Singer M. The key role of nitric oxide in hypoxia: hypoxic vasodilation and energy supply–demand matching. Antioxid Redox Signal 19: 1690–1710, 2013. doi: 10.1089/ars.2012.4979. [DOI] [PubMed] [Google Scholar]
- 8.Rowell LB, Johnson DG, Chase PB, Comess KA, Seals DR. Hypoxemia raises muscle sympathetic activity but not norepinephrine in resting humans. J Appl Physiol (1985) 66: 1736–1743, 1989. doi: 10.1152/jappl.1989.66.4.1736. [DOI] [PubMed] [Google Scholar]
- 9.Weisbrod CJ, Minson CT, Joyner MJ, Halliwill JR. Effects of regional phentolamine on hypoxic vasodilatation in healthy humans. J Physiol 537: 613–621, 2001. doi: 10.1111/j.1469-7793.2001.00613.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rosenmeier JB, Hansen J, González-Alonso J. Circulating ATP-induced vasodilatation overrides sympathetic vasoconstrictor activity in human skeletal muscle. J Physiol 558: 351–365, 2004. doi: 10.1113/jphysiol.2004.063107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rosenmeier JB, Yegutkin GG, González-Alonso J. Activation of ATP/UTP-selective receptors increases blood flow and blunts sympathetic vasoconstriction in human skeletal muscle. J Physiol 586: 4993–5002, 2008. doi: 10.1113/jphysiol.2008.155432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ellsworth ML, Forrester T, Ellis CG, Dietrich HH. The erythrocyte as a regulator of vascular tone. Am J Physiol 269: H2155–H2161, 1995. doi: 10.1152/ajpheart.1995.269.6.H2155. [DOI] [PubMed] [Google Scholar]
- 13.Gorman MW, Feigl EO, Buffington CW. Human plasma ATP concentration. Clin Chem 53: 318–325, 2007. doi: 10.1373/clinchem.2006.076364. [DOI] [PubMed] [Google Scholar]
- 14.Sprague RS, Ellsworth ML, Stephenson AH, Lonigro AJ. Participation of cAMP in a signal-transduction pathway relating erythrocyte deformation to ATP release. Am J Physiol Cell Physiol 281: C1158–C1164, 2001. doi: 10.1152/ajpcell.2001.281.4.C1158. [DOI] [PubMed] [Google Scholar]
- 15.Chiu Y-H, Jin X, Medina CB, Leonhardt SA, Kiessling V, Bennett BC, Shu S, Tamm LK, Yeager M, Ravichandran KS, Bayliss DA. A quantized mechanism for activation of pannexin channels. Nat Commun 8: 14324, 2017. doi: 10.1038/ncomms14324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dahl G. The Pannexin1 membrane channel: distinct conformations and functions. FEBS Lett 592: 3201–3209, 2018. doi: 10.1002/1873-3468.13115. [DOI] [PubMed] [Google Scholar]
- 17.Locovei S, Bao L, Dahl G. Pannexin 1 in erythrocytes: function without a gap. Proc Natl Acad Sci USA 103: 7655–7659, 2006. doi: 10.1073/pnas.0601037103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gödecke S, Roderigo C, Rose CR, Rauch BH, Gödecke A, Schrader J. Thrombin-induced ATP release from human umbilical vein endothelial cells. Am J Physiol Cell Physiol 302: C915–C923, 2012. doi: 10.1152/ajpcell.00283.2010. [DOI] [PubMed] [Google Scholar]
- 19.Lohman AW, Billaud M, Straub AC, Johnstone SR, Best AK, Lee M, Barr K, Penuela S, Laird DW, Isakson BE. Expression of pannexin isoforms in the systemic murine arterial network. J Vasc Res 49: 405–416, 2012. doi: 10.1159/000338758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sridharan M, Adderley SP, Bowles EA, Egan TM, Stephenson AH, Ellsworth ML, Sprague RS. Pannexin 1 is the conduit for low oxygen tension-induced ATP release from human erythrocytes. Am J Physiol Heart Circ Physiol 299: H1146–H1152, 2010. doi: 10.1152/ajpheart.00301.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhu H, Zennadi R, Xu BX, Eu JP, Torok JA, Telen MJ, McMahon TJ. Impaired adenosine-5′-triphosphate release from red blood cells promotes their adhesion to endothelial cells: a mechanism of hypoxemia after transfusion. Crit Care Med 39: 2478–2486, 2011. doi: 10.1097/CCM.0b013e318225754f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Seminario-Vidal L, Okada SF, Sesma JI, Kreda SM, van Heusden CA, Zhu Y, Jones LC, O’Neal WK, Penuela S, Laird DW, Boucher RC, Lazarowski ER. Rho signaling regulates pannexin 1-mediated ATP release from airway epithelia. J Biol Chem 286: 26277–26286, 2011. doi: 10.1074/jbc.M111.260562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leal Denis MF, Incicco JJ, Espelt MV, Verstraeten SV, Pignataro OP, Lazarowski ER, Schwarzbaum PJ. Kinetics of extracellular ATP in mastoparan 7-activated human erythrocytes. Biochim Biophys Acta 1830: 4692–4707, 2013[Erratum inBiochim BiophysActa1840: 1837, 2014]. doi: 10.1016/j.bbagen.2013.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Farias M, Gorman MW, Savage MV, Feigl EO. Plasma ATP during exercise: possible role in regulation of coronary blood flow. Am J Physiol Heart Circ Physiol 288: H1586–H1590, 2005. doi: 10.1152/ajpheart.00983.2004. [DOI] [PubMed] [Google Scholar]
- 25.Gorman MW, Marble DR, Ogimoto K, Feigl EO. Measurement of adenine nucleotides in plasma. Luminescence 18: 173–181, 2003. doi: 10.1002/bio.721. [DOI] [PubMed] [Google Scholar]
- 26.Dietrich HH, Ellsworth ML, Sprague RS, Dacey RG. Red blood cell regulation of microvascular tone through adenosine triphosphate. Am J Physiol Heart Circ Physiol 278: H1294–H1298, 2000. doi: 10.1152/ajpheart.2000.278.4.H1294. [DOI] [PubMed] [Google Scholar]
- 27.Sprague RS, Olearczyk JJ, Spence DM, Stephenson AH, Sprung RW, Lonigro AJ. Extracellular ATP signaling in the rabbit lung: erythrocytes as determinants of vascular resistance. Am J Physiol Heart Circ Physiol 285: H693–H700, 2003. doi: 10.1152/ajpheart.01026.2002. [DOI] [PubMed] [Google Scholar]
- 28.Sprague RS, Bowles EA, Achilleus D, Stephenson AH, Ellis CG, Ellsworth ML. A selective phosphodiesterase 3 inhibitor rescues low PO2-induced ATP release from erythrocytes of humans with type 2 diabetes: implication for vascular control. Am J Physiol Heart Circ Physiol 301: H2466–H2472, 2011[Erratum inAm J Physiol Heart Circ Physiol302: H378, 2012]. doi: 10.1152/ajpheart.00729.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bennett-Guerrero E, Veldman TH, Doctor A, Telen MJ, Ortel TL, Reid TS, Mulherin MA, Zhu H, Buck RD, Califf RM, McMahon TJ. Evolution of adverse changes in stored RBCs. Proc Natl Acad Sci USA 104: 17063–17068, 2007. doi: 10.1073/pnas.0708160104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kirby BS, Hanna G, Hendargo HC, McMahon TJ. Restoration of intracellular ATP production in banked red blood cells improves inducible ATP export and suppresses RBC-endothelial adhesion. Am J Physiol Heart Circ Physiol 307: H1737–H1744, 2014. doi: 10.1152/ajpheart.00542.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Olearczyk JJ, Stephenson AH, Lonigro AJ, Sprague RS. Heterotrimeric G protein Gi is involved in a signal transduction pathway for ATP release from erythrocytes. Am J Physiol Heart Circ Physiol 286: H940–H945, 2004. doi: 10.1152/ajpheart.00677.2003. [DOI] [PubMed] [Google Scholar]
- 32.Harboe M. A method for determination of hemoglobin in plasma by near-ultraviolet spectrophotometry. Scand J Clin Lab Invest 11: 66–70, 1959. doi: 10.3109/00365515909060410. [DOI] [PubMed] [Google Scholar]
- 33.Malinauskas RA. Plasma hemoglobin measurement techniques for the in vitro evaluation of blood damage caused by medical devices. Artif Organs 21: 1255–1267, 1997. doi: 10.1111/j.1525-1594.1997.tb00486.x. [DOI] [PubMed] [Google Scholar]
- 34.Keller AS, Diederich L, Panknin C, DeLalio LJ, Drake JC, Sherman R, Jackson EK, Yan Z, Kelm M, Cortese-Krott MM, Isakson BE. Possible roles for ATP release from RBCs exclude the cAMP-mediated Panx1 pathway. Am J Physiol Cell Physiol 313: C593–C603, 2017. doi: 10.1152/ajpcell.00178.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kirby BS, Schwarzbaum PJ, Lazarowski ER, Dinenno FA, McMahon TJ. Liberation of ATP secondary to hemolysis is not mutually exclusive of regulated export. Blood 125: 1844–1845, 2015. doi: 10.1182/blood-2014-11-609610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lopez Domowicz DA, Welsby I, Esther CR, Zhu H, Marek RD, Lee G, Shah N, Poisson JL, McMahon TJ. Effects of repleting organic phosphates in banked erythrocytes on plasma metabolites and vasoactive mediators after red cell exchange transfusion in sickle cell disease. Blood Transfus 18: 200–207, 2020. doi: 10.2450/2020.0237-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sikora J, Orlov SN, Furuya K, Grygorczyk R. Hemolysis is a primary ATP-release mechanism in human erythrocytes. Blood 124: 2150–2157, 2014. doi: 10.1182/blood-2014-05-572024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Totzeck M, Hendgen-Cotta UB, Luedike P, Berenbrink M, Klare JP, Steinhoff H-J, Semmler D, Shiva S, Williams D, Kipar A, Gladwin MT, Schrader J, Kelm M, Cossins AR, Rassaf T. Nitrite regulates hypoxic vasodilation via myoglobin-dependent nitric oxide generation. Circulation 126: 325–334, 2012. doi: 10.1161/CIRCULATIONAHA.111.087155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sonobe T, Tsuchimochi H, Schwenke DO, Pearson JT, Shirai M. Treadmill running improves hindlimb arteriolar endothelial function in type 1 diabetic mice as visualized by X-ray microangiography. Cardiovasc Diabetol 14: 51, 2015. doi: 10.1186/s12933-015-0217-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang C-H, Chen K-T, Mei H-F, Lee J-F, Cherng W-J, Lin S-J. Assessment of mouse hind limb endothelial function by measuring femoral artery blood flow responses. J Vasc Surg 53: 1350–1358, 2011. doi: 10.1016/j.jvs.2010.10.128. [DOI] [PubMed] [Google Scholar]
- 41.Forsyth AM, Wan J, Owrutsky PD, Abkarian M, Stone HA. Multiscale approach to link red blood cell dynamics, shear viscosity, and ATP release. Proc Natl Acad Sci USA 108: 10986–10991, 2011. doi: 10.1073/pnas.1101315108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wan J, Ristenpart WD, Stone HA. Dynamics of shear-induced ATP release from red blood cells. Proc Natl Acad Sci USA 105: 16432–16437, 2008. doi: 10.1073/pnas.0805779105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bao L, Locovei S, Dahl G. Pannexin membrane channels are mechanosensitive conduits for ATP. FEBS Lett 572: 65–68, 2004. doi: 10.1016/j.febslet.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 44.Wang L, Olivecrona G, Götberg M, Olsson ML, Winzell MS, Erlinge D. ADP acting on P2Y13 receptors is a negative feedback pathway for ATP release from human red blood cells. Circ Res 96: 189–196, 2005. doi: 10.1161/01.RES.0000153670.07559.E4. [DOI] [PubMed] [Google Scholar]
- 45.Billaud M, Lohman AW, Straub AC, Looft-Wilson R, Johnstone SR, Araj CA, Best AK, Chekeni F, Ravichandran K, Penuela S, Laird DW, Isakson BE. Pannexin1 regulates α1-adrenoreceptor-mediated vasoconstriction. Circ Res 109: 80–85, 2011. doi: 10.1161/CIRCRESAHA.110.237594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kirby BS, Crecelius AR, Voyles WF, Dinenno FA. Modulation of postjunctional α-adrenergic vasoconstriction during exercise and exogenous ATP infusions in ageing humans. J Physiol 589: 2641–2653, 2011. doi: 10.1113/jphysiol.2010.204081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Burnstock G. Blood cells: an historical account of the roles of purinergic signalling. Purinergic Signal 11: 411–434, 2015. doi: 10.1007/s11302-015-9462-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mortensen SP, Thaning P, Nyberg M, Saltin B, Hellsten Y. Local release of ATP into the arterial inflow and venous drainage of human skeletal muscle: insight from ATP determination with the intravascular microdialysis technique. J Physiol 589: 1847–1857, 2011. doi: 10.1113/jphysiol.2010.203034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Crecelius AR, Kirby BS, Luckasen GJ, Larson DG, Dinenno FA. ATP-mediated vasodilatation occurs via activation of inwardly rectifying potassium channels in humans. J Physiol 590: 5349–5359, 2012. doi: 10.1113/jphysiol.2012.234245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Crecelius AR, Kirby BS, Richards JC, Garcia LJ, Voyles WF, Larson DG, Luckasen GJ, Dinenno FA. Mechanisms of ATP-mediated vasodilation in humans: modest role for nitric oxide and vasodilating prostaglandins. Am J Physiol Heart Circ Physiol 301: H1302–H1310, 2011. doi: 10.1152/ajpheart.00469.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mortensen SP, González-Alonso J, Bune LT, Saltin B, Pilegaard H, Hellsten Y. ATP-induced vasodilation and purinergic receptors in the human leg: roles of nitric oxide, prostaglandins, and adenosine. Am J Physiol Regul Integr Comp Physiol 296: R1140–R1148, 2009. doi: 10.1152/ajpregu.90822.2008. [DOI] [PubMed] [Google Scholar]
- 52.Kauffenstein G, Drouin A, Thorin-Trescases N, Bachelard H, Robaye B, D’Orléans-Juste P, Marceau F, Thorin E, Sévigny J. NTPDase1 (CD39) controls nucleotide-dependent vasoconstriction in mouse. Cardiovasc Res 85: 204–213, 2010. doi: 10.1093/cvr/cvp265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mortensen SP, González-Alonso J, Nielsen J-J, Saltin B, Hellsten Y. Muscle interstitial ATP and norepinephrine concentrations in the human leg during exercise and ATP infusion. J Appl Physiol 107: 1757–1762, 2009. doi: 10.1152/japplphysiol.00638.2009. [DOI] [PubMed] [Google Scholar]
- 54.González-Alonso J, Olsen DB, Saltin B. Erythrocyte and the regulation of human skeletal muscle blood flow and oxygen delivery: role of circulating ATP. Circ Res 91: 1046–1055, 2002. doi: 10.1161/01.RES.0000044939.73286.E2. [DOI] [PubMed] [Google Scholar]
- 55.Kirby BS, Crecelius AR, Richards JC, Dinenno FA. Sources of intravascular ATP during exercise in humans: critical role for skeletal muscle perfusion. Exp Physiol 98: 988–998, 2013. doi: 10.1113/expphysiol.2012.071555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Alvarez CL, Schachter J, de Sá Pinheiro AA, Silva L de S, Verstraeten SV, Persechini PM, Schwarzbaum PJ. Regulation of extracellular ATP in human erythrocytes infected with plasmodium falciparum. PLoS ONE 9: e96216, 2014. doi: 10.1371/journal.pone.0096216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Beutler E. Red cell enzyme defects. Hematol Pathol 4: 103–114, 1990. [PubMed] [Google Scholar]
- 58.Racine ML, Dinenno FA. Reduced deformability contributes to impaired deoxygenation-induced ATP release from red blood cells of older adult humans. The J Physiol 597: 4503–4519, 2019. doi: 10.1113/JP278338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chu H, McKenna MM, Krump NA, Zheng S, Mendelsohn L, Thein SL, Garrett LJ, Bodine DM, Low PS. Reversible binding of hemoglobin to band 3 constitutes the molecular switch that mediates O2 regulation of erythrocyte properties. Blood 128: 2708–2716, 2016. doi: 10.1182/blood-2016-01-692079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Faris A, Spence DM. Measuring the simultaneous effects of hypoxia and deformation on ATP release from erythrocytes. Analyst 133: 678–682, 2008. doi: 10.1039/b719990b. [DOI] [PubMed] [Google Scholar]
- 61.Crecelius AR, Richards JC, Luckasen GJ, Larson DG, Dinenno FA. Reactive hyperemia occurs via activation of inwardly rectifying potassium channels and Na+/K+-ATPase in humans. Circ Res 113: 1023–1032, 2013. doi: 10.1161/CIRCRESAHA.113.301675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Racine ML, Crecelius AR, Luckasen GJ, Larson DG, Dinenno FA. Inhibition of Na+/K+ -ATPase and KIR channels abolishes hypoxic hyperaemia in resting but not contracting skeletal muscle of humans. J Physiol 596: 3371–3389, 2018. doi: 10.1113/JP275913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Burnstock G. Purinergic signaling in the cardiovascular system. Circ Res 120: 207–228, 2017. doi: 10.1161/CIRCRESAHA.116.309726. [DOI] [PubMed] [Google Scholar]
- 64.Segal SS, Duling BR. Flow control among microvessels coordinated by intercellular conduction. Science 234: 868–870, 1986. doi: 10.1126/science.3775368. [DOI] [PubMed] [Google Scholar]
- 65.Winter P, Dora KA. Spreading dilatation to luminal perfusion of ATP and UTP in rat isolated small mesenteric arteries. J Physiol 582: 335–347, 2007. doi: 10.1113/jphysiol.2007.135202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.McMahon TJ, Stone AE, Bonaventura J, Singel DJ, Stamler JS. Functional coupling of oxygen binding and vasoactivity in S-nitrosohemoglobin. J Biol Chem 275: 16738–16745, 2000. doi: 10.1074/jbc.M000532200. [DOI] [PubMed] [Google Scholar]