

Keywords: acute kidney injury, hypoxia-inducible factor-1α, ischemia-reperfusion, mitochondria, nuclear factor erythroid 2-related factor 2
Abstract
Nuclear factor erythroid 2-related factor 2 (Nrf2) and hypoxia-inducible factor-1α (HIF1α) transcription factors protect against ischemic acute kidney injury (AKI) by upregulating metabolic and cytoprotective gene expression. In this study, we tested the hypothesis that Nrf2 is required for HIF1α-mediated hypoxic responses using Nrf2-sufficient (wild-type) and Nrf2-deficient (Nrf2–/–) primary murine renal/kidney tubular epithelial cells (RTECs) and human immortalized tubular epithelial cells (HK2 cells) with HIF1 inhibition and activation. The HIF1 pathway inhibitor digoxin blocked hypoxia-stimulated HIF1α activation and heme oxygenase (HMOX1) expression in HK2 cells. Hypoxia-mimicking cobalt (II) chloride-stimulated HMOX1 expression was significantly lower in Nrf2–/– RTECs than in wild-type counterparts. Similarly, hypoxia-stimulated HIF1α-dependent metabolic gene expression was markedly impaired in Nrf2–/– RTECs. Nrf2 deficiency impaired hypoxia-induced HIF1α stabilization independent of increased prolyl 4-hydroxylase gene expression. We found decreased HIF1α mRNA levels in Nrf2–/– RTECs under both normoxia and hypoxia-reoxygenation conditions. In silico analysis and chromatin immunoprecipitation assays demonstrated Nrf2 binding to the HIF1α promoter in normoxia, but its binding decreased in hypoxia-exposed HK2 cells. However, Nrf2 binding at the HIF1α promoter was enriched following reoxygenation, demonstrating that Nrf2 maintains constitutive HIF1α expression. Consistent with this result, we found decreased levels of Nrf2 in hypoxia and that were restored following reoxygenation. Inhibition of mitochondrial complex I prevented hypoxia-induced Nrf2 downregulation and also increased basal Nrf2 levels. These results demonstrate a crucial role for Nrf2 in optimal HIF1α activation in hypoxia and that mitochondrial signaling downregulates Nrf2 levels in hypoxia, whereas reoxygenation restores it. Nrf2 and HIF1α interact to provide optimal metabolic and cytoprotective responses in ischemic AKI.
INTRODUCTION
Oxidative stress produced during ischemia-reperfusion (IR) contributes to acute kidney injury (AKI) (1). Nuclear factor erythroid 2-related factor 2 (Nrf2; NFE2-L2) is a b-ZIP transcription factor and confers cellular protection via the antioxidant response element (ARE) (2). Nrf2 plays key roles in preventing renal epithelial injury induced by IR-induced AKI (3). Nrf2 deficiency results in deregulated antioxidant and cytokine gene expression and worsens IR-induced AKI, whereas activation of endogenous Nrf2 or antioxidant supplementation to Nrf2-deficient mice improves renal structure and function in vivo (4, 5). These results demonstrate that insufficient levels or impaired activation of Nrf2 during IR contributes to abnormal tissue repair and renal pathogenesis. Heme oxygenase 1 (Hmox1) plays a key role in conferring cytoprotection against injury to renal epithelia (6, 7), and we have shown that hypoxia stimulates HMOX1 expression in an Nrf2-dependent manner (8). Hypoxia-inducible factor-1α (HIF1α) confers protection against IR-induced AKI (9). This transcription factor is also crucial for upregulating expression of genes controlling metabolism and cytoprotection in hypoxia (10, 11). We tested whether Nrf2 via HIF1α regulates cytoprotective and metabolic gene expression in hypoxia. Our results revealed that Nrf2, through HIF1α, promotes hypoxia-stimulated HMOX1 induction and that Nrf2 is required for HIF1α stabilization and its target metabolic gene activation in RTECs exposed to hypoxia. In addition, we found that mitochondrial complex I downregulates Nrf2 levels immediately following hypoxia, but the expression levels were restored upon reoxygenation, leading to antioxidant and cytoprotective gene expression.
METHODS
Cell Culture and Hypoxia Exposure
Human papilloma virus-transformed “normal” renal proximal tubular epithelial cells, HK2 (CRL-2190, ATCC), were cultured in DMEM/F12 medium (Life Technologies) with 10% FBS and antibiotics. Primary murine renal/kidney tubular epithelial cells (RTECs) were isolated from naïve wild-type (WT; C57BL6) and Nrf2–/– mice (3–4 mo, males), as described previously (12, 13), under the protocol guidelines approved by The Animal Care and Use Committee at University of Illinois at Chicago. Briefly, mouse kidney cortical tissue was minced in serum-free DMEM/F12 media with collagenase (1 mg/mL) (Sigma-Aldrich) and incubated at 37°C for 45 min with frequent mixing. Digested tissue was passed through a 100-μm filter, and cells were washed with medium and centrifuged at 4°C for 5 min. The cell pellet was resuspended and cultured in DMEM/F12 medium with 10% FBS and antibiotics.
Hypoxia Exposure
Cells at subconfluence were replaced with DMEM/F12 medium with 1% serum 1 h before hypoxia exposure. Cells were exposed to hypoxia infused with a gas mixture of 5% CO2 and nitrogen to obtain 1% O2 concentration. Cells were exposed to hypoxia for 2 h (referred to as acute hypoxia) or 12 h (referred to as chronic hypoxia). It is noteworthy that exposure of HK2 cells to chronic (12 h) hypoxia modestly stimulated LDH release without reducing cell viability, as assessed by the CytoTox 96 Non-Radioactive Cytotoxicity kit (G1780, Promega) and MTT (M5655, Sigma) viability assay (data not shown). Immediately after hypoxia exposure, cells were harvested, while some cells were switched to complete medium and placed in a cell culture incubator for 0–360 min (referred as reoxygenated).
Quantitative Real-Time PCR
RNA from kidney cells was isolated using TRIzol reagent (Thermo Fisher Scientific). cDNA was prepared using the qScript cDNA synthesis Kit (95047-100, Quanta Biosciences), and SYBR Green quantitative real-time PCR (qRT-PCR) assay (Thermo Fisher Scientific) was performed using gene-specific primers (Table 1). β-Actin was used as a control to normalize and calculate the expression levels of the target genes analyzed in the study.
Table 1.
Human and mouse PCR primers used in the quantitative RT-PCR analysis
| Gene | Forward | Reverse |
|---|---|---|
| Human | ||
| VEGF | CTGGTCTTGGGTGCATTG | CACCGCCTCGGCTTGTCACAT |
| EPO | GGAGGCCGAGAATATCACGAC | CCCTGCCAGACTTCTACGG |
| HMOX1 | GCCTGGAAGACACCCTAATGTG | GGCCGTGTCAACAAGGATACTT |
| NQO1 | CCATTCTGAAAGGCTGGTTTG | TACTCCGGAAGGGTCCTTTGT |
| GCLM | TGATGCCACCAGATTTGACTG | GTGGGCTTGAATGTCAGGATT |
| β-ACTIN | GCAAGCAGGAGTATGACGAG | CAAATAAAGCCATGCCAATC |
| Mouse | ||
| Vegf | ACTTGTGTTGGGAGGAGGATGTC | AATGGGTTTGTCGTGTTTCTGG |
| Epo | GGGCTCCGAAGAACTTCTGTG | TGACTTTCGTGACTCACCCTC |
| Hmox1 | CACGCATATACCCGCTACCT | CCAGAGTGTTCATTCGAGCA |
| Glut1 | CATCCTTATTGCCCAGGTGTTT | GAAGACGACACTGAGCAGCAGA |
| Phd1 | GAACCCACATGAGGTGAAGC | AACACCTTTCTGTCCCGATG |
| Phd2 | CATTGTTGGCAGAAGGTGTG | CAAAGGACTACAGGGTCTCCA |
| Phd3 | TGTCTGGTACTTCGATGCTGA | GCAAGAGCAGATTCAGTTTTTCT |
| Hif1α | TCAAGTCAGCAACGTGGAAG | TATCGAGGCTGTGTCGACTG |
| Pgk1 | ATGTCGCTTTCCAACAAGCTG | GCTCCATTGTCCAAGCAGAAT |
| Pdk1 | GGACTTCGGGTCAGTGAATGC | TCCTGAGAAGATTGTCGGGGA |
| β-Actin | GCAAGCAGGAGTACGATGAGT | AACGCAGCTCAGTAACAGTC |
Immunoblot Analysis
Cells were lysed in RIPA buffer (R0278, Sigma) with a protease cocktail inhibitor (P8340, Sigma). An aliquot of protein (∼40 µg) was separated, blotted onto the PVDF membrane, and probed with antibodies specific for Nrf2 (H-300, 13032, Santa Cruz Biotechnology), HIF1α (No. 10006421, Cayman Chemicals), HMOX1 (H-105, 10789, Santa Cruz Biotechnology), or β-actin (2Q1055, 58673, Santa Cruz Biotechnology). Immunoblots were developed using the HyGlo ECL kit (E2400, Denville Scientific Inc, NJ) and visualized by the Bio-Rad Gel Doc system.
Chromatin Immunoprecipitation Assays
Chromatin immunoprecipitation (ChIP) assays were performed using a commercially available kit (17–295, Millipore), as detailed previously (13). Briefly, HK2 cells cultured on 100-mm plates were exposed to room air, hypoxia, or hypoxia-reoxygenation, as detailed earlier, and then crosslinked immediately with 1% formaldehyde for 10 min. Chromatin was prepared, and precleared chromatin was incubated with Nrf2 antibodies or IgG antibodies at 4°C overnight, and immunoprecipitated products were amplified using human HIF1α promoter-specific primers: primer 1, forward 5′-TCAGGTGAGGCGGGCTT-3′, and primer 2, reverse 5′-CACTGTGCACTGAGGAGCTG-3′ (14, 15). Relative Nrf2 binding to the promoter was calculated using room air-exposed sample values as one unit.
Statistical Analysis
Statistical significance for multiple groups of WT cells and Nrf2–/– RTECs was calculated by two-way ANOVA using GraphPad Prism software. Student’s t test was used for comparison between the experimental groups in HK2 cells. P values equal to or less than 0.05 were considered significant.
RESULTS
HIF1α Signaling Regulates Hypoxia-Stimulated HMOX1 Expression in Renal Epithelial Cells
We previously demonstrated that Nrf2 is required for the upregulation of HMOX1 expression in human renal epithelia exposed to hypoxia and this correlated with HIF1α levels (8). To examine if HIF1α signaling directly regulates hypoxia-inducible HMOX1 expression, HK2 cells were incubated with HIF1α signaling inhibitor digoxin and then exposed to hypoxia and hypoxia-reoxygenation. As expected, HIF1α levels were elevated in cells exposed to hypoxia and decreased after reoxygenation up to 6 h (Fig. 1A). Digoxin blocked hypoxia-stimulated HIF1α activation and HMOX1 expression, consistent with the concept that HIF1α signaling regulates HMOX1 induction by hypoxia in renal epithelia.
Figure 1.

HIF1α regulates Nrf2 target HMOX1 induction by hypoxia. A: HMOX1 levels in HK2 cells treated with HIF1 pathway inhibitor digoxin (10 µM) or DMSO and exposed to RA or 12-h hypoxia (0hR) and reoxygenated for 6 h (6hR). Cellular extracts were prepared and immunoblotted with HIF1α and HMOX1 antibodies using β-actin as reference. B: primary RTECs were isolated from WT and Nrf2–/– mice and cultured for 6–7 days and then treated with 200 µM cobalt (II) chloride (CoCl2) (60818, Sigma) for 12 h. Hmox1 expression was analyzed by qRT-PCR. * Versus vehicle; § versus WT. Two-way ANOVA was used to calculate the effects of CoCl2 on Hmox1 expression (n = 3 for vehicle; and n = 4 for CoCl2-treated groups). HIF1α, hypoxia-inducible factor 1α; HMOX1, heme oxygenase 1, HK2, human immortalized renal tubular epithelial cells; Nrf2, Nuclear factor erythroid 2-related factor 2; RA, room air; RTECs, primary murine kidney tubular epithelial cells; WT, wild type.
Nrf2 Deficiency Impairs HIF1α Activation Induced by Hypoxia in Renal Epithelia
We previously reported that Nrf2 is required for Hmox1 induction by hypoxia in human and mouse RTECs, but expression of Nrf2 putative target genes Nqo1, Gclc, and Gclm was not induced by hypoxia. In contrast, their expression was suppressed in chronic hypoxia (8). Because HIF1α regulates Hmox1 induction in response to stressful stimuli, including hypoxia (16), we examined whether Nrf2 regulates hypoxia-inducible Hmox1 expression through HIF1α activation. Primary murine RTECs were isolated from Nrf2-deficient (Nrf2–/–) and Nrf2-sufficient (WT) mice and treated with HIF1α activator CoCl2, and Hmox1 mRNA expression was analyzed. As shown in Fig. 1B, CoCl2-stimulated Hmox1 expression was markedly reduced in Nrf2-deficient RTECs, suggesting a requirement for Nrf2 in the regulation of Hmox1 expression by HIF1α in hypoxia.
We next examined whether Nrf2 regulates hypoxia-induced HIF1α-dependent metabolic gene expression. WT and Nrf2–/– RTECs were exposed to hypoxia and hypoxia-reoxygenation, and HIF1α target gene induction was analyzed. HIF1α-regulated glucose transporter 1 (Glut1), vascular endothelial growth factor (Vegf), erythropoietin (Epo), pyruvate dehydrogenase kinase 1 (Pdk1), and phosphoglycerate kinase 1 (Pgk1) mRNA expression induced by acute hypoxia was markedly lower in Nrf2–/– RTECs as compared with WT counterparts (Fig. 2A). Likewise, this HIF1α target gene expression by chronic hypoxia was stronger than acute hypoxia in WT cells, but the induction was markedly lower in Nrf2–/– cells compared with WT counterparts (Fig. 2B).
Figure 2.
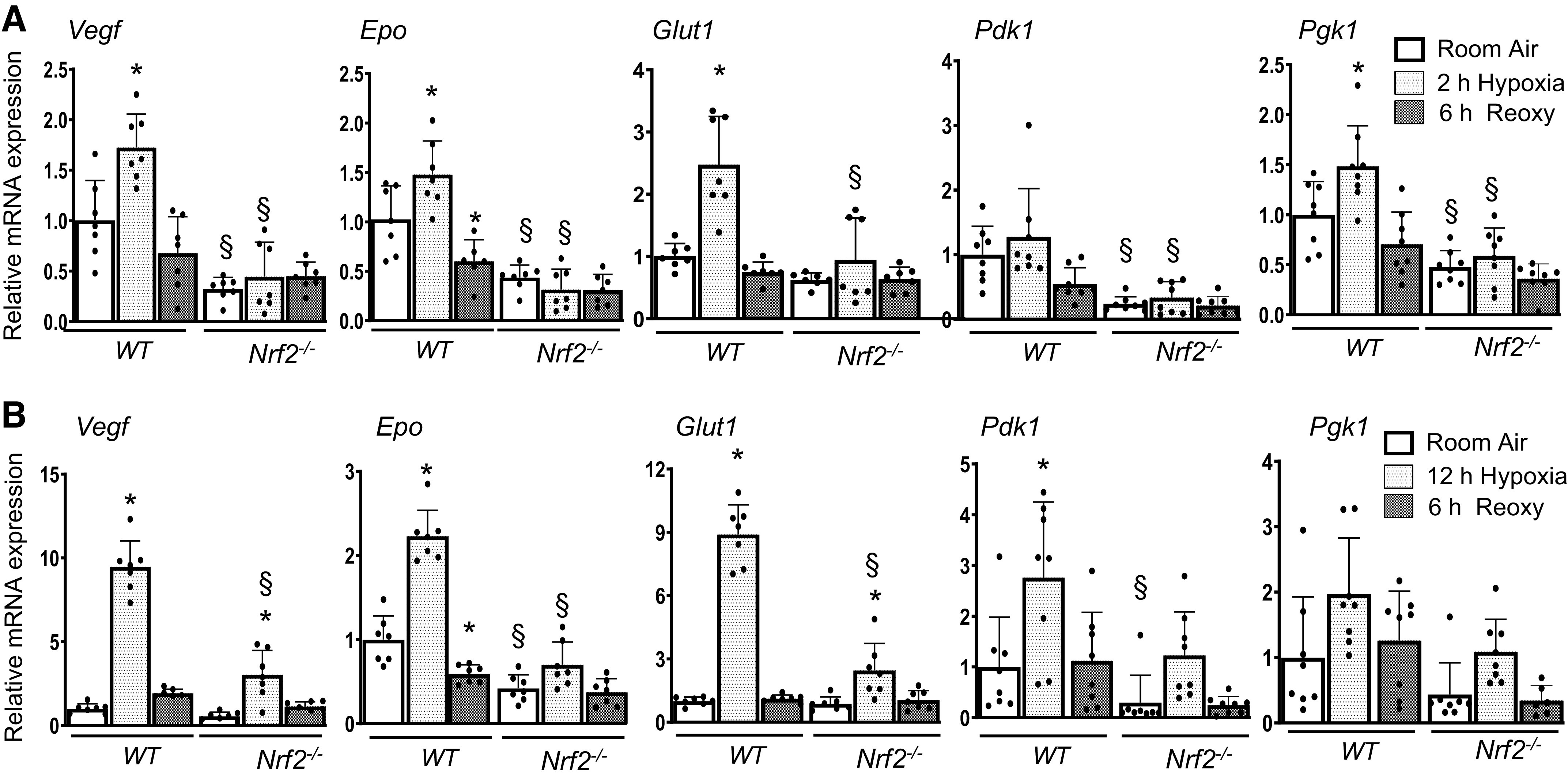
HIF1α target gene expression induced by hypoxia is impaired in Nrf2–/– RTECs. WT and Nrf2–/– RTECs exposed to 2 h (A) or 12 h (B) hypoxia and then subjected to reoxygenation for 6 h. HIF1α putative targets Vegf1, Epo, Glut1, Pdk1, and Pgk1 mRNA expression were analyzed by qRT-PCR analysis. Two-way ANOVA with Tukey’s multiple-comparisons test was used to calculate the effects of hypoxia or reoxygenation on target gene expression (n = 6–8). *Versus room air; §versus WT counterparts. Epo, erythropoietin; Glut1, glucose transporter 1; HIF1α, hypoxia-inducible factor 1α; Nrf2–/–, Nrf2-deficient; Pdk1, pyruvate dehydrogenase kinase 1; Pgk1, phosphoglycerate kinase 1; RTECs, primary murine kidney tubular epithelial cells; WT, wild type.
We then examined whether Nrf2 regulates HIF1α activation (stabilization), thereby affecting its target gene expression in hypoxia. We assessed HIF1α protein levels by immunoblot analysis in both acute and chronic hypoxia-exposed WT and Nrf2–/– RTECs. HIF1α accumulation induced by hypoxia was markedly lower in Nrf2–/– cells than in their WT counterparts (Fig. 3A). We did not find significant differences in HIF1α levels in both WT and Nrf2–/– RTECs following reoxygenation after acute hypoxia (Fig. 3B) or chronic hypoxia (Fig. 3C). Collectively, aforementioned data demonstrated that hypoxia-stimulated HIF1α activation and its target metabolic gene expression is largely regulated by Nrf2.
Figure 3.

Nrf2 deficiency impairs hypoxia-stimulated HIF1α activation. A: primary cultured RTECs isolated from WT and Nrf2–/– mice were exposed to hypoxia for 0 h, 0.5 h, 1 h, and 2 h. B: WT and Nrf2–/– RTECs were exposed to RA or acute hypoxia for 2 h (0hR) and subsequently reoxygenated for 6 h (6hR). C: WT and Nrf2–/– RTECs exposed to chronic (12 h) hypoxia and reoxygenated for 6 h (6hR). Cells were lysed and immunoblotted with HIF1α antibody β-actin antibodies. A representative blot of two independent experiments is shown. HIF1α, hypoxia-inducible factor 1α; Nrf2, nuclear factor erythroid 2-related factor 2; Nrf2–/–, Nrf2-deficient; RTECs, primary murine kidney tubular epithelial cells; WT, wild type.
Nrf2 Regulates HIF1α and PhD2/3 mRNA Steady-State Levels in Kidney Epithelia
To determine whether Nrf2 modulates HIF1α activation by regulating prolyl 4-hydroxylase (PHD) expression, we evaluated PhD1/2/3 mRNA expression levels in WT and Nrf2–/– RTECs without and with hypoxia exposure (Fig. 4). Our studies revealed modestly decreased steady-state Phd2 and PhD3 mRNA expression in Nrf2–/– cells compared with WT cells (Fig. 4A). Phd2 and Phd3 mRNA levels but not Phd1 mRNA levels were induced by acute hypoxia and restored to the basal state after reoxygenation in WT cells. Likewise, chronic hypoxia stimulated Phd2 and Phd3 expression (Fig. 4B), which was markedly higher than in cells exposed to acute hypoxia (Fig. 4A). Both acute and chronic hypoxia-stimulated Phd3 expression was significantly lower in Nrf2–/– cells than in WT counterparts (Fig. 4B). Likewise, Phd2 induction by acute hypoxia but not chronic hypoxia was significantly lower in Nrf2–/– cells than in WT cells. We next examined whether Nrf2 regulates HIF1α mRNA expression and found that Nrf2 deficiency negatively affects steady-state levels of HIF1α mRNA (Fig. 5). HIF1α mRNA expression was not stimulated by both acute and chronic hypoxia, but its mRNA expression was remained lower in Nrf2–/– cells exposed to hypoxia and following reoxygenation.
Figure 4.
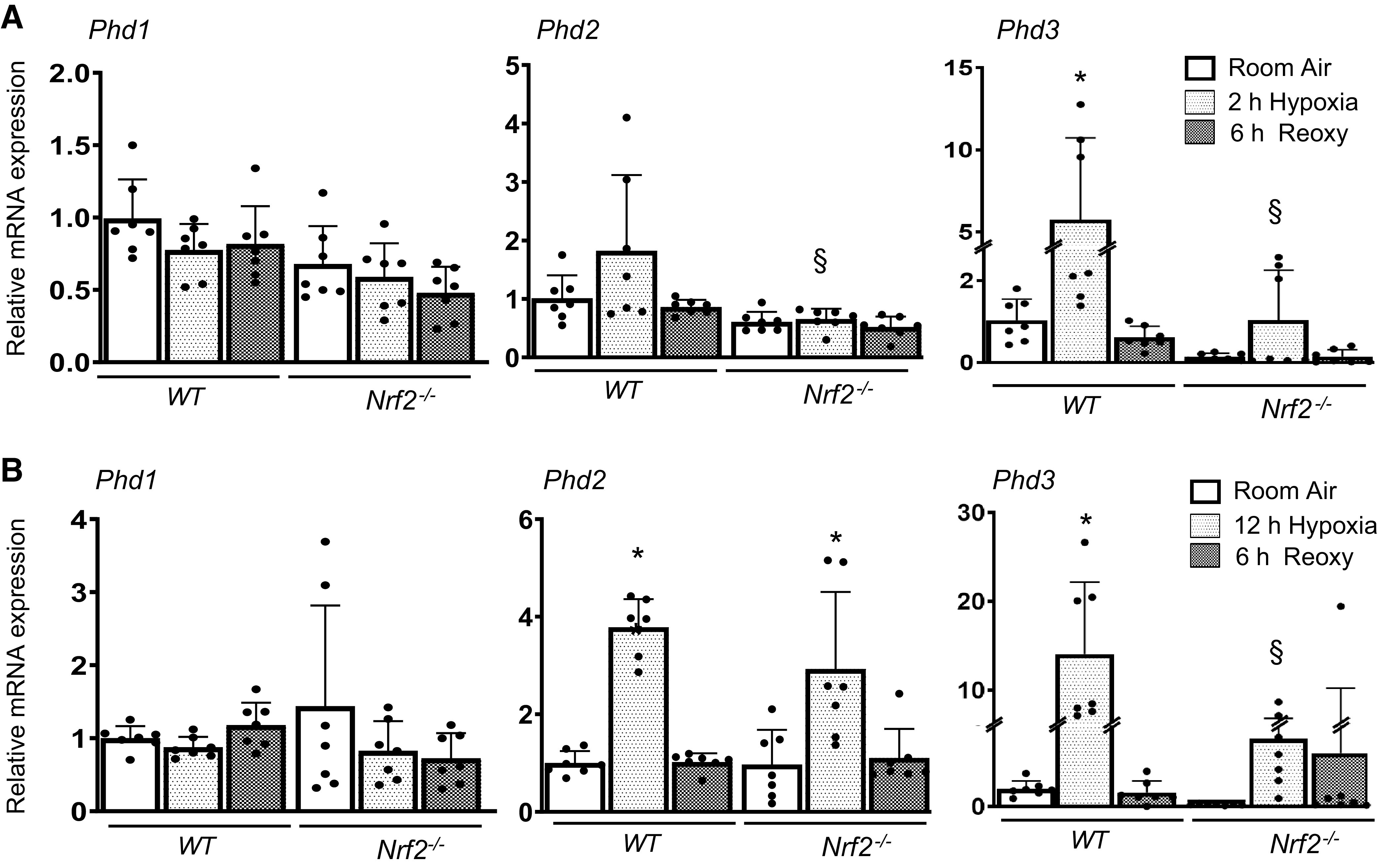
Hypoxia-stimulated PHD expression is regulated by Nrf2. WT and Nrf2–/– RTECs were exposed to 2-h or 12-h hypoxia and reoxygenated for 6 h. Phd1/2/3 mRNA expression was analyzed by qRT-PCR. A: mRNA expression induced by 2-h hypoxia and followed by reoxygenation. B: mRNA levels induced by 12-h hypoxia and followed by reoxygenation. Two-way ANOVA with Tukey’s multiple-comparisons test was used to calculate the effects of hypoxia or reoxygenation on target gene expression (n = 6–8). *Versus room air; §versus WT counterparts. Nrf2, nuclear factor erythroid 2-related factor 2; Nrf2–/–, Nrf2-deficient; PHD, prolyl 4-hydroxylase; RTECs, primary murine kidney tubular epithelial cells; WT, wild type.
Figure 5.

Nrf2 positively regulates HIF1α mRNA expression. WT and Nrf2–/– RTECs exposed to 2-h or 12-h hypoxia and/or subjected for reoxygenation. HIF1α mRNA expression was analyzed by qRT-PCR. A: expression levels in 2-h hypoxia and reoxygenation for 6 h. B: expression levels in 12-h hypoxia and reoxygenation for 6 h. Two-way ANOVA with Tukey’s multiple-comparisons test was used to calculate the effects of hypoxia or reoxygenation on target gene expression (n = 6–8). *Versus room air; §versus WT counterparts. HIF1α, hypoxia-inducible factor 1α; Nrf2, nuclear factor erythroid 2-related factor 2; Nrf2–/–, Nrf2-deficient; RTECs, primary murine kidney tubular epithelial cells; WT, wild type.
Nrf2 Binds to the HIF1α Promoter in Renal Epithelia
In silico analysis revealed the presence of putative Nrf2-binding sites, AREs, in both human and murine HIF1α promoters (Fig. 6A). To examine whether Nrf2 binds to the endogenous HIF1α promoter, we performed ChIP assays in HK2 cells exposed to acute (2 h) or chronic (12 h) hypoxia and hypoxia-reoxygenation for 1 h, 3 h, or 6 h. Nrf2 binding to the HIF1α promoter bearing AREs was decreased in acute hypoxia, but the binding restored to the basal level following reoxygenation (Fig. 6B). In chronic hypoxia, Nrf2 binding to the HIF1α promoter was modestly increased above the basal level, but its recruitment to the promoter during hypoxia-reoxygenation was markedly increased by 3 h and persisted up to 6 h (Fig. 6C). Taken together, these data suggest that reduced levels of hypoxia-stimulated HIF1α activation and its target gene expression in Nrf2–/– cells are not due to increased PHD expression but rather due to its diminished HIF1α mRNA levels regulated by Nrf2.
Figure 6.

Nrf2 binds to the endogenous HIF1α promoter in vivo. HK2 cells exposed to room air (RA), 2-h/12-h hypoxia (0hR), and hypoxia-reoxygenation for 1 h (1hR), 3 h (3hR) or 6 h (6hR). ChIP assays were performed using anti-Nrf2 antibodies. IgG antibodies were used as negative controls. A: schema of human HIF1α promoter with a transcriptional start site and Sp1 sites (11, 17). Position of putative Nrf2-binding sites, AREs, and primer 1 and primer 2 used for ChIP assay are indicated. Bottom represents murine HIF1α promoter. B: Nrf2 binding to the human HIF1α promoter in HK2 cells exposed to 2-h hypoxia and hypoxia-reoxygenation. C: Nrf2 binding to the human HIF1α promoter in HK2 cells exposed to 12-h hypoxia and hypoxia-reoxygenation. ChIP assays performed with IgG antibodies showed undetectable levels of promoter amplification (not shown). Student’s t test was used to calculate the effects of hypoxia or reoxygenation on Nrf2 binding compared with RA control (n = 8–9) or hypoxia (0hR), respectively. *Versus RA; §versus 0hR. AREs, antioxidant response elements; ChIP, chromatin immunoprecipitation; HIF1α, hypoxia-inducible factor 1α; HK2, human immortalized tubular epithelial cells; Nrf2, Nuclear factor erythroid 2-related factor 2; RA, room air.
Hypoxia Downregulates Nrf2 Expression
To examine whether hypoxia regulates Nrf2 expression levels, HK2 cells were exposed to hypoxia for different time points followed by reoxygenation for 0 h or 3 h. Cellular extracts were prepared and probed with Nrf2 antibodies (Fig. 7A). We found reduced expression of Nrf2 levels in HK2 cells exposed to hypoxia; however, reoxygenation restored its expression to the basal level. HK2 cells were then exposed to hypoxia for shorter time intervals (15, 30, or 60 min) to determine the timescale of hypoxia-induced Nrf2 downregulation (Fig. 7B). Nrf2 expression was immediately decreased by 30 min and remained low thereafter in hypoxia. We also examined the kinetics of Nrf2 restoration following acute (Fig. 7C) or chronic (Fig. 7D) hypoxia and found that its expression restored back to normal (steady-state) levels by 30 min upon reoxygenation and remained above the basal level by 3 h. The loss and restoration of Nrf2 expression in hypoxia and reoxygenation were inversely correlated with HIF1α activation and inactivation, respectively (Fig. 7A).
Figure 7.

Hypoxia downregulates Nrf2 levels. A: HK2 cells were exposed to hypoxia for 30, 60, 90, or 120 mins (0hR) followed by reoxygenation for 3 h (3hR). B: HK2 cells were exposed to room air or hypoxia for 15, 30, or 60 mins. Kinetics of Nrf2 restoration during reoxygenation following acute hypoxia (C) and chronic hypoxia (D). Cells were lysed and immunoblotted with Nrf2 and β-actin antibody. A representative blot of two independent experiments is shown. HK2, human immortalized tubular epithelial cells; Nrf2, Nuclear factor erythroid 2-related factor 2.
Proteasomal or PKC Inhibition Does Not Affect Hypoxia-Downregulated Nrf2 Expression
Kelch-like ECH-associated protein 1 (KEAP1)-mediated proteasomal degradation largely regulates Nrf2 expression in multiple cell types under the basal state (2). Various stressful stimuli affect this process largely via disruption of Keap1:Nrf2 interaction, which promotes Nrf2 stabilization and nuclear accumulation, accompanied by its target gene expression. We therefore examined whether proteasome inhibition prevents hypoxia-induced Nrf2 downregulation in renal epithelia. Cells incubated with a proteasome inhibitor MG132 were exposed to hypoxia for 1 h, and Nrf2 levels were analyzed. As anticipated, MG132 enhanced steady-state levels of Nrf2 in cells exposed to normoxia; however, it did not significantly inhibit hypoxia-decreased Nrf2 levels (Fig. 8A). Similarly, protein synthesis inhibitor cycloheximide suppressed constitutive Nrf2 expression, but it did not block hypoxia-downregulated Nrf2 levels (Fig. 8A).
Figure 8.

The effects of proteasome and protein kinase inhibition on Nrf2 levels in hypoxia. HK2 cells were exposed to room air or hypoxia for 60 min in the presence of vehicle (DMSO) (D2650, Sigma), 10 µM MG132 (M7449, Sigma), 10 µM cycloheximide (C4859, Sigma) (A), or 10 µM bisindolylmaleimide I (BIM I) (203290, Calbiochem) (B). Cell extracts were prepared and probed with Nrf2 and β-actin antibodies. A representative blot of two independent experiments is shown. HK2, human immortalized renal tubular epithelial cells; Nrf2, Nuclear factor erythroid 2-related factor 2.
PKC signaling regulates Nrf2 nuclear accumulation (18, 19). To examine whether this signaling regulates Nrf2 levels in hypoxia, cells were treated with a general PKC inhibitor bisindolylmaleimide I (BIM I) before hypoxia exposure. Cell extracts were prepared and immunoprobed with Nrf2. Decreased levels of Nrf2 in hypoxia-exposed cells were not restored to the basal state by BIM I (Fig. 8B), suggesting that PKC signaling does not regulate Nrf2 levels affected by hypoxia in kidney epithelia.
Mitochondrial Complex I Inhibition Prevents Hypoxia-Reduced Nrf2 Levels
Mitochondrial respiration is known to contribute to oxidative stress in IR-induced AKI (6). To determine whether mitochondrial signaling modulates Nrf2 levels in hypoxia, HK2 cells were exposed to hypoxia in the presence of mitochondrial complex I inhibitor rotenone and complex II inhibitor antimycin A. As shown in Fig. 9A, rotenone significantly blocked hypoxia-induced negative effects on Nrf2 levels, whereas antimycin A had no such effect. Next, we have examined the effects of rotenone on Nrf2 target gene expression. Consistent with this result, rotenone significantly upregulated HMOX1, VEGF, and EPO expression (Fig. 9B), and this was correlated with increased levels of HIF1α (Fig. 9C). However, Nrf2 putative target genes such as NQO1, GCLC, and GCLM were not upregulated with mitochondrial complex I inhibition (Fig. 9B), despite increased Nrf2 steady-state levels.
Figure 9.

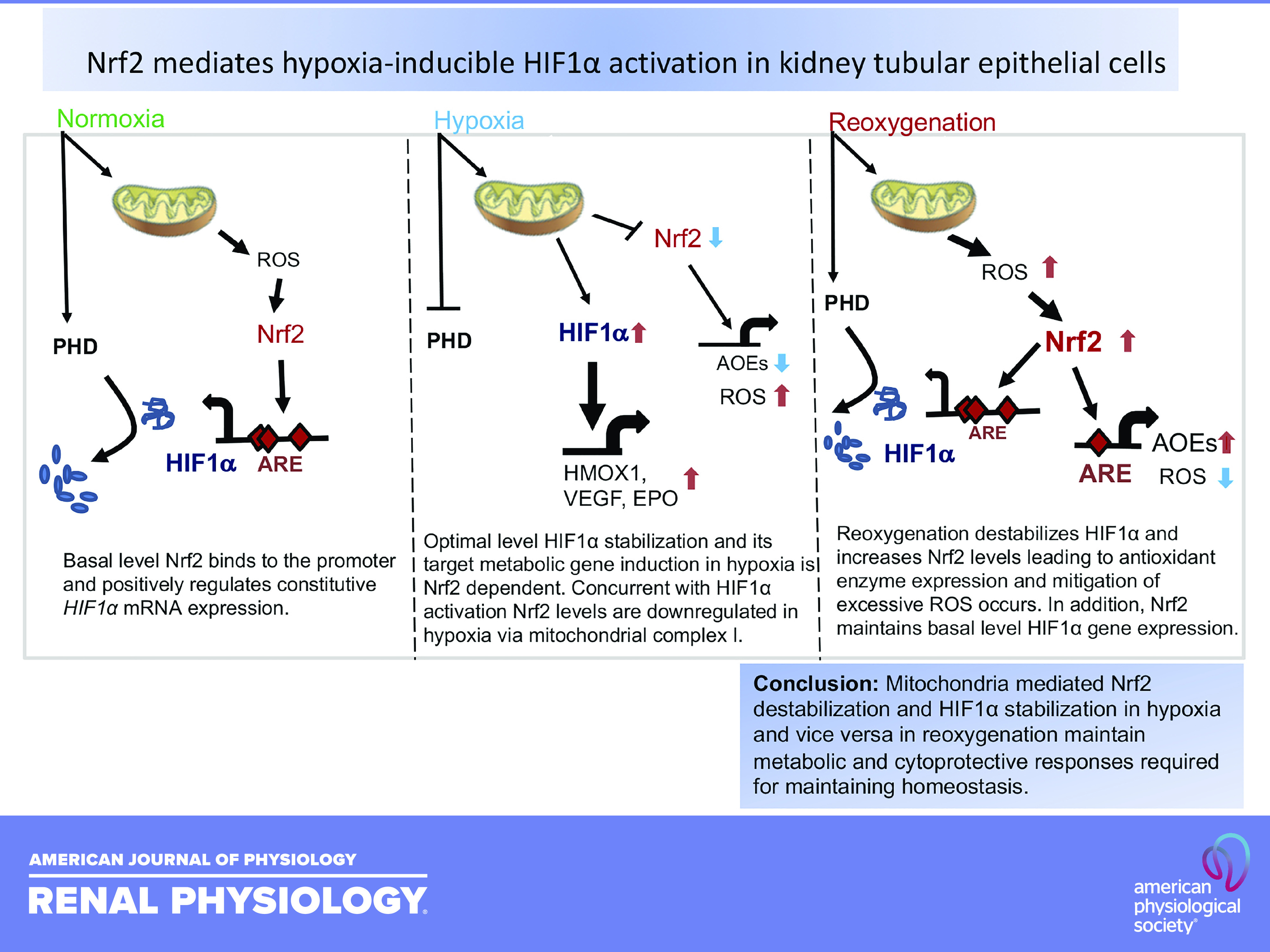
Mitochondrial complex I mediates hypoxia-induced Nrf2 downregulation and HIF1α target gene expression. A: HK2 cells treated with 10 µM rotenone (557368, Calbiochem) or 10 µM antimycin A (A8674, Sigma) or DMSO for 1 h were exposed to room air or hypoxia for 1 h, cell lysates prepared, and immunoblotted with β-actin and Nrf2 antibodies. A representative blot of at least two independent experiments is shown. B: HK2 cells treated with DMSO or 10 µM rotenone for 6 h, RNA isolated, and gene expression was analyzed by qRT-PCR. Student’s t test was used to calculate the effect of rotenone on gene expression (n = 6). * Versus DMSO. C: the effects of rotenone on HIF1α levels. Cellular extracts treated with rotenone were probed with HIF1α and β-actin antibodies. D: schema of mitochondria-mediated Nrf2 destabilization and HIF1α stabilization in hypoxia and vice versa in reoxygenation. Basal level Nrf2 binds to the promoter and positively regulates constitutive HIF1α mRNA expression (left). Optimal level HIF1α stabilization and its target metabolic gene induction in hypoxia is Nrf2 dependent. Concurrent with HIF1α activation, Nrf2 levels are downregulated in hypoxia via mitochondrial complex I (middle). Reoxygenation destabilizes HIF1α and increases Nrf2 levels leading to antioxidant enzyme expression and mitigation of excessive ROS occurs. In addition, Nrf2 maintains basal-level HIF1α gene expression (right). HIF1α, hypoxia-inducible factor 1α; HK2, human immortalized renal tubular epithelial cells; Nrf2, Nuclear factor erythroid 2-related factor 2; RNA, ribonucleic acid.
DISCUSSION
The present study demonstrates that Nrf2 is required for hypoxia-inducible HIF1α activation and its target metabolic gene expression in human and mouse renal tubular epithelial cells. We also found that along with HIF1α activation, steady-state levels of Nrf2 are immediately downregulated in hypoxia, but they are restored during reoxygenation, concurrent with HIF1α inactivation (destabilization) and cytoprotective gene induction (8, 20). Thus, both distinct biphasic and opposite activation of HIF1α and Nrf2 transcription factors in hypoxia and reoxygenation conditions seem to play key roles in selective upregulation and maintenance of renal tubular cell metabolic and cytoprotective gene expression to mitigate ischemic AKI (Fig. 9C).
HIF1α is constitutively transcribed and translated followed by subsequent E3 ubiquitin ligase dependent-proteasome pathway-mediated degradation (half-life <10 min), leading to undetectable/low levels of protein expression in normoxia (10, 21). However, a rapid activation (accumulation) of HIF1α is crucial for the transcriptional induction of genes encoding metabolic enzymes in renal epithelia exposed to hypoxia (10). HMOX1 confers cytoprotection against renal IR-induced injury (7). We have previously shown that Nrf2 is important for hypoxia-stimulated HMOX1 expression, but ChIP assays revealed decreased recruitment of this transcription factor at the endogenous HMOX1 promoter following hypoxia (8), as opposed to nonhypoxic stimuli (22, 23), suggesting that Nrf2 indirectly regulates HMOX1 expression. The present study using Nrf2-deficient primary murine RTECs demonstrated that this transcription factor is crucial for HIF1α activation and its target metabolic gene transcription including Hmox1 in acute and chronic hypoxic conditions. Other studies showed that Nrf2 regulates HIF1α activation in tumor epithelia (17, 24) and HIF1α and Hmox1 activation in endothelial progenitor cells (25). Moreover, treatment of colon cancer cells with Nrf2 inhibitor caused downregulation of HIF1α protein levels via proteasomal degradation (26). Collectively, these results suggest that Nrf2 plays a key role in HIF1α activation in epithelial cells by hypoxia under both physiological and pathological conditions.
Mitochondria-mediated signaling in hypoxia promotes HIF1α activation (stabilization) largely through PHD1 and PHD2 inhibition, although other mechanisms also contribute (21, 27). However, our studies revealed that Nrf2 does not regulate Phd1/2/3 mRNA expression in basal state, suggesting that their increased expression unlikely contributes to impaired activation of HIF1α in Nrf2–/– RTECs following hypoxia. In contrast, Phd2 and Phd3 mRNA expression was strongly induced by hypoxia in WT but not in Nrf2–/– RTECs. Other proteins such as factor inhibiting HIF (FIH) (28) and coiled-coil-helix-coiled-coil-helix domain-containing protein 4 (CHCHD4), a redox-sensitive mitochondrial protein, control HIF1α protein accumulation (29). Overexpression of Nrf2 transcriptional target NQO1 increases steady-state HIF1α protein levels by directly interacting and inhibiting proteasomal degradation of the latter in colorectal and breast cancer cells (30). However, we found downregulation of both Nrf2 and NQO1 levels in RTECs exposed to prolonged hypoxia without impairing HIF1α activation, suggesting that HIF1α protein accumulation in chronic hypoxia is upregulated in an Nrf2-independent manner, perhaps through ROS-mediated suppression of HIF1α-negative regulators.
HIF1α promoter contains a high-GC region with a TATA-less transcription start site and ubiquitous factor binding sites such as Sp1 (14, 15). Previous studies have shown that phosphatidylinositol 3-kinase (PI3K) and STAT3 signaling positively regulates HIF1α mRNA expression in human tumor epithelia (31, 32). Although HIF1α mRNA is constitutively expressed in diverse cell types, upregulation of its transcription by hypoxia is variable and occurs in a contextual manner both in vitro and in vivo (33, 34). We found that HIF1α mRNA expression was not altered in RTECs by hypoxia or following hypoxia-reoxygenation. Our findings obtained from ChIP assays revealed that Nrf2 constitutively binds to the HIF1α promoter in normoxia, but its binding decreases in hypoxia, which correlates with decreased levels of Nrf2. However, Nrf2 recruitment at the HIF1α promoter was enriched following reoxygenation, correlating with increased Nrf2 levels. These results and the fact that Nrf2 deficiency negatively affects HIF1α mRNA levels both in normoxia and hypoxia-reoxygenation conditions strongly suggest that Nrf2 controls HIF1α expression in renal epithelia. Additional studies are warranted to address the exact mechanism through which Nrf2 positively regulates constitutive HIF1α mRNA expression, and whether this decreased expression primarily contributes to reduced level of HIF1α accumulation and its downstream target gene expression in hypoxic RTECs lacking Nrf2. It is noteworthy that hypoxia-stimulated PHD2 and PHD3 but not PHD1 and HIF1α mRNA expression observed in RTECs is consistent with previous results obtained from both malignant and nonmalignant cells (35–37). However, we observed reduced levels of PHD2 and PHD3 mRNA expression induced by hypoxia in Nrf2–/– RTECs, and this is associated with lack of HIF1α activation. As previous studies have shown that PHD induction is regulated by HIF1α (35, 36), it is likely that reduced expression of PHD2 and PHD3 in Nrf2–/– RTECs could be due to impaired HIF1α activation.
The present study demonstrated that Nrf2 levels are downregulated by hypoxia as early as 30 min after exposure, and these levels remained low/undetectable even in chronic hypoxia. Inhibition of mitochondrial complex I was sufficient for preventing Nrf2 downregulation caused by hypoxia. Increased Nrf2 levels by rotenone alone were accompanied by HMOX1 induction but not that of other putative Nrf2 targets NQO1, GCLC, and GCLM. HIF1α target genes (VEGF and EPO) were also upregulated by rotenone correlating with increased levels of both Nrf2 and HIF1α. These findings suggest that immediate Nrf2 downregulation in hypoxia occurs via the mitochondrial complex I-mediated mechanism at least in the settings of acute hypoxia. Nrf2 protein half-life is ∼30 min and is constantly degraded by proteasome pathway under the steady-state condition (38, 39). Our studies revealed that proteasomal inhibition does not prevent hypoxia-promoted Nrf2 downregulation. Thus, it is possible that decreased levels of Nrf2 caused by hypoxia could be either due to decreased protein synthesis and/or altered protein modifications, and this requires a detailed investigation. Despite the fact that hypoxia is known to increase the levels of ROS, which are known to disrupt the Keap1:Nrf2 interaction and promote Nrf2 nuclear accumulation, Nrf2 levels remained low in chronic hypoxia. Previously, it was shown that hypoxia stimulates seven in absentia homolog 2 (Siah2), an E3 ubiquitin protein ligase, which then interacts and promotes Nrf2 degradation (40). It is unlikely that a Siah2-regulated mechanism contributes to the immediate downregulation of Nrf2 by hypoxia that we have observed by 30–60 min in renal epithelia, but a Siah2-mediated mechanism in Nrf2 downregulation in chronic hypoxia cannot be ruled out. Because HIF1α inhibition markedly reduced hypoxia-stimulated HMOX1 expression and Nrf2 deficiency impairs HIF1α protein stabilization and HMOX1 induction by hypoxia (8), it is likely that the initial Nrf2-dependent HIF1α activation occurs immediately following hypoxia.
By upregulating antioxidant gene expression, Nrf2 mitigates excessive ROS production, whereas excessive ROS is needed to inhibit PHD function and to stabilize HIF1α levels (27). We have previously reported diminished NQO1, GCLC, and GCLM expression in RTECs exposed to both acute and chronic hypoxia (8), and others have found reduced expression of these genes in the lungs of mice exposed to hypoxia (41). Thioredoxin reductase 1 is a transcriptional target of Nrf2 and plays an important role in scavenging H2O2, but its expression is downregulated in hypoxia (42). Thus, it is conceivable that the downregulation of Nrf2 and its putative target gene expression in hypoxia could result in increased ROS production and PHD destabilization resulting in HIF1α protein accumulation and metabolic gene transcription (21). Despite the fact that hypoxia is known to increase the levels of ROS, which are known to disrupt the Keap1:Nrf2 interaction and promote Nrf2 nuclear accumulation, Nrf2 levels remained low in chronic hypoxia. The MAFG transcription factor, a major heterodimeric partner of Nrf2, interacts with and promotes HIF1α nuclear accumulation in hepatoma cells exposed to hypoxia (41). Thus, whether Nrf2 downregulation in hypoxia is required for MAFG to interact with and facilitate HIF1α nuclear accumulation and metabolic gene induction and/or to increase ROS levels for inhibiting PHD function remains to be investigated.
The current study has a number of limitations. Based on previous work by our team and others of the important roles for Nrf2 and HIF1α during in vivo models of ischemic AKI, we focused on in vitro studies in the current article. However, by studying both murine and human kidney cells, we aimed to increase the human relevance. In addition, though these key transcription factors are important in many different kidney cell types, we only focused on the kidney tubular epithelial cell.
Significance and Perspectives
Dysregulation of cytoprotective and metabolic responses following kidney ischemic reperfusion injury leads to short-term increased morbidity and mortality and long-term, occasionally chronic kidney disease. The mechanisms underlying these processes are incompletely understood, and a better understanding will improve diagnostics and therapeutics. Several effector pathways are known to control responses during tissue injury and repair. Among these, HIF1α is an important mediator of the cellular response to hypoxia, and Nrf2 transcription factor drives cytoprotective responses to mitigate cellular stress. Our current studies demonstrated that Nrf2 is a critical positive regulator of HIF1α activation and subsequent metabolic gene induction in kidney tubular epithelial cells during both acute and chronic hypoxia. Our studies also demonstrated that destabilization and stabilization of Nrf2 and HIF1α occurs during hypoxic and reoxygenation conditions. These results suggest that a compensatory regulatory mechanism modulates both metabolic and cytoprotective gene expression to maintain cellular metabolism in hypoxia, as well as to mitigate stress produced by tissue reoxygenation in kidney epithelia. Further studies in preclinical models and humans focused on understanding the potential cross talk between these two crucial transcription factors in the pathogenesis of IRI and abnormal repair are warranted and may lead to better diagnostics and therapeutics for patients in the future.
GRANTS
This work was supported by National Institutes of Health Grant DK084445 (to H.R. and S.P.R.) and partly by National Institutes of Health Grants HL136946 (to S.P.R.) and GM124235 (to S.P.R.) as well as departmental funds.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
H.R.P., H.R., and S.P.R. conceived and designed research; H.R.P., P.M.N., C.R.T., and A.A. performed experiments; H.R.P., and P.M.N. analyzed data; H.R.P., S.N., H.R., and S.P.R. interpreted results of experiments; H.R.P., P.M.N., and S.P.R. prepared figures; P.M.N. and S.P.R. drafted manuscript; P.M.N., S.N., H.R., and S.P.R. edited and revised manuscript; H.R.P., P.M.N., C.R.T., A.A., S.N., and S.P.R. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Thomas Kensler and Masayuki Yamamoto for providing the Nrf2-null mouse used in the present study. We thank Usha Raj for allowing us to use the hypoxia chamber.
REFERENCES
- 1.Ronco C, Bellomo R, Kellum JA. Acute kidney injury. Lancet 394: 1949–1964, 2019. doi: 10.1016/S0140-6736(19)32563-2. [DOI] [PubMed] [Google Scholar]
- 2.Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol 47: 89–116, 2007. doi: 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]
- 3.Nezu M, Suzuki N. Roles of Nrf2 in protecting the kidney from oxidative damage. Int J Mol Sci 21: 2951, 2020. doi: 10.3390/ijms21082951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu M, Grigoryev DN, Crow MT, Haas M, Yamamoto M, Reddy SP, Rabb H. Transcription factor Nrf2 is protective during ischemic and nephrotoxic acute kidney injury in mice. Kidney Int 76: 277–285, 2009. doi: 10.1038/ki.2009.157. [DOI] [PubMed] [Google Scholar]
- 5.Liu M, Reddy NM, Higbee EM, Potteti HR, Noel S, Racusen L, Kensler TW, Sporn MB, Reddy SP, Rabb H. The Nrf2 triterpenoid activator, CDDO-imidazolide, protects kidneys from ischemia-reperfusion injury in mice. Kidney Int 85: 134–141, 2014. doi: 10.1038/ki.2013.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Agarwal A, Balla J, Alam J, Croatt AJ, Nath KA. Induction of heme oxygenase in toxic renal injury: a protective role in cisplatin nephrotoxicity in the rat. Kidney Int 48: 1298–1307, 1995. doi: 10.1038/ki.1995.414. [DOI] [PubMed] [Google Scholar]
- 7.Lever JM, Boddu R, George JF, Agarwal A. Heme oxygenase-1 in kidney health and disease. Antioxid Redox Signal 25: 165–183, 2016. doi: 10.1089/ars.2016.6659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Potteti HR, Tamatam CR, Marreddy R, Reddy NM, Noel S, Rabb H, Reddy SP. Nrf2-AKT interactions regulate heme oxygenase 1 expression in kidney epithelia during hypoxia and hypoxia-reoxygenation. Am J Physiol Renal Physiol 311: F1025–F1034, 2016. doi: 10.1152/ajprenal.00362.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci USA 92: 5510–5514, 1995. doi: 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schodel J, Ratcliffe PJ. Mechanisms of hypoxia signalling: new implications for nephrology. Nat Rev Nephrol 15: 641–659, 2019. doi: 10.1038/s41581-019-0182-z. [DOI] [PubMed] [Google Scholar]
- 11.Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell 148: 399–408, 2012. doi: 10.1016/j.cell.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bell CL, Tenenhouse HS, Scriver CR. Initiation and characterization of primary mouse kidney epithelial cultures. In Vitro Cell Dev Biol 24: 683–695, 1988. doi: 10.1007/BF02623606. [DOI] [PubMed] [Google Scholar]
- 13.Potteti HR, Reddy NM, Hei TK, Kalvakolanu DV, Reddy SP. The NRF2 activation and antioxidative response are not impaired overall during hyperoxia-induced lung epithelial cell death. Oxid Med Cell Longev 2013: 798401, 2013. doi: 10.1155/2013/798401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Iyer NV, Leung SW, Semenza GL. The human hypoxia-inducible factor 1alpha gene: HIF1A structure and evolutionary conservation. Genomics 52: 159–165, 1998. doi: 10.1006/geno.1998.5416. [DOI] [PubMed] [Google Scholar]
- 15.Minet E, Ernest I, Michel G, Roland I, Remacle J, Raes M, Michiels C. HIF1A gene transcription is dependent on a core promoter sequence encompassing activating and inhibiting sequences located upstream from the transcription initiation site and cis elements located within the 5'UTR. Biochem Biophys Res Commun 261: 534–540, 1999. doi: 10.1006/bbrc.1999.0995. [DOI] [PubMed] [Google Scholar]
- 16.Lee PJ, Jiang BH, Chin BY, Iyer NV, Alam J, Semenza GL, Choi AM. Hypoxia-inducible factor-1 mediates transcriptional activation of the heme oxygenase-1 gene in response to hypoxia. J Biol Chem 272: 5375–5381, 1997. doi: 10.1074/jbc.272.9.5375. [DOI] [PubMed] [Google Scholar]
- 17.Kim TH, Hur EG, Kang SJ, Kim JA, Thapa D, Lee YM, Ku SK, Jung Y, Kwak MK. NRF2 blockade suppresses colon tumor angiogenesis by inhibiting hypoxia-induced activation of HIF-1alpha. Cancer Res 71: 2260–2275, 2011. doi: 10.1158/0008-5472.CAN-10-3007. [DOI] [PubMed] [Google Scholar]
- 18.Huang HC, Nguyen T, Pickett CB. Phosphorylation of Nrf2 at Ser-40 by protein kinase C regulates antioxidant response element-mediated transcription. J Biol Chem 277: 42769–42774, 2002. doi: 10.1074/jbc.M206911200. [DOI] [PubMed] [Google Scholar]
- 19.Huang HC, Nguyen T, Pickett CB. Regulation of the antioxidant response element by protein kinase C-mediated phosphorylation of NF-E2-related factor 2. Proc Natl Acad Sci USA 97: 12475–12480, 2000. [Erratum in Proc Natl Acad Sci USA 98: 379, 2001]. doi: 10.1073/pnas.220418997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leonard MO, Kieran NE, Howell K, Burne MJ, Varadarajan R, Dhakshinamoorthy S, Porter AG, O'Farrelly C, Rabb H, Taylor CT. Reoxygenation-specific activation of the antioxidant transcription factor Nrf2 mediates cytoprotective gene expression in ischemia-reperfusion injury. FASEB J 20: 2624–2626, 2006. doi: 10.1096/fj.06-5097fje. [DOI] [PubMed] [Google Scholar]
- 21.Iommarini L, Porcelli AM, Gasparre G, Kurelac I. Non-canonical mechanisms regulating hypoxia-inducible factor 1 alpha in cancer. Front Oncol 7: 286, 2017. doi: 10.3389/fonc.2017.00286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alam J, Cook JL. How many transcription factors does it take to turn on the heme oxygenase-1 gene? Am J Respir Cell Mol Biol 36: 166–174, 2007. doi: 10.1165/rcmb.2006-0340TR. [DOI] [PubMed] [Google Scholar]
- 23.Alam J, Cook JL. Transcriptional regulation of the heme oxygenase-1 gene via the stress response element pathway. Curr Pharm Des 9: 2499–2511, 2003. doi: 10.2174/1381612033453730. [DOI] [PubMed] [Google Scholar]
- 24.Ji X, Wang H, Zhu J, Zhu L, Pan H, Li W, Zhou Y, Cong Z, Yan F, Chen S. Knockdown of Nrf2 suppresses glioblastoma angiogenesis by inhibiting hypoxia-induced activation of HIF-1alpha. Int J Cancer 135: 574–584, 2014. doi: 10.1002/ijc.28699. [DOI] [PubMed] [Google Scholar]
- 25.Zhao R, Feng J, He G. Hypoxia increases Nrf2-induced HO-1 expression via the PI3K/Akt pathway. Front Biosci (Landmark Ed) 21: 385–396, 2016. doi: 10.2741/4395. [DOI] [PubMed] [Google Scholar]
- 26.Lu Y, Wang B, Shi Q, Wang X, Wang D, Zhu L. Brusatol inhibits HIF-1 signaling pathway and suppresses glucose uptake under hypoxic conditions in HCT116 cells. Sci Rep 6: 39123, 2016. doi: 10.1038/srep39123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McElroy GS, Chandel NS. Mitochondria control acute and chronic responses to hypoxia. Exp Cell Res 356: 217–222, 2017. doi: 10.1016/j.yexcr.2017.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Masson N, Singleton RS, Sekirnik R, Trudgian DC, Ambrose LJ, Miranda MX, Tian YM, Kessler BM, Schofield CJ, Ratcliffe PJ. The FIH hydroxylase is a cellular peroxide sensor that modulates HIF transcriptional activity. EMBO Rep 13: 251–257, 2012. doi: 10.1038/embor.2012.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang J, Staples O, Thomas LW, Briston T, Robson M, Poon E, Simoes ML, El-Emir E, Buffa FM, Ahmed A, Annear NP, Shukla D, Pedley BR, Maxwell PH, Harris AL, Ashcroft M. Human CHCHD4 mitochondrial proteins regulate cellular oxygen consumption rate and metabolism and provide a critical role in hypoxia signaling and tumor progression. J Clin Invest 122: 600–611, 2012. doi: 10.1172/JCI58780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oh ET, Kim JW, Kim JM, Kim SJ, Lee JS, Hong SS, Goodwin J, Ruthenborg RJ, Jung MG, Lee HJ, Lee CH, Park ES, Kim C, Park HJ. NQO1 inhibits proteasome-mediated degradation of HIF-1alpha. Nat Commun 7: 13593, 2016. doi: 10.1038/ncomms13593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Niu G, Briggs J, Deng J, Ma Y, Lee H, Kortylewski M, Kujawski M, Kay H, Cress WD, Jove R, Yu H. Signal transducer and activator of transcription 3 is required for hypoxia-inducible factor-1alpha RNA expression in both tumor cells and tumor-associated myeloid cells. Mol Cancer Res 6: 1099–1105, 2008. doi: 10.1158/1541-7786.MCR-07-2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu Q, Briggs J, Park S, Niu G, Kortylewski M, Zhang S, Gritsko T, Turkson J, Kay H, Semenza GL, Cheng JQ, Jove R, Yu H. Targeting Stat3 blocks both HIF-1 and VEGF expression induced by multiple oncogenic growth signaling pathways. Oncogene 24: 5552–5560, 2005. doi: 10.1038/sj.onc.1208719. [DOI] [PubMed] [Google Scholar]
- 33.Wenger RH, Rolfs A, Marti HH, Guenet JL, Gassmann M. Nucleotide sequence, chromosomal assignment and mRNA expression of mouse hypoxia-inducible factor-1 alpha. Biochem Biophys Res Commun 223: 54–59, 1996. doi: 10.1006/bbrc.1996.0845. [DOI] [PubMed] [Google Scholar]
- 34.Wiener CM, Booth G, Semenza GL. In vivo expression of mRNAs encoding hypoxia-inducible factor 1. Biochem Biophys Res Commun 225: 485–488, 1996. doi: 10.1006/bbrc.1996.1199. [DOI] [PubMed] [Google Scholar]
- 35.Berra E, Benizri E, Ginouves A, Volmat V, Roux D, Pouyssegur J. HIF prolyl-hydroxylase 2 is the key oxygen sensor setting low steady-state levels of HIF-1alpha in normoxia. EMBO J 22: 4082–4090, 2003. doi: 10.1093/emboj/cdg392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Epstein AC, Gleadle JM, McNeill LA, Hewitson KS, O'Rourke J, Mole DR, Mukherji M, Metzen E, Wilson MI, Dhanda A, Tian YM, Masson N, Hamilton DL, Jaakkola P, Barstead R, Hodgkin J, Maxwell PH, Pugh CW, Schofield CJ, Ratcliffe PJ. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 107: 43–54, 2001. doi: 10.1016/S0092-8674(01)00507-4. [DOI] [PubMed] [Google Scholar]
- 37.Marxsen JH, Stengel P, Doege K, Heikkinen P, Jokilehto T, Wagner T, Jelkmann W, Jaakkola P, Metzen E. Hypoxia-inducible factor-1 (HIF-1) promotes its degradation by induction of HIF-alpha-prolyl-4-hydroxylases. Biochem J 381: 761–767, 2004. doi: 10.1042/BJ20040620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nguyen T, Sherratt PJ, Huang HC, Yang CS, Pickett CB. Increased protein stability as a mechanism that enhances Nrf2-mediated transcriptional activation of the antioxidant response element. Degradation of Nrf2 by the 26 S proteasome. J Biol Chem 278: 4536–4541, 2003. doi: 10.1074/jbc.M207293200. [DOI] [PubMed] [Google Scholar]
- 39.Stewart D, Killeen E, Naquin R, Alam S, Alam J. Degradation of transcription factor Nrf2 via the ubiquitin-proteasome pathway and stabilization by cadmium. J Biol Chem 278: 2396–2402, 2003. doi: 10.1074/jbc.M209195200. [DOI] [PubMed] [Google Scholar]
- 40.Baba K, Morimoto H, Imaoka S. Seven in absentia homolog 2 (Siah2) protein is a regulator of NF-E2-related factor 2 (Nrf2). J Biol Chem 288: 18393–18405, 2013. doi: 10.1074/jbc.M112.438762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ueda K, Xu J, Morimoto H, Kawabe A, Imaoka S. MafG controls the hypoxic response of cells by accumulating HIF-1alpha in the nuclei. FEBS Lett 582: 2357–2364, 2008. doi: 10.1016/j.febslet.2008.05.040. [DOI] [PubMed] [Google Scholar]
- 42.Naranjo-Suarez S, Carlson BA, Tsuji PA, Yoo MH, Gladyshev VN, Hatfield DL. HIF-independent regulation of thioredoxin reductase 1 contributes to the high levels of reactive oxygen species induced by hypoxia. PLoS One 7: e30470, 2012. doi: 10.1371/journal.pone.0030470. [DOI] [PMC free article] [PubMed] [Google Scholar]