

Keywords: cell-signaling, immune cells, metabolism, SIRT6, Warburg effect
Abstract
The ability to ward off pathogens with minimal damage to the host determines the immune system's robustness. Multiple factors, including pathogen processing, identification, secretion of mediator and effector molecules, and immune cell proliferation and differentiation into various subsets, constitute the success of mounting an effective immune response. Cellular metabolism controls all of these intricate processes. Cells utilize diverse fuel sources and switch back and forth between different metabolic pathways depending on their energy needs. The three most critical metabolic pathways on which immune cells depend to meet their energy needs are oxidative metabolism, glycolysis, and glutaminolysis. Dynamic switching between these metabolic pathways is needed for optimal function of the immune cells. Moreover, switching between these metabolic pathways needs to be tightly regulated to achieve the best results. Immune cells depend on the Warburg effect for their growth, proliferation, secretory, and effector functions. Here, we hypothesize that the sirtuin, SIRT6, could be a negative regulator of the Warburg effect. We also postulate that SIRT6 could act as a master regulator of immune cell metabolism and function by regulating critical signaling pathways.
IMMUNE SYSTEM OVERVIEW
The immune system comprises a complex network of cells that communicate with each other to neutralize or eliminate pathogens and molecules that would, otherwise, damage our body. Different lines of defenses exist to combat the entry of a harmful intruder (Fig. 1). The first line of defense against pathogens comprises of our skin, mucous membrane, stomach acid, and other body secretions like tears and saliva (1). Pathogens breaching the first line of defense are met with the innate immune system, the second line of defense, which comprises of macrophages, dendritic cells, natural killer cells, monocytes, neutrophils, basophils, eosinophils, mast cells, and complement proteins (1). The innate immune system is often activated by molecules present on the pathogens such as bacterial lipopolysaccharide, bacterial cell wall components, and viral DNA/RNA, often referred to as pathogen-associated molecular patterns (PAMPs) (2). Alternatively, the host tissue damage activates the innate immune system by releasing endogenous danger molecules from the damaged or dying cells. These molecules are referred to as danger-associated molecular patterns (DAMPs). Both extracellular and intracellular components, which include elements of different cellular organelles, constitute DAMPs. Some examples of extracellular component DAMPs include biglycan, fibrinogen, and tenascin C; intracellular component DAMPs include S100 protein, heat shock protein (HSP), ATP, histones, HMGB1, DNA, RNA, mitochondrial DNA, defensins, and syndecans (3). Both PAMPs and DAMPs are recognized through pattern recognition receptors (PRRs) (4). Innate immune system activation by PAMPs and DAMPs causes inflammation by generating heat, redness, pain, swelling, and loss of tissue function, which is associated with vascular permeability changes, leukocyte recruitment, and accumulation and release of inflammatory mediators (5). All these processes result in the recruitment of more immune cells to the site of injury to defend the tissue against the pathogen.
Figure 1.

Barriers to infection: immune system has several components to block the entry of pathogens. These components fundamentally belong to three categories: anatomical and physiological barriers, innate immunity, and adaptive immunity. Components of physical barriers include skin, mucous membrane, stomach acid, and other body secretions like tears and saliva. The innate immune system is a nonspecific defense mechanism comprised of neutrophil, basophil, eosinophil, monocytes, and their differentiated forms. The adaptive immune system is specific for the pathogen and is comprised of the differentiated forms of lymphocytes.
All of the innate immune system components can interact with the third line of defense, referred to as the adaptive immune system or acquired immune system. This system relies on the precision and long memory of immune cells (6). The adaptive immune system comprises lymphocytes; B lymphocytes (B cells) and T lymphocytes (T cells) are components of the humoral and cell-mediated immunity, respectively. B cells originate and develop in the bone marrow, whereas precursors of T cells originate in the bone marrow but migrate to the thymus for further development (7). These two lymphocytes also differ in their mode of action. B cells secrete antibodies to target extracellular antigen, T cells target intracellular antigens. T lymphocytes are further categorized into T helper (Th) cells, or CD4+ cells, and cytotoxic T cells, or CD8+ cells. Th cells help in B cell activation, whereas CD8+ cells kill infected cells directly. Both CD8+ and CD4 + cells secrete cytokines, promoting the recruitment of macrophages and neutrophils to the site of infection (8).
The adaptive immune system works in tandem with the innate immune system to elicit a specific immune response against a foreign agent that enters our body. In short, dendritic cells engulf pathogens to form a phagosome (Fig. 2). The digestive enzymes present in the phagosome then degrade the protein into smaller peptide fragments, which are bound to MHC class II molecules and get displayed on the cell surface of the dendritic cells (9). Immune activation of Th cells by antigen-presenting cells (APC) is a multistep process requiring three essential events. First, a naïve Th cell that has receptors specific for the same antigen must come in contact with a dendritic cell presenting the antigen. Second, the costimulatory proteins, especially the B7 proteins (CD80 and CD86), on the surface of APC should be recognized by the co-receptor protein CD28 on the surface of the Th cell. Following this, as a third signal the antigen-presenting cells will start secreting cytokines needed to proliferate Th cells into effector Th cells and memory Th cells (Fig. 2). Like dendritic cells, B cells also act as antigen-presenting cells to Th cells via class II MHC (9, 10).
Figure 2.
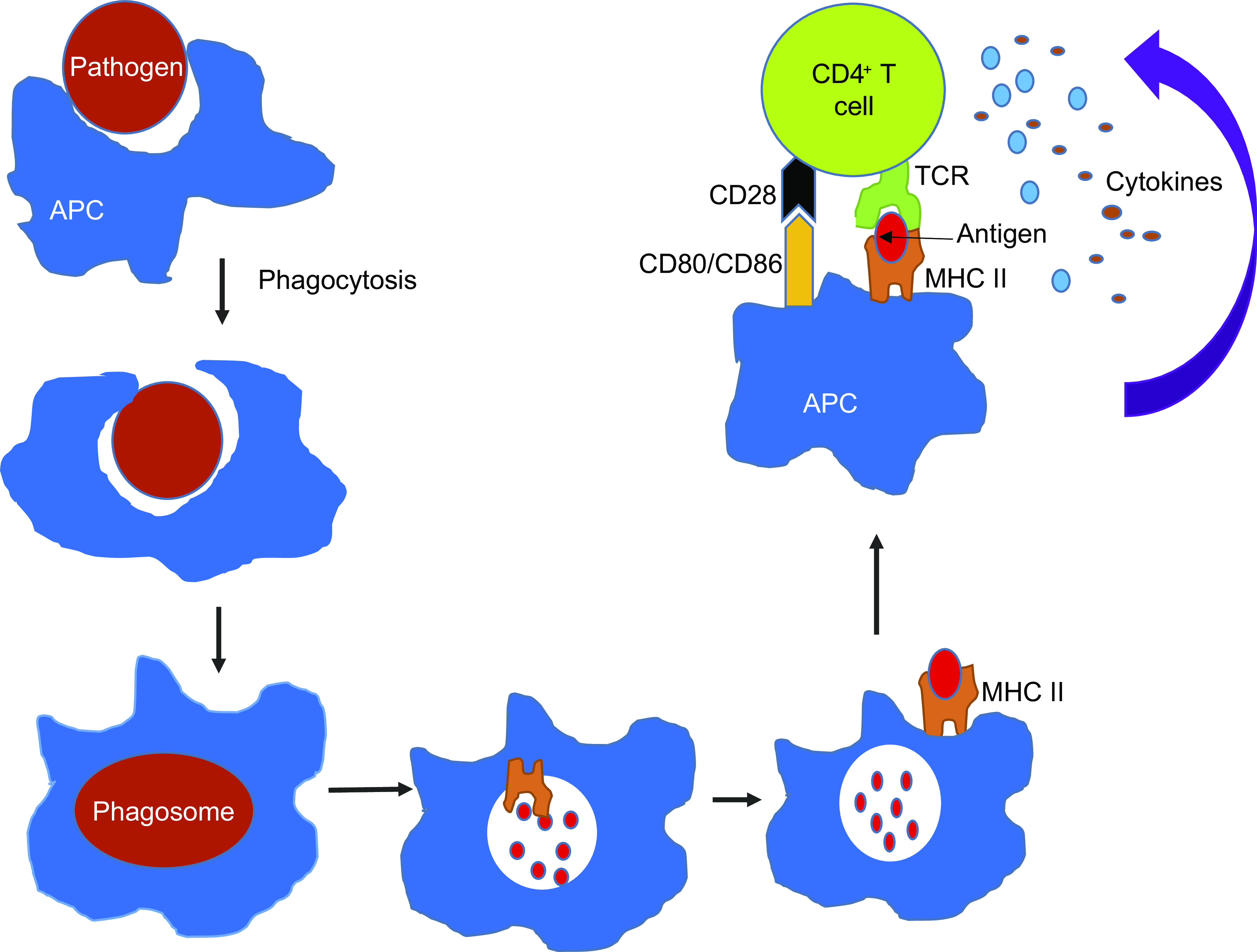
Activation of helper T cell by APC: antigen-presenting cell (APC) ingests the invading pathogen (red) and breaks them into peptides within the phagocytic vesicle. A fragment of foreign peptide gets associated with an MHC class II molecule and transported to the cell surface. CD4+ cell recognizes the antigen presented by APC via antigen-specific T-cell receptors (TCR), causing the intracellular signaling process activation. The costimulatory molecules present on the APC (CD80/CD86) interact with CD28 on the CD4+ cell and amplify the intracellular signaling process triggered by the TCR activation. These processes cause secretion of several different cytokines resulting in the proliferation and differentiation of CD4+ cells. APC, antigen-presenting cell; TCR, T-cell receptors.
In most cases, B-cell activation is dependent on Th cells. Similar to Th cell activation, B-cell activation is also a multistep process. First, an effector Th cell, specific for an antigen, should come in contact with a B cell displaying the same antigen. Second is the interaction between costimulatory molecules, CD40 ligand (CD40L) on the surface of Th cell with CD40 molecule on the surface of B cell. The third step is the secretion of cytokines by the Th cells. Activated B cells can then proliferate and differentiate into effector B cells and memory B cells (9).
Effector B cells are also called plasma cells, which secrete antibodies to neutralize the antigen. Depending on the type of pathogen and the cytokine milieu generated by antigen-presenting cells, T cells become activated and differentiate into functionally distinct T helper 1 (Th1), Th2, Th17, Th9, Treg, or T follicular helper (Tfh) cells (7). For example, viral infection favors the secretion of IFN-γ and IL-12, leading to development of Th1-polarized cells. Likewise, bacterial and parasitic infections result in the release of IL-17, leading to Th17-polarized cells (7).
IMMUNE RESPONSE AND METABOLISM
Immune cell function is intricately associated with metabolism (11). It is a two-way process, where an alteration in cellular metabolism affects immune cell response and vice versa (12). These interdependent effects are mediated through several hormones and are influenced by the nutritional status of the body. Both undernutrition and overnutrition have a deleterious effect on the immune system (13). For example, in obese mice, excessive TNF-α secretion from adipose tissue induces insulin resistance, and the deletion of TNF-α protects obese mice from developing insulin resistance (14, 15). Similar effects have been reported for IL-6, a proinflammatory cytokine secreted from immune and nonimmune cells (16). Leptin is another hormone that influences both cellular metabolism and immune cell function. Leptin-deficient mice are obese and have impaired immune response, characterized by reduced macrophagic activity and production of both IL-6 and TNF-α (17). Several other nutrition-associated hormones, including adiponectin, resistin, and visfatin, also play a critical role in immune cell regulation (13).
Overnutrition affects cells involved in both adaptive and innate immune responses (13). Obese individuals have a higher number of proinflammatory (M1) macrophages, whereas lean individuals have a higher number of anti-inflammatory macrophages (M2). Obese individuals also show an increased number of neutrophils and mast cells, but have fewer eosinophils and NK cells. In like manner, obese individuals also have a heightened number of adaptive immune cells (18, 19). Both T and B cells accumulate in adipose tissue to impose a proinflammatory environment with a concomitant reduction in regulatory T cells, a subset of T cells that maintain tolerance to self-antigens and prevent evolution of autoimmune diseases. In contrast, malnutrition is associated with a reduced number of immune cells. Malnourished children have decreased T and B lymphocytes with a generalized state of leukopenia (20).
Under basal conditions, innate and adaptive immune cells depend on oxidative phosphorylation to meet their energy demands. They use glucose or fatty acids to maintain homeostatic proliferation and survival (12). Upon activation by intrinsic or extrinsic factors, immune cells need a burst of energy for their growth, proliferation, secretory, and effector functions (21). Even though oxidative phosphorylation is the most efficient system to generate energy, it is a slow process. Therefore, once activated, immune cells rely on glycolysis and glutaminolysis to generate extra ATP and intermediary metabolites to meet their energy and substrate demand through a phenomenon called the Warburg effect, named after its discoverer, Otto Warburg, in 1924 (14). In the presence of oxygen, healthy cells break down glucose (glycolysis) into two molecules of pyruvate, yielding 2 ATP molecules. Further breakdown of pyruvate (tricarboxylic acid cycle (TCA) cycle + electron transport chain in mitochondria) into CO2 and water needs oxygen as an electron acceptor to recycle NADH to NAD+. Complete oxidation of glucose (glycolysis plus oxidative phosphorylation) will yield 36 molecules of ATP (22). When cells are deprived of oxygen, the pyruvate generated by glycolysis is converted to lactate with the recycling of NADH to NAD+. Lactate is secreted out of the cell into the bloodstream, whereas NAD+ sustains glycolysis. This method (glycolysis) of ATP generation is 10–100 times faster, but is only 5% efficient than the complete oxidation of glucose in the mitochondria (2 molecules of ATP vs. 36 molecules of ATP) (23). The Warburg effect is characterized by a profound increase in glycolysis, regardless of the presence of oxygen in the environment. Since proliferative cells cannot rely on glycolysis as the only source of energy to sustain their high energy demand, they shunt the by-products of glutaminolysis into the TCA cycle to maintain oxidative phosphorylation. The by-products of glutaminolysis also serve as substrates to synthesize macromolecules needed to construct new cells. Hence, the Warburg effect is also characterized by an increase in glutaminolysis (24).
Glutaminolysis refers to a series of biochemical reactions involved in glutamine's catabolism, the most abundant amino acid in human plasma (24). One of the by-products of glutaminolysis is α-ketoglutarate generated by the reversible transamination of glutamate, which enters the TCA cycle and progresses the TCA cycle blocked by increased glycolysis (Fig. 3) (24). Such reactions that help to replenish the TCA cycle intermediates are called anaplerotic reactions. α-Ketoglutarate entering into the TCA cycle will be further converted to malate, pyruvate, and lactate. Malate to pyruvate conversion in the presence of malate dehydrogenase generates NADPH, which is essential for nucleotide synthesis, lipid synthesis, redox balance, and cellular proliferation (24).
Figure 3.
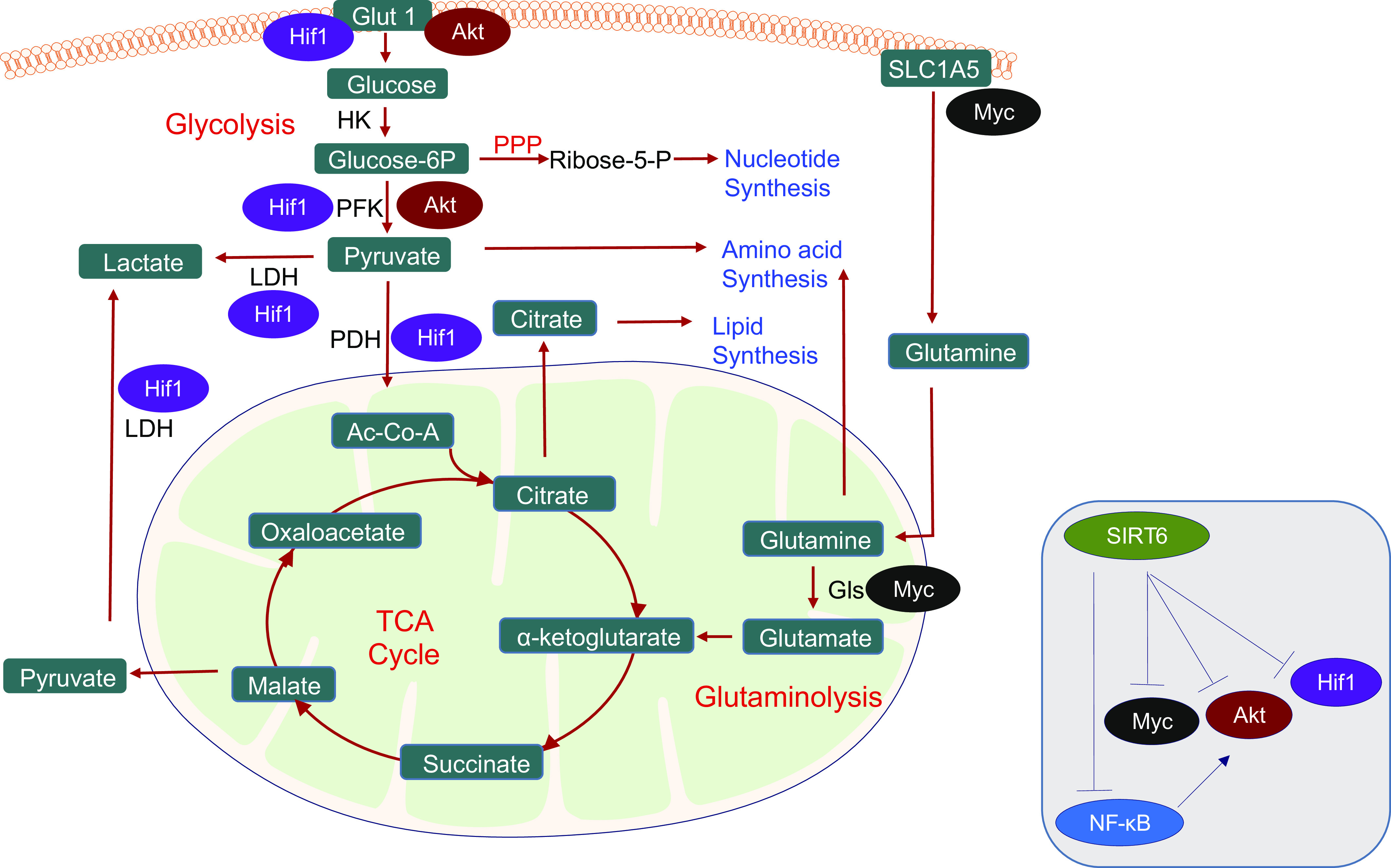
Important aspects of the Warburg effect: glycolysis, lactate fermentation, TCA cycle, pentose pyruvate pathway (PPP), and glutamine metabolism provide energy and substrates needed for cell proliferation and immune functions. Key metabolic pathways are colored red, and metabolites shown in green boxes. SIRT6 suppresses the activation of HIF-1α, PI3/Akt, NF-κB, and Myc signaling pathways involved in the metabolic shift from oxidative metabolism to glycolysis (inset). These signaling pathways target several enzymes involved in multiple metabolic pathways. HIF-1α activation induces the expression and translocation of the glucose transporter GLUT1 to the plasma membrane. Also, HIF-1α can increase the expression of lactate dehydrogenase, thus increasing the conversion of pyruvate to lactate. HIF-1α upregulates the expression of pyruvate dehydrogenase kinase (PDK), the upstream kinase of pyruvate dehydrogenase (PDH). PDK phosphorylates and inhibits PDH activity and represents another mechanism by which HIF-1α puts a brake on the TCA cycle. Similarly, Akt activation also activates several steps in the glycolysis pathway, including the expression and membrane translocation of the glucose transporter GLUT1. Akt also phosphorylates and activates phosphofructokinase-2 (PFK-2), generating fructose-2,6-bisphosphate (Fru-2,6-P2), which is an allosteric activator of the rate-limiting enzyme phosphofructokinase-1 (PFK-1), hence progressing glycolysis. NF-kB also contributes to the Warburg effect by promoting the Akt-induced translocation of GLUT1 to the plasma membrane. Myc plays a central role in the activation of glutaminolysis. Myc activates glutaminase, which converts glutamine to glutamate. Glutamate is then transaminated into α-ketoglutarate, which contributes to citrate and malate synthesis. Citrate and malate can be exported out of mitochondria to generate acetyl-CoA and pyruvate, respectively, along with NADPH. Further, Myc also upregulates the expression of glutamine transporters needed to import glutamine into the cell. Together, these metabolic pathways allow the cell to generate sufficient raw materials for the synthesis of lipids, amino acids, and nucleotides needed for the growth and proliferation of immune cells. PDH, pyruvate dehydrogenase; PDK, pyruvate dehydrogenase kinase; PFK-2, phosphofructokinase-2; PPP, pentose pyruvate pathway.
Furthermore, glutamine metabolism can generate oxaloacetic acid, which condenses with acetyl-CoA to form citrate in the mitochondria. Citrate can be exported out of mitochondria into the cytoplasm for lipid synthesis (25). Correspondingly, the TCA cycle and glutaminolysis intermediates provide fatty acids, such as acetyl-CoA, along with nitrogen molecules in nucleotides, amino acids, and amino sugars, which are necessary for cell growth, proliferation, and survival (26, 27). Likewise, glucose and glutamine together provide the carbon and nitrogen sources to synthesize all nonessential amino acids, except tyrosine (28). Reactions that utilize TCA cycle intermediates for their sustenance are termed as cataplerotic reactions. Most of the cellular signaling mechanisms in the Warburg effect converge into the following cell signaling pathways: PI3K/Akt, mTOR, HIF, NF-κB, and Myc, all of which are under the regulation of SIRT6, which is dependent on NAD+ for its activity (24, 29–33).
IMMUNE RESPONSE AND NAD+ LEVELS
NAD+ plays a central role in cellular metabolism by serving as an electron acceptor and transferring energy between different metabolic pathways, including fatty acid oxidation and TCA cycle (34). The generation and consumption of NAD+ determine cellular NAD+ levels in a cell (34). Several factors can cause reduced NAD+ levels in the cells. Oxidative stress induces the opening of connexin43 hemichannels allowing NAD+ to leak out of the cell (35). Another mechanism contributing to NAD+ depletion is the increased activity of NAD+-consuming enzymes, such as PARP1, CD38, and ADP-ribosyl cyclase (36). Further, cellular stress can inhibit the activity of NAD+ biosynthetic pathways, causing a depletion of intracellular NAD+ levels (36, 37).
The role of NAD+ in maintaining immunity became first evident from its role in curing pellagra by supplementing the diet with NAD+ precursor vitamin B3 (nicotinic acid or niacin) (38). Pellagra is characterized by depressed immune functions and frequent infections (39). Accumulating evidence suggests that NAD+ is a major modulator of immune metabolic circuits, playing a significant role in the host's innate and adaptive immune response (38, 40). The expression of Nampt (rate-limiting enzyme of the NAD+ salvage pathway) is elevated in activated T cells, neutrophils, and monocytes. Correspondingly, patients suffering from diseases with an autoimmune response, like rheumatoid arthritis, psoriasis, osteoarthritis, systemic lupus erythematosus, and inflammatory bowel disease, have high levels of circulating Nampt (41–47). Moreover, treatment with FK866, an inhibitor of Nampt, has the potential to attenuate the synthesis of several proinflammatory cytokines, including IL-6, TNF, and IFN-γ(48–50). Several studies also highlight the direct role of NAD+ in modulating immune response. Injection of mice with NAD+ facilitated the expansion of primed T cells in a model of acute inflammation induced by polyacrylamide beads (51). In another study, exogenous NAD+ induced CD4+ and CD8+ T cell apoptosis, but not B cells (52). Also, NAD+ can induce CD4 T-cell differentiation independent of MHC II and the T cell receptor signaling machinery (53). In this context, it is important to note that calorie restriction is associated with increased cellular NAD+ levels and calorie-restricted mice have reduced serum levels of IL-1β, TNF-α, and IL-6 and are resistant to LPS-induced hepatitis (54).
On the contrary, proliferating cells exhibit high ratios of ATP/ADP and NADH/NAD+, suggesting that NAD+-dependent enzymes may be downregulated during cell division (25, 55). These studies highlight NAD+'s complex role and the importance of NAD+ utilizing/synthesizing enzymes to regulate the immune response. Further, NAD+ levels in the cells influence the activity of NAD+-dependent enzymes. One class of NAD+-dependent enzymes that have a profound effect on the immune response is sirtuins (56).
Overview of Sirtuins
Sirtuins belong to a group of enzymes called histone/protein deacetylases, all of which are dependent on zinc for their deacetylase activity. There are four defined classes of histone deacetylases, and sirtuins are class III histone/protein deacetylases, which depend on NAD+ for their deacetylase activity (57). The silent information regulator 2 (Sir2) gene, a protein needed for the silencing of genes in specific loci, was first described in yeast and gained importance when it was found that Sir2 activity extends the lifespan of yeast in response to calorie restriction (58–61). The primary function of Sir2 is deacetylation, where it catalyzes the hydrolysis of acetyl-lysine in histones using NAD+ as a cosubstrate (57). There are seven mammalian sirtuin isoforms (SIRT1-SIRT7), and all of them share a catalytic domain similar to yeast Sir2 (62). Sirtuins are expressed ubiquitously in all the cells. They localize in different cellular compartments, including the nucleus, cytoplasm, and mitochondria. Though initially identified as histone deacetylases, later studies have revealed that sirtuins can deacetylate a variety of nonhistone proteins, which is why they are called protein deacetylases (63).
SIRT1 is the most studied among mammalian sirtuins and is primarily localized in the nucleus, but can also be transported to the cytoplasm and plasma membrane under stress conditions (64–66). Likewise, SIRT2 is a cytoplasmic sirtuin but can also localize to the nucleus (64). SIRT6 is exclusively localized in the nucleus, whereas SIRT7 is localized to the nucleolus (64). All the remaining sirtuins, including SIRT3, SIRT4, and SIRT5, are localized in the mitochondria (67). The primary function of SIRT1, SIRT2, SIRT6, and SIRT7 is to regulate gene expression by deacetylating intracellular signaling molecules and/or histones, whereas SIRT3, SIRT4, and SIRT5 regulate energy metabolism and oxidation-redox state of the cell in response to mitochondrial stress (68). The in vitro enzymatic activity varies drastically among different isoforms of sirtuins. Although SIRT1, SIRT2, and SIRT3 have robust deacetylase activity, SIRT4, SIRT5, SIRT6, and SIRT7 possess weak deacetylase activity (69). Sirtuins also show additional enzymatic activity other than deacetylation. All sirtuins except SIRT5 have mono-ADP-ribosyl transferase activity, whereas SIRT5 catalyzes the removal of succinyl, malonyl, and glutaryl groups from the protein targets (70). Since sirtuins are dependent on NAD+ for their enzymatic activity, any fluctuations in NAD+ levels affect their activity. An increase in NAD+ levels activates sirtuins, whereas a decrease in NAD+ levels suppresses their activity. Hence, sirtuins act as biosensors of the cellular NAD+ levels (71). SIRT6 is unique among sirtuins because of its ability to bind to NAD+ without an acetylated substrate (72).
Enzymatic Activity of SIRT6
SIRT6 is the chromatin-associated sirtuin implicated in numerous biological functions, including transcriptional repression, glucose homeostasis, DNA repair, telomeric function, cellular differentiation, mitosis, and meiosis (73). It is the only sirtuin shown to extend mammalian lifespan directly (74). Even though SIRT6 has mono-ADP-ribosylase activity, the most robust activity of SIRT6 is the deacetylation of histones. SIRT6 deacetylates histone H3 lysine 9 (H3K9), H3K18, and H3K56 to induce transcriptional repression. SIRT6 also can remove long-chain fatty acyl group from the lysine (75, 76). By recombinantly synthesizing histone H3 with fatty acyl lysine, Wang et al. found that SIRT6 actively removes acylation from H3K9, H3K18, and H3K27. SIRT6 shows relatively low activities toward H3K4 and H3K23, but sluggishly removes acylation at H3K14, H3K36, H3K56, and H3K79 (77). Besides histones, SIRT6 also deacetylates nonhistone proteins. The nonhistone protein substrates of SIRT6 include forkhead box protein O1, GCN5, pyruvate kinase M2, GATA binding protein 3, and Nampt (78–82). Unlike other sirtuins, SIRT6 has a unique structure that binds to NAD+ even in the absence of an acetylated substrate, making it an NAD+ sensor (72). Another feature that makes SIRT6 distinct from other sirtuins is its weak in vitro deacetylase activity. In vitro deacetylase activity of SIRT6 is 1000 times slower than other highly active members of the sirtuin family (72).
The weak in vitro deacetylase activity of SIRT6 led to the finding that long-chain free fatty acids (FFA) could enhance the deacetylation activity of SIRT6. Binding of free fatty acids, including myristic, oleic, and linoleic acids at physiological concentrations, increases the catalytic efficiency of SIRT6 by ∼35-fold (75). As mentioned before, SIRT6 also catalyzes long-chain free fatty acid deacylation (76). SIRT6 can hydrolyze long-chain fatty acyl groups several 100-fold more efficiently than hydrolyzing acetyl groups (76). In the context of immune functions, SIRT6 mediated lysine deacylation of TNF-α at K19, and K20 residues are necessary for its secretion (76). A separate study shows that TNF-α synthesis is increased in immune cells at increased intracellular concentrations of NAD+, and SIRT6 regulates TNF-α expression (50). These studies suggest a potential relationship between NAD+ metabolism, SIRT6 activity, and immune response.
SIRT6 and Immune Response
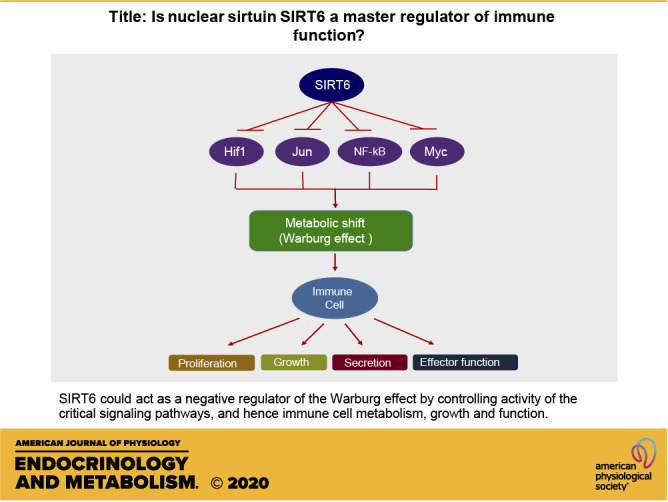
Metabolism fuels all cellular processes. Three metabolic pathways crucial for immune cell function are glycolysis, glutaminolysis, and oxidative phosphorylation (83). These pathways are interconnected by sharing or generating substrates for each other to meet the bioenergetic needs of the cells. Moreover, immune cells have to switch back and forth between different metabolic pathways to utilize different metabolic substrates (12). This dynamic switching of metabolic pathways needs to be tightly regulated for mounting an efficient effector response, and for the generation and maintenance of memory cells once the infection is cleared (21). High SIRT6 expression levels are observed in the organs and cells related to the immune system, including the thymus, appendix, spleen, lymph node, tonsil, bone marrow, and white blood cells (84). SIRT6 regulates the critical signaling pathways involved in glucose and glutamine metabolism, including PI3K/Akt/mTOR, HIF-1α, NF-κB, and cMyc (Fig. 4) (29–33). For this reason, SIRT6 can be considered as a master regulator of immune cell function. SIRT6 can exert a short-term or long-term effect on metabolism. Post-translational modifications of substrates exert short-term control, and long-term control is achieved by transcriptional regulation of the metabolic genes. Here, we will discuss the established mechanisms through which SIRT6 regulates metabolism and, hence, possibly controls immune response.
Figure 4.

Role of SIRT6 in transcriptional regulation: Under basal conditions, SIRT6 deacetylates histone H3K9 causing chromatin condensation leading to suppression of binding of transcription factors, NF-κB, HIF-1α, Jun, and Myc to their target gene promoters. Since these factors positively regulate IGF-Akt signaling, cytokine synthesis, and metabolic shift from oxidative phosphorylation to the Warburg effect characterized by increase in glycolysis and glutaminolysis, SIRT6 can block these processes and thus metabolic reprogramming in immune cells by blocking transcriptional activity of these factors.
One of the components of the innate immune system is neutrophils, which are short-lived, terminally differentiated, and most abundant leukocytes (85). At the basal level, neutrophils mostly depend on glucose metabolism for ATP production via aerobic glycolysis (85). But upon activation, neutrophils depend on the Warburg effect and pentose phosphate pathway (PPP) to generate NADPH by reducing NADP+, for their phagocytic and bacterial killing ability (83). Neutrophils neutralize pathogens via rapid release of reactive oxygen species (superoxide anion and hydrogen peroxide), referred to as oxidative burst (85). Oxidative burst is dependent on the production of NADPH via the PPP pathway. The PPP shunt utilizes the glycolytic intermediate glucose-6-phosphate to generate ribose-5-phosphate and NADPH in the cytosol. The enzyme NADPH oxidase (Nox) catalyzes the transfer of electrons from NADPH to molecular oxygen via their “Nox” catalytic subunit, generating microbicidal products, superoxide anion O2•− and H2O2 (86). After their oxidative burst and clearance of infection, neutrophils initiate apoptosis by releasing cytochrome-c to avoid collateral damage to tissues (87). Yet, another mechanism that neutrophils utilize to neutralize pathogens is called neutrophil extracellular traps (NETs) (88, 89). NETs are large, extracellular, Web-like structures comprised of nuclear and granular components assembled on a scaffold of decondensed chromatin. It acts as a trap where antimicrobial components accumulate to neutralize and kill bacteria, fungi, viruses, and parasites, thus preventing their dissemination (89). Both ROS and NET formations are dependent on the Warburg effect (90). Other granulocytes, including eosinophils and basophils, are considered metabolically similar to neutrophils because of their scanty mitochondria and terminally differentiated status (83).
Multiple studies have indicated the possible role of SIRT6 in neutrophil deactivation, a necessary step to prevent postinfection tissue damage. Fibroblast-like synoviocytes overexpressed with SIRT6 showed reduced secretion of epithelial neutrophil-activating peptide-78, a polymorphonuclear leukocyte (PMN) chemotactic cytokine, suggesting that SIRT6 has the potential to reduce neutrophil-induced inflammation (91). Similarly, SIRT6 overexpression in mice reduced TAC (transverse aortic constriction)-induced neutrophil infiltration in the heart (92). However, the direct effect of SIRT6 overexpression on neutrophil activity has not been studied so far. Since neutrophils are dependent on glycolysis and glutaminolysis for their phagocytic and bacterial killing ability, and SIRT6 is a key regulator of this metabolic switch, it is plausible that SIRT6 could be playing a key role in neutrophil function. Both ROS and NETs are considered as double-edged swords of innate immunity. Excessive ROS and NET formations are detrimental to the organism, as it leads to excessive inflammation (89). Since SIRT6 is a suppressor of the Warburg effect, it is plausible that activation of SIRT6 causes decreased bacterial killing but prevents excessive inflammation, which is often responsible for the fatal outcome (93, 94). The consequences of SIRT6 activation on neutrophils in containing infection and inflammation need to be tested.
Dead neutrophils are cleared by macrophages, cells differentiated from monocytes (95). Macrophages are large, specialized cells that recognize, phagocytose, and destruct target cells and pathogens. Monocytes can differentiate into two distinct forms, M1 and M2 macrophages. M1 macrophages are proinflammatory and are induced by intracellular pathogens, bacterial cell-wall components, lipoproteins, and cytokines such as interferon-gamma (IFN-γ) and tumor necrosis factor α (TNF-α) (96). M2 macrophages are anti-inflammatory and are induced by several factors, including parasites, immune complexes, complement proteins, apoptotic cells, macrophage colony-stimulating factor (MCSF), interleukin-4 (IL-4), IL-13, IL-10, and transforming growth factor-beta (TGFβ) (96). These two macrophages also differ in their energy source preferences. M1 macrophages depend on glycolysis for ATP generation, whereas M2 macrophages prefer fatty acid oxidation and, consequently, have higher mitochondrial mass and mitochondrial respiration (83). Several studies underscore the role of SIRT6 in macrophage polarization.
Adipose tissue-specific SIRT6 knockout mice fed with standard chow showed increased body weight and glucose intolerance, and exhibited systemic insulin resistance. Consequently, SIRT6 deletion accelerated M1 macrophage infiltration into the white adipose tissue (WAT), whereas the presence of SIRT6 in adipocytes increased the production of the cytokine IL-4 to drive M2 polarization in a paracrine manner (97, 98). There are also reports showing the role of SIRT6 in regulating fatty acid metabolism, necessary for M2 macrophage maintenance. Naiman et al. (99) reported that SIRT6 activates PPAR-α to promote fatty acid β-oxidation and inhibit pyruvate oxidation during fasting in the liver . PPAR-α regulates mitochondrial β-oxidation, peroxisomal β-oxidation, acyl-CoA binding/hydrolysis, lipid storage/transport, and ketogenesis. These studies suggest a potential role of SIRT6 in maintaining fatty acid oxidation as an energy source in M2 macrophages.
A direct role of SIRT6 in macrophage polarization was determined by knocking out SIRT6 in myeloid cells. Myeloid cell-specific SIRT6-deficient mice exhibited delayed wound healing with reduced collagen deposition, suppressed angiogenesis, and reduced expression of wound healing-related genes, compared to the wild-type mice (100). These changes were associated with increased infiltration of M1 macrophages and decreased M2 macrophages due to impaired Akt signaling. In this context, it is important to note that macrophages also function as antigen-presenting cells and are critical for the activation and proliferation of T and B lymphocytes, thereby acting as a bridge between innate and adaptive immune systems (101). Like neutrophils, macrophages also depend on the Warburg effect for their activation (102). Inhibition of the Warburg effect by SIRT6 may lead to decreased aggregation, motility, and bacterial killing of the macrophages, but it also may protect the organism from excessive inflammation. In line with this, Imtiyaz et al. (103) observed that mice lacking HIF-2α (an activator of glycolysis) in myeloid cells are resistant to lipopolysaccharide-induced endotoxemia and display a marked inability to mount inflammatory responses to cutaneous and peritoneal irritants leading to enhanced survival of HIF-2α deleted micet. Akin to macrophages, dendritic cells (DC), the key antigen-presenting cells, also depend on a similar mechanism for their activation (102). Inhibition of glycolysis in DC causes impaired DC migration and maturation (104). Defective DC maturation is characterized by lower expression of costimulatory molecules (CD80 and CD86), leading to ineffective T-cell activation, suggesting poor pathogen clearance and reduced inflammation (105, 106). Overall, activation of HIF-1α is essential for myeloid cell-mediated inflammation (107). The role of SIRT6 in the balance between pathogen clearance and undesirable inflammation needs to be investigated.
In the same line, both T-cell and B-cell proliferation and effector functions are also heavily dependent on the Warburg effect. Glycolytic activity is different in different T-cell subsets. Blocking of glycolysis inhibited Th17 development while promoting Treg cell generation, suggesting a complex role played by the Warburg effect in T-cell differentiation and function (108). Contrary to myeloid cells, in the cecal ligation and puncture (CLP)-induced bacterial sepsis model, T cell-specific deletion of HIF-1α resulted in higher levels of proinflammatory cytokines, stronger antibacterial effects of T cells and granulocytes, and better survival of mice (109). Similarly, HIF-1α-mediated activation of glycolysis promotes the expression of anti-inflammatory cytokine IL-10; the reduction of IL-10 producing B cells exacerbates collagen-induced arthritis and experimental autoimmune encephalomyelitis in mice (110). Further, glycolysis is positively associated with expression of CD11b, which has been shown to inhibit T-cell activation and differentiation as well as maintain autoreactive B cell tolerance (111, 112). These studies suggest an anti-inflammatory role of the Warburg effect in T-cell and B-cell function. Although, at present, little is known about the role of SIRT6 in adaptive immune response, we know a great deal about the immune response pathways regulated by SIRT6.
MAJOR IMMUNE RESPONSE PATHWAYS REGULATED BY SIRT6
HIF-1α
Increased metabolic demand of immune cells following pathogen activation requires an increase in oxygen consumption. Increased glycolysis also causes an increase in microenvironmental pH due to increased lactate production (113). This alteration in the metabolic environment is associated with activation of the transcription factor hypoxia-inducible factor (HIF) in the immune cells (113). HIF is a dimer protein composed of an α-subunit and a β-subunit. The stability of HIF-1α is oxygen-dependent. Under normoxic conditions, HIF-1α subunits are hydroxylated at multiple prolyl residues by the prolyl hydroxylase domain (PHD) enzymes and are targeted for proteasome-mediated degradation by Von Hippel–Lindau (pVHL), an E3 ubiquitin ligase-mediated ubiquitination (113). During hypoxia, lack of nonmitochondrial oxygen inactivates the PHD enzymes, thereby stabilizing HIF-1α. Stabilization of HIF-1α causes an increase in GLUT1 expression, the primary glucose transporter in immune cells, and also of many other genes involved in the Warburg effect, including lactate dehydrogenase (LDH), phosphoglycerate kinase (PGK-1), glucose-6-phosphate isomerase (GPI), and phosphofructokinase-1 (PFK-1) (114). Another mechanism by which HIF-1α puts a brake on the TCA cycle is by upregulating the expression of pyruvate dehydrogenase kinase (PDK) (115, 116). PDK-mediated phosphorylation inactivates pyruvate dehydrogenase (PDH), a rate-limiting enzyme that converts pyruvate to acetyl-CoA to fuel the TCA cycle. Furthermore, HIF-1α can inhibit the expression of cytochrome oxidase subunit Cox4-1 and the coactivator PGC-1β, thus diminishing mitochondrial activity (117). Other HIF-1α targets that are crucial for pathogen killing include the expression of several antimicrobial factors, such as serine proteases, neutrophil elastase, cathepsin G, cathelicidin-related antimicrobial peptide, and inducible nitric oxide synthase (118).
SIRT6 antagonizes HIF-1α activity by multiple mechanisms. SIRT6 acts as a corepressor of HIF-1α by deacetylating H3K9 at HIF-1α target gene promoters (33). Further, SIRT6 decreases gene expression, protein synthesis, and protein stability of HIF-1α (33, 119). Although no direct interaction and deacetylation of HIF-1α by SIRT6 have yet been reported, several mechanisms account for the destabilization of HIF-1α. High pyruvate and lactate levels during the Warburg effect promote pseudohypoxia, leading to the stabilization of HIF-1α (120). Further Krebs cycle metabolites, including succinate and fumarate, can leak out of the mitochondria and into the cytosol, inhibiting PHD enzymes' activity, resulting in the stabilization of HIF-1α (121). Increased levels of α-ketoglutarate, another TCA cycle intermediate, can also destabilize HIF-1α (122). These observations suggest that SIRT6-mediated regulation of the Warburg effect and preservation of mitochondrial function could be one mechanism by which SIRT6 destabilizes HIF-1α. Yet, another mechanism could be the altered NAD+/NADH levels (123). Increased NAD+/NADH levels activate several other sirtuins that can regulate HIF-1α stability. SIRT2 can directly interact and deacetylate HIF-1α to increase the binding affinity of HIF-1α for PDH (123). SIRT3 decreases the cellular ROS levels to destabilize HIF-1α (124). In contrast, the role of SIRT1 in HIF-1α stability is ambiguous, with SIRT1 having no effect on HIF-1α or serving as a positive or negative regulator depending on the cellular conditions (125–128).
HIF-1α also plays a crucial role in apoptosis. However, according to the cell type and experimental conditions, the role of HIF-1α in apoptosis is again context-dependent (129). HIF-1α induces apoptosis during severe or prolonged hypoxia, but shows prosurvival effect by activating the anti-apoptotic pathways in other conditions (129). Similar to HIF-1α, the effect of SIRT6 on apoptosis is also context-dependent, although, by convention, SIRT6 is an anti-apoptotic molecule (130). Accordingly, SIRT6-deficient mice show enhanced lymphocytic apoptosis, suggesting the anti-apoptotic role of SIRT6 in immune cells (131). These observations propose that SIRT6 may regulate the quantity and quality of the immune response by controlling the activity of HIF-1α. Further, activation of HIF-1α is associated with a proinflammatory phenotype in most of the immune cells, except B cells (88). Hence, it is plausible that activation of SIRT6 may overall have an anti-inflammatory effect on the organism. More studies are needed to understand the direct effect of SIRT6 on immune cell function in relation to HIF-1α.
PI3/Akt Pathway
The Ser and Thr kinase Akt, also known as protein kinase B (PKB), is one of the most frequently activated oncoproteins in human cancers (132, 133). Akt's role in metabolic shift through the Warburg effect is so pronounced that it is even referred to as the Warburg kinase (132). Akt activation can influence metabolic shift by increasing cellular ATP levels by twofold to threefold (132). This can be mediated through several mechanisms. In the immune cells, Akt can activate the expression and membrane localization of the glucose transporter GLUT1 (134). Akt also phosphorylates and activates phosphofructokinase-2 (PFK-2), generating fructose-2,6-bisphosphate (Fru-2,6-P2), which is an allosteric activator of the rate-limiting enzyme phosphofructokinase-1 (PFK-1), hence progressing glycolysis (135). In addition, Akt-mediated phosphorylation inhibits translocation of the transcription factor FoxO to the nucleus, and this event prevents the negative effect of FoxO on glycolytic gene expression (136). Besides, Akt-mediated phosphorylation increases mTORC1, a serine/threonine kinase activity, thereby increasing HIF-1α abundance and expression of HIF-1α-associated glycolytic enzyme and glucose transporter genes (137).
Akt and its downstream target mTORC1 are central to modulate cellular responses toward proliferation, migration, anti-apoptosis, and maintenance of metabolic homeostasis (138). Through mTORC1, Akt positively regulates immune activation in neutrophils, mast cells, monocytes, macrophages, and dendritic cells (139). Akt activation is necessary for the survival of activated neutrophils, which has a short life span of 6–10 h (140). Akt-induced phosphorylation of Gsk3β, Bax, Bad, or caspase-9 helps lose their proapoptotic potential, thus enhancing the lifespan of neutrophils (140). Furthermore, Akt also enhances the migration and cytokine secretion of the neutrophils. Like neutrophils, Akt can also improve cytokine secretions and survival of the macrophages (140).
Akt signaling also plays a central role in T-cell proliferation, function, and survival (141). Rapamycin, an mTOR inhibitor, promotes the development of regulatory T cells from naïve T cells, cells that modulate the immune system, maintain tolerance to self-antigens, and prevent autoimmune disease (142). This effect suggests that Akt activation plays a negative role in CD4+ T cell differentiation to Treg cells. However, mTOR activation is also necessary for the proper functioning of Treg cells, and loss of mTOR leads to a loss of Treg cell suppressive function and autoimmunity (143, 144). Since the development of CD8 memory cells is dependent on oxidative phosphorylation and autophagy, mTOR inhibition promotes the development of central CD8+ T memory (Tmem) cells (145). Like T cells, Akt also regulates the activation, proliferation, and survival of B cells (146). These studies suggest the crucial role played by Akt in immune cell activation and maintenance through metabolic regulation.
SIRT6 can regulate Akt activity at the transcriptional or post-translational levels. R-Ras2 lysine fatty acylation promotes the localization of R-Ras2 at the plasma membrane, promoting its interaction with phosphatidylinositol 3-kinase (PI3K) to activate Akt. SIRT6-mediated fatty acid deacylation prevents membrane localization of R-Ras2, thus blocking Akt activation (147).
Akt activation is essential for mediating effects of insulin. Insulin induces tyrosine phosphorylation of insulin receptor substrates (IRS), which then interact with PI3K to generate PIP3. PIP3, in turn, recruits Akt to the plasma membrane to get activated by PDK1 at T308 and mTORC2 at S473, resulting in a 1000-fold increase in Akt activity, from its basal level (148, 149). SIRT6 inhibits IR (insulin receptor), IRS1 (insulin receptor substrate 1), and IRS2 (insulin receptor substrate 2) to block Akt signaling. SIRT6 deficiency causes increased expression as well as increased phosphorylation of IR, IRS1, and total IRS2 levels following insulin treatment in the liver and muscle. This was associated with reduced expression of glucose transporters GLUT1 and GLUT4, suggesting the potential of SIRT6 to block immune cell activation by regulating Akt activation post-translationally (138). Consequently, SIRT6-deficient mice have increased AKT phosphorylation at Ser-473 and Thr-308 and are hypersensitive to LPS stimulation. These mice show chronic inflammation characterized by increased MCP-1, IL-6, and TNF-α expression levels in bone marrow-derived macrophages (138). Even though the exact mechanism by which SIRT6 regulates IRS phosphorylation is not known, loss of SIRT6 is associated with increased mRNA levels of key IGF-signaling-related genes including Igf1r, Igf2r, Insr, Irs2, Akt1, Akt3, Igf2, Mapk3, Ins2, Igfbp3, Pten, Gsk3b, FoxO1, and mTOR (31). In line with this, the increased median lifespan (15%) of male SIRT6.Tg mice is also attributed to alterations in IGF/Akt signaling-related genes (60). Likewise, Akt also antagonistically regulates SIRT6 protein levels. Akt-mediated phosphorylation of SIRT6 at Ser338 causes Mdm2-induced ubiquitination and subsequent proteasome-dependent degradation of SIRT6 (117). Further, SIRT6 regulates Akt at the transcriptional level too. SIRT6 physically interacts with c-Jun, a transcription factor for Akt, and gets recruited to the chromatin where it deacetylates H3K9 and inhibits the transcription of Akt (31). Accordingly, these observations suggest that the suppressive effect of SIRT6 on Akt could result from a combination of the regulation of Akt expression and activity (31, 150). It will be intriguing to see how SIRT6, Akt interplay, regulates immune cell function in different pathologies.
Together, these studies demonstrate that SIRT6 plays a pivotal role in regulating IGF/Akt signaling by establishing its control at both the transcriptional and post-translational levels. Since Akt is necessary for immune activation, its inhibition by SIRT6 may cause immune suppression. Rapamycin, an inhibitor of mTOR, has been shown to attenuate LPS-induced acute lung injury in mice (151). Further, rapamycin also protects mice from postsepsis cognitive impairment (152). These studies suggest that the immunosuppressive effect of SIRT6 can be utilized for developing therapeutic options against many immune system disorders.
In addition to SIRT6, other sirtuins have also been shown to play a role in regulating Akt activity. SIRT1 deacetylates Akt at the PH domain and enables the binding of Akt with PIP3 leading to membrane localization and activation of Akt (65). Similarly, SIRT2 also activates Akt via PH domain interaction (153). Cellular ROS activates Akt, and SIRT3 antagonizes Akt activity indirectly by suppressing ROS by enhancing mitochondrial antioxidant activity (154). Likewise, through unknown mechanisms, SIRT4 overexpression also suppresses Akt activity (155). Further, depletion of SIRT7 in breast cancer cells induced Akt hyperphosphorylation (156). Mechanistically, SIRT7-mediated deacetylation of the scaffolding protein FKBP51 enhances Akt's interaction with the phosphatase PHLPP leading to increased dephosphorylation of Akt (156). These studies show the complex role of sirtuins in Akt regulation and underscore the need for specific sirtuin inhibitors/activators in pharmacological interventions.
NF-κB Pathway
The PI3K/Akt pathway can also control cellular functions by interfacing with other signaling pathways, such as the nuclear factor κB (NF-κB) pathway (157). NF-κB, nuclear factor-κB, is a family of transcription factors regulating diverse cellular activities related to inflammation and innate and adaptive immune responses. The NF-kB family is composed of five structurally related members, including NF-κB1 (p50), NF-κB2 (p52), RelA (p65), RelB, and c-Rel. These members can form various homo- and heterodimers, and mediate target gene transcription by binding to a specific DNA element, the κB enhancer. The common form of NF-κB is a heterodimer, p65/RelA-p50. Under basal conditions, p65/RelA-p50 is sequestered in the cytoplasm by the inhibitor of κB (IκB) (158). Upon stimulation, IκB is phosphorylated at critical serine residues by a multi-subunit IκB kinase (IKK) complex. This results in the polyubiquitination and degradation of IkB by the 26S proteasome, making p65/RelA-p50 free to translocate to the nucleus. AKT can also phosphorylate the p65 subunit of NF-κB and promote NF-κB translocation to the nucleus (159). In the nucleus, NF-kB binds to specific NF-κB-sites in the enhancer regions of target genes and induces the transcription of several proinflammatory cytokines and chemokines including IL-1, IL-6, IL-8, IL-12, TNF-α, and chemokines, including MCP1, IL18, RANTES, MIP-2, CxCl1, and CXCL10 (152).
NF-κB can also regulate cellular metabolism by regulating the activity of pyruvate kinase (PK), which catalyzes the rate-limiting step of glycolysis by converting phosphoenolpyruvate and adenosine diphosphate (ADP) to pyruvate and adenosine triphosphate (ATP) (160). Pyruvate kinase isoenzyme type M1 (PKM1) and pyruvate kinase isoenzyme type M2 (PKM2) are two isoforms of PK. High ratios of PKM2/PKM1 promote glycolysis, but inhibit oxidative phosphorylation (161, 162). NF-kB contributes to the Warburg effect by the upregulation of PKM2 and promoting the AKT-induced translocation of GLUT1 to the plasma membrane (163, 164). PKM2 secreted from the cells can also induce colon cancer cell migration via PI3K/Akt activation (165). NF-kB also has a central role in regulating the expression of molecules involved in apoptosis, cell cycle regulation, and adhesion molecules (152).
SIRT6 attenuates the hyperactivation of the NF-κB pathway in two ways. First, SIRT6 physically interacts with the NF-kB RelA subunit and deacetylates histone H3 lysine 9 (H3K9) at the NF-kB target gene promoters (29). In the second mechanism, SIRT6 transcriptionally upregulates the expression of NF-κB inhibitor, IkB. Here, SIRT6 induces cysteine monoubiquitination of suppressor of variegation 3–9 homolog 1 (SUV39H1), a histone methyltransferase, enabling SUV39H1 dissociation from the NF-κB1a promoter. SUV39H1 is an inhibitor of gene transcription. Dissociation of SUV39H1 enables the transcription of IκB, which causes the relocalization of NF-κB to the cytoplasm (166). Since NF-κB is involved in the synthesis of numerous proinflammatory cytokines and chemokines, SIRT6 activation could result in the suppression of proinflammatory response. In sepsis models, suppression of NF-κB decreases the acute inflammatory process and organ dysfunction (167, 168). Similarly, deregulated activation of NF-κB is also associated with chronic inflammation of autoimmune diseases, suggesting the potential role of SIRT6 to prevent excessive immune activation (169).
Other sirtuins, including SIRT1 and SIRT2, also regulate NF-κB activity. Both SIRT1 and SIRT2 can interact with and deacetylate the p65 subunit to inhibit the NF-κB complex activity (170, 171). SIRT1-mediated deacetylation also enhances the ubiquitination-mediated degradation of NF-κB (151). Thus, when SIRT1 and SIRT2 modulate NF-κB activity by post-translational modification, SIRT6 regulates NF-κB activity by decreasing the promoter accessibility to p65, suggesting that sirtuins alleviate NF-κB-driven inflammatory and metabolic disorders via different mechanisms. The compensatory role of SIRT6 in regulating NF-κB in SIRT1-deficient macrophages has also been demonstrated, highlighting the complementary nature of SIRT1 and SIRT6 in regulating NF-κB activity (172). Further studies are needed to understand the direct consequence of SIRT6 activation on NF-κB signaling in immune cells.
Myc Pathway
Another pathway that is considered a global regulator of immune response is Myc proto-oncogene (myelocytomatosis oncogene) (173). Emerging evidence suggests that Myc family members play essential roles in regulating the development, differentiation, and activation of immune cells (173). Myc is a transcription factor regulating cell cycle, cell growth, cell survival, metabolism, protein synthesis, and differentiation (174, 175). Myc is one of the key players that coordinates metabolic reprogramming and immune function (176). Myc is essential for maintaining the quantity and quality of innate immune cells, including NK cells, dendritic cells, macrophages, and neutrophils (173). In B cells, Myc mediates B-cell receptor (BCR)-triggered cell proliferation that is necessary for clonal expansion (177). Further, Myc-deficient B cells are resistant to spontaneous and induced cell death, highlighting the critical role of Myc in B cell-mediated immune response (178). Similarly, Myc is essential for activation, proliferation, differentiation, and survival of T cells (173). Overall, these studies imply that Myc is a global regulator of the immune response.
Myc exerts its effect by regulating immune cell metabolism (173). Myc promotes the expression of several genes involved in glycolysis and PPP (179). Apart from that, Myc also plays a crucial role in glutamine metabolism for sustaining the Warburg effect. Myc regulates the absorption and metabolism of glutamine (180, 181). Glutamine transport is bidirectional. When glutamine is transported into the cell, a portion of it is also exported out of the cell (182). The amino acid transporter SLC1A5 (ASCT2) is responsible for the import of glutamine into the cell, whereas SLC7A5 is responsible for glutamine export to outside of the cell (182). Myc activation increases the expression of SLC1A5 and SLC7A5, resulting in the increased uptake of glutamine (180). Myc also induces expression of the enzyme glutaminase, which converts glutamine to glutamate. Glutamate can be incorporated into glutathione and nonessential amino acid biosynthesis pathways, or it can be deaminated to yield α-ketoglutarate an important energy source in the TCA cycle (180). Further, Myc regulates the expression of several enzymes involved in nucleotide biosynthesis using glutamine, thus promoting immune cell activation and proliferation (28, 183).
All of the sirtuins (SIRT1-7) regulate glutaminolysis by utilizing one of the three mechanisms: (a) by directly regulating the activity of Myc (SIRT1, SIRT6, SIRT7), (b) by indirectly regulating Myc stability (SIRT2, SIRT3), and (c) by controlling the activity of enzymes involved in glutaminolysis (SIRT4, SIRT5). Multiple studies have shown that SIRT1 and Myc mutually cooperate (184–186). Myc induces SIRT1 expression by binding to the SIRT1 promoter, and SIRT1, in turn, deacetylates to increase Myc stability and activity (187, 188). SIRT7 inhibits Myc transcriptional activity by H3K18 deacetylation (189).
SIRT2 stabilizes Myc by suppressing the expression of NEDD4, an E3 ubiquitin-protein ligase involved in the degradation of Myc (190). On the other hand, SIRT3 regulates Myc stability by controlling the activity of PI3/Akt signaling. SIRT3 inhibits the activation of the PI3K/Akt pathway by decreasing the ROS levels. PI3K/Akt pathway regulates Myc activity via GSK3β and mTORC1 (191). Myc is targeted for ubiquitin-mediated proteasomal degradation by its phosphorylation at T58/62(191). T58 phosphorylation is mediated through GSK3β. Akt-mediated phosphorylation suppresses GSK3β activity and reduces Myc T58 phosphorylation, hence increasing Myc stability (192). Additionally, Akt can also stimulate mTORC1, which activates the phosphatase PP2A involved in T58/S62 dephosphorylation. Inhibition of Akt by SIRT3 can thus prevent Myc dephosphorylation at T58/S62 residues leading to enhanced Myc degradation (191).
SIRT4 and SIRT5 play contrasting roles in glutaminolysis. By ADP-ribosylation, SIRT4 inactivates glutamate dehydrogenase, thereby inhibiting the synthesis of α-ketoglutarate, thus suppressing the Warburg effect (193). Likewise, SIRT5 regulates glutamine metabolism by desuccinylating glutaminase, thereby protecting it from ubiquitin-mediated degradation (194). These studies reveal the contrasting roles of these two sirtuins; SIRT4 suppresses glutaminolysis, whereas SIRT5 activates glutaminolysis.
Myc, too, is under the regulation of SIRT6 (30). SIRT6 can physically interact with Myc, but this interaction does not affect the Myc expression or protein stability; rather, it affects Myc's transcriptional activity on the promoter regions of ribosomal protein genes, including Rpl3, Rpl6, Rpl23, and Rps15a (30). In other words, SIRT6, through its interaction with Myc, gains access to histones leading to H3K56 deacetylation, thereby reducing the transcriptional output of Myc. Further, SIRT6 also regulates the glutaminase expression, thus regulating glutaminolysis (30). Even though the direct role of SIRT6 in the regulation of Myc in immune cells is not studied, these studies provide compelling evidence that SIRT6 may be a master regulator of immune functions by controlling critical proteins involved in multiple metabolic and cell signaling pathways involved in immune cell regulation.
Summary and Future Perspectives
Immune cells utilize multiple energy sources for their metabolic needs. At the basal level, immune cells mostly prefer fatty acid oxidation and oxidative phosphorylation to maintain their homeostatic proliferation and survival [97]. Once activated, the immune cells require a burst of energy and substrates needed for their growth, proliferation, secretion of immune mediatory molecules, and effector functions. In the activated cells, the increased substrate, as well as rapid and excessive energy requirement, is met by a phenomenon referred to as the Warburg effect, characterized by an increase in glycolysis and glutaminolysis in the presence of oxygen. SIRT6 regulates cell signaling pathways involved in the metabolic switch from oxidative phosphorylation to glycolysis. The key cell signaling pathways involved in immune cell metabolism include PI3K/Akt, mTOR, HIF, NF-κB, and Myc. SIRT6 regulates all these pathways at the transcriptional and/or post-translational levels. However, very little is known about the direct role of SIRT6 in immune cell regulation. Even though the direct role of SIRT6 in macrophage differentiation and the indirect role of SIRT6 in neutrophil infiltration have been studied, its effect on other immune cells, including T and B lymphocytes, is lacking. Such studies are now a necessity when the whole world is fighting pandemics such as COVID-19. The fatality of many infectious diseases, including COVID-19, flu, and sepsis, is attributed to the “cytokine storm” where the immune system goes awry, resulting in an out-of-control inflammatory response (122). Cytokine storm is also reported in many noninfectious diseases, including pancreatitis, multiple sclerosis, graft versus host disease, and multiple organ dysfunction syndromes. SIRT6, by negatively regulating the Warburg effect, may help in the resolution of heightened immune activation. It is important to study whether SIRT6 plays a role in regulating the cytokine storm, which could have implications for saving lives.
In addition, SIRT6 is a molecule implicated in aging, and immune system dysregulation is a hallmark of aging. Aging is associated with dysregulated immune cell function and inflammation. The observation that SIRT6 has a tight regulatory effect on multiple cell signaling pathways that are needed for sustaining immune cell function and inflammatory response, suggests that pharmacological or genetic interventions aimed at regulating SIRT6 activity may prevent morbidity and mortality associated with inflammation and autoimmune diseases.
GRANTS
This work was supported by NIH RO1 Grants HL136712 and HL143488.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
V.B.P. drafted manuscript; M.P.G. edited and revised manuscript.
ACKNOWLEDGMENTS
We thank Samantha Hund for proofreading this article.
REFERENCES
- 1.Liaskou E, Wilson DV, Oo YH. Innate immune cells in liver inflammation. Mediators of Inflammation 2012: 1–21, 2012. doi: 10.1155/2012/949157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mogensen TH. Pathogen recognition and inflammatory signaling in innate immune defenses. CMR 22: 240–273, 2009. doi: 10.1128/CMR.00046-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Roh JS, Sohn DH. Damage-associated molecular patterns in inflammatory diseases. Immune Netw 18: e27–e27, 2018. doi: 10.4110/in.2018.18.e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Amarante-Mendes GP, Adjemian S, Branco LM, Zanetti LC, Weinlich R, Bortoluci KR. Pattern recognition receptors and the host cell death molecular machinery. Front Immunol 9, 2018. doi: 10.3389/fimmu.2018.02379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen L, Deng H, Cui H, Fang J, Zuo Z, Deng J, Li Y, Wang X, Zhao L. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 9: 7204–7218, 2018. doi: 10.18632/oncotarget.23208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Netea MG, Schlitzer A, Placek K, Joosten LAB, Schultze JL. Innate and adaptive immune memory: an evolutionary continuum in the host’s response to pathogens. Cell Host Microbe 25: 13–26, 2019. doi: 10.1016/j.chom.2018.12.006. [DOI] [PubMed] [Google Scholar]
- 7.Luckheeram RV, Zhou R, Verma AD, Xia B. CD4+T cells: differentiation and functions. Clinical and Developmental Immunology 2012: 1–12, 2012. 2012: 925135 doi: 10.1155/2012/925135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nicholson LB. The immune system. Essays Biochem 60: 275–301, 2016. doi: 10.1042/EBC20160017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chaplin DD. Overview of the immune response. J Allergy Clin Immunol 125: S3–S23, 2010. doi: 10.1016/j.jaci.2009.12.980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gutcher I, Becher B. APC-derived cytokines and T cell polarization in autoimmune inflammation. J Clin Invest 117: 1119–1127, 2007. doi: 10.1172/JCI31720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Domblides C, Lartigue L, Faustin B. Metabolic stress in the immune function of T cells, macrophages and dendritic cells. Cells 7: 68, 2018. doi: 10.3390/cells7070068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ganeshan K, Chawla A. Metabolic regulation of immune responses. Annu Rev Immunol 32: 609–634, 2014. doi: 10.1146/annurev-immunol-032713-120236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alwarawrah Y, Kiernan K, MacIver NJ. Changes in nutritional status impact immune cell metabolism and function. Front Immunol 9: 1055, 2018. doi: 10.3389/fimmu.2018.01055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science (New York, NY) 259: 87–91, 1993. doi: 10.1126/science.7678183. [DOI] [PubMed] [Google Scholar]
- 15.Uysal KT, Wiesbrock SM, Marino MW, Hotamisligil GS. Protection from obesity-induced insulin resistance in mice lacking TNF-alpha function. Nature 389: 610–614, 1997. doi: 10.1038/39335. [DOI] [PubMed] [Google Scholar]
- 16.Yamaguchi K, Nishimura T, Ishiba H, Seko Y, Okajima A, Fujii H, Tochiki N, Umemura A, Moriguchi M, Sumida Y, Mitsuyoshi H, Yasui K, Minami M, Okanoue T, Itoh Y. Blockade of interleukin 6 signalling ameliorates systemic insulin resistance through upregulation of glucose uptake in skeletal muscle and improves hepatic steatosis in high-fat diet fed mice. Liver Int 35: 550–561, 2015. doi: 10.1111/liv.12645. [DOI] [PubMed] [Google Scholar]
- 17.Loffreda S, Yang SQ, Lin HZ, Karp CL, Brengman ML, Wang DJ, Klein AS, Bulkley GB, Bao C, Noble PW, Lane MD, Diehl AM. Leptin regulates proinflammatory immune responses. FASEB J 12: 57–65, 1998. doi: 10.1096/fsb2fasebj.12.1.57. [DOI] [PubMed] [Google Scholar]
- 18.Huh JY, Park YJ, Ham M, Kim JB. Crosstalk between adipocytes and immune cells in adipose tissue inflammation and metabolic dysregulation in obesity. Mol Cell 37: 365–371, 2014. doi: 10.14348/molcells.2014.0074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu D, Molofsky AB, Liang HE, Ricardo-Gonzalez RR, Jouihan HA, Bando JK, Chawla A, Locksley RM. Eosinophils sustain adipose alternatively activated macrophages associated with glucose homeostasis. Science 332: 243–247, 2011. doi: 10.1126/science.1201475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Savino W, Dardenne M, Velloso LA, Dayse Silva-Barbosa S. The thymus is a common target in malnutrition and infection. The. Br J Nutr 98: S11–16, 2007. Suppl 1 doi: 10.1017/S0007114507832880. [DOI] [PubMed] [Google Scholar]
- 21.Kim J. Regulation of immune cell functions by metabolic reprogramming. J Immunol Res 2018: 1–12, 2018. doi: 10.1155/2018/8605471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Scott CB. Contribution of anaerobic energy expenditure to whole body thermogenesis. Nutr Metab (Lond) 2: 14, 2005. doi: 10.1186/1743-7075-2-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liberti MV, Locasale JW. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem Sci 41: 211–218, 2016. [Erratum in Trends Biochem Sci: 287]. doi: 10.1016/j.tibs.2015.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Burns JS, Manda G. Metabolic pathways of the Warburg effect in health and disease: perspectives of choice, chain or chance. Int J Mol Sci 18: 2755, 2017. doi: 10.3390/ijms18122755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science (New York, NY) 324: 1029–1033, 2009. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.DeBerardinis RJ, Chandel NS. We need to talk about the Warburg effect. Nat Metab 2: 127–129, 2020. doi: 10.1038/s42255-020-0172-2. [DOI] [PubMed] [Google Scholar]
- 27.Metallo CM, Gameiro PA, Bell EL, Mattaini KR, Yang J, Hiller K, Jewell CM, Johnson ZR, Irvine DJ, Guarente L, Kelleher JK, Vander Heiden MG, Iliopoulos O, Stephanopoulos G. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 481: 380–384, 2012. doi: 10.1038/nature10602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wise DR, Thompson CB. Glutamine addiction: a new therapeutic target in cancer. Trends Biochem Sci 35: 427–433, 2010. doi: 10.1016/j.tibs.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kawahara TLA, Michishita E, Adler AS, Damian M, Berber E, Lin M, McCord RA, Ongaigui KCL, Boxer LD, Chang HY, Chua KF. SIRT6 links histone H3 lysine 9 deacetylation to NF-κB-dependent gene expression and organismal life span. Cell 136: 62–74, 2009. doi: 10.1016/j.cell.2008.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sebastián C, Zwaans BM, Silberman DM, Gymrek M, Goren A, Zhong L, Ram O, Truelove J, Guimaraes AR, Toiber D, Cosentino C, Greenson JK, MacDonald AI, McGlynn L, Maxwell F, Edwards J, Giacosa S, Guccione E, Weissleder R, Bernstein BE, Regev A, Shiels PG, Lombard DB, Mostoslavsky R. The histone deacetylase SIRT6 is a tumor suppressor that controls cancer metabolism. Cell 151: 1185–1199, 2012. doi: 10.1016/j.cell.2012.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sundaresan NR, Vasudevan P, Zhong L, Kim G, Samant S, Parekh V, Pillai VB, Ravindra PV, Gupta M, Jeevanandam V, Cunningham JM, Deng C-X, Lombard DB, Mostoslavsky R, Gupta MP. The sirtuin SIRT6 blocks IGF-Akt signaling and development of cardiac hypertrophy by targeting c-Jun. Nat Med 18: 1643–1650, 2012. doi: 10.1038/nm.2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Takasaka N, Araya J, Hara H, Ito S, Kobayashi K, Kurita Y, Wakui H, Yoshii Y, Yumino Y, Fujii S, Minagawa S, Tsurushige C, Kojima J, Numata T, Shimizu K, Kawaishi M, Kaneko Y, Kamiya N, Hirano J, Odaka M, Morikawa T, Nishimura SL, Nakayama K, Kuwano K. Autophagy induction by SIRT6 through attenuation of insulin-like growth factor signaling is involved in the regulation of human bronchial epithelial cell senescence. J Immunol 192: 958–968, 2014. doi: 10.4049/jimmunol.1302341. [DOI] [PubMed] [Google Scholar]
- 33.Zhong L, D'Urso A, Toiber D, Sebastian C, Henry RE, Vadysirisack DD, Guimaraes A, Marinelli B, Wikstrom JD, Nir T, Clish CB, Vaitheesvaran B, Iliopoulos O, Kurland I, Dor Y, Weissleder R, Shirihai OS, Ellisen LW, Espinosa JM, Mostoslavsky R. The histone deacetylase Sirt6 regulates glucose homeostasis via Hif1alpha. Cell 140: 280–293, 2010. doi: 10.1016/j.cell.2009.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cantó C, Menzies KJ, Auwerx J. NAD(+) metabolism and the control of energy homeostasis: a balancing act between mitochondria and the nucleus. Cell Metab 22: 31–53, 2015. doi: 10.1016/j.cmet.2015.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Elfgang C, Eckert R, Lichtenberg-Fraté H, Butterweck A, Traub O, Klein RA, Hülser DF, Willecke K. Specific permeability and selective formation of gap junction channels in connexin-transfected HeLa cells. J Cell Biol 129: 805–817, 1995. doi: 10.1083/jcb.129.3.805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pehar M, Harlan BA, Killoy KM, Vargas MR. Nicotinamide adenine dinucleotide metabolism and neurodegeneration. Antioxid Redox Signal 28: 1652–1668, 2018. doi: 10.1089/ars.2017.7145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pillai VB, Sundaresan NR, Kim G, Gupta M, Rajamohan SB, Pillai JB, Samant S, Ravindra PV, Isbatan A, Gupta MP. Exogenous NAD blocks cardiac hypertrophic response via activation of the SIRT3-LKB1-AMP-activated kinase pathway. J Biol Chem 285: 3133–3144, 2010. doi: 10.1074/jbc.M109.077271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Singhal A, Cheng CY. Host NAD+ metabolism and infections: therapeutic implications. Int Immunol 31: 59–67, 2019. doi: 10.1093/intimm/dxy068. [DOI] [PubMed] [Google Scholar]
- 39.Ns H, Suraj BM, Kanakavidu S, Pellagra KR. A forgotten ailment in current clinical practice. Med J Dr DY Patil Vidyapeeth 12: 78, 2019. doi: 10.4103/mjdrdypu.mjdrdypu_62_18. [DOI] [Google Scholar]
- 40.Billingham LK, Chandel NS. NAD-biosynthetic pathways regulate innate immunity. Nat Immunol 20: 380–382, 2019. doi: 10.1038/s41590-019-0353-x. [DOI] [PubMed] [Google Scholar]
- 41.De Sanctis JB, Zabaleta M, Bianco NE, Garmendia JV, Rivas L. Serum adipokine levels in patients with systemic lupus erythematosus. Autoimmunity 42: 272–274, 2009. doi: 10.1080/08916930902828031. [DOI] [PubMed] [Google Scholar]
- 42.Gosset M, Berenbaum F, Salvat C, Sautet A, Pigenet A, Tahiri K, Jacques C. Crucial role of visfatin/pre-B cell colony-enhancing factor in matrix degradation and prostaglandin E2 synthesis in chondrocytes: possible influence on osteoarthritis. Arthritis Rheum 58: 1399–1409, 2008. doi: 10.1002/art.23431. [DOI] [PubMed] [Google Scholar]
- 43.Ismail SA, Mohamed SA. Serum levels of visfatin and omentin-1 in patients with psoriasis and their relation to disease severity. Br J Dermatol 167: 436–439, 2012. doi: 10.1111/j.1365-2133.2012.10980.x. [DOI] [PubMed] [Google Scholar]
- 44.Jia SH, Li Y, Parodo J, Kapus A, Fan L, Rotstein OD, Marshall JC. Pre-B cell colony-enhancing factor inhibits neutrophil apoptosis in experimental inflammation and clinical sepsis. J Clin Invest 113: 1318–1327, 2004. doi: 10.1172/JCI19930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moschen AR, Kaser A, Enrich B, Mosheimer B, Theurl M, Niederegger H, Tilg H. Visfatin, an adipocytokine with proinflammatory and immunomodulating properties. J Immunol 178: 1748–1758, 2007. doi: 10.4049/jimmunol.178.3.1748. [DOI] [PubMed] [Google Scholar]
- 46.Otero M, Lago R, Gomez R, Lago F, Dieguez C, Gómez-Reino JJ, Gualillo O. Changes in plasma levels of fat-derived hormones adiponectin, leptin, resistin and visfatin in patients with rheumatoid arthritis. Ann Rheumatic Dis 65: 1198–1201, 2006. doi: 10.1136/ard.2005.046540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ye SQ, Simon BA, Maloney JP, Zambelli-Weiner A, Gao L, Grant A, Easley RB, McVerry BJ, Tuder RM, Standiford T, Brower RG, Barnes KC, Garcia JG. Pre-B-cell colony-enhancing factor as a potential novel biomarker in acute lung injury. Am J Respir Crit Care Med 171: 361–370, 2005. doi: 10.1164/rccm.200404-563OC. [DOI] [PubMed] [Google Scholar]
- 48.Bruzzone S, Fruscione F, Morando S, Ferrando T, Poggi A, Garuti A, D'Urso A, Selmo M, Benvenuto F, Cea M, Zoppoli G, Moran E, Soncini D, Ballestrero A, Sordat B, Patrone F, Mostoslavsky R, Uccelli A, Nencioni A. Catastrophic NAD+ depletion in activated T lymphocytes through Nampt inhibition reduces demyelination and disability in EAE. PLoS ONE 4: e7897, 2009. doi: 10.1371/journal.pone.0007897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Busso N, Karababa M, Nobile M, Rolaz A, Van Gool F, Galli M, Leo O, So A, De Smedt T. Pharmacological inhibition of nicotinamide phosphoribosyltransferase/visfatin enzymatic activity identifies a new inflammatory pathway linked to NAD. PLoS ONE 3: e2267, 2008. doi: 10.1371/journal.pone.0002267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Van Gool F, Gallí M, Gueydan C, Kruys V, Prevot PP, Bedalov A, Mostoslavsky R, Alt FW, De Smedt T, Leo O. Intracellular NAD levels regulate tumor necrosis factor protein synthesis in a sirtuin-dependent manner. Nat Med 15: 206–210, 2009. doi: 10.1038/nm.1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Adriouch S, Hubert S, Pechberty S, Koch-Nolte F, Haag F, Seman M. NAD+ released during inflammation participates in T cell homeostasis by inducing ART2-mediated death of naive T cells in vivo. J Immunol 179: 186–194, 2007. doi: 10.4049/jimmunol.179.1.186. [DOI] [PubMed] [Google Scholar]
- 52.Liu Z, Azhipa O, Okamoto S, Govindarajan S, Dennert G. Extracellular nicotinamide adenine dinucleotide induces T cell apoptosis in vivo and in vitro. J Immunol 167: 4942–4947, 2001. doi: 10.4049/jimmunol.167.9.4942. [DOI] [PubMed] [Google Scholar]
- 53.Rodriguez Cetina Biefer H, Heinbokel T, Uehara H, Camacho V, Minami K, Nian Y, Koduru S, El Fatimy R, Ghiran I, Trachtenberg AJ, de la Fuente MA, Azuma H, Akbari O, Tullius SG, Vasudevan A, Elkhal A. Mast cells regulate CD4(+) T-cell differentiation in the absence of antigen presentation. J Allergy Clin Immunol 142: 1894–1908, 2018. e1897. doi: 10.1016/j.jaci.2018.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Matsuzaki J, Kuwamura M, Yamaji R, Inui H, Nakano Y. Inflammatory responses to lipopolysaccharide are suppressed in 40% energy-restricted mice. J Nutr 131: 2139–2144, 2001. doi: 10.1093/jn/131.8.2139. [DOI] [PubMed] [Google Scholar]
- 55.Imai S-I, Guarente L. It takes two to tango: NAD+ and sirtuins in aging/longevity control. NPJ Aging Mech Dis 2: 16017, 2016. doi: 10.1038/npjamd.2016.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Preyat N, Leo O. Sirtuin deacylases: a molecular link between metabolism and immunity. J Leukoc Biol 93: 669–680, 2013. doi: 10.1189/jlb.1112557. [DOI] [PubMed] [Google Scholar]
- 57.Imai S, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 403: 795–800, 2000. doi: 10.1038/35001622. [DOI] [PubMed] [Google Scholar]
- 58.Haigis MC, Guarente LP. Mammalian sirtuins–emerging roles in physiology, aging, and calorie restriction. Genes Dev 20: 2913–2921, 2006. doi: 10.1101/gad.1467506. [DOI] [PubMed] [Google Scholar]
- 59.Ivy JM, Klar AJ, Hicks JB. Cloning and characterization of four SIR genes of Saccharomyces cerevisiae. Mol Cell Biol 6: 688–702, 1986. doi: 10.1128/MCB.6.2.688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kaeberlein M, McVey M, Guarente L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev 13: 2570–2580, 1999. doi: 10.1101/gad.13.19.2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sinclair DA, Guarente L. Extrachromosomal rDNA circles— a cause of aging in yeast. Cell 91: 1033–1042, 1997. doi: 10.1016/S0092-8674(00)80493-6. [DOI] [PubMed] [Google Scholar]
- 62.Finkel T, Deng CX, Mostoslavsky R. Recent progress in the biology and physiology of sirtuins. Nature 460: 587–591, 2009. doi: 10.1038/nature08197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.North BJ, Verdin E. Sirtuins: Sir2-related NAD-dependent protein deacetylases. Genome Biol 5: 224–224, 2004. doi: 10.1186/gb-2004-5-5-224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Michishita E, Park JY, Burneskis JM, Barrett JC, Horikawa I. Evolutionarily conserved and nonconserved cellular localizations and functions of human SIRT proteins. MBoC 16: 4623–4635, 2005. doi: 10.1091/mbc.e05-01-0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sundaresan NR, Pillai VB, Wolfgeher D, Samant S, Vasudevan P, Parekh V, Raghuraman H, Cunningham JM, Gupta M, Gupta MP. The deacetylase SIRT1 promotes membrane localization and activation of Akt and PDK1 during tumorigenesis and cardiac hypertrophy. Sci Signal 4: ra46–ra46, 2011. doi: 10.1126/scisignal.2001465. [DOI] [PubMed] [Google Scholar]
- 66.Tanno M, Sakamoto J, Miura T, Shimamoto K, Horio Y. Nucleocytoplasmic shuttling of the NAD+-dependent histone deacetylase SIRT1. J Biol Chem 282: 6823–6832, 2007. doi: 10.1074/jbc.M609554200. [DOI] [PubMed] [Google Scholar]
- 67.Houtkooper RH, Pirinen E, Auwerx J. Sirtuins as regulators of metabolism and healthspan. Nat Rev Mol Cell Biol 13: 225–238, 2012. doi: 10.1038/nrm3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lee IH. Mechanisms and disease implications of sirtuin-mediated autophagic regulation. Exp Mol Med 51: 1–11, 2019. doi: 10.1038/s12276-019-0302-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hirschey MD. Old enzymes, new tricks: sirtuins are NAD(+)-dependent de-acylases. Cell Metab 14: 718–719, 2011. doi: 10.1016/j.cmet.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kumar S, Lombard DB. Functions of the sirtuin deacylase SIRT5 in normal physiology and pathobiology. Crit Rev Biochem Mol Biol 53: 311–334, 2018. doi: 10.1080/10409238.2018.1458071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Anderson KA, Madsen AS, Olsen CA, Hirschey MD. Metabolic control by sirtuins and other enzymes that sense NAD(+), NADH, or their ratio. Biochim Biophys Acta Bioenerg 1858: 991–998, 2017. doi: 10.1016/j.bbabio.2017.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pan PW, Feldman JL, Devries MK, Dong A, Edwards AM, Denu JM. Structure and biochemical functions of SIRT6. J Biol Chem 286: 14575–14587, 2011. doi: 10.1074/jbc.M111.218990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chang AR, Ferrer CM, Mostoslavsky R. SIRT6, a mammalian deacylase with multitasking abilities. Physiol Rev 100: 145–169, 2020. doi: 10.1152/physrev.00030.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kanfi Y, Naiman S, Amir G, Peshti V, Zinman G, Nahum L, Bar-Joseph Z, Cohen HY. The sirtuin SIRT6 regulates lifespan in male mice. Nature 483: 218–221, 2012. doi: 10.1038/nature10815. [DOI] [PubMed] [Google Scholar]
- 75.Feldman JL, Baeza J, Denu JM. Activation of the protein deacetylase SIRT6 by long-chain fatty acids and widespread deacylation by mammalian sirtuins. J Biol Chem 288: 31350–31356, 2013. doi: 10.1074/jbc.C113.511261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jiang H, Khan S, Wang Y, Charron G, He B, Sebastian C, Du J, Kim R, Ge E, Mostoslavsky R, Hang HC, Hao Q, Lin H. SIRT6 regulates TNF-α secretion through hydrolysis of long-chain fatty acyl lysine. Nature 496: 110–113, 2013. doi: 10.1038/nature12038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wang WW, Zeng Y, Wu B, Deiters A, Liu WR. A chemical biology approach to reveal Sirt6-targeted histone H3 sites in nucleosomes. ACS Chem Biol 11: 1973–1981, 2016. doi: 10.1021/acschembio.6b00243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bhardwaj A, Das S. SIRT6 deacetylates PKM2 to suppress its nuclear localization and oncogenic functions. Proc Natl Acad Sci USA 113: E538–547, 2016. doi: 10.1073/pnas.1520045113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dominy JE, Jr,Lee Y, Jedrychowski MP, Chim H, Jurczak MJ, Camporez JP, Ruan HB, Feldman J, Pierce K, Mostoslavsky R, Denu JM, Clish CB, Yang X, Shulman GI, Gygi SP, Puigserver P. The deacetylase Sirt6 activates the acetyltransferase GCN5 and suppresses hepatic gluconeogenesis. Mol Cell 48: 900–913, 2012. doi: 10.1016/j.molcel.2012.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jang HY, Gu S, Lee SM, Park BH. Overexpression of sirtuin 6 suppresses allergic airway inflammation through deacetylation of GATA3. J Allergy Clin Immunol 138: 1452–1455, 2016. e1413,doi: 10.1016/j.jaci.2016.05.019. [DOI] [PubMed] [Google Scholar]
- 81.Sociali G, Grozio A, Caffa I, Schuster S, Becherini P, Damonte P, Sturla L, Fresia C, Passalacqua M, Mazzola F, Raffaelli N, Garten A, Kiess W, Cea M, Nencioni A, Bruzzone S. SIRT6 deacetylase activity regulates NAMPT activity and NAD(P)(H) pools in cancer cells. FASEB J 33: 3704–3717, 2019. doi: 10.1096/fj.201800321R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Song MY, Wang J, Ka SO, Bae EJ, Park BH. Insulin secretion impairment in Sirt6 knockout pancreatic β cells is mediated by suppression of the FoxO1-Pdx1-Glut2 pathway. Sci Rep 6: 30321, 2016. doi: 10.1038/srep30321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pearce EL, Pearce EJ. Metabolic pathways in immune cell activation and quiescence. Immunity 38: 633–643, 2013. doi: 10.1016/j.immuni.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Project THPA. SIRT6. 2020, June 3. https://www.proteinatlas.org/ENSG00000077463-SIRT6/tissue.
- 85.Injarabian L, Devin A, Ransac S, Marteyn BS. Neutrophil metabolic shift during their lifecycle: impact on their survival and activation. Int J Mol Sci 21: 287, 2019. doi: 10.3390/ijms21010287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Slauch JM. How does the oxidative burst of macrophages kill bacteria? Still an open question. Mol Microbiol 80: 580–583, 2011. doi: 10.1111/j.1365-2958.2011.07612.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Geering B, Simon HU. Peculiarities of cell death mechanisms in neutrophils. Cell Death Differ 18: 1457–1469, 2011. doi: 10.1038/cdd.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.McGettrick AF, O'Neill LAJ. The role of HIF in immunity and inflammation. Cell Metab 32: 524–536, 2020. doi: 10.1016/j.cmet.2020.08.002. [DOI] [PubMed] [Google Scholar]
- 89.Papayannopoulos V. Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol 18: 134–147, 2018. doi: 10.1038/nri.2017.105. [DOI] [PubMed] [Google Scholar]
- 90.Awasthi D, Nagarkoti S, Sadaf S, Chandra T, Kumar S, Dikshit M. Glycolysis dependent lactate formation in neutrophils: A metabolic link between NOX-dependent and independent NETosis. Biochim Biophys Acta Mol Basis Dis 1865: 165542, 2019. doi: 10.1016/j.bbadis.2019.165542. [DOI] [PubMed] [Google Scholar]
- 91.Lee H-S, Ka S-O, Lee S-M, Lee S-I, Park J-W, Park B-H. Overexpression of sirtuin 6 suppresses inflammatory responses and bone destruction in mice with collagen-induced arthritis. Arthritis Rheumatol 65: 1776–1785, 2013. doi: 10.1002/art.37963. [DOI] [PubMed] [Google Scholar]
- 92.Li Y, Meng X, Wang W, Liu F, Hao Z, Yang Y, Zhao J, Yin W, Xu L, Zhao R, Hu J. Cardioprotective effects of SIRT6 in a mouse model of transverse aortic constriction-induced heart failure. Front Physiol 8, 2017. doi: 10.3389/fphys.2017.00394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Guarente L. The many faces of sirtuins: sirtuins and the Warburg effect. Nat Med 20: 24–25, 2014. doi: 10.1038/nm.3438. [DOI] [PubMed] [Google Scholar]
- 94.Tisoncik JR, Korth MJ, Simmons CP, Farrar J, Martin TR, Katze MG. Into the eye of the cytokine storm. Microbiol Mol Biol Rev 76: 16–32, 2012. doi: 10.1128/MMBR.05015-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Greenlee-Wacker MC. Clearance of apoptotic neutrophils and resolution of inflammation. Immunol Rev 273: 357–370, 2016. doi: 10.1111/imr.12453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Rőszer T. Understanding the mysterious M2 macrophage through activation markers and effector mechanisms. Mediators Inflamm 2015: 1–16, 2015. doi: 10.1155/2015/816460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kanfi Y, Peshti V, Gil R, Naiman S, Nahum L, Levin E, Kronfeld-Schor N, Cohen HY. SIRT6 protects against pathological damage caused by diet-induced obesity. Aging Cell 9: 162–173, 2010. doi: 10.1111/j.1474-9726.2009.00544.x. [DOI] [PubMed] [Google Scholar]
- 98.Song M-Y, Kim SH, Ryoo G-H, Kim M-K, Cha H-N, Park S-Y, Hwang HP, Yu HC, Bae EJ, Park B-H. Adipose sirtuin 6 drives macrophage polarization toward M2 through IL-4 production and maintains systemic insulin sensitivity in mice and humans. Exp Mol Med 51: 1–10, 2019. doi: 10.1038/s12276-019-0256-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Naiman S, Huynh FK, Gil R, Glick Y, Shahar Y, Touitou N, Nahum L, Avivi MY, Roichman A, Kanfi Y, Gertler AA, Doniger T, Ilkayeva OR, Abramovich I, Yaron O, Lerrer B, Gottlieb E, Harris RA, Gerber D, Hirschey MD, Cohen HY. SIRT6 promotes hepatic beta-oxidation via activation of PPARα. Cell Rep 29: 4127–4143, 2019. e4128 doi: 10.1016/j.celrep.2019.11.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Koo J-H, Jang H-Y, Lee Y, Moon YJ, Bae EJ, Yun S-K, Park B-H. Myeloid cell-specific sirtuin 6 deficiency delays wound healing in mice by modulating inflammation and macrophage phenotypes. Exp Mol Med 51: 1–10, 2019. doi: 10.1038/s12276-019-0248-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hughes CE, Benson RA, Bedaj M, Maffia P. Antigen-presenting cells and antigen presentation in tertiary lymphoid organs. Front Immunol 7, 2016. doi: 10.3389/fimmu.2016.00481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kelly B, O'Neill LAJ. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res 25: 771–784, 2015. doi: 10.1038/cr.2015.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Imtiyaz HZ, Williams EP, Hickey MM, Patel SA, Durham AC, Yuan L-J, Hammond R, Gimotty PA, Keith B, Simon MC. Hypoxia-inducible factor 2alpha regulates macrophage function in mouse models of acute and tumor inflammation. J Clin Invest 120: 2699–2714, 2010. doi: 10.1172/JCI39506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Köhler T, Reizis B, Johnson RS, Weighardt H, Förster I. Influence of hypoxia-inducible factor 1α on dendritic cell differentiation and migration. Eur J Immunol 42: 1226–1236, 2012. doi: 10.1002/eji.201142053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bhandari T, Olson J, Johnson RS, Nizet V. HIF-1α influences myeloid cell antigen presentation and response to subcutaneous OVA vaccination. J Mol Med 91: 1199–1205, 2013. doi: 10.1007/s00109-013-1052-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Jantsch J, Chakravortty D, Turza N, Prechtel AT, Buchholz B, Gerlach RG, Volke M, Gläsner J, Warnecke C, Wiesener MS, Eckardt KU, Steinkasserer A, Hensel M, Willam C. Hypoxia and hypoxia-inducible factor-1 alpha modulate lipopolysaccharide-induced dendritic cell activation and function. J Immunol 180: 4697–4705, 2008. doi: 10.4049/jimmunol.180.7.4697. [DOI] [PubMed] [Google Scholar]
- 107.Cramer T, Yamanishi Y, Clausen BE, Förster I, Pawlinski R, Mackman N, Haase VH, Jaenisch R, Corr M, Nizet V, Firestein GS, Gerber HP, Ferrara N, Johnson RS. HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell 112: 645–657, 2003. [Erratum in Cell 113:419]. doi: 10.1016/S0092-8674(03)00154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, Chi H. HIF1α–dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med 208: 1367–1376, 2011. doi: 10.1084/jem.20110278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Thiel M, Caldwell CC, Kreth S, Kuboki S, Chen P, Smith P, Ohta A, Lentsch AB, Lukashev D, Sitkovsky MV. Targeted deletion of HIF-1alpha gene in T cells prevents their inhibition in hypoxic inflamed tissues and improves septic mice survival. PLoS ONE 2: e853-e853, 2007. doi: 10.1371/journal.pone.0000853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Meng X, Grötsch B, Luo Y, Knaup KX, Wiesener MS, Chen X-X, Jantsch J, Fillatreau S, Schett G, Bozec A. Hypoxia-inducible factor-1α is a critical transcription factor for IL-10-producing B cells in autoimmune disease. Nat Commun 9: 251, 2018. doi: 10.1038/s41467-017-02683-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ding C, Ma Y, Chen X, Liu M, Cai Y, Hu X, Xiang D, Nath S, Zhang HG, Ye H, Powell D, Yan J. Integrin CD11b negatively regulates BCR signalling to maintain autoreactive B cell tolerance. Nat Commun 4: 2813, 2013. doi: 10.1038/ncomms3813. [DOI] [PubMed] [Google Scholar]
- 112.Ehirchiou D, Xiong Y, Xu G, Chen W, Shi Y, Zhang L. CD11b facilitates the development of peripheral tolerance by suppressing Th17 differentiation. J Exp Med 204: 1519–1524, 2007. doi: 10.1084/jem.20062292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Taylor CT, Colgan SP. Regulation of immunity and inflammation by hypoxia in immunological niches. Nat Rev Immunol 17: 774–785, 2017. doi: 10.1038/nri.2017.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Nagao A, Kobayashi M, Koyasu S, Chow CCT, Harada H. HIF-1-dependent reprogramming of glucose metabolic pathway of cancer cells and its therapeutic significance. Int J Mol Sci 20: 238, 2019. doi: 10.3390/ijms20020238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab 3: 177–185, 2006. doi: 10.1016/j.cmet.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 116.Semenza GL. Oxygen-dependent regulation of mitochondrial respiration by hypoxia-inducible factor 1. Biochem J 405: 1–9, 2007. doi: 10.1042/BJ20070389. [DOI] [PubMed] [Google Scholar]
- 117.Thomas LW, Ashcroft M. Exploring the molecular interface between hypoxia-inducible factor signalling and mitochondria. Cell Mol Life Sci 76: 1759–1777, 2019. doi: 10.1007/s00018-019-03039-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Peyssonnaux C, Datta V, Cramer T, Doedens A, Theodorakis EA, Gallo RL, Hurtado-Ziola N, Nizet V, Johnson RS. HIF-1alpha expression regulates the bactericidal capacity of phagocytes. J Clin Invest 115: 1806–1815, 2005. doi: 10.1172/JCI23865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Wang J, Sheng Z, Cai Y. SIRT6 overexpression inhibits HIF1α expression and its impact on tumor angiogenesis in lung cancer. Int J Clin Exp Pathol 11: 2940–2947, 2018. [PMC free article] [PubMed] [Google Scholar]
- 120.Iommarini L, Porcelli AM, Gasparre G, Kurelac I. Non-canonical mechanisms regulating hypoxia-inducible factor 1 alpha in cancer. Front Oncol 7, 2017. doi: 10.3389/fonc.2017.00286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.King A, Selak MA, Gottlieb E. Succinate dehydrogenase and fumarate hydratase: linking mitochondrial dysfunction and cancer. Oncogene 25: 4675–4682, 2006. doi: 10.1038/sj.onc.1209594. [DOI] [PubMed] [Google Scholar]
- 122.Pan Y, Mansfield KD, Bertozzi CC, Rudenko V, Chan DA, Giaccia AJ, Simon MC. Multiple factors affecting cellular redox status and energy metabolism modulate hypoxia-inducible factor prolyl hydroxylase activity in vivo and in vitro. Mol Cell Biol 27: 912–925, 2007. doi: 10.1128/MCB.01223-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Seo KS, Park JH, Heo JY, Jing K, Han J, Min KN, Kim C, Koh GY, Lim K, Kang GY, Uee Lee J, Yim YH, Shong M, Kwak TH, Kweon GR. SIRT2 regulates tumour hypoxia response by promoting HIF-1α hydroxylation. Oncogene 34: 1354–1362, 2015. doi: 10.1038/onc.2014.76. [DOI] [PubMed] [Google Scholar]
- 124.Finley LWS, Carracedo A, Lee J, Souza A, Egia A, Zhang J, Teruya-Feldstein J, Moreira PI, Cardoso SM, Clish CB, Pandolfi PP, Haigis MC. SIRT3 opposes reprogramming of cancer cell metabolism through HIF1α destabilization. Cancer Cell 19: 416–428, 2011. doi: 10.1016/j.ccr.2011.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Dioum EM, Chen R, Alexander MS, Zhang Q, Hogg RT, Gerard RD, Garcia JA. Regulation of hypoxia-inducible factor 2α signaling by the stress-responsive deacetylase sirtuin 1. Science 324: 1289–1293, 2009. doi: 10.1126/science.1169956. [DOI] [PubMed] [Google Scholar]
- 126.Joo H-Y, Yun M, Jeong J, Park E-R, Shin H-J, Woo SR, Jung JK, Kim Y-M, Park J-J, Kim J, Lee K-H. SIRT1 deacetylates and stabilizes hypoxia-inducible factor-1α (HIF-1α) via direct interactions during hypoxia. Biochem Biophys Res Comm 462: 294–300, 2015. doi: 10.1016/j.bbrc.2015.04.119. [DOI] [PubMed] [Google Scholar]
- 127.Laemmle A, Lechleiter A, Roh V, Schwarz C, Portmann S, Furer C, Keogh A, Tschan MP, Candinas D, Vorburger SA, Stroka D. Inhibition of SIRT1 impairs the accumulation and transcriptional activity of HIF-1α protein under hypoxic conditions. PLoS ONE 7: e33433, 2012. doi: 10.1371/journal.pone.0033433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Lim JH, Lee YM, Chun YS, Chen J, Kim JE, Park JW. Sirtuin 1 modulates cellular responses to hypoxia by deacetylating hypoxia-inducible factor 1alpha. Mol Cell 38: 864–878, 2010. doi: 10.1016/j.molcel.2010.05.023. [DOI] [PubMed] [Google Scholar]
- 129.Greijer AE, van der Wall E. The role of hypoxia inducible factor 1 (HIF-1) in hypoxia induced apoptosis. J Clin Pathol 57: 1009–1014, 2004. doi: 10.1136/jcp.2003.015032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.de Céu Teixeira M, Sanchez-Lopez E, Espina M, Garcia ML, Durazzo A, Lucarini M, Novellino E, Souto SB, Santini A, Souto EB. Sirtuins and SIRT6 in carcinogenesis and in diet. Int J Mol Sci 20: 4945, 2019. doi: 10.3390/ijms20194945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Mostoslavsky R, Chua KF, Lombard DB, Pang WW, Fischer MR, Gellon L, , et al. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell 124: 315–329, 2006. doi: 10.1016/j.cell.2005.11.044. [DOI] [PubMed] [Google Scholar]
- 132.Robey RB, Hay N. Is Akt the “Warburg kinase”?-Akt-energy metabolism interactions and oncogenesis. Semin Cancer Biol 19: 25–31, 2009. doi: 10.1016/j.semcancer.2008.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Szymonowicz K, Oeck S, Malewicz NM, Jendrossek V. New insights into protein kinase B/Akt signaling: role of localized Akt activation and compartment-specific target proteins for the cellular radiation response. Cancers (Basel) 10: 78, 2018. doi: 10.3390/cancers10030078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Palmer CS, Ostrowski M, Balderson B, Christian N, Crowe SM. Glucose metabolism regulates T cell activation, differentiation, and functions. Front Immunol 6, 2015. doi: 10.3389/fimmu.2015.00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Deprez J, Vertommen D, Alessi DR, Hue L, Rider MH. Phosphorylation and activation of heart 6-phosphofructo-2-kinase by protein kinase B and other protein kinases of the insulin signaling cascades. J Biol Chem 272: 17269–17275, 1997. doi: 10.1074/jbc.272.28.17269. [DOI] [PubMed] [Google Scholar]
- 136.Zhang W, Patil S, Chauhan B, Guo S, Powell DR, Le J, Klotsas A, Matika R, Xiao X, Franks R, Heidenreich KA, Sajan MP, Farese RV, Stolz DB, Tso P, Koo SH, Montminy M, Unterman TG. FoxO1 regulates multiple metabolic pathways in the liver: effects on gluconeogenic, glycolytic, and lipogenic gene expression. J Biol Chem 281: 10105–10117, 2006. doi: 10.1074/jbc.M600272200. [DOI] [PubMed] [Google Scholar]
- 137.Brugarolas JB, Vazquez F, Reddy A, Sellers WR, Kaelin WG. TSC2 regulates VEGF through mTOR-dependent and -independent pathways. Cancer cell 4: 147–158, 2003. doi: 10.1016/S1535-6108(03)00187-9. [DOI] [PubMed] [Google Scholar]
- 138.Xue G, Zippelius A, Wicki A, Mandalà M, Tang F, Massi D, Ba H. Integrated Akt/PKB signaling in immunomodulation and its potential role in cancer immunotherapy. J Natl Cancer Inst 107: djv171, 2015. doi: 10.1093/jnci/djv171. [DOI] [PubMed] [Google Scholar]
- 139.Weichhart T, Säemann MD. The PI3K/Akt/mTOR pathway in innate immune cells: emerging therapeutic applications. Ann Rheum Dis 67, Suppl 3: iii70–74, 2008. doi: 10.1136/ard.2008.098459. [DOI] [PubMed] [Google Scholar]
- 140.Zhang Y, Wang X, Yang H, Liu H, Lu Y, Han L, Liu G. Kinase AKT controls innate immune cell development and function. Immunology 140: 143–152, 2013. doi: 10.1111/imm.12123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Sun J, Dotti G, Huye LE, Foster AE, Savoldo B, Gramatges MM, Spencer DM, Rooney CM. T cells expressing constitutively active Akt resist multiple tumor-associated inhibitory mechanisms. Mol Ther 18: 2006–2017, 2010. doi: 10.1038/mt.2010.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Chapman NM, Chi H. mTOR signaling, Tregs and immune modulation. Immunotherapy 6: 1295–1311, 2014. doi: 10.2217/imt.14.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Jones RG, Pearce EJ. MenTORing immunity: mTOR signaling in the development and function of tissue-resident immune cells. Immunity 46: 730–742, 2017. doi: 10.1016/j.immuni.2017.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Zeng H, Yang K, Cloer C, Neale G, Vogel P, Chi H. mTORC1 couples immune signals and metabolic programming to establish T(reg)-cell function. Nature 499: 485–490, 2013. doi: 10.1038/nature12297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Raud B, McGuire PJ, Jones RG, Sparwasser T, Berod L. Fatty acid metabolism in CD8(+) T cell memory: challenging current concepts. Immunol Rev 283: 213–231, 2018. doi: 10.1111/imr.12655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Jellusova J, Rickert RC. The PI3K pathway in B cell metabolism. Crit Rev Biochem Mol Biol 51: 359–378, 2016. doi: 10.1080/10409238.2016.1215288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Zhang X, Spiegelman NA, Nelson OD, Jing H, Lin H. SIRT6 regulates Ras-related protein R-Ras2 by lysine defatty-acylation. eLife 6, 2017. doi: 10.7554/eLife.25158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, Hemmings BA. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J 15: 6541–6551, 1996. doi: 10.1002/j.1460-2075.1996.tb01045.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Scheid MP, Marignani PA, Woodgett JR. Multiple phosphoinositide 3-kinase-dependent steps in activation of protein kinase B. Mol Cell Biol 22: 6247–6260, 2002. doi: 10.1128/MCB.22.17.6247-6260.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Xiao C, Wang RH, Lahusen TJ, Park O, Bertola A, Maruyama T, Reynolds D, Chen Q, Xu X, Young HA, Chen WJ, Gao B, Deng CX. Progression of chronic liver inflammation and fibrosis driven by activation of c-JUN signaling in Sirt6 mutant mice. J Biol Chem 287: 41903–41913, 2012. doi: 10.1074/jbc.M112.415182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Yang XD, Tajkhorshid E, Chen LF. Functional interplay between acetylation and methylation of the RelA subunit of NF-kappaB. Mol Cell Biol 30: 2170–2180, 2010. doi: 10.1128/MCB.01343-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Liu T, Zhang L, Joo D, Sun S-C. NF-κB signaling in inflammation. Signal Transduct Target Ther 2: 17023, 2017. doi: 10.1038/sigtrans.2017.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Ramakrishnan G, Davaakhuu G, Kaplun L, Chung W-C, Rana A, Atfi A, Miele L, Tzivion G. Sirt2 deacetylase is a novel AKT binding partner critical for AKT activation by insulin. J Biol Chem 289: 6054–6066, 2014. doi: 10.1074/jbc.M113.537266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Sundaresan NR, Gupta M, Kim G, Rajamohan SB, Isbatan A, Gupta MP. Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a-dependent antioxidant defense mechanisms in mice. J Clin Invest 119: 2758–2771, 2009. doi: 10.1172/JCI39162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Tao Y, Yu S, Chao M, Wang Y, Xiong J, Lai H. SIRT4 suppresses the PI3K/Akt/NF-κB signaling pathway and attenuates HUVEC injury induced by oxLDL. Mol Med Report 19: 4973–4979, 2019. doi: 10.3892/mmr.2019.10161. [DOI] [PubMed] [Google Scholar]
- 156.Yu J, Qin B, Wu F, Qin S, Nowsheen S, Shan S, Zayas J, Pei H, Lou Z, Wang L. Regulation of serine-threonine kinase Akt activation by NAD(+)-dependent deacetylase SIRT7. Cell Rep 18: 1229–1240, 2017. doi: 10.1016/j.celrep.2017.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Kim EH, Suresh M. Role of PI3K/Akt signaling in memory CD8 T cell differentiation. Front Immun 4: 20–20, 2013. doi: 10.3389/fimmu.2013.00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Giridharan S, Srinivasan M. Mechanisms of NF-κB p65 and strategies for therapeutic manipulation. J Inflamm Res 11: 407–419, 2018. doi: 10.2147/JIR.S140188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Bai D, Ueno L, Vogt PK. Akt-mediated regulation of NFkappaB and the essentialness of NFkappaB for the oncogenicity of PI3K and Akt. Int J Cancer 125: 2863–2870, 2009. doi: 10.1002/ijc.24748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Luo W, Semenza GL. Pyruvate kinase M2 regulates glucose metabolism by functioning as a coactivator for hypoxia-inducible factor 1 in cancer cells. Oncotarget 2: 551–556, 2011. doi: 10.18632/oncotarget.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Christofk HR, Vander Heiden MG, Wu N, Asara JM, Cantley LC. Pyruvate kinase M2 is a phosphotyrosine-binding protein. Nature 452: 181–186, 2008. doi: 10.1038/nature06667. [DOI] [PubMed] [Google Scholar]
- 162.Vander Heiden MG, Locasale JW, Swanson KD, Sharfi H, Heffron GJ, Amador-Noguez D, Christofk HR, Wagner G, Rabinowitz JD, Asara JM, Cantley LC. Evidence for an alternative glycolytic pathway in rapidly proliferating cells. Science 329: 1492–1499, 2010. doi: 10.1126/science.1188015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Sommermann TG, O'Neill K, Plas DR, Cahir-McFarland E. IKKβ and NF-κB transcription govern lymphoma cell survival through AKT-induced plasma membrane trafficking of GLUT1. Cancer Res 71: 7291–7300, 2011. doi: 10.1158/0008-5472.CAN-11-1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Yang W, Xia Y, Cao Y, Zheng Y, Bu W, Zhang L, You MJ, Koh MY, Cote G, Aldape K, Li Y, Verma IM, Chiao PJ, Lu Z. EGFR-induced and PKCε monoubiquitylation-dependent NF-κB activation upregulates PKM2 expression and promotes tumorigenesis. Mol Cell 48: 771–784, 2012. [Erratum in Mol Cell 69:347, 2018]. doi: 10.1016/j.molcel.2012.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Yang P, Li Z, Wang Y, Zhang L, Wu H, Li Z. Secreted pyruvate kinase M2 facilitates cell migration via PI3K/Akt and Wnt/β-catenin pathway in colon cancer cells. Biochem Biophys Res Comm 459: 327–332, 2015. doi: 10.1016/j.bbrc.2015.02.112. [DOI] [PubMed] [Google Scholar]
- 166.Santos-Barriopedro I, Bosch-Presegué L, Marazuela-Duque A, de la Torre C, Colomer C, Vazquez BN, Fuhrmann T, Martínez-Pastor B, Lu W, Braun T, Bober E, Jenuwein T, Serrano L, Esteller M, Chen Z, Barceló-Batllori S, Mostoslavsky R, Espinosa L, Vaquero A. SIRT6-dependent cysteine monoubiquitination in the PRE-SET domain of Suv39h1 regulates the NF-κB pathway. Nat Commun 9: 101, 2018. doi: 10.1038/s41467-017-02586-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Abraham E. Nuclear factor-kappaB and its role in sepsis-associated organ failure. J INFECT DIS 187: S364–369, 2003. Suppl 2 doi: 10.1086/374750. [DOI] [PubMed] [Google Scholar]
- 168.Park SD, Cheon SY, Park TY, Shin BY, Oh H, Ghosh S, Koo BN, Lee SK. Intranuclear interactomic inhibition of NF-κB suppresses LPS-induced severe sepsis. Biochem Biophys Res Comm 464: 711–717, 2015. doi: 10.1016/j.bbrc.2015.07.008. [DOI] [PubMed] [Google Scholar]
- 169.Pai S, Thomas R. Immune deficiency or hyperactivity-Nf-kappab illuminates autoimmunity. J Autoimmunity 31: 245–251, 2008. doi: 10.1016/j.jaut.2008.04.012. [DOI] [PubMed] [Google Scholar]
- 170.Rothgiesser KM, Erener S, Waibel S, Lüscher B, Hottiger MO. SIRT2 regulates NF-κB dependent gene expression through deacetylation of p65 Lys310. J Cell Sci 123: 4251–4258, 2010. [Erratum in J Cell Sci 132, 2019]. doi: 10.1242/jcs.073783. [DOI] [PubMed] [Google Scholar]
- 171.Yeung F, Hoberg JE, Ramsey CS, Keller MD, Jones DR, Frye RA, Mayo MW. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J 23: 2369–2380, 2004. doi: 10.1038/sj.emboj.7600244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Schug TT, Xu Q, Gao H, Peres-da-Silva A, Draper DW, Fessler MB, Purushotham A, Li X. Myeloid deletion of SIRT1 induces inflammatory signaling in response to environmental stress. Mol Cell Biol 30: 4712–4721, 2010. doi: 10.1128/MCB.00657-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Jnr G, Wang R. MYC in regulating immunity: metabolism and beyond. Genes (Basel) 8: 88, 2017. doi: 10.3390/genes8030088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Dang CV. c-Myc target genes involved in cell growth, apoptosis, and metabolism. Mol Cell Biol 19: 1–11, 1999. doi: 10.1128/MCB.19.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Dang CV. MYC on the path to cancer. Cell 149: 22–35, 2012. doi: 10.1016/j.cell.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Casey SC, Baylot V, Felsher DW. The MYC oncogene is a global regulator of the immune response. Blood 131: 2007–2015, 2018. doi: 10.1182/blood-2017-11-742577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Murn J, Mlinaric-Rascan I, Vaigot P, Alibert O, Frouin V, Gidrol X. A Myc-regulated transcriptional network controls B-cell fate in response to BCR triggering. BMC Genomics 10: 323–323, 2009. doi: 10.1186/1471-2164-10-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.de Alborán IM, Baena E, Martinez AC. c-Myc-deficient B lymphocytes are resistant to spontaneous and induced cell death. Cell Death Differ 11: 61–68, 2004. [Erratum in Cell Death Differ: 690, 2004]. doi: 10.1038/sj.cdd.4401319. [DOI] [PubMed] [Google Scholar]
- 179.Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, McCormick LL, Fitzgerald P, Chi H, Munger J, Green DR. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 35: 871–882, 2011. doi: 10.1016/j.immuni.2011.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Gao P, Tchernyshyov I, Chang TC, Lee YS, Kita K, Ochi T, Zeller KI, De Marzo AM, Van Eyk JE, Mendell JT, Dang CV. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 458: 762–765, 2009. doi: 10.1038/nature07823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang XY, Pfeiffer HK, Nissim I, Daikhin E, Yudkoff M, McMahon SB, Thompson CB. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci USA 105: 18782–18787, 2008. doi: 10.1073/pnas.0810199105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Zwaans BMM, Lombard DB. Interplay between sirtuins, MYC and hypoxia-inducible factor in cancer-associated metabolic reprogramming. Disease Models Mechanisms 7: 1023–1032, 2014. doi: 10.1242/dmm.016287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Dang CV. Rethinking the Warburg effect with Myc micromanaging glutamine metabolism. Cancer Res 70: 859–862, 2010. doi: 10.1158/0008-5472.CAN-09-3556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Jang KY, Noh SJ, Lehwald N, Tao GZ, Bellovin DI, Park HS, Moon WS, Felsher DW, Sylvester KG. SIRT1 and c-Myc promote liver tumor cell survival and predict poor survival of human hepatocellular carcinomas. PLoS ONE 7: e45119, 2012. doi: 10.1371/journal.pone.0045119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Kriegl L, Vieth M, Kirchner T, Menssen A. Up-regulation of c-MYC and SIRT1 expression correlates with malignant transformation in the serrated route to colorectal cancer. Oncotarget 3: 1182–1193, 2012. doi: 10.18632/oncotarget.628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Menssen A, Hydbring P, Kapelle K, Vervoorts J, Diebold J, Lüscher B, Larsson L-G, Hermeking H. The c-MYC oncoprotein, the NAMPT enzyme, the SIRT1-inhibitor DBC1, and the SIRT1 deacetylase form a positive feedback loop. Proc Natl Acad Sci USA 109: E187–E196, 2012. doi: 10.1073/pnas.1105304109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Mao B, Zhao G, Lv X, Chen HZ, Xue Z, Yang B, Liu DP, Liang CC. Sirt1 deacetylates c-Myc and promotes c-Myc/Max association. Int J Biochem Cell Biol 43: 1573–1581, 2011. doi: 10.1016/j.biocel.2011.07.006. [DOI] [PubMed] [Google Scholar]
- 188.Yuan J, Minter-Dykhouse K, Lou Z. A c-Myc-SIRT1 feedback loop regulates cell growth and transformation. J Cell Biol 185: 203–211, 2009. doi: 10.1083/jcb.200809167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Shin J, He M, Liu Y, Paredes S, Villanova L, Brown K, Qiu X, Nabavi N, Mohrin M, Wojnoonski K, Li P, Cheng H-L, Murphy AJ, Valenzuela DM, Luo H, Kapahi P, Krauss R, Mostoslavsky R, Yancopoulos GD, Alt FW, Chua KF, Chen D. SIRT7 represses Myc activity to suppress ER stress and prevent fatty liver disease. Cell Rep 5: 654–665, 2013. [Erratum in Cell Rep: 1479, 2013]. doi: 10.1016/j.celrep.2013.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Liu PY, Xu N, Malyukova A, Scarlett CJ, Sun YT, Zhang XD, Ling D, Su SP, Nelson C, Chang DK, Koach J, Tee AE, Haber M, Norris MD, Toon C, Rooman I, Xue C, Cheung BB, Kumar S, Marshall GM, Biankin AV, Liu T. The histone deacetylase SIRT2 stabilizes Myc oncoproteins. Cell Death Differ 20: 503–514, 2013. doi: 10.1038/cdd.2012.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Gustafson WC, Weiss WA. Myc proteins as therapeutic targets. Oncogene 29: 1249–1259, 2010. doi: 10.1038/onc.2009.512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Quan Y, Wang N, Chen Q, Xu J, Cheng W, Di M, Xia W, Gao W-Q. SIRT3 inhibits prostate cancer by destabilizing oncoprotein c-MYC through regulation of the PI3K/Akt pathway. Oncotarget 6: 26494–26507, 2015. doi: 10.18632/oncotarget.4764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Haigis MC, Mostoslavsky R, Haigis KM, Fahie K, Christodoulou DC, Murphy AJ, Valenzuela DM, Yancopoulos GD, Karow M, Blander G, Wolberger C, Prolla TA, Weindruch R, Alt FW, Guarente L. SIRT4 inhibits glutamate dehydrogenase and opposes the effects of calorie restriction in pancreatic beta cells. Cell 126: 941–954, 2006. doi: 10.1016/j.cell.2006.06.057. [DOI] [PubMed] [Google Scholar]
- 194.Greene KS, Lukey MJ, Wang X, Blank B, Druso JE, Lin M-cJ, Stalnecker CA, Zhang C, Negrón Abril Y, Erickson JW, Wilson KF, Lin H, Weiss RS, Cerione RA. SIRT5 stabilizes mitochondrial glutaminase and supports breast cancer tumorigenesis. Proc Natl Acad Sci USA 116: 26625–26632, 2019. doi: 10.1073/pnas.1911954116. [DOI] [PMC free article] [PubMed] [Google Scholar]