

Keywords: diabetes, nicotine, podocytes, reactive oxygen species, smoking
Abstract
Diabetic nephropathy (DN) is the leading cause of end-stage kidney disease. Besides glycemic and blood pressure control, environmental factors such as cigarette smoking (CS) adversely affect the progression of DN. The effects of CS on DN progression have been attributed to combustion-generated molecules without consideration to the role of nicotine (NIC), responsible for the addictive properties of both CS and electronic cigarettes (ECs). Podocytes are essential to preserve the structure and function of the glomerular filtration barrier, and strong evidence indicates that early podocyte loss promotes DN progression. We performed experiments in human podocytes and in a mouse model of diabetes that develops nephropathy resembling human DN. We determined that NIC binding to podocytes in concentrations achieved with CS and ECs activated NADPH oxidase, which sets in motion a dysfunctional molecular network integrated by cyclooxygenase 2, known to induce podocyte injury; downregulation of AMP-activated protein kinase, important for maintaining cellular energy stores and antioxidation; and upregulation of CD36, which increased lipid uptake and promoted apoptosis. In diabetic mice, NIC increased proteinuria, a recognized marker of chronic kidney disease progression, accompanied by reduced glomerular podocyte synaptopodin, a crucial stabilizer of the podocyte cytoskeleton, and increased fibronectin expression. This novel study critically implicates NIC itself as a contributor to DN progression in CS and EC users.
NEW & NOTEWORTHY In this study, we demonstrate that nicotine increases the production of reactive oxygen species, increases cyclooxygenase-2 expression, and upregulates Cd36 while inducing downregulation of AMP-activated protein kinase. In vivo nicotine increases proteinuria and fibronectin expression in diabetic mice. This study demonstrates that effects of nicotine on podocytes are responsible, at least in part, for the deleterious effects of smoking in the progression of chronic kidney disease, including diabetic nephropathy.
INTRODUCTION
Diabetes is the leading cause of end-stage kidney disease (ESKD). Hypertension, hyperglycemia, dyslipidemia, and cigarette smoking (CS) are major risk factors that contribute to the progression of diabetic nephropathy (DN). The impact of CS on the progression of DN is preventable by smoking cessation; however, and despite concerted tobacco control efforts, the prevalence of smoking in the general population remains close to 20% (1). Central to tobacco control efforts has been the strategy of denormalization of smoking behavior through limiting tobacco advertisement, smoke-free air laws, and excise taxes. This strategy was considerably challenged with the introduction of electronic cigarettes (ECs), which are marketed as nontoxic alternatives for traditional cigarettes (TCs) because of their novel technology of nicotine (NIC) delivery. Although there are reports of individual smokers who replaced TCs with ECs, the benefit of ECs as a smoking cessation aid remains unproven; moreover, most smokers continue using both products (2, 3). Importantly, the potentially toxic effects of ECs in general and on the progression of DN have not been thoroughly investigated (4, 5). This is a critical issue since the blood concentration of cotinine, a stable metabolite of NIC, achieved in smokers and EC users is similar (6).
The mechanisms by which CS contributes to the progression of DN remain poorly understood. In TCs, tobacco combustion generates a large number of biologically active stable and unstable compounds (7). We have shown that stable reactive aldehydes in CS induce endothelial injury via mechanisms that involve activation of NADPH oxidase and increased oxidative stress (8). NIC is a stable biologically active compound present in large amounts in CS and is responsible for the addictive properties of tobacco smoking (7, 9). NIC administration via skin patches and chewing gum has been used with variable success as a smoking cessation strategy (10). In subjects enrolled in smoking cessation programs of relative short duration, lasting 6–12 mo, unwanted cardiovascular and renal effects have not been reported (11).
Studies by Cooke et al. (12) demonstrated that NIC has direct actions upon vascular smooth muscle cells that promote plaque formation. In our laboratory, studies in vivo and in vitro by Zhou et al. (13) have shown that NIC potentiates the proatherogenic effects of oxidized LDL; moreover, we have reported that NIC worsens renal injury in several preclinical models of chronic kidney disease (CKD) associated with glomerular injury (14–16).
Glomerular podocytes are indispensable gatekeepers of the selective permeability of the glomerular basement membrane. A reduction in podocyte population beyond 30%–40% cannot be compensated by podocyte proliferation and results in scarring, persistent proteinuria, continuous podocyte loss, and CKD progression (17). Clinically, it is well documented that smoking plays an important role in promoting worsening of proteinuria and progression of diabetic nephropathy, particularly in type I diabetes (TIDM) (18, 19).
Here, we hypothesized that the effects of NIC on glomerular renal disease are at least in part mediated by direct injurious effects of NIC on glomerular podocytes, including increased generation of reactive oxygen species (ROS), apoptosis, and reduced synaptopodin (SYNPO), a major molecule involved in the stabilization of the glomerular basement membrane.
We tested our hypothesis in vitro using human podocytes in culture (17) and in vivo in a mouse model of type I diabetes that develops renal injury (20), accepted by the Animal Models of Diabetic Complications Consortium as a model that closely resembles DN in humans with TIDM (20).
METHODS
Human Podocyte Culture
Human podocytes were cultured and differentiated in RPMI-1640 culture medium containing 10% FCS, 1% insulin-transferrin-selenium, 10 U/mL glucose, 200 mg/dL human interferon-γ, and 1% penicillin-streptomycin as previously described (21). Terminally differentiated podocytes were starved in 0.1% FBS containing RPMI-1640 medium for 24 h before the experiments were performed.
Determination of Superoxide Anion Production
The production of superoxide anion (O2−) was measured using lucigenin-enhanced chemiluminescence as previously described (22, 23) and validated as a method to measure O2− production (24). To determine the time-course response to NIC, human podocytes were treated with NIC (Sigma-Aldrich, 100 nmol/L for 0, 1, 2, 4, or 6 h). To characterize the source of O2−, cells were preincubated with the following compounds for 30 min before incubation with NIC (100 nmol/L): 1) the NADPH oxidase inhibitor diphenyleneiodonium (DPI; 10 µmol/L, Sigma-Aldrich); 2) AICAR, an agonist of AMP-activated protein kinase (AMPK) (25); or 3) the cyclooxygenase-2 (COX2) inhibitor NS-398 (100 nmol/L). Results were adjusted for protein content and expressed as counts/min (cpm)/µg protein (8).
Immunoblot Analyses
Cells were harvested with lysis buffer containing a cocktail of protein inhibitors (26). Total protein content was quantified using the Bio-Rad assay, and 30 µg of total protein was first subjected to SDS-PAGE and then transferred to nitrocellulose membranes. Membranes were incubated with primary rabbit anti-COX2, CD36, or anti-SYNPO (Santa Cruz Biotechnology) or nicotinic acetylcholine receptor (nAChR) α2-, α3-, α4-, α5-, α6-, α7-, β2-, β3-, and β4-subunits (Sigma) followed by incubation with peroxidase-conjugated secondary antibody for 1 h. Relative quantities of each protein were normalized to β-actin (Santa Cruz Biotechnology) and expressed as the fold increase versus control (27).
siRNA for CD36
Human podocytes (5 × 105 cells) were cultured in RPMI-1640 medium with 10% FCS and grown to 70% confluence. Twenty-four hours before transfection, cells were washed, and the medium was changed to RPMI-1640 medium with 3% FBS with no antibiotics. Cells were transfected using transfection medium and 50 nmol/L CD36 siRNA and then incubated for 24 h according to the manufacturer’s instructions (Santa Cruz Biotechnology). Nonspecific scrambled RNA was used as a control. The efficiency of siRNA transfection for CD36 was confirmed by the over 70% reduction in CD36 mRNA expression as assessed by real-time PCR compared with the scrambled control RNA.
Determination of Total Cellular Cholesterol Content
Human podocytes grown in six-well dishes were incubated with or without NIC (100 nmol/L) for 24 h followed by incubation with oxidized LDL (oxLDL; 50 µg/mL) for an additional 24 h. In some experiments, cells were transfected with CD36 siRNA to knockdown CD36 before incubation with NIC and oxLDL. Total cellular cholesterol was extracted by adding 1 mL of hexane-isopropanol [3:2 (vol/vol)] to the wells. Cholesterol content was determined with an enzyChrom AF cholesterol assay kit according to the manufacturer’s instructions (Bioassay Systems). The sample total protein content was used to normalize the results.
Apoptosis Assay
A total of 2 × 106 cells were seeded in 60-mm petri dishes and treated with one of following conditions: 1) vehicle, 2) NIC (100 nmol/L) for 6 h, 3) preincubation with the NADPH oxidase inhibitor DPI (10 µmol/L) for 30 min followed by incubation with NIC for 6 h, 4) preincubation with the COX2 inhibitor NS-398 (100 nmol/L) for 30 min followed with NIC for 24 h, 5) oxLDL (50 µg/mL) for 24 h, 6) incubation with NIC for 6 h followed by oxLDL (50 µg/mL) for 24 h, and 7) cells transfected with siRNA CD36 and then treated with oxLDL for 6 h followed by NIC for 24 h. Apoptosis was measured using the annexin V-fluorescein isothiocyanate apoptosis detection kit (Roche Diagnostics) followed by flow cytometry.
In Vivo Experiments
For these experiments, we used the model of streptozotocin-induced diabetes as previously described by Kanetsuna et al. (28) (50 mg/kg ip for 5 days) in endothelial nitric oxide synthase (eNOS)-deficient (eNOS−/−) mice (Jackson Laboratory). Once diabetes was confirmed, mice were given NIC in the drinking water (100 µg/mL) for 10 wk. As controls, we used nondiabetic wild-type mice on either tap water or NIC and diabetic mice on tap water (n = 8 per group). Blood pressure was measured by tail cuff as previously described (29), and urine was collected weekly for protein excretion measured by Bio-Rad.
Immunohistochemistry
The avidin-biotin-peroxidase immunohistochemical technique (ABC kit, Vector) was used to detect SYNPO using primary antibodies against SYNP (ProGen Biotechnik, Heidelberg, Germany) or fibronectin (Sigma). After deparaffinization and heat-mediated antigen retrieval, SYNPO- or fibronectin-positive areas were immunolocalized by incubation with the respective primary antibody followed by application of biotinylated goat anti-rabbit secondary antibody (1:200) for 30 min. A semi-automated image-analysis algorithm was used for quantification of the extent of SYNPO- or fibronectin-positive signal intensity as previously described (30).
Immunofluorescence
Formalin-fixed, paraffin-embedded rat kidney cortex sections (5 μm) were deparaffinized, and antigen retrieval was performed on all sections. Sections used to identify proximal tubules were incubated with biotinylated Lotus tetragonolobus lectin (1:200); to identify distal tubules and collecting duct, sections were incubated with biotinylated peanut agglutinin (1:1,000). Sections were blocked with PBS + 1% bovine serum and 5% goat serum for 1 h at room temperature and then incubated with primary antibody to α2-, α3-, α4-, α5-, α6-, α7-, β2-, β3-, and β4-subunits (Sigma). Sections were then washed three times for 5 min with PBS and incubated with secondary antibody (Alexa Fluor 488-labeled goat anti-rabbit, Molecular Probes, Portland, OR) at 1:400 dilution in blocking buffer for 1 h at room temperature. Image acquisition was performed on a Leica DM6000 epifluorescence microscope (Leica Microsystems, Bannockburn, IL). DAPI stain was used to identify nuclei. Images were adjusted appropriately to remove background fluorescence.
Renal Injury
Severity of renal injury was measured in periodic acid-Schiff-stained slides by one of the coauthors (S. Seshan) purposely blinded to the experimental conditions. All glomeruli available and the interstitium in each slide were analyzed, and the severity of injury was described.
Statistics
Results are expressed as means ± SE. Statistical analyses were performed by one-way ANOVA with Bonferroni’s correction for multiple comparisons followed by a post hoc Scheffe’s test. Significance was assumed at P < 0.05.
RESULTS
Expression of nAChRs in Human Podocytes
In our previous studies, we identified the expression of several nAchR subunits in human mesangial cells (MCs) (23); here, we used Western blot analysis to determine the expression of nAchR subunits in human podocytes. As shown in Fig. 1, nAchR α2-, α3-, α4-, and β3-subunits were expressed in human podocyte. We did not detect the expression of nAchR α5-, α6-, α7-, or β2-subunits in these cells (not shown).
Figure 1.

Western blot analysis demonstrating expression of nicotinic acetylcholine receptor α2-, α3-, α4-, and β3-subunits in cultured human podocytes.
NIC Increases ROS Production and COX2 Expression in Human Podocytes
We have previously shown that the stimulation of nAchRs with NIC increased ROS production in MCs (23) and macrophages (13) via NADPH oxidase. Therefore, we evaluated the effects of NIC on ROS production in human podocytes. As shown in Fig. 2, NIC significantly increased ROS production in a time-dependent manner, with an initial effect at 2 h and a maximal effect at 4–6 h. The observed increase in ROS induced by NIC was prevented by the NADPH oxidase inhibitor DPI; by AICAR, a cell-permeable activator of AMKP; and by the COX2 inhibitor NS-398. We also determined that NIC increased COX2 expression in a time-dependent manner in human podocytes and that this upregulation of COX2 by NIC was significantly attenuated by the antioxidant enzyme catalase and the NADPH oxidase inhibitor DPI (Fig. 3), suggesting that ROS play a role in NIC-induced upregulation of COX2 in human podocytes. Because in this study the COX2 inhibitor NS-398 significantly reduced NIC-stimulated ROS production (Fig. 2B), we surmise that there is an interdependent relationship between ROS and COX2 expression by NIC. Of interest, COX2 has been shown to be associated with podocyte injury (31), and in an in vivo model of CKD, we have shown that COX2 inhibition protects against the deleterious effects of NIC on renal injury (15).
Figure 2.

Production of reactive oxygen species. A: dose response of reactive oxygen species production in response to nicotine (Nic) in cultured human podocytes. *P < 0.05 vs. control. n = 6. B: the production of reactive oxygen species induced by nicotine in cultured podocytes was inhibited by NADPH oxidase inhibition [diphenyleneiodonium (DPI)], AICAR, catalase (Cat), and cyclooxygenase-2 inhibition [NS-398 (NS)]. *P < 0.05 vs. control; #P < 0.05 vs. nicotine. n = 6. CPM, counts per minute.
Figure 3.
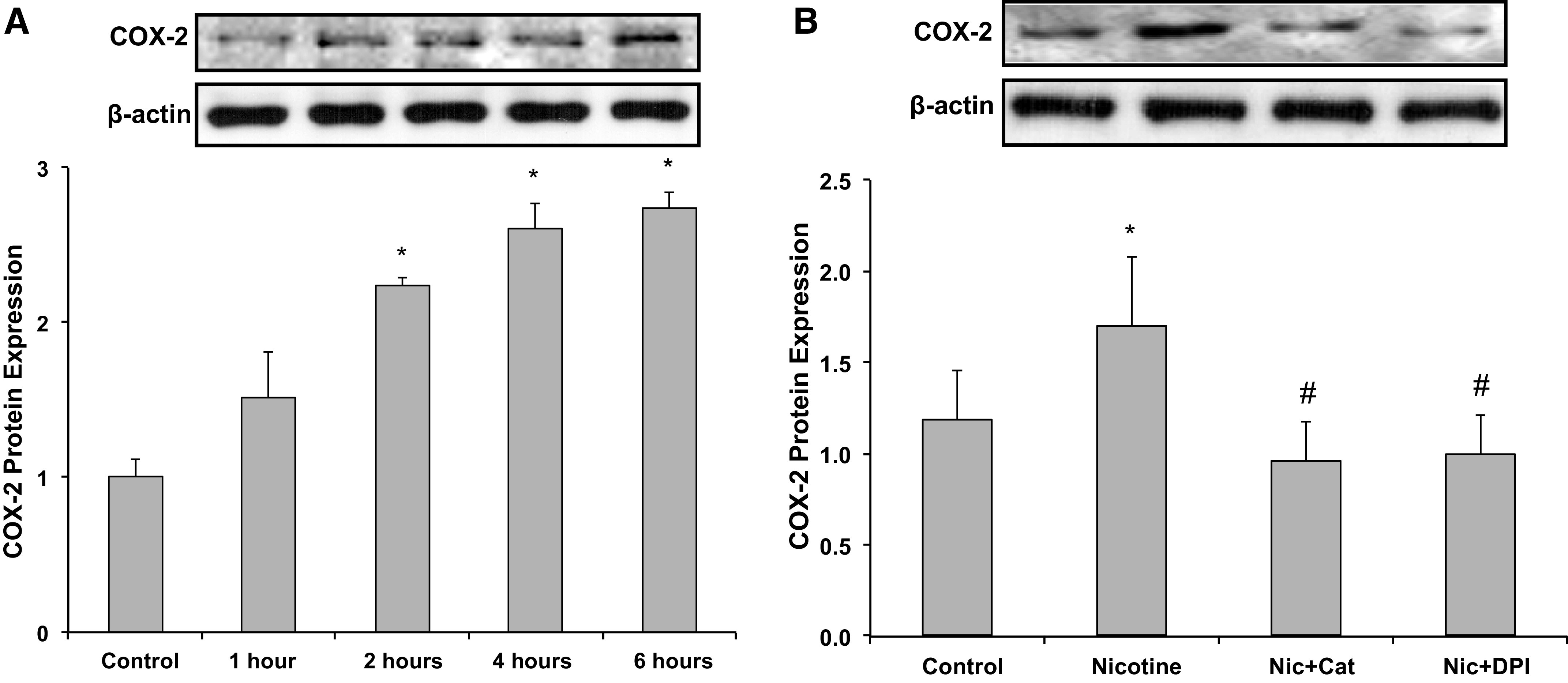
Cyclooxygenase-2 (COX2) expression in response to nicotine (Nic). A: time course for the expression of COX2 in response to nicotine. *P < 0.05 vs. control. n = 6. B: expression of COX2 in response to nicotine was inhibited by diphenyleneiodonium (DPI) and catalase (Cat). *P < 0.05 vs. control; #P < 0.05 vs. nicotine. n = 6.
NIC Increases CD36 Expression and Promotes Lipid Deposits in Human Podocytes
Recently, we have shown that NIC upregulates the expression of CD36 in human THP1-differentiated macrophages (13). Here, we examined the effect of NIC on CD36 expression in human podocytes. As shown in Fig. 4, NIC significantly increased the expression of CD36, which was significantly attenuated by either the COX2 inhibitor NS-398 or by catalase, suggesting that NIC increased CD36 expression via activation of the COX2/ROS pathway. Hexamethonium (100 µmol/L), a nonselective nAchR inhibitor, also blocked NIC-induced CD36 expression, thereby confirming that in human podocytes, NIC increases the expression of CD36 through stimulation of nAchRs.
Figure 4.

CD36 protein expression in response to nicotine (Nic). Expression of CD36 was increased by nicotine and prevented by cyclooxygenase-2 inhibition [NS-398 (NS)], nicotinic acetylcholine receptor blockade [hexamethonium (Hex)], and catalase (Cat). *P < 0.05 vs. control; #P < 0.05 vs. nicotine. n = 6.
CD36 is a major receptor that mediates oxLDL uptake by macrophages (32). We have shown that upregulation of CD36 by NIC increases atherogenic lipid uptake as well as the accumulation and foam cell formation in THP-1/macrophages (13). However, it is still unclear whether CD36 also participates in mediating oxLDL uptake and lipid deposition in podocytes. As shown in Fig. 5, podocytes treated with oxLDL showed a significant increase in cholesterol accumulation, an effect that was prevented by pretreatment with CD36 siRNA. NIC in the presence of oxLDL further increased podocyte cholesterol accumulation, which was reduced by podocyte pretreatment with CD36 siRNA. In the aggregate, these results indicate that CD36 is involved in the regulation of lipid metabolism associated with oxLDL in podocytes and that NIC enhances CD36-mediated lipid accumulation in human podocytes through upregulation of CD36.
Figure 5.

Cholesterol uptake in response to nicotine (Nic) and oxidized LDL (oxLDL). Both nicotine and oxLDL increased podocyte cholesterol uptake, which was prevented by knockdown of CD36. *P < 0.05 vs. control; #P < 0.05 vs. Nic/oxLDL/control (Ctrl)-siRNA. n = 6.
NIC Potentiates oxLDL-Induced Podocyte Apoptosis Through a CD36-Dependent Mechanism
It is well established that the loss of glomerular podocytes is a key feature in the progression of renal diseases (33). Experimental and clinical studies have demonstrated that apoptosis is an important contributor to reduced podocyte number, exacerbating proteinuria, and/or promoting the development of segmental glomerulosclerosis (34). Here, we investigated by flow cytometry the effect of NIC independently and/or combined with oxLDL on podocyte apoptosis. As shown in Fig. 6, NIC exposure slightly but significantly increased podocyte apoptosis, which was prevented by pretreatment with DPI. In addition, oxLDL also induced podocyte apoptosis to a level similar to NIC-treated cells. The combination of NIC and oxLDL further augmented podocyte apoptosis. Knockdown of CD36 by CD36 siRNA significantly reduced podocyte apoptosis in combined NIC + oxLDL-treated cells, suggesting that NIC potentiates oxLDL-induced podocyte apoptosis via a CD36-dependent mechanism.
Figure 6.

Apoptosis induced by nicotine (Nic) and oxidized LDL (oxLDL). Nicotine and oxLDL have additive effects on apoptosis in human podocytes that were prevented by knockdown of CD36 and diphenyleneiodonium (DPI).
NIC Reduced the Expression of SYNPO Through Oxidative Stress
SYNPO is an actin-associated protein that is expressed exclusively by podocytes (35). SYNPO is a stabilizer of the podocyte cytoskeleton that plays a critical role in the support of podocyte-matrix interaction and the prevention of podocyte effacement and detachment (35). To determine whether NIC affects SYNPO expression in human podocytes, we determined the protein expression of SYNPO in NIC-treated cells. As shown in Fig. 7, NIC exposure for 6 h significantly reduced the expression of SYNPO, which was prevented by pretreatment with the antioxidant DPI, suggesting that NIC reduces SYNPO via increased oxidative stress.
Figure 7.

Effects of nicotine (Nic) on synaptopodin expression. Exposure to nicotine reduced synaptopodin expression in human podocytes, which was prevented by diphenyleneiodonium (DPI), AICAR, and cyclooxygenase-2 inhibition [NS-398 (NS)]. *P < 0.05 vs. control; #P < 0.05 vs. nicotine. n = 6.
Expression of NIC Receptors in Mouse Kidneys
By immunofluorescence, we measured the cortical expression of the different nicotine receptor units. We determined that mice express α4-, α5-, α6-, α7-, β2-, β3-, and β4-nAChRs. The subunits α1, α2, α3, and β1 were undetectable, indicating that the normal mouse kidney is endowed with neuronal nAChRs but not muscle nAChRs (Fig. 8). By using specific colocalization, we determined that proximal tubules express α5-, α7-, β2-, β3-, and β4-subunits and distal tubules express α5-, α7-, β3-, and β4-subunits. In addition, we observed that the normal mouse glomerulus expresses α4-, α5-, β3-, and β4-subunits. The α6-subunit was observed only in the interstitium, and, as shown in Fig. 9, expression of the α4-subunit colocalized with SYNPO.
Figure 8.

Expression of nicotinic acetylcholine receptors (nAChRs) in normal mice. Normal mice express several nAChRs (green) in the kidney cortex, as assessed by immunofluorescence including proximal tubular (PT; red) and distal tubular (DT; red) expression as well as glomerular expression. DAPI was used as the nuclear stain. We determined that mice express α4-, α5-, α6-, α7-, β2-, β3-, and β4-nAChRs. Of these, α4 and α5 had predominantly glomerular expression, whereas the rest had predominantly tubular expression.
Figure 9.
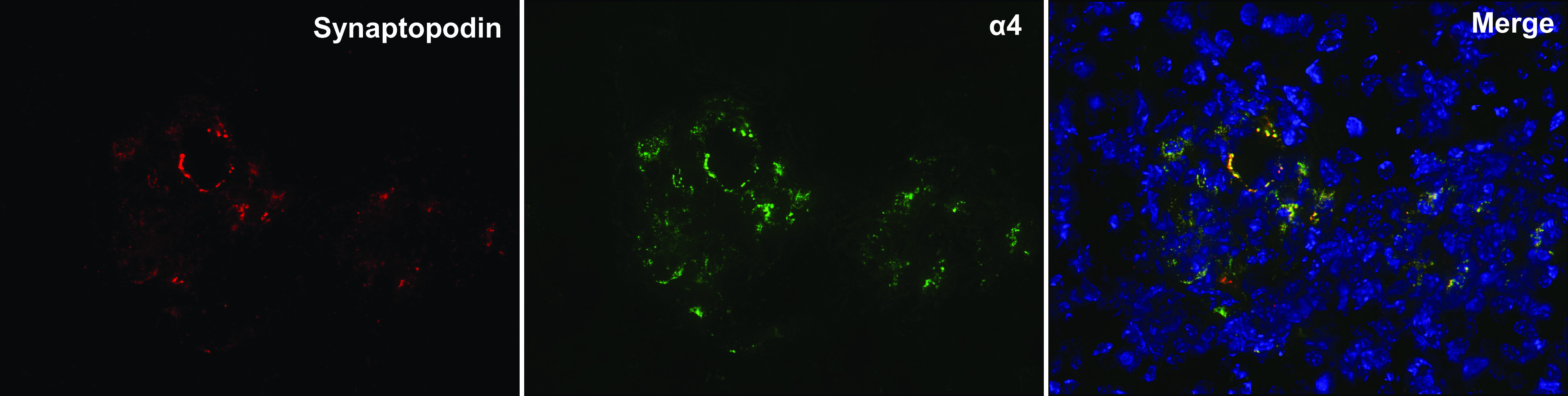
Expression of α4-nicotinic acetylcholine receptor (green) and synaptopodin (red) colocalized in glomeruli of normal mice, suggesting podocyte expression of this receptor in vivo.
Effects of NIC on Proteinuria and Renal Injury in Diabetic Mice
We next determined the effects of chronic administration of NIC for 10 wk on the severity of renal injury in diabetic mice. The administration of NIC to wild-type nondiabetic mice did not result in significant changes in urinary protein excretion. In contrast, diabetic mice had a significant increase in proteinuria that was further increased by the administration of NIC in the drinking water. As shown in Table 1, both groups of mice on NIC had levels of cotinine similar to those found in the plasma of smokers (36). In addition, all diabetic mice had similar levels of glucose that was not affected by the administration of NIC. As expected, all diabetic mice were hypertensive, as these mice lack eNOS. The administration of NIC did not produce any increases in blood pressure in wild-type mice and did not induce further increases in blood pressure in diabetic mice (Table 1).
Table 1.
Levels of glucose, proteinuria, cotinine, and systolic blood pressure in the various groups of mice
| Glucose, mg/dL | Proteinuria, mg/24 h | Cotinine, ng/mL | Systolic Blood Pressure, mmHg | |
|---|---|---|---|---|
| Control | 180.1 ± 9.7 | 21 ± 5.8 | ND | 124.2 ± 1.25 |
| Nicotine | 187.2 ± 18.3 | 25.0 ± 5.7 | 44 ± 1 | 124.7 ± 5.2 |
| Diabetes/eNOS−/− | 539 ± 31* | 73 ± 23* | ND | 149.7 ± 7* |
| Diabetes/eNOS−/− + nicotine | 571 ± 18* | 157.3 ± 44*† | 63 ± 1.8 | 146.2 ± 9.5* |
Values are means ± SE. eNOS, endothelial nitric oxide synthase; ND, not determined. *P < 0.05 vs. control and nicotine; †P < 0.05 vs. diabetes.
To assess the effects of NIC on fibrosis in these mice, we measured the expression of fibronectin by immunohistochemistry. As shown in Fig. 10, the administration of NIC to wild-type mice had no significant effect on fibronectin expression but resulted in a significant increase in fibronectin in diabetic mice. Although none of the control mice either on tap water or NIC had evidence of glomerulosclerosis, diabetic mice on tap water and diabetic mice on NIC had a similar percent of glomeruli with global sclerosis (2.5%). However, while foci of cortical scarring were observed in 50% of diabetic mice on tap water, 100% of diabetic mice on NIC had evidence of cortical scarring.
Figure 10.
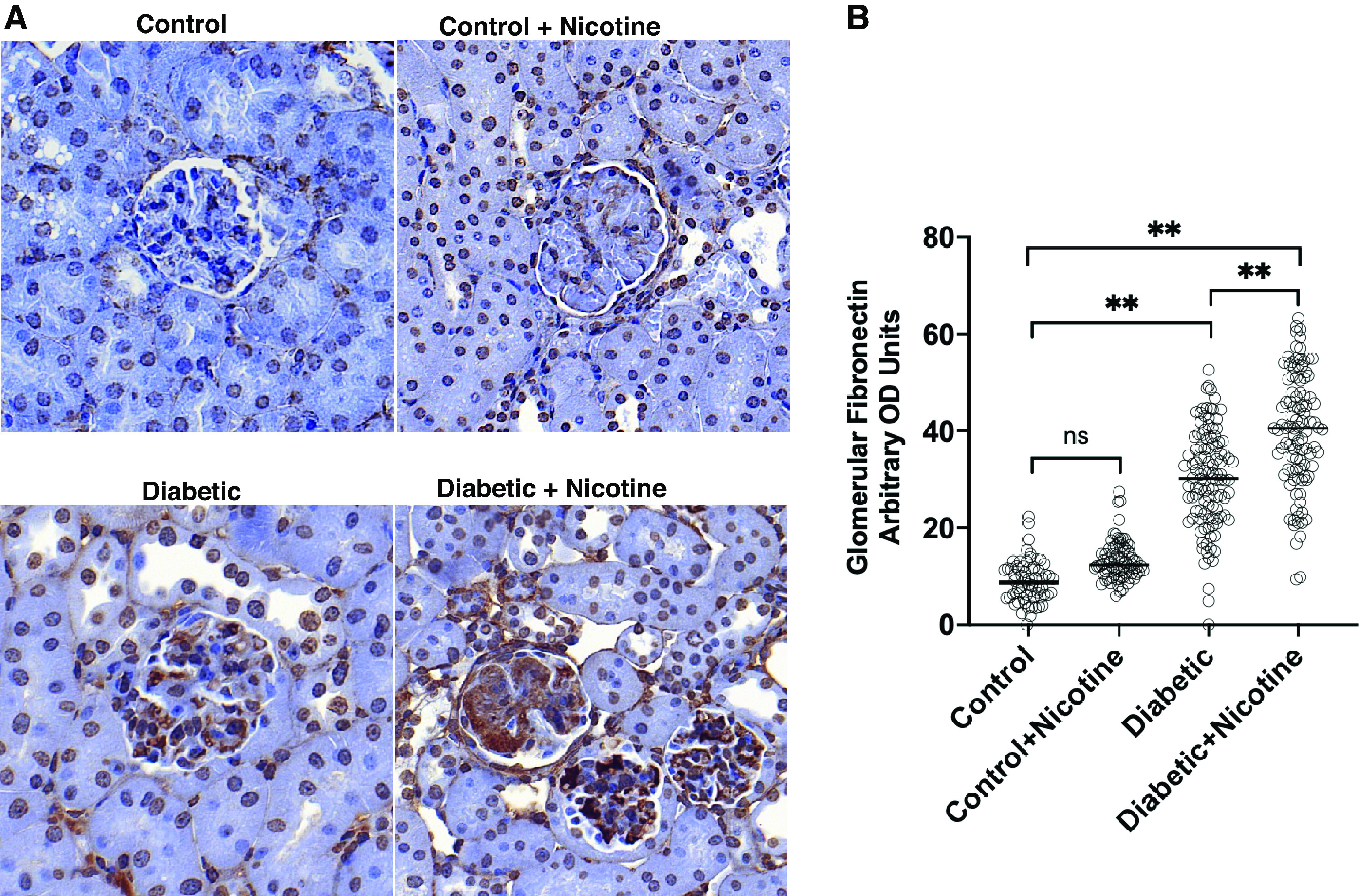
Fibronectin expression. Glomerular expression of fibronectin was not modified by the administration of nicotine to control mice. Diabetic mice had an increase in glomerular expression of fibronectin, which was further increased by nicotine. A: representative photomicrograph of fibronectin expression from the different groups as assessed by immunohistochemistry. B: quantitation of glomerular expression of fibronectin for the different groups studied. OD: optical density. **P < 0.01.
NIC Reduces the Expression of SYNPO In Vivo
As shown in Fig. 11, diabetic mice had reduced expression of SYNPO. The administration of NIC to diabetic mice resulted in further reductions in SYNPO expression. Unexpectedly, in control mice, the administration of NIC resulted in a modest albeit significant increase in SYNPO expression. These findings support our in vitro findings demonstrating that NIC worsens renal injury in diabetic mice, as assessed by urinary protein excretion, and that this renal injury is linked to a reduction in SYNPO expression.
Figure 11.
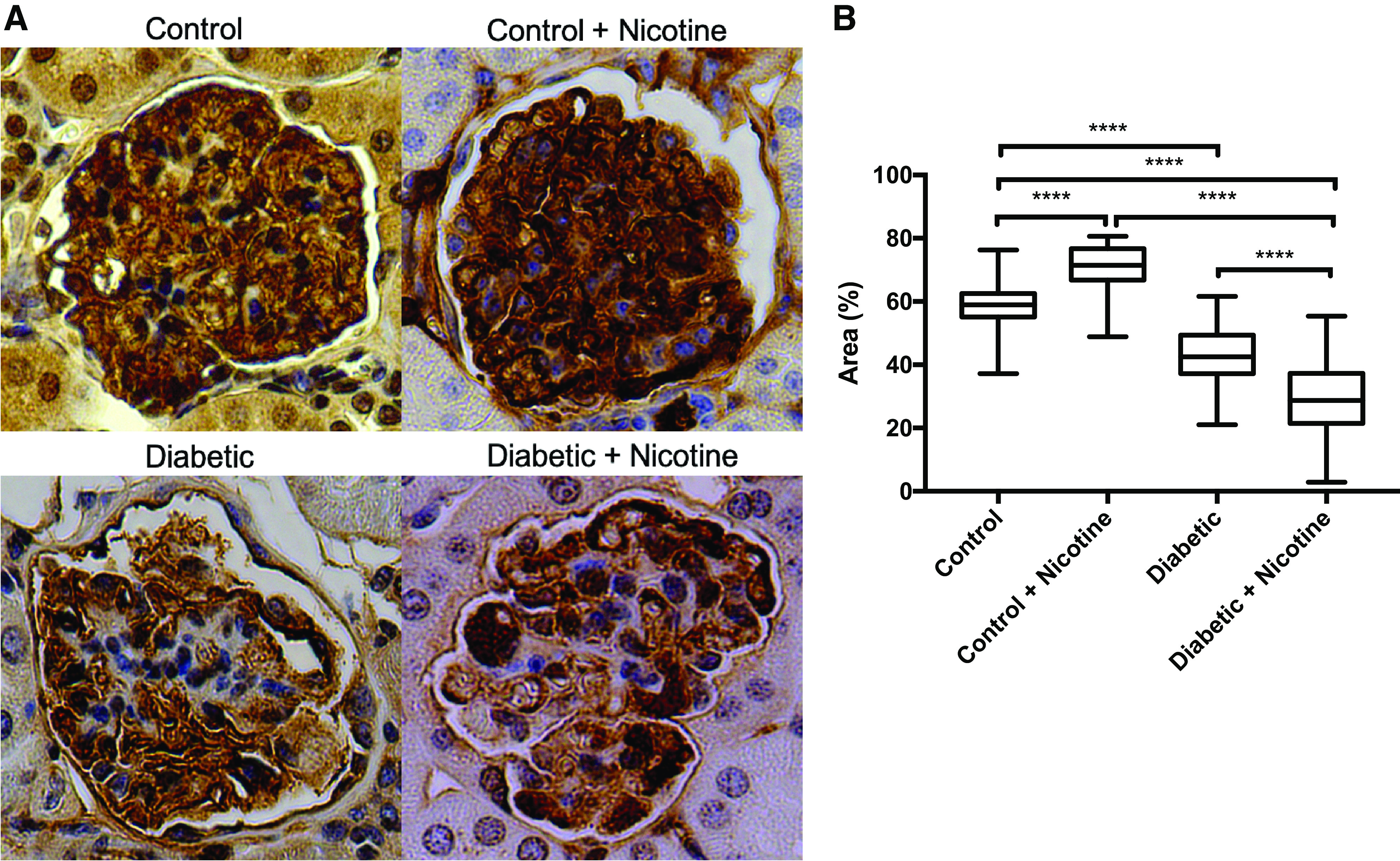
Expression of synaptopodin in vivo. Expression of synaptopodin was slightly increased by nicotine in normal mice but reduced in diabetic mice and further reduced in diabetic mice on nicotine. A: representative photomicrograph of synaptopodin expression from the different groups as assessed by immunohistochemistry. B: percentage of the glomerular area positive for synaptopodin for the different groups studied. ***P < 0.05.
DISCUSSION
Large epidemiological studies have demonstrated the strong association between CS and the development of proteinuria and renal impairment in the normal population and faster progression of CKD, especially in patients with CKD secondary to hypertension and with diabetes, particularly type I diabetes but also in type II diabetes (37–44).
NIC, which is the habituating agent in tobacco smoking, is a stable biologically active compound present in large concentrations in tobacco. Experimental studies have shown that NIC via activation of nAchRs elicits pathophysiological responses in vascular (45–47) and extravascular tissues (16, 23, 48–50). In previous studies, we have shown that human MCs are endowed with nAChRs (23) and that exposure of MCs to NIC, in concentrations similar to those found in smokers, increased the production of ROS and induced fibronectin production and MC proliferation (14, 23). Because of the pivotal role of podocytes in maintaining the functionality of the kidney filter in DN and the results of the in vitro and in vivo NIC studies reported by us and others (13, 16, 23, 46, 51), we concluded that it was critical to investigate whether podocytes are a target for NIC-mediated injury.
In the current study, we demonstrated that as we have previously reported in human MCs (23), human podocytes express several nAchRs, including α2, α3, α4, and β3. Next, we determined that NIC produced a significant increase in the production of the ROS O2−, which in podocytes originates principally from mitochondrial NOX4 (52) and that was prevented by DPI, an inhibitor of NADPH oxidase.
AMPK is a serine/threonine kinase expressed in podocytes that acts as an energy sensor (53, 54). Active AMPK requires phosphorylation (pAMPK) of a critical threonine residue (55) that enables cells to reduce NADPH oxidase-dependent ROS formation (56–59). AMPK plays a critical role in glomerular permeability and is a key regulator of albuminuria likely by modulating oxidative stress at the level of the podocytes (60). In our study, the exposure of podocytes to NIC reduced pAMPK (Fig. 3B), and AICAR, an activator of AMPK (56–58), normalized the pAMPK-to-AMPK ratio and concomitantly inhibited NIC-driven O2− production (Fig. 3A).
Increased COX2 expression has been shown to be associated with podocyte injury in models of proteinuric glomerulopathies (55), including DN (61). Moreover, as we have recently shown, the deleterious effects of NIC on renal injury in a rat model of 5/6 nephrectomy (Nx) are prevented by the administration of a COX2 inhibitor, demonstrating that these effects are in large part mediated by COX2-derived prostaglandins (15). Here, we determined that NIC increased COX2 expression in human podocytes, which was significantly attenuated by the NADPH oxidase inhibitor DPI (Fig. 3), confirming that ROS play an important role in NIC upregulation of COX2 in human podocytes. As shown in Fig. 4, catalase inhibited COX2 upregulation, implying that H2O2 is the link for NIC-mediated upregulation of COX2 in podocytes. Moreover, since the COX2 inhibitor NS-398 significantly reduced NIC-stimulated ROS production (Fig. 2B), we infer that in podocytes there is a critical interdependent relationship between the regulation of ROS and COX2 expression. It remains to be determined the role of COX2-derived prostaglandins as mediators of the effects of NIC on renal injury in diabetes, and this is the subject of ongoing studies in our laboratory.
In recent studies, we reported that NIC increases the expression of CD36 in macrophages resulting in increased cholesterol uptake by atherosclerotic vessels (13). In the kidney, CD36 is expressed in tubular cells, podocytes, and MCs and is markedly upregulated in the setting of CKD (62). In podocytes, enhanced free fatty acid (FFA) uptake is regulated by increased expression of CD36 and decreased fatty acid β-oxidation, resulting in intracellular lipid accumulation leading to the production of ROS, lipid peroxidation, and mitochondrial damage and dysfunction (63). In this study, we demonstrated that NIC upregulates CD36 expression and cholesterol uptake by podocytes. Importantly, we also demonstrated that NIC promotes podocyte apoptosis via CD36-mediated oxLDL uptake (64). We determined that the pathophysiological pathways associated with podocyte injury are linked with ROS acting directly as well indirectly via upregulation of COX2 and of CD36. In addition to inducing podocyte apoptosis, we have demonstrated, for the first time, to our knowledge, that NIC reduces the expression of SYNPO, a critical protein involved in the maintenance of normal function and structure of the podocyte (35).
In our in vivo experiments, we determined that the cortical kidney of the normal mouse expresses neuronal nAChRs α4, α5, α6, α7, β2, β3, and β4. By using specific immunofluorescence colocalization methods, we showed the expression of neuronal nicotinic receptors in both proximal and distal tubules as well as in the glomerulus. Importantly, we demonstrated that expression of the neuronal α4-receptor colocalized with SYNPO, a specific marker of glomerular podocytes (35).
Since CS worsens the progression of DN (43, 65–68), we carried out in vivo experiments to determine whether NIC administered in drinking water to achieve similar levels to those reported to circulate in smokers of conventional cigarettes and of EC users in a model of TIDM in Adkison et al. (4). We determined that the exposure to NIC in these mice results in significant increases in albuminuria, cortical fibrosis, and glomerular fibronectin expression consistent with a higher severity of renal injury. Moreover, and consistent with our in vitro experiments, the administration of NIC produced further reductions in SYNPO expression compared with diabetes alone. A limitation of our study is that the model of diabetes used for these experiments is on an eNOS−/− background, and therefore, at baseline, these mice are hypertensive. Additional studies will be needed to characterize the role of eNOS deficiency on the effects of NIC in the setting of hypertension.
Importantly, blood levels of cotinine, the predominant metabolite of NIC, observed in diabetic mice were similar to those found in the plasma of active smokers (69).
Consistent with the abovementioned findings, we have previously demonstrated that chronic NIC administration significantly increases glomerular injury and proteinuria in rats with 5/6 Nx, an experimental model of CKD accompanied by glomerulosclerosis and podocyte injury (16), as well as in Db/Db mice (14), a model of type II diabetes. In our previous study, we have shown that blockade of α7-nAchR, which in vivo is exclusively expressed in proximal tubules (16), reduces the severity of injury in response to NIC in a model of 5/6 Nx, which supports the notion that NIC may be exerting biological effects via more than one receptor and highlights the existence of a feedback loop between proximal tubules and glomerular injury.
Our findings unveil novel mechanisms of injury induced by NIC that may explain, at least in part, the deleterious effects of CS in renal injury. These findings are particularly relevant given the growing popularity of noncombustible tobacco products, including ECs, for NIC delivery (70). In addition, this study supports the notion that podocytes are a major target of NIC in concentrations achieved in smokers of both conventional cigarettes and ECs (6). The prevalence of EC use has been doubling in youth, including middle and high school students, with a greater proportion (compared with adults) of previous nonsmokers trying ECs (71). Moreover, EC use has not proven to be an effective smoking cessation intervention (72). The evidence that use of ECs results in reduced cardiovascular consequences compared with CS is still lacking, and evidence of a reduced risk of DN progression is unavailable in preclinical as well as clinical studies. Future studies in humans will be essential to characterize the renal effects of these products and their safety in subjects with chronic renal disease and especially in diabetes.
GRANTS
This work was supported by the Miami Veterans Affairs Foundation for Research and Education (to L.R.) and by National Institutes of Health Grants RO1ES014948 and P30CA008748 (to E.A.J.); R01DK117599, R01DK104753, and R01CA227493 (to A.F. and S.M.); and U54DK083912, UM1DK100846, U01DK116101, and UL1TR000460 (to A.F.).
DISCLAIMERS
E.A.J. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
DISCLOSURES
E.A.J. is the co-founder, shareholder, and chief medical officer of Goldilocks Therapeutics, Inc. A.F. is the Vice-President of L&F Health LLC and is a consultant for ZyVersa Therapeutics, Inc. A.F. is the founder of LipoNexT LLC. S.M. is a consultant for Kintai Therapeutics Inc. and holds equity interest in L&F Research. A.F. and S.M. are supported by Hoffman-La Roche and by Boehringer Ingelheim.
AUTHOR CONTRIBUTIONS
E.A.J. and L.R. conceived and designed research; M.S., G.R., and R.T. performed experiments; M.S., G.R., S.V.S., A.N.M., and N.J.W. analyzed data; E.A.J., L.R., M-S.Z., A.N.M., N.J.W., and E.U.A. interpreted results of experiments; E.A.J. and R.T. prepared figures; E.A.J., L.R., and M-S.Z. drafted manuscript; E.A.J., A.F., S.M., L.R., A.N.M., N.J.W., and E.U.A. edited and revised manuscript; E.A.J. and L.R. approved final version of manuscript.
ACKNOWLEDGMENTS
We acknowledge the excellent technical support of Philip Chumley and Ping Hua.
REFERENCES
- 1.Centers for Disease Control and Prevention. Current cigarette smoking prevalence among working adults–United States, 2004-2010. MMWR Morbid Mortal Week Rep 60: 1305–1309, 2011. [PubMed] [Google Scholar]
- 2.Akinboro O, Nwabudike S, Elias R, Balasire O, Ola O, Ostroff JS. Electronic cigarette use among survivors of smoking-related cancers in the United States. Cancer Epidemiol Biomarkers Prev 28: 2087–2094, 2019. doi: 10.1158/1055-9965.EPI-19-0105. [DOI] [PubMed] [Google Scholar]
- 3.Kalkhoran S, Kruse GR, Rigotti NA, Rabin J, Ostroff JS, Park ER. Electronic cigarette use patterns and reasons for use among smokers recently diagnosed with cancer. Cancer Med 7: 3484–3491, 2018. doi: 10.1002/cam4.1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Adkison SE, O'Connor RJ, Bansal-Travers M, Hyland A, Borland R, Yong HH, Cummings KM, McNeill A, Thrasher JF, Hammond D, Fong GT. Electronic nicotine delivery systems: international tobacco control four-country survey. Am J Prev Med 44: 207–215, 2013. doi: 10.1016/j.amepre.2012.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vickerman KA, Carpenter KM, Altman T, Nash CM, Zbikowski SM. Use of electronic cigarettes among state tobacco cessation quitline callers. Nicotine Tob Res 15: 1787–1791, 2013. doi: 10.1093/ntr/ntt061. [DOI] [PubMed] [Google Scholar]
- 6.Marsot A, Simon N. Nicotine and cotinine levels with electronic cigarette: a review. Int J Toxicol 35: 179–185, 2016. doi: 10.1177/1091581815618935. [DOI] [PubMed] [Google Scholar]
- 7.Stedman RL. The chemical composition of tobacco and tobacco smoke. Chem Rev 68: 153–207, 1968. doi: 10.1021/cr60252a002. [DOI] [PubMed] [Google Scholar]
- 8.Jaimes EA, DeMaster EG, Tian RX, Raij L. Stable compounds of cigarette smoke induce endothelial superoxide anion production via NADPH oxidase activation. Arterioscler Thromb Vasc Biol 24: 1031–1036, 2004. doi: 10.1161/01.ATV.0000127083.88549.58. [DOI] [PubMed] [Google Scholar]
- 9.Jain G, Jaimes EA. Nicotine signaling and progression of chronic kidney disease in smokers. Biochem Pharmacol 86: 1215–1223, 2013. doi: 10.1016/j.bcp.2013.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schuit E, Panagiotou OA, Munafo MR, Bennett DA, Bergen AW, Sp D. Pharmacotherapy for smoking cessation: effects by subgroup defined by genetically informed biomarkers. Cochrane Database Syst Rev 9: CD011823, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 11.Glantz SA, Bareham DW. E-cigarettes: use, effects on smoking, risks, and policy implications. Annu Rev Public Health 39: 215–235, 2018. doi: 10.1146/annurev-publhealth-040617-013757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee J, Cooke JP. The role of nicotine in the pathogenesis of atherosclerosis. Atherosclerosis 215: 281–283, 2011. doi: 10.1016/j.atherosclerosis.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou MS, Chadipiralla K, Mendez AJ, Jaimes EA, Silverstein RL, Webster K, Raij L. Nicotine potentiates proatherogenic effects of oxLDL by stimulating and upregulating macrophage CD36 signaling. Am J Physiol Heart Circ Physiol 305: H563–H574, 2013. doi: 10.1152/ajpheart.00042.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hua P, Feng W, Ji S, Raij L, Jaimes EA. Nicotine worsens the severity of nephropathy in diabetic mice: implications for the progression of kidney disease in smokers. Am J Physiol Renal Physiol 299: F732–F739, 2010. doi: 10.1152/ajprenal.00293.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rangarajan S, Rezonzew G, Chumley P, Fatima H, Golovko M, Feng W, Hua P, Jaimes EA. COX-2 derived prostaglandins as mediators of the deleterious effects of nicotine in chronic kidney disease. Am J Physiol Renal Physiol 318: F475–F485, 2020. doi: 10.1152/ajprenal.00407.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rezonzew G, Chumley P, Feng W, Hua P, Siegal GP, Jaimes EA. Nicotine exposure and the progression of chronic kidney disease: role of the alpha7-nicotinic acetylcholine receptor. Am J Physiol Renal Physiol 303: F304–F312, 2012. doi: 10.1152/ajprenal.00661.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Raij L, Tian R, Wong JS, He JC, Campbell KN. Podocyte injury: the role of proteinuria, urinary plasminogen, and oxidative stress. Am J Physiol Renal Physiol 311: F1308–F1317, 2016. doi: 10.1152/ajprenal.00162.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feodoroff M, Harjutsalo V, Forsblom C, Thorn L, Waden J, Tolonen N, Lithovius R, Groop PH. Smoking and progression of diabetic nephropathy in patients with type 1 diabetes. Acta Diabetol 53: 525–533, 2016. doi: 10.1007/s00592-015-0822-0. [DOI] [PubMed] [Google Scholar]
- 19.Liao D, Ma L, Liu J, Fu P. Cigarette smoking as a risk factor for diabetic nephropathy: a systematic review and meta-analysis of prospective cohort studies. PLoS One 14: e0210213, 2019. doi: 10.1371/journal.pone.0210213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brosius FC, 3rd, Alpers CE, Bottinger EP, Breyer MD, Coffman TM, Gurley SB, Harris RC, Kakoki M, Kretzler M, Leiter EH, Levi M, McIndoe RA, Sharma K, Smithies O, Susztak K, Takahashi N, Takahashi T; Animal Models of Diabetic Complications C. Mouse models of diabetic nephropathy. J Am Soc Nephrol 20: 2503–2512, 2009. doi: 10.1681/ASN.2009070721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Villarreal R, Mitrofanova A, Maiguel D, Morales X, Jeon J, Grahammer F, Leibiger IB, Guzman J, Fachado A, Yoo TH, Busher Katin A, Gellermann J, Merscher S, Burke GW, Berggren PO, Oh J, Huber TB, Fornoni A. Nephrin contributes to insulin secretion and affects mammalian target of rapamycin signaling independently of insulin receptor. J Am Soc Nephrol 27: 1029–1041, 2016. doi: 10.1681/ASN.2015020210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jaimes EA, Galceran JM, Raij L. Angiotensin II induces superoxide anion production by mesangial cells. Kidney Int 54: 775–784, 1998. doi: 10.1046/j.1523-1755.1998.00068.x. [DOI] [PubMed] [Google Scholar]
- 23.Jaimes EA, Tian RX, Raij L. Nicotine: the link between cigarette smoking and the progression of renal injury? Am J Physiol Heart Circ Physiol 292: H76–H82, 2007. doi: 10.1152/ajpheart.00693.2006. [DOI] [PubMed] [Google Scholar]
- 24.Kopprasch S, Pietzsch J, Graessler J. Validation of different chemilumigenic substrates for detecting extracellular generation of reactive oxygen species by phagocytes and endothelial cells. Luminescence 18: 268–273, 2003. doi: 10.1002/bio.737. [DOI] [PubMed] [Google Scholar]
- 25.Camici M, Allegrini S, Tozzi MG. Interplay between adenylate metabolizing enzymes and AMP-activated protein kinase. FEBS J 285: 3337–3352, 2018. doi: 10.1111/febs.14508. [DOI] [PubMed] [Google Scholar]
- 26.Jaimes EA, Tian RX, Pearse D, Raij L. Up-regulation of glomerular COX-2 by angiotensin II: role of reactive oxygen species. Kidney Int 68: 2143–2153, 2005. doi: 10.1111/j.1523-1755.2005.00670.x. [DOI] [PubMed] [Google Scholar]
- 27.Jaimes EA, Zhou MS, Pearse DD, Puzis L, Raij L. Upregulation of cortical COX-2 in salt-sensitive hypertension: role of angiotensin II and reactive oxygen species. Am J Physiol Renal Physiol 294: F385–F392, 2008. doi: 10.1152/ajprenal.00302.2007. [DOI] [PubMed] [Google Scholar]
- 28.Kanetsuna Y, Takahashi K, Nagata M, Gannon MA, Breyer MD, Harris RC, Takahashi T. Deficiency of endothelial nitric-oxide synthase confers susceptibility to diabetic nephropathy in nephropathy-resistant inbred mice. Am J Pathol 170: 1473–1484, 2007. doi: 10.2353/ajpath.2007.060481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Feng W, Chumley P, Prieto MC, Miyada K, Seth DM, Fatima H, Hua P, Rezonzew G, Sanders PW, Jaimes EA. Transcription factor avian erythroblastosis virus E26 oncogen homolog-1 is a novel mediator of renal injury in salt-sensitive hypertension. Hypertension 65: 813–820, 2015. doi: 10.1161/HYPERTENSIONAHA.114.04533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Calizo RC, Bhattacharya S, van Hasselt JGC, Wei C, Wong JS, Wiener RJ, Ge X, Wong NJ, Lee JJ, Cuttitta CM, Jayaraman G, Au VH, Janssen W, Liu T, Li H, Salem F, Jaimes EA, Murphy B, Campbell KN, Azeloglu EU. Disruption of podocyte cytoskeletal biomechanics by dasatinib leads to nephrotoxicity. Nat Commun 10: 2061, 2019. doi: 10.1038/s41467-019-09936-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang L, Sha Y, Bai J, Eisner W, Sparks MA, Buckley AF, Spurney RF. Podocyte-specific knockout of cyclooxygenase 2 exacerbates diabetic kidney disease. Am J Physiol Renal Physiol 313: F430–F439, 2017. doi: 10.1152/ajprenal.00614.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kunjathoor VV, Febbraio M, Podrez EA, Moore KJ, Andersson L, Koehn S, Rhee JS, Silverstein R, Hoff HF, Freeman MW. Scavenger receptors class A-I/II and CD36 are the principal receptors responsible for the uptake of modified low density lipoprotein leading to lipid loading in macrophages. J Biol Chem 277: 49982–49988, 2002. doi: 10.1074/jbc.M209649200. [DOI] [PubMed] [Google Scholar]
- 33.Pavenstadt H. Roles of the podocyte in glomerular function. Am J Physiol Renal Physiol 278: F173–F179, 2000. doi: 10.1152/ajprenal.2000.278.2.F173. [DOI] [PubMed] [Google Scholar]
- 34.Kim YH, Goyal M, Kurnit D, Wharram B, Wiggins J, Holzman L, Kershaw D, Wiggins R. Podocyte depletion and glomerulosclerosis have a direct relationship in the PAN-treated rat. Kidney Int 60: 957–968, 2001. doi: 10.1046/j.1523-1755.2001.060003957.x. [DOI] [PubMed] [Google Scholar]
- 35.Chalovich JM, Schroeter MM. Synaptopodin family of natively unfolded, actin binding proteins: physical properties and potential biological functions. Biophys Rev 2: 181–189, 2010. doi: 10.1007/s12551-010-0040-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wagenknecht LE, Cutter GR, Haley NJ, Sidney S, Manolio TA, Hughes GH, Jacobs DR. Racial differences in serum cotinine levels among smokers in the coronary artery risk development in (young) adults study. Am J Public Health 80: 1053–1056, 1990. doi: 10.2105/AJPH.80.9.1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hall ME, Wang W, Okhomina V, Agarwal M, Hall JE, Dreisbach AW, Juncos LA, Winniford MD, Payne TJ, Robertson RM, Bhatnagar A, Young BA. Cigarette smoking and chronic kidney disease in African Americans in the Jackson Heart Study. J Am Heart Assoc 5: e003280, 2016. doi: 10.1161/JAHA.116.003280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hallan S, de Mutsert R, Carlsen S, Dekker FW, Aasarod K, Holmen J. Obesity, smoking, and physical inactivity as risk factors for CKD: are men more vulnerable? Am J Kidney Dis 47: 396–405, 2006. doi: 10.1053/j.ajkd.2005.11.027. [DOI] [PubMed] [Google Scholar]
- 39.Hallan SI, Orth SR. Smoking is a risk factor in the progression to kidney failure. Kidney Int 80: 516–523, 2011. doi: 10.1038/ki.2011.157. [DOI] [PubMed] [Google Scholar]
- 40.Haroun MK, Jaar BG, Hoffman SC, Comstock GW, Klag MJ, Coresh J. Risk factors for chronic kidney disease: a prospective study of 23,534 men and women in Washington County, Maryland. J Am Soc Nephrol 14: 2934–2941, 2003. doi: 10.1097/01.ASN.0000095249.99803.85. [DOI] [PubMed] [Google Scholar]
- 41.Ishani A, Grandits GA, Grimm RH, Svendsen KH, Collins AJ, Prineas RJ, Neaton JD. Association of single measurements of dipstick proteinuria, estimated glomerular filtration rate, and hematocrit with 25-year incidence of end-stage renal disease in the multiple risk factor intervention trial. J Am Soc Nephrol 17: 1444–1452, 2006. doi: 10.1681/ASN.2005091012. [DOI] [PubMed] [Google Scholar]
- 42.Klag MJ, Whelton PK, Randall BL, Neaton JD, Brancati FL, Ford CE, Shulman NB, Stamler J. Blood pressure and end-stage renal disease in men. N Engl J Med 334: 13–18, 1996. doi: 10.1056/NEJM199601043340103. [DOI] [PubMed] [Google Scholar]
- 43.Xu H, Suo J, Lian J. Cigarette smoking and risk of albuminuria in patients with type 2 diabetes: a systematic review and meta-analysis of observational studies. Int Urol Nephrol 50: 911–922, 2018. doi: 10.1007/s11255-018-1825-x. [DOI] [PubMed] [Google Scholar]
- 44.Yamagata K, Ishida K, Sairenchi T, Takahashi H, Ohba S, Shiigai T, Narita M, Koyama A. Risk factors for chronic kidney disease in a community-based population: a 10-year follow-up study. Kidney Int 71: 159–166, 2007. doi: 10.1038/sj.ki.5002017. [DOI] [PubMed] [Google Scholar]
- 45.Heeschen C, Chang E, Aicher A, Cooke JP. Endothelial progenitor cells participate in nicotine-mediated angiogenesis. J Am Coll Cardiol 48: 2553–2560, 2006. doi: 10.1016/j.jacc.2006.07.066. [DOI] [PubMed] [Google Scholar]
- 46.Heeschen C, Jang JJ, Weis M, Pathak A, Kaji S, Hu RS, Tsao PS, Johnson FL, Cooke JP. Nicotine stimulates angiogenesis and promotes tumor growth and atherosclerosis. Nat Med 7: 833–839, 2001. doi: 10.1038/89961. [DOI] [PubMed] [Google Scholar]
- 47.Mayhan WG, Patel KP. Effect of nicotine on endothelium-dependent arteriolar dilatation in vivo. Am J Physiol 272: H2337–H2342, 1997. doi: 10.1152/ajpheart.1997.272.5.H2337. [DOI] [PubMed] [Google Scholar]
- 48.Dasgupta C, Xiao D, Xu Z, Yang S, Zhang L. Developmental nicotine exposure results in programming of alveolar simplification and interstitial pulmonary fibrosis in adult male rats. Reprod Toxicol 34: 370–377, 2012. doi: 10.1016/j.reprotox.2012.05.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vang A, Clements RT, Chichger H, Kue N, Allawzi A, O'Connell K, Jeong EM, Dudley SC, Jr,Sakhatskyy P, Lu Q, Zhang P, Rounds S, Choudhary G. Effect of alpha7 nicotinic acetylcholine receptor activation on cardiac fibroblasts: a mechanism underlying RV fibrosis associated with cigarette smoke exposure. Am J Physiol Lung Cell Mol Physiol 312: L748–L759, 2017. doi: 10.1152/ajplung.00393.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vinet R, Cortes M, Luxoro M, Delpiano MA. Nicotine-evoked cytosolic Ca(2+) increase and cell depolarization in capillary endothelial cells of the bovine adrenal medulla. Biol Res 42: 111–119, 2009. [Erratum in Biol Res 42: 261, 2009]. [PubMed] [Google Scholar]
- 51.Arany I, Clark J, Reed DK, Juncos LA. Chronic nicotine exposure augments renal oxidative stress and injury through transcriptional activation of p66shc. Nephrol Dial Transplant 28: 1417–1425, 2013. doi: 10.1093/ndt/gfs596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen S, Meng XF, Zhang C. Role of NADPH oxidase-mediated reactive oxygen species in podocyte injury. Biomed Res Int 2013: 839761, 2013. doi: 10.1155/2013/839761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cammisotto PG, Bendayan M. Adiponectin stimulates phosphorylation of AMP-activated protein kinase alpha in renal glomeruli. J Mol Histol 39: 579–584, 2008. doi: 10.1007/s10735-008-9198-6. [DOI] [PubMed] [Google Scholar]
- 54.Matsusaka T, Sandgren E, Shintani A, Kon V, Pastan I, Fogo AB, Ichikawa I. Podocyte injury damages other podocytes. J Am Soc Nephrol 22: 1275–1285, 2011. doi: 10.1681/ASN.2010090963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stein SC, Woods A, Jones NA, Davison MD, Carling D. The regulation of AMP-activated protein kinase by phosphorylation. Biochem J 345: 437–443, 2000. doi: 10.1042/0264-6021:3450437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Eid AA, Ford BM, Block K, Kasinath BS, Gorin Y, Ghosh-Choudhury G, Barnes JL, Abboud HE. AMP-activated protein kinase (AMPK) negatively regulates Nox4-dependent activation of p53 and epithelial cell apoptosis in diabetes. J Biol Chem 285: 37503–37512, 2010. doi: 10.1074/jbc.M110.136796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Eid AA, Gorin Y, Fagg BM, Maalouf R, Barnes JL, Block K, Abboud HE. Mechanisms of podocyte injury in diabetes: role of cytochrome P450 and NADPH oxidases. Diabetes 58: 1201–1211, 2009. doi: 10.2337/db08-1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ruderman NB, Carling D, Prentki M, Cacicedo JM. AMPK, insulin resistance, and the metabolic syndrome. J Clin Invest 123: 2764–2772, 2013. doi: 10.1172/JCI67227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Towler DA. Mitochondrial ROS deficiency and diabetic complications: AMP[K]-lifying the adaptation to hyperglycemia. J Clin Invest 123: 4573–4576, 2013. doi: 10.1172/JCI72326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sharma K, Ramachandrarao S, Qiu G, Usui HK, Zhu Y, Dunn SR, Ouedraogo R, Hough K, McCue P, Chan L, Falkner B, Goldstein BJ. Adiponectin regulates albuminuria and podocyte function in mice. J Clin Invest 118: 1645–1656, 2008. doi: 10.1172/JCI32691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cheng H, Fan X, Moeckel GW, Harris RC. Podocyte COX-2 exacerbates diabetic nephropathy by increasing podocyte (pro)renin receptor expression. J Am Soc Nephrol 22: 1240–1251, 2011. doi: 10.1681/ASN.2010111149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yang X, Okamura DM, Lu X, Chen Y, Moorhead J, Varghese Z, Ruan XZ. CD36 in chronic kidney disease: novel insights and therapeutic opportunities. Nat Rev Nephrol 13: 769–781, 2017. doi: 10.1038/nrneph.2017.126. [DOI] [PubMed] [Google Scholar]
- 63.Soetikno V, Sari FR, Lakshmanan AP, Arumugam S, Harima M, Suzuki K, Kawachi H, Watanabe K. Curcumin alleviates oxidative stress, inflammation, and renal fibrosis in remnant kidney through the Nrf2-keap1 pathway. Mol Nutr Food Res 57: 1649–1659, 2013. doi: 10.1002/mnfr.201200540. [DOI] [PubMed] [Google Scholar]
- 64.Mitrofanova A, Sosa MA, Fornoni A. Lipid mediators of insulin signaling in diabetic kidney disease. Am J Physiol Renal Physiol 317: F1241–F1252, 2019. doi: 10.1152/ajprenal.00379.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chase HP, Garg SK, Marshall G, Berg CL, Harris S, Jackson WE, Hamman RE. Cigarette smoking increases the risk of albuminuria among subjects with type I diabetes. JAMA 265: 614–617, 1991. doi: 10.1001/jama.1991.03460050068022. [DOI] [PubMed] [Google Scholar]
- 66.Christiansen JS. Cigarette smoking and prevalence of microangiopathy in juvenile-onset insulin-dependent diabetes mellitus. Diabetes Care 1: 146–149, 1978. doi: 10.2337/diacare.1.3.146. [DOI] [PubMed] [Google Scholar]
- 67.Christiansen JS, Nerup J. Smoking and diabetic nephropathy. Lancet 1: 605, 1978. doi: 10.1016/S0140-6736(78)91046-2. [DOI] [PubMed] [Google Scholar]
- 68.Sawicki PT, Didjurgeit U, Mühlhauser I, Bender R, Heinemann L, Berger M. Smoking is associated with progression of diabetic nephropathy. Diabetes Care 17: 126–131, 1994. doi: 10.2337/diacare.17.2.126. [DOI] [PubMed] [Google Scholar]
- 69.Heinrich J, Holscher B, Seiwert M, Carty CL, Merkel G, Schulz C. Nicotine and cotinine in adults' urine: the German Environmental Survey 1998. J Expo Anal Environ Epidemiol 15: 74–80, 2005. doi: 10.1038/sj.jea.7500373. [DOI] [PubMed] [Google Scholar]
- 70.Kasza KA, Ambrose BK, Conway KP, Borek N, Taylor K, Goniewicz ML, Cummings KM, Sharma E, Pearson JL, Green VR, Kaufman AR, Bansal-Travers M, Travers MJ, Kwan J, Tworek C, Cheng YC, Yang L, Pharris-Ciurej N, van Bemmel DM, Backinger CL, Compton WM, Hyland AJ. Tobacco-product use by adults and youths in the United States in 2013 and 2014. N Engl J Med 376: 342–353, 2017. [Erratum in N Engl J Med 378: 492, 2018]. doi: 10.1056/NEJMsa1607538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dutra LM, Glantz SA. Electronic cigarettes and conventional cigarette use among U.S. adolescents: a cross-sectional study. JAMA Pediatr 168: 610–617, 2014. [Erratum in JAMA Pediatr 168: 684, 2014]. doi: 10.1001/jamapediatrics.2013.5488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hajek P, Phillips-Waller A, Przulj D, Pesola F, Myers Smith K, Bisal N, Li J, Parrott S, Sasieni P, Dawkins L, Ross L, Goniewicz M, Wu Q, McRobbie HJ. A randomized trial of E-cigarettes versus nicotine-replacement therapy. N Engl J Med 380: 629–637, 2019. doi: 10.1056/NEJMoa1808779. [DOI] [PubMed] [Google Scholar]