

Abstract
The spa transgenic mouse displays spasticity and hypertonia that develops during the early postnatal period, with motor impairments that are remarkably similar to symptoms of human cerebral palsy. Previously, we observed that spa mice have fewer phrenic motor neurons innervating the diaphragm muscle (DIAm). We hypothesize that spa mice exhibit increased susceptibility to neuromuscular transmission failure (NMTF) due to an expanded innervation ratio. We retrogradely labeled phrenic motor neurons with rhodamine and imaged them in horizontal sections (70 µm) using confocal microscopy. Phrenic nerve–DIAm strip preparations from wild type and spa mice were stretched to optimal length, and force was evoked by phrenic nerve stimulation at 10, 40, or 75 Hz in 330-ms duration trains repeated each second (33% duty cycle) across a 120-s period. To assess NMTF, force evoked by phrenic nerve stimulation was compared to force evoked by direct DIAm stimulation superimposed every 15 s. Total DIAm fiber number was estimated in hematoxylin and eosin-stained strips. Compared to wild type, spa mice had over twofold greater NMTF during the first stimulus train that persisted throughout the 120 s period of repetitive activation. In both wild type and spa mice, NMTF was stimulation-frequency dependent. There was no difference in neuromuscular junction morphology or the total number of DIAm fibers between wild type and spa mice, however, there was an increase innervation ratio (39%) in spa mice. We conclude that early-onset developmental neuromotor disorders impair the efficacy of DIAm neuromuscular transmission, likely to contribute to respiratory complications.
NEW & NOTEWORTHY Individuals with motor control deficits, including cerebral palsy (CP) often have respiratory impairments. Glycine-receptor mutant spa mice have early-onset hypertonia, and limb motor impairments, similar to individuals with CP. We hypothesized that in the diaphragm of spa mice, disruption of glycinergic inputs to MNs would result in increased phrenic–DIAm neuromuscular transmission failure. Pathophysiologic abnormalities in neuromuscular transmission may contribute to respiratory dysfunction in conditions where early developmental MN loss or motor control deficits are apparent.
Keywords: cerebral palsy, innervation ratio, muscle specific force, neuromuscular junction, spasticity
INTRODUCTION
Early-onset hypertonia and spasticity are common phenotypes observed in a variety of developmental neuromuscular disorders, including hereditary spastic paraplegia, leukoencephalopathy and notably, cerebral palsy (CP), the most common motor disability of childhood (1, 2). The most common type of CP is spastic CP, where marked spasticity, hypertonia, and hyperreflexia co-exist with neuromuscular impairments (1, 3). Hypertonia and spasticity are related to disinhibition of the motor neuron (MN), with symptoms unequivocally involving dysfunction of the spinal cord and motor unit (1, 4).
For humans with CP, clinical interventions and research has focused on locomotor dysfunction, yet, respiratory difficulties occur even when overall motor dysfunction is mild (5, 6). Respiratory dysfunction is one of the most common causes of death for individuals with CP (5, 7, 8). Furthermore, children with CP appear to be particularly vulnerable to adverse respiratory side effects of botulinum toxin, an intervention reducing hypertonia by temporarily blocking neuromuscular signaling (9, 10). Despite the importance of respiratory muscle involvement in CP, including the increased risk for respiratory adverse effects due to botulinum toxin administered to limb muscles (9, 10) and a leading cause of CP mortality (7), very little work has been done to advance the understanding of diaphragm muscle (DIAm) dysfunction in this and similar conditions.
The spa mouse ([B6.Cg-Glrbspa/J]) displays symptoms of spontaneous hypertonicity and motor impairments (including abnormal gait) (1, 3) similar to those physical symptoms observed in human CP (11–14), due to a glycine receptor mutation (11, 13, 15, 16). In hypoglossal MNs of spa mutants, the input resistance, decay time constant, and rheobase is higher than wild type hypoglossal MNs (16). Additionally, glycinergic currents have reduced amplitudes and frequencies in spa MNs compared to wild type (15, 16), a phenomenon that appears to depend on αβ heteromeres in adults (15). Although glycine receptor or receptor clustering mutations are not a known cause of human CP, they may provide insight into the mechanisms underlying spasticity and neuromotor impairments in this congenital condition (1, 17). In adult spa mice, we previously observed marked reductions in the number of phrenic (18) and tibialis anterior MNs (12) compared to wild type control mice. In spa mice, the reduced number of tibialis anterior MNs was concomitant with an unchanged number of tibialis anterior muscle fibers with a resultant increase in the innervation ratio compared to wild type mice (19).
With aging, there is also a reduction in the number of phrenic MNs and an increase in innervation ratio, which leads to increased neuromuscular transmission failure (NMTF) (20, 21). In other cases of increased innervation ratio, NMTF is related to the greater number of axonal bifurcations required to innervate the full complement of muscle fibers, with each branch point a potential site for action potential propagation failure (22, 23). Therefore, we hypothesize that in spa mice there is an increase in the innervation ratio of DIAm motor units that results in increased susceptibility to NMTF compared to wild type mice. We do not expect any changes in DIAm fiber density and DIAm fiber-type proportions, as developmental denervation did not alter the proportion of myosin heavy chain expression in DIAm (24). We do not expect any neuromuscular junction degeneration or abnormalities at the age of the mice used in the present study (∼6 to 12 weeks old), as previous studies of early MN loss due to glycine neurotransmission defects reported no perturbations of pre- or postsynaptic neuromuscular junctions (25–29).
MATERIALS AND METHODS
Ethical Approval
All procedures were approved by the Institutional Animal Care and Use Committee at Mayo Clinic (protocols #A23215 and #A00003598) and complied with National Institutes of Health and The Physiological Society guidelines (30). Prior to experimentation, animals received an intraperitoneal injection of ketamine (100 mg/kg) and xylazine (10 mg/kg), and were euthanized via exsanguination.
Experimental Animals and Anesthesia
This study used 23 wild type and 16 homozygous spa knockout mice (B6.Cg-Glrbspa/J), ages 6 to 12 weeks, of both sexes. Previous rodent studies have shown that hyperpolarizing glycinergic neurotransmission (31, 32) to MNs is mature by ∼4 weeks of age in spa mice MNs (15, 16) and spinal cord (33). The spa knockout mice have a homozygous insertion of LINE-1 in Glrb, the β subunit of the glycine receptor gene resulting in a splicing error of this subunit (34) and exhibit symptoms by the second week of life including abnormal gait, muscle rigidity, myoclonic jerks, exaggerated startle response, smaller size, and spasticity (11, 13, 35, 36). Founder heterozygous mice were obtained from Jackson Labs (Bar Harbor, ME) and used in a heterozygote × heterozygote breeding scheme. Mice were housed in identical conditions, following recommended housing and care guidelines (37), in a pathogen-free facility. Mice were exposed to 12:12-h light:dark cycle year round. The room was kept at ∼21–23°C. Mouse chow (PicoLab Rodent Diet 5053, LabDiet, St. Louis, MO) and tap water (via water bottles) were freely accessible. Genotyping was done on tail snips as previously described (15).
Retrograde Labeling, Imaging, and Counting of Phrenic Motor Neurons
For the initial nerve-dip survival surgery, animals received carprofen (5 mg/kg) in their water bottle starting at least 48 h before surgery for analgesia. Animals were anesthetized with intraperitoneal injections of diazepam (5 mg/kg) and droperidol (15 mg/kg) for survival surgery. Animals also received an intraperitoneal injection of fentanyl (0.3 mg/kg) and subcutaneous injection of buprenorphine SR (0.5 to 1.0 mg/kg) for analgesia. All procedures were performed using an aseptic technique, and insulated heating pads maintained body temperature at 38°C. In anesthetized mice, the phrenic nerve was identified in the ventral aspect of the neck, the fascia was then incised and dissected to expose as much of the phrenic nerve as possible. The phrenic nerve was then cut as far distal as possible and the proximal end of the nerve was placed in a sterile microdish with 1–3 μL of a 5% solution of tetramethylrhodamine (rhodamine) (Molecular Probes, Life Technologies, Grand Island, NY), previously validated for the accurate labeling of phrenic MNs (21, 38, 39). Petroleum jelly was placed on the surrounding tissues to prevent exposure to errant rhodamine solution. The retrograde labeling procedure was performed for 45 min during which the microdish was checked at 5-min intervals, with rhodamine solution added as required (up to 9 μL total). Following this, the phrenic nerve end was removed from the microdish and the surgical site irrigated, cleansed, and sutured. A post-surgery period of 24 h provided ample time for retrograde transport to MNs, based on distance between where the nerve was transected and spinal cord and the speed of retrograde transport (40). It is important to note that spa mice have reduced body mass compared to wild type at this age (35), and mortality following surgery is higher (∼95% survival for wild type and ∼85% survival in spa). In past studies, there were no differences in body weight loss between spa and wild type mice following nerve dip procedures (12, 18).
At 24 h post phrenic nerve dip, animals were anesthetized and euthanized by exsanguination. Following transcardial perfusion with phosphate-buffered saline (PBS; pH 7.4) and 4% paraformaldehyde in PBS, the cervical spinal cord was post fixed in 4% paraformaldehyde overnight, and then transferred to 24% sucrose in PBS for 72 h or until sunk, before flash-freezing and cryosectioning. A cryostat (Leica Biosystems, Buffalo Grove, IL) was used to cut the samples in 70-µm longitudinal (horizontal) sections. These sections were ordered, with the ventral sections preceding the more dorsal sections. Sections were placed on gelatin-coated slides, treated with graded ethanols and xylenes, and cover-slipped with DPX mounting media (Fluka, Sigma-Aldrich, St. Louis, MO) in a manner identical to past reports (12, 38, 41, 42).
Cervical spinal cord sections were then imaged using an Olympus FluoView 1200 laser scanning confocal microscope (Olympus America Inc., Melville, NY) mounted on an upright Olympus BX50WI microscope. Within the ventral portion of the spinal cord rhodamine-labeled phrenic MNs were imaged with a ×40 oil immersion lens (NA 1.35), and three dimensional image stacks were collected in a 1024 × 1024 array (pixel dimensions 0.50 µm × 0.50 µm) with a 2.0-µm step size. Laser intensity was 3.0–9.0% with confocal aperture and photomultiplier gain kept fixed across samples. Optical slices containing the nucleus of a rhodamine-labeled phrenic MN were identified and used to quantify the number of phrenic MNs. In a previous study (18), we found that wild type mice have a total of ∼424 phrenic MNs, whereas spa mice have a total of ∼294 phrenic MNs, a result used to assess the number MNs required to sample in the present study to observe 25% or greater loss of phrenic MNs.
Estimation of Total Number of Diaphragm Muscle Fibers and Innervation Ratio
The total number of fibers in the DIAm was estimated via extrapolations from density measurements in DIAm strips and total DIAm mass. Prior to flash-freezing, the entire DIAm was weighed and ∼2 to 3–mm wide strips were dissected and weighed. DIAm strips were stretched to 150% of resting length, which approximates the optimal length for isometric force generation by the DIAm (24, 43, 44), and rapidly frozen in melting isopentane. Using a cryostat, serial 10-μm transverse sections of frozen DIAm were cut and then stained with hematoxylin and eosin (45). Bright-field mosaic images (Olympus IX71, Olympus America, Melville, NY) were obtained using a ×20 objective, and these images were used to quantify the total number of DIAm fibers, determined using morphometric tools in FIJI (46) for a known weight of DIAm. The total number of DIAm fibers was estimated by multiplying the fiber/weight ratio of the individual DIAm strip by total weight of the DIAm. The innervation ratio was calculated by dividing the number of DIAm fibers by the mean number of phrenic MNs, as assessed above. In a subset of mice (n = 3 wild type, n = 1 spa), there was a within-animal assessment of phrenic MN number, the total number of DIAm fibers, and the consequent innervation ratio.
Estimation of Fiber Type-specific Percentage of Diaphragm Muscle Fibers
Serial 10-μm transverse sections were cut using a Reichert Jung Frigocut 2800 Cryostat from fresh-frozen DIAm samples. Sections were fixed in acetone for 10 min before commencing immunofluorescence staining protocols. Sections were blocked for 30 min in 10% goat serum and incubated overnight at 4°C in primary antibodies for the following MyHC isoforms: MyHCSlow (BA-F8, 1:3 dilution; Developmental Studies Hybridoma Bank, Iowa City, IA) and MyHC2A (SC-71, 1:3 dilution; Developmental Studies Hybridoma Bank). Fluorescently conjugated secondary antibodies were then applied at a 1:200 dilution, using Alexa Fluor 488 to visualize MyHCSlow and Alexa Fluor 568 to visualize MyHC2A. Transverse DIAm sections were imaged using a ×20 oil-immersion objective (NA 1.0) on an Olympus FV2000 laser confocal microscope capable of simultaneous multilabel fluorescence imaging. Images were captured in a 1,200 × 1,200 pixel array, with similar acquisition parameters across preparations. Based on the staining pattern, DIAm fibers were classified as Type I, Type IIa, and Type IIx and/or IIb as outlined previously using morphometric tools in ImageJ (47–50).
Diaphragm Neuromuscular Junction Morphology
The entire DIAm was dissected and pinned on a sylgard-coated dish with the thoracic side up and processed to visualize presynaptic and postsynaptic domains of the neuromuscular junction as previously described (51–54). Briefly, the DIAm was fixed in 4% paraformaldehyde and incubated with α-bungarotoxin–conjugated to Alexa Fluor 488 (0.1 μg/mL; B35451, Invitrogen Corp., Carlsbad CA) to label the postsynaptic cholinergic receptors. Anti-SV2A antibody was used to label presynaptic terminals (1 mg/mL; Developmental Studies Hybridoma Bank, Iowa City, IA) with an Alexa 594-conjugated goat anti-mouse IgG secondary antibody (1:200; 715605159, Jackson ImmunoResearch Laboratories Inc., West Grove, PA). Images were acquired using an Olympus FV1200 confocal microscope and a ×60 water immersion objective (NA 1.3). Images were digitized in a 1,200 × 1,200 xy pixel array in a Z-stack series with a 1-μm step. Images of labeled axon terminals and motor endplates were circumscribed using Image J software with thresholding of the fluorescence intensities of α-bungarotoxin–labeled motor endplates and SV2-labeled axon terminals that were then converted into binary images. The planar area of the presynaptic terminals that were co-localized to the postsynaptic endplate was expressed as a percentage of the total planar postsynaptic area (i.e., % invasion), in a manner similar to past reports (44, 53).
Phrenic Nerve-diaphragm Muscle Preparation for Neuromuscular Transmission
The DIAm, with the intact phrenic nerve attached, was excised from the body, placed in a tissue bath containing Rees-Simpson’s solution (pH 7.4) at 26°C, and gassed with carbogen (95% O2:5% CO2). The rib insertions of the DIAm were secured by a clamp, and the central tendon was attached to a force transducer (Aurora Scientific, 6350, Cambridge Technology, MA) using non-compliant sutures. Readouts of the force transducer were digitized and recorded using LabChart software (ADInstruments, Dunedin, New Zealand). Optimal DIAm length and supramaximal stimulus settings were established in a manner identical to previous reports (20, 44, 55–57). Neuromuscular transmission was assessed during supramaximal stimulation (701 C, Aurora Scientific, ON, Canada) at rates of 10, 40, and 75 Hz. Assessment of NMTF at 40 Hz phrenic nerve stimulation was conducted in a separate set of animals, whereas assessment at 10 and 75 Hz stimulation was conducted in the same animals in a manner identical to past reports (20, 56). At all stimulation rates, the train duration was set to 330 ms providing a 33% duty cycle but with varying numbers of stimulus pulses (3, 13, or 25, respectively). At all stimulation frequencies, the phrenic nerve was stimulated via a suction electrode using 0.05-ms duration supramaximal (∼10 mA) current pulses. Every 15 s during the 120-s period of nerve stimulation, direct muscle stimulation was superimposed using platinum plate electrodes placed on either side of the muscle strip. Direct muscle stimulation used supramaximal (∼150 mA) current pulses of 0.5-ms duration.
Assessment of Diaphragm Neuromuscular Transmission Failure
With NMTF, the affected muscle fibers are not activated by nerve stimulation, and thus during repetitive nerve stimulation, these fibers are spared from muscle-derived fatigue (58, 59). Accordingly, increased NMTF reflects greater differences between forces evoked by nerve compared to direct muscle stimulation. Based on the differences in DIAm forces evoked by phrenic nerve versus direct muscle stimulation, the extent of NMTF was calculated in a manner identical to past reports (20, 23, 44, 55–57). The initial DIAm forces evoked by phrenic nerve stimulation (NFinit) and direct muscle stimulation (MFinit) were measured and the NF and MF measures were repeated every 15 s during the 120-s stimulation period. The extent of NMTF was calculated using the following equation:
Diaphragm muscle-specific force (N/cm2) was calculated in a manner identical to past reports (20, 44, 47, 60, 61) by normalizing DIAm force to the cross-sectional area of the DIAm strip (strip weight/[length × 1.056 (muscle specific gravity)]).
Statistical Methods
All statistical analyses were performed using standard software (GraphPad PRISM Version 8.04, La Jolla, CA). All data was assessed for normality with D’Agostino and Pearson’s tests. For comparisons between two groups, Student’s unpaired two-tailed t tests were used. Where both genotype and stimulation time were factors, two-way ANOVAs with repeated measures were used to compare groups and factors, with Bonferroni post hoc tests when warranted. Statistical significance was established at the P < 0.05 level. The n was the number of mice assessed, except for the neuromuscular junction morphology analysis, where n was the number of junctions assessed. All experimental data are presented as means ± 95% confidence intervals, unless otherwise specified. All percentage comparisons across genotypes are related to the mean of wild type control mice. Investigators were not blind to genotypes during experiments, and post hoc analyses were done in a blinded fashion.
RESULTS
Reduced Number of Phrenic Motor Neurons in Spa Mice
First, we confirmed that there was a reduction in the number of phrenic MNs that survive until young adulthood in spa mice. Maximum intensity projection images through the Z-stack of the phrenic motor pool of wild type and spa mice illustrate the effective labeling of phrenic MNs using nerve-dip (Fig. 1, A and B). In this study, the number of phrenic MNs per hemi-spinal cord was reduced by 32% in spa (140 ± 58) compared to wild type mice (207 ± 18; Student’s unpaired t test, P = 0.003; Fig. 1C).
Figure 1.

Reduced number of phrenic motor neurons in spa mice. Pictomicrographs show maximum intensity projections of confocal stacks of the phrenic motor pool, illustrating specific labeling of phrenic motor neurons via nerve dip in wild type (A) and spa mice (B). Scatterplot (C) shows reduced phrenic motor neuron (PhMNs) numbers in spa (n = 4) compared to wild type (n = 8) mice (P = 0.003). Student’s unpaired t test. All data presented as means ± 95% CI. *P < 0.05.
Average Diaphragm Muscle Motor Unit Innervation Ratio Is Greater in Spa Compared to Wild Type Mice
The DIAm dissected from wild type and spa mice appeared similar (Fig. 2, A and B). There was no difference in the DIAm mass of spa (0.0490 ± 0.0240 g) compared to wild type (0.0619 ± 0.0096 g; P = 0.0918; Student’s unpaired t test) mice. The estimated total number of DIAm fibers was unchanged in spa (10,343 ± 1,999) compared to wild type mice (10,735 ± 2,055; P = 0.76, Student’s unpaired t test; Fig. 2C).
Figure 2.

Unchanged number of DIAm fibers and increased DIAm motor unit innervation ratio in spa mice. Wild type (A) and spa (B) DIAm fibers stained with hematoxylin and eosin. C: no difference in the total DIAm fiber number between wild type and spa mice (P = 0.7554, Student’s unpaired t-test). The DIAm innervation ratio (muscle fibers per MN) (D) was greater in spa mice compared to wild type (P = 0.0123, Student’s unpaired t test), with within-animal subset denoted by gray-shaded data points. All data presented as means ± 95% CI, n = 6 wild type and n = 3 spa. *P < 0.05. Scale bar: 100 μm.
The mean innervation ratio (the number of DIAm muscle fibers innervated by an individual phrenic MN) was calculated by dividing the total number of DIAm fibers by the number of phrenic MNs, as established for spa (280 phrenic MNs) and wild type (414 phrenic MNs) mice in this study. The average DIAm motor unit innervation ratio was 39% greater in spa (35.2 ± 6.8) compared to wild type mice (25.3 ± 4.8; P = 0.0123, Student’s unpaired t test; Fig. 2D). In the figure, the animals in which we determined the within-animal innervation ratio (i.e., phrenic MN number and DIAm total fibers in the same animal) are indicated in the scatter-plot (Fig. 2D) by shaded circles (wild type) or squares (spa).
Diaphragm Fiber Type % Is Similar between Spa and Wild Type Mice
The proportion of DIAm fiber types was examined in wild type and spa mice. In both groups, fiber types were not evenly distributed (F2,27 = 451.5; P < 0.0001), with Type IIa DIAm fibers being the most prevalent (∼77%), with no difference between spa and wild type mice (F1,21 < 0.1; P > 0.99, two-way ANOVA). The proportion of Type I fibers (∼12%) and Type IIx and/or IIb fibers (∼11%) was also similar in wild type and spa mice.
Diaphragm Neuromuscular Junction Morphology Is Similar between spa and Wild Type Mice
A total of 64 wild type and 73 spa neuromuscular junctions from 9 different animals (4 wild type and 5 spa) were analyzed morphologically (Fig. 3, A and B). The relative area of neuromuscular junction postsynaptic endplates occupied by presynaptic axon terminals (% invasion) was unchanged between wild type (55 ± 3%) and spa mice (53 ± 3%; P = 0.3046, Student’s unpaired t test; Fig. 3C). Fragmentation of the motor-endplate area of the neuromuscular junctions, a measure of complexity of the neuromuscular junctions, did not differ between wild type (8.30 ± 0.83 μm2) and spa mice (8.64 ± 1.01 μm2; P = 0.6034, Student’s unpaired t test; Fig. 3D). The area of presynaptic axon terminals within a motor endplate did not differ between wild type (143 ± 15 μm2) and spa mice (139 ± 14 μm2; P = 0.6972, Student’s unpaired t test). Similarly, the endplate area did not differ between wild type (261 ± 22 μm2) and spa mice (263 ± 20 μm2; P = 0.8565, Student’s unpaired t test). The percent of DIAm fibers with confirmed neuromuscular junctions did not differ between wild type (83 ± 8%) and spa mice (87 ± 11%; P = 0.4530, Student’s unpaired t test; Fig. 3E).
Figure 3.
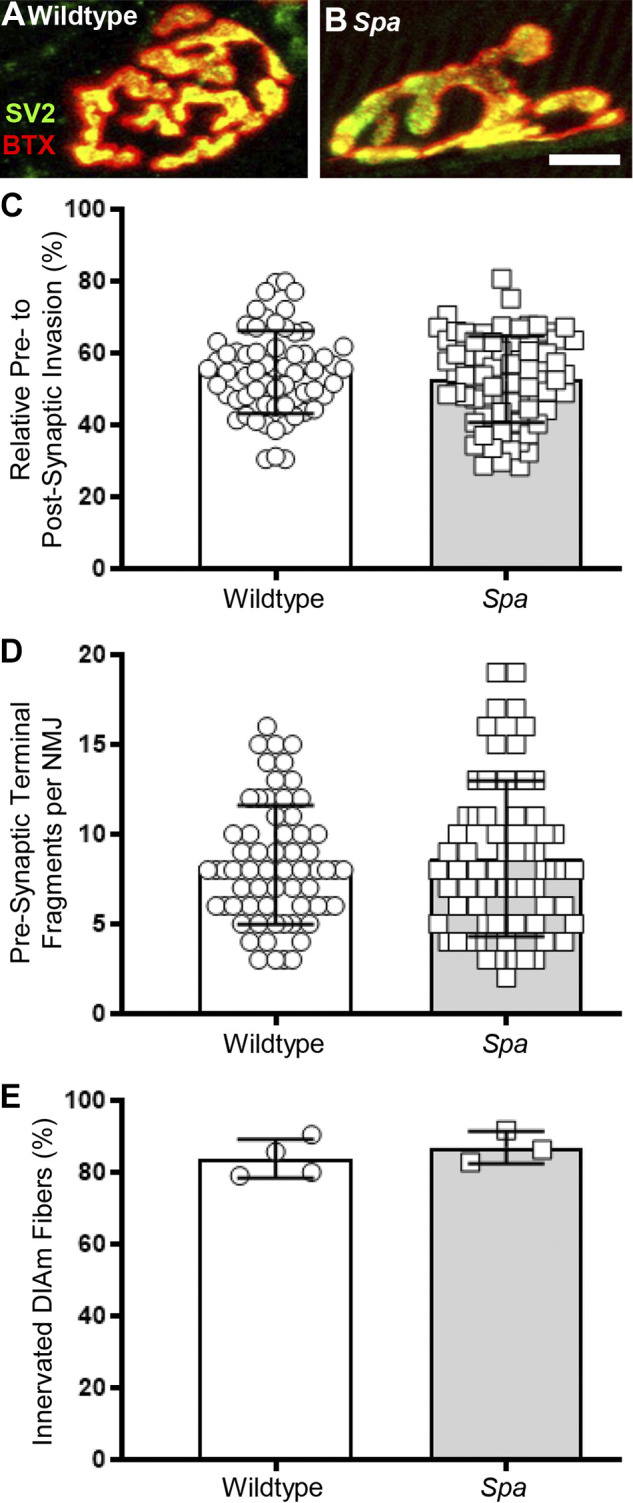
Invasion of DIAm neuromuscular junction endplates by phrenic nerve axon terminals is unchanged between wild type and spa mice. Wild type (A) and spa (B) DIAm neuromuscular junction axon terminals labeled by SV2 (green) and endplate acetylcholine receptors labeled by α-bungarotoxin (red). The axonal invasion of the neuromuscular junction is co-localized to yellow. C shows no difference in axonal invasion % (co-localization) of DIAm neuromuscular junctions of wild type (○) and spa mice (□; P = 0.3046). D shows no difference in fragmentation of the motor-endplate area of the neuromuscular junctions of wild type (○) and spa mice (□; P = 0.6034). E shows no difference in the percent of DIAm fibers innervated by neuromuscular junctions of wild type (○) and spa mice (□; P = 0.4530, Student’s unpaired t test). All data presented as means ± 95% CI, n = 64 neuromuscular junctions from 4 wild type mice and n = 73 neuromuscular junctions from 5 spa mice. Scale bar: 10 μm.
Diaphragm Neuromuscular Transmission Failures during the First Stimulus Train Are Frequency Dependent in Spa Mice
In wild type and spa mice, the extent of NMTF during the initial train of phrenic nerve stimulation was assessed at 10, 40, and 75 Hz. Representative tracings of the DIAm forces evoked by 40 Hz phrenic nerve stimulation compared to 40 Hz direct muscle stimulation in the first stimulus train are shown for wild type and spa mice in Fig. 4A. In spa mice, the DIAm-specific force generated by phrenic nerve stimulation in the initial stimulus train was significantly less than that evoked in wild type mice (F3,56 = 0.3, P = 0.0004; two-way ANOVA) and was stimulation frequency dependent (F2,56 = 5.4, P < 0.0001; two-way ANOVA).
Figure 4.

Increased failure of neuromuscular transmission during first stimulus train at 40 or 75 Hz in spa mice. A: representative traces of DIAm force evoked by phrenic nerve or superimposed direct muscle stimulation at 40 Hz. B: increased NMTF in the initial (first train) comparison of DIAm forces evoked by phrenic nerve stimulation (means ± 95% CI) in spa (□) compared to wild type (○) mice at 10 (wild type, n = 5; spa, n = 5), 40 (wild type, n = 9; spa, n = 5), and 75 Hz (wild type, n = 5; spa, n = 5). Post tests: 10 Hz, P = 0.82; 40 Hz, P = 0.006; 75 Hz, P = 0.016. Two-way ANOVA with Bonferroni post hoc tests. All data presented as means ± 95% CI. *P < 0.05.
Within the first stimulus train, NMTF was negligible for wild type DIAm at 10 (−9.4 ± 5.9%), 40 (−7.0 ± 4.5%), and 75 Hz (−6.8 ± 4.2%) stimulations (Fig. 4B). There was markedly more NMTF in the initial stimulus train of spa mice compared to wild type (F1,29 = 9.3, P = 0.0001; two-way ANOVA), with an ∼2-fold increase in initial NMTF in spa mice during 40 Hz (−18.5 ± 11.7%; P = 0.006, Bonferroni post hoc test) and 75 Hz (−18.6 ± 5.6%; P = 0.016, Bonferroni post hoc test) stimulations (Fig. 4B).
Susceptibility to NMTF during 120 s of Repeated Stimulation Is Greater in DIAm of Spa Compared to Wild Type Mice and Is Frequency Dependent
Representative tracings of the DIAm forces evoked by repetitive phrenic nerve stimulation compared to superimposed direct muscle stimulation are shown for wild type and spa mice at 10 (Fig. 5A), 40 (Fig. 5B), and 75 Hz (Fig. 5C) stimulation. During the 120-s period of repeated 10 Hz stimulation, there was an effect of time (F8,64 = 5.0; P < 0.0001), but not genotype (F1,8 = 0.2; P = 0.67) on the extent of NMTF (two-way ANOVA; Fig. 5D). At repeated 40 Hz stimulation, there was also an effect of time (F8,96 = 36.6; P < 0.0001) as well as genotype (F1,12 = 49.2; P < 0.0001) on the extent of NMTF (two-way ANOVA; Fig. 5E). Similarly, at repeated 75 Hz stimulation, there was an effect of time (F8,64 = 57.8; P < 0.0001) and genotype (F1,8 = 6.6; P = 0.034) on the extent of NMTF (two-way ANOVA; Fig. 5F).
Figure 5.

Increased NMTF during repetitive stimulation at 40 or 75 Hz in DIAm of spa mice compared to wild type. Example traces show DIAm force evoked by phrenic nerve stimulation across a 120-s period at 10 (A), 40 (B), and 75 Hz (C), with direct muscle stimulations superimposed every 15 s in wild type (top row) and spa (bottom row) mice. D: unchanged extent of NMTF (means ± 95% CI) in spa mice (□; n = 5) compared to wild type (○; n = 5) mice during 120 s of stimulation at 10 Hz (two-way ANOVA with Bonferroni post hoc test, P = 0.67). E: a greater extent of NMTF (means ± 95% CI) in spa mice (squares; n = 5) compared to wild type (○; n = 9) mice during 120 s of stimulation at 40 Hz (two-way ANOVA with Bonferroni post hoc test, P = 0.0001). F: a greater extent of NMTF (means ± 95% CI) in spa mice (□; n = 5) compared to wild type (○; n = 5) mice during 120 s of stimulation at 75 Hz (two-way ANOVA with Bonferroni post hoc test, P = 0.03). All data presented as means ± 95% CI. In all cases, *P < 0.05.
The final extent of NMTF following 120 s of repeated nerve stimulation was dependent on stimulation frequency (F2,27 = 115.6; P < 0.0001) and genotype (F1,27 = 68.1; P < 0.0001; two-way ANOVA; Fig. 6). Final NMTF in wild type mice is greater with increasing frequency of stimulation in rank order: 10 (−19.8 ± 13.6%), 40 (−40.3 ± 6.9%), and 75 Hz (−52.3 ± 12.1%) (P ≤ 0.001 for all comparisons, Bonferroni post hoc tests). Following repeated 10 Hz stimulation, there was no significant difference in final NMTF between wild type and spa mice (P > 0.99, Bonferroni post hoc test; Fig. 6). However, in spa mice following repeat stimulation at 40 (−81.9 ± 6.7%; P < 0.0001, Bonferroni post hoc test) and 75 Hz (−82.4 ± 6.2%; P = 0.0008, Bonferroni post hoc test), there was a marked increase in final NMTF when compared to wild type (Fig. 6).
Figure 6.

Increased final NMTF in spa mice compared to wild type following 120 s of 40 or 75 Hz stimulation. Scatterplot shows greater extent of NMTF in spa (□) compared to wild type (○) mice, expressed as % of final muscle-evoked force at 10 (wild type, n = 5; spa, n = 5), 40 (wild type, n = 9; spa, n = 5), and 75 Hz (wild type, n = 5; spa, n = 5) stimulation (P < 0.0001, two-way ANOVA with Bonferroni post hoc tests). Post-tests: 10 Hz, P > 0.99; 40 Hz, P = 0. 0001; 75 Hz, P = 0.0008. All data presented as means ± 95% CI. *P < 0.05.
DISCUSSION
The present study, which examined DIAm NMTF in an animal model of early onset hypertonia, produced seven main findings: 1) the number of phrenic MNs was reduced in spa compared to wild type mice; 2) the number of DIAm fibers is not different between spa and wild type mice; 3) all DIAm fibers are innervated (displayed neuromuscular junctions) in both spa and wild type mice; 4) average DIAm motor unit innervation ratio is greater in spa compared to wild type mice; 5) the extent of DIAm NMTF is greater in spa compared to wild type mice; 6) the extent of DIAm NMTF worsens with time during repeated phrenic nerve stimulation with this effect more pronounced in spa compared to wild type mice; and 7) the extent of DIAm NMTF is dependent on stimulation frequency in both genotypes.
Spa mice are known to have motor neuron loss in a variety of different motor pools, with the current study showing a ∼32% loss of phrenic MNs, with the results within and between genotypes similar to our past report on phrenic motor neuron numbers of slightly older spa and wild type mice (18). Previously in adult spa mice, we found a reduced number of tibialis anterior MNs in the lumbar spinal cord (12) concomitant with an increased innervation ratio and NMTF in the tibialis anterior muscle (19). In human CP, there is evidence for MN loss based on electrophysiology (62), although MN loss has not been directly assessed. In the present study, we observed no difference in the total number of DIAm fibers and no difference in the proportion of different muscle fiber types between spa and wild type mice. However, due to fewer phrenic MNs in spa mice, the average innervation ratio of DIAm motor units in spa mice is greater. Importantly, all DIAm fibers in spa mice had neuromuscular junctions that appeared morphologically normal. Thus, the reduced DIAm force evoked by phrenic nerve stimulation compared to direct muscle stimulation in spa mice was most likely due to gross changes in axonal branching rather than innervation of muscle fibers. This also suggests that the loss of phrenic MNs in spa mice occurred early in development, perhaps at the time of synapse elimination or before.
In the present study, we did not directly determine the innervation ratio of different types of DIAm motor units or when innervation ratio expanded in spa mice. Although in a subset of animals, we did observe that within-animal estimates of DIAm innervation ratio closely resemble those of population-based estimates. Regardless, the age of onset of spa symptoms between 2 and 4 weeks old (63, 64) is coincident with the emergence of FInt and FF DIAm motor units (39, 65, 66) and the expression of 2X and 2B isoforms of myosin heavy chain (MyHC2X and MyHC2B) in DIAm fibers (67, 68). In spa mice, larger phrenic MNs are disproportionately lost, with these larger MNs most likely comprising FInt and FF units (65, 69–72). In the case of spa mice, it is unclear if changes in innervation ratio are restricted to a motor unit type or if they are homogeneous across all motor units. In the cat, the innervation ratio of DIAm motor units is similar across type S, FR, FInt, and FF motor units (73, 74), a phenomenon unlike limb muscles, where innervation ratios are greater in FInt and FF units compared to type S and FR (75, 76). In spa mice there are fewer large phrenic MNs that typically comprise FInt and FF motor units (18). Thus, we speculate that the innervation ratios of these motor units increase, whereas the innervation ratios of type S and FR units, comprising the spared smaller phrenic MNs, are unaffected. In support, we saw no shift in the relative proportions of DIAm MyHC isoform expression between spa and wild type, suggesting that the same number of Type IIx and/or IIb DIAm fibers must be innervated by a dwindling number of phrenic MNs. This interpretation is consistent with what we observed with aging, where there is also a selective loss of larger phrenic MNs (21), and only expulsive/straining maneuvers necessitating the recruitment of FInt and FF units are impaired (77), with preservation of ventilatory behaviors (47, 77). A limitation of the present study is that we cannot rule out that the innervation ratio of all DIAm motor unit types is increased, although we find this interpretation to be unlikely as we were unable to observe substantial differences in NMTF between wild type and spa mice during the low-range of stimulation, 10 Hz, that approximated the discharge rate of S and FR DIAm motor units (78), recruited to perform eupneic behaviors.
Loss of neuromuscular junction morphological integrity may, in some cases, lead to increased NMTF (20, 23, 79). Despite a substantially greater innervation ratio of DIAm motor units, due to reduced phrenic MNs in spa mice (18), we observed that all DIAm fibers were innervated with no evidence of morphological differences of pre- or postsynaptic elements of neuromuscular junctions between spa and wild type mice. Our results show that presynaptic terminal invasion of the postsynaptic endplate was similar in wild type and spa mice, albeit over a wide range (∼30–70%). These results are similar to past studies in wild type mice (44), and in other mutant models of perturbed inhibitory neuromuscular transmission, where the ultrastructure of mutant neuromuscular junctions was reported to be unchanged compared to controls when assessed with electron microscopy (27, 28). Despite observing no anatomical differences at the light microscopy level, we did not investigate neuromuscular ultrastructure, nor any specific molecular markers for acetylcholine receptor clustering or Schwann cell integration, factors that could conceivably result in altered neuromuscular transmission without being observed with fluorescent assessment of neurofilament/synaptophysin and α-bungarotoxin. Previously, in a mouse model of spinal muscle bulbar atrophy (a progressive neuromotor disease), confocal techniques were insufficient to determine neuromuscular junction synaptic cleft alterations and changes in synaptic vesicle docking (80). Indeed, anatomical assessment in some scenarios may be totally unrelated to the fidelity of neuromuscular transmission (81). In studies of amyotrophic lateral sclerosis, the Schwann cell support of neuromuscular junctions is highly impaired (82), with recent studies suggesting that this reduction in glial activity may occur before denervation (83, 84). In early-onset developmental conditions, such as in spa mice, studies of Schwann cells have been relatively rare, despite their known effect on postnatal synaptic elimination (85). In future studies, comprehensive fiber-type specific assessment of neuromuscular junctions may shed light on any “hidden” deficits. Indeed, there is some limited evidence of neuromuscular junction abnormalities in children with CP with respect to distribution of acetylcholinesterase relative to acetylcholine receptors (86, 87), it is unknown whether these individuals had previous exposure to botulinum toxins, which could alter neuromuscular junction morphology (88). One study evaluated presynaptic terminal invasion of the postsynaptic endplate of neuromuscular junctions and found no difference between children with CP and those who did not have CP (89).
The extent of NMTF during the first stimulus train, as measured by the difference between DIAm force evoked by phrenic nerve versus direct muscle stimulation was negligible (∼9%) across all stimulation frequencies in wild type mice, consistent with past rodent studies (20, 22, 56). In spa mice, the extent of DIAm NMTF during the first stimulus train was ∼20% at 40 and 75 Hz, indicating that neuromuscular transmission is impaired at higher activation rates in spa mice even without continued repetitive stimulation. Our observation of a greater extent of DIAm NMTF during the first stimulus train at 40 and 75 Hz in spa mice is consistent with results from other conditions of MN loss, including aging (21, 90) and amyotrophic lateral sclerosis (91, 92), where NMTF during the first stimulus train is readily apparent (20, 93). During the 120-s period of repetitive phrenic nerve stimulation at 40 Hz, the extent of DIAm NMTF worsened in wild type mice (∼39%), but to a much greater extent in spa mice (∼82%). The results for wild type mice are commensurate with past results in young adult rodents (20, 22, 44, 56). The greater extent of DIAm NMTF in spa mice may occur at multiple levels of neuromuscular communication including a general failure of axonal action potential propagation, a specific failure of propagation at axonal branch points, a failure of presynaptic vesicle release, and/or a derangement in the integrity of presynaptic or post synaptic neuromuscular junction components (23, 56). In addition, DIAm NMTF is more likely to occur in fast fatigue-intermediate (type FInt) and fast-fatigable (type FF) motor units compared to slow (type S) and fast fatigue-resistant (type FR) motor units (20, 23, 56).
To indirectly assess possible differences across motor unit types in the DIAm, we compared the extent of DIAm NMTF during 10 Hz stimulation to that during 75 Hz phrenic nerve stimulation. In previous studies, indirect evidence suggests that type S and FR DIAm motor units are less susceptible to NMTF across a range of low (10 Hz) and high frequencies of stimulation, whereas at higher stimulation frequencies, Type FInt and FF motor units are more susceptible to NMTF (20, 56). In the present study, the extent of DIAm NMTF was greater at 75 Hz phrenic nerve stimulation compared to 10 Hz stimulation in both wild type and spa mice. The reduction of DIAm force evoked by phrenic nerve stimulation at 75 Hz in spa mice was 68% greater than the reduction in DIAm force evoked in wild type mice. It is likely that at 75 Hz stimulation, NMTF occurred predominantly at Type IIx and/or IIb DIAm fibers that comprise FInt and FF DIAm motor units (65, 69, 70, 94). These FInt and FF DIAm motor units generate greater force per cross-sectional area compared to Type I and IIa fibers (72, 95).
With an expansion of DIAm motor unit innervation ratio in spa mice, there is increased axonal branching that increases susceptibility to NMTF via axonal branch point failure. With repetitive stimulation, Type FInt and FF units are more susceptible to axonal propagation failure, particularly at higher stimulation frequencies (56, 96, 97). In mature rodents, MN loss such as that with aging (21) and amyotrophic lateral sclerosis (91, 98, 99) results in increased axonal branching and re-innervation of vacant endplates on muscle fibers by spared motor axons (100–102). In spa mice, it is currently unknown whether the restrictions in phrenic MN size and increased NMTF are related to excessive programmed cell death during embryonic development or early postnatal loss of MNs due to an imbalance of excitatory/inhibitory input. The former is more likely for three main reasons. First, in other mutant mice with impaired glycinergic neurotransmission, reduced survival of phrenic MNs is evident in the last embryonic trimester (27, 29). Although these mice do not survive birth, these changes occur when polyneuronal innervation of DIAm fibers would likely compensate for any phrenic MN loss and precedes the elimination of polyneuronal innervation of DIAm fibers by ∼2–4 weeks of age (66). Second, in cases where there is excitatory/inhibitory synaptic input imbalances or intrinsic hyper-excitability of MNs, such as in the SOD1 amyotrophic lateral sclerosis models (98, 103, 104), severe neuromuscular junction degeneration is apparent in adults (105, 106). In spa mice, there are no readily apparent morphological neuromuscular junction abnormalities, making degeneration following the elimination of polyneuronal innervation less likely. Third, the onset of symptoms at ∼2–4 weeks old is coincident with the rapid growth of MNs and muscle fibers of type FInt and FF motor units (39, 65, 66). In SOD1 models where MN loss and degeneration occurs by ∼30 days of age (the very earliest reports) (107), overt symptoms are not detectable until ∼50–60 days (98, 107). Taken together in the context of other mutant mice with MN loss phenotypes, it is likely that the spa phenotype is a product of pre- or postnatal developmental derangement, rather than degeneration. Indeed, the phenotype and time of onset of hypertonia in spa mice is highly analogous to that of humans with CP.
The respiratory phenotype in CP is often not the first concern for families and guardians of children and individuals with CP. However, respiratory dysfunction, not gait is the prime determinant of morbidity and mortality in individuals with CP (7, 108–110), with an increased risk of respiratory disease, associated with increased severity (based on the Gross Motor Functional Classification System (GMFCS)) of CP symptoms (111–114). For children with CP, ∼60% had daily episodes of dyspnea (cough or wheeze), ∼10% had obstructive sleep apnea, 40% had a cough with drinking, and 20% had abnormal pulmonary function on clinical exam (115). With regard to pulmonary function, ∼35% of children with CP cannot perform breath holding tasks, and those that can do so for <50% of the time as a typically developing child (6). Individuals with mild CP do exhibit reductions in forced vital capacity, forced expiratory volume at 1 s, peak expiratory flow, slow volume capacity (i.e., maximum volume of air that can be exhaled slowly after slow maximum inhalation), and tidal volume (5). The degree of pulmonary impairment increases with severity of CP, based on GMFCS criteria (8, 116). Notably, treatment with botulinum toxin A causes an immediate adverse reaction risk of ∼1% for all individuals with CP, with those with higher GMFCS levels (indicating greater physical impairment) having risks of ∼6% for immediate adverse reactions and ∼20% for later follow-up reactions (10, 117). These adverse reactions include lower respiratory tract infections (including aspiration pneumonia), dysphagia, and death. An important distinction must be made between pulmonary function and coordination of breathing and swallowing/drinking and airway defense maneuvers. For pulmonary function, the negative thoracic pressures required are relatively low (∼10% of maximum trans-diaphragmatic pressure) (48, 77). The aforementioned impairments in pulmonary tests (5) and ∼35–40% reduction of maximum inspiratory and expiratory pressures in individuals with CP (118) may involve substantial contributions from lungs (although this is often secondary to repeated aspiration episodes (119)) and weakness or stiffness (120) of the chest wall and upper airway muscles in addition to the DIAm. Despite these confounders, it is inarguable that deficits in the neuromotor control and coordination of DIAm are responsible for the increased risk of aspiration in CP (121) and contribute to ineffective expulsive maneuvers (e.g., coughing and sneezing) that require almost compete recruitment of DIAm motor units to sufficiently clear the respiratory tract (65, 122). Indeed, it is likely defects in the ability to perform the latter that contribute most to the increased risk of infection and subsequent morbidity and mortality in the CP population (123).
The present study provides compelling data showing striking deficits in neuromuscular transmission in spa mice. In spa mice, glycine receptor abnormalities lead to a spastic phenotype that closely mirrors that of individuals with hypertonic conditions, including CP. Our results suggest that pathophysiological abnormalities in neuromuscular transmission may contribute to neuromotor symptoms. The pathophysiology underlying the greater susceptibility of spa mice to DIAm neuromuscular transmission failure is likely multifactorial and is yet to be systematically evaluated. Using spa mice, we may explore the relationships between motor neuron and muscle fiber development in association with hypertonic symptoms. These examinations, applied to the respiratory system, are of extreme clinical importance in expanding our understanding of the pathogenesis of hypertonic conditions, such as CP and in characterizing the respiratory deficits that are linked to the high morbidity and mortality in this patient cohort.
GRANTS
This work was supported by National Institutes of Health Grants R01-AG044615 (to G.C.S.) and R01-HL146114 (to G.C.S.), an Australian National Health & Medical Research Council CJ Martin Early Career Fellowship (to M.J.F.), Mayo Clinic Children’s Research Center Pediatric Team Science Award (to J.E.B.), Mayo Clinic CTSA UL1 TR000135 (to J.E.B.), a Mayo Clinic Office of Research Diversity and Inclusion Research Career Support and Advancement Award (to J.E.B.), and a grant from Richard and Rosemary Crandall (to J.E.B.).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author.
AUTHOR CONTRIBUTIONS
M.J.F., J.E.B., and G.C.S. conceived and designed research; M.J.F. and J.E.B. performed experiments; M.J.F., J.E.B., and G.C.S. analyzed data; M.J.F., J.E.B., and G.C.S. interpreted results of experiments; M.J.F. and J.E.B. prepared figures; M.J.F., J.E.B., and G.C.S. drafted manuscript; M.J.F., J.E.B., and G.C.S. edited and revised manuscript; M.J.F., J.E.B., and G.C.S. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Yun-Hua Fang, Jeffrey Bailey, Rebecca Macken, and Chris Duffey for technical assistance in the completion of this project.
REFERENCES
- 1.Brandenburg JE, Fogarty MJ, Sieck GC. A critical evaluation of current concepts in cerebral palsy. Physiology (Bethesda) 34: 216–229, 2019. doi: 10.1152/physiol.00054.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huntsman R, Lemire E, Norton J, Dzus A, Blakley P, Hasal S. The differential diagnosis of spastic diplegia. Arch Dis Child 100: 500–504, 2015. doi: 10.1136/archdischild-2014-307443. [DOI] [PubMed] [Google Scholar]
- 3.Rosenbaum P, Paneth N, Leviton A, Goldstein M, Bax M, Damiano D, Dan B, Jacobsson B. A report: the definition and classification of cerebral palsy April 2006. Dev Med Child Neurol Suppl 109: 8–14, 2007. [Erratum in Dev Med Child Neurol 49: 480, 2007]. [PubMed] [Google Scholar]
- 4.Katz RT, Rymer WZ. Spastic hypertonia: mechanisms and measurement. Arch Phys Med Rehabil 70: 144–155, 1989. [PubMed] [Google Scholar]
- 5.Kwon YH, Lee HY. Differences of respiratory function in children with spastic diplegic and hemiplegic cerebral palsy, compared with normally developed children. J Pediatr Rehabil Med 6: 113–117, 2013. doi: 10.3233/PRM-130246. [DOI] [PubMed] [Google Scholar]
- 6.McPherson KA, Kenny DJ, Koheil R, Bablich K, Sochaniwskyj A, Milner M. Ventilation and swallowing interactions of normal children and children with cerebral palsy. Dev Med Child Neurol 34: 577–588, 1992. doi: 10.1111/j.1469-8749.1992.tb11488.x. [DOI] [PubMed] [Google Scholar]
- 7.Durufle-Tapin A, Colin A, Nicolas B, Lebreton C, Dauvergne F, Gallien P. Analysis of the medical causes of death in cerebral palsy. Ann Phys Rehabil Med 57: 24–37, 2014. doi: 10.1016/j.rehab.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 8.Kwon YH, Lee HY. Differences of respiratory function according to level of the gross motor function classification system in children with cerebral palsy. J Phys Ther Sci 26: 389–391, 2014. doi: 10.1589/jpts.26.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Albavera-Hernández C, Rodríguez JM, Idrovo AJ. Safety of botulinum toxin type A among children with spasticity secondary to cerebral palsy: a systematic review of randomized clinical trials. Clin Rehabil 23: 394–407, 2009. doi: 10.1177/0269215508099860. [DOI] [PubMed] [Google Scholar]
- 10.Naidu K, Smith K, Sheedy M, Adair B, Yu X, Graham HK. Systemic adverse events following botulinum toxin A therapy in children with cerebral palsy. Dev Med Child Neurol 52: 139–144, 2010. doi: 10.1111/j.1469-8749.2009.03583.x. [DOI] [PubMed] [Google Scholar]
- 11.Becker CM, Hermans-Borgmeyer I, Schmitt B, Betz H. The glycine receptor deficiency of the mutant mouse spastic: evidence for normal glycine receptor structure and localization. J Neurosci 6: 1358–1364, 1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brandenburg JE, Gransee HM, Fogarty MJ, Sieck GC. Differences in lumbar motor neuron pruning in an animal model of early onset spasticity. J Neurophysiol 120: 601–609, 2018. doi: 10.1152/jn.00186.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heller AH, Hallett M. Electrophysiological studies with the spastic mutant mouse. Brain Res 234: 299–308, 1982. doi: 10.1016/0006-8993(82)90870-8. [DOI] [PubMed] [Google Scholar]
- 14.Simon ES. Phenotypic heterogeneity and disease course in three murine strains with mutations in genes encoding for alpha 1 and beta glycine receptor subunits. Mov Disord 12: 221–228, 1997. doi: 10.1002/mds.870120213. [DOI] [PubMed] [Google Scholar]
- 15.Graham BA, Schofield PR, Sah P, Margrie TW, Callister RJ. Distinct physiological mechanisms underlie altered glycinergic synaptic transmission in the murine mutants spastic, spasmodic, and oscillator. J Neurosci 26: 4880–4890, 2006. doi: 10.1523/JNEUROSCI.3991-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tadros MA, Farrell KE, Schofield PR, Brichta AM, Graham BA, Fuglevand AJ, Callister RJ. Intrinsic and synaptic homeostatic plasticity in motoneurons from mice with glycine receptor mutations. J Neurophysiol 111: 1487–1498, 2014. doi: 10.1152/jn.00728.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.NINDS and NICHD. NINDS/NICHD Strategic Plan for Cerebral Palsy Research. 2017. https://www.ninds.nih.gov/sites/default/files/NINDS_NICHD_2017_StrategicPlanCerebralPalsyResearch_508C.pdf.
- 18.Brandenburg JE, Fogarty MJ, Brown AD, Sieck GC. Phrenic motor neuron loss in an animal model of early onset hypertonia. J Neurophysiol 123: 1682–1690, 2020. doi: 10.1152/jn.00026.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fogarty MJ, Sieck GC, Brandenburg JE. Impaired neuromuscular transmission of the tibialis anterior in a rodent model of hypertonia. J Neurophysiol, 123: 1864–1869, 2020. doi: 10.1152/jn.00095.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fogarty MJ, Gonzalez Porras MA, Mantilla CB, Sieck GC. Diaphragm neuromuscular transmission failure in aged rats. J Neurophysiol 122: 93–104, 2019. doi: 10.1152/jn.00061.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fogarty MJ, Omar TS, Zhan WZ, Mantilla CB, Sieck GC. Phrenic motor neuron loss in aged rats. J Neurophysiol 119: 1852–1862, 2018. doi: 10.1152/jn.00868.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fournier M, Alula M, Sieck GC. Neuromuscular transmission failure during postnatal development. Neurosci Lett 125: 34–36, 1991. doi: 10.1016/0304-3940(91)90124-c. [DOI] [PubMed] [Google Scholar]
- 23.Sieck GC, Prakash YS. Fatigue at the neuromuscular junction: branch point vs. presynaptic vs. postsynaptic mechanisms. Adv Exp Med Biol 384: 83–100, 1995. [PubMed] [Google Scholar]
- 24.Zhan WZ, Miyata H, Prakash YS, Sieck GC. Metabolic and phenotypic adaptations of diaphragm muscle fibers with inactivation. J Appl Physiol (1985) 82: 1145–1153, 1997. doi: 10.1152/jappl.1997.82.4.1145. [DOI] [PubMed] [Google Scholar]
- 25.Banks GB, Chau TN, Bartlett SE, Noakes PG. Promotion of motoneuron survival and branching in rapsyn-deficient mice. J Comp Neurol 429: 156–165, 2001. doi:. [DOI] [PubMed] [Google Scholar]
- 26.Banks GB, Choy PT, Lavidis NA, Noakes PG. Neuromuscular synapses mediate motor axon branching and motoneuron survival during the embryonic period of programmed cell death. Dev Biol 257: 71–84, 2003. doi: 10.1016/s0012-1606(03)00056-3. [DOI] [PubMed] [Google Scholar]
- 27.Banks GB, Kanjhan R, Wiese S, Kneussel M, Wong LM, O’Sullivan G, Sendtner M, Bellingham MC, Betz H, Noakes PG. Glycinergic and GABAergic synaptic activity differentially regulate motoneuron survival and skeletal muscle innervation. J Neurosci 25: 1249–1259, 2005. [Erratum in J Neurosci 25: 3018–3021, 2005]. doi: 10.1523/JNEUROSCI.1786-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fogarty MJ, Smallcombe KL, Yanagawa Y, Obata K, Bellingham MC, Noakes PG. Genetic deficiency of GABA differentially regulates respiratory and non-respiratory motor neuron development. PLoS One 8: e56257, 2013. doi: 10.1371/journal.pone.0056257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fogarty MJ, Yanagawa Y, Obata K, Bellingham MC, Noakes PG. Genetic absence of the vesicular inhibitory amino acid transporter differentially regulates respiratory and locomotor motor neuron development. Brain Struct Funct 220: 525–540, 2015. doi: 10.1007/s00429-013-0673-9. [DOI] [PubMed] [Google Scholar]
- 30.Drummond GB. Reporting ethical matters in the Journal of Physiology: standards and advice. J Physiol 587: 713–719, 2009. doi: 10.1113/jphysiol.2008.167387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Becker CM, Schmieden V, Tarroni P, Strasser U, Betz H. Isoform-selective deficit of glycine receptors in the mouse mutant spastic. Neuron 8: 283–289, 1992. doi: 10.1016/0896-6273(92)90295-o. [DOI] [PubMed] [Google Scholar]
- 32.Singer JH, Talley EM, Bayliss DA, Berger AJ. Development of glycinergic synaptic transmission to rat brain stem motoneurons. J Neurophysiol 80: 2608–2620, 1998. doi: 10.1152/jn.1998.80.5.2608. [DOI] [PubMed] [Google Scholar]
- 33.Graham BA, Schofield PR, Sah P, Callister RJ. Altered inhibitory synaptic transmission in superficial dorsal horn neurones in spastic and oscillator mice. J Physiol 551: 905–916, 2003. doi: 10.1113/jphysiol.2003.049064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.White WF, Heller AH. Glycine receptor alteration in the mutant mouse spastic. Nature 298: 655–657, 1982. doi: 10.1038/298655a0. [DOI] [PubMed] [Google Scholar]
- 35.Brandenburg JE, Fogarty MJ, Sieck GC. Growth and survival characteristics of spa mice. Anim Models Exp Med 1–6, 2020. doi: 10.1002/ame2.12137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Molon A, Di Giovanni S, Hathout Y, Natale J, Hoffman EP. Functional recovery of glycine receptors in spastic murine model of startle disease. Neurobiol Dis 21: 291–304, 2006. doi: 10.1016/j.nbd.2005.05.030. [DOI] [PubMed] [Google Scholar]
- 37.National Research Council. Guide for the Care and Use of Laboratory Animals. Washington, DC: The National Academies Press, 1996, p. 140. [Google Scholar]
- 38.Mantilla CB, Zhan WZ, Sieck GC. Retrograde labeling of phrenic motoneurons by intrapleural injection. J Neurosci Methods 182: 244–249, 2009. doi: 10.1016/j.jneumeth.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Prakash YS, Mantilla CB, Zhan WZ, Smithson KG, Sieck GC. Phrenic motoneuron morphology during rapid diaphragm muscle growth. J Appl Physiol (1985) 89: 563–572, 2000. doi: 10.1152/jappl.2000.89.2.563. [DOI] [PubMed] [Google Scholar]
- 40.Ogilvy CS, Borges LF. A quantitative analysis of the retrograde axonal transport of 4 different fluorescent dyes in peripheral sensory and motor neurons and lack of anterograde transport in the corticospinal system. Brain Res 475: 244–253, 1988. doi: 10.1016/0006-8993(88)90612-9. [DOI] [PubMed] [Google Scholar]
- 41.Gransee HM, Gonzalez Porras MA, Zhan WZ, Sieck GC, Mantilla CB. Motoneuron glutamatergic receptor expression following recovery from cervical spinal hemisection. J Comp Neurol 525: 1192–1205, 2017. doi: 10.1002/cne.24125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gransee HM, Zhan W-Z, Sieck GC, Mantilla CB. Targeted delivery of TrkB receptor to phrenic motoneurons enhances functional recovery of rhythmic phrenic activity after cervical spinal hemisection. PLoS One 8: e64755, 2013. doi: 10.1371/journal.pone.0064755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Prakash YS, Fournier M, Sieck GC. Effects of prenatal undernutrition on developing rat diaphragm. J Appl Physiol (1985) 75: 1044–1052, 1993. doi: 10.1152/jappl.1993.75.3.1044. [DOI] [PubMed] [Google Scholar]
- 44.Sieck DC, Zhan WZ, Fang YH, Ermilov LG, Sieck GC, Mantilla CB. Structure-activity relationships in rodent diaphragm muscle fibers vs. neuromuscular junctions. Respir Physiol Neurobiol 180: 88–96, 2012. doi: 10.1016/j.resp.2011.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Greising SM, Mantilla CB, Gorman BA, Ermilov LG, Sieck GC. Diaphragm muscle sarcopenia in aging mice. Exp Gerontol 48: 881–887, 2013. doi: 10.1016/j.exger.2013.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. Fiji: an open-source platform for biological-image analysis. Nat Methods 9: 676–682, 2012. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fogarty MJ, Mantilla CB, Sieck GC. Impact of sarcopenia on diaphragm muscle fatigue. Exp Physiol 104: 1090–1099, 2019. doi: 10.1113/EP087558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Khurram OU, Fogarty MJ, Rana S, Vang P, Sieck GC, Mantilla CB. Diaphragm muscle function following midcervical contusion injury in rats. J Appl Physiol (1985) 126: 221–230, 2019. doi: 10.1152/japplphysiol.00481.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Prakash YS, Sieck GC. Age-related remodeling of neuromuscular junctions on type-identified diaphragm fibers. Muscle Nerve 21: 887–895, 1998. doi:. [DOI] [PubMed] [Google Scholar]
- 50.Tyagi S, Beqollari D, Lee CS, Walker LA, Bannister RA. Semi-automated analysis of mouse skeletal muscle morphology and fiber-type composition. J Vis Exp 126: 56024, 2017. doi: 10.3791/56024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gonzalez Porras MA, Fogarty MJ, Gransee HM, Sieck GC, Mantilla CB. Frequency-dependent lipid raft uptake at rat diaphragm muscle axon terminals. Muscle Nerve 59: 611–618, 2019. doi: 10.1002/mus.26421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mantilla CB, Rowley KL, Zhan WZ, Fahim MA, Sieck GC. Synaptic vesicle pools at diaphragm neuromuscular junctions vary with motoneuron soma, not axon terminal, inactivity. Neuroscience 146: 178–189, 2007. doi: 10.1016/j.neuroscience.2007.01.048. [DOI] [PubMed] [Google Scholar]
- 53.Prakash YS, Smithson KG, Sieck GC. Growth-related alterations in motor endplates of type-identified diaphragm muscle fibres. J Neurocytol 24: 225–235, 1995. doi: 10.1007/BF01181536. [DOI] [PubMed] [Google Scholar]
- 54.Sieck GC, Van Balkom RH, Prakash YS, Zhan WZ, Dekhuijzen PN. Corticosteroid effects on diaphragm neuromuscular junctions. J Appl Physiol (1985) 86: 114–122, 1999. doi: 10.1152/jappl.1999.86.1.114. [DOI] [PubMed] [Google Scholar]
- 55.Greising SM, Ermilov LG, Sieck GC, Mantilla CB. Ageing and neurotrophic signalling effects on diaphragm neuromuscular function. J Physiol 593: 431–440, 2015. doi: 10.1113/jphysiol.2014.282244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Johnson BD, Sieck GC. Differential susceptibility of diaphragm muscle fibers to neuromuscular transmission failure. J Appl Physiol (1985) 75: 341–348, 1993. doi: 10.1152/jappl.1993.75.1.341. [DOI] [PubMed] [Google Scholar]
- 57.Kuei JH, Shadmehr R, Sieck GC. Relative contribution of neurotransmission failure to diaphragm fatigue. J Appl Physiol (1985) 68: 174–180, 1990. doi: 10.1152/jappl.1990.68.1.174. [DOI] [PubMed] [Google Scholar]
- 58.Rowley KL, Mantilla CB, Ermilov LG, Sieck GC. Synaptic vesicle distribution and release at rat diaphragm neuromuscular junctions. J Neurophysiol 98: 478–487, 2007. doi: 10.1152/jn.00251.2006. [DOI] [PubMed] [Google Scholar]
- 59.Rowley KL, Mantilla CB, Sieck GC. Respiratory muscle plasticity. Respir Physiol Neurobiol 147: 235–251, 2005. doi: 10.1016/j.resp.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 60.Fogarty MJ, Marin Mathieu N, Mantilla CB, Sieck GC. Aging reduces succinate dehydrogenase activity in rat type IIx/IIb diaphragm muscle fibers. J Appl Physiol (1985) 128: 70–77, 2020. doi: 10.1152/japplphysiol.00644.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lewis MI, Sieck GC, Fournier M, Belman MJ. Effect of nutritional deprivation on diaphragm contractility and muscle fiber size. J Appl Physiol (1985) 60: 596–603, 1986. doi: 10.1152/jappl.1986.60.2.596. [DOI] [PubMed] [Google Scholar]
- 62.Marciniak C, Li X, Zhou P. An examination of motor unit number index in adults with cerebral palsy. J Electromyogr Kinesiol 25: 444–450, 2015. doi: 10.1016/j.jelekin.2015.02.007. [DOI] [PubMed] [Google Scholar]
- 63.Chai CK. Hereditary spasticity in mice. J Heredity 52: 241–243, 1961. doi: 10.1093/oxfordjournals.jhered.a107083. [DOI] [Google Scholar]
- 64.Chai CK, Roberts E, Sidman RL. Influence of aminooxyacetic acid, a gamma-aminobutyrate transaminase inhibitor, on hereditary spastic defect in the mouse. Proc Soc Exp Biol Med 109: 491–495, 1962. doi: 10.3181/00379727-109-27245. [DOI] [PubMed] [Google Scholar]
- 65.Fogarty MJ, Sieck GC. Evolution and functional differentiation of the diaphragm muscle of mammals. Compr Physiol 9: 715–766, 2019. doi: 10.1002/cphy.c180012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mantilla CB, Fahim MA, Brandenburg JE, Sieck GC. Functional development of respiratory muscles. In: Fetal and Neonatal Physiology, edited by Polin RA, Abman SH, Rowitch DH, Beneithz WE, and WW Fox. Philadelphia, PA: Elsevier, 2017, p. 690–702. [Google Scholar]
- 67.Geiger PC, Bailey JP, Mantilla CB, Zhan WZ, Sieck GC. Mechanisms underlying myosin heavy chain expression during development of the rat diaphragm muscle. J Appl Physiol (1985) 101: 1546–1555, 2006. doi: 10.1152/japplphysiol.00221.2006. [DOI] [PubMed] [Google Scholar]
- 68.Geiger PC, Cody MJ, Macken RL, Bayrd ME, Fang Y-H, Sieck GC. Selected contribution: mechanisms underlying increased force generation by rat diaphragm muscle fibers during development. J Appl Physiol (1985) 90: 380–388, 2001. doi: 10.1152/jappl.2001.90.1.380. [DOI] [PubMed] [Google Scholar]
- 69.Burke RE, Levine DN, Tsairis P, Zajac FE 3rd. Physiological types and histochemical profiles in motor units of the cat gastrocnemius. J Physiol 234: 723–748, 1973. doi: 10.1113/jphysiol.1973.sp010369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Burke RE, Levine DN, Zajac FE 3rd. Mammalian motor units: physiological-histochemical correlation in three types in cat gastrocnemius. Science 174: 709–712, 1971. [DOI] [PubMed] [Google Scholar]
- 71.Fogarty MJ, Mantilla CB, Sieck GC. Breathing: motor control of diaphragm muscle. Physiology (Bethesda) 33: 113–126, 2018. doi: 10.1152/physiol.00002.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fournier M, Sieck GC. Mechanical properties of muscle units in the cat diaphragm. J Neurophysiol 59: 1055–1066, 1988. doi: 10.1152/jn.1988.59.3.1055. [DOI] [PubMed] [Google Scholar]
- 73.Fournier M, Sieck GC. Topographical projections of phenic motoneurons and motor unit territories in the cat diaphragm. In: Respiratory Muscles and Their Neuromotor Control, edited by Sieck GC, Gandevia SC, and Cameron WE.. New York: Alan R. Liss, Inc., 1987, p. 215–226. [Google Scholar]
- 74.Sieck G. Neural control of the inspiratory pump. Physiology 6: 260–264, 1991. doi: 10.1152/physiologyonline.1991.6.6.260. [DOI] [Google Scholar]
- 75.Chamberlain S, Lewis DM. Contractile characteristics and innervation ratio of rat soleus motor units. J Physiol 412: 1–21, 1989. doi: 10.1113/jphysiol.1989.sp017601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kanda K, Hashizume K. Factors causing difference in force output among motor units in the rat medial gastrocnemius muscle. J Physiol 448: 677–695, 1992. doi: 10.1113/jphysiol.1992.sp019064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Khurram OU, Fogarty MJ, Sarrafian TL, Bhatt A, Mantilla CB, Sieck GC. Impact of aging on diaphragm muscle function in male and female Fischer 344 rats. Physiol Rep 6: e13786, 2018. doi: 10.14814/phy2.13786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Seven YB, Mantilla CB, Zhan WZ, Sieck GC. Non-stationarity and power spectral shifts in EMG activity reflect motor unit recruitment in rat diaphragm muscle. Respir Physiol Neurobiol 185: 400–409, 2013. doi: 10.1016/j.resp.2012.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lee KM, Chand KK, Hammond LA, Lavidis NA, Noakes PG. Functional decline at the aging neuromuscular junction is associated with altered laminin-α4 expression. Aging (Albany NY) 9: 880–899, 2017. doi: 10.18632/aging.101198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Poort JE, Rheuben MB, Breedlove SM, Jordan CL. Neuromuscular junctions are pathological but not denervated in two mouse models of spinal bulbar muscular atrophy. Hum Mol Genet 25: 3768–3783, 2016. doi: 10.1093/hmg/ddw222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Willadt S, Nash M, Slater CR. Age-related fragmentation of the motor endplate is not associated with impaired neuromuscular transmission in the mouse diaphragm. Sci Rep 6: 24849, 2016. doi: 10.1038/srep24849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Arbour D, Vande Velde C, Robitaille R. New perspectives on amyotrophic lateral sclerosis: the role of glial cells at the neuromuscular junction. J Physiol 595: 647–661, 2017. doi: 10.1113/JP270213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Arbour D, Tremblay E, Martineau E, Julien JP, Robitaille R. Early and persistent abnormal decoding by glial cells at the neuromuscular junction in an ALS model. J Neurosci 35: 688–706, 2015. doi: 10.1523/JNEUROSCI.1379-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Keller AF, Gravel M, Kriz J. Live imaging of amyotrophic lateral sclerosis pathogenesis: disease onset is characterized by marked induction of GFAP in Schwann cells. Glia 57: 1130–1142, 2009. doi: 10.1002/glia.20836. [DOI] [PubMed] [Google Scholar]
- 85.Darabid H, St-Pierre-See A, Robitaille R. Purinergic-dependent glial regulation of synaptic plasticity of competing terminals and synapse elimination at the neuromuscular junction. Cell Rep 25: 2070–2082, 2018. e2076. doi: 10.1016/j.celrep.2018.10.075. [DOI] [PubMed] [Google Scholar]
- 86.Theroux MC, Akins RE, Barone C, Boyce B, Miller F, Dabney KW. Neuromuscular junctions in cerebral palsy: presence of extrajunctional acetylcholine receptors. Anesthesiology 96: 330–335, 2002. doi: 10.1097/00000542-200202000-00017. [DOI] [PubMed] [Google Scholar]
- 87.Theroux MC, Oberman KG, Lahaye J, Boyce BA, DuHadaway D, Miller F, Akins RE. Dysmorphic neuromuscular junctions associated with motor ability in cerebral palsy. Muscle Nerve 32: 626–632, 2005. doi: 10.1002/mus.20401. [DOI] [PubMed] [Google Scholar]
- 88.Rogozhin AA, Pang KK, Bukharaeva E, Young C, Slater CR. Recovery of mouse neuromuscular junctions from single and repeated injections of botulinum neurotoxin A. J Physiol 586: 3163–3182, 2008. doi: 10.1113/jphysiol.2008.153569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Robinson KG, Mendonca JL, Militar JL, Theroux MC, Dabney KW, Shah SA, Miller F, Akins RE. Disruption of basal lamina components in neuromotor synapses of children with spastic quadriplegic cerebral palsy. PLoS One 8: e70288, 2013. doi: 10.1371/journal.pone.0070288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hepple RT. When motor unit expansion in ageing muscle fails, atrophy ensues. J Physiol 596: 1545–1546, 2018. doi: 10.1113/JP275981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Dukkipati SS, Garrett TL, Elbasiouny SM. The vulnerability of spinal motoneurons and soma size plasticity in a mouse model of amyotrophic lateral sclerosis. J Physiol 596: 1723–1745, 2018. doi: 10.1113/JP275498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hegedus J, Putman CT, Tyreman N, Gordon T. Preferential motor unit loss in the SOD1 G93A transgenic mouse model of amyotrophic lateral sclerosis. J Physiol 586: 3337–3351, 2008. doi: 10.1113/jphysiol.2007.149286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Rizzuto E, Pisu S, Nicoletti C, Del Prete Z, Musaro A. Measuring neuromuscular junction functionality. J Vis Exp 126: 55227, 2017. doi: 10.3791/55227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sieck GC, Fournier M, Enad JG. Fiber type composition of muscle units in the cat diaphragm. Neurosci Lett 97: 29–34, 1989. doi: 10.1016/0304-3940(89)90134-1. [DOI] [PubMed] [Google Scholar]
- 95.Geiger PC, Cody MJ, Macken RL, Sieck GC. Maximum specific force depends on myosin heavy chain content in rat diaphragm muscle fibers. J Appl Physiol (1985) 89: 695–703, 2000. doi: 10.1152/jappl.2000.89.2.695. [DOI] [PubMed] [Google Scholar]
- 96.Krnjevic K, Miledi R. Failure of neuromuscular propagation in rats. J Physiol 140: 440–461, 1958. [PMC free article] [PubMed] [Google Scholar]
- 97.Krnjevic K, Miledi R. Presynaptic failure of neuromuscular propagation in rats. J Physiol 149: 1–22, 1959. doi: 10.1113/jphysiol.1959.sp006321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Fogarty MJ. Driven to decay: excitability and synaptic abnormalities in amyotrophic lateral sclerosis. Brain Res Bull 140: 318–333, 2018. doi: 10.1016/j.brainresbull.2018.05.023. [DOI] [PubMed] [Google Scholar]
- 99.Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 264: 1772–1775, 1994. [Erratum in Science 14: 269: 149, 1995]. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- 100.Clark JA, Southam KA, Blizzard CA, King AE, Dickson TC. Axonal degeneration, distal collateral branching and neuromuscular junction architecture alterations occur prior to symptom onset in the SOD1(G93A) mouse model of amyotrophic lateral sclerosis. J Chem Neuroanat 76: 35–47, 2016. doi: 10.1016/j.jchemneu.2016.03.003. [DOI] [PubMed] [Google Scholar]
- 101.Fisher TJ, Vrbova G, Wijetunge A. Partial denervation of the rat soleus muscle at two different developmental stages. Neuroscience 28: 755–763, 1989. doi: 10.1016/0306-4522(89)90020-1. [DOI] [PubMed] [Google Scholar]
- 102.Gordon T, Hegedus J, Tam SL. Adaptive and maladaptive motor axonal sprouting in aging and motoneuron disease. Neurol Res 26: 174–185, 2004. doi: 10.1179/016164104225013806. [DOI] [PubMed] [Google Scholar]
- 103.Kuo JJ, Schonewille M, Siddique T, Schults AN, Fu R, Bar PR, Anelli R, Heckman CJ, Kroese AB. Hyperexcitability of cultured spinal motoneurons from presymptomatic ALS mice. J Neurophysiol 91: 571–575, 2004. doi: 10.1152/jn.00665.2003. [DOI] [PubMed] [Google Scholar]
- 104.van Zundert B, Peuscher MH, Hynynen M, Chen A, Neve RL, Brown RH JrConstantine-Paton MBellingham MC. Neonatal neuronal circuitry shows hyperexcitable disturbance in a mouse model of the adult-onset neurodegenerative disease amyotrophic lateral sclerosis. J Neurosci 28: 10864–10874, 2008. doi: 10.1523/JNEUROSCI.1340-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Dupuis L, Loeffler JP. Neuromuscular junction destruction during amyotrophic lateral sclerosis: insights from transgenic models. Curr Opin Pharmacol 9: 341–346, 2009. doi: 10.1016/j.coph.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 106.Steyn FJ, Lee K, Fogarty MJ, Veldhuis JD, McCombe PA, Bellingham MC, Ngo ST, Chen C. Growth hormone secretion is correlated with neuromuscular innervation rather than motor neuron number in early-symptomatic male amyotrophic lateral sclerosis mice. Endocrinology 154: 4695–4706, 2013. doi: 10.1210/en.2013-1570. [DOI] [PubMed] [Google Scholar]
- 107.Ngo ST, Baumann F, Ridall PG, Pettitt AN, Henderson RD, Bellingham MC, McCombe PA. The relationship between Bayesian motor unit number estimation and histological measurements of motor neurons in wild-type and SOD1(G93A) mice. Clin Neurophysiol 123: 2080–2091, 2012. doi: 10.1016/j.clinph.2012.01.028. [DOI] [PubMed] [Google Scholar]
- 108.Hutton JL. Cerebral palsy life expectancy. Clin Perinatol 33: 545–555, 2006. doi: 10.1016/j.clp.2006.03.016. [DOI] [PubMed] [Google Scholar]
- 109.Maudsley G, Hutton JL, Pharoah PO. Cause of death in cerebral palsy: a descriptive study. Arch Dis Child 81: 390–394, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Pilla M, Langlois NEI, Byard RW. Causes of death in a series of decedents with cerebral palsy in a medicolegal context. Aust J Forensic Sci 50: 428–434, 2018. doi: 10.1080/00450618.2016.1259432. [DOI] [Google Scholar]
- 111.Proesmans M. Respiratory illness in children with disability: a serious problem? Breathe (Sheff) 12: e97–e103, 2016. doi: 10.1183/20734735.017416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ryan JM, Peterson MD, Matthews A, Ryan N, Smith KJ, O’Connell NE, Liverani S, Anokye N, Victor C, Allen E. Noncommunicable disease among adults with cerebral palsy: a matched cohort study. Neurology 93: e1385–e1396, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ryan JM, Peterson MD, Ryan N, Smith KJ, O’Connell NE, Liverani S, Anokye N, Victor C, Allen E. Mortality due to cardiovascular disease, respiratory disease, and cancer in adults with cerebral palsy. Dev Med Child Neurol 61: 924–928, 2019. doi: 10.1111/dmcn.14176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Seddon PC, Khan Y. Respiratory problems in children with neurological impairment. Arch Dis Child 88: 75–78, 2003. doi: 10.1136/adc.88.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Reddihough DS, Baikie G, Walstab JE. Cerebral palsy in Victoria, Australia: mortality and causes of death. J Paediatr Child Health 37: 183–186, 2001. doi: 10.1046/j.1440-1754.2001.00644.x. [DOI] [PubMed] [Google Scholar]
- 116.Kwon H-Y. Comparison of differences in respiratory function and pressure as a predominant abnormal movement of children with cerebral palsy. J Phys Ther Sci 29: 261–265, 2017. doi: 10.1589/jpts.29.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Swinney CM, Bau K, Burton KLO, O’Flaherty SJ, Bear NL, Paget SP. Severity of cerebral palsy and likelihood of adverse events after botulinum toxin A injections. Dev Med Child Neurol 60: 498–504, 2018. doi: 10.1111/dmcn.13686. [DOI] [PubMed] [Google Scholar]
- 118.Wang H-Y, Chen C-C, Hsiao S-F. Relationships between respiratory muscle strength and daily living function in children with cerebral palsy. Res Dev Disab 33: 1176–1182, 2012. doi: 10.1016/j.ridd.2012.02.004. [DOI] [PubMed] [Google Scholar]
- 119.Drubach LA, Zurakowski D, Palmer EL, Tracy DA, Lee EY. Utility of salivagram in pulmonary aspiration in pediatric patients: comparison of salivagram and chest radiography. AJR Am J Roentgenol 200: 437–441, 2013. [DOI] [PubMed] [Google Scholar]
- 120.Brandenburg JE, Eby SF, Song P, Kingsley-Berg S, Bamlet W, Sieck GC, An KN. Quantifying passive muscle stiffness in children with and without cerebral palsy using ultrasound shear wave elastography. Dev Med Child Neurol 58: 1288–1294, 2016. doi: 10.1111/dmcn.13179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Karatas AF, Miller EG, Miller F, Dabney KW, Bachrach S, Connor J, Rogers K, Holmes Jr L. Cerebral palsy patients discovered dead during sleep: experience from a comprehensive tertiary pediatric center. J Pediatr Rehabil Med 6: 225–231, 2013. [DOI] [PubMed] [Google Scholar]
- 122.Sieck GC, Fournier M. Diaphragm motor unit recruitment during ventilatory and nonventilatory behaviors. J Appl Physiol (1985) 66: 2539–2545, 1989. doi: 10.1152/jappl.1989.66.6.2539. [DOI] [PubMed] [Google Scholar]
- 123.Brandenburg JE, Fogarty MJ, Sieck GC. Why individuals with cerebral palsy are at higher risk for respiratory complications from COVID-19. J Pediatr Rehabil Med 13: 317–327, 2020. doi: 10.3233/PRM-200746. [DOI] [PubMed] [Google Scholar]