

Keywords: calcineurin inhibitors, calcineurin isoforms, renal fibrosis
Abstract
Recently, research has directed its interests into identifying molecular pathways implicated in calcineurin inhibitor (CNI)-induced renal fibrosis. An emerging body of studies investigating calcineurin (CnA) activity has identified distinct actions of two main ubiquitously expressed isoforms: CnAα and CnAβ. CNIs have the capacity to inhibit both of these CnA isoforms. In the kidney, CnAα is required for development, whereas CnAβ predominantly modulates the immune response and glomerular hypertrophic signaling powered by activation of the transcription factor, nuclear factor of activated T lymphocytes (NFAT). Interestingly, data have shown that loss of CnAα activity contributes to the expression of profibrotic proteins in the kidney. Although this finding is of great significance, follow-up studies are needed to identify how loss of the CnAα isoform causes progressive renal damage. In addition, it is also necessary to identify downstream mediators of CnAα signaling that assist in upregulation of these profibrotic proteins. The goal of this review is to provide insight into strides taken to close the gap in elucidating CnA isoform-specific mechanisms of CNI-induced renal fibrosis. It is with hope that these contributions will lead to the development of newer generation CNIs that effectively blunt the immune response while circumventing extensive renal damage noted with long-term CNI use.
INTRODUCTION
The use of calcineurin inhibitors (CNIs) in the prevention of organ graft rejection posttransplantation has dramatically improved graft and patient survival since its introduction into clinical practice in 1980 (1, 2). Yet despite the advantages of graft and patient survival, solid organ transplant recipients face consequences of long-term immunosuppression on a variety of organ systems, particularly the kidneys. Detailed histopathological analyses describe irreversible damage by CNI treatment with fibrotic lesions noted within the vessels (arteriolar hyalinosis) (3), glomeruli (glomerulosclerosis) (4, 5), and between tubular epithelial cells (tubulointerstitial fibrosis). Mechanisms for this phenomenon remain unclear, and there are currently no therapeutic strategies in place to mitigate this phenomenon. Even though physiological roles of calcineurin signaling have been investigated extensively, less is known about specific functions of the main isoforms. Particularly, it remains better to be understood how calcineurin signaling and fibrotic pathways intersect in the kidneys and which calcineurin isoforms serve as targets of CNI therapy.
This review provides clues for future studies that address this gap in knowledge. First, this article briefly provides an overview of CNI discovery and integration into clinical practice, followed by relevant studies demonstrating CNIs to be nephrotoxic. Then, the focus will address the growing body of knowledge characterizing the presence of multiple active calcineurin isoforms, which have been found to signal independently of each other. Next, evidence will be introduced demonstrating that loss of the alpha catalytic isoform of calcineurin (CnAα) promotes expression of key fibrogenic proteins in the kidney. Lastly, this review article will conclude with insight inspiring future studies identifying the renal CnA isoform responsible for CNI nephropathy. Moreover, identification of downstream mediators of CnAα signaling will advance understanding of how calcineurin signaling and fibrogenic pathways intersect in the kidneys. This endeavor, once completed, will advocate for the development of future CNIs that preserve CnAα function, ultimately reducing renal side effects due to CNI toxicity.
CALCINEURIN INHIBITORS: MEDICINE OF THE EARTH
With its incorporation into clinical practice in the early 1980s, CNIs quickly became the cornerstone of immunosuppressive therapy post organ transplantation. The first and prototype of its class, cyclosporine A (CsA), was isolated from the soil fungus Tolypocladium inflatum in 1970 by Sandoz Laboratory (now Novartis) scientists, Dr. Sandor Lazary and Dr. Jean-Francois Borel (6). Successful preliminary human studies fast-tracked its immediate widespread use in renal transplant recipients in 1980 (7). Upon dosage adjustments, initial reports citing its lethal side effects decreased with posttransplantation survival rates increasing dramatically. In 1985, Streptomyces tsukubaensis, another soil microbe, provided an additional immunosuppressive compound for scientists at Fujisawa Pharmaceuticals in Japan. Its initial name FK-506 was later renamed Tacrolimus (acronym Tsukuba macrolide immunosuppressive). Owing to its milder range of side effects and better efficacy, it largely outpaced CsA in success, with majority of solid organ transplant recipients placed on tacrolimus postoperatively (8, 9).
CALCINEURIN INHIBITORS: MECHANISM OF IMMUNOSUPPRESSION
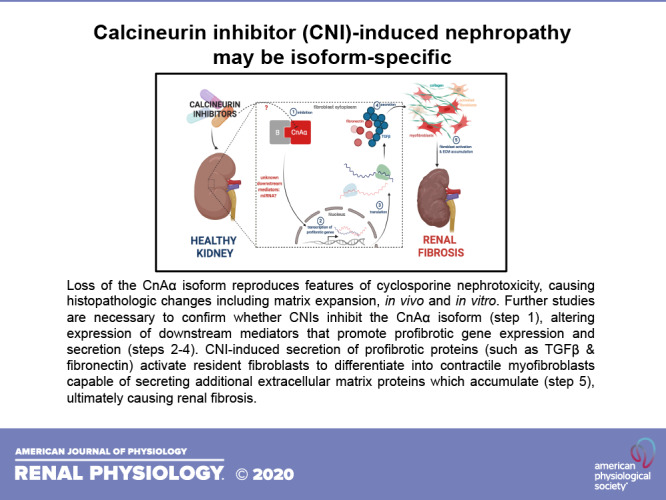
Studies attributed CsA and tacrolimus’ immunosuppressive effects to diminished T cells activity via inhibition of calcineurin (6, 10). Calcineurin is a Ca2+-dependent serine/threonine protein phosphatase that is a critical component of immune system regulation. To achieve their immunosuppressive effect, CNIs bind with high affinity to a class of cytosolic protein receptors called immunophilins: cyclophilins in the case of CsA and the FK-binding protein-12 in the case of tacrolimus (Fig. 1) (11). CNI-immunophilin complexes then bind calcineurin to sterically inhibit its intrinsic phosphatase activity (11). In T cells, calcineurin inhibition prevents dephosphorylation and subsequent activation of the transcription factor nuclear factor of activated T cells (NFAT1c). NFAT inactivity leads to decreased synthesis of interleukin-2 (IL-2), a key cytokine required for T cell activation in amplification of the immune response (9, 12).
Figure 1.

Calcineurin inhibitors (CNIs) blunt the immune response by inactivating calcineurin phosphatase activity. CNIs bind to cytosolic proteins called immunophilins (I) (step 1) enabling the CNI-I complex to bind calcineurin active site, thereby inhibiting phosphatase activity (step 2). Upon inhibition, transcription factors such as nuclear factor of activated T cells (NFAT) are unable to become activated by dephosphorylation (step 3). This prevents NFAT translocation into the nucleus to increase IL-2 transcription (step 4), thereby blunting immune activation. [Image created with BioRender.com.]
Figure 2.
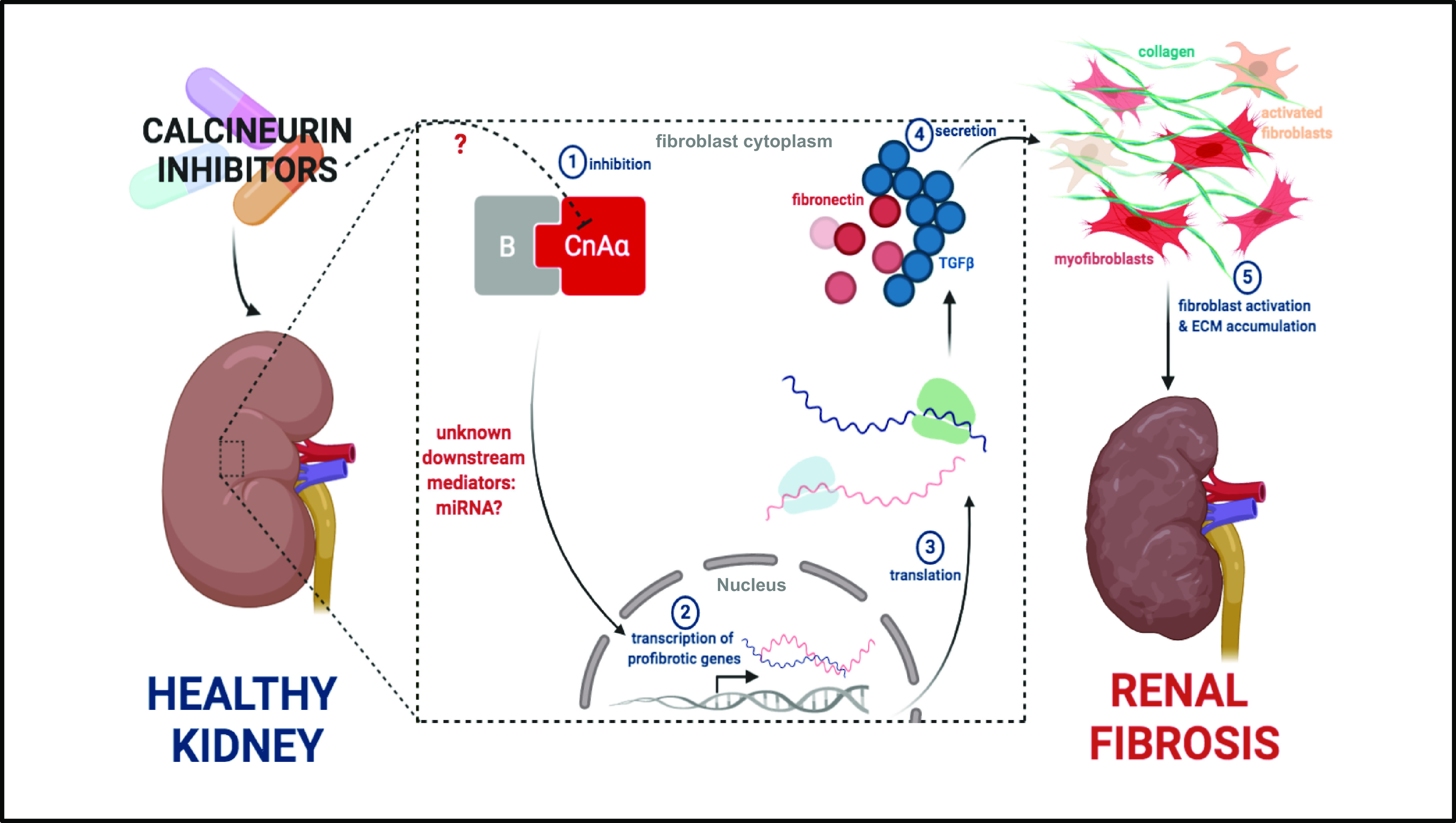
Calcineurin inhibitor (CNI)-induced nephropathy may be isoform-specific. Loss of the CnAα isoform reproduces features of cyclosporine nephrotoxicity, causing histopathological changes including matrix expansion. Further studies are necessary to confirm whether CNIs specifically inhibit the CnAα isoform (step 1), altering expression of downstream mediators that promote profibrotic gene expression and secretion (steps 2–4). CNI-induced secretion of profibrotic proteins (such as TGFβ and fibronectin) activates resident fibroblasts to differentiate into contractile myofibroblasts capable of secreting additional extracellular matrix proteins that accumulate (step 5), ultimately causing renal fibrosis. [Image created with BioRender.com.]
TROUBLE IN PARADISE: RENAL DAMAGE
The initial success of CNIs in transplant medicine was quickly followed up with early clinical and animal studies supporting evidence of CNI nephropathy, leading to renal failure in some cases (2, 13). In one group of patients, CNI nephropathy was observed as early as 3 mo postoperatively (14), with increased prevalence in 20%–25% of patients at 1 yr (14, 15) and 50% at 2 yr. In a cohort of patients assessed using a chronic allograft damage index, fibrosis was present in 70%–85% of grafts at 1 yr, making it the most common feature of nephropathy (16, 17). A histological analysis of kidneys from extrarenal transplant recipients demonstrated that many areas of the kidneys were irreversibly affected by CNI treatment, including the tubulointerstitium (interstitial fibrosis) and glomeruli (fibrosis of Bowman’s capsule and focal segmental/global glomerulosclerosis) (18). These fibrotic lesions, at least partially attributable to CsA nephrotoxicity, were seen in virtually all histological sections 10 yr after transplantation (19). These structural and functional changes were not only exclusive to long-term use of CsA but also were seen with tacrolimus treatment. CNI-induced nephropathy was later characterized as progressive and irreversible deterioration of renal function associated with fibrosis and tubular atrophy (13, 18).
A MOLECULAR CULPRIT: TRANSFORMING GROWTH FACTOR-β
Upregulation of the proinflammatory cytokine and potent stimulator of fibrosis transforming growth factor-β (TGF-β) is known to be a major contributor to this fibroproliferative disease (20, 21). Anti-TGFβ antibody staining of renal biopsy samples revealed higher levels of active TGF-β in patients on CsA therapy than in patients receiving tacrolimus (17, 22). Khanna et. al. (23) found that renal biopsy samples from patients diagnosed with tacrolimus nephrotoxicity also had increased mRNA expression of TGF-β, fibronectin, and collagen, additional contributors to fibrosis. CNIs have also been reported to increase in vitro and in vivo TGF-β expression and receptor activity in experimental models (24, 25). Molecular mechanisms of TGF-β regulation have been proposed, with microRNA expression emerging as suspected culprits. MicroRNAs, short noncoding RNAs that regulate protein-coding RNA expression, have been previously linked to renal fibrosis (26). Differential regulation of microRNAs regulating TGF-β signaling genes have also been identified by Gooch et al. (27). Among 76 differentially expressed microRNAs, 16 microRNA/mRNA clusters were identified that regulate genes involved in the TGF-β signaling pathway in CsA-treated mice (27). This study identified microRNAs previously linked to renal fibrosis that includes let-7d, miR-21, miR-29, miR-30, miR-130, miR-192, and miR-200, as well as microRNAs that have not been reported to be related to nephropathy or immune suppression. Pathway analysis of microRNA/mRNA changes highlights the TGF-β, Wnt, mTOR, and VEGF signaling pathways (27). It is unclear if tacrolimus alters microRNA levels in a similar manner as CsA. Additional mechanisms of TGF-β regulation, extracellular matrix accumulation and fibrosis following calcineurin inhibition remain better to be understood.
A TALE OF TWO MAIN ISOFORMS: CNAα AND CNAβ
Investigation of the distinct calcineurin isoforms that signal independently of one another will offer additional insight into CNI nephrotoxicity. Full catalytic activation of calcineurin requires formation of a Ca2+/calmodulin-dependent subunit heterodimer consisting of catalytic subunit A (CnA) and regulatory subunit B (CnB) (28). In 1989, Kuno et al and Kincaid (11, 31) published reports discerning the existence of three major calcineurin A isoforms expressed in humans: CnAα, CnAβ, and CnAγ (29, 30). CnAγ is mainly enriched in the testis and brain, whereas CnAα and CnAβ exist in most, if not all, organs and tissues (29, 30).
Although separate genes control the expression of these isoforms, PPP3CA (CnAα), PPP3CB (CnAβ), and PPP3CC (CnAγ), it is unknown whether these genes arose from duplication. Amino acid sequences of CnAα and CnAβ are 81% identical, with these sequences being highly conserved throughout evolution (28). The most striking difference between the catalytic subunit CnAα and CnAβ isoforms is the amino acid proline-rich region located in the N-terminus of CnAβ. It was later revealed that the proline-rich N-terminal sequence of CnAβ promotes substrate binding (31).
Although these isoforms share a high degree of sequence homology, they contribute to a more precise regulation of diverse calcineurin functions in different tissues (26). To distinguish roles of CnAα and CnAβ in T cell activation, Zhang (32) utilized CnAα−/− mice lacking a functional CnAα gene. When this isoform was ablated in mice, no substantial deficiency in T cell receptor (TCR)-mediated IL-2 production was observed, raising the suspicion that CnAβ mediates TCR signaling (32). CnAβ was later confirmed to play a critical role in T cell development, whereas CnAα was shown to mediate the antigen-specific T cell response (32, 33).
Data have shown CnAα to be the predominant active isoform expressed in the kidneys (34). In primary CnAα−/− renal fibroblasts, nuclear translocation of the calcineurin substrate NFATc is unaffected, but it fails to occur in CnAβ−/− fibroblasts (35). In addition, Gooch et al. (34) found CnAα to be essential in renal development, as CnAα−/− mice on a mixed genetic background experienced kidney failure, with death occurring within weeks after birth. In addition to the kidney, there are isoform-specific differences in several other systems including the skin, where loss of CnAα results in increased apoptosis of squamous epithelial cells and salivary glands, where CnAα is required for vesicle transport (5, 36).
Upregulation of the CnAβ isoform was shown to promote kidney hypertrophy, a feature of diabetic nephropathy (37, 38). This evidence was provided when Reddy et al. (37) induced type I diabetes in both wild-type and CnAβ−/− mice. They found that there was a significant increase in kidney hypertrophy in wild-type mice as early as 1 wk after the induction of diabetes. However, there was no increase in whole kidney hypertrophy in diabetic CnAβ−/− mice. After several weeks, both wild-type and CnAβ−/− mice demonstrated significant whole kidney hypertrophy. However, the degree of hypertrophy was significantly less in diabetic CnAβ−/− mice compared with diabetic wild-type. Using kidney fibroblasts lacking CnAα or CnAβ, in vitro findings confirmed that the CnAβ isoform is required for high glucose-induced cellular hypertrophy (38). Taken together, findings indicate that CnA isoforms possess distinct functions.
In addition to studies demonstrating these isoforms having distinct tissue-specific roles, Kilka et al. provided evidence that CnAα, CnAβ, and CnAγ each confer substrate specificities (31). Utilizing a comparative kinetic analysis of the dephosphorylation of five specific calcineurin substrates: NFAT, DARPP-32, Elk-1, Tau, and RII peptide, their results revealed that the substrate preferences of the isoforms may contribute to distinct physiological functions. Additional supporting data demonstrate that CnAβ utilizes its downstream target NFAT in kidney hypertrophy signaling in response to hyperglycemia (38).
Gooch et al. (39) identified CnAα as a potential key player in CNI-induced renal fibrosis. They found that loss of this isoform reproduces features of CsA nephrotoxicity in vivo and in vitro. Particularly, loss of CnAα in vivo results in histopathological changes including matrix expansion, whereas loss of the β isoform does not (39). Consistent with their in vivo findings, CnAα−/− renal fibroblasts exhibited increased fibronectin and TGF-β protein expression also similar to what is seen in CsA nephrotoxicity (39). They found that CnAα−/− cells had higher basal levels of fibronectin transcription activity compared with wild type cells and that administration of neutralizing TGF-β antibodies did not reduce fibronectin protein levels in CnAα−/− renal fibroblasts. Another study demonstrated that both CsA treatment and loss of CnAα are accompanied by a significant increase in metalloproteinase-9 (MMP-9) expression and activity in renal fibroblasts (40). MMP-9 works in conjunction with TGF-β to promote extracellular matrix remodeling, which also contributes to fibrosis. The additional molecules acting upstream of TGF-β and MMP-9 to induce renal fibrosis currently remain a mystery. Although these pivotal studies paved the way into a better understanding of calcineurin signaling and fibrosis, unexpected findings leave important questions to be answered.
FUTURE DIRECTIONS
CNI-induced hypertension is a well-known adverse effect of CNI therapy that indirectly contributes to renal damage. Vasoconstriction, sympathetic excitation, and sodium retention by the kidney have all been proposed in contributing to this form of hypertension (41). Hoorn et al. (42) have shown that CNIs increase the activity of the thiazide-sensitive sodium chloride cotransporter through an effect on the kinases WNK and SPAK. Future studies are needed to conclusively determine which calcineurin isoform contributes to this phenomenon. Recently, Borschewski et al. (43) demonstrated that loss of CnAβ activity increases the abundance and activity of the Na+-K+-2Cl−- cotransporter (NKCC2) in rats. NKCC2 overactivity is known to promote electrolyte (and water) retention, leading to hypertension (43). Further, CNIs downregulate the sodium-bicarbonate cotransporter-1 (NBCn1) expression in the epithelial cells of the medullary thick ascending limb, thereby causing acid-base disturbances that also promote renal dysfunction (44). It is currently unknown which calcineurin isoform is involved in regulating NBCn1 expression or activity, underscoring the need for further investigation.
There has been no published information regarding sex differences in outcomes using CNIs, which currently remains a gap in the field. In addition, it remains to be known whether there exist any single-nucleotide polymorphisms in any of the CnA isoforms that correlate with patient outcomes. Taken together, these data generate a compelling need to investigate the distinct functional roles of CnAα and CnAβ, despite their structural similarities. Although more studies point to loss of CnAα activity in directly reproducing features of CNI-nephrotoxicity, more studies are necessary to rule out any indirect contribution from CnAβ to this phenomenon. Strategies and therapies to preserve CnAα function could, therefore, reduce side effects and prevent subsequent kidney transplants because of CNI toxicity. The key to taking advantage of the established experimental findings is designing studies that investigate how CNI impacts regulation of these isoforms, particularly CnAα. Moreover, it will be quite interesting to learn additional molecular signals altered by the loss of CnAα, ultimately providing clues into potential downstream mediators promoting renal fibrosis. The emergence of systemic CsA analogues, such as voclosporin in long-term treatment of autoimmune diseases such as lupus nephritis, highlights the real need to investigate this phenomenon further (1, 45–48). Clinical trials are currently underway to assess its efficacy and safety compared with CsA and tacrolimus (48).
As strides have been made in untangling the complexities of calcineurin signaling, researchers have provided clues as a road map for future work to elucidate exact mechanisms of chronic CNI-induced nephropathy. This matter is quite complex, as both direct and indirect mechanisms contribute to CNI nephropathy. It is crucial that researchers continue in the pursuit to understand molecular mechanisms underlying CNI-induced renal fibrosis. Until then, managing optimum immune suppression while minimizing renal side effects will be an ongoing challenge.
GRANTS
This study was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grant R21DK119879 (to C.R.W.) and American Heart Association Grant 16SDG27080009 (to C.R.W.).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
A.C.U. prepared figures; A.C.U. drafted manuscript; A.C.U., T.-Y.W., and C.R.W. edited and revised manuscript; T.-Y.W. and C.R.W. approved final version of manuscript.
REFERENCES
- 1.Calne RY, Thiru S, Mcmaster P, Craddock GN, White DJG, Evans DB, Dunn DC, Pentlow BD, Rolles K. Cyclosporin A in patients receiveing renal allografts from cadaver donors. Lancet 312: 1323–1327, 1978. doi: 10.1016/S0140-6736(78)91970-0. [DOI] [PubMed] [Google Scholar]
- 2.Borel JF. History of the discovery of cyclosporin and of its early pharmacological development. Wien Klin Wochenschr 114: 433–437, 2002. [PubMed] [Google Scholar]
- 3.Kahan B. Cyclosporine. N Engl J Med 321: 1725–1738, 1989. doi: 10.1056/NEJM198912213212507. [DOI] [PubMed] [Google Scholar]
- 4.Williams CR, Gooch JL. Calcineurin inhibitors and immunosuppression – a tale of two isoforms. Expert Rev Mol Med 14: e14, 2012. doi: 10.1017/erm.2012.8. [DOI] [PubMed] [Google Scholar]
- 5.Klintmalm G, Sundelin B, Bohman SO, Wilczek H. Interstitial fibrosis in renal allografts after 12 to 46 months of cyclosporin treatment: beneficial effect of low doses in early post-transplantation period. The Lancet, 324: 950–954, 1984. doi: 10.1016/S0140-6736(84)91166-8. [DOI] [PubMed] [Google Scholar]
- 6.Borel JF, Feurer C, Gubler HU, Stahelin H. Biological Effects of Cyclosporin A: A New Antilymphocytic Agent. Agents Actions 6: 468–475, 1976. doi: 10.1007/BF01973261. [DOI] [PubMed] [Google Scholar]
- 7.Calne RY, White DJG, Rolles K, Smith DP, Herbertson BM. Prolonged survival of pig orthotopic heart grafts treated with cyclosporin A. The Lancet, 311: 1183–1185, 1978. doi: 10.1016/S0140-6736(78)90971-6. [DOI] [PubMed] [Google Scholar]
- 8.Hooks MA. Tacrolimus, a New Immunosuppressant—A Review of the Literature. Ann Pharmacother 28: 501–511, 1994. doi: 10.1177/106002809402800414. [DOI] [PubMed] [Google Scholar]
- 9.Starzl TE, Fung J, Jordan M, Shapiro R, Tzakis A, McCauley J, Johnston J, Iwaki Y, Jain A, Alessiani M. Kidney transplantation under FK 506. JAMA, 264: 63–67, 1990. doi: 10.1001/jama.1990.03450010067032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fruman DA, Klee CB, Bierer BE, Burakoff SJ. Calcineurin phosphatase activity in T lymphocytes is inhibited by FK 506 and cyclosporin A. Proc Natl Acad Sci U S A 89: 3686–3690, 1992. doi: 10.1073/pnas.89.9.3686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rusnak F, Mertz P. Calcineurin: Form and Function. Physiol Rev 80: 1483–1521, 2000. doi: 10.1152/physrev.2000.80.4.1483. [DOI] [PubMed] [Google Scholar]
- 12.Kung L, Halloran PF. Immunophilins may limit calcineurin inhibition by cyclosporine and tacrolimus at high drug concentrations. Transplantation 70: 327–335, 2000. doi: 10.1097/00007890-200007270-00017. [DOI] [PubMed] [Google Scholar]
- 13.Naesens M, Kuypers DRJ, Sarwal M. In-Depth Review Calcineurin Inhibitor Nephrotoxicity. Clin J Am Soc Nephrol 4: 481–508, 2009. doi: 10.2215/CJN.04800908. [DOI] [PubMed] [Google Scholar]
- 14.Lehtonen SRK, Taskinen EI, Isoniemi HM. Histological alterations in implant and one-year protocol biopsy specimens of renal allografts. Transplantation 72: 1138–1144, 2001. doi: 10.1097/00007890-200109270-00026. [DOI] [PubMed] [Google Scholar]
- 15.Margreiter R. Efficacy and safety of tacrolimus compared with ciclosporin microemulsion in renal transplantation: A randomised multicentre study. Lancet 359: 741–746, 2002. doi: 10.1016/S0140-6736(02)07875-3. [DOI] [PubMed] [Google Scholar]
- 16.Mohamed MAS, Robertson H, Booth TA, Balupuri S, Kirby JA, Talbot D. TGF-β expression in renal transplant biopsies: A Comparative Study Between Cyclosporin-A and Tacrolimus. Transplantation 69: 1002–1005, 2000. doi: 10.1097/00007890-200003150-00059. [DOI] [PubMed] [Google Scholar]
- 17.Legendre C, Thervet E, Skhiri H, Mamzer-Bruneel MF, Cantarovich F, Noël LH, Kreis H. Histologic features of chronic allograft nephropathy revealed by protocol biopsies in renal transplant recipients. Transplantation 65: 1506–1509, 1998. doi: 10.1097/00007890-199806150-00020. [DOI] [PubMed] [Google Scholar]
- 18.Myers BD. Cyclosporine nephrotoxicity. Kidney Int 30: 964–974, 1986. doi: 10.1038/ki.1986.280. [DOI] [PubMed] [Google Scholar]
- 19.Randhawa PS, Shapiro R, Jordan ML, Starzl TE, Demetris AJ. The histopathological changes associated with allograft rejection and drug toxicity in renal transplant recipients maintained on FK506. Clinical significance and comparison with cyclosporine. Am J Surg Pathol 17: 60–68, 1993. doi: 10.1097/00000478-199301000-00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matl I, Viklický O, Voska L, Lodererová A, Vítko Š. The effect of different immunosuppressive regimens on TGF-β1 expression in kidney transplant patients. Transplant International 18: 668–671, 2005. doi: 10.1111/j.1432-2277.2005.00115.x. [DOI] [PubMed] [Google Scholar]
- 21.Campistol JM, Igo PIÄ, Larios S, Nica Bescos MÂ, Oppenheimer F. Role of transforming growth factor-b 1 in the progression of chronic allograft nephropathy. Nephrol Dial Transplant 16: 114–116, 2001. doi: 10.1093/ndt/16.suppl_1.114. [DOI] [PubMed] [Google Scholar]
- 22.Mohamed MAS, Robertson H, Booth TA, Balupuri S, Kirby JA, Talbot D. TGF-β expression in renal transplant biopsies: A Comparative Study Between Cyclosporin-A and Tacrolimus. Transplantation 69: 1002–1005, 2000. doi: 10.1097/00007890-200003150-00059. [DOI] [PubMed] [Google Scholar]
- 23.Khanna A, Plummer M, Bromberek C, Bresnahan B, Hariharan S. Expression of TGF-β and fibrogenic genes in transplant recipients with tacrolimus and cyclosporine nephrotoxicity. Kidney Int 62: 2257–2263, 2002. doi: 10.1046/j.1523-1755.2002.00668.x. [DOI] [PubMed] [Google Scholar]
- 24.Bennett J, Cassidy H, Slattery C, Ryan MP, Mcmorrow T. Tacrolimus Modulates TGF-β Signaling to Induce Epithelial-Mesenchymal Transition in Human Renal Proximal Tubule Epithelial Cells. JCM 5: 50, 2016. doi: 10.3390/jcm5050050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kern G, Mair SM, Noppert SJ, Jennings P, Schramek H, Rudnicki M, Eller K. Tacrolimus Increases Nox4 Expression in Human Renal Fibroblasts and Induces Fibrosis-Related Genes by Aberrant TGF-Beta Receptor Signalling. PLoS ONE 9: e96377, 2014. doi: 10.1371/journal.pone.0096377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gooch JL, King C, Francis CE, Garcia PS, Bai Y. Cyclosporine A alters expression of renal microRNAs: New insights into calcineurin inhibitor nephrotoxicity. PLOS ONE 12: e0175242, 2017. doi: 10.1371/journal.pone.0175242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang G, Kwan BCH, Lai FMM, Chow KM, Li PKT, Szeto CC. Urinary miR-21, miR-29, and miR-93: Novel Biomarkers of Fibrosis. American Journal of Nephrology 36: 412–418, 2012. doi: 10.1159/000343452. [DOI] [PubMed] [Google Scholar]
- 28.Mumby MC, Walter G. Protein Serine/Threonine Phosphatases: Structure, Regulation, and Functions in Cell Growth. Physiol Rev 73: 673–699, 1993. doi: 10.1152/physrev.1993.73.4.673. [DOI] [PubMed] [Google Scholar]
- 29.Kuno T, Takeda T, Hirai M, Ito A, Mukai H, Tanaka C. Evidence for a second isoform of the catalytic subunit of calmodulin-dependent protein phosphatase (calcineurin A). Biochem Biophys Res Commun 165: 1352–1358, 1989. doi: 10.1016/0006-291X(89)92752-6. [DOI] [PubMed] [Google Scholar]
- 30.Kincaid RL, Giri PR, Higuchi S, Tamura J, Dixon SC, Marietta CA, Amorese DA, Martin BM. Cloning and Characterization of Molecular Isoforms of the Catalytic Subunit of Calcineurin Using Nonisotopic Methods. Journal of Biological Chemistry 265: 11312–11319, 1990. doi: 10.1016/S0021-9258(19)38593-X. [DOI] [PubMed] [Google Scholar]
- 31.Kilka S, Erdmann F, Migdoll A, Fischer G, Weiwad M. The Proline-Rich N-Terminal Sequence of Calcineurin Aβ Determines Substrate Binding. Biochemistry 48: 1900–1910, 2009. doi: 10.1021/bi8019355. [DOI] [PubMed] [Google Scholar]
- 32.Zhang BW, Zimmer G, Chen J, Ladd D, Li E, Alt FW, Seidman JG. T cell responses in calcineurin A alpha-deficient mice. J Exp Med 183: 413–420, 1996. doi: 10.1084/jem.183.2.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bueno OF, Brandt EB, Rothenberg ME, Molkentin JD. Defective T cell development and function in calcineurin A-deficient mice. Proc Natl Acad Sci U S A 99: 9398–9403, 2002. doi: 10.1073/pnas.152665399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gooch JL, Toro JJ, Guler RL, Barnes JL. Calcineurin A-alpha but not A-beta is required for normal kidney development and function. The American Journal of Pathology 165: 1755–1765, 2004. doi: 10.1016/s0002-9440(10)63430-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gooch JL. An emerging role for calcineurin A in the development and function of the kidney. Am J Physiol Renal Physiol 290: 769–776, 2006. doi: 10.1152/ajprenal.00281.2005. [DOI] [PubMed] [Google Scholar]
- 36.Pena JA, Losi-Sasaki JL, Gooch JL. Loss of Calcineurin Aa Alters Keratinocyte Survival and Differentiation. J Invest Dermatol 130: 135–140, 2010. doi: 10.1038/jid.2009.222. [DOI] [PubMed] [Google Scholar]
- 37.Reddy RN, Knotts TL, Roberts BR, Molkentin JD, Price SR, Gooch JL. Calcineurin Aβ is required for hypertrophy but not matrix expansion in the diabetic kidney. J Cell Mol Med 15: 414–422, 2011. doi: 10.1111/j.1582-4934.2009.00910.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Williams CR, Gooch JL. Calcineurin Aβ regulates NADPH oxidase expression and activity via NFAT in response to high glucose. J Biol Chem 289: 4896–4905, 2014. doi: 10.1074/jbc.M113.514869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gooch JL, Roberts BR, Cobbs SL, Tumlin JA. Loss of the α-Isoform of Calcineurin Is Sufficient to Induce Nephrotoxicity and Altered Expression of Transforming Growth Factor-β. Ttansplantation 83: 439–447, 2007. doi: 10.1097/01.tp.0000251423.78124.51. [DOI] [PubMed] [Google Scholar]
- 40.Francis CE, Bai Y. Differential expression of cyclosporine A-Induced calcineurin isoform-specific matrix metalloproteinase 9 (MMP-9) in renal fibroblasts. Biochem Biophys Res Commun 503: 2549–2554, 2018. doi: 10.1016/j.bbrc.2018.07.014. [DOI] [PubMed] [Google Scholar]
- 41.Hoorn EJ, Walsh SB, Mccormick JA, Zietse R, Unwin RJ, Ellison DH. Pathogenesis of calcineurin inhibitor-induced hypertension. J Nephrol 25: 269–275, 2012. doi: 10.5301/jn.5000174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hoorn EJ, Walsh SB, McCormick JA, Fürstenberg A, Yang CL, Roeschel T, Author NM. The calcineurin inhibitor tacrolimus activates the renal sodium chloride cotransporter to cause hypertension. Nat Med 17: 1304–1309, 2011. doi: 10.1038/nm.2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Borschewski A, Himmerkus N, Boldt C, Blankenstein KI, McCormick JA, Lazelle R, Mutig K. Calcineurin and Sorting-Related Receptor with A-Type Repeats Interact to Regulate the Renal Na+-K+-2Cl− Cotransporter. J Am Soc Nephrol 27: 107–119, 2016. doi: 10.1681/asn.2014070728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gill HS, Roush ED, Dutcher L, Patel S. Direct evidence for calcineurin binding to the exon-7 loop of the sodium-bicarbonate cotransporter NBCn1. Int J Biol Sci 10: 771–776, 2014. doi: 10.7150/ijbs.9539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Aspeslet L, Freitag D, Trepanier D, Abel M, Naicker S, Kneteman N, Foster R, Yatscoff R. ISA TX 247: A Novel Calcineurin Inhibitor. Transplant Proc 33: 1048–1051, 2001. doi: 10.1016/s0041-1345(00)02325-3. [DOI] [PubMed] [Google Scholar]
- 46.En Sin F, Isenberg D. Expert Opinion on Pharmacotherapy An evaluation of voclosporin for the treatment of lupus nephritis An evaluation of voclosporin for the treatment of lupus nephritis. Expert Opin Pharmacother 19: 1613–1621, 2018. doi: 10.1080/14656566.2018.1516751. [DOI] [PubMed] [Google Scholar]
- 47.Rovin BH, Solomons N, Pendergraft Iii WF, Dooley MA, Tumlin J, Romero-Diaz J, Huizinga RB. A randomized, controlled double-blind study comparing the efficacy and safety of dose-ranging voclosporin with placebo in achieving remission in patients with active lupus nephritis. Kidney Int 95: 219–231, 2019. doi: 10.1016/j.kint.2018.08.025. [DOI] [PubMed] [Google Scholar]
- 48.Camille B, Derrick D, Randall F, Yatscoff W, Morris R, Tudor G. The novel calcineurin inhibitor ISA247: a more potent immunosuppressant than cyclosporine in vitro. Transpl Int 17: 767–771, 2005. doi: 10.1007/s00147-004-0799-z. [DOI] [PubMed] [Google Scholar]