

Abstract
Prostaglandins are critical lipid mediators involved in the wound healing response, with prostaglandin E2 (PGE2) being the most complex and exhibiting the most diverse physiological outputs. PGE2 signals via four G protein-coupled receptors, termed EP-receptors 1-4 that induce distinct signaling pathways upon activation and lead to an array of different outputs. Recent studies examining the role of PGE2 and EP receptor signaling in wound healing following various forms of tissue damage are discussed in this review.
Keywords: cell signaling, prostaglandins, repair, tissue damage, wound healing
INTRODUCTION
Wound healing is a highly complex and coordinated process that has three sequential but overlapping phases: 1) hemostasis/inflammation, consisting of vascular changes, immune infiltration, and inflammatory signaling; 2) proliferation, focusing on re-epithelialization and angiogenesis; and 3) remodeling, aiding in maturation and strengthening of the newly generated tissue (1). Following a wounding event, these processes must be tightly regulated to ensure that proper healing occurs. Dysregulation of any stage of the response can lead to either insufficient repair or chronic inflammation and excessive tissue damage. Therefore, coordinated inflammation is critical for a wound to heal properly, which requires a number of different cell types and inflammatory signals. Here, we focus on the role of prostaglandin E2 (PGE2) in the response to tissue injury and the important roles of this signaling mediator to induce proper wound healing. In addition, we discuss recent discoveries related to PGE2 and wound repair.
PROSTAGLANDIN E2 SYNTHESIS AND SIGNALING
Prostaglandins (PGs) are a family of bioactive lipids that exert a wide range of physiological effects throughout the body and are commonly involved in vasodilation, tissue repair/homeostasis, and inflammation. PGs are enzymatically synthesized from plasma membrane-derived phospholipids. These phospholipids are cleaved by the action of phospholipase A2 (PLA2), which releases fatty acids into the cytosol to be acted upon by a variety of enzymes (2). Arachidonic acid (AA) is the predominant fatty acid that is released, which is converted to prostaglandin G2 (PGG2) and PGH2 via a two-step reaction by cyclooxygenase (COX) enzymes, including the constitutively active isoform, COX-1, and the inflammation-inducible isoform, COX-2. Prostaglandin H2 (PGH2) is then subsequently acted upon by PGE synthase enzymes to generate PGE2. Although some prostaglandins are preferentially synthesized from COX-1-specific or COX-2-specific products, PGE2 can be generated from both COX-1- and COX-2-derived PGH2 (3). PGH2 is acted on by one of three isoforms of PGE synthase enzymes: microsomal PGE synthase-1 (mPGES-1), mPGES-2, and cytosolic PGE synthase (cPGES). Generally, mPGES-1 is considered the inflammation-activated isoform, whereas mPGES-2 and cPGES are both constitutively expressed in most cell types (4). PGE2 is secreted by most cells throughout the body and is inactivated by the enzyme 15-hydroxyprostaglandin dehydrogenase (15-PGDH), which oxidizes PGE2 to form 15-keto-PGE2 (5).
PGE2 exerts varying effects depending on the concentration of PGE2 present, the cell/tissue type, and the receptor isoform (EP 1–4) of which it interacts with (2). For these reasons, PGE2-EP receptor signaling is highly complex yet crucially important for an appropriate inflammatory response (Fig. 1). EP receptors are PGE2-specific G protein-coupled receptors (GPCRs) that are widely expressed throughout the body and induce distinct intracellular signaling cascades via activation of their respective G proteins: EP1 utilizes Gq, EP3 utilizes Gi, and EP2 and EP4 utilize Gs (6). These G proteins activate different downstream signaling pathways that lead to either pro- or anti-inflammatory responses (7). EP1 activates phospholipase C (PLC), leading to increased diacylglycerol (DAG) and inositol 1,4,5-trisphosphate (IP3), increasing intracellular calcium levels, and subsequent protein kinase C (PKC) activation (2). EP2 and EP4 receptors both activate two identical signaling cascades: The first entails activation of adenylate cyclase causing increased adenosine 3′,5′-cyclic monophosphate (cAMP) production and subsequent protein kinase A (PKA) and cAMP response element binding protein (CREB) activity; the second includes activation of phosphoinositol-3-kinase (PI3K) and Akt via the β-arrestin pathway following EGFR transactivation (8). EP3 receptor activation leads to inhibition of adenylate cyclase and reduced cAMP levels, contradicting EP2/4 signaling. In general, EP1-induced signaling and EP3-induced signaling are considered anti-inflammatory, whereas EP2 and EP4 are considered pro-inflammatory (2). EP receptor distribution varies across tissues with EP3 and EP4 being the most commonly expressed subtypes found almost ubiquitously throughout cell types and EP1 and EP2 being present in only select tissues (9). Tissue mRNA and protein content of EP receptors can be evaluated via the Human Protein Atlas (6), and a more detailed description of arachidonic acid metabolism and prostaglandin signaling can be found in previously published reviews (2, 4, 10).
Figure 1.
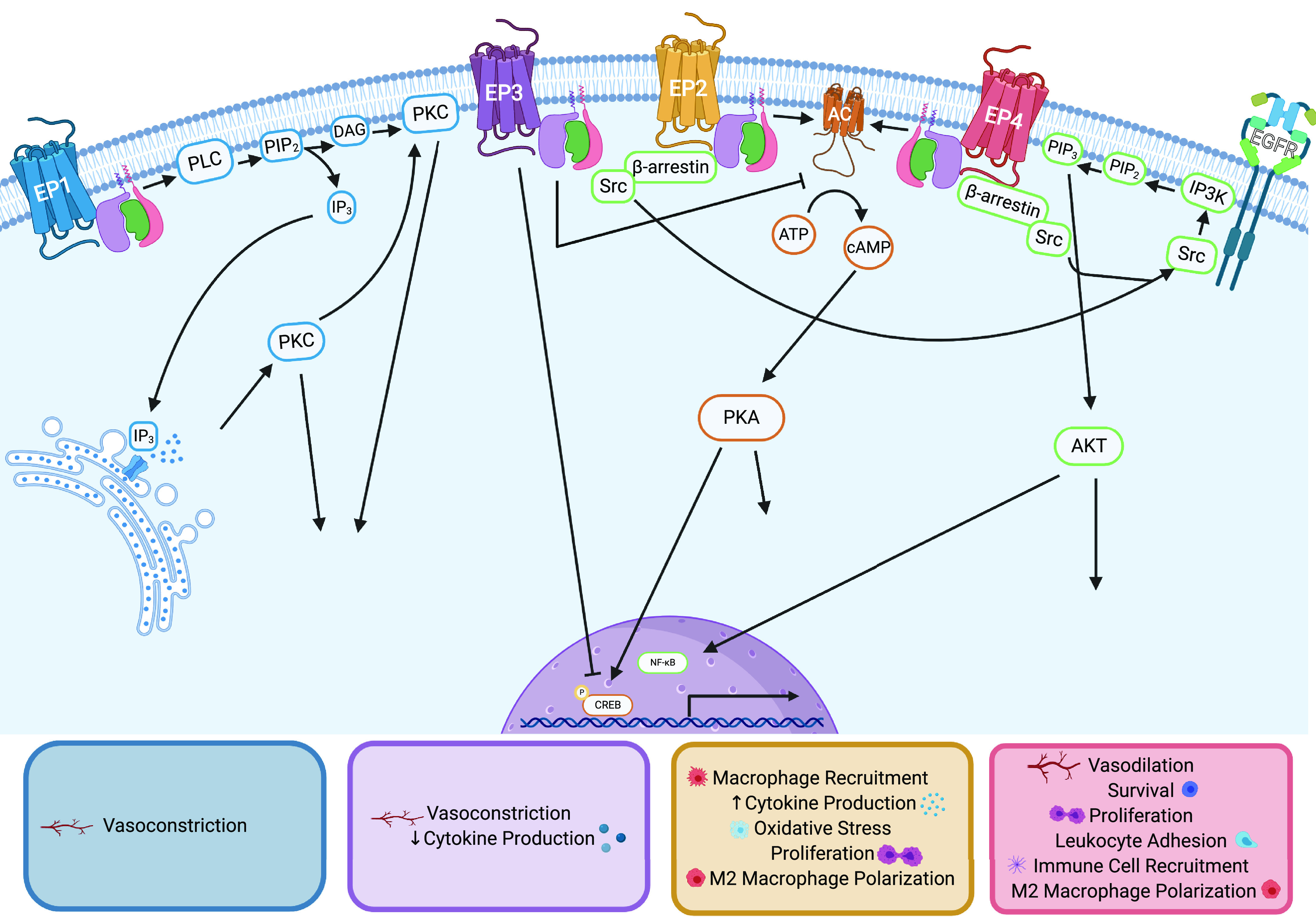
EP receptor signaling and PGE2-mediated wound healing phenotypes. EP1 (blue) associates with Gq G protein, which activates phospholipase C (PLC), leading to cleavage of phosphatidylinositol 4,5-bisphosphate (PIP2), increasing intracellular levels of diacylglycerol (DAG) and inositol 1,4,5-trisphosphate (IP3). IP3 binding to IP3-gated calcium channels on the endoplasmic reticulum induces release of calcium into the cytosol, which acts in concert with DAG to activate protein kinase C (PKC), leading to phosphorylation of various proteins. EP1-regulated wound healing outputs are listed in the blue box. EP2 (yellow) and EP4 (red) activate two identical signaling pathways. First, Gs G protein activates adenylate cyclase (AC, orange), which increases intracellular adenosine 3′,5′-cyclic monophosphate (cAMP), activating protein kinase A (PKA), leading to phosphorylation of downstream targets including the transcription factor cAMP response element binding protein (CREB). The second pathway (green) occurs via β-arrestin/Src-induced EGFR transactivation. This activates IP3 kinase (IP3K) to phosphorylate PIP2 to become phosphatidylinositol 3,4,5-trisphosphate (PIP3), which recruits Akt to the plasma membrane to be phosphorylated and activated. Activated Akt phosphorylates various downstream targets and often induces NF-κB nuclear translocation. EP2-regulated wound healing outputs are listed in the yellow box. EP4-regulated wound healing outputs are listed in the red box. EP3 (purple) associates with the inhibitory G protein (Gi) blocking the activity of AC and reducing intracellular cAMP levels. EP3-regulated wound healing outputs are listed in the purple box. PGE2, prostaglandin E2.
PGE2 IS PRODUCED FOLLOWING TISSUE INJURY
PGE2 is the most complex prostaglandin due to the multiple receptor isoforms that each induce distinct signaling cascades and outputs. The opposing signaling pathways initiated by PGE2 make it difficult to determine whether PGE2 is beneficial or detrimental to the healing response. Many types of tissue injury are described below, with the commonality of each of the studies described being that PGE2 is produced following injury. For example, in DSS-induced colitis, PGE2 levels were elevated during the induction phase, at day 4 post-DSS initiation, that coincided with increased PLA2 and COX-2 expression, which was sustained for 2 wk (11). PGE2 levels were also increased following dermal excisional wounding, with the highest concentration observed days 1–3 following injury with levels remaining elevated above baseline through day 12 (12). Although these studies suggest PGE2 is important for the wound healing process, more details describing how PGE2 exerts its effects are necessary to fully understand the potentially beneficial and/or detrimental pathways induced by PGE2-EP signaling.
PGE2 MODULATES CELL PHYSIOLOGY
EP1 signaling and EP3 signaling have been shown to induce vasoconstriction, whereas EP2 signaling and EP4 signaling lead to vasodilation (13). In general, vascular alterations are necessary for tissue repair and these observations alone are not indicative of promoting or impeding wound healing. In the murine renal vasculature system, EP3 activation by PGE2 led to constriction of the vasculature and EP3-deficient mice showed a reduced ability to vasoconstrict (14). Interestingly, varying levels of PGE2 have been shown to regulate cerebral blood flow in a dichotomous fashion; low-level PGE2-induced EP4 activation led to vasodilation, whereas high-level PGE2-induced EP1 activation led to vasoconstriction in cerebral parenchymal arterioles, with both phenotypes inhibited by antagonism of EP4 and EP1, respectively (15). Further, in vascular smooth muscle cells, deletion or inhibition of EP4 led to increased vascular constriction and increased blood pressure following stimulation with angiotensin II, although EP4 agonism inhibited this phenotype (16). PGE2-EPR regulation of vascular flux is therefore necessary for the beginning stages of wound healing to prevent blood loss upon immediate injury and to allow for increased blood flow and immune infiltration at later time points. Pharmacological modulation of PGE2 concentration could provide a critical therapeutic benefit for individuals needing alterations in vascular pressure following injury.
PGE2 is also able to induce differentiation of cell subtypes. Tendon-derived stem cells isolated from rats that were treated with PGE2 display osteogenic differentiation by increasing protein content of bone-specific proteins, bone morphogenetic protein-2 (BMP2), runt-related transcription factor 2 (Runx2), osterix (OSX), and osteocalcin (OCN) along with increased alkaline phosphatase activity and calcium deposition (17). Additionally, in zebrafish intestinal tissue and human-derived intestinal organoids, PGE2 treatment increased mucus production, indicative of intestinal differentiation toward a goblet cell phenotype (18). PGE2 can also influence macrophage polarization and metabolism. In bone marrow-derived macrophages (BMDM), IL-4 and PGE2 cotreatment induced M2 macrophage polarization, an anti-inflammatory macrophage phenotype that is represented an increase by M2 markers, arginase (Arg1), mannose receptor C type 1 (Mrc1), resistin-like α (Fizz1), and Chitinase type protein 3 (Ym1) due to CREB-Kruppel-like factor 4 (KLF4)-mediated transcription (19). In addition, PGE2 treatment following IL-4-induced M2 macrophage polarization modulated metabolic pathways by reducing mitochondrial membrane potential and subsequent ATP production due to reduced transcription of malate-aspartate shuttle components that led to an overall decrease in intracellular reactive oxygen species (20). Further, in mice with ear hole punch injuries, EP3 and EP4 were shown to influence M2 macrophage recruitment and polarization, with EP3- and EP4-deficient mice exhibiting altered ratios of M1 to M2 macrophages (21). These studies illustrate how PGE2 treatment can alter specific cell physiology in a manner that could be either beneficial or detrimental to an injured site, depending on the context.
PGE2 IN PROMOTING WOUND HEALING
EP Receptor-Mediated Phenotypes
PGE2-EP receptor signaling positively influences wound healing by altering immune migration, increasing proliferation, and promoting survival. In a rodent model of myocardial infarction (MI), EP2 activation in macrophages was critical for macrophage recruitment to the injured site with EP2-deficient mice exhibiting increased infarct size, reduced immune cell mobilization, and poorer tissue regeneration than wild-type counterparts (22). In addition, mice deficient in EP3 or EP4 exhibit reduced wound closure following ear hole punch injury that correlated with reduced macrophage recruitment to the injured site, an overall reduction in M2 macrophages and a reduction in new lymph and blood vessel formation that coincided with reduced mRNA of vascular endothelial growth factor receptor-3 (VEGFR3) and lymphatic vessel endothelial hyaluronan receptor-1 (LYVE-1) in granulation tissue compared to wild-type mice (21). Corroborating these results, mice receiving bone marrow transplants exhibited increased hematopoiesis, chemokine production, colony formation, and immune cell homing posttransplant that correlated with increased survival, which was greatly reduced with either EP2 or EP4 antagonism (23). Interestingly, inhibiting EP4 signaling has also been shown to increase immune infiltration at sites of injury. Mice lacking EP4 on vascular smooth muscle cells (VSMCs) present with increased immune infiltration, aortic size, and oxidative stress following angiotensin II (ANG II)-induced damage to the vasculature (16). Similarly, in angioplasty wire-injured mice, EP4-specific deletion on endothelial cells increased immune infiltration and impeded tissue repair, whereas EP4 agonism improved endothelial repair and reduced neointima formation in wild-type mice (24). Likewise, zebrafish with amputated tail fins required EP4 signaling to eliminate neutrophils from the injured site to allow for continued tissue repair postinjury (25). These studies suggest a dual role for EP receptor signaling, especially EP4, in mediating immune infiltration and egress during tissue repair.
EP2 and EP4 also induce proliferation postinjury to aid in tissue regeneration. EP4 signaling following angioplasty wire injury was shown to be important for re-endothelialization because of EP4-cAMP-PKA-induced proliferation, which occurred to a lesser extent following EP2 activation (24). In mice with DSS-induced colitis, PGE2 treatment increased expression of EP4 that led to β-arrestin-PI3K-Akt signaling, which increased proliferation and reduced disease severity and apoptosis levels (26). Interestingly, samples from human colitis patients or DSS-treated mice show a reduction in EP4, COX-1, and PGE2 content but increased COX-2 levels compared to healthy controls (26). Together, these studies show the importance of EP4, and to a lesser extent EP2 signaling, in proliferation induction to allow for re-epithelialization at the injured site to enhance tissue repair.
Modulation of PGE2 Concentration
Modifying PGE2 levels via treatment with PGE2, PGE analogs, or by impeding PGE2 inactivation can improve wound healing phenotypes. Supporting this, mice deficient in the PGE2 inactivating enzyme, 15-PGDH, have elevated PGE2 levels in bone marrow and liver and have improved regenerative abilities and reduced markers of tissue damage following bone marrow transplantation, or hepatectomy-induced liver regeneration, respectively, that was also observed following pharmacological 15-PGDH blockade (23). In a mouse model of DSS-induced colitis, PGE2 treatment reduced symptom severity and apoptosis levels while increasing colonic repair and proliferation via upregulation of EP4-dependent β-arrestin-PI3K-Akt signaling (26). Corroborating these results, genetic or pharmacological inhibition of 15-PGDH led to increased colonic PGE2 content that correlated with reduced histological markers of colitis and increased proliferation of intestinal tissue 7 days following DSS treatment in mice (23). Further, in a zebrafish model of DSS-induced intestinal injury, PGE2 treatment increased mucus production, barrier integrity, autophagy and cell viability, reduced Escherichia coli uptake, and improved overall survival following injury (18). Interestingly, treatment of cutaneous wounds with PGE2-incorporated hydrogels improved speed of recovery by enhancing angiogenesis, reducing inflammation and immune infiltration, and shortening the time of wound closure (27). In addition, in mice with bleomycin-induced pulmonary fibrosis, 15-PGDH inhibition reduced markers of fibrosis, inflammatory cytokine expression in lung and serum, and improved airway function that correlated with increased survival (28). Finally, following angioplasty wire injury, treatment of mice with a PGE analog, misoprostol, increased re-endothelialization and improved healing, whereas loss of mPGES-1 blocked tissue repair (24). Combined, these studies illustrate the essential role of PGE2-induced reparative tissue responses following injury and suggest that increasing PGE2 concentration at sites of damage may improve wound resolution.
PGE2 treatment can also alter macrophage polarization and metabolism, leading to an M2, anti-inflammatory macrophage phenotype (20), which has been observed in white adipose tissue in an obesity model (19), in skin following cutaneous wounding (22) and in microglia following traumatic brain injury (29). As mentioned previously, macrophages treated with PGE2 present markers of M2 macrophages, including Arg1, Mrc1, Fizz1, and Ym1 that is dependent on CREB-KLF4-induced transcription (19) and can modify mitochondrial membrane potential and transcription of malate-aspartate shuttle components to reduce ATP production (20). Interestingly, macrophage proliferation is influenced by PGE2 signaling in a dose-dependent manner, with the highest rate observed following 1 µM PGE2 treatment in vitro, that leads to M2 macrophage polarization and increased expression of arginase, interleukin (IL)-1 receptor a, IL-10, CD68, and CD206 with reduced M1 markers, IL-1β, IL-6, and TNFα (27). Mice with skin wounds that are treated with PGE2 incorporated hydrogels have increased M2 macrophages present at the site of injury that correlated with increased angiogenesis, faster wound closure, and reduced scarring (27). These signaling pathways are critical during a healing response, as the transition from a pro-inflammatory to an anti-inflammatory immune cell phenotype will directly dictate a tissue’s ability to begin the remodeling phase of the response.
Inhibiting PGE2 production with COX inhibitors can be detrimental to wound healing. After mouse ear hole punch injury, treatment with the selective COX-2 inhibitor, celecoxib, reduced wound closure due to decreased macrophage recruitment and M2 macrophage numbers, and reduced blood and lymph vessel formation at the injured site due to decreased transcriptional content of LYVE-1, VEGFR3, VEGF-C, and VEGF-D (21). In a rodent model of bone injury, treatment with the nonsteroidal anti-inflammatory drug, naproxen, led to a reduction in serum PGE2 levels, which correlated with poorer woven bone formation following mechanical loading, indicating a need for PGE2 signaling in bone remodeling postinjury (30). Together, these studies underline the importance of PGE2-mediated signaling following injury, but more studies defining the role of EP receptors in these responses would be highly beneficial.
PGE2 IN IMPAIRED WOUND HEALING
EP Receptor-Mediated Phenotypes
Regulating EP-mediated signaling is essential to allow for adequate repair following injury. Excessive EP2 signaling can increase cytokine production and lead to dysregulated inflammation. PGE2-EP2 signaling has been shown to impede blood-brain barrier integrity and exacerbate hippocampal injury induced by seizures by increasing oxidative stress and inflammatory cytokine and chemokine production, which was reversed by EP2 antagonism (31). EP2 blockade also reduced markers of gliosis and neuronal injury that correlated with improvements in behavior scores postinjury (31). Furthermore, following spinal cord injury, there is increased COX-2, mPGES-1, and PGE2 content in astrocytes and astrocyte-mediated PGE2 secretion increased production of pro-inflammatory cytokines IL-1β, and IL-6 in macrophages via EP2, which was inhibited by antagonizing EP2 or blocking COX-2 enzyme function (32). Conversely, in human epidermal keratinocytes, EP3 agonism reduced cytokine production following Toll-like receptor stimulation, which is thought to influence symptom severity in patients with toxic epidermal necrolysis, as there is typically reduced EP3 expression and concomitant chronic inflammation in conjunctival epithelial tissue of patients (33). Although cytokine production is generally necessary during an injury response, having excessive or dysregulated inflammation can exacerbate tissue damage and lead to inadequate repair. Modifying EP signaling to alter cytokine production may prove to be a valuable therapeutic option for highly inflamed tissues.
Modulation of PGE2 Concentration
Although PGE2 signaling has been found to be critical for adequate wound healing to occur in some injury models, there have also been situations where PGE2 signaling was detrimental to the healing response. In radiation-induced salivary gland damage, elevated PGE2 levels correlated with salivary gland dysfunction, whereas reduced PGE2 levels correlated with improved salivary gland function postdamage (34). In an impaired wound healing model utilizing LIGHT−/− mice, PGE2 levels were greatly elevated days 1–7 postdermal excisional wounding when compared to wild-type mice, which correlated with increased elastase activity, platelet aggregation, and reduced hemostasis following injury (12). In a rodent model of tendinopathy, PGE2 treatment increased the activity of the osteoblast marker alkaline phosphatase, leading to increased ossification of tendon-derived stem cells, which is indicative of poorer recovery. Interestingly, treatment with the COX-2-selective inhibitor, celecoxib, attenuated this phenotype and improved tendon repair post injury (17). Furthermore, reducing PGE2 levels via celecoxib treatment improved pressure ulcer healing by reducing iNOS, hydroperoxidase, matrix metalloprotease-1, and TNF-α levels. In addition, celecoxib treatment improved re-endothelialization, shortened time to wound closure, and strengthened scar formation postinjury (3). These studies suggest that fine-tuning of PGE2 signaling is necessary for sufficient healing to occur and that dysregulation of PGE2-mediated inflammation can impair healing following an injury. More detailed descriptions of the studies outlined above can be found in Table 1.
Table 1.
PGE2-EP receptor-mediated wound healing phenotypes in different damage models
| Model | EP Receptor | [PGE2] | Major Findings | Ref |
|---|---|---|---|---|
| PGE2 synthesis or signaling in humans | ||||
| Ex vivo human cerebral parenchymal arterioles | EP1EP4 | 1–10 µM PGE21–100 nM PGE2 |
|
(15) |
| In vitro human acute epidermal inflammation | EP3 | 100 µg/mL PGE2 |
|
(33) |
| Effects of PGE2 signaling on cell physiology | ||||
| Ex vivo and in vivo mouse renal arterial pressure | EP3EP2/EP4 | 0.3 µM PGE2 |
|
(14) |
| In vitro rat tendon-derived stem cells (TDSCs) | Not evaluated | 50 ng/mL PGE2 |
|
(17) |
| In vitro human-derived enteroid monolayers | Not evaluated | 1 µM PGE2 |
|
(18) |
| In vivo Zebrafish DSS-induced colitis | Not evaluated | 0.1-10 µM PGE2 |
|
(18) |
| In vitro mouse bone marrow-derived macrophage (BMDM) activation | Not evaluated | 10 µM PGE2 |
|
(20) |
| In vitro and in vivo mouse BMDM activation | Not evaluated | 10 nM PGE2 |
|
(19) |
| In vivo mouse ear-hole punch injury and macrophage polarization | EP3/EP4 | Not quantified |
|
(21) |
| PGE2 signaling leading to promotion of wound healing | ||||
| In vivo mouse myocardial infarction (MI) induced injury | EP2 | Not quantified |
|
(22) |
| In vivo mouse in vitro human DSS-induced colitis | EP4 | 20 µM PGE2 |
|
(26) |
| In vivo zebrafish tailfin amputation | EP4 | 0.001-1 µM PGE2 |
|
(25) |
| In vivo mouse angiotensin II (ANG II)-induced vascular damage | EP4 | Not quantified |
|
(16) |
| In vivo mouse angioplasty wire injury-induced vascular damage | EP2/EP4 | 100 µg/kg misoprostol (a PGE analog) |
|
(24) |
| In vivo mouse bone marrow alterations/efficiency of bone marrow transplantation | EP2/EP4 | 2–10 ng/mg protein PGE2 |
|
(23) |
| In vivo mouse DSS-induced colitis | Not evaluated | 10–30 ng/mg protein PGE2 |
|
(23) |
| In vivo mouse hepatectomy-induced liver regeneration | Not evaluated | 5–20 ng/mg protein PGE2 |
|
(23) |
| In vivo mouse ear-hole punch injury and macrophage polarization | EP3/EP4 | Not quantified |
|
(21) |
| In vivo mouse mechanical loading and stress fractured bone | Not evaluated | 0.75–1.25 pg/mL PGE2 in serum |
|
(30) |
| In vivo zebrafish DSS-induced colitis | Not evaluated | 0.1–10 µM PGE2 |
|
(18) |
| In vivo mouse skin excision wounding In vitro macrophage polarization |
Not evaluated | 0.5–2 µM; 0-1 ng/mL PGE2 |
|
(27) |
| In vivo mouse bleomycin-induced pulmonary fibrosis | Not evaluated | Not quantified |
|
(28) |
| In vitro mouse BMDM activation | Not evaluated | 10 µM PGE2 |
|
(20) |
| In vitro and in vivo mouse BMDM activation |
Not evaluated | 10 nM PGE2 |
|
(19) |
| PGE2 signaling leading to inhibition of wound healing | ||||
| In vivo mouse seizure-induced neuronal inflammation | EP2 | Not quantified |
|
(31) |
| In vivo rat spinal cord contusion injury | EP2 | 200–1,000 pg/mg protein; 100–6,000 pg/mL media; 0.1–10 µM PGE2 |
|
(32) |
| In vivo and in vitro rat Achilles tendinopathy | Not evaluated | 50 ng/mL PGE2 |
|
(17) |
| In vivo mouse cutaneous pressure ulcer wounding | Not evaluated | 200–500 pg/mg tissue PGE2 |
|
(3) |
| In vivo mouse DSS-induced colitis | Not evaluated | 35–75 pg/mg tissue PGE2 |
|
(11) |
| In vitro and In vivo mouse radiation-induced salivary gland damage | Not evaluated | 200–16,000 pg/mL media PGE2 |
|
(34) |
| In vivo mouse impaired dermal excisional wound healing | Not evaluated | Relative values; twofold to sixfold increase in WT, 1.3- to 1.8-fold increase in LIGHT−/− |
|
(12) |
| In vitro and in vivo mouse BMDM activation and high-fat feeding-induced insulin resistance | Not evaluated | 10 nM PGE2 |
|
(19) |
CONCLUSIONS
Taken together, these studies illustrate that PGE2 is a crucial eicosanoid in all three phases of the wound healing response. Regulated PGE2 signaling is important for proper repair to occur and treatment with nonsteroidal anti-inflammatory drugs may be detrimental to a healing tissue. More clearly deciphering the roles of upstream mediators of PGE2 production, including PLA2, COX-1, COX-2, mPGES-1, mPGES-2, and cPGES with future studies will help delinate the inflammatory pathways leading to elevated PGE2 levels and provide more direct therapeutic targets for modulating PGE2 production. Additionally, defining the regulation and activity of the PGE2 inactivating enzyme 15-PGDH will more precisely outline the timeframe that PGE2 is able to induce EP-mediated signaling and may unveil opportunities to activate or inhibit 15-PGDH to improve wound resolution. A better understanding of these components of the PGE2 pathway will allow for more precise drug targeting to modify PGE2-induced signaling.
EP receptor signaling plays a major role in dictating the transition from pro- to anti-inflammatory state, either allowing for or inhibiting completion of wound healing. The physiological outputs of EP receptor activation have been more clearly defined for EP2 and EP4 receptors, whereas the importance of EP1 and EP3 signaling is insufficiently studied in the context of wound healing (Fig. 1). Uncovering different components that regulate EP receptor distribution, such as signaling components that modulate transcription and/or translation, would be highly useful to better understand the importance of each EP receptor subtype. Finally, a clearer understanding how EP receptor activation is PGE2 concentration-dependent across isoforms and tissue types could help guide the use of NSAIDs or 15-PGDH inhibitors to modulate PGE2-mediated outputs. Further evaluation of EP-specific signaling and generation of EP-selective agonists and antagonists may provide novel therapeutic treatment options to enhance wound recovery following an injury.
GRANTS
The authors are supported by the National Institute of Dental and Craniofacial Research Grants R01DE029166 (to K. H. Limesand) and F31DE028737 (to K. E. Gilman).
DISCLAIMER
The funding agency had no role in preparation of this manuscript.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
K.E.G. drafted manuscript; K.E.G. and K.H.L. edited and revised manuscript; K.E.G. and K.H.L. approved final version of manuscript.
REFERENCES
- 1.Gonzalez ACdO, Costa TF, Andrade ZA, Medrado ARAP. Wound healing - A literature review. An Bras Dermatol 91: 614–620, 2016. doi: 10.1590/abd1806-4841.20164741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ricciotti E, FitzGerald GA. Prostaglandins and Inflammation. Arterioscler Thromb Vasc Biol 31: 986–1000, 2011. doi: 10.1161/ATVBAHA.110.207449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Romana-Souza B, Santos JS, Bandeira LG, Monte-Alto-Costa A. Selective inhibition of COX-2 improves cutaneous wound healing of pressure ulcers in mice through reduction of iNOS expression. Life Sci 153: 82–92, 2016. doi: 10.1016/j.lfs.2016.04.017. [DOI] [PubMed] [Google Scholar]
- 4.Park JY, Pillinger MH, Abramson SB. Prostaglandin E2 synthesis and secretion: the role of PGE2 synthases. Clinical Immunology 119: 229–240, 2006. doi: 10.1016/j.clim.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 5.Yao L, Chen W, Song K, Han C, Gandhi CR, Lim K, Wu T. 15-Hydroxyprostaglandin dehydrogenase (15-PGDH) prevents lipopolysaccharide (LPS)-induced acute liver injury. PLOS ONE 12: e0176106-e0176106, 2017. doi: 10.1371/journal.pone.0176106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, , et al. Proteomics. Tissue-based map of the human proteome. Science 347: 1260419, 2015. doi: 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- 7.Nakanishi M, Rosenberg DW. Multifaceted roles of PGE2 in inflammation and cancer. Semin Immunopathol 35: 123–137, 2013. doi: 10.1007/s00281-012-0342-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buchanan FG, Wang D, Bargiacchi F, DuBois RN. Prostaglandin E2 regulates cell migration via the intracellular activation of the epidermal growth factor receptor. J Biol Chem 278: 35451–35457, 2003. doi: 10.1074/jbc.M302474200. [DOI] [PubMed] [Google Scholar]
- 9.Sugimoto Y, Narumiya S. Prostaglandin E receptors. J Biol Chem 282: 11613–11617, 2007. doi: 10.1074/jbc.R600038200. [DOI] [PubMed] [Google Scholar]
- 10.Dennis EA, Norris PC. Eicosanoid storm in infection and inflammation. Nat Rev Immunol 15: 511–523, 2015. [Erratum in Nat Rev Immunol 15: 724, 2015] doi: 10.1038/nri3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hamabata T, Nakamura T, Masuko S, Maeda S, Murata T. Production of lipid mediators across different disease stages of dextran sodium sulfate-induced colitis in mice. J Lipid Res 59: 586–595, 2018. doi: 10.1194/jlr.M079095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dhall S, Wijesinghe DS, Karim ZA, Castro A, Vemana HP, Khasawneh FT, Chalfant CE, Martins-Green M. Arachidonic acid-derived signaling lipids and functions in impaired healing. Wound Rep and Reg 23: 644–656, 2015. doi: 10.1111/wrr.12337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hata AN, Breyer RM. Pharmacology and signaling of prostaglandin receptors: Multiple roles in inflammation and immune modulation. Pharmacol Ther 103: 147–166, 2004. doi: 10.1016/j.pharmthera.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 14.Liu B, Wu X, Zeng R, Yin Y, Guo T, Xu Y, Zhang Y, Leng J, Ge J, Yu G, Guo J, Zhou Y. Prostaglandin E(2) sequentially activates E-prostanoid receptor-3 and thromboxane prostanoid receptor to evoke contraction and increase in resistance of the mouse renal vasculature. FASEB J 34: 2568–2578, 2020. doi: 10.1096/fj.201901611R. [DOI] [PubMed] [Google Scholar]
- 15.Czigler A, Toth L, Szarka N, Szilágyi K, Kellermayer Z, Harci A, Vecsernyes M, Ungvari Z, Szolics A, Koller A, Buki A, Toth P. Prostaglandin E 2, a postulated mediator of neurovascular coupling, at low concentrations dilates whereas at higher concentrations constricts human cerebral parenchymal arterioles. Prostaglandins Other Lipid Mediat 146: 106389, 2020. doi: 10.1016/j.prostaglandins.2019.106389. [DOI] [PubMed] [Google Scholar]
- 16.Xu H, Du S, Fang B, Li C, Jia X, Zheng S, Wang S, Li Q, Su W, Wang N, Zheng F, Chen L, Zhang X, Gustafsson J-Å, Guan Y. VSMC-specific EP4 deletion exacerbates angiotensin II-induced aortic dissection by increasing vascular inflammation and blood pressure. Proc Natl Acad Sci USA 116: 8457–8462, 2019. doi: 10.1073/pnas.1902119116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou W, Lin X, Chu J, Jiang T, Zhao H, Yan B, Zhang Z. Magnolol prevents ossified tendinopathy by inhibiting PGE2-induced osteogenic differentiation of TDSCs. Int Immunopharmacol 70: 117–124, 2019. doi: 10.1016/j.intimp.2019.02.010. [DOI] [PubMed] [Google Scholar]
- 18.Chuang L-S, Morrison J, Hsu N-Y, Labrias PR, Nayar S, Chen E, Villaverde N, Facey JA, Boschetti G, Giri M, Castillo-Martin M, Thin TH, Sharma Y, Chu J, Cho JH. Zebrafish modeling of intestinal injury, bacterial exposures and medications defines epithelial in vivo responses relevant to human inflammatory bowel disease. Dis Model Mech 12: dmm037432, 2019. doi: 10.1242/dmm.037432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Luan B, Yoon Y-S, Lay JL, Kaestner KH, Hedrick S, Montminy M. CREB pathway links PGE2 signaling with macrophage polarization. Proc Natl Acad Sci USA 112: 15642–15647, 2015. doi: 10.1073/pnas.1519644112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sanin DE, Matsushita M, Klein Geltink RI, Grzes KM, van Teijlingen Bakker N, Corrado M, Kabat AM, Buck MD, Qiu J, Lawless SJ, Cameron AM, Villa M, Baixauli F, Patterson AE, Hässler F, Curtis JD, O'Neill CM, O'Sullivan D, Wu D, Mittler G, Huang SC, Pearce EL, Pearce EJ. Mitochondrial membrane potential regulates nuclear gene expression in macrophages exposed to prostaglandin E2. Immunity 49: 1021–1033.e1026, 2018. doi: 10.1016/j.immuni.2018.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hosono K, Isonaka R, Kawakami T, Narumiya S, Majima M. Signaling of prostaglandin E receptors, EP3 and EP4 facilitates wound healing and lymphangiogenesis with enhanced recruitment of M2 macrophages in mice. PLOS ONE 11: e0162532-e0162532, 2016. doi: 10.1371/journal.pone.0162532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu JMF, Cheng YY, Tang TWH, Shih C, Chen JH, Hsieh PCH. Prostaglandin E(2) receptor 2 modulates macrophage activity for cardiac repair. J Am Heart Assoc 7: e009216, 2018. [Erratum in J Am Heart Assoc 7: e002261, 2018]. doi: 10.1161/JAHA.118.009216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang Y, Desai A, Yang SY, Bae KB, Antczak MI, Fink SP, , et al. Tissue regeneration. Inhibition of the prostaglandin-degrading enzyme 15-PGDH potentiates tissue regeneration. Science 348: aaa2340, 2015. doi: 10.1126/science.aaa2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hao H, Hu S, Wan Q, Xu C, Chen H, Zhu L, Xu Z, Meng J, Breyer RM, Li N, Liu D-P, FitzGerald GA, Wang M. A protective role of microsomal prostaglandin E synthase-1 derived PGE2 and the endothelial EP4 receptor in vascular responses to injury. Arterioscler Thromb Vasc Biol 38: 1115–1124, 2018. doi: 10.1161/ATVBAHA.118.310713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Loynes CA, Lee JA, Robertson AL, Steel MJ, Ellett F, Feng Y, Levy BD, Whyte MKB, Renshaw SA. PGE(2) production at sites of tissue injury promotes an anti-inflammatory neutrophil phenotype and determines the outcome of inflammation resolution in vivo. Sci Adv 4: eaar8320, 2018. doi: 10.1126/sciadv.aar8320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Peng X, Li J, Tan S, Xu M, Tao J, Jiang J, Liu H, Wu B. COX-1/PGE2/EP4 alleviates mucosal injury by upregulating beta-arr1-mediated Akt signaling in colitis. Sci Rep 7: 1055, 2017. doi: 10.1038/s41598-017-01169-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang S, Liu Y, Zhang X, Zhu D, Qi X, Cao X, Fang Y, Che Y, Han Z-C, He Z-X, Han Z, Li Z. Prostaglandin E(2) hydrogel improves cutaneous wound healing via M2 macrophages polarization. Theranostics 8: 5348–5361, 2018. doi: 10.7150/thno.27385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smith JNP, Witkin MD, Jogasuria AP, Christo KF, Raffay TM, Markowitz SD, Desai AB. Therapeutic targeting of 15-PGDH in murine pulmonary fibrosis. Sci Rep 10: 11657, 2020. doi: 10.1038/s41598-020-68336-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu C, Fu F, Li X, Zhang S. Mesenchymal stem cells maintain the microenvironment of central nervous system by regulating the polarization of macrophages/microglia after traumatic brain injury. Int J Neurosci 127: 1124–1135, 2017. doi: 10.1080/00207454.2017.1325884. [DOI] [PubMed] [Google Scholar]
- 30.Park J, Fertala A, Tomlinson RE. Naproxen impairs load-induced bone formation, reduces bone toughness, and diminishes woven bone formation following stress fracture in mice. Bone 124: 22–32, 2019. doi: 10.1016/j.bone.2019.04.009. [DOI] [PubMed] [Google Scholar]
- 31.Jiang J, Yu Y, Kinjo ER, Du Y, Nguyen HP, Dingledine R. Suppressing pro-inflammatory prostaglandin signaling attenuates excitotoxicity-associated neuronal inflammation and injury. Neuropharmacology 149: 149–160, 2019. doi: 10.1016/j.neuropharm.2019.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang Y, Zhou Y, Chen S, Hu Y, Zhu Z, Wang Y, Du N, Song T, Yang Y, Guo A, Wang Y. Macrophage migration inhibitory factor facilitates prostaglandin E(2) production of astrocytes to tune inflammatory milieu following spinal cord injury. J Neuroinflammation 16: 85–85, 2019. doi: 10.1186/s12974-019-1468-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mieno H, Ueta M, Yamada K, Yamanaka Y, Nakayama T, Watanabe A, Kinoshita S, Sotozono C. Expression of prostaglandin E(2) receptor 3 in the eyelid epidermis of patients with Stevens-Johnson syndrome/toxic epidermal necrolysis. Br J Ophthalmol 104: 1022–1027, 2020. doi: 10.1136/bjophthalmol-2018-313587. [DOI] [PubMed] [Google Scholar]
- 34.Gilman KE, Camden JM, Klein RR, Zhang Q, Weisman GA, Limesand KH. P2X7 receptor deletion suppresses γ-radiation-induced hyposalivation. Am J Physiol Regul Integr Comp Physiol 316: R687–R696, 2019. doi: 10.1152/ajpregu.00192.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]