

Abstract
We aimed to determine 1) the mechanism(s) that enables glucose-6-phosphate dehydrogenase (G6PD) to regulate serum response factor (SRF)- and myocardin (MYOCD)-driven smooth muscle cell (SMC)-restricted gene expression, a process that aids in the differentiation of SMCs, and 2) whether G6PD-mediated metabolic reprogramming contributes to the pathogenesis of vascular diseases in metabolic syndrome (MetS). Inhibition of G6PD activity increased (>30%) expression of SMC-restricted genes and concurrently decreased (40%) the growth of human and rat SMCs ex vivo. Expression of SMC-restricted genes decreased (>100-fold) across successive passages in primary cultures of SMCs isolated from mouse aorta. G6PD inhibition increased Myh11 (47%) while decreasing (>50%) Sca-1, a stem cell marker, in cells passaged seven times. Similarly, CRISPR-Cas9-mediated expression of the loss-of-function Mediterranean variant of G6PD (S188F; G6PDS188F) in rats promoted transcription of SMC-restricted genes. G6PD knockdown or inhibition decreased (48.5%) histone deacetylase (HDAC) activity, enriched (by 3-fold) H3K27ac on the Myocd promoter, and increased Myocd and Myh11 expression. Interestingly, G6PD activity was significantly higher in aortas from JCR rats with MetS than control Sprague-Dawley (SD) rats. Treating JCR rats with epiandrosterone (30 mg/kg/day), a G6PD inhibitor, increased expression of SMC-restricted genes, suppressed Serpine1 and Epha4, and reduced blood pressure. Moreover, feeding SD control (littermates) and G6PDS188F rats a high-fat diet for 4 mo increased Serpine1 and Epha4 expression and mean arterial pressure in SD but not G6PDS188F rats. Our findings demonstrate that G6PD downregulates transcription of SMC-restricted genes through HDAC-dependent deacetylation and potentially augments the severity of vascular diseases associated with MetS.
NEW & NOTEWORTHY This study gives detailed mechanistic insight about the regulation of smooth muscle cell (SMC) phenotype by metabolic reprogramming and glucose-6-phosphate dehydrogenase (G6PD) in diabetes and metabolic syndrome. We demonstrate that G6PD controls the chromatin modifications by regulating histone deacetylase (HDAC) activity, which deacetylates histone 3-lysine 9 and 27. Notably, inhibition of G6PD decreases HDAC activity and enriches H3K27ac on myocardin gene promoter to enhance the expression of SMC-restricted genes. Also, we demonstrate for the first time that G6PD inhibitor treatment accentuates metabolic and transcriptomic reprogramming to reduce neointimal formation in coronary artery and large artery elastance in metabolic syndrome rats.
Keywords: G6PD, Mediterranean variant, metabolic syndrome, PPP, smooth muscle cell phenotype
The primary function of differentiated smooth muscle cells (SMCs) in the medial layer of the blood vessel wall is to contract and relax to regulate blood flow throughout the circulatory system. However, SMCs can switch from a differentiated to a dedifferentiated phenotype and promote vascular remodeling in response to strain caused by physical stress (e.g., very high pressure and turbulent flow), chemical insult, or iatrogenic intervention (1). Similarly, SMCs in cell culture dedifferentiate when they are passaged (2). These dedifferentiated SMCs are highly proliferative, synthetic, and migratory (1). SMCs also transdifferentiate to endothelial-, macrophage-, fibroblast-, or osteoblast-like cells (3–7). The de/transdifferentiated SMCs contribute to the pathogenic remodeling of blood vessels that eventually impedes the flow of blood to vital organs (1).
SMC plasticity is controlled by several transcription factors, among which serum response factor (SRF) and myocardin (MYOCD), a transcriptional coactivator of SRF (8), maintain adult SMCs in a differentiated state (9). Overexpression of MYOCD transforms non-smooth muscle cells into differentiated SMC-like cells (10–13). SRF-MYOCD modulates these effects by promoting expression of SMC-restricted genes and proteins. Conversely, decreasing MYOCD expression elicits the downregulation of SMC-restricted genes, a process that precedes dedifferentiation of SMCs, and vascular remodeling (1). Our laboratory recently demonstrated that increased activity of glucose-6-phosphate dehydrogenase (G6PD), the rate-limiting enzyme in the pentose phosphate pathway (PPP), downregulates the expression of Myocd and SRF-MYOCD-driven SMC-restricted genes (14, 15). Although the PPP produces the ribose sugar used for the synthesis of the nucleotides needed for de novo synthesis of RNA (gene expression) and DNA (DNA replication), silencing G6pd expression or inhibiting G6PD activity paradoxically increases expression of SMC-restricted genes (15). In an earlier study, we suggested that inhibiting G6PD activity increases expression of Myocd and SMC-restricted genes in part via microRNA (miRNA)- and protein kinase G-dependent mechanisms (15). In addition, we and others showed that G6PD-derived NADPH increases histone deacetylase (HDAC) activity in cancer cells (16) and that inhibiting G6PD and/or silencing G6pd decreases proliferation of cancer cells and SMCs (15, 16). From these studies, we hypothesized that G6PD directly or indirectly regulates HDAC-dependent histone deacetylation and, through this mechanism, downregulates the transcription of Myocd and SMC-restricted genes in SMCs.
In recent years, significant progress has been made in the prevention and management of cardiovascular diseases. Nevertheless, the incidence of vascular diseases such as hypertension, coronary artery disease, and peripheral vascular disease continues to increase unabated without adequate treatments, and they remain to this day the leading cause of mortality in the United States and worldwide. This is explained, at least in part, by the fact that vascular diseases are multifactorial disorders and are caused by multiple genetic and modifiable risk factors (17). For example, type 2 diabetes and metabolic syndrome are risk factors for vascular diseases. In patients with these disorders, cells of various organs and tissues, including vascular tissue, undergo metabolic reprogramming that manifests as notable alterations in lipid and glucose metabolism (18). In particular, both glycolysis and G6PD activity (which participates in both glucose and lipid oxidation) are increased in the liver, the pancreas, adipose tissue, vascular tissue, and the heart (19–24). However, it remains unknown whether this increased G6PD activity and metabolic reprogramming contribute to the downregulated expression of Myocd and SRF-MYOCD gene targets in SMCs affected by diabetes or metabolic syndrome. Here, we set out to determine whether increased G6PD activity contributes to vascular remodeling and, in so doing, to the etiology of the hypertension and occlusive vascular diseases associated with diabetes and metabolic syndrome.
MATERIALS AND METHODS
Experimental Animals
All animal experimental protocols (IACUC no. 30-1-0517) were approved by the New York Medical College Institutional Animal Care and Use Committee (IACUC), and all procedures conformed to the guidelines from the NIH Guide for the Care and Use of Laboratory Animals. The animals were housed at ambient room temperature and pressure and were exposed to a 12:12-h light-dark cycle. They were given food and water ad libitum. At the end of the study period, hemodynamic parameters were measured through cardiac catheterization in animals anesthetized through inhalation of isoflurane (USP, 1-chloro-2,2,2-trifluoroethyl difluoromethyl ether; induced at 3% and maintained at 1.5%), after which the animals were euthanized and various organs were collected for biochemical analysis and metabolomic and transcriptomic studies.
Animal Models
We used Myh11-CreERT2 mice (32 wk old; male, because Myh11 is an X-linked gene), Sprague-Dawley rats (SD; 28–36 wk old; male and female), Mediterranean G6PD variant rats (G6PDS188F; 28–36 wk old; male and female), and JCR rats (32–36 wk old; male and female), a model of metabolic syndrome. SD rats were used as the control for JCR rats and to mimic the normal heterogeneous human population. JCR rats were provided by P. Rocic, who acquired them from Dr. Spencer Proctor (University of Alberta, Edmonton, AB, Canada). These rats were treated with either epiandrosterone (EPI), a G6PD inhibitor, or the vehicle (DMSO) for 28 days. A detailed description of the methods used to generate and genotype G6PDS188F rats and Myh11-CreERT2 mice was provided in previous studies (25, 26).
High-Fat Diet Feeding
SD and G6PDS188F rats were divided randomly into two groups: one received normal chow (NC) and the other received a high-fat diet (HFD) for 4 mo. The HFD (no. D12492, Research Diets Inc., USA) contained 20% protein, 60% fat, and 20% carbohydrate.
Echocardiography and Hemodynamics
Echocardiography and hemodynamics were performed as described previously (27–29). All animals were anesthetized and placed on a heated table for performance of echocardiography and cardiac catheterization. Briefly, echocardiography was performed in 2% isoflurane-anesthetized rats with a Vevo 770 imaging system (VisualSonics, Toronto, ON, Canada). Two-dimensional parasternal short-axis views were obtained, M-mode assessment of left ventricular (LV) function was performed, and LV parameters were measured. At the end of the experimental protocol, rats were anesthetized with 4% isoflurane, after which 1–2% isoflurane was used to maintain anesthesia during the entire duration of the surgery and data acquisition. Body temperature of the animal during the surgery was maintained with a heating pad. About 3 cm2 of skin over the ventral neck region was exposed to locate the right common carotid artery. After the artery was carefully isolated, a 2.0 F Millar Micro-Tip pressure catheter was inserted and pushed to the left ventricle, where hemodynamic parameters were recorded. Data were acquired and analyzed with PowerLab (ADInstruments).
Histology
The hearts were removed from the chest and flushed retrogradely with 1% neutral buffered formalin (NBF) in 0.5% agarose at 20 cmH2O pressure and fixed in 10% NBF. The fixed hearts cut in midplane section were blocked and embedded in paraffin, after which 5-μm transverse sections were cut and stained with hematoxylin-eosin and Verhoeff-Van Gieson (VVG). Images were captured on an Axio imager M1 microscope attached to an Axiocam MRm camera with AxioVision microscopy software (Carl Zeiss, Jena, Germany), and data were analyzed with ImageJ software.
G6PD Mutagenesis
Human full-length G6PD cDNA was cloned into the pCMV6-XL5 vector (Origene Technologies, Inc.) previously in our laboratory at the University of South Alabama (30). The 1,638-bp full-length/wild-type cDNA sequence of G6PD (NM_000402.3) was isolated by polymerase chain reaction and cloned into the p-Ds-RedN1 expression vector (Clontech) between the NheI and XhoI sites (G6PD-RFP-wt). Single (S210A) site-directed G6PD-Red-wt mutants were constructed with a Quikchange Multi Site-Directed Mutagenesis Kit (Stratagene). SMCs (106) plated in 24-well plates for 48 h and then transfected with 1) G6PD-RFP-wt alone or 2) 0.8 μg of p-Ds-Red or G6PD-RFP-S210A with 2 μg of FUGENE 6 reagent (Roche) for 48 h were used for cell growth studies.
Adenovirus Preparation
Working in collaboration with Dr. Fabio Recchia at New York Medical College, we previously developed adenoviral vectors to deliver G6PD-specific short hairpin (sh)RNA into cultured cells (30–32). Briefly, a G6PD-shRNA gene sequence (CGGAAACGUCGUACACUUtt) that specifically and efficaciously downregulated G6PD and a scrambled sequence (negative control) were custom cloned by GeneScript in an adenoviral vector under the H1 promoter to drive shRNA expression. To monitor transfection efficiency, the vector also carried a green fluorescent protein (GFP) marker (coral GPF, cGFP) under the control of the cytomegalovirus (CMV) promoter. These vector-based shRNAs were packaged in adenoviruses by Welgen Laboratories. Stocks of adenoviral vector [3 × 1013 plaque-forming units (PFU)] encoding the G6PD (Ad-G6PD-shRNA) or scrambled shRNA were diluted (1012 PFU) and used for transfecting cells.
Primary Aortic Smooth Muscle Cell Extraction Protocol
Before the rats and mice were euthanized, the following solutions were prepared on the day of dissection: buffer A: 39.5 mL of DMEM-F-12 medium + 10 mL of FBS (final 20%) (catalog no. A3160601, Thermo Fisher) + 0.5 mL of Pen-Strep (final 1%) warmed to 37°C; buffer B: 10 mL of Hanks’ buffered saline solution (HBSS) + 10 mg of collagenase II (catalog no. LS004174, Worthington) + 10 mg of soybean trypsin inhibitor (catalog no. LS003570, Worthington) filtered with a 0.2-μm filter; and buffer C: 3 mL of buffer B + 67 μL of elastase (catalog no. LS002279, Worthington) at 24.8 mg/mL. Aortas from three or four mice were pooled and used per preparation. Rats and mice were anesthetized with 4–4.5% isoflurane and placed in supine position with the dorsal part lying on a flat, movable surface. Abdomen and thorax regions were wiped with 70% alcohol. With surgical scissors, the skin was cut to expose the abdomen and thorax, with a puncture made in the diaphragm to euthanize the animal. After this, the lungs and other organs were removed to expose the entire aorta until the renal bifurcation. Working rapidly, we carefully dissected out the aortas and placed them in 3 mL of buffer B in a 15-mL conical tube at 37°C for 20 min. After this digestion for 20 min, the aortas were cleaned with fine pairs of forceps by peeling off the fuzzy outer layer of adventitia. They were then cut longitudinally and scraped to remove the endothelial cell layer, the plate was then covered and transferred to a tissue culture biosafety hood, and they were cut into 1- to 2-mm pieces with scissors and placed in buffer C in one well of a six-well plate. This was then incubated for 30 min with intermittent pipetting of pieces every 10–15 min. After ∼30 min, tissue chunks disappeared and thin strings were seen, which were centrifuged at 1,500 rpm for 10 min. Supernatant was then resuspended in 5 mL of buffer A and plated in one well of a six-well plate. This was placed in the incubator at 37°C for 3 days without disturbance. After ∼3 days, two-thirds of the medium was replaced with fresh medium, and once the cells were 100% confluent they were trypsinized with 0.25% trypsin-EDTA (catalog no. 25200, Gibco) for subculturing.
Cell Culture
Rat aortic smooth muscle cells (A7r5, ATCC CRL-1444TM) were purchased from American Type Culture Collection (ATCC, Virginia). A7r5 cells, human coronary artery (HSMCs; Cell Application, California), rat aorta (RSMCs, freshly isolated), and mouse aorta (MSMCs, freshly isolated) smooth muscle cells were maintained at 37°C under 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with l-glutamine, 4.5 g/L glucose serum (Gibco, catalog no. 11995-065), and 10% fetal bovine serum (Gibco, catalog no. 10082-147). Once cells reached ∼70–80% confluence, they were subcultured with 0.05% trypsin-EDTA (Gibco, catalog no. 25300-054; Thermo Fisher Scientific, Grand Island, NY) into six-well plates at ∼3 × 105 cells/well. The next day, the cells were incubated for 48 h with Ad-G6PD-shRNA (1012 PFU) or Ad-scramble-shRNA-GFP (1012 PFU; Welgen Inc., catalog no. V1050). DMEM with 25 mM glucose (Gibco, catalog no. 11995) or 5.5 mM glucose (HyClone, catalog no. SH30002-04) was used for high-glucose (HG) and low-glucose (LG) conditions, respectively. Cells were incubated with G6PD inhibitor epiandrosterone (EPI; 50 μmol/L), a selective small-molecule inhibitor, N-ethyl-N′-[(3β,5α)-17-oxoandrostan-3-yl]urea (NEOU; 1 μmol/L), a derivative of dihydroepiandrosterone, or trichostatin A (TSA; 5 μmol/L) for 24 h, after which cells were washed three times in ice-cold PBS and harvested in NP-40 lysis buffer (50 mmol/L Tris·HCl pH 7.4, 150 mmol/L NaCl, 0.5% NP-40) containing EDTA-free complete protease and phosphatase inhibitors (1×; Roche Life Sciences) for protein analysis or collected in QIAzol lysis buffer (Qiagen, catalog no. 79306) and subjected to RT-PCR for analysis of various genes. We used different methods and sets of genes/proteins tested for rigor and reproducibility. To make sure we see what we see in cell culture and confirm our observation we relied on multiple gene/protein markers. Beyond that, we used RT-PCR and Western blot to overcome the limitations of nonspecificity of some antibodies to detect proteins and to identify the trend by two different methods.
Histone Deacetylase Activity Measurement
A7r5 cells were cultured with DMEM medium containing 10% FBS with or without NEOU (1 μmol/L) or EPI (50 μmol/L) for 24 h and Ad-G6PD-shRNA or Ad-scramble-shRNA for 48 h. Trichostatin A (TSA; 5 μmol/L), a histone deacetylase (HDAC) inhibitor, was used as positive control, and HDAC activity was measured with the HDAC Activity Colorimetric Assay Kit (MBL International Corporation, catalog no. JM-K331) per the manufacturer’s protocol. Briefly, 50–200 μg of cell lysate was diluted to a 85-μL volume with deionized H2O in a flat-bottom 96-well plate. To this 10 μL of 10× HDAC assay buffer and 5 μL of HDAC colorimetric substrate were added. This mixture was incubated at 37°C for 1 h, followed by addition of 10 μL of lysine developer to stop the reaction. This was again incubated at 37°C for 30 min. After this incubation period, the Synergy HT Microplate Reader (BioTek, Winooski, VT) was used to measure the absorbance at 400 nm.
Western Blot
Rat aortas were homogenized and cells lysed in lysis buffer (50 mmol/L Tris·HCl pH 7.4, 150 mmol/L NaCl, 0.5% Nonidet P-40) containing EDTA-free complete protease and phosphatase inhibitor (Sigma-Aldrich/Roche). Protein levels were measured with Bradford assays (Bio-Rad), after which 20- to 40-μg samples were run on SDS-PAGE gels, transferred to nitrocellulose membranes, blocked with either 5% bovine serum albumin (BSA) or 4% milk, and subsequently incubated with primary and secondary antibodies and detected with SuperSignal West Pico Chemiluminescent Substrate (catalog no. 34080, ThermoFisher Scientific) on autoradiography film. The following previously validated primary antibodies were used: anti-α-tubulin (TUBA1A; catalog no. sc5286, Santa Cruz), H3K9 (catalog no. 9649 P, Cell Signaling), H3K18 (catalog no. 13998, Cell Signaling), H3K56 (catalog no. 4243, Cell Signaling), total H3 (catalog no. 4499, Cell Signaling), HDAC5 (catalog no. A00430, GenScript), MYH11 (catalog no. BT562, Biomedical technology, Inc.), TAGLN (catalog no. ab14106, Abcam), KLF4 (catalog no. 4038p, Cell Signaling), ACTA2 (catalog no. A2547, Sigma), and ACTB (catalog no. sc47778, Santacruz). TUBA1A or ACTB was used as a loading control, and protein levels were determined through densitometric analysis using ImageJ software.
Measurement of G6PD Activity
G6PD activity was measured spectrophotometrically. The standard assay buffer contained 1 mmol/L MgCl2, 50 mmol/L Tris, pH 8.10 (carefully adjusted with concentrated HCl), 0.1 mmol/L NADP+, and 0.2 mmol/L glucose-6-phosphate. Five-microliter aliquots (containing 5 μg of protein) of homogenate were pipetted into the wells of a 96-well plate (Fisher Scientific), followed by addition of 200 μL of the standard assay buffer. The absorbance at 339 nm was then measured with a plate reader (Synergy HT, BioTek) immediately and for up to 25 min at 50-s intervals. Background absorbance was corrected by subtracting the value of a blank containing no homogenate from all sample readings, and G6PD activity was determined quantitatively with a molar extinction coefficient of 6220 M−1cm−1.
RT-PCR
mRNA levels were analyzed by RT-PCR. Briefly, total RNA was extracted with a Qiagen miRNEasy Kit (catalog no. 217004). Input RNA quality and concentration were measured with a Synergy HT Take3 Microplate Reader (BioTek, Winooski, VT), and cDNA was prepared with SuperScript IV VILO Master Mix (catalog no. 11756500, Invitrogen) for mRNA. RT-PCR was performed in triplicates with TaqManFast Advanced Master Mix for mRNA (catalog no. 44-445-57) in a Mx3000p Real-Time PCR System (Stratagene, Santa Clara, CA). The primers for Myocd (Assay ID Rn01786178), Myh11 (Assay ID Rn1530321_m1), Cnn1 (Assay ID Rn00582058), Tagln (Assay ID Rn01642285), Msx1 (Assay ID Rn00667535), Msx2 (Assay ID Rn01448617_m1), Sca1 (Assay ID Mm03957019_s1), Oct4 (Assay ID Mm03053917_g1), c-Myc (Assay ID Mm01192721_m1), Serpine1 (Assay ID Rn01481341_m1), Epha4 (Assay ID Rn02114236_s1), Tuba1a (Assay ID Rn0153218_g1), and Gapdh (Assay ID Rn01775763), were purchased from ThermoFisher Scientific/TaqMan. Results for mRNA were normalized to internal control Gapdh or Tuba1a, and relative mRNA expression was determined with the ΔCt method (where Ct is threshold cycle).
RNA Sequencing Analysis
Whole genome RNA sequencing (RNA-seq) analysis was performed at the University of Rochester Genomics Research Center. After aortas were collected from rats, total RNA was isolated from tissue with the Qiagen All Prep DNA/RNA/miRNA Universal Kit according to manufacturer’s instructions. RNA was quantified with the NanoDrop (ThermoFisher), and quality was assessed with the Agilent Bioanalyzer 2100. RNA-seq library construction was performed with the TruSeq Stranded Total RNA Preparation Kit (Illumina) with 200 ng of RNA as input according to manufacturer’s instructions. Libraries were sequenced on the HiSeq2500 with single-end reads of 100 nt. Single-end sequencing was done at a depth of 10 million reads per replicate (n = 3). Quantitative analysis, including statistical analysis of differentially expressed genes, was done with Cufflinks 2.0.2 and Cuffdiff2 (http://cufflinks.cbcb.umd.edu). The Benjamini–Hochberg method was applied for multiple test correction [false discovery rate (FDR) < 0.05].
Chromatin Cross-Linking
After culturing under experimental conditions, A7r5 cells were washed with PBS and immediately cross-linked for 10 min with 1% formaldehyde in PBS. Cross-linking was terminated with the addition of glycine, and cells were transferred from flasks, washed with PBS, flash frozen as cell pellets, and stored at −80°C.
Chromatin Immunoprecipitation Sequencing and Differential Binding Analysis
The results of chromatin immunoprecipitation (ChIP) assays were analyzed as described previously (33–35). The following procedures were performed by the Genomics Research Center at the University of Rochester. For each cell pellet, 50-μL aliquots of Protein G Dynabeads (ThermoFisher Scientific) were prepared according to manufacturer’s instructions and subsequently incubated with 7.5 µg of anti-acetyl histone H3 lysine 27 (H3K27Ac; Abcam ab177178) for 6 h at 4°C with rotation. During antibody-bead incubation, cross-linked cell pellets were thawed on ice and subsequently processed through cell lysis (10 mmol/L Tris, 10 mmol/L NaCl, 0.2% Nonidet P-40) and nuclear lysis (50 mmol/L Tris, 10 mmol/L EDTA, 1% SDS) and resuspended in immunoprecipitation (IP) dilution buffer (20 mmol/L Tris, 2 mmol/L EDTA, 150 mmol/L NaCl, 1% Triton X-100, 0.01% SDS), to isolate chromatin. All buffers were kept ice cold and supplemented with protease inhibitor (Roche). Chromatin was fragmented to ∼200 bp by sonication with a Covaris S2 for 10 min at 4°C with the following settings: duty cycle 5%, intensity 4, 200 cycles per burst. Eighty percent of the fragmented chromatin was then incubated with antibody-bead complexes overnight at 4°C, with the remaining 20% set aside at 4°C as the input fraction. Chromatin was then immunoprecipitated, washed, and eluted from antibody-bead complexes in 100 mmol/L NaHCO3, 1% SDS. Immunoprecipitated chromatin and the paired input fractions were then incubated overnight at 65°C to reverse cross-linking. After proteinase K (Sigma) treatment at 45°C for 2 h, DNA from immunoprecipitated chromatin and input chromatin fractions was purified according to manufacturer’s instructions with the Qiagen PCR Purification Kit. Quantification of chromatin immunoprecipitated (ChIP) DNA was determined with the Qubit Fluorometer (ThermoFisher). Quality of ChIP DNA size (200–600 bp) was assessed with the Agilent Bioanalyzer 2100 (Agilent). Library construction was performed with a modified protocol for the TruSeq RNA Sample Preparation v2 Kit (Illumina). Briefly, 5 ng of DNA from ChIP and input samples was subjected to end repair with subsequent 3′ adenylation to create 3′dA overhang suitable for adaptor ligation. Illumina adaptors were ligated to both ends of the DNA, purified with the Pippin Prep (Sage Science) with size selection of the 225- to 500-bp fragments, and amplified with 15 cycles of PCR with primers specific to the adaptor sequences to generate the final library. Libraries were sequenced on a HiSeq2500 (Illumina) with single-end reads of 100 nt. Raw reads generated from the Illumina HiSeq2500 sequencer were demultiplexed with bcl2fastq version 2.19.0. Raw reads were processed with Trimmomatic (SLIDINGWINDOW:4:20 TRAILING:13 LEADING:13 ILLUMINACLIP:/scratch/grc_group/screening/trimmomatic_adapters.fasta:2:30:10 MINLEN:15). Quality reads were aligned to the rat genome (rn6) with Bowtie2. Read alignments were filtered to exclude multimapping reads using samtools (view –q 10). Enriched regions were identified with MACS2 using histone specific parameters (–broad –broad-cutoff 0.1 –c –g 2.15e9). Bedgraph files were converted to bigwig for viewing in the Integrative Genomics Viewer with the UCSC utility called bedGraphToBigWig and the rn6 chromosome sizes. MACS2 bdgcmp was used to subtract treatment-specific input signal from the ChIP bedgraph for each replicate. UCSC bedGraphToBigWig was used to convert the bedgraph files to bigwig. Biological replicate bigwig files were merged by using UCSC bigWigMerge to generate treatment bedgraph files. UCSC bedGraphToBigWig was used to convert the treatment bedgraph file to bigwig for viewing in IGV. SVG file was exported around the Myocd locus for publication quality. Enrichments for all H3K27Ac for NEOU, vehicle control (Veh-Ctrl), Epi, and TSA were used to define differentially bound regions by DiffBind version 2.6.6 within the R version 3.4.1 environment (a language and environment for statistical computing; R Foundation for Statistical Computing, Vienna, Austria. URL https://www.R-project.org/) and DiffBind: differential binding analysis of ChIP sequencing (ChIP-seq) peak data (http://bioconductor.org/packages/release/bioc/vignettes/DiffBind/inst/doc/DiffBind.pdf). A consensus peak set (77,730 peaks) was identified by DiffBind requiring the peak to be present in at least 2 out of 8 samples. Reads were counted within the consensus intervals and library size normalized during differential binding that was performed by DeSeq2 (v2.1.18.1) within DiffBind.
Metabolomics
Details of the methods used to perform metabolomics and the analysis of metabolomic results were described previously (36, 37). Briefly, snap-frozen tissues were ground to powder (GenoGrinder; SPEX, Metuchen, NJ) and extracted in ice-cold methanol-acetonitrile-water (5:3:2 vol/vol/vol) at a 10 mg/mL of extraction solution/tissue ratio or at 1 million cell/ml ratio. Samples were vortexed for 30 min at 4°C, followed by centrifugation at 15,000 g for 10 min at 4°C. Supernatants (20 µL) were collected from each extract for metabolomics analyses. Analyses were performed with a Vanquish UHPLC system coupled online to a Q Exactive mass spectrometer (Thermo Fisher, Bremen, Germany). Samples were resolved over a Kinetex C18 column (2.1 × 150 mm, 1.7 µm; Phenomenex, Torrance, CA) at 25°C with a 3-min isocratic condition of 5% acetonitrile, 95% water, and 0.1% formic acid flowing at 250 µL/min or with a 9-min gradient at 400 µL/min from 5–95% B (A: water, B: acetonitrile; both phases coupled with either 0.1% formic acid or 10 mmol/L ammonium acetate for positive and negative ion mode, respectively). Metabolite assignments were performed with MAVEN (Princeton, NJ).
Statistical Analysis
Graphs and statistical analyses were prepared with GraphPad Prism 5.0 (GraphPad Software, Inc, La Jolla, CA) and Metaboanalyst 3.0 (38). Hierarchical clustering analyses and heat maps were plotted with GENE-E (Broad Institute, Boston, MA). Values are presented as means ± SE for the number of samples (n) from different animals and cell culture experiments. One-way ANOVA with post hoc Bonferroni correction was used to compare multiple groups. Unpaired Student’s t tests were used to compare two groups. Values of P < 0.05 were considered significant.
RESULTS
G6PD Inhibition or G6pd Knockdown Increases Expression of SMC-Restricted Genes and Reduces Proliferation of SMCs Isolated from Different Species
Our laboratory recently proposed that G6PD contributes to the regulation of SMC phenotype (14, 15). In those studies, we used a combination of nonspecific inhibitors of G6PD activity and genetic manipulation (knockdown) of G6pd in SMCs to show that G6PD expression and activity regulate expression of SMC-restricted genes. We further showed that G6PD activity was higher (25 ± 2%; P < 0.05) in proliferating than quiescent SMCs. Here, we set out to determine whether G6PD has a critical role in modulating SMC-restricted gene expression across different species. To accomplish that, we used shRNA to genetically manipulate G6PD in SMCs from rodents and humans. In parallel, we used two G6PD inhibitors, NEOU [1 µmol/L; a newly developed selective inhibitor (39)] and epiandrosterone (EPI, 100 µmol/L; a selective but less potent inhibitor than NEOU), to suppress G6PD activity in SMCs from mice (MSMCs), rats (RSMCs), and humans (HSMCs). After application of G6PD shRNA, which decreased G6PD expression and activity by 50–60% in SMCs (14, 15, 32, 40), or inhibitors to SMCs for 48 h, expression of SMC-restricted genes and proteins was determined. Application of G6PD shRNA or inhibitors to the A7r5 rat aortic SMC line decreased glucose-6-phosphate (G-6-P) metabolism through the PPP (Fig. 1A) and increased protein expression of myosin heavy chain 11 (MYH11; Fig. 1, B and C). At the same time, G6PD inhibitors increased transgelin (TAGLN) and smooth muscle α-actin (ACTA2) expression in A7r5 cells and decreased Krüppel-like factor 4 (KLF4; a marker of dedifferentiated SMC phenotype) expression (Fig. 1B). In addition, NEOU increased expression of Myh11 (Fig. 1D) and Myocd (Fig. 1E) in A7r5 cells and in primary cultures of RSMCs and MSMCs, and application of NEOU to HSMCs and RSMCs for 48 h also decreased cell numbers (Fig. 1F). Similarly, ectopic expression of functionally compromised G6PD mutants (activity <70% of the wild type), generated previously in our laboratory (30), in A7r5 cells visibly reduced the cell numbers, and the cells grew slower than the cells expressing wild-type G6PD (Fig. 1G). The reduction in growth induced by ectopic expression of G6PD mutants was not due to accelerated cell death, as there was no change in the release of lactate dehydrogenase into the culture medium, as previously observed with HEK293 17 T cells (30).
Figure 1.
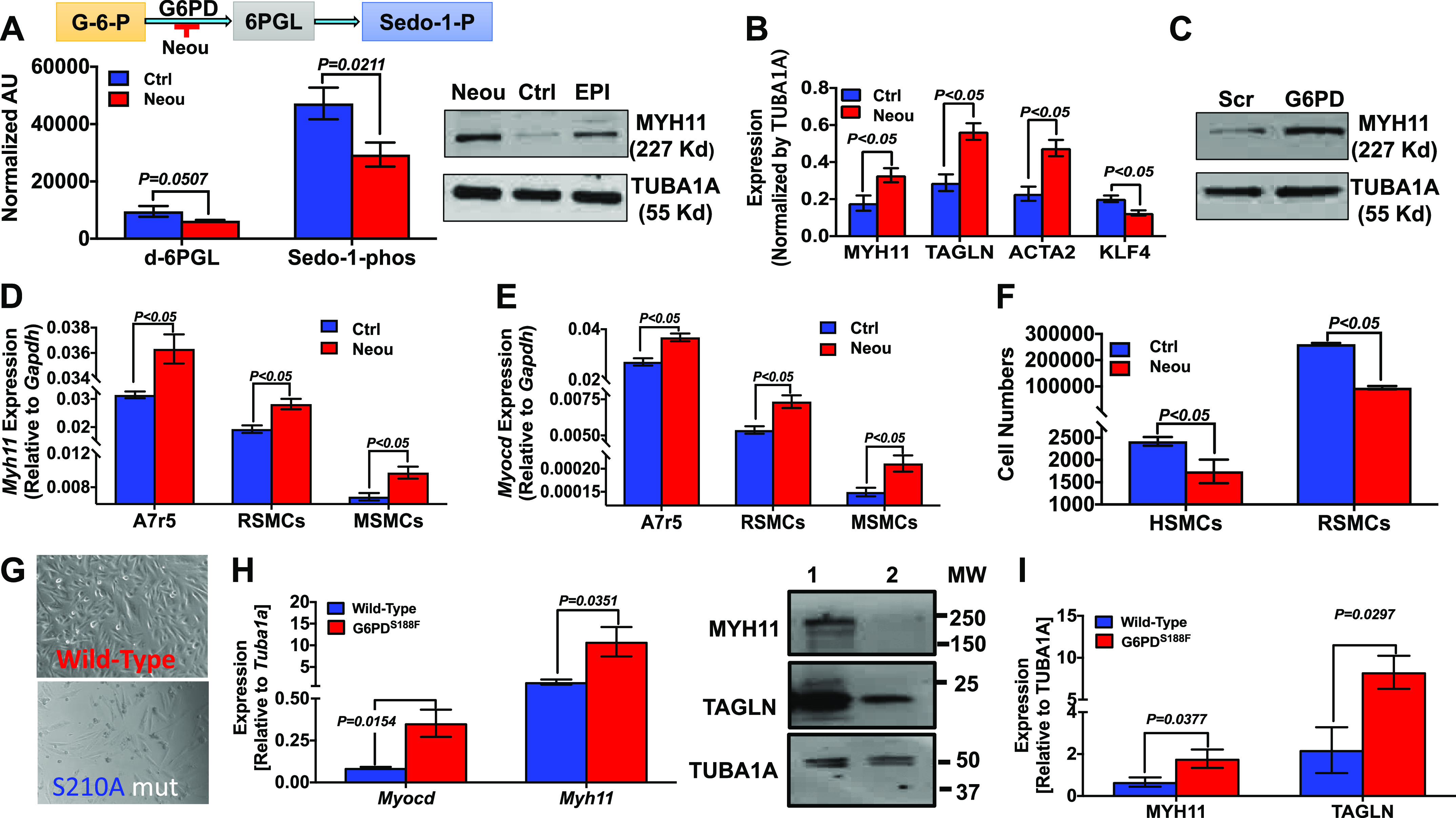
Glucose-6-phosphate dehydrogenase (G6PD) inhibition, G6pd knockdown, and ectopic expression of the loss-of-function Mediterranean G6PD variant increase expression of smooth muscle cell (SMC)-restricted genes and decrease cell growth. A: glucose metabolism through the pentose phosphate pathway (PPP) (schematic, top) and levels of 6-phospho-d-gluconate (6PGL) and sedoheptulose-1-phosphate (Sedo-1-phos), 2 metabolites in the PPP, are decreased in SMCs treated with N-ethyl-N′-[(3β,5α)-17-oxoandrostan-3-yl]urea (NEOU, 1 µmol/L; bottom). AU, arbitrary units. B: Western blot data representative of 5 experiments demonstrating that G6PD inhibition with epiandrosterone (EPI; 50 µmol/L) or NEOU (1 µmol/L) increases expression of myosin heavy chain 11 (MYH11) in A7r5 cells. Summary data showing that expression of MYH11, transgelin (TAGLN, also known as SM22α), and α-actin (ACTA2) is increased and Krüppel-like factor 4 (KLF4) decreased in A7r5 cells treated for 48 h with vehicle control (Ctrl; n = 5 samples) or NEOU (1 µmol/L; n = 5 samples). C: Western blot data representative of 5 experiments demonstrating G6pd knockdown with Ad-G6PD-shRNA [1012 plaque-forming units (PFU]. Scrambled (Scr) RNA was used as the control for G6PD-shRNA, and α-tubulin (TUBA1A) served as a loading control. D and E: expression of smooth muscle myosin heavy chain (Myh11; D) and myocardin (Myocd; E) mRNA in A7r5 cells and primary rat (RSMCs) and mouse (MSMCs) aortic SMCs is higher after treatment for 48 h with NEOU (1 µmol/L; n = 5 samples) vs. vehicle control (n = 5 samples). F: numbers of human coronary artery SMCs (HSMCs) and RSMCs, determined in CyQUANT Cell Proliferation Assays, are decreased after treatment for 48 h with NEOU (1 µmol/L; n = 6 samples) vs. vehicle control (n = 6 samples). G: ectopic expression of enzymatically active human G6PD (wild type) and inactive human G6PD (S210A) mutant in A7r5 cells is shown. A7r5 cells expressing inactive human G6PD mutant grew slower than those expressing active human G6PD 48 h after transfection. H: RT-PCR results showing that relative expression of Myocd and Myh11 was higher in aorta from Mediterranean G6PD variant (S188F) than wild-type rats. I: representative Western blot and summary results demonstrating that relative expression of MYH11 and TAGLN were higher in aortas from Mediterranean G6PD variant (lane 1) than wild-type (lane 2) rats. MW, molecular weight.
Loss-of-Function Mediterranean G6PD Variant Increases SMC-Restricted Genes
We sought to confirm that pharmacological or genetic inhibition of G6PD activity enhances expression of Myocd and Myh11 in cultured SMCs by determining whether inhibiting G6PD activity through genetic manipulation would yield similar results in vivo. To accomplish that, we used a G6PDS188F rat model of the Mediterranean G6PD variant generated through CRISPR-mediated single-nucleotide polymorphism (SNP) modeling. These G6PDS188F mutant rats express a functionally compromised G6PD protein exhibiting 80% less activity than the wild-type enzyme (25). Aortas from these mutants expressed SMC-restricted Myocd and Myh11 genes (Fig. 1H) and MYH11 and TAGLN proteins (Fig. 1I) more strongly than their wild-type littermates.
G6PD Inhibition with NEOU Increases Expression of SMC-Restricted Genes Lost from Cultured Primary Cells through Passaging
Since the first report in the early 1900s suggesting that SMCs lose their contractile properties when they are cultured and passaged in vitro (2), numerous studies have confirmed this phenomenon and have shown that expression of SMC-restricted genes/proteins is downregulated (2). We therefore isolated SMCs from the aortas of tamoxifen-treated Myh11-CreERT2 mice (MSMCs) and performed lineage tracking of GFP-enriched (Myh11) MSMCs cultured for up to 7 passages in the presence and absence of NEOU (1 µmol/L). We added NEOU or vehicle to the culture medium from passage 1 to passage 7 (Fig. 2A) and then measured Myh11 mRNA levels. Consistent with our earlier study (26), we observed that numbers of GFP-enriched cells gradually declined from passage 3 and >80% of GFP-enriched cells were lost by passage 7. In addition, expression of Myh11 and Myocd mRNA was ∼100-fold lower in vehicle-treated passage 7 cells than passage 1 cells, and this decline was attenuated by the presence of NEOU in culture medium. It is noteworthy that expression of Myh11 and Myocd mRNA was higher (P < 0.05) in passage 7 cells treated with NEOU than vehicle (Fig. 2, B and C). Application of NEOU also increased Myh11 (1.5-fold) and Myocd (1.5-fold) mRNA compared with vehicle in passage 1 cells. Conversely, expression of Msx2, which encodes MSX2, a transcriptional repressor of Myocd (41), was reduced by NEOU (Fig. 2D). Although expression of SMC-restricted genes was lower in passage 7 than passage 1 cells, expression of genes encoding stem cell-restricted marker (Sca-1) and transcription factors (Oct-4 and cMyc) were higher in passage 7 cells, and their expression was reduced (P < 0.05) by NEOU (Fig. 2, E–G).
Figure 2.
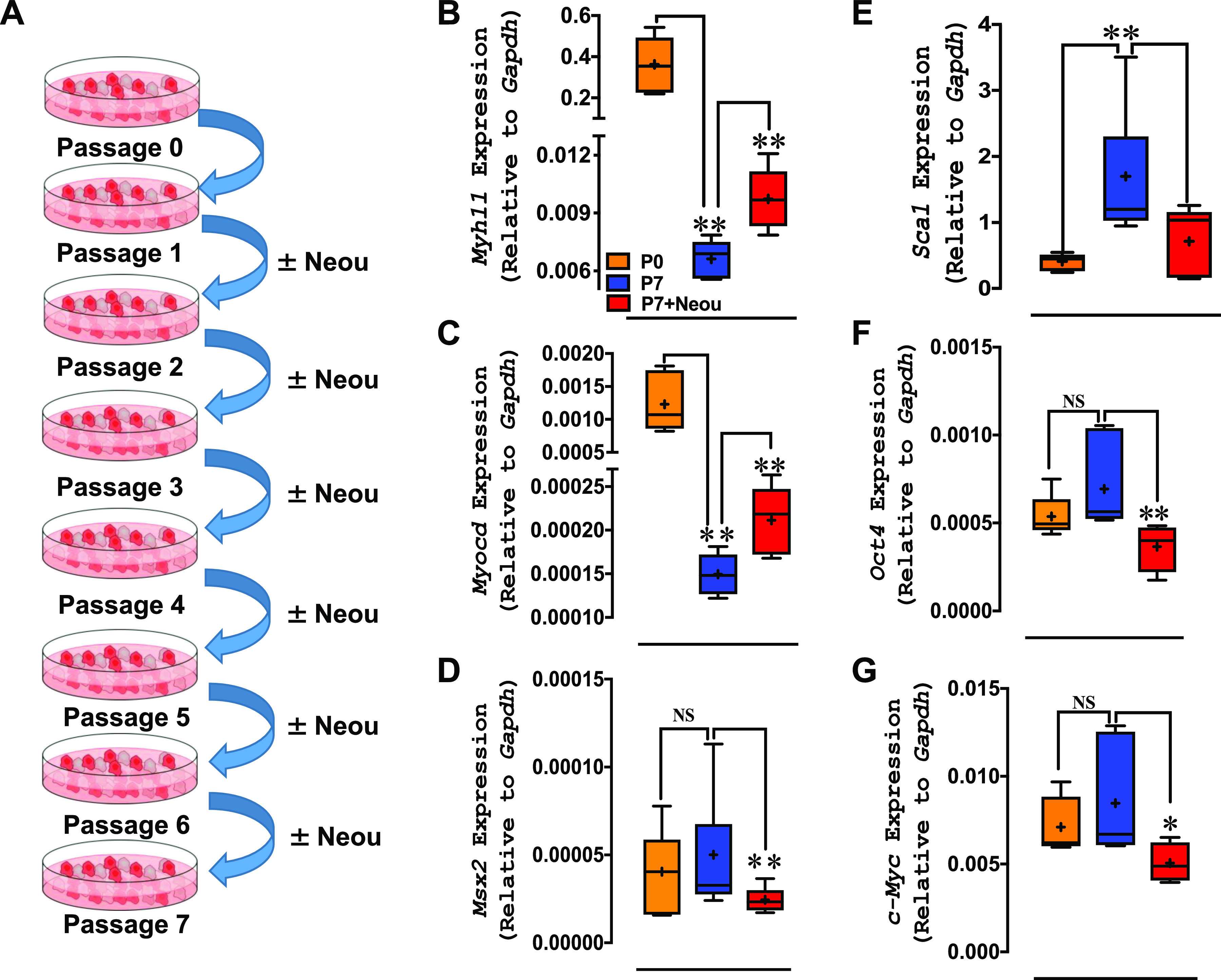
Application of N-ethyl-N′-[(3β,5α)-17-oxoandrostan-3-yl]urea (NEOU) to primary mouse aortic smooth muscle cells (SMCs) prevents downregulation of SMC-restricted genes and upregulation of stem cell marker genes. A: schematic showing SMCs isolated from mouse aorta cultured in DMEM with 10% FBS from passage 0 (P0) to passage 7 (P7) in the absence and presence of the glucose-6-phosphate dehydrogenase (G6PD) inhibitor NEOU (1 µmol/L). B–G: graphical representation of RT-PCR results showing the relative expression levels of the indicated genes in mouse aorta SMCs at P0, P7, and P7 treated with NEOU (P7+NEOU) (n = 6 samples/group). RT-PCR results were normalized to the level of Gapdh expression. Data are presented as means ± SE. *P < 0.05 and **P < 0.01. NS, not significant.
Knockdown or Inhibition of G6PD-Histone Deacetylase Enhances Expression of Myocd and SRF-MYOCD-Driven Myh11 Genes
G6PD interacts with HDACs, and G6PD-derived NADPH is required for HDAC1 and 2 activity (16). In that context, immunoprecipitation and proteomic analysis revealed that G6PD interacts directly with histone 3 (Fig. 3A, top) and forms a complex with HDAC-SAP30 and HDAC-SAP18 (Table 1). Since the importance of HDAC to cell biology and phenotype is established, and epigenetic modifications have been implicated in SMC phenotypic modulation (42), we postulated that epigenetic modification via a HDAC pathway may be instrumental in mediating the observed effects of G6PD inhibition and knockdown on gene expression in SMCs. As expected, application of trichostatin A (TSA; 5 µmol/L) or a G6PD inhibitor [NEOU (1 µmol/L) or EPI (100 µmol/L)] to A7r5 cells decreased HDAC activity, and the decrease elicited by the G6PD inhibitors was comparable to that elicited by TSA (Fig. 3A, bottom). Moreover, NEOU increased H3K9ac/H3K18ac/H3K56ac (Fig. 3B) and G6PD-shRNA increased H3K9ac expression (Fig. 3C) compared with vehicle and scrambled-shRNA controls, respectively. In addition to decreasing HDAC activity, G6PD-shRNA also attenuated HDAC5 expression (Fig. 3D). Given that G6PD inhibition increased acetylation of histone 3 at various lysine residues, including the open chromatin mark H3K9ac, we performed whole genome ChIP-seq analysis using H3K27ac, another commonly used open chromatin mark, as bait in A7r5 cells treated with vehicle, TSA, EPI, or NEOU. We used H3K27ac as the bait instead of H3K9ac because we knew from Western blotting that G6PD inhibition robustly increases H3K9ac, a mark indicative of active genes, but we did not know the status of H3K27ac. A7r5 cells treated with a HDAC (TSA) or G6PD (EPI or NEOU) inhibitor exhibited differential enrichment of H3K27ac regions compared with vehicle throughout the genome. Differentially bound region (DBR) analysis revealed genes common to both the TSA and G6PD inhibitor groups (Fig. 3E). For example, 16 significant (FDR < 0.05) DBRs (shown in magenta in the MA plot in Fig. 3F and in the heat map in Fig. 3G) were identified on the basis of binding affinity and hierarchical clustering analysis in NEOU- versus vehicle-treated A7r5 cells. Gene Ontology (GO) analysis revealed that those 16 DBRs belong to a cluster of genes that function in biological regulation, cell biogenesis and processes, cell signaling, metabolic process, localization, and development process (Fig. 3H). Interestingly, the development cluster included 3.5-fold (FDR < 0.05) more H3K27ac enrichment of Myocd (Fig. 3I) within the intergenic region and near the promoter-transcription start site (TSS). At the same time, application of NEOU, TSA, or NEOU + TSA to A7r5 cells upregulated expression of Myocd and SRF-MYOCD-dependent Myh11 mRNA (Fig. 3, J and K). This suggests that G6PD inhibition in SMCs decreases removal of acetyl groups from the H3 tail.
Figure 3.
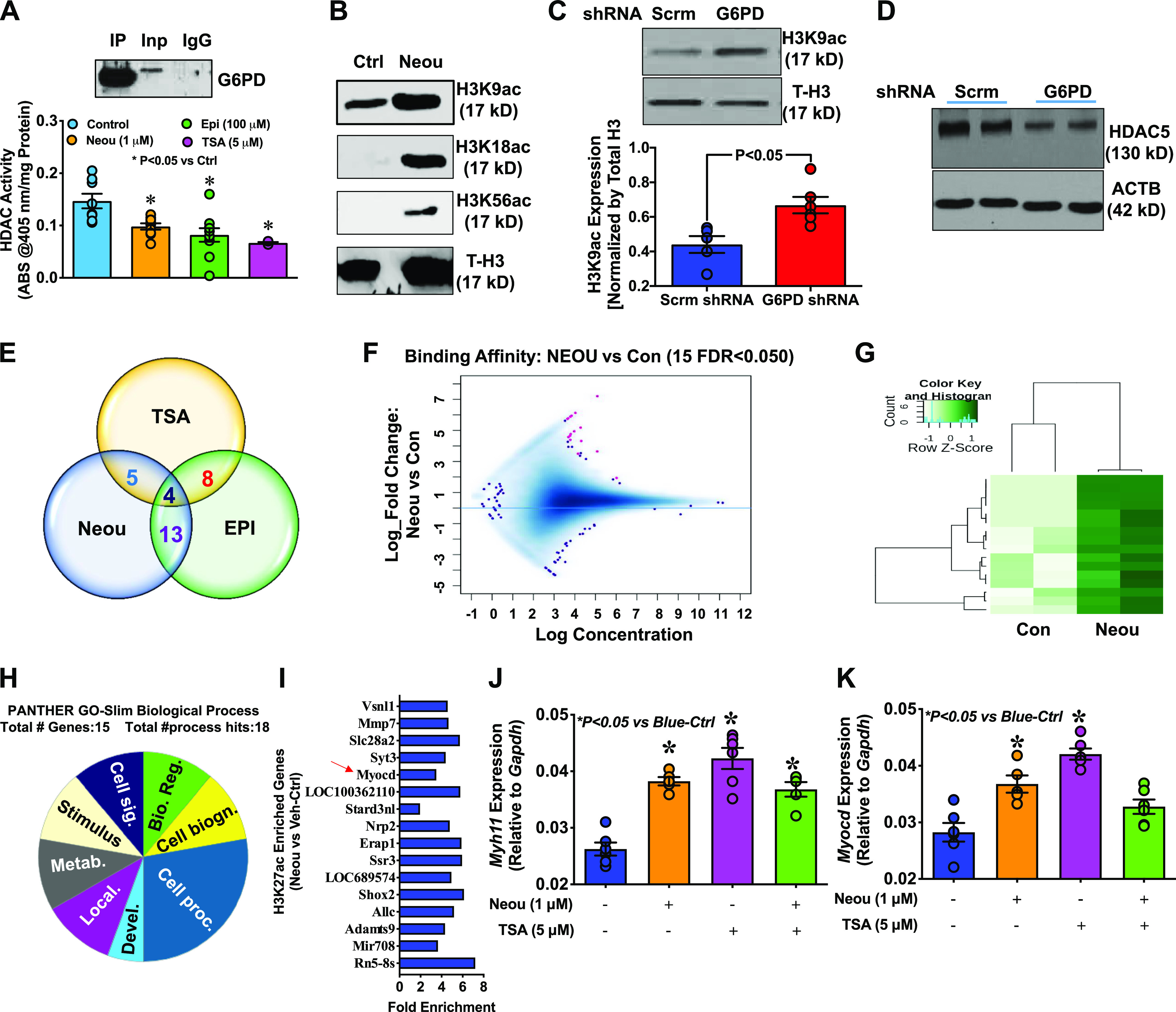
Glucose-6-phosphate dehydrogenase (G6PD) inhibition and G6pd knockdown decrease histone deacetylase (HDAC) activity and increase acetylation of histone 3 (H3) in smooth muscle cells (SMCs). A, top: immunoprecipitation (IP) of the G6PD-H3 complex. Bottom: HDAC activity in A7r5 cells was decreased by application of a G6PD inhibitor or trichostatin A (TSA) for 24 h [Control, n = 10 samples; epiandrosterone (EPI, 100 µmol/L), n = 12 samples; N-ethyl-N′-[(3β,5α)-17-oxoandrostan-3-yl]urea (NEOU, 1 µmol/L), n = 12 samples); TSA (5 µmol/L), n = 5 samples]. B: Western blot data representative of 5 experiments demonstrating that H3K9ac, H3K18ac, and H3K56ac, but not total H3 (T-H3), expression was increased in A7r5 cells treated with NEOU for 24 h. C, top: H3K9ac expression normalized to T-H3 was higher in A7r5 cells treated for 48 h with Ad-G6PD shRNA [1012 plaque-forming units (PFU); n = 6 samples] than with scrambled shRNA (Scrm, 1012 PFU; n = 6 samples). Bottom: summary results demonstrate that H3K9ac increased in A7r5 cells treated with Ad-G6PD shRNA. D: Western blot data representative of 5 experiments showing that HDAC5 expression was lower in SMCs treated for 48 h with Ad-G6PD shRNA (1012 PFU) than with scrambled shRNA. E: Venn diagram summarizing the results of a genomewide chromatin immunoprecipitation sequencing (ChIP-seq) analysis that show differentially bound regions of H3K27ac in A7r5 cells that are common between G6PD inhibitor (EPI and NEOU) and HDAC inhibitor (TSA) treatment compared with vehicle control. F: MA plot displaying the log read concentration for binding sites (x-axis) vs. logFold_Change between NEOU and Vehicle control (Con; y-axis). Significantly [false discovery rate (FDR) < 0.05]differentially bound regions are shown in magenta. G: hierarchically clustered, row scaled, significantly differentially bound regions (16) identified by comparison of NEOU with vehicle control (Con). H: Gene Ontology (GO) analysis of 16 differentially bound regions revealing genes associated with 8 biological processes, including biological regulation (Bio. Reg.), cell biogenesis (Cell biogn.), cell processes (Cell proc.), development process (Devel.), localization (Local.), metabolic process (Metab.), stimulus, and cell signaling (Cell sig.). I: fold enrichment of 16 differentially bound regions annotated by using Homer annotatePeaks.pl to provide proximity to and the name of the nearby rn6 gene. Red arrow: Myocd gene is enriched 3.5-fold upstream of the transcription start site (TSS) and promoter-TSS region. J and K: expression of Myh11 (J) and Myocd (K) was increased by NEOU (1 µmol/L), TSA (5 µmol/L), and NEOU+TSA. Data are presented as means ± SE.
Table 1.
Histone deacetylase-associated proteins in the immunocomplex with G6PD detected after immunoprecipitation followed by LC-MALDI-TOF
| Top-Ranked Protein Name | Accession No. | Protein MW | Protein pI | Peptide Count | Total Ion Score | Total Ion CI, % |
|---|---|---|---|---|---|---|
| Histone deacetylase complex subunit SAP30 | gi 300798346 | 23,262 | 9.3 | 1 | 35 | 90 |
| Histone deacetylase complex subunit SAP18 | gi 215490085 | 19,562 | 9.8 | 1 | 33 | 83 |
G6PD, glucose-6-phosphate dehydrogenase; LC-MALDI-TOF, liquid chromatography-matrix-assisted laser desorption/ionization-time of flight; MW, molecular weight; pI, isoelectric point; CI, chemical ionization.
G6PD Activity and Glucose Metabolism through the PPP Are Enhanced in Aortas from JCR Rats, and G6PD Inhibition Suppresses the Metabolic Reprogramming
The functional and metabolic phenotypes of SMCs change in angio-proliferative diseases like peripheral vascular disease, coronary artery disease, and pulmonary hypertension (1). In addition, G6PD expression and activity are increased in metabolic diseases (20). Although G6PD activity is protective against oxidant stress in cells devoid of mitochondria, such as red blood cells, G6PD activity is the rate-limiting enzyme in the synthesis of NADPH, a cofactor that also fuels generation of reactive oxygen species by NADPH oxidases. Indeed, we and others have previously shown that increased G6PD activity and G6PD-derived NADPH fuel production of free radicals and inflammation in various tissue types, including vascular tissue, in patients and animal models of diabetes and metabolic syndrome (19, 21, 22, 24). It is known that inhibition of G6PD-derived NADPH-dependent generation of free radicals improves endothelial function (23). Since the effect of elevated G6PD activity on glucose metabolism through the PPP within arteries impacted by metabolic syndrome is unknown, we determined whether G6PD activity is enhanced and/or metabolic reprogramming occurs within blood vessels from JCR rats, a rodent model of metabolic syndrome. Blood chemistry showed that JCR rats have increased insulin and hyperlipidemia (Table 2). We found that G6PD activity and glucose metabolism through the PPP were higher in aortas from JCR than SD rats, and treating JCR rats with the G6PD inhibitor EPI (30 mg/kg/day) reduced G6PD activity (Fig. 4A). In aortas from SD rats and control SMCs, G6PD inhibitors decreased activity (by 20.5 ± 1.5% EPI vs. Ctrl; n = 5) and reduced glucose flux through the PPP (Fig. 1A). Similarly, EPI-suppressed metabolites included mannitol, trihydroxy-cholanate, pregnenolone, nucleosides, fatty acids, and the PPP, among others (Fig. 4B and Table 3). Moreover, in aortas from JCR rats, reduced glucose flux through the PPP, as determined by tracing experiments with 13C glucose, led to lower levels of nucleosides (Fig. 4C) and various metabolites of the PPP (Fig. 4D) and higher levels of glutathione (M + 4) (Fig. 4D).
Table 2.
Blood chemistry results
| SD | JCR | JCR+EPI | |
|---|---|---|---|
| BUN, mg/dL | 20.0 ± 0.6 | 18.0 ± 1.2 | 19.0 ± 2.6 |
| CREA, mg/dL | 0.5 ± 0.1 | 0.4 ± 0.2 | 0.3 ± 0.1 |
| GLU, mg/dL | 353 ± 77 | 282 ± 38 | 274 ± 26 |
| INS, μg/L | 3.64 ± 1.32 | 6.34 ± 1.77* | 12.49 ± 9.16 |
| Na, mmol/L | 143.0 ± 1.5 | 147.0 ± 1.5 | 146.0 ± 2.9 |
| K, mmol/L | 4.7 ± 0.1 | 4.9 ± 1.2 | 4.5 ± 0.69 |
| Cl, mmol/L | 104.0 ± 1.5 | 104.0 ± 1.7 | 105.0 ± 1.5 |
| ALP, U/L | 146.0 ± 8.5 | 104.0 ± 22.0 | 84.0 ± 4.7 |
| ALT, U/L | 37.0 ± 4.9 | 27.0 ± 5.1 | 25.0 ± 5.9 |
| AST, U/L | 52.0 ± 3.6 | 45.0 ± 13.9 | 39.0 ± 2.9 |
| TBIL, mg/dL | 0.1 ± 0 | 0.1 ± 0 | 0.1 ± 0 |
| DBIL, mg/dL | 0 ± 0 | 0.1 ± 0.23 | 0 ± 0 |
| LDH, U/L | 50.0 ± 2.1 | 76.0 ± 40.2 | 113.0 ± 40.2 |
| CPK, U/L | 84.0 ± 30.1 | 109.0 ± 93.6 | 130.0 ± 42.6 |
| GGT, U/L | 0 ± 0.6 | 1 ± 0.6 | 0 ± 0 |
| TPRO, g/dL | 4.7 ± 0.1 | 5.1 ± 0.4 | 4.5 ± 0.5 |
| ALB, g/dL | 2.3 ± 0.1 | 2.1 ± 0.1 | 2.0 ± 0.2 |
| Ca, mg/dL | 8.5 ± 0.1 | 9.2 ± 0.6 | 8.9 ± 0.7 |
| Phos, mg/dL | 6.5 ± 0.6 | 6.5 ± 0.6 | 7.2 ± 1.2 |
| Mg, mg/dL | 1.6 ± 0.1 | 1.8 ± 0.2 | 1.8 ± 0.2 |
| CHOL, mg/dL | 38 ± 2 | 126 ± 79* | 137 ± 40 |
| TRIG, mg/dL | 35.0 ± 0.6 | 186.0 ± 94.1* | 237.0 ± 85.1 |
| AMY, U/L | 466.0 ± 39.2 | 593.0 ± 52.1 | 562.0 ± 71.2 |
| LIP, U/L | 7 ± 1 | 41 ± 32* | 31 ± 18 |
Values are means ± SD. ALB, albumin; ALP, alkaline phosphatase; ALT, alanine amino transferase; AMY, amylase; AST, aspartate amino transferase; BUN, blood urea nitrogen; CHOL, cholesterol; CPK, creatine phosphokinase; CREA, creatinine; DBIL, direct bilirubin; EPI, epiandrosterone; GGT, gamma-glutamyl transferase; GLU, glucose; INS, insulin; LDH, lactate dehydrogenase; SD, Sprague-Dawley; TBIL, total bilirubin; TPRO, total protein; TRIG, triglycerides; LIP, lipase. *P < 0.05 vs. SD.
Figure 4.

Epiandrosterone (EPI) decreases glucose-6-phosphate dehydrogenase (G6PD) activity and glucose flux through the pentose phosphate pathway (PPP) and increases glutathione in aortas from JCR rats. A, top: 6-phospho-d-gluconate (6PGL), pentose phosphate isobars (PP-isobars), and adenosine (nucleoside) are increased in aortas from JCR vs. Sprague-Dawley (SD) rats (n = 5–7 samples in each group). Bottom: G6PD activity in aortas from SD rats (n = 5 samples), JCR rats treated with vehicle (n = 3 samples), and JCR rats treated with EPI (30 mg/kg/day; JCR-Epi, n = 5 samples) is shown. EPI decreased G6PD activity. B: heat map (blue to red = low to high levels of metabolites in that pathway across all replicates) showing that some metabolites in purine, other sugars, bile, and fatty acid metabolic pathways are reduced by inhibition of G6PD by EPI (n = 3 samples/group). C: 13C-glucose pulse chase tracing experiments in aortas from JCR rats treated with vehicle (n = 3 samples) or EPI (n = 3 samples) show that glucose flux through the PPP and nucleoside levels are decreased. D: graphical representation of the effects of G6PD inhibition on the indicated PPP intermediates and glutathione (GSH) homeostasis tracked by 13C-glucose pulse chase tracing experiments in JCR rats treated with vehicle or EPI (n = 3 samples/group). AU, arbitrary units. *P < 0.05 and **P < 0.01.
Table 3.
Steady-state metabolites of glycolytic and pentose phosphate pathway in aorta of SD, JCR+Veh, and JCR+EPI rats
| Compound | JCR-Veh vs. SD |
JCR-Epi vs. JCR-Veh |
||
|---|---|---|---|---|
| Fold change | P value | Fold change | P value | |
| d-Glucose | 1.94 | 0.38 | 0.79 | 0.57 |
| d-Fructose 1-6-bisphosphate | 1.59 | 0.72 | 0.43 | 0.57 |
| d-Glyceraldehyde 3-phosphate/glycerone phosphate | 1.75 | 0.41 | 0.49 | 0.69 |
| Pyruvate | 1.01 | 0.83 | 1.32 | 0.43 |
| d-Glucono-1-5-lactone 6-phosphate | 2.34 | 0.13 | 1.29 | 0.79 |
| Sedoheptulose 7-phosphate | 0.93 | 0.65 | 0.82 | 0.91 |
| Pentose phosphates (isobars) | 1.19 | 0.47 | 0.53 | 0.56 |
Epi, epiandrosterone; SD, Sprague-Dawley; Veh, vehicle.
Inhibition of G6PD Activity Decreases Expression of Maladaptive Genes and Increases Expression of SMC-Restricted Genes in Aortas from JCR Rats
On the basis of our findings that G6PD is involved in altering the gene expression profile in cultured SMCs, we speculated that increased G6PD might contribute to the reduction in SMC-restricted gene expression, which initiates the transition of SMCs from a differentiated to a dedifferentiated phenotype in metabolic diseases. To test that idea, we determined whether increases in G6PD activity affect SMC gene expression and vascular function in metabolic syndrome. Because EPI (30 mg/kg/day) treatment decreased G6PD activity and glucose metabolism through the PPP in aortas from JCR rats (Fig. 4), we used genomewide RNA-seq analysis to determine whether G6PD inhibition affects gene expression in the same tissues.
A transcriptomics analysis revealed that expression levels of numerous genes were higher (2.6% of 14,194 with nonzero total read counts; adjusted P < 0.05) or lower (2.3% of 14,194 with nonzero total read counts; adjusted P < 0.05) in aortas from JCR versus SD rats. Moreover, hierarchical clustering analysis of the transcriptomics data (heat maps shown in Fig. 5A) as well as unsupervised principal component analysis (PCA; Fig. 5B) showed that this phenomenon was reduced by inhibition of G6PD. However, among the differentially expressed genes only 30 were significantly (adjusted P < 0.05) upregulated >1.5 log2_fold, whereas 26 genes were significantly downregulated (Fig. 5C). GO analysis of the upregulated genes revealed them to encode plasminogen activator inhibitor-1 and proteins in the blood coagulation pathway (Serpine1 gene), Gi and Gq signaling pathway (Rgs4 gene), transforming growth factor (TGF)-β signaling pathway (Bambi gene), and p53 signaling pathway (Serpine1 gene) (Fig. 5D). The downregulated genes encoded plasminogen activator (Plau gene) and proteins in the Alzheimer disease-presenilin pathway (Fzd9 gene); the ANG II (Agt gene), apoptosis (Bcl2i11 gene), cadherin (Fzd9 gene), interferon γ (Socs2 gene) WTN (Fzd9 gene), and p53 signaling pathway (Hmgb1 gene); and the T cell activation pathway (RT1-Ba and Cd74 genes) (Fig. 5E). G6PD inhibition suppressed abnormal expression of genes encoding proteins that contribute to vascular contraction (GPCR signaling pathway) and thrombosis (plasminogen activator inhibitor-1) (43–45) in aortas from JCR rats. For example, genes such as Abra, which encodes actin binding Rho activating protein; Serpine1, which encodes endothelial plasminogen activator inhibitor-1; and Epha4, which encodes ephrin receptor, were all decreased by G6PD inhibition in aortas from JCR rats (Fig. 5F). By contrast, G6PD inhibition did not significantly decrease expression of Abra (1.84-fold EPI vs. control), Serpine1 (0.92-fold EPI vs. control), or Epha4 (1.13 EPI vs. control) in aortas from SD rats. However, G6PD inhibition upregulated expression of SMC-restricted genes (Myh11, Cnn1, and Tagln) in aortas from both JCR (Fig. 5G) and SD (data not shown) rats.
Figure 5.
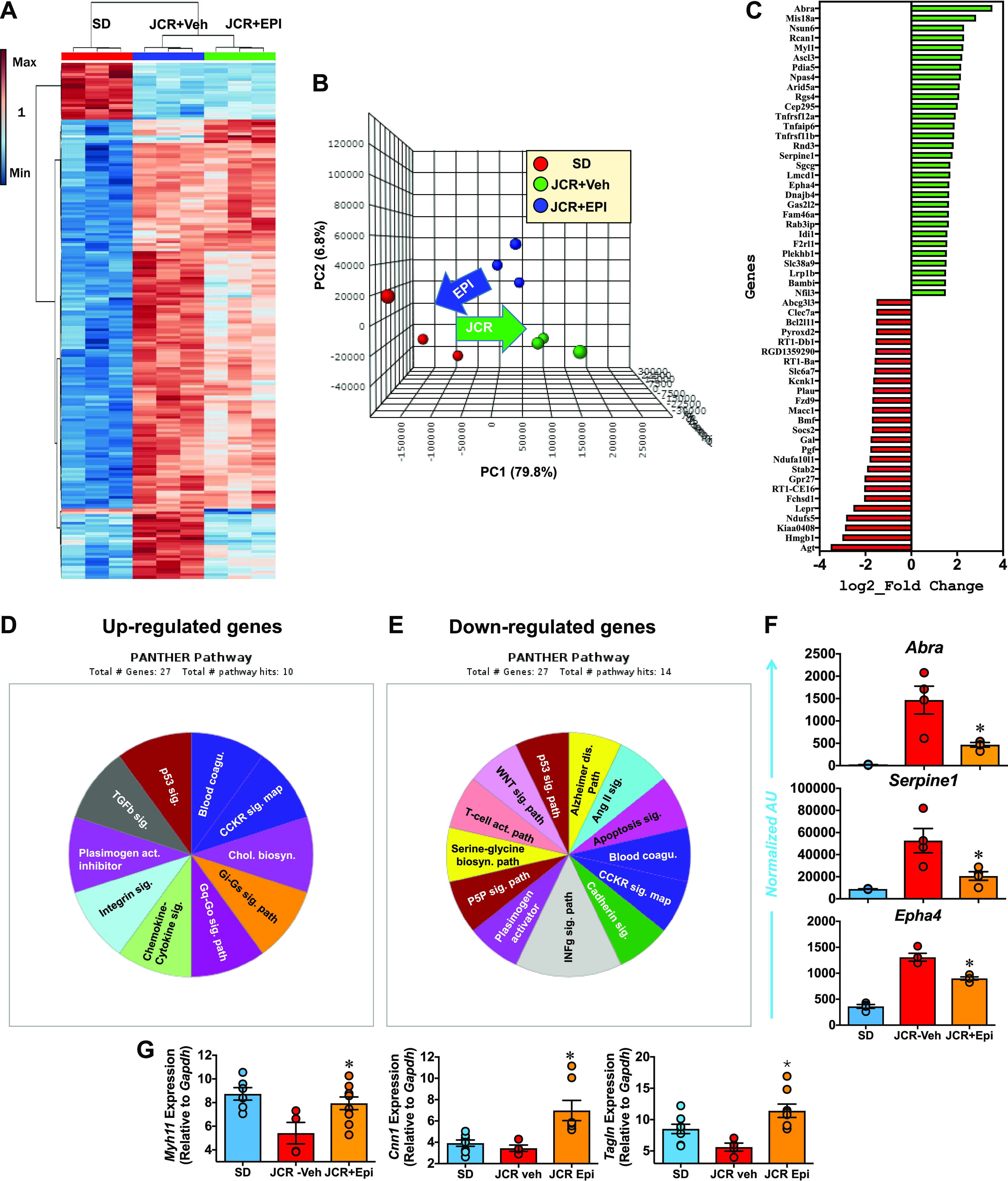
Epiandrosterone (EPI) decreases gene expression in aortas from JCR rats. A and B: heat map produced through whole genome RNA sequencing (RNA-seq) analysis and next-generation sequencing (NGS) and principal component (PC) analyses reveal that differences between the aortic transcriptomes of JCR and Sprague-Dawley (SD) rats are decreased by treating JCR rats with the G6PD inhibitor EPI (30 mg/kg/day) (n = 3 samples in each group). C: in aorta of JCR vs. SD rats, 30 genes are increased and 26 genes are decreased >1.5 log2_fold [false discovery rate (FDR < 0.05). D and E: Gene Ontology (GO) analysis annotation of increased and decreased genes revealed in blood coagulation and signaling pathways that affect contractile functions of smooth muscle cells (SMCs) are altered. F: RNA-seq analysis showing that aortic expression of Abra, Serpine1, and Epha4 was lower in JCR rats treated with EPI (30 mg/kg/day; n = 3 samples) than JCR-Veh control rats (n = 4 samples) but not SD rats (n = 3 samples). AU, arbitrary units. G: RT-PCR data showing that expression of Myh11, Cnn1, and Tagln (SD, n = 5 samples; JCR-veh, n = 3 samples; JCR+Epi, n = 8 samples) was increased by EPI treatment. RT-PCR data were normalized to the expression levels of Gapdh. *P < 0.05 vs. JCR-Veh.
An integrated analysis of metabolomics and transcriptomics data revealed a strong correlation between the levels of metabolites and expression of genes that encode enzymes that regulate the affected metabolic pathways in aortas from JCR rats. Pathway enrichment analysis highlighted the Krebs cycle as the most significantly affected pathway, followed by propanoate metabolism, glycolysis, gluconeogenesis, and the PPP (Fig. 6A). For example, levels of both succinate and Sdha mRNA were lower in aortas from JCR rats treated with EPI compared with vehicle (ethanol; Fig. 6, B and C).
Figure 6.
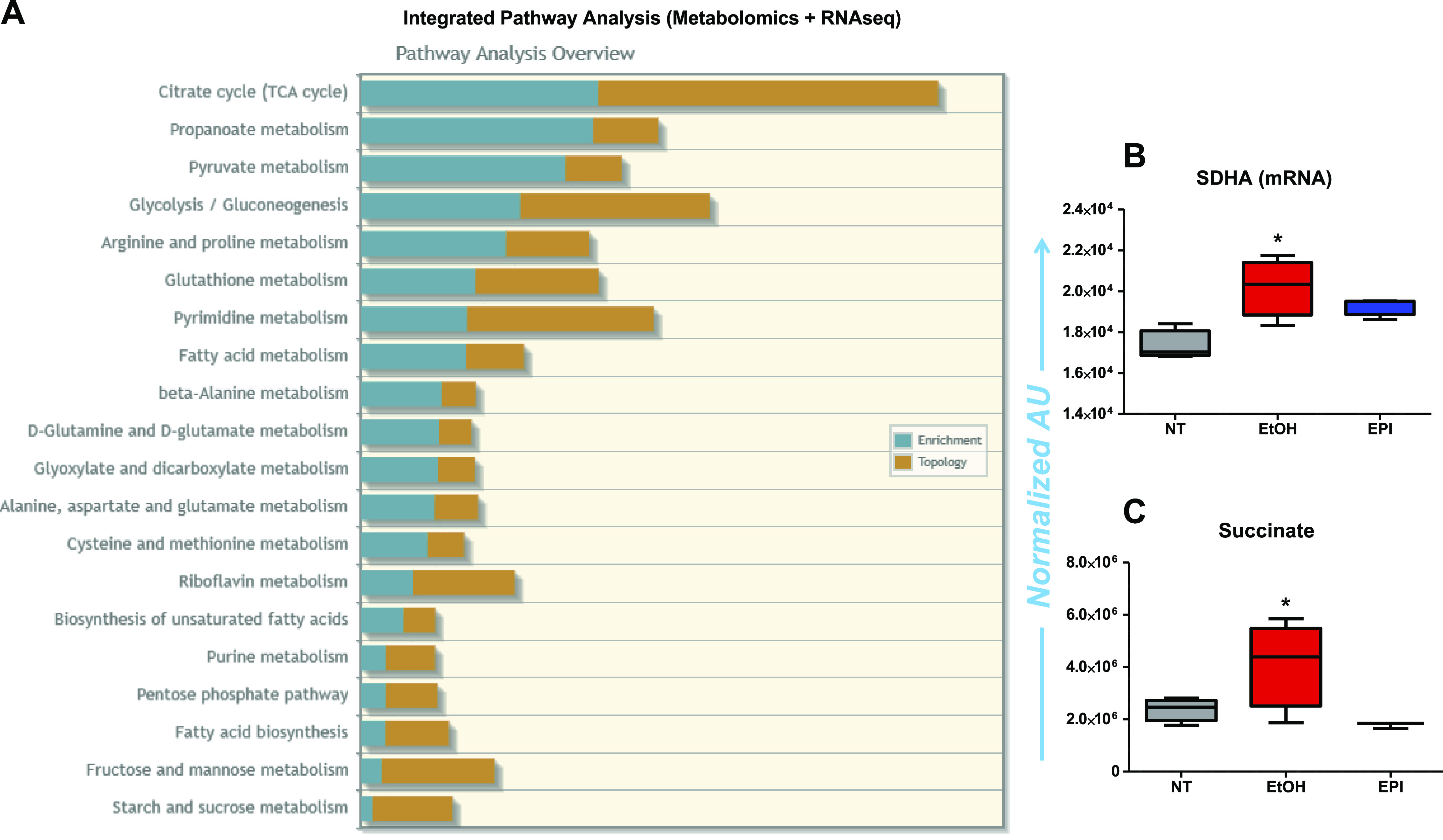
Integrated pathway analysis of metabolomics + RNA sequencing (RNA-seq) results in aorta of JCR rats. A: analyses show enrichment of genes that encode metabolic enzyme and metabolites of metabolic pathways (such as TCA cycle, propanoate metabolism, pyruvate metabolism, glycolysis, gluconeogenesis, arginine and proline metabolism, glutathione metabolism, etc.) in aorta of JCR rats. B and C: Sdha mRNA (B) and succinate (C) levels increased in the aorta of JCR rats are decreased by epiandrosterone (EPI). AU, arbitrary units. B and C: NT, SD normal rats; EtOH, JCR rats treated with vehicle; EPI, JCR rats treated with epiandrosterone (30 mg/kg/day). *P < 0.05 vs. NT.
G6PD Inhibition Decreases Blood Pressure and Arterial Elastance and Reduces Occlusive Growth in JCR Rats
Apart from reducing expression of differentially upregulated genes that encode proteins involved in mediating vascular dysfunction and thrombosis, G6PD inhibition by EPI (30 mg/kg/day) also decreased systolic and diastolic blood pressure, total peripheral resistance (TPR), and arterial elastance [Ea; determined as described previously (46)] in JCR rats (Fig. 7, A and B). Likewise, G6PD inhibition reduced mean arterial pressure in SD rats (SD: 88 ± 4 and SD + EPI: 77 ± 2 mmHg; n = 5; P < 0.05). Because JCR rats spontaneously develop vascular lesions (47), we histologically assessed coronary artery pathology. As expected, Verhoeff-Van Gieson staining for elastin revealed broken internal lamina (with growth) in both conduit and small resistant coronary arteries in transverse midplane sections from the hearts of all three JCR rats examined (Fig. 7C), whereas G6PD inhibition suppressed this growth in all three JCR + EPI rats.
Figure 7.
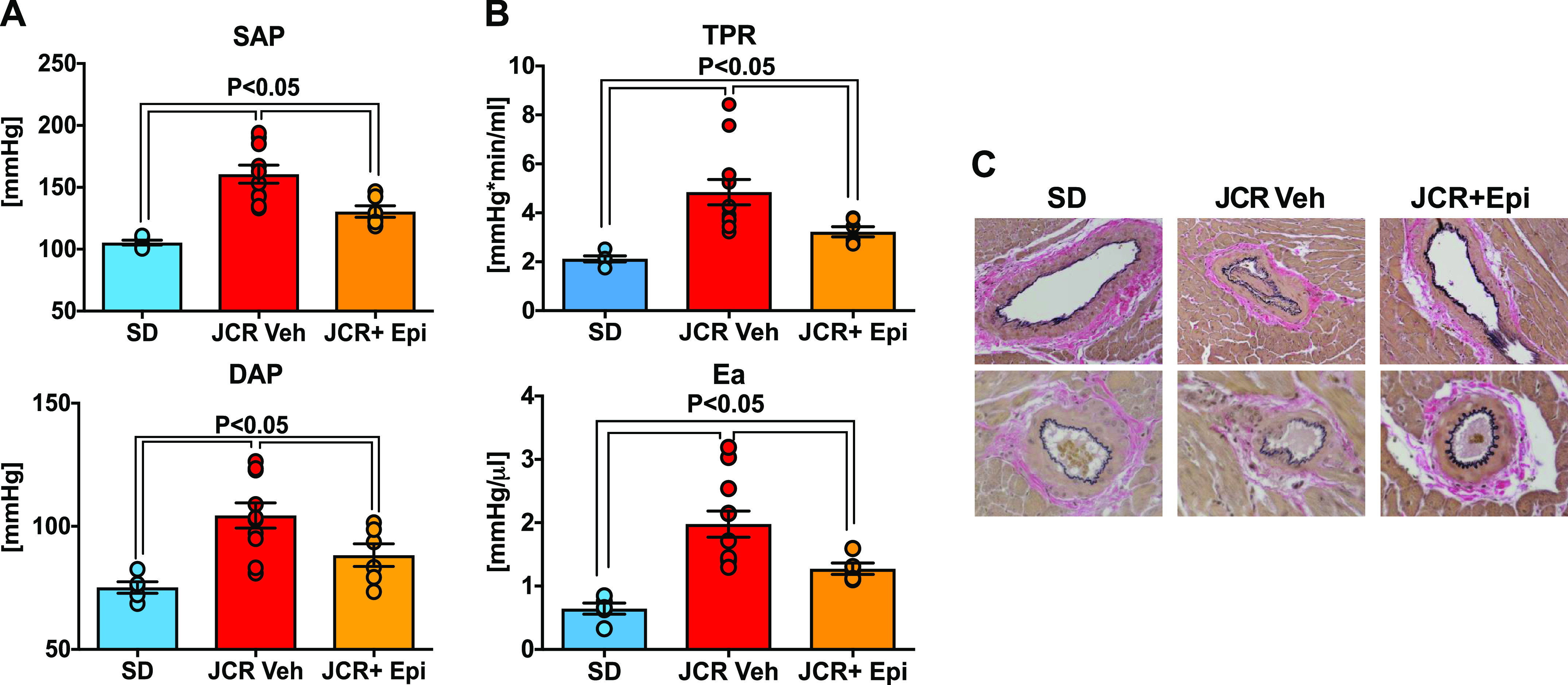
Epiandrosterone (EPI) decreases blood pressure, arterial elastance (Ea), and neointimal growth in coronary arteries from JCR rats. Systolic (SAP; A, top) and diastolic (DAP; A, bottom) blood pressures, total peripheral resistance (TPR; B, top), and Ea (B, bottom) were higher in JCR than Sprague-Dawley (SD) rats. EPI administration to JCR rats decreased blood pressure, resistance, and elastance (SD, n = 5 samples; JCR-veh, n = 11 samples; JCR-Epi, n = 6 samples). C: representative images of Verhoeff-Van Gieson staining of elastin in transverse midplane sections of heart showing large (epicardial) and small coronary arteries. Occlusive growth and rupture/break of the internal elastin laminae are seen in sections from JCR rats (n = 3 samples) but not JCR rats treated with EPI (n = 3 samples) or SD rats (n = 3 samples) rats. Data are presented as means ± SE.
The Mediterranean G6PD (S188F) Variant Decreases SMC-Nonrestricted Gene Expression and Reduces Vascular Resistance in Rats Fed a High-Fat Diet
Because we compared changes in aortic gene expression between JCR and SD rats, we sought to determine the effects of a high-fat diet (HFD) versus normal chow on gene expression and vascular function in SD rats. We found that feeding SD rats a HFD for 4 mo led to increased expression of Serpine1 and Epha4 (Fig. 8A) as well as increases in blood pressure and vascular resistance similar to those observed in JCR rats (Fig. 8B). Next, given that G6PD inhibition suppressed expression of Serpine1 and Epha4 and reduced hypertension in JCR rats, we sought to confirm those findings by determining whether G6PD inhibition induced through genetic manipulation would yield similar results. We therefore fed G6PDS188F variant rats a HFD for 4 mo. Intriguingly, the HFD decreased expression of Abra in the variant rats (Fig. 8A) and failed to increase expression of Serpine1 and Epha4 (Fig. 8A) or blood pressure and vascular resistance (Fig. 8B) compared with their wild-type littermates.
Figure 8.
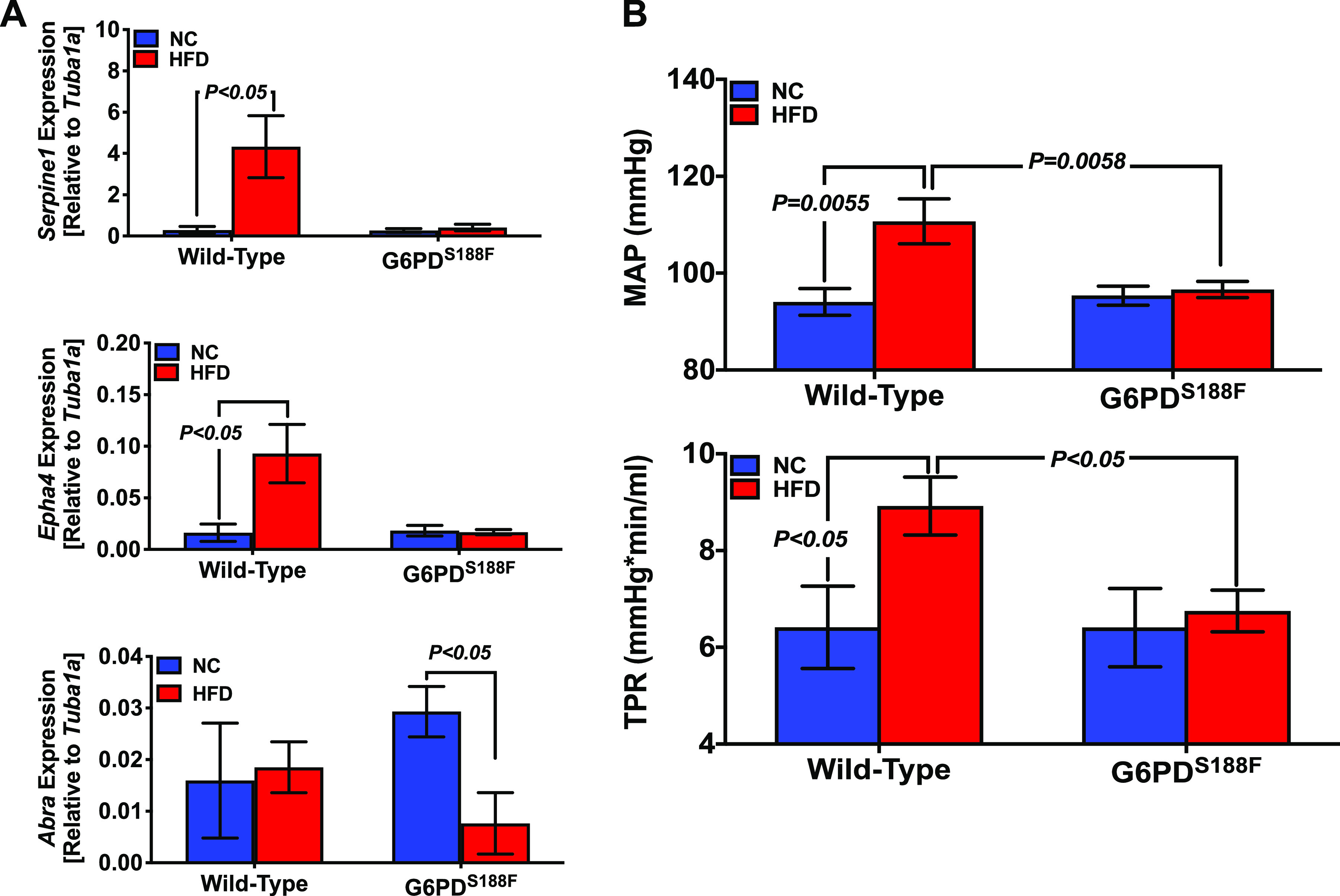
Loss-of-function Mediterranean G6PD variant prevents high-fat diet (HFD)-induced increases in Sperine1 and Epha4 expression and vascular resistance in rats. A: RT-PCR results showing that a HFD (4 mo) increased relative expression of Sperine1, Epha4, and Abra in aortas from wild-type rats but not Mediterranean G6PD variant rats. B: eating a HFD (4 mo) increased mean arterial pressure (MAP) and total peripheral resistance (TPR) in wild-type but not Mediterranean G6PD variant rats. n = 5 samples in each group. NC, normal chow.
DISCUSSION
We found that the increased G6PD activity and upregulated glucose flux through the PPP pathway were associated with altered gene expression in arterial tissue from rats with metabolic syndrome and in cultured SMCs. These findings demonstrate the heretofore unrecognized role of adaptive metabolic reprogramming in directing transcriptomic reprogramming within arteries of rats with metabolic diseases. Conversely, altered transcriptomic reprogramming regulated expression of genes that encode metabolic enzymes (e.g., SDHA) and controlled metabolic homeostasis beyond the PPP. Cellular metabolism is essential for life, and cells undergo adaptive changes in metabolism during growth. The PPP produces the majority of cellular redox potential, which is used by cells for various anabolic and synthetic transactions for pentose/ribose sugar, which is utilized to synthesize the nucleotides needed for de novo synthesis of RNA and DNA during cell proliferation and growth (48). Because G6PD is >10-fold more active in metabolically and replicatively active liver cells and reproductive system, respectively, than in differentiated heart and skeletal muscle (49), we anticipated that G6PD inhibition or G6pd knockdown would downregulate gene expression. Unexpectedly, they selectively increased expression of SMC-restricted genes (e.g., Myocd, Myh11, Tagln, and Cnn1) and downregulated Serpine1 and Epha4, which encode proteins linked to SMC differentiation, vascular dysfunction, and thrombosis (43, 44, 50). These results suggest that blockade of G6PD and the PPP does not merely reduce the supply of nucleotides to the replicative and transcriptional machinery but also controls the transcriptional regulatory process. Although considerable work has been done to date to determine the mechanism underlying phenotypic remodeling of SMCs (1, 42), our present results connect a fundamental metabolic enzyme and metabolism to the regulation of SMC gene expression and phenotypic fate.
The progressive loss (>100-fold) of SMC-restricted genes (Myh11 and Myocd) and significant increase of Sca-1 and upward trend in Oct4 and c-Myc in cultured MSMCs over passages 1−7 suggest that there is a gradual disappearance of differentiated MSMCs and emergence of stem cell-like cells across successive passages of primary cells isolated from mouse aorta. Inhibition of G6PD activity in cell cultures decreased Sca-1/Oct4/c-Myc and increased Myh11/Myocd at passage 7. There are two likely interpretations of these findings: 1) G6PD inhibition differentially regulates Sca-1/Oct4 and Mycod/Myh11 in the same cell and/or 2) G6PD contributes to both the increase in Sca-1+-mediated cell growth and the reduction in MSMC numbers. The upregulation of SMC-nonrestricted genes and downregulation of SMC-restricted genes observed in aortas from JCR rats and SD rats fed a HFD suggest that, in situ, SMCs respond to metabolic changes as well as physical (shear) stress by altering their gene expression profile or phenotype. G6PD inhibition in JCR rats increased SMC-restricted genes, also known as differentiation markers (Myh11, Cnn1, and Tagln), and reduced expression of Serpine1, encoding plasminogen activator inhibitor-1, which promotes thrombosis (51) and the risk of atherosclerosis (43), and Epha4, encoding ephrin type-A receptor 4, which has been implicated in the regulation of SMC contractility and calcification (44). These results were confirmed by the findings that a HFD significantly increased (3-fold) expression of Serpine1 and Epha4 in aortas from SD rats but not G6PDS188F (Mediterranean G6PD variant) mutants. Since G6PD inhibition and knockdown reduced HDAC activity that leads to increase acetylation of histones, we propose that this could have potentially regulated expression of genes in JCR rats and HFD-fed rats.
Our findings have significant clinical implications, as the proteins encoded by the aforementioned genes play key roles in thrombosis and contractility and in the calcification of SMCs/blood vessels. Along those lines, increases in Serpine1 and Epha4 were associated with the pathogenesis of large artery stiffness, vascular resistance, and occlusive growth within coronary arteries in a rat model of metabolic syndrome (JCR rats). A similar association between Serpine1 and Epha4 expression and elevated blood pressure and vascular resistance was observed in SD rats fed a HFD. In addition, adult SMCs de/transdifferentiate into bone and stem cell-like lineages in response to growth factors and scratch injuries in vitro and in situ (3–7, 52) and in angio-proliferative diseases and wire-injury models in vivo (1, 42, 53). In the context of metabolic disease, angio-proliferative and hypertrophic remodeling are pathognomonic of perivascular and coronary artery disease and hypertension, respectively. Studies, including ours, previously showed that increased G6PD activity in the PPP fuels free radical generation (20, 23, 54), which promotes inflammation in vascular and other tissues in patients and animal models with diabetes or metabolic syndrome (19, 21, 22, 24). Our present findings demonstrate that phenotypic transition of SMCs can be stopped and vascular remodeling/growth can be slowed or even reversed by inhibiting G6PD and activity in the PPP.
These findings also revealed that G6PD is a determinant of SMC-restricted (Myocd, Myh11, Tagln, and Cnn1) gene expression under normal and pathogenic conditions. Inhibition of G6PD activity increased expression of SMC-restricted contractile proteins (MYH11, TAGLN, and ACTA2) and decreased expression of KLF4. Transcription factors such as SRF and cofactors such as MYOCD, which forms a ternary complex with SRF and binds to CArG boxes to transactivate SMC contractile gene expression (55), and KLF4/5 precisely regulate expression of SMC-restricted genes to maintain a balance between the differentiated and de/transdifferentiated phenotypes. MYOCD is the master regulator of SMC-restricted genes and plays a critical role in directing and maintaining SMC differentiation through embryogenesis and postnatal life (9, 56). Emerging evidence also suggests that the MYOCD family of transcription coactivators play a role in lipid and glucose hemeostasis (57). Because pharmacological or genetic inhibition of G6PD activity enhanced expression of Myocd mRNA in A7r5 cells, MSMCs, RSMCs, and the Mediterranean G6pd variant, we suggest that it activated Myocd gene transcription and/or stabilized Myocd mRNA posttranscriptionally. Transcriptional control of Myocd is complex and incompletely understood (9, 58). Increased HDAC activity signals heterochromatin formation and repression of gene expression (59). Because HDAC inhibition increased Myocd transcription, while both G6PD inhibition and G6pd knockdown increased H3K9ac and enriched H3K27ac on the Myocd promoter near the TSS, we suggest that G6PD-elicited histone 3 modifications contribute to Myocd transcriptional activation. Since G6PD inhibition did not enrich H3K27ac on Myh11 or Tagln, these results suggest that G6PD does not directly modify chromatin on these contractile genes or activate them in SMCs. Moreover, G6pd knockdown decreased expression of HDAC5, which interacts with Myocd (60). This suggests that reduction of class II HDAC5 may have acted to reduce HDAC activity and enrich H3K9ac and H3K27ac on the Myocd promoter in SMCs. We therefore propose that increased G6PD activity and metabolic reprogramming-mediated epigenomic (histone 3/chromatin) modifications contribute to repress SMC-restricted genes and proteins, thereby inducing SMC dedifferentiation ex vivo in cell cultures and in vivo under pathological conditions.
It was recently shown that G6PD-derived NADPH is required for HDAC2/3 (16) and HDAC2 activities in C2 myoblasts (61) and that G6PD inhibition increases acetylation of histone 3 and histone 4 in MDA-MB-453 breast cancer cells (16). We therefore suggest that G6PD acting at specific genomic loci to produce NADPH locally for HDAC activation and histone deacetylation modified chromatin structure and impaired Myocd transcription. Activation of class II HDAC inhibits, while p300 enhances, the promyogenic activity of MYOCD and plays a key role in modifying the phenotypes of SMCs and cardiac myocytes (62). Additionally, binding of KLF4/5 to their binding elements in the Myocd promoter powerfully represses Myocd transcription and mediates PDGF-induced repression of Myocd gene expression in RSMCs (9, 58, 63, 64). What is more, binding of KLF4 to the SRF-MYOCD complex suppresses SMC-restricted gene activation (65–68). We therefore suggest that downregulation of KLF4 through G6PD inhibition may attenuate KLF4-induced repression of Myocd transcription or increase SRF-MYOCD activity to upregulate SMC-restricted contractile proteins. In another recent study, we observed that G6PD inhibition leads to demethylation of Myocd in the lungs of hypoxic mice and rats (unpublished), and others suggest that ten-eleven translocation-2 (TET2)-mediated demethylation of Myocd is a critical regulator of SMC phenotypic fate (69). In addition to KLF4- and DNA methylation-mediated repression, we showed in the present study that G6PD controls Myocd expression via histone remodeling driven by NADPH and redox signaling. We previously proposed that G6PD regulates expression of Myocd mRNA posttranscriptionally via a miRNA-dependent mechanism (15). Taken together, these observations strongly suggest that G6PD is a crucial transcriptional and posttranscriptional regulator of Myocd.
In summary, the salient findings of the present study are that 1) inhibition of G6PD activity and glucose flux through the PPP contributes to switching off Klf4 and Sca-1 and turning on Myocd and Myh11 in adult arterial SMCs in vitro and in vivo and 2) increased G6PD activity is a driver of metabolic and transcriptomic reprogramming and a contributor to the pathogenesis of vascular disease in rat models of diabetes and metabolic syndrome. Mechanistically, we revealed that G6PD-dependent acetylation/deacetylation of H3K9 and H3K27, two open chromatin marks, on the Myocd promoter mediated at least in part changes in expression of SMC-restricted genes. More importantly, pharmacological or genetic inhibition of G6PD activity increased expression of SMC-restricted genes and suppressed transcriptomic reprogramming in vascular tissues and concurrently reduced occlusive lesions in coronary arteries, arterial elastance, vascular resistance, and hypertension, in rats with metabolic syndrome. Finally, although the effect of G6PD inhibition on other mechanisms, including regulation of ion channel activity, pressure sensing, neurohormonal control of blood pressure, and the renin-angiotensin system, cannot be eliminated, our present findings suggest that G6PD is a key determinant of SMC-restricted gene expression in various species under both normal and pathogenic conditions and that G6PD could potentially be a useful pharmacotherapeutic target to slow or even stop progression of metabolic syndrome-associated vascular diseases.
A limitation of the study is that, considering that EPI decreases blood pressure (40), one cannot dissect the effects of G6PD per se on gene/metabolite expression from an effect simply due to lowering of blood pressure. Moreover, since high blood pressure induces vascular hypertrophy, growth-inhibiting effect of EPI could be mediated by blood pressure reduction. Therefore, more studies on metabolism and blood pressure are warranted.
GRANTS
This study was supported by National Institutes of Health (NIH) Grant RO1 HL-132574 (S.A.G.), American Heart Association Grant-in-Aid 17GRNT33670454 (S.A.G.), NIH Grants R01 HL-146442 and R01 HL-148151, and the Boettcher Webb-Waring Early Career Award (A.D.).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
V.D. and S.A.G. conceived and designed research; V.D., A.K., C.J., and C.Z. performed experiments; V.D., A.K., C.Z., A.D., R.G., and S.A.G. analyzed data; V.D., A.D., J.G.E., P.R., R.G., and S.A.G. interpreted results of experiments; V.D., A.D., and S.A.G. prepared figures; V.D. and S.A.G. drafted manuscript; V.D., A.K., C.J., A.D., J.G.E., P.R., R.G., and S.A.G. edited and revised manuscript; V.D., C.J., C.Z., A.D., R.G., and S.A.G. approved final version of manuscript.
REFERENCES
- 1.Frismantiene A, Philippova M, Erne P, Resink TJ. Smooth muscle cell-driven vascular diseases and molecular mechanisms of VSMC plasticity. Cell Signal 52: 48–64, 2018. doi: 10.1016/j.cellsig.2018.08.019. [DOI] [PubMed] [Google Scholar]
- 2.Chamley-Campbell J, Campbell GR, Ross R. The smooth muscle cell in culture. Physiol Rev 59: 1–61, 1979. doi: 10.1152/physrev.1979.59.1.1. [DOI] [PubMed] [Google Scholar]
- 3.Azuma K, Ichimura K, Mita T, Nakayama S, Jin WL, Hirose T, Fujitani Y, Sumiyoshi K, Shimada K, Daida H, Sakai T, Mitsumata M, Kawamori R, Watada H. Presence of alpha-smooth muscle actin-positive endothelial cells in the luminal surface of adult aorta. Biochem Biophys Res Commun 380: 620–626, 2009. doi: 10.1016/j.bbrc.2009.01.135. [DOI] [PubMed] [Google Scholar]
- 4.Haust MD, More RH, Movat HZ. The role of smooth muscle cells in the fibrogenesis of arteriosclerosis. Am J Pathol 37: 377–389, 1960. [PMC free article] [PubMed] [Google Scholar]
- 5.Rong JX, Shapiro M, Trogan E, Fisher EA. Transdifferentiation of mouse aortic smooth muscle cells to a macrophage-like state after cholesterol loading. Proc Natl Acad Sci USA 100: 13531–13536, 2003. doi: 10.1073/pnas.1735526100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Speer MY, Yang HY, Brabb T, Leaf E, Look A, Lin WL, Frutkin A, Dichek D, Giachelli CM. Smooth muscle cells give rise to osteochondrogenic precursors and chondrocytes in calcifying arteries. Circ Res 104: 733–741, 2009. doi: 10.1161/CIRCRESAHA.108.183053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Steitz SA, Speer MY, Curinga G, Yang HY, Haynes P, Aebersold R, Schinke T, Karsenty G, Giachelli CM. Smooth muscle cell phenotypic transition associated with calcification: upregulation of Cbfa1 and downregulation of smooth muscle lineage markers. Circ Res 89: 1147–1154, 2001. doi: 10.1161/hh2401.101070. [DOI] [PubMed] [Google Scholar]
- 8.Wang D, Chang PS, Wang Z, Sutherland L, Richardson JA, Small E, Krieg PA, Olson EN. Activation of cardiac gene expression by myocardin, a transcriptional cofactor for serum response factor. Cell 105: 851–862, 2001. doi: 10.1016/s0092-8674(01)00404-4. [DOI] [PubMed] [Google Scholar]
- 9.Miano JM. Myocardin in biology and disease. J Biomed Res 29: 3–19, 2015. doi: 10.7555/JBR.29.20140151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen J, Kitchen CM, Streb JW, Miano JM. Myocardin: a component of a molecular switch for smooth muscle differentiation. J Mol Cell Cardiol 34: 1345–1356, 2002. doi: 10.1006/jmcc.2002.2086. [DOI] [PubMed] [Google Scholar]
- 11.Du KL, Ip HS, Li J, Chen M, Dandre F, Yu W, Lu MM, Owens GK, Parmacek MS. Myocardin is a critical serum response factor cofactor in the transcriptional program regulating smooth muscle cell differentiation. Mol Cell Biol 23: 2425–2437, 2003. doi: 10.1128/mcb.23.7.2425-2437.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang Z, Wang DZ, Pipes GC, Olson EN. Myocardin is a master regulator of smooth muscle gene expression. Proc Natl Acad Sci USA 100: 7129–7134, 2003. doi: 10.1073/pnas.1232341100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yoshida T, Sinha S, Dandré F, Wamhoff BR, Hoofnagle MH, Kremer BE, Wang DZ, Olson EN, Owens GK. Myocardin is a key regulator of CArG-dependent transcription of multiple smooth muscle marker genes. Circ Res 92: 856–864, 2003. doi: 10.1161/01.RES.0000068405.49081.09. [DOI] [PubMed] [Google Scholar]
- 14.Chettimada S, Gupte R, Rawat D, Gebb SA, McMurtry IF, Gupte SA. Hypoxia-induced glucose-6-phosphate dehydrogenase overexpression and -activation in pulmonary artery smooth muscle cells: implication in pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 308: L287–L300, 2015. doi: 10.1152/ajplung.00229.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chettimada S, Joshi SR, Dhagia V, Aiezza A 2nd, Lincoln TM, Gupte R, Miano JM, Gupte SA. Vascular smooth muscle cell contractile protein expression is increased through protein kinase G-dependent and -independent pathways by glucose-6-phosphate dehydrogenase inhibition and deficiency. Am J Physiol Heart Circ Physiol 311: H904–H912, 2016. doi: 10.1152/ajpheart.00335.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vogelauer M, Krall AS, McBrian MA, Li JY, Kurdistani SK. Stimulation of histone deacetylase activity by metabolites of intermediary metabolism. J Biol Chem 287: 32006–32016, 2012. doi: 10.1074/jbc.M112.362467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schwartz SM, Schwartz HT, Horvath S, Schadt E, Lee SI. A systematic approach to multifactorial cardiovascular disease: causal analysis. Arterioscler Thromb Vasc Biol 32: 2821–2835, 2012. doi: 10.1161/ATVBAHA.112.300123. [DOI] [PubMed] [Google Scholar]
- 18.Rajas F, Gautier-Stein A, Mithieux G. Glucose-6 phosphate, a central hub for liver carbohydrate metabolism . Metabolites 9: 282, 2019. doi: 10.3390/metabo9120282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carley AN, Severson DL. Fatty acid metabolism is enhanced in type 2 diabetic hearts. Biochim Biophys Acta 1734: 112–126, 2005. doi: 10.1016/j.bbalip.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 20.Gupte RS, Floyd BC, Kozicky M, George S, Ungvari ZI, Neito V, Wolin MS, Gupte SA. Synergistic activation of glucose-6-phosphate dehydrogenase and NAD(P)H oxidase by Src kinase elevates superoxide in type 2 diabetic, Zucker fa/fa, rat liver. Free Radic Biol Med 47: 219–228, 2009. doi: 10.1016/j.freeradbiomed.2009.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ham M, Choe SS, Shin KC, Choi G, Kim JW, Noh JR, Kim YH, Ryu JW, Yoon KH, Lee CH, Kim JB. Glucose-6-phosphate dehydrogenase deficiency improves insulin resistance with reduced adipose tissue inflammation in obesity. Diabetes 65: 2624–2638, 2016. doi: 10.2337/db16-0060. [DOI] [PubMed] [Google Scholar]
- 22.Schneider AM, Rawat D, Weinstein LS, Gupte SA, Richards WO. Effects of laparoscopic Roux-en-Y gastric bypass on glucose-6 phosphate dehydrogenase activity in obese type 2 diabetics. Surg Endosc 26: 823–830, 2012. doi: 10.1007/s00464-011-1959-8. [DOI] [PubMed] [Google Scholar]
- 23.Serpillon S, Floyd BC, Gupte RS, George S, Kozicky M, Neito V, Recchia F, Stanley W, Wolin MS, Gupte SA. Superoxide production by NAD(P)H oxidase and mitochondria is increased in genetically obese and hyperglycemic rat heart and aorta before the development of cardiac dysfunction. The role of glucose-6-phosphate dehydrogenase-derived NADPH. Am J Physiol Heart Circ Physiol 297: H153–H162, 2009. doi: 10.1152/ajpheart.01142.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yamashita A, Zhao Y, Matsuura Y, Yamasaki K, Moriguchi-Goto S, Sugita C, Iwakiri T, Okuyama N, Koshimoto C, Kawai K, Tamaki N, Zhao S, Kuge Y, Asada Y. Increased metabolite levels of glycolysis and pentose phosphate pathway in rabbit atherosclerotic arteries and hypoxic macrophage. PLoS One 9: e86426, 2014. doi: 10.1371/journal.pone.0086426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kitagawa A, Kizub I, Jacob C, Michael K, D’Alessandro A, Reisz JA, Grzybowski M, Geurts AM, Rocic P, Gupte R, Miano JM, Gupte SA. CRISPR-mediated single nucleotide polymorphism modeling in rats reveals insight into reduced cardiovascular risk associated with Mediterranean G6PD variant. Hypertension 76: 523–532, 2020. doi: 10.1161/HYPERTENSIONAHA.120.14772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lyu Q, Dhagia V, Han Y, Guo B, Wines-Samuelson ME, Christie CK, Yin Q, Slivano OJ, Herring P, Long X, Gupte SA, Miano JM. CRISPR-Cas9-mediated epitope tagging provides accurate and versatile assessment of myocardin—brief report. Arterioscler Thromb Vasc Biol 38: 2184–2190, 2018. doi: 10.1161/ATVBAHA.118.311171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ciuclan L, Bonneau O, Hussey M, Duggan N, Holmes AM, Good R, Stringer R, Jones P, Morrell NW, Jarai G, Walker C, Westwick J, Thomas M. A novel murine model of severe pulmonary arterial hypertension. Am J Respir Crit Care Med 184: 1171–1182, 2011. doi: 10.1164/rccm.201103-0412OC. [DOI] [PubMed] [Google Scholar]
- 28.Gao S, Ho D, Vatner DE, Vatner SF. Echocardiography in mice. Curr Protoc Mouse Biol 1: 71–83, 2011. doi: 10.1002/9780470942390.mo100130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thibault HB, Kurtz B, Raher MJ, Shaik RS, Waxman A, Derumeaux G, Halpern EF, Bloch KD, Scherrer-Crosbie M. Noninvasive assessment of murine pulmonary arterial pressure: validation and application to models of pulmonary hypertension. Circ Cardiovasc Imaging 3: 157–163, 2010. doi: 10.1161/CIRCIMAGING.109.887109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gupte RS, Ata H, Rawat D, Abe M, Taylor MS, Ochi R, Gupte SA. Glucose-6-phosphate dehydrogenase is a regulator of vascular smooth muscle contraction. Antioxid Redox Signal 14: 543–558, 2011. doi: 10.1089/ars.2010.3207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chettimada S, Joshi SR, Alzoubi A, Gebb SA, McMurtry IF, Gupte R, Gupte SA. Glucose-6-phosphate dehydrogenase plays a critical role in hypoxia-induced CD133+ progenitor cells self-renewal and stimulates their accumulation in the lungs of pulmonary hypertensive rats. Am J Physiol Lung Cell Mol Physiol 307: L545–L556, 2014. doi: 10.1152/ajplung.00303.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gupte RS, Rawat DK, Chettimada S, Cioffi DL, Wolin MS, Gerthoffer WT, McMurtry IF, Gupte SA. Activation of glucose-6-phosphate dehydrogenase promotes acute hypoxic pulmonary artery contraction. J Biol Chem 285: 19561–19571, 2010. doi: 10.1074/jbc.M109.092916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods 9: 357–359, 2012. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ross-Innes CS, Stark R, Teschendorff AE, Holmes KA, Ali HR, Dunning MJ, Brown GD, Gojis O, Ellis IO, Green AR, Ali S, Chin SF, Palmieri C, Caldas C, Carroll JS. Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 481: 389–393, 2012. doi: 10.1038/nature10730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, Liu XS. Model-based analysis of ChIP-Seq (MACS). Genome Biol 9: R137, 2008. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.D’Alessandro A, Nemkov T, Yoshida T, Bordbar A, Palsson BO, Hansen KC. Citrate metabolism in red blood cells stored in additive solution-3. Transfusion 57: 325–336, 2017. doi: 10.1111/trf.13892. [DOI] [PubMed] [Google Scholar]
- 37.Nemkov T, Hansen KC, D’Alessandro A. A three-minute method for high-throughput quantitative metabolomics and quantitative tracing experiments of central carbon and nitrogen pathways. Rapid Commun Mass Spectrom 31: 663–673, 2017. doi: 10.1002/rcm.7834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xia J, Sinelnikov IV, Han B, Wishart DS. MetaboAnalyst 3.0—making metabolomics more meaningful. Nucleic Acids Res 43: W251–W257, 2015. doi: 10.1093/nar/gkv380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hamilton NM, Dawson M, Fairweather EE, Hamilton NS, Hitchin JR, James DI, Jones SD, Jordan AM, Lyons AJ, Small HF, Thomson GJ, Waddell ID, Ogilvie DJ. Novel steroid inhibitors of glucose 6-phosphate dehydrogenase. J Med Chem 55: 4431–4445, 2012. doi: 10.1021/jm300317k. [DOI] [PubMed] [Google Scholar]
- 40.Gupte R, Dhagia V, Rocic P, Ochi R, Gupte SA. Glucose-6-phosphate dehydrogenase increases Ca2+ currents by interacting with Cav1.2 and reducing intrinsic inactivation of the L-type calcium channel. Am J Physiol Heart Circ Physiol 319: H144–H158, 2020. doi: 10.1152/ajpheart.00727.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hayashi K, Nakamura S, Nishida W, Sobue K. Bone morphogenetic protein-induced MSX1 and MSX2 inhibit myocardin-dependent smooth muscle gene transcription. Mol Cell Biol 26: 9456–9470, 2006. doi: 10.1128/MCB.00759-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gomez D, Swiatlowska P, Owens GK. Epigenetic control of smooth muscle cell identity and lineage memory. Arterioscler Thromb Vasc Biol 35: 2508–2516, 2015. doi: 10.1161/ATVBAHA.115.305044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ji Y, Weng Z, Fish P, Goyal N, Luo M, Myears SP, Strawn TL, Chandrasekar B, Wu J, Fay WP. Pharmacological targeting of plasminogen activator inhibitor-1 decreases vascular smooth muscle cell migration and neointima formation. Arterioscler Thromb Vasc Biol 36: 2167–2175, 2016. doi: 10.1161/ATVBAHA.116.308344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ogita H, Kunimoto S, Kamioka Y, Sawa H, Masuda M, Mochizuki N. EphA4-mediated Rho activation via Vsm-RhoGEF expressed specifically in vascular smooth muscle cells. Circ Res 93: 23–31, 2003. doi: 10.1161/01.RES.0000079310.81429.C8. [DOI] [PubMed] [Google Scholar]
- 45.Wallace MA, Lamon S, Russell AP. The regulation and function of the striated muscle activator of rho signaling (STARS) protein. Front Physiol 3: 469, 2012. doi: 10.3389/fphys.2012.00469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rawat DK, Alzoubi A, Gupte R, Chettimada S, Watanabe M, Kahn AG, Okada T, McMurtry IF, Gupte SA. Increased reactive oxygen species, metabolic maladaptation, and autophagy contribute to pulmonary arterial hypertension-induced ventricular hypertrophy and diastolic heart failure. Hypertension 64: 1266–1274, 2014. doi: 10.1161/HYPERTENSIONAHA.114.03261. [DOI] [PubMed] [Google Scholar]
- 47.Russell JC, Proctor SD. Small animal models of cardiovascular disease: tools for the study of the roles of metabolic syndrome, dyslipidemia, and atherosclerosis. Cardiovasc Pathol 15: 318–330, 2006. doi: 10.1016/j.carpath.2006.09.001. [DOI] [PubMed] [Google Scholar]
- 48.Stincone A, Prigione A, Cramer T, Wamelink MM, Campbell K, Cheung E, Olin-Sandoval V, Grüning NM, Krüger A, Tauqeer Alam M, Keller MA, Breitenbach M, Brindle KM, Rabinowitz JD, Ralser M. The return of metabolism: biochemistry and physiology of the pentose phosphate pathway. Biol Rev Camb Philos Soc 90: 927–963, 2015. doi: 10.1111/brv.12140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hecker PA, Mapanga RF, Kimar CP, Ribeiro RF Jr, Brown BH, O’Connell KA, Cox JW, Shekar KC, Asemu G, Essop MF, Stanley WC. Effects of glucose-6-phosphate dehydrogenase deficiency on the metabolic and cardiac responses to obesogenic or high-fructose diets. Am J Physiol Endocrinol Metab 303: E959–E972, 2012. doi: 10.1152/ajpendo.00202.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Komori T. Signaling networks in RUNX2-dependent bone development. J Cell Biochem 112: 750–755, 2011. doi: 10.1002/jcb.22994. [DOI] [PubMed] [Google Scholar]
- 51.Law RH, Zhang Q, McGowan S, Buckle AM, Silverman GA, Wong W, Rosado CJ, Langendorf CG, Pike RN, Bird PI, Whisstock JC. An overview of the serpin superfamily. Genome Biol 7: 216, 2006. doi: 10.1186/gb-2006-7-5-216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Majesky MW, Horita H, Ostriker A, Lu S, Regan JN, Bagchi A, Dong XR, Poczobutt J, Nemenoff RA, Weiser-Evans MC. Differentiated smooth muscle cells generate a subpopulation of resident vascular progenitor cells in the adventitia regulated by Klf4. Circ Res 120: 296–311, 2017. doi: 10.1161/CIRCRESAHA.116.309322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Regan CP, Adam PJ, Madsen CS, Owens GK. Molecular mechanisms of decreased smooth muscle differentiation marker expression after vascular injury. J Clin Invest 106: 1139–1147, 2000. doi: 10.1172/JCI10522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gupte SA, Levine RJ, Gupte RS, Young ME, Lionetti V, Labinskyy V, Floyd BC, Ojaimi C, Bellomo M, Wolin MS, Recchia FA. Glucose-6-phosphate dehydrogenase-derived NADPH fuels superoxide production in the failing heart. J Mol Cell Cardiol 41: 340–349, 2006. doi: 10.1016/j.yjmcc.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 55.Wang Z, Wang DZ, Hockemeyer D, McAnally J, Nordheim A, Olson EN. Myocardin and ternary complex factors compete for SRF to control smooth muscle gene expression. Nature 428: 185–189, 2004. doi: 10.1038/nature02382. [DOI] [PubMed] [Google Scholar]
- 56.Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev 84: 767–801, 2004. doi: 10.1152/physrev.00041.2003. [DOI] [PubMed] [Google Scholar]
- 57.Swärd K, Stenkula KG, Rippe C, Alajbegovic A, Gomez MF, Albinsson S. Emerging roles of the myocardin family of proteins in lipid and glucose metabolism. J Physiol 594: 4741–4752, 2016. doi: 10.1113/JP271913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wamhoff BR, Bowles DK, Owens GK. Excitation-transcription coupling in arterial smooth muscle. Circ Res 98: 868–878, 2006. doi: 10.1161/01.RES.0000216596.73005.3c. [DOI] [PubMed] [Google Scholar]
- 59.Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet 33, Suppl: 245–254, 2003. doi: 10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
- 60.Long X, Creemers EE, Wang DZ, Olson EN, Miano JM. Myocardin is a bifunctional switch for smooth versus skeletal muscle differentiation. Proc Natl Acad Sci USA 104: 16570–16575, 2007. doi: 10.1073/pnas.0708253104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cacchiarelli D, Martone J, Girardi E, Cesana M, Incitti T, Morlando M, Nicoletti C, Santini T, Sthandier O, Barberi L, Auricchio A, Musarò A, Bozzoni I. MicroRNAs involved in molecular circuitries relevant for the Duchenne muscular dystrophy pathogenesis are controlled by the dystrophin/nNOS pathway. Cell Metab 12: 341–351, 2010. doi: 10.1016/j.cmet.2010.07.008. [DOI] [PubMed] [Google Scholar]
- 62.Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet 10: 32–42, 2009. doi: 10.1038/nrg2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Deaton RA, Gan Q, Owens GK. Sp1-dependent activation of KLF4 is required for PDGF-BB-induced phenotypic modulation of smooth muscle. Am J Physiol Heart Circ Physiol 296: H1027–H1037, 2009. doi: 10.1152/ajpheart.01230.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Turner EC, Huang CL, Govindarajan K, Caplice NM. Identification of a Klf4-dependent upstream repressor region mediating transcriptional regulation of the myocardin gene in human smooth muscle cells. Biochim Biophys Acta 1829: 1191–1201, 2013. doi: 10.1016/j.bbagrm.2013.09.002. [DOI] [PubMed] [Google Scholar]
- 65.Lincoln TM, Wu X, Sellak H, Dey N, Choi CS. Regulation of vascular smooth muscle cell phenotype by cyclic GMP and cyclic GMP-dependent protein kinase. Front Biosci 11: 356–367, 2006. doi: 10.2741/1803. [DOI] [PubMed] [Google Scholar]
- 66.Liu N, Olson EN. MicroRNA regulatory networks in cardiovascular development. Dev Cell 18: 510–525, 2010. doi: 10.1016/j.devcel.2010.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Liu Y, Sinha S, McDonald OG, Shang Y, Hoofnagle MH, Owens GK. Kruppel-like factor 4 abrogates myocardin-induced activation of smooth muscle gene expression. J Biol Chem 280: 9719–9727, 2005. doi: 10.1074/jbc.M412862200. [DOI] [PubMed] [Google Scholar]
- 68.Miano JM, Long X, Fujiwara K. Serum response factor: master regulator of the actin cytoskeleton and contractile apparatus. Am J Physiol Cell Physiol 292: C70–C81, 2007. doi: 10.1152/ajpcell.00386.2006. [DOI] [PubMed] [Google Scholar]
- 69.Liu R, Jin Y, Tang WH, Qin L, Zhang X, Tellides G, Hwa J, Yu J, Martin KA. Ten-eleven translocation-2 (TET2) is a master regulator of smooth muscle cell plasticity. Circulation 128: 2047–2057, 2013. doi: 10.1161/CIRCULATIONAHA.113.002887. [DOI] [PMC free article] [PubMed] [Google Scholar]